
Abstract
Retinitis pigmentosa (RP), affecting more than 20 million people worldwide, refers to a group of inherited retinal dystrophies characterized by progressive photoreceptor degeneration and vision loss. However, the underlying genetic causes of substantial RP cases remain unidentified. In this report, we identified a novel homozygous splicing variant, c.219-1delG, which introduced skipping of exon 4 of the ZNF124 gene in a large RP pedigree by whole-exome sequencing analysis. To elucidate the pathogenesis of the mutation, we generated a retina-specific knockout mouse model of ZNF124 murine homologous gene Gm20541, which manifested RP-like phenotypes characterized by reduced electroretinogram response and progressive retinal degeneration. Integrated analysis using CUT&Tag, ChIP-exo, and RNA-seq data further revealed that ZNF124 regulated MSX2 expression through binding its promoter region. Moreover, deletion of Msx2in the retina led to thinning of retina owing to progressive degeneration of rod cells. Integrated analysis of RNA-seq data from both Gm20541 and Msx2 mutant retinas indicated that ZNF124 is essential for maintaining normal retinal function by regulating Msx2 transcription, which in turn controls the expression of murine homologues of retinal dystrophy genes Rs1, Pde6g, and Pdc. Taken together, our study identified a novel mechanism of transcriptional regulation for retinal homeostasis via ZNF124-MSX2 axis and ZNF124 as a novel candidate gene for RP.
Similar content being viewed by others
Introduction
Retinitis pigmentosa (RP; OMIM:26800) refers to a group of hereditary degenerative diseases of the retina characterized by night blindness, a gradual constriction of peripheral visual field initially, followed by impairment of central vision and ultimately legal blindness [1]. Progressive loss of photoreceptors (including both rods and cones) is the primary underlying cause for the progressive vision loss in RP pathologies [2, 3]. As a genetically heterogeneous disease, RP is linked to various genes and can be inherited in different ways, such as autosomal-dominant (adRP, 30-40%), autosomal-recessive (arRP, 50-60%) or X-linked (5-15%) manners [4]. Moreover, non-Mendelian inheritance patterns, including digenic and maternal inheritances, have also been reported to account for very few cases [5, 6]. Until present, according to the RetNet database (https://sph.uth.edu/retnet/), over 80 genes have been implicated in non-syndromic forms of RP, most of which are preferentially expressed in retinal photoreceptors and encode various proteins involved in the phototransduction cascade, the metabolic pathway, the photoreceptor structure, or transcription regulation. However, these identified genes can only account for approximately 50-60% of existing RP patients, and the genetic causes for nearly 40% of RP cases remain undetermined. To this end, more unknown RP associated pathogenic genes and underlying mechanisms are expected to be uncovered.
Zinc-finger (ZNF) proteins containing the Krüppel-associated box (KRAB-containing proteins) are the largest single family of transcription factors (TFs) in mammals, which play an essential role in cell differentiation, cell proliferation, apoptosis, and neoplastic transformation [7, 8]. Here we identified a novel candidate gene associated with autosomal-recessive RP, ZNF124, encoding a KRAB-related zinc-finger protein of 351 amino acids that is predicted to harbor seven C2H2-type finger binding domains thought to be implicated in DNA binding [9]. ZNF124 gene consisted of 4 exons and 3 introns, which mapped to chromosome 1q44, and alternative splicing results in multiple transcript variants encoding distinct proteins. Research on the function of ZNF124 remains limited at present. A previous study cloned ZNF124 and identified it as a transcription factor, which was involved in the inhibitory effect of VEGF on apoptotic cell death in human hematopoietic cells [10]. In terms of pathogenesis, ZNF124 has been associated with the progression of head and neck squamous cell carcinoma (HNSCC) [11] as well as highly aggressive medulloblastomas [12]. In the nervous system, ZNF124 was reported as a candidate gene for the Dandy-Walker complex (DWC) in human [13]. Regrettably, its exact functions have not been elucidated in vivo yet.
Muscle segment homeobox 2 (MSX2), a homeobox-containing transcription factor, which dysregulation has been implicated in developmental anomalies of several types of tissues, including craniofacial tissues, hair follicles, teeth, heart, and brain [14,15,16]. Intriguingly, MSX2 appears to have dual functions in regulating apoptosis and retinal fate determination. For example, overexpression of Msx2 in mice has been shown to induce microphthalmia and optic nerve aplasia by suppressing Bmp4 expression in the optic vesicle before lens induction. Increased retinal apoptosis combined with reduced proliferation finally leads to retinal thinning and microphthalmia [17]. Conversely, disruption of Msx2 during early neurulation also results in hypoplasia of the maxillary, mandibular, and frontonasal prominences, as well as abnormalities in the somite, neural tube, and eye. Eye defects are characterized by enlarged optic vesicles, which may ultimately contribute to microphthalmia. This conflict suggested that the normal activity of MSX2 may establish a balance between survival and apoptosis of neural crest-derived cells required for proper craniofacial morphogenesis [18]. Additionally, Msx2 overexpression reduces the expression of retinal ganglion cell (RGC)-specific differentiation markers Math5 and Brn3b, indicating that Msx2 influences RGC fate commitment and differentiation by delaying the timing of cell cycle exit of retinal progenitors [19]. Previous findings also indicated that Msx2 gene is critically involved in anterior segment development and lens formation during early eye development, which finally contributes to the failure of retina folding and microphthalmia. [20, 21]. These evidences highlighted that the key regulating effect of MSX2 in the developmental process of the vertebrate eye. However, whether MSX2 plays a role in neuroretina at adult stage is still unclear.
In the current study, we report a novel homozygous splicing mutation (c.219-1delG) of ZNF124 in a family suffering from RP. Detailed cDNA analysis demonstrated a 9 bp skipping across exon 3 and 4 in the patient’s ZNF124 transcript. To investigate the in vivo roles of ZNF124 in retina, we first confirmed Gm20541 as the homologous gene of ZNF124 in mice, and subsequent knockout study of this murine homologous gene manifested an RP-like phenotypes characterized by reduced electroretinogram response and retinal degeneration in mice. By using CUT&Tag, ChIP-exo, and RNA-seq studies, we further revealed that ZNF124 regulated MSX2 expression through a direct binding of its promoter region. Moreover, rod-specific knockout of Msx2 also led to apparently thinning of retinal outer nuclear layer (ONL) owing to comprehensive degeneration of rod cells. Integrated analysis of bulk RNA-seq data from both Gm20541 and Msx2 mutant retinas reveals that ZNF124 is essential for maintaining normal retinal function by regulating Msx2 transcription, which in turn controls the expression of retinal disease homologous genes Rs1, Pde6g, and Pdc. Collectively, our findings identify ZNF124 as a novel candidate gene associated with retinitis pigmentosa (RP) and provide molecular insights into its underlying mechanisms, highlighting its functional significance in the retina.
Results
Identification of a novel pathogenic splicing variant in ZNF124 in a large arRP consanguineous family
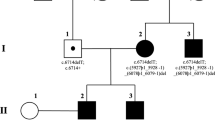
In the present study, two probands affected with RP from one consanguineous family were recruited (Fig. 1A). Both RP probands exhibited typical RP clinical features, including early-onset night blindness, gradually decreased vision acuity, and loss of peripheral visual field. No abnormalities were observed in other organs or systems, and the overall physical development of the probands was normal. The clinical examinations of patients IV:4 (12 years old) and IV:5 (14 years old) as well as their parents III:3 and III:4 were shown in Fig. 1B. Fundus examination showed the loss of pigment epithelial with narrowed arterioles, pale optic disk and irregular pigment clumps with both peripheral retina and macula involved in both eyes, while their parents exhibited normal structure of fundus (Fig. 1B). Optical coherence tomography (OCT) examination, which provides an indirect measure of axonal and neuronal injury in the anterior visual pathways, revealed disorganized photoreceptor layers and severely thinner retina compared to their unaffected parents (Fig. 1B). Visual field test on patient IV:4 further revealed loss of peripheral vision in both eyes (Fig. 1C), and weak response to light stimulus in both eyes was presented upon electroretinogram examination (Fig. 1D), indicating significant impairment of visual function.

A Pedigrees of the family with arRP. Arrows indicated the proband patient, and they were denoted with black symbols. B Left panel shows fundus photographs of unaffected controls III:3 and III:4 and patients IV:4 and IV:5. pigmentary deposits were presented in retinas of two patients. The right panel shows representative images of OCT examination. Both probands presented disorganized and thinner retinal layers due to atrophy. Visual field test (C) and ERG examination (D) of the proband IV:4, which revealed loss of peripheral vision and weak response to light stimulus in both eyes. E Sanger sequencing of the splicing mutation c.219-1G>- in ZNF124 gene identified in this family, which was co-segregated with the phenotype. Arrows indicate the position of altered nucleotide. Both patients IV:4 and IV:5 harboured the homozygous splicing mutation, and their parents III: 3 and III:4 were heterozygous carriers. F Diagram showing the location of the identified ZNF124 splicing mutation in current study. The mutant nucleotide locates at the 3’ splice site before exon 4 of ZNF124 gene. G Nucleotide sequence alignment of human ZNF124 and orthologues from other species, depicting a high conservation of c.219-1 G > - (encircled in red). H Sequence chromatogram of the cDNA samples from probands (IV:4 and IV:5) and control subject, depicting a 10 bp deletion across exon 3–4 of ZNF124 transcript in probands (IV:4 and IV:5). The deleted bases are marked by red. I Diagram showing the location of the deleted base in ZNF124 transcript, which causes a frameshift variation. J Predicted structure of ZNF124. Mutation lies between the KRAB and the first zinc-finger motif. The red sequence represents the mis-translated amino acid residues affected by the missense mutation. OS, oculus sinister/left eye; OD, oculus dexter/right eye.
