
Abstract
The STING pathway plays a central role in immune activation; however, STING protein levels decline during the progression of various cancers, including lung cancer, thereby limiting the efficacy of immunotherapies. Our study uncovers a previously unrecognized mechanism whereby ISGylation stabilizes STING by preventing its autophagic degradation, thereby enhancing its immunostimulatory function. Moreover, we demonstrate USP18 as a negative regulator that removes ISGylation from STING, and identify Tanshinone IIA sulfonate (TST) as a potent USP18 inhibitor that enhances STING ISGylation and stabilizes STING protein levels. When combined with the STING agonist diABZi, TST exhibits a synergistic effect, eliciting a potent antitumor immune response by increased infiltration of NK1.1⁺ cells and pronounced suppression of tumor growth in lung cancer models. These findings underscore the therapeutic potential of targeting STING ISGylation, particularly in patients with low STING expression who often respond poorly to current STING-targeted therapies.
Similar content being viewed by others

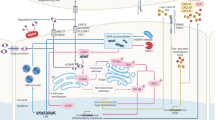

Introduction
The cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway is a key sensor of aberrant cytoplasmic DNA, driving both innate and adaptive immune responses [1]. Upon DNA binding, cGAS catalyzes the production of the second messenger cGAMP, which subsequently activates STING [2,3,4]. This activation recruits TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), promoting type I interferon (IFN-I) production [5]. The resulting immune response includes dendritic cell maturation, antitumor macrophage polarization, T cell activation, and natural killer (NK) cell activation [6,7,8].
The cGAS-STING signaling pathway is often suppressed in various cancers, with reduced expression of cGAS or STING in tumor cells linked to poorer clinical outcomes [9,10,11,12]. The downregulation is associated with tumor immune evasion, hindering effective immune surveillance [13]. Therefore, enhancing STING stability is pivotal for optimizing STING mediated anticancer immunotherapy and presents a promising target for chemical intervention. Despite the therapeutic promise of targeting this pathway, the clinical translation of STING agonists have faced significant hurdles. Various STING agonists, including the non-cyclic dinucleotide (CDN) agonist DMXAA [14], the synthetic CDN agonist MK-1454 [15], the small molecule diABZi [16], and MSA-2 [17], have been discovered to activate the STING pathway for cancer therapy, showing promising results in preclinical mouse models. However, current clinical trials of STING agonists, either as monotherapy or in combination with immune checkpoint PD-1/PD-L1 inhibitors, have shown less than optimal antitumor effects [18,19,20]. These findings suggest unknown mechanisms may be limiting their therapeutic efficacy.
Restoring effective immunosurveillance in STING-deficient tumors requires a clear understanding of the mechanisms regulating STING protein stability. STING homeostasis is governed by a multilayered network of post-translational modifications (PTMs) that determine its activation, trafficking, and degradation [21,22,23]. For example, palmitoylation of STING at Cys88/91 is indispensable for signal initiation [23], whereas phosphorylation of STING by TBK1 [5] or ULK1 [24] modulates downstream its activity and turnover. Multiple E3 ligases, including RNF5 [25] and TRIM29 [26], could promote K48-linked ubiquitination and proteasomal degradation. However, accumulating evidence indicates that the autophagy-lysosome system is the dominant route for STING clearance, mediated by selective autophagy adaptors such as p62/SQSTM1 [27] and AP-1/ESCRT-dependent lysosomal processing [28,29,30,31]. Although the mechanisms initiating STING degradation are increasingly documented, how specific PTMs coordinate to preserve STING abundance in tumor cells remains unclear.
ISGylation, a ubiquitination-like process, involves the covalent attachment of interferon-stimulated gene 15 (ISG15) protein to lysine residues on target protein through the coordinated action of E1 activating enzyme, E2 conjugating enzymes, and E3 ligases [32]. This modification is reversed by ubiquitin specific peptidase 18 (USP18), which hydrolyzes the isopeptide bond between ISG15 and target proteins [33]. ISGylation has been studied primarily in the context of antiviral immunity. Host cells dramatically upregulate ISG15 upon pathogen exposure, where it functions to restrict viral replication through diverse mechanisms, such as disrupting the assembly of viral proteins like the Ebola virus VP40 [34, 35] or inhibiting the nuclear import of Influenza A virus NS1A protein [36]. Furthermore, ISGylation modifies key host sensor proteins, including MDA5 [37] and cGAS [38], to amplify type I interferon signaling pathways, thereby orchestrating a robust antiviral state. However, beyond its established antiviral functions, the role of ISGylation in cancer biology is far more complex, ISGylation can drive oncogenesis by stabilizing oncoproteins like EGFR to sustain proliferative signaling [39] or by destabilizing tumor suppressors such as p53 [40] and PTEN [41] via the ubiquitin-proteasome pathway. On the other hand, ISGylation can exert potent anti-tumor effects. For instance, free ISG15 has been shown to recruit NK cells to the tumor microenvironment [42], and ISGylation of PD-L1 [43] promotes its degradation, thereby enhancing T-cell mediated cytotoxicity. Recent studies indicate that STING can also be ISGylated. Lin et al. [44] reported that ISG15 deficiency impairs the activation of the STING-IFN-I pathway during HIV-1 infection,while Qin et al. (2024) demonstrated that STING ISGylation stabilizes its protein expression and enhances the antiviral innate immune response [45]. These studies highlight the important regulatory role of STING ISGylation in viral infection models, leaving unresolved whether ISGylation regulates STING turnover in the tumor microenvironment and whether this process can be leveraged therapeutically.
Here, we define a tumor-specific role for STING ISGylation in regulating antitumor immunity. Using ISGylation-deficient mutants, we show that ISGylation prevents autophagic degradation of STING, thereby maintaining its protein stability and sustaining downstream IFN-I responses. We further identify USP18 as the deISGylase that counteracts this process and demonstrate that pharmacological inhibition of USP18 enhances STING ISGylation, boosts innate immune activation and synergizes with the STING agonist diABZi to suppress lung tumor growth. These findings reveal ISGylation as a previously unrecognized mechanism that safeguards STING abundance in tumor cells and offer a therapeutic strategy to overcome poor responsiveness to STING agonists in cancers with diminished STING protein levels.
Results
ISGylation deficiency impairs antitumor immunity in lung cancer mouse models treated with STING agonist
To investigate the role of ISGylation in modulating the immune response of lung cancer to STING agonist treatment, we first generated the ISG15-knockout (KO-) mouse Lewis lung carcinoma (LLC) cell line. Cells were further complemented with either wild-type ISG15 (ISG15-WT) or a non-ISGylatable ISG15 mutant (ISG15-C2), in which the C-terminal GG motif required for ISGylation was truncated (Fig. 1A–C). We then assessed the in vitro viability of LLC cells stably expressing ISG15-WT, ISG15-C2, or empty vector, all cell lines exhibited comparable viability (Fig. 1D). We then injected both cell lines into immunocompetent C57BL/6 mice. Once tumors became palpable, the mice received intravenous administration of diABZi (3 mg/kg) once every 3 days, and tumors were collected on day 14 post-injection (Fig. 1E). Compared with ISG15-WT, the introduction of ISG15-C2 significantly enhanced tumor growth in the LLC model and resulted in a 52% increase in tumor weight (Fig. 1F–H). Fluorescence-activated cell sorting (FACS) analysis indicated that ISG15-C2 expression slightly reduced the percentage of CD45+ cells in the tumors or spleens (Supplementary Fig. 1A–D). While ISG15-C2 expression reduced the percentage of tumor-infiltrating NK1.1+ cells among the CD45+ cells population (Fig. 1I, J). Similarly, the percentage of infiltrating CD8+ T cells was lower in the ISG15-C2 expression group (Fig. 1K, L). In contrast, ISG15-C2 expression decreased the percentage of CD8+ T cells but had statistically minimal effect on NK1.1+ cells in spleen (Supplementary Fig. 1E–H).

