
Abstract
Rat sarcoma virus homolog (Rho) guanosine triphosphatases (GTPases) function as “molecular switch” in cellular signaling regulation processes and are associated with the pathogenesis of inflammatory bowel disease (IBD). This chronic intestinal tract inflammation primarily encompasses two diseases: Crohn’s disease and ulcerative colitis. The pathogenesis of IBD is complex and considered to include four main factors and their interactions: genetics, intestinal microbiota, immune system, and environment. Recently, several novel pathogenic components have been identified. In addition, potential therapies for IBD targeting Rho GTPases have emerged and proven to be clinically effective. This review mainly focuses on Rho GTPases and their possible mechanisms in IBD pathogenesis. The therapeutic possibility of Rho GTPases is also discussed.
Similar content being viewed by others
Facts
-
Rho GTPases are vital in numerous cellular events and associated with IBD pathogenesis.
-
Rho GTPases participate in traditionally believed pathogenic factors of IBD.
-
Roles of Rho GTPases in several novel pathogenic factors of IBD are also proposed.
Open Questions
-
Exactly what roles Rho GTPases play in IBD pathogenesis and what are the specific mechanisms?
-
How to optimize the experimental design to explore the connections between Rho GTPases and IBD?
-
Can we invent more secure and effective methods for IBD treatment by targeting Rho GTPases?
Introduction
Characterized by progressive and chronic relapsing intestinal inflammation, inflammatory bowel disease (IBD) is likely to occur early in life and is unfortunately incurable [1]. Despite having an unclear pathogenesis, studies have indicated the involvement of genetic variants, environmental changes, abnormal intestinal microbiota, and immune response dysregulation, and the interactions between these factors ultimately leads to the onset of IBD [2]. In recent years, many novel pathogenic factors have been identified that provide new insights into the disease [2].
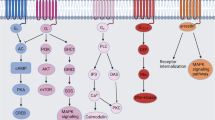
Rat sarcoma virus (Ras) homolog (Rho) guanosine triphosphatases (GTPases), a subgroup of the Ras superfamily, are guanosine triphosphate (GTP)-bound proteins [3]. The regulation of Rho GTPase activity depends on guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs). GEFs enhance the switch of Rho GTPases from guanosine diphosphate (GDP)-bound forms to GTP-bound forms to activate them, whereas GAPs and GDIs inactivate them by increasing bound GTP hydrolysis and binding directly to them, respectively [4] (Fig. 1). Rho GTPases are vital in the cytoskeleton regulation and gene expression [3, 5]. Recently, studies have found that Rho GTPases are associated with numerous cellular events, therefore being important in chronic diseases such as IBD. Within the Rho GTPase family, cell division cycle 42 (Cdc42), Ras-related C3 botulinum toxin substrate 1/2 (Rac1/2), and Rho family member A (RhoA) are the most thoroughly studied proteins.

Rho GTPases are members of the Ras superfamily, acting as molecular switches and consisting of inactive GDP-bound and active GTP-bound forms. In the inactive GDP-bound form, GEFs promote the activation of Rho GTPases by replacing GDP with GTP, thus Rho GTPases change its conformation and can bind and activate various downstream effectors in order to trigger multiple cellular functions. By contrast, GAPs increase the hydrolysis of GTP to GDP and inhibit the activation of Rho GTPases. GDIs interacts with the GDP-bound Rho GTPases, preventing its localization to the cell membrane and maintaining Rho GTPases in the inactive form. When GDIs released, GDP-bound Rho GTPases will anchor to the cell membrane and be further activated by binding to GEFs. GEFs guanine nucleotide exchange factors, GAPs GTPase-activating proteins, GDIs guanine nucleotide dissociation inhibitors.
In this review, the functions of Rho GTPases in the pathogenesis of IBD and their possible mechanisms are summarized. The therapeutic strategies associated with Rho GTPases that may be beneficial to IBD patients and their clinical applied prospect are also discussed.
Genetics
In recent years, over 200 loci have been found to be implicated in IBD. Among these loci, most increase the risk of both IBD subtypes, whereas several are unique to either CD or UC [6]. Rho GTPases are involved in this pathogenic process (Table 1).
Genes encoding Rho GTPases
Rac2 deficiency is associated with worsening colitis in mice treated with Citrobacter rodentium, implying a possible effect of this protein on human intestinal diseases development [7]. This hypothesis is supported by the fact that several Rac2 single nucleotide polymorphisms (SNPs) may function in the predisposition and susceptibility to CD [8, 9]. Interestingly, whether the association between Rac2 and IBD is based on the function of Rac2 in the NADPH complex is controversial [7, 8]. The Rac1 SNPs rs10951982 and rs4720672 are associated with UC [10], and another Rac1 SNP, rs34932801, was found to lead to poorer thiopurine treatment effect in adult CD patients [11]; however, the association between Rac1 SNPs and UC was not observed in another study [12]. In addition, mice with a genetic deletion of RhoA in intestinal epithelial cells (IECs) exhibited spontaneous chronic intestinal inflammation, and RhoA signaling was found to be downregulated in CD patients [13].
Other genes associated with Rho GTPases
Myo9B
Genetic variations in myosin IXB (Myo9B), a Rho GAP encoding gene, are associated with IBD [14, 15]. This association might be partly explained by the dysfunction of Myo9B, leading to an abnormal level of active Rho, subsequently influencing intestinal epithelial wound healing and disrupting the formation of tight junctions (TJs) [16].
Pggt1b
Protein geranylgeranyltransferase type-I (GGTase-I) transfers a 20-carbon geranylgeranyl lipid to substrate proteins, including Rho GTPases [17]. This process is a post-translational modification of Rho GTPases called prenylation. Genetic deletion of Pggt1b, a gene encoding GGTase-I, in murine IECs results in epithelial injuries and spontaneous gut inflammation, which can be significantly ameliorated by activating RhoA signaling [13]. Loss of Pggt1b in the T cells of mice also causes spontaneous colitis via impaired RhoA signaling, and the intestinal T cells of IBD patients have lower Pggt1b level [18].
ATG16L1
The SNP T300 of autophagy-related protein 16-like protein 1 (ATG16L1) contributes to CD. In mice, defective autophagy stimulates Rac1 to restrain dendritic cells (DCs) migration [19] and inhibits RhoA activity to affect IECs migration to restore the intestinal mucosa [20].
NCF2
The neutrophil cytosolic factor 2 (NCF2) gene is considered a very early onset IBD-specific susceptibility gene. A missense variant of this gene causes abnormal binding of its p67phox product to Rac2, resulting in a damaged function of oxidase and thus contributing to the development of IBD [8].
TTC7A
Lemoine et al. [21] identified biallelic missense mutations in tetratricopeptide repeat domain 7 A (TTC7A) that inappropriately activated RhoA signaling in IBD patients with an immune deficiency.
TAGAP
A study showed that T cell activation of the Rho GTPase-activating protein (TAGAP) SNP rs212388 negatively correlated with the severity of anal disease in CD, and hypothesized that the protective effect may be attributed to impaired Rho GTPase inactivation [22]. This supposition requires more studies to authenticate.
Intestinal microbiota
Studies have shown for decades that the intestinal microbiota is essential to IBD development [23]. Intestinal homeostasis partly depends on the mutual influence of intestinal microbiota and the immune system [24] and Rho GTPases are involved in the interaction. For example, segmented filamentous bacteria (SFP), which are commensal gut bacteria in several animal species, induce the differentiation of T helper 17 (Th17) cells in the mouse intestine, which requires Cdc42-dependent endocytosis [25].
Compared to normal person, IBD patients intestinal microbiota communities are markedly different, with decreased abundance of Firmicutes and Saccharomyces cerevisiae and increased abundance of Proteobacteria (especially Enterobacteriaceae) and Candida albicans [26, 27]. Additionally, the microorganisms expanded in IBD patients can experimentally induce colitis, whereas those contracted are able to ameliorate it [28]. Escherichia coli, especially adherent-invasive E. coli, participate in IBD, and adherent-invasive E. coli strains share similarities with extraintestinal pathogenic E. coli such as uropathogenic E. coli [29]. Cytotoxic necrotizing factor 1, a protein produced by uropathogenic E. coli, modifies Rac2 to restrain it in an active state. Active Rac2 then induces receptor-interacting proteins 1 and 2 (RIP1 and RIP2) to trigger nuclear factor-kappa B (NF-κB) and interferon regulatory factor (IRF) pathways, driving a protective immune response in mammalian cells [30]. Although the connection between enteropathogenic and enterohemorrhagic E. coli and IBD has seldom been studied, it has been disclosed that their pathogenesis involves the activation of downstream substrates Cdc42, Rac1, and RhoA as RhoGEFs [31]. Interestingly, with a similar mechanism, C. rodentium infects colon cells, inducing IBD-linked inflammation in mice [32]. Moreover, Salmonella spp. (family Enterobacteriaceae) produce the pro-inflammatory protein SopE to activate Rac1 and Cdc42, subsequently inducing nucleotide oligomerization domain-containing protein 1 (NOD1)/RIP2/NF-κB signaling, causing an inflammatory response in host cells [33, 34]. Taken together, these studies suggest that Rho GTPases participate in inflammation induced by expanded Enterobacteriaceae in the gut mucosa, thus contributing to the onset of IBD. Additionally, Clostridium difficile influences the deterioration and recurrence of IBD [35] probably because its enterotoxins inactivate Rho, Rac, and Cdc42 [ref. [36]]; however, whether this bacterium is associated with IBD development remains unknown. Accordingly, the role and mechanism of action of aforementioned bacteria in IBD require further research.