To determine the causative mutations in this RP family, we conducted whole-exome sequencing (WES) on genomic DNA from the proband patients IV:4 and IV:5, along with their unaffected father III:3. In total, we identified 9,366 and 9,535 nonsynonymous SNPs, as well as 452 and 449 coding indels in subject IV:4 and IV:5, respectively, which may affect amino acid sequences (Additional file 1). Among these, four homozygous regions shared by two patients (IV:4 and IV:5) were identified (Figure S1A). We then compared the variants in these homozygous regions with well-known RP-causing genes (https://sph.uth.edu/Retnet/), and no variants of these RP genes were found in these patients. By filtering these variants against the SNP database dbSNP137, and 1000 Genome Project as well as our in-house WES control database [22, 23], we exclude the variants with high frequency (>1%). After excluding heterozygous variants and synonymous variants, and considering the pathogenicity of variant and their retinal expression profile, a homozygous variant, c.219-1delG, in ZNF124 gene was identified as the most likely candidate for the cause of RP disease in this family (Additional file 2). Subsequent Sanger sequencing analysis confirmed complete co-segregation of the mutations with the disease phenotype in the consanguineous family: the homozygous splicing mutation c.219-1delG in the two affected patients (IV:4 and IV:5) was verified (Fig. 1E). The parents (III:3 and III:4) were unaffected carriers, each harboring a heterozygous variant (Fig. 1E). In contrast, the unaffected sister of the two probands did not carry this mutation (Figure S1B). Importantly, this splicing variant was not found in the Genome Aggregation Database (https://gnomad.broadinstitute.org/) as well as the human gene mutation database (http://www.hgmd.org/), indicating that this variant was novel. Moreover, the c.219-1delG variant located at the 3’ border before Exon 4 in ZNF124 gene (Fig. 1F), which may disrupt the typical AG ends of eukaryotic introns recognized by the U2 spliceosome [24, 25], and this site is evolutionarily highly conserved from human to zebrafish (Fig. 1G). Thus, these data revealed that a novel homozygous splicing variant c.219-1delG in ZNF124 gene as candidate gene for this arRP family.
ZNF124 c.219-1delG mutant disrupted splicing and led to truncated protein
To assess the effect of the c.219-1delG mutation on splicing, we collected peripheral blood from patient IV:4 and generated cDNA by reverse transcription using extracted RNA from the leukocyte. Primers were designed for ZNF124 transcript to amplify the cDNA fragment between exon 3 and exon 4 by reverse transcription (RT)-PCR. Sanger sequencing of the PCR products from the patients and controls revealed that there was a deletion spanning exon 3 and exon 4 (10 bp) of the ZNF124 cDNA (Fig. 1H), which demonstrated that the c.219-1delG splicing mutation in ZNF124 gene disrupted normal splicing and led to a frameshift deletion variant of ZNF124 transcript (Fig. 1I). In contrast, the ZNF124 cDNA sequence of their unaffected sister appeared normal (Figure S1B). In terms of ZNF124 protein, this transcriptional deletion could result in frameshift change at the 73rd amino acid residue, Serine, and a termination codon occurred after twenty amino acid (p.S73fs20*) (Fig. 1J). The ZNF124 protein contains distinct functional domains: a KRAB-A box at its NH2 terminal and seven consecutively repeated Kruppel-type (C2H2) zinc-finger motifs at carboxy terminus, which bind to DNA in a sequence-specific manner. Therefore, the premature termination of ZNF124 protein sequence induced by the identified splicing mutant could abolish the capacity of DNA binding (Fig. 1J), supporting that this splicing mutation in ZNF124 is the causative mutation for this arRP family.
Construction of knockout mouse lines of ZNF124 homologous gene Gm20541
ZNF124 has been reported as a transcriptional regulator linked to cell survival and tumorigenesis in other cellular contexts [10, 11], while its roles in the retina remain unknown. To investigate the potential pathogenesis of these RP patients, we first detected the retinal expression of ZNF124 by immunostaining on sections of monkey retinas, which manifested that ZNF124 was distinctly co-localized with DAPI-labeled nuclear in the retinal pigment epithelium (RPE), outer nuclear layer (ONL), inner nuclear layer (INL), and ganglion cell layer (GCL) of the retina (Figure S2A). To gain more insight into the expression profile of ZNF124 in human retinas, we utilized publicly available data from the Human Protein Atlas (HPA) databases (https://www.proteinatlas.org) [26] to assess the translational expression levels of ZNF124 in different retinal cell types. The single-cell data from HPA revealed that ZNF124 was broadly expressed in whole retina and various retinal cell types, especially highly enriched in rod photoreceptors (Figure S2B, C), implying the critical roles of ZNF124 in retinal rod cells.
In the mouse genome, there are eight homologous genes (Loc102637111, Gm20422, Gm14322, Zfp81, Zfp952, Zfp101, Gm20541, Zfp709) aligned to the human ZNF124 gene. After alignment, Gm20541 was considered as the closest homolog of the human counterpart, which shared 59.43% sequence similarity and approximate molecular size compared with ZNF124 (Figure S3A). Hereby, we generated Gm20541-knockout mice to explore the role of ZNF124 in the retina. In the constructed Gm20541 allele, an FRT-flanked internal ribosome entry site (IRES)-GFP reporter with an upstream splicing acceptor and a neomycin (Neo) resistance cassette was inserted into intron 2, and the FlAG-tagged exon 3 was flanked by two loxP sites, thus generating “knockout (KO) first” (Gm20541KO first) mice (Fig. 2A). After removal of the IRES-GFP reporter and Neo cassette between FRT sites by FLP recombinase, the KO first was converted into a conditional knockout allele (Gm20541flox/flox). Then, the Gm20541flox/flox mice can be crossed with a Six3-Cre driver line [27] to eliminate the exon 3 within the Gm20541 gene, which finally yields the retinal neural deletion mutant model (Gm20541flox/flox, Six3-cre, hereafter named Gm20541SKO) (Fig. 2A). Thereafter, we studied the expression pattern of Gm20541, which is transcriptionally expressed in mouse brain, cerebellum, liver, retina, intestine, muscle, heart, kidney and spleen (Figure S3B, C). Western blot of tissue lysis from Gm20541f/f mouse using an antibody specific for the FLAG tag confirmed the robust protein expression in the heart, liver and retina (Figure S3D, E). We, subsequently, examined the phenotypes of Gm20541KO first mice. Strikingly, while normal litter sizes and body weights (Figure S4A) were shown, the Gm20541KO first mice displayed defects in limb development: their hindlimbs presented oligodactyly with fewer digits (Figure S4BA). In addition, Gm20541 pan-excision in Gm20541KO first mice also resulted in degeneration of retina layers at 8 months of age (Figure S4C, D), highlighting its crucial roles in retina.

A Schematic showing the targeting strategy for the disruption of the Gm20541 gene. The knockout (KO)-first allele design is shown a nd a Flag tag was added to the end of exon 3. After recombination by flippase enzyme, the exon 3 is flanked by two loxP sites and finally excised by the Six3-Cre. B–D Representative ERG traces corresponding to responses elicited by scotopic (D) and photopic (E, F) conditions at different flash intensity (FI) in mice at 12 months of age. Statistical analysis was performed for the amplitudes of a-wave (E), b-wave (F) in scotopic conditions (n = 4 from 4 mice, two-way ANOVA followed by Tukey’s post hoc test). G Statistical analysis was performed for the photopic ERG amplitudes and flicker amplitudes in photopic conditions (n = 4 from 4 mice, two-way ANOVA followed by Tukey’s post hoc test). *P < 0.05; **P < 0.01; ***P < 0.001. ns, no significance. The data are represented as means ± SEM.
Functional deficit and progressive retinal dystrophy in Gm20541 mutants
To investigate the specific roles of Gm20541 in neuroretina, we next focus on the visual function and pathologies in conditional neural knockout Gm20541SKO mice. Scotopic electroretinograms (ERGs) were first performed on Gm20541SKO mice and controls at 12 months of age. As expected, remarkable decreases of amplitudes of both a-waves and b-waves were detected in scotopic ERG recordings in Gm20541SKO at each flash intensity compared with controls (Fig. 2B, E, F), reflecting compromised rod function. Likewise, the photopic a/b-wave and flicker amplitudes, which mainly reflect the circuits from cones, were also significantly reduced in Gm20541SKO mice (Fig. 2C, D, G), suggesting deficits in cone photoreceptors.
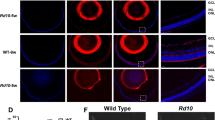
To determine the pathological changes underlying the impairment of visual function, histologic analysis was performed in Gm20541SKO mice at different ages, and data revealed that the thickness of both the outer and inner nuclear layers were gradually decreased from 8 to 21 months of age (Fig. 3A-F), accompanied by a shortened outer segment (OS). By 21-month-old, a nearly 57.34% decrease in ONL thinness was detected compared to that in control retinas (Fig. 3C, F), indicating massive loss of photoreceptors. Consistent with ONL thinning, quantification of OS length also revealed a significant reduction at 12 months and near-complete loss by 21 months (Fig. 3E, F). We, however, did not detect any alterations neither in thickness of ONL/INL nor OS length in heterozygous Gm20541SKO (Gm20541flox/+; Six-Cre) mouse retina, compared to those in control littermates (Figure S4), in line with the unaffected parents in described RP family.

H&E staining of paraffin sections of the Gm20541SKO and corresponding Gm20541f/f control retinas at the ages of 8 (A), 12 (B), and 21 months (C). Scar bar: 100 μm. The lower panels show high-magnification images of the boxed areas. Scale bars: 25 μm. Quantification analysis of the ONL, INL and OS thickness of the Gm20541f/f (n = 7 from 3 mice) and Gm20541SKO (n = 6 from 3 mice) retinas from mice at the ages of 8 (D), 12 (E), and 21 months (F). OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. *P < 0.05; **P < 0.01; ***P < 0.001. ns, no significance. The data are represented as means ± SEM.