A, B Schematic representation of the ISGylation process and the importance of the C-terminal di-glycine (C2) motif for this modification. C Stable expression of ISG15-WT and ISG15-C2 in Lewis lung carcinoma (LLC) cells as shown in western blot. D Cell viability assays showing comparable proliferation rates in vitro of ISG15-WT and ISG15-C2 in LLC cells. E Workflow for the construction of lung tumor mouse model and drug treatment. C57BL/6 mice were injected with LLC cells expressing ISG15-WT or ISG15-C2 and then intravenously (i.v.) injected with diABZi (3 mg/kg) or a control injection-solution containing 1% dimethyl sulfoxide (DMSO) and 2% polyethylene glycol 300 (PEG300) in sterile water, at the indicated time points. Tumors were collected as indicated. F–H Tumors expressing ISG15-WT or ISG15-C2 were dissected and imaged F, tumor volume G and weighed H was recorded, n = 4. I–L The effect of ISG15-WT or ISG15-C2 on tumor-infiltrating NK1.1+ cells and the CD8+ T cells among CD45+ cells were analyzed using flow cytometry. Representative scatterplots I, K and frequency J, L, n = 4. The data presented is from a representative selected from three independent experiments. Data are presented as mean ± SEM, n = 4. Statistical differences were analyzed using a unpaired t-test (two-tailed), with * indicating p < 0.05, *** indicating p < 0.001.
These results support the crucial role of ISGylation in mediating tumor-suppressive function in vivo when treated with a STING agonist. Thus, we hypothesize that ISGylation may modulate STING-mediated IFN-I immune signaling.
ISGylation at four key lysine residues (K236, K338, K347, and K370) promotes STING stability
To investigate the ISGylation of STING using cell-based and biochemical assays, we co-expressed STING with UBE1L-Myc (E1), UBCH8-Myc (E2), Myc-ISG15, and either ARIH1-Myc-His, EFP-Myc-His, or HERC5-Myc-His in HEK293T cells. Among the E3 ligases tested, HERC5 significantly enhanced the ISGylation of STING (Fig. 2A). The deISGylating enzyme USP18, efficiently cleaved ISG15 from STING, whereas the catalytically inactive USP18 mutant C64S did not (Fig. 2B). Using HERC5 as the E3 ligase, ISG15 conjugation to STING was observed with ISG15-WT, but not with the conjugation-defective mutant ISG15-C2 (Fig. 2C). Furthermore, we confirmed that ISGylation of endogenous STING can be detected in human non-small-cell lung cancer H1299 cells with diABZi (Fig. 2D) and in vitro ISGylation assays further confirmed STING as a substrate for ISGylation, using recombinant ISG15-WT and ISG15-C2 proteins (Supplementary Fig. 2A).

A Detection of STING ISGylation and screening of E3 ligases. Flag-STING was co-transfected with UBE1L-Myc (E1), UBCH8-Myc (E2), Myc-ISG15, and either ARIH1-Myc-His, EFP-Myc-His, or HERC5-Myc-His into HEK-293T cells. Cells were harvested and lysed 24 h post-transfection, and then lysates were subjected to immunoprecipitation using Flag-beads. Western blot analysis with an ISG15 antibody was performed to detect ISG15-modified STING bands. B De-ISGylation of STING by USP18. Flag-STING along with UBE1L-Myc-His (E1), UBCH8-Myc-His (E2), HERC5-Myc-His (E3), and Myc-ISG15 were co-transfected with GFP-tagged USP18-WT or the catalytically inactive USP18-C64S mutant into HEK-293T cells. Twenty-four hours post-transfection, cells were subjected to an ISGylation assay, performed as described above. C Verification of STING ISGylation using an ISGylation-deficient mutant ISG15-C2. Flag-STING along with E1, E2, and E3 plasmids as in (B) were co-transfected with either Myc-ISG15-WT or Myc-ISG15-C2 into HEK-293T cells. ISGylation experiments were performed as described in B. D ISGylation of endogenous STING was analyzed by immunoprecipitation with STING antibody followed by western blot with anti-ISG15 antibody and STING antibody in H1299 cells in the presence of diABZi (10 µM for 4 h). E The effect of ISG15-C2 on STING protein stability. H1299 cells were transfected with plasmids expressing ISG15-WT or ISG15-C2. After 24 h, cells were treated with diABZi (1 μM for 12 h). ISGylation experiments were performed as described in B. F The impact of the ISG15-C2 on IFN-β transcription. Transfection was performed as described above and changes in IFN-β mRNA were measured by RT-qPCR. G Screening for ISGylation sites on STING. Flag-STING WT or STING-K only mutant plasmid was co-transfected with E1, E2, E3, and Myc-ISG15. ISGylation experiments were performed as described in B. H The effect of specific lysine retention on STING degradation. H1299 cells were transfected with plasmids expressing single retention of nine lysine (K-only) residues of STING, along with Myc-ISG15. Twenty-four hours post-transfection, cells were treated with diABZi (1 μM for 12 h), and western blot was used to assess the degradation differences of STING protein. I The effect of STING-4KR mutation on STING and p-IRF3 protein stability. H1299 cells were transfected with either STING-WT or STING-4KR plasmid. Twenty-four hours post-transfection, cells were treated with diABZi (1 μM) at various time points (3, 6, and 12 h). Western blot analysis was conducted to detect differences in STING protein degradation, followed by densitometric analysis of the bands. J Schematic representation of the regulation of STING protein stability by ISGylation. ISGylation at K236, K338, K347, and K370 are essential for STING protein stability at rest and post-diABZi treatment. The data presented is from a representative experiment, selected from three independent experiments. Data were presented as mean ± SEM, comparisons were conducted using two-way ANOVA for multiple comparisons, with **** indicating p < 0.0001 and n.s indicating not significant.
To investigate the role of STING ISGylation in lung cancer cells, we compared empty vector, ISG15-WT and ISG15-C2. After 12 h of diABZi treatment (STING agonist), immunoblotting revealed that STING protein levels were preserved in ISG15-WT-overexpressing cells but not with vector or ISG15-C2 (Fig. 2E). While p-IRF3 was sustained in ISG15-WT, it was notably diminished in the vector and ISG15-C2 groups. RT-qPCR confirmed greater induction of IFNB1 mRNA with ISG15-WT than with vector or ISG15-C2 (Fig. 2F). Functionally, ELISA showed that ISG15-WT, but not ISG15-C2, significantly enhanced the secretion of IFN-β following diABZi stimulation (Supplementary Fig. 2B). These findings suggest that ISGylation enhances the protein stability of STING in lung cancer cells, and may promote the anti-lung cancer immune responses.
To systematically identify the ISGylation sites within STING, we first generated a global STING-K0 mutant in which all lysine (K) residues were substituted with arginine (R). Subsequently, we created nine single-site mutants (K-only) by reintroducing lysine at nine specific positions K20, K137, K150, K224, K236, K289, K338, K347, and K370, as previously described [46]. The ISGylation experiment demonstrated that each of these residues could be ISGylated (Fig. 2G). Consistently, ISGylation persisted in single lysine-to-arginine (K-R) mutants but was abolished in the STING-K0 mutant (Supplementary Fig. 2C).
To determine which specific residues contribute to protein stability via ISGylation, we screened the stability of these nine K-only mutants and found that ISG15 overexpression selectively increased the protein abundance of K236, K338, K347, and K370 mutants before (lane 1 vs. lane 3) and after-diABZi stimulation (lane 2 vs. lane 4) (Fig. 2H). Subsequently, we generated a STING-4KR mutant by simultaneously substituting these four critical lysines (K236, K338, K347, and K370) with arginine. As a result, the STING-4KR mutation significantly accelerated STING protein turnover upon diABZi stimulation, reducing the protein half-life from approximately 6 h (STING-WT) to 3 h (Fig. 2I). Additionally, the total IRF3 protein levels were unchanged, whereas STING-4KR elicited markedly less IRF3 phosphorylation (p-IRF3) than STING-WT (Fig. 2I). Mechanistically, ISGylation at K236, K338, K347, and K370 was individually promoted by the E1-E2-E3 (UBE1L-UBCH8-HERC5) cascade and removed by USP18 in an enzyme-activity-dependent manner (Supplementary Fig. 2D–G). Correspondingly, STING-4KR exhibited significantly diminished overall ISGylation levels compared to STING-WT (Supplementary Fig. 2H).
Taken together, these findings demonstrate that ISGylation at K236, K338, K347, and K370 is essential for maintaining STING protein stability. The removal of these modifications leads to rapid degradation and a significant reduction in downstream STING signaling activation (Fig. 2J).