Immune system
The innate immune system
The innate immune system includes non-classical (intestinal epithelial barrier) and classical factors (DCs, macrophages, monocytes, and neutrophils). Dysregulation of these protective components by Rho GTPases is associated with IBD [2, 37] (Table 2).
Intestinal epithelial barrier
Dysregulation of the intestinal epithelial barrier, including abnormal mucous layer [38], damaged structures and functions of the IECs [36], and decreased expression of antimicrobial peptides [39, 40], have been reported in IBD.
Increased TJs permeability, altered cytoskeletal rearrangement, and abnormal cell death in IECs are strongly associated with IBD [13] and Rho GTPases influence IECs through all these aspects. Interestingly, both the overactivation and inactivation of Rho GTPases damage TJs by directly affecting TJ proteins and indirectly affecting the F-actin cytoskeleton intimately associated with TJs [36, 41]. Reduction of cortactin, an actin-binding protein reduced in IBD patients, upregulates RhoA/Rho-associated protein kinase (ROCK) signaling to phosphorylate myosin light chain, which contracts actomyosin leading to altered TJs and increased epithelial permeability [42]. As mentioned above, MYO9B deficiency probably results in the abnormal activation of Rho to interrupt the formation of TJs [16]. Reduced RhoB levels are also associated with impaired TJs that contribute to UC [43] and increased RhoB expression combined with decreased Cdc42 expression ameliorates dextran sulfate sodium (DSS)-induced increased intestinal permeability and IEC apoptosis [44]. Impaired RhoA prenylation and signaling in IECs found in IBD patients drive altered cytoskeletal rearrangements [13]. The cytokines interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) are increased in IBD patients [45], inducing TJ proteins endocytosis and IECs apoptosis through the RhoA/ROCK pathway and the activation of Rac1 triggering the c-Jun N-terminal kinase (JNK) pathway, respectively [46, 47]. Clostridium difficile toxins induce IEC apoptosis in a Rho GTPase-dependent manner [48, 49]. Defective mucosal healing is also a characteristic of IBD. RhoA deficiency in IECs is related to CD, with defective IECs migration and intestinal injuries repairment [20], while Rac1 inhibition suppresses IECs migration and inhibits the recovery of colonic wounds in DSS-induced mice [50]. Cdc42 also contributed to mucosal healing in DSS-treated mice [51].
Compared to UC, reduced expression of inducible β- and α-defensins secreted by Paneth cells has been observed in patients with colonic CD [39, 40]. Studies also identify that α-defensins inhibit Clostridium difficile toxins by protecting Rac1 from glucosylating [52, 53], while inducible β-defensins improve intestinal wound healing in mice by activating RhoA/ROCK signaling [54].
DCs and macrophages
Dendritic cells and macrophages play both similar and different roles in maintaining intestinal homeostasis and inducing an immune response. DCs mainly function as antigen-presenting cells (APCs), as they uptake and present antigens to regulate immune responses, and participate in lymphocyte gut homing [55]. Although macrophages are also considered APCs, their major functions are engulfing and clearing pathogens and apoptotic cells to resolve inflammation [56]. The onset of IBD can be partly attributed to DCs and macrophages incorrect recognition of commensal microorganisms, their abnormal activation and migration, and the subsequent imbalance between immune tolerance and activation [55, 57]. Impaired clearance of microorganisms and apoptotic cells and inflammatory resolution have also reported at the onset of IBD [58].
Endocytosis is associated with antigen uptake and engulfing, which have been shown to depend on Cdc42 and Rac regulation in DCs [59, 60]. In macrophages, this process varies according to the receptor. Cdc42 and Rac1 are recruited to the plasma membrane where Fc gamma receptor (FcγR) is clustered, which then recruits Wiskott-Aldrich syndrome protein to activate the actin-related protein 2/3 complex. The latter activates p21-activated kinase (PAK) and phosphatidylinositol-4-phosphate 5-kinase to regulate FcγR-mediated endocytosis in macrophages [61, 62]. Rac2, RhoG, and RhoC may also participate in this process [63, 64]. Studies have demonstrated that Rac1 promotes toll-like receptor (TLR) 4-mediated bacterial phagocytosis [65] while RhoA and RhoG are required for complement receptor 3-mediated phagocytosis [66]. Furthermore, apoptotic cell engulfing mediated by integrin is enhanced by Rac1, Cdc42, and RhoG, and inhibited by RhoA [67, 68]. DCs require Rac, Cdc42, and Rho involvement to accomplish migration and antigen presentation; the former requires the dynamic regulation and cooperation of Rho GTPases. Rac1/2 and Cdc42 are indispensable in forming lamellipodia and filopodia at the leading edge, respectively, while RhoA controls contractility at the cell rear and migration [69, 70]. In macrophages, the maturation of phagosomes is promoted by Rho/ezrin-radixin-moesin (ERM) protein [71] and macrophages then pretend to exert a microbicidal function. Rac2 is an essential component of the NADPH complex [8] but Rac1 has also been shown to be involved. After phagocytosis, Rac1 is activated and recruited to the plasma membrane, causing the NADPH complex activation and reactive oxygen species generation to remove microorganisms[72, 73]. Furthermore, macrophage complement is a process that includes monocyte adhesion, migration, and differentiation. RhoA and Cdc42 are required for monocyte adhesion, whereas migration requires Rac, RhoA, and Cdc42 participation [74]. The migration mechanism is similar to that aforementioned. Another study found that RhoA functioned in both the front and back of cells, contributing to leading edge construction and governing turning while stimulating actomyosin contractility, respectively [75].
In brief, these studies imply that Rho GTPases may contribute to abnormal DCs and macrophage functions induce IBD pathogenesis; however, direct evidence is lacking to confirm the connection between them.
Neutrophils
Abnormal activation, recruitment, and function of neutrophils have been identified in IBD pathogenesis [76, 77]. In neutrophils, Rho GTPases play roles similar to that in DCs and macrophages, i.e., they regulate migration, phagocytosis, and microbe clearing [78, 79], in addition to the degranulation process [80]. A few findings directly demonstrate that dysfunction of Rho GTPases in neutrophils contributes to IBD. Rac1 deficiency in mouse neutrophils ameliorated DSS-induced colitis, probably by inhibiting neutrophil recruitment and migration to the inflammation site [10]. Failure of Rac2 to bind the NADPH complex in neutrophils results in susceptibility to CD [8].
The adaptive immune system
The adaptive immune system includes T cell-induced cellular immunity and B cell-induced humoral immunity, and the involvement of the former in IBD has been extensively explored [2]. Rho GTPases are crucial for regulating lymphocyte biology [81].
T cells
Abnormal T cell activation, accumulation, differentiation and inappropriate apoptosis are considered IBD immunopathogenesis [2]. Rho GTPases are crucial in adaptive immune response because of their wide control of T cell basic functions (Fig. 2).

Abnormal T cells activation, accumulation, differentiation of different T cells subsets and inappropriate apoptosis are linked to IBD immunopathogenesis. Rho GTPases are involved in T cells basic functions. Migration of T cell initiates with enhanced adhension mediated by Rac and RhoA, the former also participates in lymphocyte homing. Cdc42, Rac and RhoA regulate T cell movement with different mechanisms. Effective T cell activation requires stable conjunction between T cell and APC and the signaling transduction. RhoA and Rac contribute to both the process while Cdc42 enhances the former process and inhibits the latter. RhoH and RhoG are also involved in signaling transduction, mainly exerting a positive and negative function, respectively. Rho GTPases also help lymphocytes differentiate into diverse effector or regulatory T cell subsets to participate in inflammatory and anti-inflammatory processes. Cdc42, Rac and RhoA are involved in the Fas/FasL induced apoptosis of T cell, both in the induction and late stage. WASp Wiskott-Aldrich syndrome protein, Arp2/3 actin-related protein 2/3, IS immunological synapse, ERM protein ezrin-radixin-moesin protein, JNK c-jun N-terminal kinase, AP-1 activate protein-1, NFAT nuclear factor of activated T cells, NF-κB nuclear factor of kappa B, IL-12 interluekin-12.
Migration
Migration of T cells is a sophisticated process. The mechanism of this event is similar, requiring cooperation between Rac, Cdc42, and RhoA [75, 82]. In addition, there is accumulating evidence that Rho GTPases contribute to T cell recruitment. C-X-C motif chemokine ligand 12 (CXCL12) stimulates the Rac1 guanine nucleotide exchange factor (Vav1)/Rac1 pathway to upregulate integrin α4β1 expression to promote T cell adhesion [83], a process that might be RhoA-dependent [84]. Without Rac2, T cells show defective response to CXCL12 and chemokine ligands 19 and 21 and are less efficient at homing to lymph nodes [85]. Once T cells are effectively activated, Rac and RhoA start to inhibit migration. The function of Rac here depends at least partly on the dephosphorylation of ERM proteins [82, 83]. RhoH also negatively regulates T-cell migration [86]. In IBD patients, T cells isolated from gut tissues showed decreased expression of Pggt1b, which led to dysfunction of RhoA in IECs and T cells in mice, promoting CD4 + T cell accumulating at the intestine and aggravating colitis [13, 18].