Given that impaired visual function and significant thinning of ONL were detected in Gm20541SKO mice, we next focused on profiling the pathological details of retinal photoreceptors. Immunostaining using retinal sections from 12-month-old mice was performed against antibodies of Rhodopsin (rod OS marker) and NaK pump (rod IS marker) to analyze the subcellular changes in rods. Although the Rhodopsin labeled OS was well organized, the Gm20541SKO mice showed significantly decreased OS length in central retina, with a nearly 40% reduction compared to that of controls (10.13 ± 0.32 vs 16.76 ± 0.27 mm, P = 0.00065 by t-test) (Fig. 4A). To further characterize the ultrastructural features of the rod cells in Gm20541SKO retinas, transmission electron microscopy (TEM) was conducted on 12-month-old retinas and revealed a compact and well-organized OS discs in Gm20541SKO retinas (Fig. 4B). Moreover, both overall morphology and relative positioning of connecting ciliary structures appeared normal, and no statistic difference (P = 0.98 by t-test) was detected in the average length of the connecting cilium (CC) and basal body (BB) between Gm20541SKO rods and Gm20541f/f controls (Fig. 4C). Consistent with immunofluorescent results, Western blot analysis further confirmed an overall reduction in protein levels of various key molecules in rod OS, including RHO, cGMP-gated cation channel alpha-1 (CNGA1), GPCR kinase 1 (GRK1), protein peripherin 2 (PRPH2), and phosphodiesterase-6-β (PDE6B), in Gm20541SKO retinas compared to that of controls (Fig. 4D, E), indicating severe degeneration of rods.

A Retinal cryosections from 12-month-old mice were labeled with rod-specific markers rhodopsin (outer segment) and Na-K ATPase (inner segment). DAPI was used to counterstain the nuclei. Scale bars, 25 μm. The panels on the right show high-magnification images of the boxed areas. Scale bars: 10 μm. B Representative transmission electron microscope (TEM) images of photoreceptor outer segments in 12-month-old Gm20541f/f and Gm20541SKO mice. Scale bar: 1 µm. Higher-magnification images of representative ciliary region are shown in the lower of each genotype. Scale bars: 500 nm. C Quantitative analyses of the length of BB and CC between Gm20541f/f and Gm20541SKO photoreceptors (n = 10 from 3 mice). D Western blot analysis of typical OS proteins in 12-month-old Gm20541f/f and Gm20541SKO mouse retinas. GAPDH was used as a loading control. E Quantification analysis of the protein expression levels (n = 4 for each genotype). The expression of each protein was normalized to that of GAPDH. F Retinal cryosections from 12-month-old mice were labeled with cone markers PNA and M-opsin. DAPI was used to counterstain the nuclei; scale bars, 25 μm. G Quantification analysis of the M-opsin marked cone OS length (n = 12 from 3 mice). H Immunostaining of flatmount retinas from 12-month-old mice for M-opsin and PNA markers. Scale bars, 50 μm. Representitive image from dorsal retinal quadrant were shown in the lower panel. Scale bars, 25 µm. I Quantification of M-opsin positive cone number per 1 mm2 field in Gm20541f/f and Gm20541SKO retinas (n = 8 from 3 mice). schematic of retinal flat mount indicating the two sectors (radii: 1 and 2 mm) that were used to count cones. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. *P < 0.05; **P < 0.01; ***P < 0.001. ns, no significance. The data are represented as means ± SEM.
To further examine the pathological changes of cone cells in Gm20541SKO mice, we stained retinal sections with the cone OS marker M-opsin and Alexa Fluor-594-conjugated peanut agglutinin (PNA, which labels the cone matrix sheaths). Despite M-opsin was normal localized in the PNA labeled sheaths, a dramatically decrease (16.12 ± 0.32 vs 6.09 ± 0.27 mm, P < 0.0001 by t-test) in the length of M-opsin marked cone OS was observed in Gm20541SKO mice compared to controls (Fig. 4F, G), indicating the occurrence of cone cell degeneration. To precisely characterize the cone amounts in Gm20541SKO mice, retinal flat mounts from 12-month-old animals were prepared and coimmunostained with M-opsin antibody and Alexa Fluor-594-conjugated PNA (Fig. 4H). Considering that the M-opsin-containing cones were mainly distributed in the dorsal retina, with very few M-opsin-positive cones in the ventral retina [28], we thus selected the dorsal area for quantitation. Consistent with the immunolabeling results of retinal cryosections, a shortened and misshapen cone OS was observed, accompanied by a slight decrease in the number of M-cones in different dorsal regions were quantified (Fig. 4I), suggesting mild loss of cones in degenerating Gm20541SKO retinas. We further examined the retinal phenotype of 12-month-old heterozygous Gm20541SKO mice (Gm20541f/+; Six3-Cre) and found no statistically significant differences in retinal layer thickness compared to controls (Figure S5A, B), with little degenerative features in rod and cone cells (Figure S5C, D).
Given that degenerative features were also observed in other retinal layers in degenerating Gm20541SKO retinas, phenotypes of the major cell types in the inner retina were also analyzed here. Specifically, immunostaining using retinal bipolar cell (RBC)-specific marker PKCα revealed loss of RBCs in Gm20541SKO retina compared to that of controls (Figure S6A). The mean numbers of PKCα-positive cells in the control and Gm20541SKO mice were 18.58 ± 0.75 and 15.08 ± 0.67 cells/200 μm of retinal length, respectively (Figure S6B), suggesting a nearly 19% reduction of PKCα-labeled RBCs. However, immunostaining of retinal sections with anti-Brn3a antibody, an RGC-specific marker, (Figure S6C), showed that RGC number was not statistically significant (P = 0.26 by t-test) in Gm20541SKO retinas (Figure S6D).
Accompanied by the degenerative process, activation of Müller cells, as an indicator of retinal injury [29, 30], was also detected by immunostaining with Glial fibrillary acidic protein (GFAP) antibody, which showed a remarkable increase of GFAP signals across from the ganglion cell layer (GCL) to ONL in the degenerating Gm20541SKO retinas (Figure S7A). Western blot analysis further confirmed the increased GFAP level in the Gm20541SKO retina (Figure S7B, C). Moreover, TUNEL-positive nuclei were occasionally observed mainly in the ONL of degenerating Gm20541SKO retina, while this is devoid in their control littermates (Figure S7D, E), demonstrating slow apoptosis of photoreceptor cells. Together, these data indicated that ZNF124 homologous gene Gm20541 plays essential roles in the function and homeostasis of the retina, especially in photoreceptors.
ZNF124 regulates the transcription of MSX2 through binding its promotor region
Next, we set out to investigate the potential mechanisms underlying the retinal degeneration and RP occurrence induced by the ZNF124 deficit. As depicted earlier, ZNF124 is a KRAB-ZNF gene containing an array of 7 zinc fingers motifs that are known to binding to specific DNA sequences. Therefore, we aimed to identify the genomic targets bound and directly regulated by ZNF124 using the Cleavage Under Targets and Tagmentation (CUT&Tag) approach. Wildtype ZNF124 proteins bearing a C-terminal FLAG tag were overexpressed in HEK293T cells, followed by CUT&Tag assay after harvesting (Fig. 5A). In total, we identified 30,453 peaks of ZNF124 binding in HEK293T cells (Additional file 3), which showed a clear tendency for ZNF124 enriched regions within 1 kb of the transcriptional start site (TSS) and weak enrichments at transcriptional end sites (TES) (Fig. 5B, C). Consistently, analysis of the genic peaks indicates that ZNF124 binds preferentially to promoters (Fig. 5D): a total of 49.9% (15190) of the peaks map within 3 kb upstream of the TSS, and over 86.2% (13095) of these promoter peaks localized within 1 kb upstream of the TSS. A significant number of peaks, however, map to other genome regions, including intron and intergenic regions (Fig. 5D). To characterize the DNA consensus sequence recognized by ZNF124, all identified peaks validated by the MACS program were subjected to de novo and known motif analysis using HOMER, and the top consensus of ZNF124 binding motif was listed in Fig. 5E. Notably, the typical “TGASTCA” (S=G/C) binding motif was shared between both groups (Fig. 5E, arrows). We then carried out RNA-seq transcriptomic profiling of retinas from 8-month-old Gm20541SKO mice, which present no obvious retinal degenerative features during this time. Comparing to control retinas, a total of 375 significantly (|log2 fold change | >0.5; P < 0.05) differentially expressed genes (DEGs) were identified upon Gm20541 deficiency (Fig. 5F, Additional file 4), consisting of 192 up-regulated and 183 down-regulated genes (Fig. 5F). GO enrichment analysis revealed that the DEGs were primarily associated with the regulation of eye development, eye morphogenesis, photoreceptor cell differentiation, and transcription factor activity, implying the essential roles of Gm20541 in retina.

A Schematic summary for analysis of ZNF124 target genes. B, C Distribution of ZNF124-binding peaks showed strong enrichment within 1 kb upstream of the transcription start sites (TSS). D The distribution of ZNF124-binding sites within different gene regions. E The consensus sequences of ZNF124-binding sites detected by HOMER motif analysis with CUT&Tag data. The triangle arrows indicate the shared motif between de novo and known groups. F Volcano plots displaying the differentially expressed genes (DEGs) between Gm20541f/f and Gm20541SKO retinas and their statistical significance. Red represents up-regulated genes, blue represents down-regulated genes. G Venn diagram of DEGs identified by RNA-seq analysis along with ZNF124 binding genes by CUT&Tag and CHIP-exo data. Three genes were selected according to the overlaps. The RNA-seq and CUT&Tag data was acquired from present study, while the CHIP-exo data were obtained from GEO datasets (GSE78099), which was conducted in published paper. H RNA-seq results show that the filtered 3 genes are downregulated in Gm20541SKO retinas. I Integrative Genomics Viewer (IGV) tracks displaying the distribution of ZNF124-binding peaks among the filtered genes according to CUT&Tag data. J The lower diagram indicates the location of the binding site of ZNF124 in the MSX2 gene. The upper panel shows the ChIP-qPCR data of ZNF124 binding to the MSX2 promoter using primers as indicated. The HEK293T cells were transfected with the empty vector (used as negative control) or pcDNA3.1-ZNF124-Flag plasmid. Results are presented as the percentage of ChIP/Input (n = 4 technical replicates). K The upper panel shows the typical consensus sequences of ZNF124-binding sites. The lower panel illustrates the sequences of ZNF124-binding sites aligned to the promoter region of the MSX2 gene. The ZNF124-binding sites for ZNF124 are indicated in red letters. The green line refers to the promoter region of MSX2 gene. L Schematic diagram of the construction of the pGL luciferase reporter plasmid. The pRL-TK-hRluc plasmid was used as the baseline control. “WT” indicates the aligned ZNF124-binding sites in the MSX2 promoter region, and “Mut” refers to the mutant version of the sequences. M The luciferase assay shows that ZNF124 overexpression causes an increase in fluorescence in the wild-type MSX2 promoter group but not in the mutant group. Data were normalized with Renilla luciferase values used as an internal control. N RT-qPCR verified the RNA-seq results for the indicated genes (n = 5 technical replicates). O Western blot analysis of the expression of MSX2 in Gm20541f/f and Gm20541SKO retinas. GAPDH was used as a loading control. O Quantitative analysis of the expression levels of each subunit, with the statistical comparison between groups (n = 4 technical replicates). ***P < 0.001. The data are represented as means ± SEM.