ISGylation inhibits autophagic degradation of STING
To identify the pathway underlying ISGylation-mediated STING stabilization, we first examined whether ISGylation affects K48-linked ubiquitination. As a result, neither ISG15-WT overexpression nor the conjugation-defective ISG15-C2 significantly altered K48-linked polyubiquitination on STING (Fig. 3A). Next, we used the autophagy inhibitor chloroquine (CQ) and the proteasome inhibitor MG132 to dissect the degradation route. In the absence of exogenous ISG15, both CQ (Fig. 3B, top, lane 1 vs. 4) and MG132 (Fig. 3B, bottom, lane 1 vs. 4) treatment increased STING levels, confirming that basal STING turnover is regulated by both autophagy and the proteasome. However, ISG15 overexpression further increased STING protein level even with proteasome inhibition by MG132 (Fig. 3B, bottom, lanes 4-6), whereas its stabilizing effect was abolished by autophagy blockade with CQ (Fig. 3B, top, lanes 4-6). Thus, ISGylation stabilizes STING by suppressing autophagic degradation, but not proteasomal turnover.

A The effect of ISG15-WT or ISG15-C2 on STING ubiquitination. Flag-STING was co-transfected with UBE1L-Myc-His (E1), UBCH8-Myc-His (E2), HERC5-Myc-His (E3), and either Myc-vector, Myc-ISG15 or Myc-ISG15-C2. Twenty-four hours post-transfection, cells were treated with 20 μM MG132 for 4 h before collection for ubiquitination assays. Western blot analysis using a ubiquitin antibody was conducted to assess the effects on STING ubiquitination. B The effects of ISG15 on STING autophagy degradation and proteasomal degradation processes. Twenty-four hours after co-transfection with Myc-vector or Myc-ISG15, cells were treated with either DMSO, CQ (20 μM), or MG132 (20 μM) for 4 h. The levels of LC3I/II were also examined as a marker of autophagy occurrence. C–E The impact of ISGylation on STING’s interactions with p62 and LC3I/II. H1299 cells were transfected with plasmids overexpressing ISG15-WT or ISG15-C2 C, USP18-WT or USP18-MT (C64S) D, and STING-WT or STING-4KR E. Co-immunoprecipitation (co-IP) was then used to detect variations in STING’s interactions with p62 and LC3I/II. F, H The effect of ISG15-WT or ISG15-C2 on the colocalization of STING with the autophagy marker LC3. In H1299 cells, after stimulation with diABZi (10 μM) for 3 h and CQ (50 μM) for 4 h, the colocalization of STING with LC3 was assessed by confocal microscopy for cells overexpressing ISG15-WT or ISG15-C2, scale bar: 10 μm F, relative fluorescence intensity per cell was quantified H. G, I The colocalization of STING-WT or STING-4KR with the autophagy marker LC3 in WT or ISG15-deficient cells. In LLC cells, following the expression of Flag-STING-WT or STING-4KR for 24 h and subsequent treatment with CQ (50 μM) for 4 h, the colocalization of STING with LC3 was compared between the two groups, scale bar: 10 μm G, relative fluorescence intensity per cell was quantified I. Data are presented as mean ± SEM, n = 6, comparisons were conducted using two-way ANOVA for multiple comparisons, with ** indicating p < 0.01 and n.s indicating not significant.
Since selective autophagy relies on cargo receptors such as p62 (SQSTM1) to bridge targets to LC3 [27], we investigated whether ISGylation affects the interaction between STING and these autophagic factors. Co-IP assays demonstrated that STING interacts with both p62 and LC3, overexpression of ISG15-WT markedly reduced these interactions. Importantly, the ISG15-C2 mutant failed to disrupt the STING-p62 and LC3 complex, confirming that ISGylation is required to block this association (Fig. 3C). Conversely, overexpression of the deISGylase USP18-WT enhanced the binding of STING to p62 and LC3 compared to the catalytically inactive USP18-C64S mutant (Fig. 3D), further supporting the role of ISGylation in shielding STING from autophagic recognition. To validate that four specific lysine residues are critical for this effect, we utilized the STING-4KR mutant, which exhibited significantly stronger binding affinity for p62 and LC3 compared to STING-WT (Fig. 3E), suggesting that ISGylation at these sites sterically hinders the recruitment of autophagy receptors.
Next, we visualized the subcellular localization of STING and the autophagosome marker LC3 using confocal microscopy in H1299 cells. Treatment with the STING agonist diABZi (10 μM) for 3 h activated the STING pathway, and initiated the autophagic degradation of STING (Fig. 3F and H). We used CQ to block autophagosome-lysosome fusion and capture STING -LC3 interactions under non-degradative conditions. Notably, ISG15-WT, but not ISG15-C2, significantly reduced the STING-LC3 co-localization, while modulating LC3 puncta fluorescence intensity (Fig. 3F and H). Consistent with the biochemical data, STING-4KR displayed increased colocalization with LC3 compared with STING-WT (Fig. 3G and I). In ISG15-knockout (KO) cells, STING-WT and STING-4KR showed similar LC3 colocalization, abolishing the difference between them (Fig. 3G and I), which indicates that the difference in STING-LC3 co-localization between STING-4KR and STING-WT is primarily dependent on ISGylation.
These data collectively indicate that ISGylation acts as a regulatory checkpoint, preventing STING from entering into autophagosomes by disrupting its interaction with p62 and LC3, thereby stabilizing STING and maintaining its cellular levels.
ISGylation of STING promotes antitumor immunity in lung cancer models
To further investigate whether the ISGylation of STING promotes antitumor immunity, we generated LLC stable cell lines expressing STING-WT or STING-4KR. Upon diABZi stimulation, LLC cells expressing STING-WT exhibited a sustained increase in IFN-β mRNA levels within 6 h, whereas STING-4KR expressing cells exhibited significantly lower levels, especially at 12 h (Fig. 4A). Consistent with the mRNA data, IFN-β secretion was significantly reduced in STING-4KR-expressing cells compared with STING-WT at 3, 6, and 12 h post-stimulation (Supplementary Fig. 4A).

A The impact of STING-4KR on IFN-β transcription. H1299 cells were transfected with either STING-WT or STING-4KR plasmid. Twenty-four hours post-transfection, cells were treated with diABZi (1 μM) at various time points (3, 6, and 12 h). The mRNA levels of IFN-β were then measured using RT-qPCR. B Workflow for the generation of Lung tumor model and drug treatment. C57BL/6 mice were injected with LLC cells expressing STING-WT or STING-4KR. Tumors were collected at indicated time points. C–E In vivo assessment of tumor growth for celling expressing STING-WT or STING-4KR. Subcutaneous tumors were generated in C57BL/6 mice using LLC cells expressing either STING-WT or STING-4KR, and analyses included tumor imaging C, tumor growth curves D, and tumor mass measurements E. F–I Tumor-infiltrating CD8+ T cells and the NK1.1+ cells within the CD45+ cells population were analyzed using flow cytometry. Representative scatterplots F, H and cell frequencies are shown G, I (n = 4 per group). J Schematic representation of ISGylation at four specific lysine residues on STING, a key regulatory mechanism within the tumor microenvironment that orchestrates immune responses. This process is essential for the maintenance of the IFN-I pathway, which in turn activates NK1.1+ cells and CD8+ T cells, bolstering their tumor recognition capabilities and contributing to a robust antitumor immune response. Data are presented as mean ± SEM, n = 4. Statistical differences were analyzed using a unpaired t-test (two-tailed), with * indicating p < 0.05 and **** indicating p < 0.0001.
To assess the impact of STING ISGylation on tumor progression in vivo, we injected the above two cell lines into C57BL/6 J mice (Fig. 4B). Mice harboring tumors expressing STING-4KR exhibited significantly accelerated tumor growth, with increased tumor size and weight compared to those expressing STING-WT (Fig. 4C–E). Flow cytometric analysis of the tumor microenvironment revealed a profound defect in immune infiltration in STING-4KR (Fig. 4F–I and Supplementary Fig. 3A, B). Specifically, the frequency of tumor-infiltrating NK1.1+ cells and CD8+ T cells (gated on CD45+cells) were compromised by over 60% in STING-4KR tumors (Fig. 4F–I). Additionally, a marked decrease in CD8+ T cells was observed in the spleens of tumor-bearing mice (Supplementary Fig. 3E), while other immune parameters remained comparable between the two groups (Supplementary Fig. 3C, D and G, H).
Collectively, these data demonstrate that ISGylation at these key residues is essential for maintaining an immunogenic tumor microenvironment and mounting an effective antitumor response (Fig. 4J).