Activation
The interaction between T cells and APCs, an essential process for triggering T cell activity, is induced by T cell antigen receptor (TCR) and co-stimulatory molecules, followed by an immunological synapse formation, where active Rac1, Cdc42, and RhoA are observed [87]. Additionally, Rac enhances the connection between T cells and APCs by participating in the dephosphorylation of ERM proteins [88], while inactive Cdc42, Rac, and RhoA impair the activity of co-stimulatory molecules [89, 90]. The upcoming signal transduction process also requires Rho GTPases involvement. The activation of JNKs and transcription factors activator protein-1, nuclear factor of activated T cells, and NF-κB is associated with Rac1[refs. [91, 92]]. Rac1 also influences Ca2+ influx into T cells [93], an event involved in the maintenance of this interaction [94] and in cytoskeleton reorganization during signal transduction [94, 95]. The latter probably depends on the phosphoinositide 3 kinase (PI3K)/Vav1/Rac or Rho pathway [95]. Inhibition of RhoA leads to damaged Ca2+ influx [96] and RhoA is involved in T cell activation through the regulation of mitochondrial function [97]. Cdc42 deficiency results in the overactivation of murine T cells [98]. Moreover, RhoG and RhoH have been reported to regulate the TCR signaling pathway, mainly as the suppressor and enhancer, respectively [86, 99].
Differentiation
After activation, T cells differentiate into various subtypes that perform different functions. Generally, CD was considered a Th1 condition and UC an atypical Th2 condition. With the observation of other subsets engagement, such as Th17 and regulatory T cells (Tregs), this view has changed [2]. As mentioned above, Cdc42 is required for SFB-induced Th17 differentiation in mice [25]. Deficiency of Cdc42 promotes the differentiation and pathogenicity of Th17 and disrupts Tregs differentiation and stability, aggravating DSS-induced colitis [100]. Deficiency of Cdc42 in mice also interrupts Th2 differentiation [101]. RhoA induces Th2 and Th17 differentiation, and the former occurs partly via defective T cell metabolism [97, 102]. However, another study suggested that decreased activation of RhoA, because of its abnormal membrane location, contributes to a shift from Th1- to Th2-modulated immunity through the inhibition of p38 [103]. In Th1 cells, Rac2 triggers the NF-κB and p38 signaling pathways to upregulate IFN-γ expression [104]. As for RhoH, a study found a remarkably higher differentiation level of Th1 than Th2 [105]. Additionally, decreased RhoB activity promotes Tregs differentiation [106].
Apoptosis
Cdc42 activates JNK and upregulates Fas and Fas ligand (FasL) expressions to induce T cell apoptosis in secondary lymphoid organs [107]. Cdc42 and Rac are associated with both the induction and late stages of Fas/FasL-induced apoptosis but do not directly participate in it [108], and activated Rho/ROCK signaling is also involved in the process by phosphorylating ERM proteins [109]. Interestingly, decreased RhoA activation has been reported to contribute to T-cell apoptosis [110]. Activated Rac1 with Vav1 restrains the NF-κB and signal transducer and activator of transcription 3 (STAT3) signaling pathways to induce T cell apoptosis to alleviate clinical symptoms in IBD patients [111, 112].
Cytokines
Cytokines include interleukins (ILs), colony stimulating factors (CSFs), IFNs, TNFs, transforming growth factor-β family members, growth factors, and chemokines. The role of these molecules has been gradually explored in IBD [113]. Cdc42 has been shown to downregulate IL-4, IL-10, TNF-α, and IFN-γ expression both in the colon tissues and serum of 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced mouse models to regulate inflammation [114]. Conversely, TNF-α downregulated Cdc42, while CdC42 overexpression reversed the intestinal epithelial barrier damage due to TNF-α in Caco2 cells [115]. RhoA/ROCK pathway activation stimulates the production of TNF-α and IL-1β, and there is a positive correlation between RhoA and TNF-α in the intestinal inflammatory tissues of CD patients [116]. Inhibition of the RhoA/ROCK pathway also disrupted TGF-β-induced fibrosis in human [117]. RhoA dysfunction in T cells promoted IL-1β, IL-17A, TNF-α, and IFN-γ expression in mice developing spontaneous colitis [18] and blocking RhoA activation reduced IL-2 production [96]. In addition, Horowitz et al. [118] identified that TNF-α/lipopolysaccharide could activate the RhoA/ROCK pathway and contribute to IBD. Rac1 activity is upregulated by TGF-β in Caco2 cells to promote migration and epithelial restitution [119]. Rac1 can also be activated by CSF, IL-6, IL-8, and TNF-α [120]. Inhibition of Rac1 decreases IL-8 level, thus regulating immunocyte migration to the intestinal tract to maintain local remission [120, 121]. Rac1 also suppresses tryptophan-induced reduction of TNF-α, which damages intestinal barrier [122]. In DSS-treated mice, Rac1 deficiency in macrophages and neutrophils results in lower levels of IL-1β, IL-12, and TNF-α [10] whereas a Rac1 inhibitor blocks engulfment and cell motility protein 1-mediated downregulation of IL-1α, IL-6, IL-12B, IL33, TNF-α, and TGF-β to disrupt its protective function in DSS-induced mice [50]. Furthermore, Rac2 is associated with IFN-γ and IL-17A production, suggesting that impaired Rac2 function leads to IBD development [7, 104], and Rac2 inhibition can be used to treat IBD patients [112].
Environment
Smoking has been considered as a risk factor for CD but a protective factor for UC [123, 124]. Nicotine, a component of cigarettes, activates α7 nicotinic acetylcholine receptors (α7nAChRs) on plasmacytoid DCs to disrupt their migration into colonic mucosa. This protects cells from UC via activated JAK2-STAT3 signaling and the subsequent degradation of Rac1 induced by non-apoptotic caspase-3 [ref. [124]]. However, whether the mechanism of smoking-induced CD involves Rho GTPases remains unclear. The connections between Rho GTPases and other environmental factors contributing to the onset of IBD [124] have been scarcely explored. A study identified that the activation of vitamin D receptor via the RhoA/Rho kinase pathway partly inhibited the inflammatory response in benign prostatic hyperplasia [125] and myocardial ischemia-reperfusion injury [126]. In addition, vitamin D deficiency is associated with IBD [123], suggesting a possible involvement of Rho GTPases in vitamin D-deficiency-induced IBD. Therefore, there is an urgent need to investigate this association.
Novel pathogenic components
In addition to the aforementioned classical pathogenic factors, several novel components in the pathogenesis of IBD, such as pattern recognition receptors (PRRs), non-coding RNAs (ncRNAs), angiogenesis, and fatty acids, have gradually drawn researchers’ attention [2, 127, 128]. Rho GTPases are also participants in these pathogenic processes.
PRRs
Pattern recognition receptors, including receptor families such as TLRs and NOD-like receptors (NLRs), recognize pathogen-associated molecular patterns and damage-associated molecular patterns to initiate an immune response and regulate other events [129].
TLRs
TLRs are transmembrane proteins that participate in innate immunity. Upon activation, TLRs trigger downstream signaling cascades to activate NF-κB, which results in pro-inflammatory mediators secretion to regulate adaptive immunity. The expression and function of TLRs are irregular in IBD patients, suggesting that TLRs are both positively and negatively associated with IBD [130]. Rho GTPases take part in TLR-mediated immune response. In monocytes, Rac1 and RhoA are vital in TLR2-induced NF-κB activation via a Rac1/PI3K/protein kinase B pathway [131] and at least partially through atypical protein kinase C [132]. Rac1 also participates in the TLR1-mediated signaling pathway in epithelial cells [133]. Rac, RhoA, and Cdc42 are all activated after TLR4 is stimulated to promote uptake by macrophages [134, 135]. TLR7/9-mediated production of type I IFN requires Rac involvement in DCs [136]. In macrophages, following TLR activation, Rac acts as a positive regulator of phagocytosis and oxidative burst because of the downregulation of TNF-α-induced protein 8-like 2 [ref. [137]] whereas RhoB enhances the expression of pro-inflammatory mediators by binding to major histocompatibility complex class II [138]. In addition, Cdc42 is involved in TLR-activated DCs-T cells interactions [139].
NLRs
NLRs are a group of cytoplasmic proteins whose downstream signaling pathways mainly result in NF-κB and caspase-1 activation [140]. Mutations and dysfunction of NLRs are associated with IBD [140]. Rho GTPases are implicated in the functions exerted by NLRs. After activation, NOD1/2 starts to form a complex (the nodosome) containing Rho GTPases, which is necessary for the activation of the complex itself and NF-κB eventually [34, 141]. Interestingly, Rac1 negatively regulates the NOD2-mediated NF-κB activation and IL-8 induction [142]. In mouse macrophages, the NLR family pyrin domain containing protein 3 (NLRP3) inflammasome senses Rac2 activation and is then phosphorylated by p21-activated kinase 1/2, the substrate of Rac2, to induce the secretion of IL-1β, which contributes to microorganisms clearance [143]. Rho A is also a positive regulator of NLRP3 in Caco2 cells [144]. Rac1 and RhoB are associated with the NLRP3 inflammasome as well [145, 146].
ncRNAs
MicroRNAs (miRNAs, miR) are among the most extensively studied ncRNAs, as they can silence gene expression by regulating messenger RNAs (mRNAs) translation or degradation [147]. Associations between miRNAs and IBD have been proposed [148,149,150]. For instance, MiR-15a inhibits Cdc42 expression in pediatric IBD patients, resulting in a damaged intestinal epithelial barrier [116]. MiR-21 targeting and regulation of RhoB expression, which is upregulated in UC, disrupts the intestinal epithelial barrier by damaging TJs [43]. Cdc42 is also regulated by miR-21 in the intestinal epithelial barrier regulation [44]. MiR-31-3p alleviates colitis in IBD mouse models, possibly by targeting and negatively regulating RhoA expression, thus reducing the levels of several cytokines in colonic epithelial cells [151]. Circular RNAs (circRNAs), another class of ncRNAs with covalently closed circles, interact with miRNAs and proteins to perform their functions [152]. The circRNA homeodomain-interacting protein kinase 3 (circHIPK3) was recently identified as a sustainer of intestinal epithelium homeostasis and was reported to be decreased in patients with IBD [153]. CircHIPK3 is, at least partially, directly connected to and thus inhibits miR-29b availability to enhance the expression of Rac1 and Cdc42, promoting epithelial recovery in wounded IECs as well as intestinal epithelium renewal in mice [153].