To determine which of these are transcriptionally modulated by ZNF124 in our experimental conditions, we further correlated the CUT&Tag data with RNA-seq results, and found that 46 genes simultaneously differentially expressed in Gm20541SKO retinas and bound by ZNF124 in the promoter region (Fig. 5G). For reliability, we further employed a previous published ChIP-exo data of 230 KRAB-ZNF proteins (GEO database: GSE78099) [31], which identified a total of 115 gene promoters (<3 kb) bound by ZNF124 (Additional file 5). Overlapping genes through the CUT &Tag, RNA-seq and ChIP-exo data finally indicated that only three genes (MSX2, GJA1, WLS) that are bound by ZNF124 simultaneously showed significantly altered expression in Gm20541SKO retinas (Fig. 5G). Interestingly, all these three targets were significantly downregulated when Gm20541 was deleted in our RNA sequencing experiment (Fig. 5H). CUT&Tag data further showed ZNF124 binding to the promoter regions of these three candidate genes (Fig. 5I). Among the three candidate genes, MSX2 drew our attention due to its fundamental role in early eye development—deficiency of which could lead to arrested eye development and microphthalmia [14, 17, 32]. We, therefore, selected the MSX2 gene for the following investigation. As such, we further carried out chromatin immunoprecipitation (ChIP)-qPCR to investigate evidence of ZNF124 binding on MSX2 gene promoters, and data showed that the predicted MSX2 promoter fragment (-3317/-2995) is the efficient region for direct ZNF124 interaction (Fig. 5J), suggesting that ZNF124 could specifically bind this region for transcriptional regulation. Next, we compared the identified “TGASTCA” (S = G/C) binding motif with the predicted MSX2 promoter region (-3317/-2995). As expected, a typical sequence of “TGAGTCA” (-3069/-3063) in MSX2 promoter region was aligned with the binding motif (Fig. 5K). To verify whether ZNF124 activated the MSX2 promoter, we then constructed reporter plasmids which contain the predicted MSX2 promoter fragment (-3317/-2995) upstream of the Luciferase gene using the pGL4.23 luciferase system (Fig. 5L). Meanwhile, mutant plasmids were also generated in which the typical “TGAGTCA” (-3069/-3063) binding sequence was substituted with “GTCTGAC” (Fig. 5L). Consistent with the observed downregulation of Msx2 mRNA in Gm20541SKO retina, the luciferase reporter assay demonstrated that ZNF124 activated the MSX2 promoter fragment (-3317/-2995) (Fig. 5M). However, the introduction of the “GTCTGAC” mutation in the mutant MSX2 promoter fragments abolished the activation by ZNF124. Thus, these data demonstrated that the “TGAGTCA” (-3069/-3063) region of the MSX2 promoter is critical for its activation by ZNF124. Thereafter, to validate whether ZNF124 binding could regulate MSX2 expression in vivo, we applied RT-qPCR to analyze the expression changes in Gm20541SKO retinas. In line with omics results, the mRNA expression level of Msx2 in Gm20541SKO retinas was reduced by over 50% compared to that in controls (Fig. 5N) and this reduction was further validated in protein levels by Western blot analysis using MSX2 antibody (Fig. 5O, P). Taken together, we proposed that MSX2 is an important downstream target of ZNF124 in the retina, which could be modulated by ZNF124 through binding its promotor region.
MSX2 is essential for function and survival of rod photoreceptors
Msx2 encodes for a transcription factor involved in early tissue patterning and dysregulation of which can induce developmental anomalies of several types of tissues, including optic vessel, anterior segment and lens formation during early eye development [33]. To further address the optic abnormalities upon MSX2 depletion, we first constructed a homozygous Msx2-deficient (Msx2-/-) model (Figure S8A) and then verified the precise excision using Sanger sequencing (Figure S8B). The deletion was confirmed by RT-PCR (Figure S8C). Analysis of Msx2-/- mice at 2-month postnatal ages revealed that these mice displayed dramatic growth retardation when compared with WT littermates, as reflected by significantly smaller body size (Figure S8D) and lower body weight (Figure S8E). Besides, Msx2-/- mice presented progressive hair loss and developed patches of hairiness and baldness (Figure S8D). Regarding optic features, consistent with previous studies, homozygous Msx2-/- mice in present study also showed cornea opacity and edema with microphthalmia compared with that of the WT littermates. Enucleated eyes of Msx2-deficient mice at 3 postnatal months were consistently microphthalmia and 50% smaller in size than eyes of WT littermates. Overall, narrowed palpebral fissures, smaller eyeballs, and cornea edema were detected in the Msx2-deficient mice. Such phenotypes became more apparent after the eyelids opened (Figure S8F). Histologic analysis of the eyes at 3 postnatal weeks disclosed thickened cornea, iridocorneal adhesion, iris hyperplasia, and smaller lens in the anterior segment compared with that of the WT littermates (Figure S8G). Meanwhile, we also found that the cerebellum morphology of Msx2-/- mice was markedly smaller than their age matched littermates, presenting severe atrophy compared to other brain regions (Figure S8H). This observation was confirmed by histologic study of cerebellar sections (Figure S8I), suggesting impeditive cerebellar development.
These data highlighted key regulating effect of MSX2 in the development of the vertebrate eye and other tissues. We, subsequently, asked whether the retinal dystrophy observed upon ZNF124 abrogation in human and mice could also be detected in Msx2 deficiency mice. However, the microphthalmia observed in Msx2-/- mice presents a challenge in investigating the role of MSX2 in the neuroretina. We, therefore, further generated Msx2 rod conditional knockout (Msx2RKO) mice by mating Msx2f/f with Rhodopsin promoter-driven Cre transgenic mice (Fig. 6A), and genotyped them using RT-PCR (Fig. 6B). The excision efficiency of Msx2 in rods was valeted by Western blot using protein lysis extracted from retinal samples, which revealed that the expression level of MSX2 in Msx2RKO mouse retina was reduced to ~43% of that of control (Fig. 6C). Considering that MSX2 may also be expressed in other retinal cell types, the excision efficiency of Msx2 in rod cells was fairly robust. Thereafter, histologic analysis was performed to examine the retinal morphology in Msx2RKO retinas at different ages. At 2 and 4 months of age, Msx2RKO retina appeared normal compared to their corresponding Msx2f/f littermates (Figure S9A, B), while a slightly thinning of the ONL began to present in the retina of 5- (Fig. 6D) and 6-month-old (Figure S9C) Msx2RKO mice. This phenotype became pronounced in Msx2RKO mice at 9 months of age, with an average 43.5% decrease in the thickness of ONL accompanied by shorted OS, while no obvious difference was observed in the inner retinal layers between Msx2RKO and their control Msx2f/f littermates (Fig. 6E). Along with the reduction in ONL thickness, the length of the OS also decreased in 5-month-old Msx2RKO mice, with a more pronounced shortening observed at 9 months compared to controls (Fig. 6D, E). Immunostaining data showed that RHO and PDE6B were correctly localized to the shortened OSs of 9-month-old Msx2RKO mice (Fig. 6F). Moreover, reactive astrogliosis were also detected in degenerating Msx2RKO retinas by immunostaining with GFAP antibody (Fig. 6G), indicating extensive retinal damage. The retinal morphology of heterozygous Msx2RKO mice (Msx2loxP/+; Rho-Cre) was also assessed here. However, unlike significant retinal degeneration in homozygous Msx2RKO mice (Figure S9D), no visible retinal degenerative feature was observed by 11 months of age (Figure S10). Together, these data suggest that MSX2 also plays roles in the neuroretina, and the excision of which in rod photoreceptors could lead to retinal degeneration, mimicking the effects of ZNF124 (Gm20541) deficiency.

A Schematic showing the strategy for generation of Msx2 rod conditional knockout mice. In the Msx2 conditional knockout allele, Exon 3 is flanked by two loxP sites. When the floxed allele is crossed with the Rho-Cre expressing line, exon3 is deleted, resulting in specific disruption of Msx2 gene expression in rod cells. B Genotyping of Msx2RKO mice. Genomic DNA from mouse-tail lysate of wildtype (Msx2+/+), Msx2flox/flox (Msx2f/f) and Msx2flox/flox; Rho-Cre (Msx2RKO) mice were amplified using specific primers. C Immunoblotting and quantitative comparison of MSX2 protein expression in retinas from Msx2f/f (used as control) and Msx2RKO mice (n = 3 technical replicates). GAPDH was used as a loading control. H&E staining of paraffin sections of Msx2RKO and corresponding controls at the ages of 5 (D) and 9 (E) months. Scale bar: 25 μm. The lower panel shows the quantitative analysis of the ONL and OS thickness of retinas from mice at different ages (n = 8 from 4 mice). Two-way ANOVA was used for statistical analysis, followed by Tukey’s post hoc test. F Retinal cryosections from 5-month-old Msx2f/f and Msx2RKO mice were labeled with the OS marker Rhodopsin or PDE6B (green) and IS marker Na-K ATPase (red), respectively. DAPI was used to counterstain the nuclei. Scale bars, 25 μm. G Cryosections from the retinas of 5-month-old Msx2f/f and Msx2RKO mice were immunostained for the activated astrocyte marker GFAP (green). Nuclei were counter-stained with DAPI (blue). Scale bars: 250 μm. Higher-magnification images of boxed areas were shown in the right panel of each image. Scale bars: 25 μm. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. *P < 0.05; **P < 0.01; ***P < 0.001. ns, no significance. The data are represented as means ± SEM.