USP18 expression was upregulated and inversely correlated with STING levels in lung cancer patient samples
In H1299 cells, we observed that overexpression of the deISGylating enzyme USP18 negatively regulated STING protein levels. This effect is dependent on USP18’s enzymatic function, as the enzymatic mutant USP18 C64S did not alter STING protein levels (Fig. 5A). We therefore assessed the prognostic role of USP18 in lung cancer, using the Kaplan-Meier Plotter online tool. The results showed that patients with high USP18 expression had poorer overall survival (OS) than those with low USP18 expression in LUAD (Fig. 5B), while patients with low STING expression had poorer OS than those with high STING expression (Fig. 5C).

A The effect of USP18 on STING protein levels in lung cancer cells. H1299 cells were transfected with either GFP-USP18-WT or GFP-USP18-C64S for 36 h followed by assessment of endogenous STING protein levels via Western blot analysis. B, C Kaplan-Meier curves of overall survival in lung cancers patients stratified by USP18 expression (high vs. low, B) and STING expression (high vs. low, C). Data were obtained from The Cancer Genome Atlas (TCGA). D, E Quantification of immunohistochemical staining of STING (D) and USP18 (E) in a tissue array comparing 60 pairs of lung cancer and adjacent normal tissues. F Correlation analysis of STING and USP18 expression in 60 pairs of lung cancer and adjacent normal tissues in a tissue array. G, H The representative IHC staining images for USP18 (G) and STING (H) in lung cancer and adjacent tissues. The area within the black square is enlarged below. The data presented is from a representative experiment, selected from three independent experiments. Data were presented as mean ± SEM, comparisons were conducted using two-way ANOVA for multiple comparisons, with ** indicating p < 0.01.
In addition, we assessed the expression of USP18 and STING and their clinical relevance using Immunohistochemistry (IHC) staining of 60 pairs of lung cancer tissues and adjacent non-cancerous tissues from patients (Fig. 4A, B). We observed significant downregulation of STING in lung cancer tissues compared to adjacent non-cancerous tissues (Fig. 5D). In contrast, USP18 expression was relatively high in the majority of lung cancer tissues (Fig. 5E). Pearson correlation analysis revealed a significant negative correlation between the USP18 and STING protein levels in lung cancer tissues (R = -0.1843, P = 0.0487) (Fig. 5F). Representative images of high USP18 staining and low STING staining are shown (Fig. 5G, H). Taken together, these results suggest that aberrantly high USP18 expression can act as an oncogenic factor by destabilizing STING. Therefore, we hypothesize that small molecular inhibitors of USP18 could restore STING ISGylation and enhance the antitumor immunogenicity of STING.
Targeting USP18-mediated STING deISGylation as a therapeutic strategy for lung cancer
To identify small molecular inhibitors of USP18, three different compound libraries containing a total of 17,280 small molecules were screened against murine USP18 (mUSP18) using a FRET-based enzyme assay (Supplementary Fig. 5A–D). The recombinant His6-mUSP18 (residues: 16-372) was expressed in Spodoptera frugiperda 9 (Sf9) cells and fluorogenic substrate Arg-Leu-Arg-Gly-Gly-AMC, reflecting the C-terminal residues of ISG15, was utilized to measure USP18’s enzymatic activity as previously described [47]. The first round of screening provided 121 compounds with over 50% inhibition at 100 μM and were selected for further validation. After removing compounds with poor solubility, strong reactivity, or high intrinsic fluorescence, 11 relatively potent compounds were measured for IC50 (Supplementary Fig. 5E), including Tanshinone IIA sulfonate (TST) with an IC50 of 3.41 µM (Fig. 6A). To confirm the direct interaction between TST and USP18, we employed nuclear magnetic resonance-saturation transfer difference (NMR-STD) assays. Positive signals observed in the NMR-STD assays (Fig. 6B) indicate a direct binding of TST with USP18. In the STING ISGylation assay, we found that USP18 overexpression led to a decrease in STING ISGylation. However, TST treatment attenuated this effect in a dose-dependent manner, enhancing STING ISGylation (Fig. 6C).

A The inhibitory activity of TST against USP18 was measured using the peptide RLRGG-AMC as a substrate. IC50 was presented as mean ± SEM, n = 3 independent experiments. The chemical structure of TST is shown. B STD-NMR binding analysis for TST and USP18. The top spectrum is the 1H NMR of USP18 (10 μM) in a mixture with TST (400 μM), while the bottom spectrum displays the STD-NMR of the same sample. C USP18-mediated de-ISGylation of STING examined by Western blot. Flag-STING, UBE1L-Myc-His (E1), UBCH8-Myc-His (E2), HERC5-Myc-His (E3), and Myc-ISG15 were co-transfected with GFP-USP18-WT into HEK-293T cells. Twenty-four hours post-transfection, cells were treated with TST (5, 25,125 μM) for 12 h, followed by an ISGylation assay. ISGylation experiments were performed as described in (Fig. 2B). D CHX chase assay was performed to analyze the effect of TST on STING protein stability in both USP18-WT (Negative Control, si-NC) and USP18-KD cells. LLC cells were treated with 100 µg/mL cycloheximide (CHX) for the indicated time points. The graph represents quantification of the STING protein levels. E CHX chase assay was used to analyze the effect of TST on STING protein stability in cells expressing either STING-WT or STING-4KR. The LLC cells were treated with 100 µg/mL cycloheximide (CHX) for the indicated time points. The graph represents quantification of the STING protein levels. F–G The synergistic antitumor effect of combined treatment of TST (10 mg/kg) and diABZi (3 mg/kg) in a mouse model. Tumors were dissected, photographed F and weighed G. H–K Tumor-infiltrating CD8+ T cells and NK1.1+ cells among CD45+ cells were analyzed using flow cytometry. Representative scatter plots H, J and frequencies I, K are shown (n = 5 per group). The data presented is from a representative experiment, selected from three independent experiments. Data were presented as mean ± SEM, comparisons were conducted using two-way ANOVA for multiple comparisons, with ** indicating p < 0.01, *** indicating p < 0.001, **** indicating p < 0.0001 and n.s indicating not significant.
Subsequently, we first knocked down USP18 in LLC cells using siRNA. Then we treated WT and USP18-KD LLC cells with TST (50 µM). The cycloheximide (CHX)-chase experiment showed that STING protein degradation was accelerated in USP18-KD tumor cells, and TST treatment delayed the degradation of STING protein in USP18-WT cells, but not in USP18-KD cells (Fig. 6D). These findings indicate that TST may stabilize STING protein levels by inhibiting USP18. To ensure that TST’s regulation of STING protein stability is associated with STING ISGylation, we further treated STING-WT and STING-4KR LLC cells with TST (50 µM). CHX chase experiments indicated that the STING-4KR protein degraded more rapidly than STING-WT, and TST treatment delayed the degradation of STING-WT protein, but not STING-4KR (Fig. 6E). These results demonstrate the on-target effect of TST’s regulation on STING stability through ISGylation.
To investigate the effect of TST on tumor inhibition in vivo through STING ISGylation, we first established tumor models using LLC cells. TST was then administered intravenously every 3 days at doses of 2.5 mg/kg, 5 mg/kg or 10 mg/kg. Treatment of mice with TST resulted in significant dose-dependent inhibition of tumor growth (Supplementary Fig. 6A). Next, we treated LLC-derived lung cancer tumors with TST and the STING agonist diABZi, either alone or in combination. While treatment with either diABZi or TST alone delayed tumor growth, the combination of diABZi and TST more effectively inhibited tumor growth, as evidenced by the reduced tumor weight in drug-treated mice compared to the control group (Fig. 6F–G). Consistently, FACS analyses of the tumor tissues showed that TST or diABZi treatment alone increased the proportion of tumor-infiltrating CD8+ T cells and NK1.1+ cells within the CD45+ cells population. Notably, in tumors treated with the combination of TST and diABZi, while 43.9% of tumor-infiltrating NK1.1+ cells were detected, compared with only 12.5% of those in the DMSO-treated control group (Fig. 6H–K), surpassing the effects observed with either drug treatment alone. These findings indicate that the combination therapy of TST and diABZi elicited a more robust antitumor immune response. In addition, the combined treatment induced a higher proportion of CD8+ T cells in spleen compared to monotherapy (Supplementary Fig. 6F–G). All other measured parameters were comparable across the four treatment groups (Supplementary Fig. 6B–E and H, I).