Angiogenesis
One crucial event in chronic inflammatory diseases is angiogenesis, a process enhanced in the intestinal inflamed site of IBD patients, and vascular endothelial growth factor (VEGF) and IL-8 seem to act as inducers of this process [154]. Rho GTPases are implicated in this process, mainly in controlling cell migration [155]. Activated Rac1, Cdc42, and Rho, probably via steroid receptor coactivator/focal adhesion kinase signaling, participate in VEGF-induced angiogenesis [156, 157]. Inhibition of Rac1 and RhoA cause defected IL-8-induced endothelial cell migration [158]. Nevertheless, future studies should address the direct evidence of Rho GTPases as participants in angiogenesis, contributing to disclose the molecular development of IBD through more mechanistic studies.
Fatty acids
Whether the intake of fatty acids is beneficial or detrimental in IBD is still controversial, as several studies have achieved different conclusions [127, 128]. Turk et al. [159] found that n-3 polyunsaturated fatty acids damaged epidermal growth factor receptor-mediated Rac1 and Cdc42 activation to delay intestinal wound healing in DSS-treated mice.
Clinical Prospect of Rho GTPases
Emerging studies have shown that Rho GTPases act as therapeutic targets in many diseases, including cancer [160], cardiovascular disease [161], and Alzheimer’s disease [162]. As Rho GTPases have a great influence on IBD pathogenesis, their role in IBD treatment has been gradually explored (Table 3).
Cdc42
Antrum mucosal protein 18 (AMP-18), which is produced by antral mucosal epithelial cells [163], positively regulates intestinal epithelial TJs stability and mucosa recovery [164]. Cdc42 is activated after treating with AMP-18 and contributes to the formation of TJs [165]. The anti-TNF antibody infliximab, for example, has been proven to be effective in the induction and maintenance of clinical remission in IBD patients [166,167,168,169]. Recently, a study found that Cdc42 is upregulated in patients with UC in response to infliximab [170], but the mechanism underlying this upregulation still requires further studies.
Rac1
Nsc23766 is a Rac1-specific inhibitor that targets Rac1 binding and activation by RhoGEFs [171]. By moderately blocking Rac1 activation, Nsc23766 enhances phagocyte function in CD patients regarding the ability to clear the microorganisms [120]. Thiopurines is an effective drug to treat IBD [172] and 6-thioguanine triphosphate, one of the main metabolic products that exerts such function, directly binds to Rac1 to inhibit its activation [173]. A clinical trail found that IBD patients response to thiopurine therapy had a lower Rac1 level or activity [174]. Inactivated Rac1 has distinct effects on various cells. Inactivated Rac1 improves the defective migration of DCs in CD patients with the ATG16L1 mutation [19]. In monocytes, when Rac1 activity is inhibited within a certain range, phagocytic function is enhanced [120] but in T cells this change results in a reduced immune response and increased T cell apoptosis due to the suppression of T cell-APC conjugation and impaired NF-κB and STAT-3 signaling, respectively [111, 112]. In Caco2 cells, the migration of macrophages and neutrophils to the gut is suppressed by the downregulation of IL-8 expression [121]. In addition, NK cells, whose role in IBD has been recently discovered, undergo enhanced induction of apoptosis mediated by caspase-9 [ref. [175]]. Overall, thiopurines seems to function as Rac1 inhibitors in IBD treatment. Another kind of clinical drug statins are also demonstrated to suppress Rac1 and RhoA [176] and alleviate CD patients’ inflammation [177]. Moreover, a few clinical trials, working on estimating the efficacy and safety of statins in UC patients, are about to start. However, the activation of Rac1 also shows a possible benefit in IBD patients because active Rac1 improves IECs migration and mucosal healing [50, 119].
RhoA
Inhibitors of RhoA and its downstream effector ROCK are C3 exoenzyme [178] and Y-27632 [179], respectively. Both C3 exoenzyme and Y-27632 disrupt arginase expression in human intestinal microvascular endothelial cells, which likely promotes intestinal microvascular endothelial function [118]. Y-27632 has been widely used in previous studies. By interrupting the RhoA/ROCK pathway, Y-27632 has been shown to improve TNBS-induced colitis in rat by inhibiting NF-κB activation [116], alleviating DSS-induced intestinal inflammation by regulating cell function and cytokine secretion [180], and repress fibrogenesis by blocking RhoA/ROCK/MYLK/SRF signaling in colonic myofibroblasts, representing a new therapy for intestinal fibrosis in IBD patients [117]. Furthermore, several substances have shown therapeutic potential by affecting RhoA/ROCK signaling. Oxymatrine, a Chinese herb extract [181], and umbilical cord- and placenta-derived mesenchymal stem cells [182] suppress this pathway and thus alleviate intestinal inflammation in DSS-treated mice [181] and inhibit intestinal fibrosis [182], respectively. However, blockade of signaling can also be detrimental. Partially hydrolyzed guar gum, a product of guar gum, was observed to ameliorate colitis in mice [183]. Recently, a study has found that it improves wound healing in colonic epithelial cells, which is inhibited by Y-27632 [ref. [184]]. Thus, RhoA/ROCK signaling pathway regulation might provide a new insight into IBD treatment. β-glucan, which upregulates the expression of Cdc42, Rac1, and RhoA, enhances cell migration and proliferation, and ultimately promotes intestinal mucosal wound recovery, also represents a therapeutic potential for IBD via mucosal healing [185].
Although so far Rho GTPases targeted drugs haven’t been widely applied and tested in clinic, many studies have proven that several IBD therapies influence the signal transduction pathway of Rho GTPases and their regulators are effective in improving intestinal inflammation in animal experiments. Additionally, inhibition of Rho GTPases are used in other immunological disease treatments, such as rheumatoid arthritis[186] and asthma [187]. Therefore, regulation of Rho GTPases might be clinically useful in treating IBD but still waits for further researches and larger clinical trials.
Conclusions
Accumulating evidence has revealed that Rho GTPases are associated with IBD pathogenesis. According to the aforementioned studies, whether Rho GTPases have a beneficial or detrimental effect on IBD development remains unclear. Studies of Rho GTPases in IBD conducted so far have been mostly based on static conditions using cells and/or animal models, and have been limited by the deficiency of clinical samples. As numerous cell events depend on Rho GTPase dynamics, it is difficult to draw a connection between Rho GTPases and the development of IBD.
Nevertheless, therapies targeting Rho GTPases and their regulators are promising. Rho GTPase inhibitors have been widely studied and have been shown to be beneficial in alleviating disease. In addition, the mechanism of thiopurine therapy, one of the most used treatments in the clinical setting, requires Rho GTPase inactivation. However, several studies pointed out that Rho GTPase activation could also be a promising therapy. Together with the fact that extensive basic fundamental events require Rho GTPases involvement, general inhibition of Rho GTPases might not only provide the desired therapeutic effect, but also be detrimental to the individual [188]. Additionally, thiopurine therapy is limited by its long-term treatment and potential side effects. Thus, new drug discovery and fewer adverse reactions are future research directions for therapies targeting Rho GTPases. Furthermore, future studies should also focus on other Rho GTPases that have been implicated in the onset of IBD. In summary, Rho GTPases are associated with the pathogenesis of IBD, and further clinical studies are required to confirm and explore the underlying mechanisms. The Clinical Prospect of Rho GTPases is promising and require further studies on safety improvements and novel drug research development.
Data availability
All data included in this review are available upon request by contact with the corresponding author.
References
Agrawal M, Spencer EA, Colombel JF, Ungaro RC. Approach to the Management of Recently Diagnosed Inflammatory Bowel Disease Patients: A User’s Guide for Adult and Pediatric Gastroenterologists[J/OL]. Gastroenterology. 2021;161:47–65.
De Souza HSP, Fiocchi C. Immunopathogenesis of IBD: current state of the art[J/OL]. Nat Rev Gastroenterol Hepatol. 2016;13:13–27.
Jaffe AB, Hall A. Rho GTPases: biochemistry and biology[J/OL]. Annu Rev Cell Dev Biol. 2005;21:247–69.
Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators[J/OL]. Nat Rev Mol Cell Biol. 2016;17:496–510.
Hall A. Rho GTPases and the actin cytoskeleton[J/OL]. Science. 1998;279:509–14.
De Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease[J/OL]. Nat Genet. 2017;49:256–61.
Fattouh R, Guo CH, Lam GY, Gareau MG, Ngan BY, Glogauer M, et al. Rac2-deficiency leads to exacerbated and protracted colitis in response to Citrobacter rodentium infection[J/OL]. PloS One. 2013;8:e61629.
Muise AM, Xu W, Guo CH, Walters TD, Wolters VM, Fattouh R, et al. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2[J/OL]. Gut. 2012;61:1028–35.
Sironi M, Guerini FR, Agliardi C, Biasin M, Cagliani R, Fumagalli M, et al. An evolutionary analysis of RAC2 identifies haplotypes associated with human autoimmune diseases[J/OL]. Mol Biol Evol. 2011;28:3319–29.
Muise AM, Walters T, Xu W, Shen-Tu G, Guo CH, Fattouh R, et al. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase RAC1 are associated with ulcerative colitis[J/OL]. Gastroenterology. 2011;141:633–41.
Koifman E, Karban A, Mazor Y, Chermesh I, Waterman M, Almog R, et al. Thiopurine effectiveness in patients with Crohn’s disease: a study of genetic and clinical predictive factors[J/OL]. Inflamm Bowel Dis. 2013;19:1639–44.