ZNF124 maintains normal photoreceptor function by regulating expressions of Rs1, Pde6g and Pdc via MSX2 axis
To dissect the molecular mechanism underlying MSX2 regulation of photoreceptor survival, we performed bulk RNA-seq analysis on total retinal RNAs extracted from the 4-month-old Msx2RKO and Msx2f/f mice to compare the gene-expression profiles. Among the 17838 genes expressed, transcripts of 958 (5.4%) genes were upregulated, whereas transcripts of 496 (2.8%) genes were downregulated in Msx2RKO mice compared to the controls (Fig. 7A, Additional file 6). Gene ontology (GO) enrichment analysis demonstrated the top most significantly affected categories in genes that were downregulated as a result of Msx2 depletion. In keeping with the rod dysfunction after Msx2 deletion, among the top downregulated gene clusters were associated with photoreceptor inner and outer segments, photoreceptor cell maintenance, phototransduction and visual perception process (Fig. 7B). Strikingly, a subset of retinal dystrophy associated genes was downregulated in Msx2RKO cells (Fig. 7B). Among them, several known regulatory genes of visual perception, such as Prph2, Pde6g, Reep6, Rgs9bp and Gnat1, were identified. In order to elucidate how loss of ZNF124 (Gm20541) could cause retinal degeneration through regulation of MSX2, we compared downregulated transcripts that were enriched in visual perception process upon MSX2 depletion to those simultaneously down-regulated (|log2 fold change | <0; P < 0.05) in Gm20541SKO retinas, and revealed that four genes (Rs1, Pde6g, Ush2a, and Pdc) were shared (Fig. 7C). Gene-expression heatmaps and statistics of the RNA-seq data from both Msx2RKO and Gm20541SKO strains revealed that mRNA expressions of these selected genes were significantly downregulated in the Msx2RKO retinas, while showing varying degrees of reduction in Gm20541SKO retinas (Fig. 7D, E). RT-qPCR further validated these findings, showing a significant decrease (P < 0.001) in mRNA levels of Rs1, Pde6g, and Pdc in Msx2RKO retinas, while Ush2a exhibited no statistically significant change (Fig. 7F). Consistent with the transcriptional results, Western blot analysis confirmed a 45%, 46%, and 47% reduction in RS1, PDE6G, and PDC protein levels, respectively, in Msx2RKO retinas compared to those of controls (Fig. 7G, H). These findings suggested that the reduced MSX2 content due to Gm20541 deficiency subsequently downregulated expression levels of murine homologues of retinal dystrophy associated genes Rs1, Pde6g, and Pdc in the retina. This dysregulation ultimately contributes to progressive retinal degeneration and impaired visual function, providing a molecular basis for retinal dystrophy associated with ZNF124 deficiency in both humans and mice.

A Volcano plots displaying the differentially expressed genes (DEGs) between 4-month-old Msx2f/f and Msx2RKO retinas and their statistical significance. Red represents up-regulated genes, blue represents down-regulated genes. B Pathway analysis and significantly enriched GO terms in Biological process (BP), Cellular component (CC) and Molecular function (MF) categories for down-regulated genes in Msx2RKO retinas. C Venn diagram of enriched down-regulated genes in visual perception pathway of Msx2RKO retinas and total down-regulated genes in Gm20541SKO retinas. Four genes were overlapped. D The heatmap of the expression changes of the filtered four genes in Msx2f/f and Msx2RKO retinas, as well as in Gm20541f/f and Gm20541SKO retinas, as determined by RNA-seq analysis. E RNA-seq results showing that the filtered 4 genes were down-regulated in Msx2RKO and Gm20541SKO retinas to varying degrees, compared to corresponding controls. F RT-qPCR validated the RNA-seq results for the indicated genes (n = 4 technical replicates). Representative immunoblots (G) and quantification (H) of protein levels in retinas from 4-month-old Msx2f/f and Msx2RKO mice. Protein expression data were normalized according to GAPDH (n = 3 technical replicates). ***P < 0.001. ns, no significance. The data are represented as means ± SEM.
Discussion
In the current study, we report a novel homozygous splicing mutation (c.219-1delG) of ZNF124 gene in a family suffering from RP. Detailed cDNA analysis demonstrated a 9 bp skipping across exon 3 and 4 in the patient’s ZNF124 transcript. To investigate the in vivo roles of ZNF124 in retina, we first confirmed Gm20541 as the homologous gene of ZNF124 in mice, and following knockout study of this murine homologous gene manifested an RP-like phenotypes characterized by reduced electroretinogram response and late-onset retinal degeneration in mice, highlighting the essential roles of ZNF124 in both human and mouse retina and RP pathogenesis. As one of the members of this family, ZNF124 contains 7 predicted C2H2-type fingers thought to be implicated in DNA binding, and act as a transcriptional regulator linked to cell survival and tumorigenesis in several cellular contexts. Increased expression of ZNF124 was detected after vascular endothelial growth factor (VEGF) stimulation in human leukemia cell lines and results in inhibition of apoptotic cell death [10]. In the nervous system, loss of ZNF124 has been linked to defects in cerebellar development [13]. Here, we identified a novel homozygous splicing mutation c.219-1delG in the ZNF124 gene associated with RP pathologies (Fig. 1E, F). This splicing mutation resulted in a frameshift change at the 73rd Serine and a termination codon occurred after twenty amino acid (p.S73fs20*), which finally caused truncation of the ZNF124 protein from its C-terminal (Fig. 1J). Thus, our data implicated, for the first time, the crucial roles of ZNF124 in human RP pathologies. A limitation of the current study is the identification of the novel ZNF124 variant solely within a single consanguineous family. While the co-segregation, functional studies, and mouse model strongly support the pathogenicity of this gene, further investigation and validation are required to confirm the broader involvement of ZNF124 in RP pathogenesis, involving screening a larger cohort of independent families or sporadic cases to identify ZNF124 mutations and more comprehensively establish its association with inherited retinal dystrophies.
After alignment, Gm20541 was selected as the closest homolog of the human counterpart in murine genome (Figure S2A). So far, regrettably, no literature report elaborates on the biological function of Gm20541 as a stand-alone gene, and only few omics datasets point towards the association of Gm20541 with various biological processes. Interestingly, Gm20541 is controversially considered as a pseudogene between different databases. Here, we detected distinct transcriptional expression of Gm20541 in mouse brain, cerebellum, liver, retina, intestine, muscle, heart, kidney and spleen (Figure S2B, C). Subsequent western blot analysis of tissue lysis from Gm20541f/f mouse further confirmed the robust protein expression in the heart, liver and retina (Figure S2B), demonstrating the Gm20541 could be translated as a functional protein. We, subsequently, revealed that Gm20541 paly essential role in retinas, as showed by the progressive degeneration of Gm20541 deficient retina, especially in the photoreceptor layers (Fig. 4A–C), mimicking the human RP pathologies. Interestingly, compared to the clinical phenotypes of patients carrying mutations in ZNF124, the phenotype of our Gm20541SKO mice is rather mild. Actually, it is not uncommon to observe differences in mouse knockout phenotypes and human patient clinical phenotypes. One such example is the phenotypes of CRB1 patients and Rd8 mice. Mutations in CRB1 lead to LCA8, retinitis pigmentosa 12 or childhood- and juvenile-onset rod-cone and cone-rod dystrophy [34,35,36]. Most patients exhibited early-onset severe vision loss. Interestingly, a new mouse model, retinal degeneration-8 (rd8), with a single base deletion in the mouse Crb1 gene was generated [37]. rd8 mutation resulted in a frameshift and premature stop codon, and the truncated protein lost the transmembrane and cytoplasmic domains of CRB1. However, rd8 homozygous mutant mice only exhibited mild retinal degeneration, and photoreceptor degeneration was observed only within spotted regions of the retina, which implies that the severity of retinal pathology is dependent on genetic backgrounds [38, 39]. Taking these into consideration, there might be some discrepancy between the phenotypes of ZNF124 mutation carrying patients and those of Gm2054 knockout mice.
Indeed, ZNF124 should simultaneously regulates a subset of genes in different retinal cell types, as our observation that varying degrees of loss in other retinal cells in Gm20541SKO retinas, such as cones, RBCs, and RGCs (Figure S6). Although some literature indicated that prolonged degeneration of rod photoreceptors could induce the comprehensive degeneration of other retinal cells [40, 41], considering that the phenotype of photoreceptor dystrophy in the 12-month-old Gm20541SKO mice is still relatively mild (Fig. 4B), the loss of other retinal cells observed at this stage should be directly attributed to the absence of ZNF124 instead of the secondary effect of rod loss. This notion can be further supported by the evidence from our immunofluorescent data and the single cell database that ZNF124 is broadly expressed in different retinal cells (Figure S2A, B). In addition, we also observed the defects in limb development in pan-knockout Gm20541KO first mice (Figure S4B), hinting at the possibility of a more general role for ZNF124 in morphogenetic process. However, histological examination of 8-month-old Gm20541KO first retinas revealed no detectable retinal developmental abnormalities (Figure S4C), indicating that ZNF124 deficiency does not affect retinal maturation. Indeed, whether subtle abnormalities exist in other systems remains unknown, thus warranting further investigation.