Collectively, these data show that simultaneous stabilization of the STING protein cellular level by TST and the activation of STING using agonist diABZi synergistically enhanced antitumor immune responses and inhibited lung tumor growth (Fig. 7).

ISGylation at key lysine residues (K236, K338, K347, and K370) on STING prevents its autophagic degradation by disrupting interactions with p62 and LC3, thereby maintaining STING protein stability and enhancing type I interferon (IFN-I) production. USP18, a deISGylating enzyme, is overexpressed in lung cancer and negatively correlates with STING protein levels, promoting tumor progression by reducing STING ISGylation. Tanshinone IIA sulfonate (TST), a novel USP18 inhibitor, promotes STING ISGylation and stability, leading to increased IFN-I production and enhanced antitumor immunity. Combined treatment with TST and STING agonist diABZi synergistically strengthens antitumor immune responses and inhibits lung tumor growth in mouse models, offering a potential therapeutic strategy for lung cancer patients with low STING expression.
Discussion
The cGAS-STING pathway plays a pivotal role in bridging innate and adaptive immunity, transforming immunologically “cold” tumors into “hot” environments susceptible to T cell attack [48,49,50]. However, a major hurdle in current immunotherapies is the frequent downregulation of STING expression observed in advanced malignancies, including lung cancer, which greatly diminishes the responsiveness to STING agonists [51,52,53]. Reduced STING availability compromises the production of type I interferons (IFNs), central orchestrators of antiviral and antitumor immunity that shape immune gene expression and activate effector populations such as T and NK cells [13, 54,55,56]. In this context, our study uncovers an important and previously underappreciated role of ISGylation in regulating STING stability. We demonstrate that targeting the deISGylase USP18 is a promising strategy to stabilize STING and restore immune activation in lung cancers.
STING signaling must be tightly controlled to prevent autoimmunity, with the autophagy-lysosome pathway serving as a key negative‑feedback mechanism. Upon activation, STING is sorted into autophagosomes via cargo receptors such as p62/SQSTM1 [27]. In the current study, we focused on the autophagic route based on the observed accumulation of LC3 II and the interaction between STING and p62. Our findings refine this model by introducing ISGylation as a checkpoint that delays this process. We demonstrated that ISGylation at four specific lysine residues (K236, K338, K347, and K370) suppress the interaction between STING and p62/LC3. Consequently, the removal of this modification, either by USP18 overexpression or by mutation (STING-4KR), exposes STING to rapid p62-mediated capture and autophagic clearance. Our findings are consistent with an autophagy-mediated mechanism, but we cannot exclude parallel degradation pathways such as AP-1 or ESCRT-dependent lysosomal sorting [28,29,30]. Future work involving targeted knockout or knockdown of these essential components will be valuable for defining STING ISGylation coordinates with alternative lysosomal sorting mechanisms.
A critical question in ISG15 biology is whether its functions arise from free ISG15 (acting as a cytokine or scaffold) or from protein conjugation (ISGylation) [42, 57,58,59]. Our study provides definitive evidence supporting the latter in the context of STING regulation. By utilizing the ISG15-C2 mutant, which cannot form covalent bonds but retains the structural integrity of free ISG15, we observed that the non-ISGylation presence of ISG15 failed to rescue STING stability or promote antitumor immunity. Consistently, our analysis of the STING-4KR mutant provides the crucial substrate-specific validation. The STING-4KR mutant exhibited accelerated turnover, enhanced binding to autophagy receptors, reduced immune cell infiltration and faster tumor growth, underscoring the critical role of STING ISGylation at these four sites for effective antitumor immunity.
ISGylation is traditionally associated with antiviral defense [60,61,62], and recent studies show that STING ISGylation potentiates antiviral IFN responses [44, 45]. ISGylation also exerts context-dependent effects in tumor immunity, either suppressing or promoting tumor growth [42, 63, 64]. These conflicting observations underscore that the functional outcome of ISGylation is highly context-dependent, varying significantly across different cell types and pathological stages. In this study, we expand the understanding of this PTM by elucidating a novel protective mechanism of STING ISGylation specifically within the lung cancer context. Importantly, in vivo analyses indicate that the enhanced tumor growth observed in ISG15-C2 and STING-4KR models is driven by markedly reduced immune infiltration. Notably, both mutations converged on a shared phenotype marked by a > 60% reduction in tumor-infiltrating NK1.1+ cells, highlighting ISGylation as a critical determinant of tumor immune surveillance.
In this study, we identified the deISGylase USP18 as a clinically actionable target in STING-low lung cancers, its inverse correlation with STING and poor prognosis in patient samples. By repurposing TST as a USP18 inhibitor to stabilize STING, and combining it with the STING agonist diABZi, we achieved robust synergistic activation of the pathway and superior anti-tumor efficacy. A recently discovered covalent inhibitor selective for mouse USP18 (mUSP18) [65] may inform and accelerate drug discovery for human USP18, a promising therapeutic target.
Together, our findings define ISGylation as a critical modulator of STING signaling, ensuring sustained cytokine production before degradation occurs and promoting antitumor immunity in lung cancer. By mapping four essential ISGylation sites and demonstrating their role in preventing p62-dependent autophagic degradation, we reveal a regulatory mechanism that sustains STING activity in tumor cells. Our identification of TST as a direct USP18 inhibitor further provides a readily translatable strategy to re-sensitize STING-low tumors to agonist therapy. These findings offer a foundation for the rational development of USP18-targeting agents and combination immunotherapies aimed at restoring STING-dependent antitumor immunity.
Materials and methods
Cell culture
HEK293T (ATCC) and LLC (Hycyte) cells were maintained in Dulbecco’s modified Eagle medium (HyClone) with 10% fetal bovine serum (FBS, Yeasen), 1% penicillin/streptomycin (Basal media), 1×non-essential amino acids solution (Basal media), 10 mM HEPES (Solarbio). H1299 cells (Hycyte) were maintained in RPMI 1640 (HyClone) with 10% FBS, 1% penicillin/streptomycin (Basal media), 1×non-essential amino acids solution (Basal media), 10 mM HEPES (Solarbio). All cells were cultured at 37 °C in a humidified incubator containing 5% CO2.
Cell viability assay
Cell viability was determined using the CellTiter-Glo® 2.0 Luminescent Cell Viability Assay (Promega, Cat. #G9241), according to the manufacturer’s instructions. This homogeneous method quantifies ATP, which is present in metabolically active cells, to determine the number of viable cells in culture. Briefly, cells were seeded in a white-walled, clear-bottomed 96-well plate, per well in 100 µL of complete growth medium. After a 36-h incubation, the CellTiter-Glo reagent was added to each well at a 1:1 (v/v) ratio. Plates were incubated for 10 min at room temperature to stabilize the luminescent signal, and luminescence was recorded using a microplate reader.
Plasmids and transfection
ISG15-WT, ISG15 C2 mutant, STING-WT, and STING-4KR mutant were cloned into the lentivirus expression pLVX vector (Hanbio) for stable cell line generation. ISG15-WT, ISG15 C2 mutant, ISG15 AA mutant, UBE1L, UBCH8, HERC5, ARIH1 and EFP were cloned into pCMV-Myc vector. STING-WT, STING domain mutant (1-160aa), STING domain mutant (151-379aa), STING K-only mutants, STING-KR mutants and STING-4KR mutant were cloned into pCMV-Flag vector. USP18-WT and USP18 C64S mutant were cloned into pCDH-GFP vector. mUSP18 was cloned into pBac-His-SUMO vector for expression in Sf9 cells. ISG15-WT and ISG15 C2 mutant were cloned into pET28a-His-SUMO vector for expression in Escherichia coli. For transient transfection experiments, cells at 70% confluency were transfected with plasmid DNA constructs using Hieff Trans Liposomal Transfection Reagent (Yeasen, 40802).