Taher M, Ebrahimi Daryani N, Hedayat M, Eslamian M, Farhadi E, Mahmoudi M, et al. Association analysis of RAC1 single nucleotide polymorphisms with ulcerative colitis[J/OL]. Clin Res Hepatol Gastroenterol. 2017;41:487–9.
López-Posadas R, Becker C, Günther C, Tenzer S, Amann K, Billmeier U, et al. Rho-A prenylation and signaling link epithelial homeostasis to intestinal inflammation[J/OL]. J Clin Investig. 2016;126:611–26.
Van Bodegraven AA, Curley CR, Hunt KA, Monsuur AJ, Linskens RK, Onnie CM, et al. Genetic variation in myosin IXB is associated with ulcerative colitis[J/OL]. Gastroenterology. 2006;131:1768–74.
Latiano A, Palmieri O, Valvano MR, D’incà R, Caprilli R, Cucchiara S, et al. The association of MYO9B gene in Italian patients with inflammatory bowel diseases[J/OL]. Aliment Pharmacol Ther. 2008;27:241–8.
Chandhoke SK, Mooseker MS. A role for myosin IXb, a motor-RhoGAP chimera, in epithelial wound healing and tight junction regulation[J/OL]. Mol Biol Cell. 2012;23:2468–80.
Zhang FL, Casey PJ. Influence of metal ions on substrate binding and catalytic activity of mammalian protein geranylgeranyltransferase type-I[J/OL]. Biochem J. 1996;320:925–32.
López-Posadas R, Fastancz P, Martínez-Sánchez LDC, Panteleev-Ivlev J, Thonn V, Kisseleva T, et al. Inhibiting PGGT1B Disrupts Function of RHOA, Resulting in T-cell Expression of Integrin α4β7 and Development of Colitis in Mice[J/OL]. Gastroenterology. 2019;157:1293–309.
Wildenberg ME, Koelink PJ, Diederen K, Te Velde AA, Wolfkamp SCS, Nuij VJ, et al. The ATG16L1 risk allele associated with Crohn’s disease results in a Rac1-dependent defect in dendritic cell migration that is corrected by thiopurines[J/OL]. Mucosal. Immunology. 2017;10:352–60.
Prins MMC, Giugliano FP, Van Roest M, Van De Graaf SFJ, Koelink PJ, Wildenberg ME. Thiopurines correct the effects of autophagy impairment on intestinal healing - a potential role for ARHGAP18/RhoA[J/OL]. Dis Model Mech. 2021;14:dmm047233.
Lemoine R, Pachlopnik-Schmid J, Farin HF, Bigorgne A, Debré M, Sepulveda F, et al. Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency[J/OL]. The. J Allergy Clin Immunol. 2014;134:1354–64. e6.
Connelly TM, Sehgal R, Berg AS, Hegarty JP, Deiling S, Stewart DB, et al. Mutation in TAGAP is protective of anal sepsis in ileocolic Crohn’s disease[J/OL]. Dis Colon Rectum. 2012;55:1145–52.
Lee M, Chang EB. Inflammatory Bowel Diseases (IBD) and the Microbiome-Searching the Crime Scene for Clues[J/OL]. Gastroenterology. 2021;160:524–37.
Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease[J/OL]. Nature. 2011;474:298–306.
Ladinsky MS, Araujo LP, Zhang X, Veltri J, Galan-Diez M, Soualhi S, et al. Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis[J/OL]. Science. 2019;363:eaat4042.
Nishino K, Nishida A, Inoue R, Kawada Y, Ohno M, Sakai S, et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease[J/OL]. J Gastroenterol. 2018;53:95–106.
Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, et al. Fungal microbiota dysbiosis in IBD[J/OL]. Gut. 2017;66:1039–48.
Sartor RB, Wu GD. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches[J/OL]. Gastroenterology. 2017;152:327–39. e4.
Palmela C, Chevarin C, Xu Z, Torres J, Sevrin G, Hirten R, et al. Adherent-invasive Escherichia coli in inflammatory bowel disease[J/OL]. Gut. 2018;67:574–87.
Boyer L, Magoc L, Dejardin S, Cappillino M, Paquette N, Hinault C, et al. Pathogen-derived effectors trigger protective immunity via activation of the Rac2 enzyme and the IMD or Rip kinase signaling pathway[J/OL]. Immunity. 2011;35:536–49.
Wong ARC, Clements A, Raymond B, Crepin VF, Frankel G. The interplay between the Escherichia coli Rho guanine nucleotide exchange factor effectors and the mammalian RhoGEF inhibitor EspH[J/OL]. mBio. 2012;3:e00250–11.
Macdonald TT, Frankel G, Dougan G, Goncalves NS, Simmons C. Host defences to Citrobacter rodentium[J/OL]. Int J Med Microbiol. 2003;293:87–93.
Hardt WD, Chen LM, Schuebel KE, Bustelo XR, Galán JES. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells[J/OL]. Cell. 1998;93:815–26.
Keestra AM, Winter MG, Auburger JJ, Frässle SP, Xavier MN, Winter SE, et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1[J/OL]. Nature. 2013;496:233–7.
Rodríguez C, Romero E, Garrido-Sanchez L, Alcaín-Martínez G, Andrade RJ, Taminiau B, et al. MICROBIOTA INSIGHTS IN CLOSTRIDIUM DIFFICILE INFECTION AND INFLAMMATORY BOWEL DISEASE[J/OL]. Gut Microbes. 2020;12:1725220.
Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation[J/OL]. Gut. 2003;52:439–51.
Guan Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease[J/OL]. J Immunol Res. 2019;2019:7247238.
Nyström EEL, Martinez-Abad B, Arike L, Birchenough GMH, Nonnecke EB, Castillo PA, et al. An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function[J/OL]. Science. 2021;372:eabb1590.
Wehkamp J, Harder J, Weichenthal M, Mueller O, Herrlinger KR, Fellermann K, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn’s disease and ulcerative colitis[J/OL]. Inflamm Bowel Dis. 2003;9:215–23.
Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease[J/OL]. Proc Natl Acad Sci USA. 2005;102:18129–34.
Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function[J/OL]. J Cell Sci. 2003;116:725–42.
Citalán-Madrid AF, Vargas-Robles H, García-Ponce A, Shibayama M, Betanzos A, Nava P, et al. Cortactin deficiency causes increased RhoA/ROCK1-dependent actomyosin contractility, intestinal epithelial barrier dysfunction, and disproportionately severe DSS-induced colitis[J/OL]. Mucosal. Immunology. 2017;10:1237–47.
Yang Y, Ma Y, Shi C, Chen H, Zhang H, Chen N, et al. Overexpression of miR-21 in patients with ulcerative colitis impairs intestinal epithelial barrier function through targeting the Rho GTPase RhoB[J/OL]. Biochem Biophys Res Commun. 2013;434:746–52.
Shi C, Liang Y, Yang J, Xia Y, Chen H, Han H, et al. MicroRNA-21 knockout improve the survival rate in DSS induced fatal colitis through protecting against inflammation and tissue injury[J/OL]. PloS One. 2013;8:e66814.
Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms[J/OL]. J Immunol. 2003;171:6164–72.
Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, et al. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane[J/OL]. Mol Biol Cell. 2005;16:5040–52.
Jin S, Ray RM, Johnson LR. Rac1 mediates intestinal epithelial cell apoptosis via JNK[J/OL]. American Journal of Physiology. Gastrointest Liver Physiol. 2006;291:G1137–47.
Brito GAC, Fujji J, Carneiro-Filho BA, Lima AAM, Obrig T, Guerrant RL. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells[J/OL]. The. J Infect Dis. 2002;186:1438–47.
Fiorentini C, Fabbri A, Falzano L, Fattorossi A, Matarrese P, Rivabene R, et al. Clostridium difficile toxin B induces apoptosis in intestinal cultured cells[J/OL]. Infect Immun. 1998;66:2660–5.
Zheng XB, Liu HS, Zhang LJ, Liu XH, Zhong XL, Zhou C, et al. Engulfment and Cell Motility Protein 1 Protects Against DSS-induced Colonic Injury in Mice via Rac1 Activation[J/OL]. J Crohn’s Colitis. 2019;13:100–14.
Liu L, Zhuang R, Xiao L, Chung HK, Luo J, Turner DJ, et al. HuR Enhances Early Restitution of the Intestinal Epithelium by Increasing Cdc42 Translation[J/OL]. Mol Cell Biol. 2017;37:e00574–16.
Giesemann T, Guttenberg G, Aktories K. Human alpha-defensins inhibit Clostridium difficile toxin B[J/OL]. Gastroenterology. 2008;134:2049–58.
Fischer S, Ückert AK, Landenberger M, Papatheodorou P, Hoffmann-Richter C, Mittler AK, et al. Human peptide α-defensin-1 interferes with Clostridioides difficile toxins TcdA, TcdB, and CDT[J/OL]. FASEB J. 2020;34:6244–61.
Sheng Q, Lv Z, Cai W, Song H, Qian L, Mu H, et al. Human β-defensin-3 promotes intestinal epithelial cell migration and reduces the development of necrotizing enterocolitis in a neonatal rat model[J/OL]. Pediatr Res. 2014;76:269–79.
Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation[J/OL]. Nat Rev Immunol. 2008;8:435–46.
Na YR, Stakenborg M, Seok SH, Matteoli G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD[J/OL]. Nature Reviews. Gastroenterol Hepatol. 2019;16:531–43.
Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology[J/OL]. Lancet. 2007;369:1627–40.