Another key finding from our study is that ZNF124 could regulate MSX2 expression through a direct binding of its specific promoter region (Fig. 5I-P). MSX2 belongs to the MSX gene family, which acts as both a transcriptional repressor and activator whose normal activity may establish a balance between survival and apoptosis [42]. For instance, previous reports demonstrated that germline knockouts Msx2–/– mice exhibited microphthalmia commit with small lens formation or even aphakia, as well as a persistent lens stalk and iris hyperplasia in the anterior segment [20], while overexpression of which in mice could also lead to optic nerve aplasia and microphthalmia [17], suggesting that the precise regulation of MSX2 expression is critical for ocular development. Consistent with these findings, our homozygous Msx2−/− mice also showed narrowed palpebral fissure, cornea opacity and edema with microphthalmia compared with that of the WT littermates (Figure S8F). By constructing Msx2 rod-specific knockout mouse, we further revealed an apparent thinning of retinal ONL owing to comprehensive degeneration of rod cells in this mouse line (Fig. 6D, E). Notably, the degenerative phenotype of Msx2RKO mice manifested much earlier than that of the Gm20541SKO mice. This discrepancy in the age of onset likely stems from the partial loss (∼60% reduction) of Msx2 expression in the Gm20541SKO retina (Fig. 5N–O), which is insufficient to trigger early-onset pathogenesis. However, our data further showed that no observable retinal phenotype was detected in heterozygous Msx2RKO (Msx2loxP/+; Rho-Cre) mice at 6 or 11 months of age (Figure S10), suggesting a potential dosage-compensation effect of MSX2 as a transcription factor in the retina. Moreover, since Gm20541SKO mice begin to show mild retinal degeneration at 12 months (Fig. 3), it is plausible that ZNF124 also regulates the expression of other retinal proteins, such as GJA1 and WLS identified through our multi-omics analysis (Fig. 5G, H), which may collectively contribute to the late-onset retinal degeneration phenotype. Mechanistically, combined analysis of bulk RNA-seq data from Gm20541 and Msx2 mutant retinas finally unveils that MSX2 act as the transcriptional activator of Rs1, Pde6g and Pdc genes in retina (Fig. 7). Intriguingly, in humans, RS1, PDE6G and PDC are associated with severe retinal dystrophy diseases, with both PDE6G and PDC are well-known RP-causing genes [4], and mutations in RS1 cause X-linked retinoschisis (XLRS), an early-onset macular degeneration characterized by a splitting of the retina [43]. Specifically, RS1 is expressed and secreted from photoreceptors and bipolar cells as a homo-octameric complex, and its functions are related to cell adhesion and maintenance of retinal structure integrity [44, 45]. Currently, seven mouse models have been established and effectively mimicked key features of XLRS patients, including typical retinal abnormalities [46]. Pde6g encodes the gamma subunit of Rod-specific phosphodiesterase 6 (PDE6) complex, which is composed of alpha- and beta-catalytic subunits and two inhibitory gamma subunits [47, 48], serve as an essential component of the visual transduction cascade. Animal model studies also indicated that Pde6g depletion could lead to very early and rapid degeneration of rod photoreceptor cells, with only a single row of cone photoreceptor nuclei remaining by P16 [49], highlighting the crucial roles of PDE6G in retinas, especially in rod photoreceptors. The third targeted gene PDC is associated with RP and Usher syndrome type II [50], and PDC protein is located in the outer and inner segments of rod cells and participates in the regulation of visual phototransduction and photoreceptor metabolism [51]. In mice, loss of Pdc gene causes a reduction in the cellular levels and mislocalization of the Transducin in rod photoreceptors, which finally leads to retina degeneration [52, 53]. Our findings, coupled with the above published observations, provide evidence that disruption of MSX2 regulation on Rs1, Pde6g and Pdc could influence the retinal function and stability, demonstrating, for the first time, the pivotal roles of MSX2 in the function and survival of neuroretinas. The elucidation of the ZNF124-MSX2 axis in our study also provides a promising way for the development of targeted therapeutic strategies for ZNF124-associated RP. Restoring the expression of downstream target genes, such as MSX2 or its regulated retinal dystrophy-associated genes, may mitigate photoreceptor degeneration and preserve retinal function. Gene therapy approaches, including AAV-mediated overexpression of ZNF124 or MSX2 could potentially re-establish protective transcriptional networks disrupted in RP. Additionally, pharmacological interventions aimed at modulating the activity of this axis may offer alternative therapeutic options. Thus, our work provides the theoretical basis for gene therapy and pharmacological intervention for the ZNF124-associated IRDs.
In summary, our study identifies ZNF124 as a novel gene associated with arRP in human patients and demonstrates that its deficiency leads to comprehensive retinal degeneration and vision loss in knockout mouse models, supporting that ZNF124 plays essential functions in the human retina. Through multi-omics analysis, we further uncovered that MSX2 is a novel downstream player of ZNF124 to mediate normal retinal homeostasis and visual function through modulating the expression of retinal dystrophy genes RS1, PDE6G, and PDC. In summary, our study discovered a novel ZNF124 mutation linked to human RP and demonstrated a novel regulating mechanism for retinal dystrophy pathogenesis.
Materials and methods
Patient recruitment and whole-exome sequencing (WES)
This research was carried out in accordance with the tenets of the Declaration of Helsinki, and approved by the Institutional Ethics Review Boards of the Sichuan Provincial People’s Hospital and Aravind Eye Hospital (Madurai, Tamil Nadu, India). Written informed consents were obtained from the patients and their family members involved in this study. The probands and the related family members were clinically diagnosed at Sichuan Provincial People’s Hospital. Peripheral blood samples were collected from probands and their family members. Ophthalmic examinations were executed, including peripheral vision test, fundus photographic examination, and visual electrophysiological analysis.
For whole-exome sequencing (WES), peripheral blood from the patient and unaffected relatives were collected in EDTA anticoagulant tubes and then genomic DNA was isolated using DNA extraction kits according to the manufacturer’s instructions (TianGen, Beijing, China). The subsequent details of library construction, sequencing analysis of DNA samples to identify candidate pathogenic genes for RP patients were carried out as previously described [22, 23]. Validation of the mutations in patients and their relatives was performed by Sanger sequencing.
Sanger sequencing analysis
Specific PCR primers (Supplementary Table S1) were designed and synthesized by Sangon Biotech (Shanghai, China) to amplify genomic DNA or cDNA fragments. The PCR products were purified by FastAP Thermosensitive Alkaline Phosphatase (Thermo Scientific Fermentas, Waltham, MA, USA), and then were directly sequenced using BigDye version 3.1 and an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. The sequencing results were collated using Chromas software.
Mouse models
All animal experiment protocols were approved by the Institutional Animal Care and Use Committee of Sichuan Provincial People’s Hospital (Chengdu, Sichuan, China) (Approval number: LS-20180063) and conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All mice were housed in a controlled environment with a temperature of 24 ± 2 °C and a 12-hour light/dark cycle, with free access to food and water. Mice were maintained on a C57BL/6 J background.
The Gm20541-knockout first mice were generated by traditional method. Briefly, the Gm20541-knockout (KO) first targeting vector, which contains a gene trap (GT) cassette inserted into intron 2 and floxed exons 3 with a Flag tag (See Fig. 2A for diagram) was constructed. The targeting vector was transfected by electroporation of mouse 129S4 embryonic stem cells. Homologously recombined positive clones were selected by neomycin, and screened by PCR. After that, two independent clones of the targeted stem cells were microinjected into C57BL/6 J blastocysts. Resulting chimaera male mice were mated to C57BL/6 J female mice to generate F1 heterozygous founders. Heterozygous F1 mice were crossed to generate F2 off-spring. To generate a conditional allele of Gm20541, heterozygous F1 mice were crossed to flippase (FLP) mice (strain name: B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ, JAX stock number: 009086) to remove the FRT-flanked IRES-GFP reporter and neomycin cassette. The resulting conditional allele of Gm20541 was backcrossed to C57BL/6 J for 5 generations and designated as B6.129S4-Gm2054em1Zxj (Gm20541flox/flox, Gm20541f/f). Thereafter, Gm20541f/f mice were mated to Six3-Cre line (Jackson Laboratory Stock Tg(Six3-cre)69Frty/GcoJ, stock no. 019755) to generate retina-specific knockout Gm20541f/f; Six-Cre mice (hereafter referred to as Gm20541SKO).
Msx2 KO strain was purchased from Cyagen (Suzhou, China, Strain S-KO-17369). Homozygous KO mice were generated by intercrossed heterozygous Msx2 mice. Mice with Msx2 deletion specifically in retinal rods were generated using the Cre-loxP system. Briefly, Msx2flox/flox mice were purchased from GemPharmatech (Nanjing, China), which were crossed with B6.Cg-Pde6b < +> Tg(Rho-icre)1Ck/Boc (Jackson Laboratory, stock no. 015850) transgenic mice to yield progeny with the genotype Msx2flox/+; Rho-Cre. Then, the Msx2flox/+; Rho-Cre mice were crossed with Msx2flox/flox animals to generate Msx2flox/flox; Rho-Cre (Msx2RKO) mice. Msx2flox/flox (Msx2f/f) littermates were used as controls. All the mice were housed in ventilated, specific pathogen-free, and thermostatic conditions with a 12-h light-dark cycle at 24 °C.
Genotyping
Mouse genomic DNA samples were extracted from mouse tails and genotyped using PCR. The knockout alleles of Gm20541 or Msx2 were genotyped using the corresponding primers (Supplementary Table S1). All amplification reactions were performed using a master mix (Invitrogen, CA, USA) according to the manufacturer’s instructions. The first cycle of PCR consisted of holding at 95 °C for 5 min, followed by 32 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 30 seconds. The PCR products were separated by DNA electrophoresis on a 3% agarose gel.
Histological analysis
For hematoxylin and eosin staining (H&E), enucleated eyes were fixed overnight in 1.22% glutaraldehyde and 0.8% paraformaldehyde in 0.08 M phosphate buffer, embedded in paraffin and then cut into 5 μm sections. To ensure that sections used for quantification came from the same eccentricity, the globe was embedded in the same orientation. Sections that encompassed the optic nerve (ON) were selected for staining with H&E according to a standard protocol. The slices were scanned by Pannoramic DESK (3D HISTECH) and visualized or measured using CaseViewer 2.4 software (3D HISTECH). The thickness of ONL was measured every 250 μm from ON. Measurements were obtained from at least three sections from three mice of each genotype and averaged.
Electroretinography (ERG)
For electroretinographic evaluation, following overnight dark adaptation, mice were anesthetized with an intraperitoneal injection of xylazine (80 mg/kg) and ketamine (16 mg/kg) in normal saline. Thereafter, the pupils were dilated using one drop of 0.5% tropicamide phenylephrine, and one drop of 0.5% oxybuprocaine hydrochloride was administered for local anesthesia. The animals were placed on a heating pad during the ERG procedure. All handling, preparation, and electrode placements were performed under dim red light to maintain dark adaptation. ERG was performed by using an instrument set of Roland electroretinogram recorder and Ganzfeld Q450 stimulator (Roland Consult. Heidelberger. Germany). Dark (scotopic) ERG was performed firstly, with a serial of 0.03, 0.3, 3.0, and 20.0 cds/m2 stimulating luminance used in order. After that, a light adaptation period lasting for 10 min was instituted before the photopic response assessments. Photopic 3.0 ERG, 20.0 ERG, and photopic 20.0 flicker responses were recorded under corresponding stimulations.