Antibodies and reagents
The antibodies used for western blotting included anti-Flag (Sigma-Aldrich, F7425 and Cell Signaling Technology, 14793), anti-Myc (Cell Signaling Technology, 18583), anti-HA (Cell Signaling Technology, 3724), anti-STING (Cell Signaling Technology, 13647), anti-ISG15 (Invitrogen, 7H29L24), anti-IRF3 (Cell Signaling Technology, 4302), anti-pIRF3 (Cell Signaling Technology, 79945), anti-LC3I/II (Cell Signaling Technology, 3868), anti-p62 (Cell Signaling Technology, 39749), anti-Ub (Cell Signaling Technology, 20326), anti-K48-Ub (Cell Signaling Technology, 8081), anti-GAPDH (Cell Signaling Technology, 5174), anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology, 7074) and anti-mouse IgG, HRP-linked antibody (Cell Signaling Technology, 7076). The antibodies used for flow cytometry assay included anti-mouse-CD45-FITC (BioLegend, 103107), anti-mouse-CD8a-APC (BioLegend, 100711) and anti-mouse-NK1.1-PE (BioLegend, 156503).
diABZi (S8796) was purchased from Selleck, tanshinone IIA sulfonate sodium (54516ES20) was purchased from Yeasen, MG132 (T2154) and cycloheximide (T1225) were purchased from Topscience, chloroquine (14774) was purchased from Cell Signaling Technology.
Western blot and immunoprecipitation
For western blot, cells were lysed in lysis buffer (20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, 10 mM NaF, 10% glycerol, 1% Triton X-100, and 0.1% SDS), supplemented with a protease inhibitor cocktail (Roche Diagnostics, 5892791001) by incubation on ice for 30 min, and the lysates were then centrifuged at 15,000 rpm for 10 min. Proteins in the supernatant (20–40 μg) were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; ExpressPlus PAGE Gel, 4–20%, GenScript, M00657) and transferred to a nitrocellulose membrane using the eBlot-L1 system (GenScript, L00686). The membrane was blocked using 5% skim milk in TBST buffer for 1 h and incubated overnight with primary antibodies at 4 °C. Following incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 h and detected by the e-BLOT western blot imager system. Protein levels were quantified from the western blot bands using ImageJ software.
For immunoprecipitation, cells were lysed in lysis buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, and 0.5% NP-40 and protease inhibitor cocktail (Roche Diagnostics, 5892791001)) by incubation on ice for 30 min, and the lysates were then centrifuged at 15,000 rpm for 10 min. The supernatant was incubated with anti-Flag M2 Affinity Gel (Sigma-Aldrich, A2220) with rotation overnight at 4 °C. The beads were washed thrice with lysis buffer, boiled in 2× SDS sample buffer at 95 °C for 5 min and subjected to SDS-PAGE followed by western blot analysis.
ISGylation and ubiquitination
For the in cells ISGylation assay, HEK293T cells were harvested after transfected with the appropriate constructs for 36 h. Cells were lysed by the denaturing buffer (10 mM Tris-HCl (pH 8.0)), 150 mM NaCl, 2% SDS, 2 mM Na3VO4, 50 mM NaF, and protease inhibitor cocktail (Roche Diagnostics, 5892791001) and boiled at 95 °C for 10 min. Cell lysates were diluted ten-fold with buffer (10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100) and incubated at 4 °C for 60 min with rotation. The diluted samples were centrifuged at 13,000 rpm for 30 min and the supernatants were immunoprecipitated with anti-Flag M2 Affinity Gel (Sigma-Aldrich, A2220) overnight at 4 °C. The beads were washed thrice with the washing buffer (10 mM Tris-HCl (pH 8.0), 1 M NaCl, 1 mM EDTA, and 1% NP-40), boiled in 2× SDS sample buffer at 95 °C for 5 min and subjected to SDS polyacrylamide gel electrophoresis (SDS-PAGE) followed by western blot analysis.
For the in vitro ISGylation assay, purified STING was incubated with UBE1L (E1), UBCH8 (E2), HERC5 (E3) and ISG15 WT or ISG15 C2 in a reaction buffer (50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 0.1% Triton X-100, 1 mM DTT and 10 mM ATP) at 37 °C for 2 h. The mixtures were boiled in 5× SDS sample buffer at 95 °C for 5 min and subjected to SDS-PAGE followed by western blot analysis.
For the in cells Ubiquitination assay, HEK293T cells were treated with 20 μM MG132 for 4 h before harvesting. Cells were lysed by the denaturing buffer (10 mM Tris-HCl (pH 8.0)), 150 mM NaCl, 2% SDS, 2 mM Na3VO4, 5 mM NaF, 5 mM N-Ethylmaleimide (NEM) and protease inhibitor cocktail (Roche Diagnostics, 5892791001) and boiled at 95 °C for 10 min. Cell lysates were diluted ten-fold with buffer (10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100) and incubated at 4 °C for 60 min with rotation. The diluted samples were centrifuged at 13,000 rpm for 30 min and the supernatants were immunoprecipitated with anti-Flag M2 Affinity Gel (Sigma-Aldrich, A2220) overnight at 4 °C. The beads were washed thrice with the washing buffer (10 mM Tris-HCl (pH 8.0), 1 M NaCl, 1 mM EDTA, and 1% NP-40), boiled in 2× SDS sample buffer at 95 °C for 5 min and subjected to SDS-PAGE followed by western blot analysis.
Protein stability assay
The half-life of endogenous or ectopically expressing STING was determined using a Cycloheximide (CHX) chase assay. Cells were treated with TST (50 µM) for 12 h and then 50 µg/mL of CHX (T1225, Topscience) at specific time intervals. Subsequently, cells were harvested and lysed, and protein levels were quantified using western blotting.
Quantitative reverse transcription PCR (RT-qPCR)
Total RNA was extracted from various cells using the TRIeasy total RNA extraction reagent (Yeasen, 19221ES60) according to the manufacturer’s instructions. cDNA was synthesized through reverse transcription using the Hifair II first-strand cDNA synthesis kit (Yeasen, 11119ES60). Real-time PCR was performed using the cDNA samples and Hieff qPCR SYBR green master mix (No Rox) (Yeasen, 11201ES08).
The primers for human IFNB:
5’-ATGACCAACAAGTGTCTCCTCC-3’ (Forward Primer);
5’-GGAATCCAAGCAAGTTGTAGCTC-3’ (Reverse Primer).
The primers for mouse Ifnb:
5’-AGCTCCAAGAAAGGACGAACA-3’ (Forward Primer);
5’-GCCCTGTAGGTGAGGTTGAT-3’ (Reverse Primer).
The primers for human GAPDH:
5’-TGTGGGCATCAATGGATTTGG-3’ (Forward Primer);
5’-ACACCATGTATTCCGGGTCAAT-3’ (Reverse Primer).
The primers for Mouse Gapdh:
5’-AATGGATTTGGACGCATTGGT-3’ (Forward Primer);
5’-TTTGCACTGGTACGTGTTGAT-3’ (Reverse Primer).
Immunofluorescence
Cells cultured on coverslips were fixed in 4% paraformaldehyde for 15–20 min at room temperature, followed by three washes with PBS. Cells were then permeabilized with 0.1% Triton X-100 in PBS for 10 min and subsequently blocked in a solution of 5% bovine serum albumin (BSA) in PBS for 1 h at room temperature. Primary antibodies (anti-STING, Cell Signaling Technology, 13647 and anti-LC3I/II Cell Signaling Technology, 3868), diluted at 1:100 in the blocking solution, were applied overnight at 4 °C. After three washes with PBS, samples were incubated with fluorescently labeled secondary antibodies (Beyotime, A0468, A0423), diluted at 1:200 in blocking solution, for 1 h at room temperature in the dark. Following another round of PBS wash, nuclei were stained with DAPI for 5 min, and samples were mounted using antifade mounting medium. Images were captured using a fluorescence microscope (Nikon, A1R), and fluorescence intensity was quantified using appropriate image analysis software.
Stable cell line generation
To generate stable knockout (KO) cell lines for ISG15 and STING in LLC mouse cells, we utilized lenti-CRISPR vectors containing specific sgRNAs and selected with puromycin (2 μg/mL) for 7 days. Following selection, the cells were plated in 96-well plates to isolate single clones. The expression of the target proteins was assessed using western blotting. To confirm successful gene editing in the positive clones, we performed DNA sequencing of the genomic regions surrounding the targeted sgRNA sequences.
The sgRNA sequences targeting mouse ISG15:
5’-CACCGGTCCGTGACTAACTCCATGA-3’.
The sgRNA sequence targeting human STING:
5’-CACCGCACCTAGCCTCGCACGAACT-3’.
To generate stable expression of ISG15-WT, ISG15-C2, STING-WT and STING-4KR LLC cell lines, the HEK293T cells were transfected with the lentiviral expression plasmids together with pSPAX2 (12260, Addgene), pMD2G. At 48 h after transfection, lentiviruses were collected and used to infect the indicated cells in the presence of polybrene (4 µg/mL). The infected cells were selected with puromycin (2 µg/mL) for 7 days and the expression of the target protein was detected by western blot.