Steinbach EC, Plevy SE. The role of macrophages and dendritic cells in the initiation of inflammation in IBD[J/OL]. Inflamm Bowel Dis. 2014;20:166–75.
Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, Trombetta S, et al. Developmental control of endocytosis in dendritic cells by Cdc42[J/OL]. Cell. 2000;102:325–34.
West MA, Prescott AR, Eskelinen EL, Ridley AJ, Watts C. Rac is required for constitutive macropinocytosis by dendritic cells but does not control its downregulation[J/OL]. Curr Biol. 2000;10:839–48.
Zhang J, Guo J, Dzhagalov I, He YW. An essential function for the calcium-promoted Ras inactivator in Fcgamma receptor-mediated phagocytosis[J/OL]. Nat Immunol. 2005;6:911–9.
Niedergang F, Chavrier P. Regulation of phagocytosis by Rho GTPases[J/OL]. Curr Top Microbiol Immunol. 2005;291:43–60.
Mao Y, Finnemann SC. Regulation of phagocytosis by Rho GTPases[J/OL]. Small GTPases. 2015;6:89–99.
Egami Y, Kawai K, Araki N. RhoC regulates the actin remodeling required for phagosome formation during FcγR-mediated phagocytosis[J/OL]. J Cell Sci. 2017;130:4168–79.
Geng J, Shi Y, Zhang J, Yang B, Wang P, Yuan W, et al. TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection[J/OL]. Nature. Communications. 2021;12:3519.
Tzircotis G, Braga VMM, Caron E. RhoG is required for both FcγR- and CR3-mediated phagocytosis[J/OL]. J Cell Sci. 2011;124:2897–902.
Leverrier Y, Ridley AJ. Requirement for Rho GTPases and PI 3-kinases during apoptotic cell phagocytosis by macrophages[J/OL]. Curr Biol. 2001;11:195–9.
Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S. Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages[J/OL]. J Biol Chem. 2006;281:8836–42.
Burns S, Thrasher AJ, Blundell MP, Machesky L, Jones GE. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation[J/OL]. Blood. 2001;98:1142–9.
Swetman CA, Leverrier Y, Garg R, Gan CHV, Ridley AJ, Katz DR, et al. Extension, retraction and contraction in the formation of a dendritic cell dendrite: distinct roles for Rho GTPases[J/OL]. Eur J Immunol. 2002;32:2074–83.
Erwig LP, Mcphilips KA, Wynes MW, Ivetic A, Ridley AJ, Henson PM. Differential regulation of phagosome maturation in macrophages and dendritic cells mediated by Rho GTPases and ezrin-radixin-moesin (ERM) proteins[J/OL]. Proc Natl Acad Sci USA. 2006;103:12825–30.
Wu W, Hsu YMS, Bi L, Songyang Z, Lin X. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex[J/OL]. Nat Immunol. 2009;10:1208–14.
Li Y, Kim JG, Kim HJ, Moon MY, Lee JY, Kim J, et al. Small GTPases Rap1 and RhoA regulate superoxide formation by Rac1 GTPases activation during the phagocytosis of IgG-opsonized zymosans in macrophages[J/OL]. Free Radic Biol Med. 2012;52:1796–805.
Ridley AJ. Rho proteins, PI 3-kinases, and monocyte/macrophage motility[J/OL]. FEBS Lett. 2001;498:168–71.
Infante E, Ridley AJ. Roles of Rho GTPases in leucocyte and leukaemia cell transendothelial migration[J/OL]. Philos Trans R Soc Lond Ser B, Biol Sci. 2013;368:20130013.
Levine AP, Segal AW. What is wrong with granulocytes in inflammatory bowel diseases?[J/OL]. Dig Dis. 2013;31:321–7.
Wéra O, Lancellotti P, Oury C. The Dual Role of Neutrophils in Inflammatory Bowel Diseases[J/OL]. J Clin Med. 2016;5:118.
Petri B, Sanz MJ. Neutrophil chemotaxis[J/OL]. Cell Tissue Res. 2018;371:425–36.
Werner E. GTPases and reactive oxygen species: switches for killing and signaling[J/OL]. J Cell Sci. 2004;117:143–53.
Lacy P. The role of Rho GTPases and SNAREs in mediator release from granulocytes[J/OL]. Pharmacol Ther. 2005;107:358–76.
Tybulewicz VLJ, Henderson RB. Rho family GTPases and their regulators in lymphocytes[J/OL]. Nat Rev Immunol. 2009;9:630–44.
Cernuda-Morollón E, Millán J, Shipman M, Marelli-Berg FM, Ridley AJ. Rac activation by the T-cell receptor inhibits T cell migration[J/OL]. PloS One. 2010;5:e12393.
García-Bernal D, Wright N, Sotillo-Mallo E, Nombela-Arrieta C, Stein JV, Bustelo XR, et al. Vav1 and Rac control chemokine-promoted T lymphocyte adhesion mediated by the integrin alpha4beta1[J/OL]. Mol Biol Cell. 2005;16:3223–35.
Vielkind S, Gallagher-Gambarelli M, Gomez M, Hinton HJ, Cantrell DA. Integrin regulation by RhoA in thymocytes[J/OL]. J Immunol. 2005;175:350–7.
Faroudi M, Hons M, Zachacz A, Dumont C, Lyck R, Stein JV, et al. Critical roles for Rac GTPases in T-cell migration to and within lymph nodes[J/OL]. Blood. 2010;116:5536–47.
Baker CM, Comrie WA, Hyun YM, Chung HL, Fedorchuk CA, Lim K, et al. Opposing roles for RhoH GTPase during T-cell migration and activation[J/OL]. Proc Natl Acad Sci USA. 2012;109:10474–9.
Singleton KL, Roybal KT, Sun Y, Fu G, Gascoigne NRJ, Van Oers NSC, et al. Spatiotemporal patterning during T cell activation is highly diverse[J/OL]. Sci Signal. 2009;2:ra15.
Faure S, Salazar-Fontana LI, Semichon M, Tybulewicz VLJ, Bismuth G, Trautmann A, et al. ERM proteins regulate cytoskeleton relaxation promoting T cell-APC conjugation[J/OL]. Nat Immunol. 2004;5:272–9.
Delaguillaumie A, Lagaudrière-Gesbert C, Popoff MR, Conjeaud H. Rho GTPases link cytoskeletal rearrangements and activation processes induced via the tetraspanin CD82 in T lymphocytes[J/OL]. J Cell Sci. 2002;115:433–43.
Salazar-Fontana LI, Barr V, Samelson LE, Bierer BE. CD28 engagement promotes actin polymerization through the activation of the small Rho GTPase Cdc42 in human T cells[J/OL]. J Immunol. 2003;171:2225–32.
Jacinto E, Werlen G, Karin M. Cooperation between Syk and Rac1 leads to synergistic JNK activation in T lymphocytes[J/OL]. Immunity. 1998;8:31–41.
Marinari B, Costanzo A, Viola A, Michel F, Mangino G, Acuto O, et al. Vav cooperates with CD28 to induce NF-kappaB activation via a pathway involving Rac-1 and mitogen-activated kinase kinase 1[J/OL]. Eur J Immunol. 2002;32:447–56.
Arrieumerlou C, Randriamampita C, Bismuth G, Trautmann A. Rac is involved in early TCR signaling[J/OL]. J Immunol. 2000;165:3182–9.
Oh-Hora M, Rao A. Calcium signaling in lymphocytes[J/OL]. Curr Opin Immunol. 2008;20:250–8.
Acuto O, Cantrell D. T cell activation and the cytoskeleton[J/OL]. Annu Rev Immunol. 2000;18:165–84.
Angkachatchai V, Finkel TH. ADP-ribosylation of rho by C3 ribosyltransferase inhibits IL-2 production and sustained calcium influx in activated T cells[J]. J Immunol. 1999;163:3819–25.
Yang JQ, Kalim KW, Li Y, Zhang S, Hinge A, Filippi MD, et al. RhoA orchestrates glycolysis for TH2 cell differentiation and allergic airway inflammation[J/OL]. J Allergy Clin Immunol. 2016;137:231–45. e4.
Guo F, Hildeman D, Tripathi P, Velu CS, Grimes HL, Zheng Y. Coordination of IL-7 receptor and T-cell receptor signaling by cell-division cycle 42 in T-cell homeostasis[J/OL]. Proc Natl Acad Sci USA. 2010;107:18505–10.
Ahmad Mokhtar AM, Salikin NH, Haron AS, Amin-Nordin S, Hashim IF, Mohd Zaini Makhtar M, et al. RhoG’s Role in T Cell Activation and Function[J/OL]. Front Immunol. 2022;13:845064.
Kalim KW, Yang JQ, Li Y, Meng Y, Zheng Y, Guo F. Reciprocal Regulation of Glycolysis-Driven Th17 Pathogenicity and Regulatory T Cell Stability by Cdc42[J/OL]. J Immunol. 2018;200:2313–26.
Yang JQ, Kalim KW, Li Y, Duan X, Nguyen P, Khurana Hershey GK, et al. Rational targeting Cdc42 restrains Th2 cell differentiation and prevents allergic airway inflammation[J/OL]. Clin Exp Allergy. 2019;49:92–107.
Yang JQ, Kalim KW, Li Y, Zheng Y, Guo F. Ablation of RhoA impairs Th17 cell differentiation and alleviates house dust mite-triggered allergic airway inflammation[J/OL]. J Leukoc Biol. 2019;106:1139–51.
Dunn SE, Youssef S, Goldstein MJ, Prod’homme T, Weber MS, Zamvil SS, et al. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin[J/OL]. J Exp Med. 2006;203:401–12.