Immunohistochemistry
For immunohistochemistry, eyes were removed from euthanized mice by intraperitoneal injection of pentobarbital (75 mg/kg) and by cervical dislocation. and fixed in 4% paraformaldehyde in 100 mM phosphate buffer (pH|7.4) for 1 h at 4 °C, followed by cryoprotection in 30% sucrose for 2 h. The lenses were removed, and the eyes were embedded in optimal cutting temperature (OCT) solution and sectioned at a 10 μm thickness. After blocking and permeabilization with 10% normal donkey serum and 0.2% Triton X-100 in phosphate buffer for 1 h, the sections were labeled with the primary antibody overnight at 4 °C. The primary antibodies used are shown in Supplementary Table S2. The sections were rinsed in PBS three times, Alexa Fluor-594/488-conjugated goat anti-mouse/rabbit secondary antibody (Cat# A11005 and A11008, Invitrogen, Waltham, MA, USA, 1:500 dilution) was applied, and nuclei were counterstained with DAPI (Cat# D8417, Sigma, St Louis, MO, USA) for 1 h at room temperature. Images were captured on a Zeiss LSM 800 confocal scanning microscope. In addition, the fluorescence intensity was quantified using ZEN 2.3 imaging software.
Retinal flat mounts and quantification of cone cells
Retinal whole mounts were prepared as previously described [30, 54]. Briefly, retinas were dissected from enucleated eyes and flattened, which were immersed in 4% PFA for 24 h at 4 °C, then blocked in PBS containing 1% bovine serum albumin and 0.5% Triton X-100 and incubated with polyclonal rabbit anti-Brn3a antibody for 12 h at 4 °C. After several washes, the retinas were incubated with the secondary antibody, AlexaFluor 488-conjugated goat anti-rabbit secondary antibody (Cat#A11008, Invitrogen, Waltham, MA, USA, 1:250 dilution) for 4 h at room temperature, and subsequently washed with PBS. To precisely quantitate the loss of Cone cells, the dorsal-ventral orientation of each retina was determined using previously described instructions [30, 54], and the retina was then subdivided into 2 sectors with radii of 1 mm and 2 mm. Cones were counted in the dorsal squares in each sector to determine the average cone density (cones/mm2).
Transmission electron microscopy (TEM)
TEM used for detecting rod degenerative characterization was performed as previously described [30]. The sections were imaged under a Philips CM120 scanning transmission electron microscope.
TUNEL assay
The terminal deoxynucleotidyl transferase-mediated biotinylated UTP nickend labeling (TUNEL) assay was used to detect the apoptotic cell death according to the manufacturer’s instruction (Roche Diagnostics, Indianapolis, IN, USA) on retinal frozen sections. Images were captured by Zeiss LSM 900 confocal scanning microscope.
Western blot analysis
Protein samples were prepared and subjected to SDS-PAGE analysis as previously described [30]. The primary antibodies used are shown in Supplementary Table S2. The relative intensity of the immunoreactive bands was quantified using the gel analysis tool provided in ImageJ software. The intensity of the proteins of interest was normalized to that of GAPDH. At least three independent Western blots were conducted, and one typical blot is presented. The uncropped images were shown in Additional file 7.
RNA extraction and RT-qPCR
Retinal total RNA was extracted using TRIzol reagent (Cat# T9424, Sigma, Saint Louis, MO, USA) as recommended by the manufacturer. First-strand cDNA was synthesized using an iScript cDNA Synthesis Kit (Cat# 170-8890, Bio-Rad, Hercules, California, USA). Quantitative PCR was carried out using iTaq SYBRMix (Cat# 1725120, Bio-Rad, Hercules, California, USA) and a CFX384 Touch Real-Time PCR Detection System (Cat# BJ005303, Bio-Rad, Hercules, California, USA). Primers were designed using Primer3Plus or obtained from published resources. Supplementary Table S1 shows the specific primer sequences used. All target genes were normalized to actin mRNA levels, and the fold change was calculated by performing delta-delta Ct analysis.
RNA‑sequencing
RNA sequencing was performed as before [30]. After harvest, two retinas from one mouse were collected as a sample, which was frozen immediately and sent to Seqhealth Corporation Inc. (Wuhan, China) for high-throughput RNA seq on an Illumina HiSeq 2500 platform generating 150 bp paired-end reads. All gene-expression values from RNA-seq were converted to related log2 values for further analyzed. A P-value no more than 0.05 was considered to be significant.
CUT&Tag sequencing
In order to study the targeted genes of ZNF124, we employed CUT&Tag sequencing using ZNF124 overexpressed HEK293T cells. 1×105 HEK293T cells with pcDNA-ZNF124-Flag transfected were prepared and sent to Personalbio Inc. (Shanghai, China) for CUT&Tag assay, which was performed as described previously with modifications [55]. In brief, cells were prepared and immobilized on concanavalin A beads. Beads are incubated with a Flag primary antibody (F1804, Sigma), followed by incubation with a secondary antibody anti-Mouse IgG (Millipore cat.no. 12-371). Beads were washed and incubated with pA-Tn5. Tn5 was activated by addition of Mg2+ and incubated at 37 °C for 1 h. Then cells were resuspended in tagmentation buffer (10 mM MgCl2 in Dig-med Buffer) and incubated at 37 °C for 1 h. DNA was purified with phenol-chloroform-isoamyl alcohol extraction and constructed CUT&Tag library according to the manufacturer’s instructions. Library was quantified by Equalbit dsDNA HS Assay Kit (Vazyme, EQ111-01) using Qubit™ 4 Fluorometer (Invitrogen, Q33226). Libraries were subjected to paired-end 150 bp sequencing in the Illumina Novaseq 6000 platform following the manufacturer’s instructions.
Cell culture and transfection
HEK293T cells were purchased from National Infrastructure of cell line Resource (Wuhan, China) and were recently authenticated by STR profiling. They were cultured in DMEM with high glucose (Hyclone, South Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and 100 U/ml penicillin/streptomycin (Invitrogen, Waltham, MA, USA) in an incubator set to 37 °C with 5% CO2. For transfection, cells were seeded in plates (Corning, NY, USA) and transiently transfected with constructed plasmid using Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer’s instructions, and the cell lysis were harvested after 48 h.
Chromatin immunoprecipitation (ChIP) qPCR
A chromatin immunoprecipitation (ChIP) assay was performed using a Magna ChIP Kit (Millipore, Billerica, MA) under manufacturer’s instructions. In brief, the suspension containing DNA fragments was sonicated from the empty vector (used as control) or pcDNA3.1-ZNF124-Flag transfected HEK293T cells and collected by centrifugation. A part of suspension soluble chromatin without immunoprecipitation was saved as input DNA. This suspension containing soluble chromatin was then incubated with anti-Flag or IgG antibodies and protein-G/A magnetic beads at 4 °C overnight. Subsequently, DNA fragment binding with the above antibodies was purified by DNA-purifying slurry and detected by qPCR using primers targeting the specific promoter regions (primer sequences are presented in the Supplementary Table S1).
Plasmids and luciferase reporter assays
A human MSX2 promoter fragment was amplified and cloned into the pGL4.23 basic vector. Mutations in the MSX2 promoter were generated by site-directed mutagenesis. The pcDNA3.1-ZNF124-Flag, pcDNA3.1-MSX2-Flag, and the empty vector were also constructed. All plasmids used in this study were constructed and purchased from Youbao Biotechnology (Changsha, China). The nucleotide sequences of all promoter fragments and mutants were confirmed by DNA sequencing. A Dual-Luciferase Reporter Assay System (Promega, WI) was used to measure luciferase activity. Briefly, cells were harvested 48 hours post-transfection and lysed using cell lysis buffer, followed by incubation on a shaker at room temperature for 5 minutes. The lysates were then centrifuged at 16,000 g for 10 minutes at 4°C. A 96-well microplate was used for the assay. Subsequently, 20 µL of the supernatant was added to each well, followed by 50 µL of Firefly luciferase substrate for activity measurement. Afterward, 50 µL of Renilla luciferase substrate was added for activity measurement. The luciferase activity of each sample was determined by calculating the ratio of Firefly luciferase to Renilla luciferase activity. All luciferase values are shown as mean ± SEM of three independent experiments.
Statistical analysis
Each experimental group was repeated three times, yielding at least three independent data points. Statistical analysis was performed using GraphPad Prism 6 software. The data sets were tested for a normal distribution using the Shapiro-Wilk test. For normally distributed data, statistical significance was determined by Student’s t test or ANOVA. If the data were not normally distributed, a nonparametric statistic was used. p-values were calculated with Student’s t test, one-way or two-way ANOVA followed by Tukey’s, Dunnett’s or Sidak’s multiple comparisons test as appropriate. P < 0.05 was considered to indicate statistical significance.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809.
Chang S, Vaccarella L, Olatunji S, Cebulla C, Christoforidis J. Diagnostic challenges in retinitis pigmentosa: genotypic multiplicity and phenotypic variability. Curr Genom. 2011;12:267–75.
Narayan DS, Wood JP, Chidlow G, Casson RJ. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016;94:748–54.
Daiger S, Sullivan L, Bowne S. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84:132–41.
Sandberg MA, Rosner B, Weigel-DiFranco C, Dryja TP, Berson EL. Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Investig Ophthalmol Vis Sci. 2007;48:1298–304.
Neveling K, Collin RW, Gilissen C, van Huet RA, Visser L, Kwint MP, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012;33:963–72.
Thomas JH, Emerson RO. Evolution of C2H2-zinc finger genes revisited. BMC Evolut Biol. 2009;9:1–8.
Huntley S, Baggott DM, Hamilton AT, Tran-Gyamfi M, Yang S, Kim J, et al. A comprehensive catalog of human KRAB-associated zinc finger genes: insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 2006;16:669–77.
Yang P, Wang Y, Macfarlan TS. The role of KRAB-ZFPs in transposable element repression and mammalian evolution. Trends Genet. 2017;33:871–81.
Kuramoto K, Uesaka T, Kimura A, Kobayashi M, Watanabe H, Katoh O. ZK7, a novel zinc finger gene, is induced by vascular endothelial growth factor and inhibits apoptotic death in hematopoietic cells. Cancer Res. 2000;60:425–30.
Miyake N, Katoh O, Hirata S, Kimura S, Watanabe H, Yajin K. Expression of the Krüppel-type zinc finger gene, ZK7, in head and neck squamous cell carcinoma and normal mucosa. Cancer Lett. 2002;185:111–8.