RNA interference
For the siRNA-mediated knockdown assay, the cells were transfected with the appropriate siRNAs using Lipofectamine 3000 (Thermo Fisher, L3000015), and scrambled siRNA was used as a control. After 48 h, the cells were harvested, and the efficiency of the knockdown was verified by western blot. The siRNAs targeting USP18 were a mixture of four siRNAs purchased from Dharmacon.
The sense sequences of siRNAs were as follows: 5’-CUGCAUAUCUUCUGGUUUA-3’; 5’-GGAAGAAGACAGCAACAUG-3’; 5’-GGACUACCCUCAUGGCCUG-3’;
5’-GCAAAUCUGUCAGUCCAUC-3’.
Enzyme-Linked Immunosorbent Assay (ELISA)
To quantify the secretion of IFN-β, cells were seeded in 6-well plates and transfected with the indicated plasmids (i.e., STING-WT, STING-4KR, ISG15-WT, or ISG15-C2) for 24 h. Subsequently, cells were stimulated with diABZi (1 μM) for the indicated time (0, 3, 6, and 12 h). Culture supernatants were harvested and centrifuged at 1000 × g for 10 min at 4 °C to remove cell debris. The concentration of secreted IFN-β was measured using a Human/Mouse IFN-β ELISA kit (Elabscience, E-EL-M0033L) according to the manufacturer’s instructions.
Xenograft animal model
C57BL/6 mice were purchased from GemPharmatech Co., Ltd. Six-week-old BALB/c male nude mice were used in the xenograft experiments. C57BL/6 mice were housed under conventional laboratory conditions at a room temperature maintained at 25 ± 2 °C with a relative humidity range of 55 ± 5% and a regular 12 h light/12 h dark cycle. The mice were fed with a standard animal pellet diet and allowed free access to water. LLC stable cells were implanted into the dorsal flanking sites of C57BL/6 mice at 5 × 106 cells in 200 µl PBS. To evaluate the efficacy of drugs, diABZi (3 mg/kg), TST (2.5 mg/kg, 5 mg/kg, 10 mg/kg) or a combination were intravenously injected when tumors reached 200 mm³, and this treatment was continued every 3 days until the end of the experiment. Mice were euthanized using carbon dioxide when tumors reached 1000 mm³, tumors and spleens were extracted for flow cytometry assay. Tumor volume was calculated using the formula: Volume = (longer diameter) × (shorter diameter) ²/2. Mice with poorly engrafting tumors were excluded from analysis.
Flow cytometry
Tumors were cut into small pieces (around 2 mm in diameter) and incubated with 1 mg/mL Collagenase D (Sigma-Aldrich, C5138), 0.2 mg/mL hyaluronidase (Yeasen, 20426ES60) and 0.1 mg/mL DNase I (Yeasen, 10608ES25) in RPMI1640 medium supplemented with 2% FBS for 0.5 h with continuous agitation. The digestion mixture was homogenized using repetitive pipetting and then passed through a 40 μm nylon filter. The single-cell suspensions were washed twice with PBS. The spleens were then gently mashed through a 40 μm nylon filter into a collection tube containing RPMI1640 medium to create a single-cell suspension. Red blood cells were lysed using RBC lysis buffer (Yeasen, 40401ES60) lysis buffer, followed by a thorough wash with PBS to remove any residual lysis buffer. After washing, the cells from tumors or spleens were stained with the corresponding antibodies, incubated at room temperature in darkness for 20 min. Flow cytometry data were acquired on the Attune NxT analyser (Thermo Fisher) and analyzed using FlowJo software.
Tissue microarray and immunohistochemistry (IHC)
The tissue arrays of Lung cancer samples were purchased from Biotechwell (ZL-LUC1601), and contained 60 lung cancer tissues and 60 paracancerous tissues. IHC staining was performed with anti-USP18 (Thermo Fisher, PA5-110555, dilution: 1:100) and anti-STING (Cell Signaling Technology, 13647, dilution: 1:100) antibodies. The ethics committee approved the IHC experiments on NSCLC patient tissues, and informed consent was obtained from all individual participants in the study (approval ID number: SHYJS-CP-1904008).
Screening of inhibitory fragments against USP18
USP18 activity was monitored using the substrate peptide-AMC (Z-Arg-Leu-Arg-Gly-Gly-AMC, Cat. No. 4027158, Bachem Bioscience). Experiments were performed in 384-well black non-binding plates (Cat. No. 3575, Corning) with a final reaction volume of 30 μL. The assay buffer contained 50 mM HEPES, pH 7.4, 0.01% Triton X-100 (v/v), 0.1 mg/mL BSA, and 2 mM DTT. USP18 was added to the plates at a final concentration of 50 nM. Enzyme reactions were initiated with 5 μL of peptide-AMC (final 100 nM) dissolved in the above assay buffer. Upon addition of peptide substrate, the fluorescence signals were monitored at 340 nm (excitation) and 450 nm (emission) with 3 min intervals in a 2104 EnVision Multilabel Plate Reader (PerkinElmer). To determine the IC50 values of TST, a series of 8-point, 1: 2 serial dilutions was performed from a highest starting concentration of 50 μM. The data were fitted using GraphPad Prism.
NMR-STD spectroscopy
Using STD-NMR to validate the binding of TST and USP18 and the TST (400 μM) was incubated with 10 μM protein (mUSP18) in solution containing 20 mM sodium phosphate, 100 mM NaCl, 2 mM DTT, pH 7.5, and 5% d6-DMSO. STD-NMR spectra were acquired on a Bruker Avance III-600 MHz spectrometer equipped with a cryogenically cooled probe (Bruker Biospin) at 25 °C. The saturation time was set at 2 sec. NMR data were acquired at 25 °C on a 600 MHz Bruker AVANCE III spectrometer. The 600 MHz spectrometer was equipped with a 5 mm TCI Cryoprobe.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 9.5.1 software. Quantitative data from multiple replicate experiments were analyzed by unpaired two-tailed Student’s t test or one-way ANOVA, and the data are presented as the mean ± SEM. Differences were considered significant when the P value was less than 0.05 (*P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001; ns, not significant). Each experiment was repeated independently with similar results.
Data availability
All of the source data supporting the findings of this study are available within this paper and/or from the corresponding author upon reasonable request. The human lung cancer data were derived from The Cancer Genome Atlas Research Network (https://cancergenome.nih.gov/). Source data are provided with this paper.
References
Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657–74.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91.
Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–7.
Ergun SL, Fernandez D, Weiss TM, Li L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell. 2019;178:290–301.e210.
Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92.
Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity. 2018;49:754–63.e754.
Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci Transl Med. 2020;12:eaay9013.
Jneid B, Bochnakian A, Hoffmann C, Delisle F, Djacoto E, Sirven P, et al. Selective STING stimulation in dendritic cells primes antitumor T cell responses. Sci Immunol. 2023;8:eabn6612.
Bu Y, Liu F, Jia QA, Yu SN. Decreased Expression of TMEM173 Predicts Poor Prognosis in Patients with Hepatocellular Carcinoma. PLoS ONE. 2016;11:e0165681.
Falahat R, Berglund A, Perez-Villarroel P, Putney RM, Hamaidi I, Kim S, et al. Epigenetic state determines the in vivo efficacy of STING agonist therapy. Nat Commun. 2023;14:1573.
Hines JB, Kacew AJ, Sweis RF. The Development of STING Agonists and Emerging Results as a Cancer Immunotherapy. Curr Oncol Rep. 2023;25:189–99.
Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114:E4612–20.
Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell. 2021;39:96–108.e106.
Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, et al. Structure-function analysis of STING activation by c[G(2’,5’)pA(3’,5’)p] and targeting by antiviral DMXAA. Cell. 2013;154:748–62.
Chang W, Altman MD, Lesburg CA, Perera SA, Piesvaux JA, Schroeder GK, et al. Discovery of MK-1454: A Potent Cyclic Dinucleotide Stimulator of Interferon Genes Agonist for the Treatment of Cancer. J Med Chem. 2022;65:5675–89.
Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439–43.
Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An orally available non-nucleotide STING agonist with antitumor activity. Science. 2020;369:eaba6098.
Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. 2018;29:viii712.
van der Zanden SY, Luimstra JJ, Neefjes J, Borst J, Ovaa H. Opportunities for Small Molecules in Cancer Immunotherapy. Trends Immunol. 2020;41:493–511.
Meric-Bernstam F, Sweis RF, Hodi FS, Messersmith WA, Andtbacka RHI, Ingham M, et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clin Cancer Res. 2022;28:677–88.
Wang C, Wang X, Veleeparambil M, Kessler PM, Willard B, Chattopadhyay S, et al. EGFR-mediated tyrosine phosphorylation of STING determines its trafficking route and cellular innate immunity functions. EMBO J. 2020;39:e104106.
Ni G, Konno H, Barber GN. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol. 2017;2:eaah7119.
Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING requires palmitoylation at the Golgi. Nat Commun. 2016;7:11932.
Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–98.
Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30:397–407.
Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. 2017;8:945.
Prabakaran T, Bodda C, Krapp C, Zhang BC, Christensen MH, Sun C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018;37:e97858.
Balka KR, Venkatraman R, Saunders TL, Shoppee A, Pang ES, Magill Z, et al. Termination of STING responses is mediated via ESCRT-dependent degradation. EMBO J. 2023;42:e112712.
Kuchitsu Y, Mukai K, Uematsu R, Takaada Y, Shinojima A, Shindo R, et al. STING signalling is terminated through ESCRT-dependent microautophagy of vesicles originating from recycling endosomes. Nat Cell Biol. 2023;25:453–66.
Gentili M, Liu B, Papanastasiou M, Dele-Oni D, Schwartz MA, Carlson RJ, et al. ESCRT-dependent STING degradation inhibits steady-state and cGAMP-induced signalling. Nat Commun. 2023;14:611.
Liu Y, Xu P, Rivara S, Liu C, Ricci J, Ren X, et al. Clathrin-associated AP-1 controls termination of STING signalling. Nature. 2022;610:761–7.
Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. J Interferon Cytokine Res. 2011;31:119–30.
Basters A, Geurink PP, Rocker A, Witting KF, Tadayon R, Hess S, et al. Structural basis of the specificity of USP18 toward ISG15. Nat Struct Mol Biol. 2017;24:270–8.
Malakhova OA, Zhang DE. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J Biol Chem. 2008;283:8783–7.
Okumura A, Pitha PM, Harty RN. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc Natl Acad Sci USA. 2008;105:3974–9.
Zhao C, Hsiang TY, Kuo RL, Krug RM. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc Natl Acad Sci USA. 2010;107:2253–8.
Liu G, Lee JH, Parker ZM, Acharya D, Chiang JJ, van Gent M, et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat Microbiol. 2021;6:467–78.
Xiong TC, Wei MC, Li FX, Shi M, Gan H, Tang Z, et al. The E3 ubiquitin ligase ARIH1 promotes antiviral immunity and autoimmunity by inducing mono-ISGylation and oligomerization of cGAS. Nat Commun. 2022;13:5973.
Bolado-Carrancio A, Lee M, Ewing A, Muir M, Macleod KG, Gallagher WM, et al. ISGylation drives basal breast tumour progression by promoting EGFR recycling and Akt signalling. Oncogene. 2021;40:6235–47.
Ren Y, Yang J, Ding Z, Zheng M, Qiu L, Tang A, et al. NFE2L3 drives hepatocellular carcinoma cell proliferation by regulating the proteasome-dependent degradation of ISGylated p53. Cancer Sci. 2023;114:3523–36.
Du X, Sheng J, Chen Y, He S, Yang Y, Huang Y, et al. The E3 ligase HERC5 promotes antimycobacterial responses in macrophages by ISGylating the phosphatase PTEN. Sci Signal. 2023;16:eabm1756.
D’Cunha J, Knight E Jr., Haas AL, Truitt RL, Borden EC. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc Natl Acad Sci USA. 1996;93:211–5.
Qu T, Zhang W, Yan C, Ren D, Wang Y, Guo Y, et al. ISG15 targets glycosylated PD-L1 and promotes its degradation to enhance antitumor immune effects in lung adenocarcinoma. J Transl Med. 2023;21:341.
Lin C, Kuffour EO, Fuchs NV, Gertzen CGW, Kaiser J, Hirschenberger M, et al. Regulation of STING activity in DNA sensing by ISG15 modification. Cell Rep. 2023;42:113277.
Qin Y, Wang M, Meng X, Wang M, Jiang H, Gao Y, et al. ISGylation by HERCs facilitates STING activation. Cell Rep. 2024;43:114135.
Cao D, Duan L, Huang B, Xiong Y, Zhang G, Huang H. The SARS-CoV-2 papain-like protease suppresses type I interferon responses by deubiquitinating STING. Sci Signal. 2023;16:eadd0082.
Fu Z, Huang B, Tang J, Liu S, Liu M, Ye Y, et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat Commun. 2021;12:488.
Wu B, Zhang B, Li B, Wu H, Jiang M. Cold and hot tumors: from molecular mechanisms to targeted therapy. Signal Transduct Target Ther. 2024;9:274.
Samson N, Ablasser A. The cGAS-STING pathway and cancer. Nat Cancer. 2022;3:1452–63.
Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21:548–69.
Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019;9:34–45.
Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016;76:6747–59.
Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016;14:282–97.
Dvorkin S, Cambier S, Volkman HE, Stetson DB. New frontiers in the cGAS-STING intracellular DNA-sensing pathway. Immunity. 2024;57:718–30.
Zhang Y, Yang Q, Zeng X, Wang M, Dong S, Yang B, et al. MET Amplification Attenuates Lung Tumor Response to Immunotherapy by Inhibiting STING. Cancer Discov. 2021;11:2726–37.
Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L, et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell. 2021;39:109–21.e105.
Mirzalieva O, Juncker M, Schwartzenburg J, Desai S. ISG15 and ISGylation in Human Diseases. Cells. 2022;11:538.
Swaim CD, Scott AF, Canadeo LA, Huibregtse JM. Extracellular ISG15 Signals Cytokine Secretion through the LFA-1 Integrin Receptor. Mol Cell. 2017;68:581–90.e585.
Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun. 2016;7:11496.
Sarkar L, Liu G, Gack MU. ISG15: its roles in SARS-CoV-2 and other viral infections. Trends Microbiol. 2023;31:1262–75.
Kang JA, Kim YJ, Jeon YJ. The diverse repertoire of ISG15: more intricate than initially thought. Exp Mol Med. 2022;54:1779–92.
Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. 2018;16:423–39.
Chen Z, Li Z, Wang Y, Dushimova Z, Gulnara K, Takeda S, et al. ISGylation: is our genome yearning for such a modification? Acta Biochim Biophys Sin. 2025;57:1743–57.
Ali MS, Tang YS, Lee HHY, Baker SC, Mok CKP. ISG-15, beyond its functions in the cell: a mini review. Cell Mol Life Sci. 2025;82:289.
Kooij R, Pol V, Gan J, van Doodewaerd BR, Sapmaz A, Campos Alonso M, et al. High-Throughput Synthesis and Screening of a Cyanimide Library Identifies Selective Inhibitors of ISG15-Specific Protease mUSP18. Angew Chem Int Ed Engl. 2025:e202510941.
Acknowledgements
Research reported in this publication was supported in whole or in part by the Shenzhen Science and Technology Program (KQTD20190929174023858), the National Natural Science Foundation of China (22577007) and the Guang Dong Basic and Applied Basic Research Foundation (2025A1515011104). All NMR-STD experiments were conducted at the Beijing NMR Center and the NMR facility of the National Center for Protein Sciences at Peking University.
Author information
Authors and Affiliations
Contributions
HH, DC and HZ conceived this project. HH, DC and BH designed the experiments. DC performed cell-based assays. DC and BH performed animal experiment. DC and XN performed NMR-STD experiments. ML and HZ conducted inhibitor screening. XF performed enzymatic kinetic assays. DC, HH and HB wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All animal experiments were performed according to the guidelines of the Animal Care and Use Committee of Peking University (AP0038001). This study was compliant with all of the relevant ethical regulations regarding animal research.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Boris Zhivotovsky
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, D., Huang, B., Fu, X. et al. ISGylation prevents autophagic degradation of STING and promotes antitumor immunity in lung cancer. Cell Death Dis 17, 271 (2026). https://doi.org/10.1038/s41419-026-08527-1
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-026-08527-1