Li B, Yu H, Zheng W, Voll R, Na S, Roberts AW, et al. Role of the guanosine triphosphatase Rac2 in T helper 1 cell differentiation[J/OL]. Science. 2000;288:2219–22.
Li X, Bu X, Lu B, Avraham H, Flavell RA, Lim B. The hematopoiesis-specific GTP-binding protein RhoH is GTPase deficient and modulates activities of other Rho GTPases by an inhibitory function[J/OL]. Mol Cell Biol. 2002;22:1158–71.
Tang HC, Lai YY, Zheng J, Jiang HY, Xu G. miR-223-3p Inhibits Antigen Endocytosis and Presentation and Promotes the Tolerogenic Potential of Dendritic Cells through Targeting Mannose Receptor Signaling and Rhob[J/OL]. J Immunol Res. 2020;2020:1379458.
Na S, Li B, Grewal IS, Enslen H, Davis RJ, Hanke JH, et al. Expression of activated CDC42 induces T cell apoptosis in thymus and peripheral lymph organs via different pathways[J/OL]. Oncogene. 1999;18:7966–74.
Subauste MC, Von Herrath M, Benard V, Chamberlain CE, Chuang TH, Chu K, et al. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas[J/OL]. J Biol Chem. 2000;275:9725–33.
Hébert M, Potin S, Sebbagh M, Bertoglio J, Bréard J, Hamelin J. Rho-ROCK-dependent ezrin-radixin-moesin phosphorylation regulates Fas-mediated apoptosis in Jurkat cells[J/OL]. J Immunol. 2008;181:5963–73.
Brinkkoetter PT, Gottmann U, Schulte J, Van Der Woude FJ, Braun C, Yard BA. Atorvastatin interferes with activation of human CD4(+) T cells via inhibition of small guanosine triphosphatase (GTPase) activity and caspase-independent apoptosis[J/OL]. Clin Exp Immunol. 2006;146:524–32.
Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D, et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes[J/OL]. J Clin Investig. 2003;111:1133–45.
Poppe D, Tiede I, Fritz G, Becker C, Bartsch B, Wirtz S, et al. Azathioprine suppresses ezrin-radixin-moesin-dependent T cell-APC conjugation through inhibition of Vav guanosine exchange activity on Rac proteins[J/OL]. J Immunol. 2006;176:640–51.
Neurath MF. Cytokines in inflammatory bowel disease[J/OL]. Nat Rev Immunol. 2014;14:329–42.
Dong LM, Chen XW, He XX, Jiang XP, Wu F. Cell division cycle protein 42 regulates the inflammatory response in mice bearing inflammatory bowel disease[J/OL]. Artif Cells, Nanomed, Biotechnol. 2019;47:1833–8.
Tang WJ, Peng KY, Tang ZF, Wang YH, Xue AJ, Huang Y. MicroRNA-15a - cell division cycle 42 signaling pathway in pathogenesis of pediatric inflammatory bowel disease[J/OL]. World J Gastroenterol. 2018;24:5234–45.
Segain JP, Raingeard DE, La Blétière D, Sauzeau V, Bourreille A, Hilaret G, et al. Rho kinase blockade prevents inflammation via nuclear factor kappa B inhibition: evidence in Crohn’s disease and experimental colitis[J/OL]. Gastroenterology. 2003;124:1180–7.
Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PDR. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-β-induced fibrogenesis in human colonic myofibroblasts[J/OL]. Inflamm Bowel Dis. 2014;20:154–65.
Horowitz S, Binion DG, Nelson VM, Kanaa Y, Javadi P, Lazarova Z, et al. Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease[J/OL]. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1323–1336.
Seidelin JB, Larsen S, Linnemann D, Vainer B, Coskun M, Troelsen JT, et al. Cellular inhibitor of apoptosis protein 2 controls human colonic epithelial restitution, migration, and Rac1 activation[J/OL]. Am J Physiol Gastrointest Liver Physiol. 2015;308:G92–99.
Parikh K, Zhou L, Somasundaram R, Fuhler GM, Deuring JJ, Blokzijl T, et al. Suppression of p21Rac signaling and increased innate immunity mediate remission in Crohn’s disease[J/OL]. Sci Transl Med. 2014;6:233ra53.
Marinković G, Hamers AAJ, De Vries CJM, De Waard V. 6-Mercaptopurine reduces macrophage activation and gut epithelium proliferation through inhibition of GTPase Rac1[J/OL]. Inflamm Bowel Dis. 2014;20:1487–95.
Liu G, Gu K, Wang F, Jia G, Zhao H, Chen X, et al. Tryptophan Ameliorates Barrier Integrity and Alleviates the Inflammatory Response to Enterotoxigenic Escherichia coli K88 Through the CaSR/Rac1/PLC-γ1 Signaling Pathway in Porcine Intestinal Epithelial Cells[J/OL]. Front Immunol. 2021;12:748497.
Piovani D, Danese S, Peyrin-Biroulet L, Nikolopoulos GK, Lytras T, Bonovas S. Environmental Risk Factors for Inflammatory Bowel Diseases: An Umbrella Review of Meta-analyses[J/OL]. Gastroenterology. 2019;157:647–59. e4.
Kanauchi Y, Yamamoto T, Yoshida M, Zhang Y, Lee J, Hayashi S, et al. Cholinergic anti-inflammatory pathway ameliorates murine experimental Th2-type colitis by suppressing the migration of plasmacytoid dendritic cells[J/OL]. Sci Rep. 2022;12:54.
Penna G, Fibbi B, Amuchastegui S, Corsiero E, Laverny G, Silvestrini E, et al. The vitamin D receptor agonist elocalcitol inhibits IL-8-dependent benign prostatic hyperplasia stromal cell proliferation and inflammatory response by targeting the RhoA/Rho kinase and NF-kappaB pathways[J/OL]. Prostate. 2009;69:480–93.
Qian X, Zhu M, Qian W, Song J. Vitamin D attenuates myocardial ischemia-reperfusion injury by inhibiting inflammation via suppressing the RhoA/ROCK/NF-ĸB pathway[J/OL]. Biotechnol Appl Biochem. 2019;66:850–7.
Aslan A, Triadafilopoulos G. Fish oil fatty acid supplementation in active ulcerative colitis: a double-blind, placebo-controlled, crossover study[J]. Am J Gastroenterol. 1992;87:432–7.
Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature[J/OL]. Am J Gastroenterol. 2011;106:563–73.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation[J/OL]. Cell. 2010;140:805–20.
Kordjazy N, Haj-Mirzaian A, Haj-Mirzaian A, Rohani MM, Gelfand EW, Rezaei N, et al. Role of toll-like receptors in inflammatory bowel disease[J/OL]. Pharmacol Res. 2018;129:204–15.
Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, et al. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway[J/OL]. Nat Immunol. 2000;1:533–40.
Teusch N, Lombardo E, Eddleston J, Knaus UG. The low molecular weight GTPase RhoA and atypical protein kinase Czeta are required for TLR2-mediated gene transcription[J/OL]. J Immunol. 2004;173:507–14.
Schmeck B, Huber S, Moog K, Zahlten J, Hocke AC, Opitz B, et al. Pneumococci induced TLR- and Rac1-dependent NF-kappaB-recruitment to the IL-8 promoter in lung epithelial cells[J/OL]. Am J Physiol Lung Cell Mol Physiol. 2006;290:L730–L737.
Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, et al. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake[J/OL]. Circ Res. 2009;104:1355–63.
Kong L, Ge BX. MyD88-independent activation of a novel actin-Cdc42/Rac pathway is required for Toll-like receptor-stimulated phagocytosis[J/OL]. Cell Res. 2008;18:745–55.
Gotoh K, Tanaka Y, Nishikimi A, Nakamura R, Yamada H, Maeda N, et al. Selective control of type I IFN induction by the Rac activator DOCK2 during TLR-mediated plasmacytoid dendritic cell activation[J/OL]. J Exp Med. 2010;207:721–30.
Wang Z, Fayngerts S, Wang P, Sun H, Johnson DS, Ruan Q, et al. TIPE2 protein serves as a negative regulator of phagocytosis and oxidative burst during infection[J/OL]. Proc Natl Acad Sci USA. 2012;109:15413–8.
Liu S, Huang L, Lin Z, Hu Y, Chen R, Wang L, et al. RhoB induces the production of proinflammatory cytokines in TLR-triggered macrophages[J/OL]. Mol Immunol. 2017;87:200–6.
Pulecio J, Petrovic J, Prete F, Chiaruttini G, Lennon-Dumenil AM, Desdouets C, et al. Cdc42-mediated MTOC polarization in dendritic cells controls targeted delivery of cytokines at the immune synapse[J/OL]. J Exp Med. 2010;207:2719–32.
Rubino SJ, Selvanantham T, Girardin SE, Philpott DJ. Nod-like receptors in the control of intestinal inflammation[J/OL]. Curr Opin Immunol. 2012;24:398–404.
Keestra AM, Bäumler AJ. Detection of enteric pathogens by the nodosome[J/OL]. Trends Immunol. 2014;35:123–30.
Eitel J, Krüll M, Hocke AC, N’guessan PD, Zahlten J, Schmeck B, et al. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2[J/OL]. J Immunol. 2008;181:2664–71.
Dufies O, Doye A, Courjon J, Torre C, Michel G, Loubatier C, et al. Escherichia coli Rho GTPase-activating toxin CNF1 mediates NLRP3 inflammasome activation via p21-activated kinases-1/2 during bacteraemia in mice[J/OL]. Nat Microbiol. 2021;6:401–12.