Luo Z, Dong X, Yu J, Xia Y, Berry KP, Rao R, et al. Genomic and transcriptomic analyses reveals ZNF124 as a critical regulator in highly aggressive medulloblastomas. Front Cell Dev Biol. 2021;9:634056.
Poot M, Kroes HY, V D Wijst SE, Eleveld MJ, Rooms L, Nievelstein RAJ, et al. Dandy-Walker complex in a boy with a 5 Mb deletion of region 1q44 due to a paternal t (1; 20)(q44; q13. 33). Am J Med Genet Part A. 2007;143:1038–44.
Han J, Ishii M, Bringas P Jr, Maas RL, Maxson RE Jr, Chai Y. Concerted action of Msx1 and Msx2 in regulating cranial neural crest cell differentiation during frontal bone development. Mech Dev. 2007;124:729–45.
Antonopoulou I, Mavrogiannis LA, Wilkie AO, Morriss-Kay GM. Alx4 and Msx2 play phenotypically similar and additive roles in skull vault differentiation. J Anat. 2004;204:487–99.
Zhang Q, Huang Z, Zuo H, Lin Y, Xiao Y, Yan Y, et al. Chromatin accessibility predetermines odontoblast terminal differentiation. Front Cell Dev Biol. 2021;9:769193.
Wu L-Y, Li M, Hinton DR, Guo L, Jiang S, Wang JT, et al. Microphthalmia resulting from MSX2-induced apoptosis in the optic vesicle. Investig Ophthalmol Vis Sci. 2003;44:2404–12.
Foerst-Potts L, Sadler T. Disruption of Msx-1 and Msx-2 reveals roles for these genes in craniofacial, eye, and axial development. Dev Dyn Off Publ Am Assoc Anat. 1997;209:70–84.
Jiang S-Y, Wang J-T. Msx2 alters the timing of retinal ganglion cells fate commitment and differentiation. Biochem Biophys Res Commun. 2010;395:524–9.
Zhao J, Kawai K, Wang H, Wu D, Wang M, Yue Z, et al. Loss of Msx2 function down-regulates the FoxE3 expression and results in anterior segment dysgenesis resembling Peters anomaly. Am J Pathol. 2012;180:2230–9.
Yu Z, Yu W, Liu J, Wu D, Wang C, Zhang J, et al. Lens-specific deletion of the Msx2 gene increased apoptosis by enhancing the caspase-3/caspase-8 signaling pathway. J Int Med Res. 2018;46:2843–55.
Zhou Y, Li S, Huang L, Yang Y, Zhang L, Yang M, et al. A splicing mutation in aryl hydrocarbon receptor associated with retinitis pigmentosa. Hum Mol Genet. 2018;27:2563–72.
Zhang L, Sun Z, Zhao P, Huang L, Xu M, Yang Y, et al. Whole-exome sequencing revealed HKDC1 as a candidate gene associated with autosomal-recessive retinitis pigmentosa. Hum Mol Genet. 2018;27:4157–68.
Corrionero A, Raker VA, Izquierdo JM, Valcárcel J. Strict 3′ splice site sequence requirements for U2 snRNP recruitment after U2AF binding underlie a genetic defect leading to autoimmune disease. RNA. 2011;17:401–11.
Clancy S. RNA splicing: introns, exons and spliceosome. Nat Educ. 2008;1:31.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. 2015;347:1260419.
Furuta Y, Lagutin O, Hogan BL, Oliver GC. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. genesis. 2000;26:130–2.
Eldred KC, Avelis C, Johnston RJ Jr, Roberts E. Modeling binary and graded cone cell fate patterning in the mouse retina. PLoS Comput Biol. 2020;16:e1007691.
Vidal L, Díaz F, Villena A, Moreno M, Campos JG, Pérez de Vargas I. Reaction of Müller cells in an experimental rat model of increased intraocular pressure following timolol, latanoprost and brimonidine. Brain Res Bull. 2010;82:18–24.
Yang Y, Shuai P, Li X, Sun K, Jiang X, Liu W, et al. Mettl14-mediated m6A modification is essential for visual function and retinal photoreceptor survival. BMC Biol. 2022;20:140.
Imbeault M, Helleboid P-Y, Trono D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature. 2017;543:550–4.
Plaisancié J, Collet C, Pelletier V, Perdomo Y, Studer F, Fradin M, et al. MSX2 gene duplication in a patient with eye development defects. Ophthalmic Genet. 2015;36:353–8.
Markitantova Y, Simirskii V. Inherited eye diseases with retinal manifestations through the eyes of homeobox genes. Int J Mol Sci. 2020;21:1602.
Bujakowska K, Audo I, Mohand-Saïd S, Lancelot ME, Antonio A, Germain A, et al. CRB1 mutations in inherited retinal dystrophies. Hum Mutat. 2012;33:306–15.
den Hollander AI, Davis J, van der Velde-Visser SD, Zonneveld MN, Pierrottet CO, Koenekoop RK, et al. CRB1 mutation spectrum in inherited retinal dystrophies. Hum Mutat. 2004;24:355–69.
den Hollander AI, Heckenlively JR, van den Born LI, de Kok YJ, van der Velde-Visser SD, Kellner U, et al. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet. 2001;69:198–203.
Mehalow AK, Kameya S, Smith RS, Hawes NL, Denegre JM, Young JA, et al. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet. 2003;12:2179–89.
Aleman TS, Cideciyan AV, Aguirre GK, Huang WC, Mullins CL, Roman AJ, et al. Human CRB1-associated retinal degeneration: comparison with the rd8 Crb1-mutant mouse model. Invest Ophthalmol Vis Sci. 2011;52:6898–910.
Luhmann UF, Carvalho LS, Holthaus SM, Cowing JA, Greenaway S, Chu CJ, et al. The severity of retinal pathology in homozygous Crb1rd8/rd8 mice is dependent on additional genetic factors. Hum Mol Genet. 2015;24:128–41.
Jones BW, Marc RE. Retinal remodeling during retinal degeneration. Exp Eye Res. 2005;81:123–37.
Strettoi E, Pignatelli V, Rossi C, Porciatti V, Falsini B. Remodeling of second-order neurons in the retina of rd/rd mutant mice. Vis Res. 2003;43:867–77.
Ekker M, Akimenko MA, Allende ML, Smith R, Drouin G, Langille RM, et al. Relationships among msx gene structure and function in zebrafish and other vertebrates. Mol Biol Evol. 1997;14:1008–22.
Tolun G, Vijayasarathy C, Huang R, Zeng Y, Li Y, Steven AC, et al. Paired octamer rings of retinoschisin suggest a junctional model for cell–cell adhesion in the retina. Proc Natl Acad Sci. 2016;113:5287–92.
Heymann JB, Vijayasarathy C, Huang RK, Dearborn AD, Sieving PA, Steven AC. Cryo-EM of retinoschisin branched networks suggests an intercellular adhesive scaffold in the retina. J Cell Biol. 2019;218:1027–38.
Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31:195–212.
Vijayasarathy C, Pasha SPBS, Sieving PA. Of men and mice: Human X-linked retinoschisis and fidelity in mouse modeling. Prog Retin Eye Res. 2022;87:100999.
Baehr W, Devlin MJ, Applebury ML. Isolation and characterization of cGMP phosphodiesterase from bovine rod outer segments. J Biol Chem. 1979;254:11669–77.
Hurley J, Stryer L. Purification and characterization of the gamma regulatory subunit of the cyclic GMP phosphodiesterase from retinal rod outer segments. J Biol Chem. 1982;257:11094–9.
Farber DB, Tsang SH. Stationary night blindness or progressive retinal degeneration in mice carrying different alleles of PDE gamma. Front Biosci. 2003;8:s666–s675.
Nishiguchi KM, Berson EL, Dryja TP. Mutation screening of the phosducin gene PDC in patients with retinitis pigmentosa and allied diseases. Mol Vis. 2004;10:62–4.
Lee RH, Whelan JP, Lolley RN, McGinnis JF. The photoreceptor-specific 33 kDa phosphoprotein of mammalian retina: generation of monospecific antibodies and localization by immunocytochemistry. Exp Eye Res. 1988;46:829–40.
Sokolov M, Strissel KJ, Leskov IB, Michaud NA, Govardovskii VI, Arshavsky VY. Phosducin facilitates light-driven transducin translocation in rod photoreceptors: evidence from the phosducin knockout mouse. J Biol Chem. 2004;279:19149–56.
Krispel CM, Sokolov M, Chen YM, Song H, Herrmann R, Arshavsky VY, et al. Phosducin regulates the expression of transducin βγ subunits in rod photoreceptors and does not contribute to phototransduction adaptation. J Gen Physiol. 2007;130:303–12.
Yang Y, Li X, Wang J, Tan J, Fitzmaurice B, Nishina PM, et al. A missense mutation in Pitx2 leads to early-onset glaucoma via NRF2-YAP1 axis. Cell Death Dis. 2021;12:1017.
Henikoff S, Henikoff JG, Kaya-Okur HS, Ahmad K. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019;10:1930.
Acknowledgements
This study was supported by the National Key R&D Program of China (grant no. 2024YFC2510900/2024YFC2510902), the National Natural Science Foundation of China (82371083, 82471100, 82121003, 82271084), the Department of Science and Technology of Sichuan Province (2023ZYD0172, 23ZDYF2057), research grant from Jinfeng Laboratory (JFLKYXM202403AZ-101). The funders had no role in the study design, data collection and analysis, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
XZ, ZY, LZ, LH, and PS conceived the project. XZ and ZY designed experiments. YY, XJ, SL, KS, RZ, CC, YS, and WL acquired data. YY, LH, and XZ analyzed data. YY, PS and XZ interpreted data and drafted the manuscript. XZ, ZY, LZ, and SL revised the manuscript. All authors approved submission for publication.
Corresponding authors
Ethics declarations
Competing interests
All authors declare that there are no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Wei-Na Jin
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Y., Jiang, X., Li, S. et al. Unveiling ZNF124 as a novel determinant in neurodegeneration: orchestration of photoreceptor homeostasis through MSX2 transcriptional regulation. Cell Death Dis 17, 234 (2026). https://doi.org/10.1038/s41419-026-08487-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-026-08487-6