Xie S, Yang T, Wang Z, Li M, Ding L, Hu X, et al. Astragaloside IV attenuates sepsis-induced intestinal barrier dysfunction via suppressing RhoA/NLRP3 inflammasome signaling[J/OL]. Int Immunopharmacol. 2020;78:106066.
Ying C, Zhou Z, Dai J, Wang M, Xiang J, Sun D, et al. Activation of the NLRP3 inflammasome by RAC1 mediates a new mechanism in diabetic nephropathy[J/OL]. Inflamm Res. 2022;71:191–204.
Yan Y, Lu K, Ye T, Zhang Z. MicroRNA‑223 attenuates LPS‑induced inflammation in an acute lung injury model via the NLRP3 inflammasome and TLR4/NF‑κB signaling pathway via RHOB[J/OL]. Int J Mol Med. 2019;43:1467–77.
Rebane A, Akdis CA. MicroRNAs: Essential players in the regulation of inflammation[J/OL]. J Allergy Clin Immunol. 2013;132:15–26.
Wu F, Zikusoka M, Trindade A, Dassopoulos T, Harris ML, Bayless TM, et al. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha[J/OL]. Gastroenterology. 2008;135:1624–35. e24.
Wu F, Zhang S, Dassopoulos T, Harris ML, Bayless TM, Meltzer SJ, et al. Identification of microRNAs associated with ileal and colonic Crohn’s disease[J/OL]. Inflamm Bowel Dis. 2010;16:1729–38.
Pekow JR, Dougherty U, Mustafi R, Zhu H, Kocherginsky M, Rubin DT, et al. miR-143 and miR-145 are downregulated in ulcerative colitis: putative regulators of inflammation and protooncogenes[J/OL]. Inflamm Bowel Dis. 2012;18:94–100.
Fang K, Law IKM, Padua D, Sideri A, Huang V, Kevil CG, et al. MicroRNA-31-3p Is Involved in Substance P (SP)-Associated Inflammation in Human Colonic Epithelial Cells and Experimental Colitis[J/OL]. Am J Pathol. 2018;188:586–99.
Zhou WY, Cai ZR, Liu J, Wang DS, Ju HQ, Xu RH. Circular RNA: metabolism, functions and interactions with proteins[J/OL]. Mol Cancer. 2020;19:172.
Xiao L, Ma XX, Luo J, Chung HK, Kwon MS, Yu TX, et al. Circular RNA CircHIPK3 Promotes Homeostasis of the Intestinal Epithelium by Reducing MicroRNA 29b Function[J/OL]. Gastroenterology. 2021;161:1303–17. e3.
Danese S, Sans M, De La Motte C, Graziani C, West G, Phillips MH, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis[J/OL]. Gastroenterology. 2006;130:2060–73.
Merajver SD, Usmani SZ. Multifaceted role of Rho proteins in angiogenesis[J/OL]. J Mammary Gland Biol Neoplasia. 2005;10:291–8.
Sulpice E, Ding S, Muscatelli-Groux B, Bergé M, Han ZC, Plouet J, et al. Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells[J/OL]. Biol Cell. 2009;101:525–39.
Pang X, Zhang L, Lai L, Chen J, Wu Y, Yi Z, et al. 1’-Acetoxychavicol acetate suppresses angiogenesis-mediated human prostate tumor growth by targeting VEGF-mediated Src-FAK-Rho GTPase-signaling pathway[J/OL]. Carcinogenesis. 2011;32:904–12.
Lai Y, Shen Y, Liu XH, Zhang Y, Zeng Y, Liu YF. Interleukin-8 induces the endothelial cell migration through the activation of phosphoinositide 3-kinase-Rac1/RhoA pathway[J/OL]. Int J Biol Sci. 2011;7:782–91.
Turk HF, Monk JM, Fan YY, Callaway ES, Weeks B, Chapkin RS. Inhibitory effects of omega-3 fatty acids on injury-induced epidermal growth factor receptor transactivation contribute to delayed wound healing[J/OL]. Am J Physiol Cell Physiol. 2013;304:C905–917.
Maldonado MDM, Dharmawardhane S. Targeting Rac and Cdc42 GTPases in Cancer[J/OL]. Cancer Res. 2018;78:3101–11.
Budzyn K, Marley PD, Sobey CG. Targeting Rho and Rho-kinase in the treatment of cardiovascular disease[J/OL]. Trends Pharmacol Sci. 2006;27:97–104.
Aguilar BJ, Zhu Y, Lu Q. Rho GTPases as therapeutic targets in Alzheimer’s disease[J/OL]. Alzheimer’s Res Ther. 2017;9:97.
Martin TE, Powell CT, Wang Z, Bhattacharyya S, Walsh-Reitz MM, Agarwal K, et al. A novel mitogenic protein that is highly expressed in cells of the gastric antrum mucosa[J/OL]. Am J Physiol Gastrointest Liver Physiol. 2003;285:G332–43.
Chen P, Bakke D, Kolodziej L, Lodolce J, Weber CR, Boone DL, et al. Antrum Mucosal Protein-18 Peptide Targets Tight Junctions to Protect and Heal Barrier Structure and Function in Models of Inflammatory Bowel Disease[J/OL]. Inflamm Bowel Dis. 2015;21:2393–402.
Chen P, Kartha S, Bissonnette M, Hart J, Toback FG. AMP-18 facilitates assembly and stabilization of tight junctions to protect the colonic mucosal barrier[J/OL]. Inflamm Bowel Dis. 2012;18:1749–59.
D’haens G, Van Deventer S, Van Hogezand R, Chalmers D, Kothe C, Baert F, et al. Endoscopic and histological healing with infliximab anti-tumor necrosis factor antibodies in Crohn’s disease: A European multicenter trial[J/OL]. Gastroenterology. 1999;116:1029–34.
Rutgeerts P, D’haens G, Targan S, Vasiliauskas E, Hanauer SB, Present DH, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease[J/OL]. Gastroenterology. 1999;117:761–9.
Bouguen G, Roblin X, Bourreille A, Feier L, Filippi J, Nancey S, et al. Infliximab for refractory ulcerative proctitis[J/OL]. Aliment Pharmacol Ther. 2010;31:1178–85.
Caviglia R, Ribolsi M, Rizzi M, Emerenziani S, Annunziata ML, Cicala M. Maintenance of remission with infliximab in inflammatory bowel disease: efficacy and safety long-term follow-up[J/OL]. World J Gastroenterol. 2007;13:5238–44.
Liu L, Liu Q, Chang J, Dong X, Ma W. Cell division control 42 elevates during infliximab therapy, and its increment relates to treatment response in ulcerative colitis patients[J/OL]. J Clin Lab Anal. 2022;36:e24477.
Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor[J/OL]. Proc Natl Acad Sci USA. 2004;101:7618–23.
Nielsen OH, Vainer B, Rask-Madsen J. Review article: the treatment of inflammatory bowel disease with 6-mercaptopurine or azathioprine[J/OL]. Aliment Pharmacol Ther. 2001;15:1699–708.
De Boer NKH, Peyrin-Biroulet L, Jharap B, Sanderson JD, Meijer B, Atreya I, et al. Thiopurines in Inflammatory Bowel Disease: New Findings and Perspectives[J/OL]. J Crohn’s Colitis. 2018;12:610–20.
Seinen ML, Van Nieuw Amerongen GP, De Boer NKH, Mulder CJJ, Van Bezu J, Van Bodegraven AA. Rac1 as a Potential Pharmacodynamic Biomarker for Thiopurine Therapy in Inflammatory Bowel Disease[J/OL]. Ther Drug Monit. 2016;38:621.
Yusung S, Mcgovern D, Lin L, Hommes D, Lagishetty V, Braun J. NK cells are biologic and biochemical targets of 6-mercaptopurine in Crohn’s disease patients[J/OL]. Clin Immunol. 2017;175:82–90.
Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results[J/OL]. Trends Mol Med. 2008;14:37–44.
Grip O, Janciauskiene S, Bredberg A. Use of atorvastatin as an anti-inflammatory treatment in Crohn’s disease[J/OL]. Br J Pharmacol. 2008;155:1085–92.
Chardin P, Boquet P, Madaule P, Popoff MR, Rubin EJ, Gill DM. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells[J/OL]. EMBO J. 1989;8:1087–92.
Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension[J/OL]. Nature. 1997;389:990–4.
Guzman JR, Koo JS, Goldsmith JR, Mühlbauer M, Narula A, Jobin C. Oxymatrine prevents NF-κB nuclear translocation and ameliorates acute intestinal inflammation[J/OL]. Sci Rep. 2013;3:1629.
Choi YJ, Koo JB, Kim HY, Seo JW, Lee EJ, Kim WR, et al. Umbilical cord/placenta-derived mesenchymal stem cells inhibit fibrogenic activation in human intestinal myofibroblasts via inhibition of myocardin-related transcription factor A[J/OL]. Stem Cell Res Ther. 2019;10:291.
Naito Y, Takagi T, Katada K, Uchiyama K, Kuroda M, Kokura S, et al. Partially hydrolyzed guar gum down-regulates colonic inflammatory response in dextran sulfate sodium-induced colitis in mice[J/OL]. J Nutr Biochem. 2006;17:402–9.
Wang Y, Shou Z, Fan H, Xu M, Chen Q, Tang Q, et al. Protective effects of oxymatrine against DSS-induced acute intestinal inflammation in mice via blocking the RhoA/ROCK signaling pathway[J/OL]. Biosci Rep. 2019;39:BSR20182297.
Horii Y, Uchiyama K, Toyokawa Y, Hotta Y, Tanaka M, Yasukawa Z, et al. Partially hydrolyzed guar gum enhances colonic epithelial wound healing via activation of RhoA and ERK1/2[J/OL]. Food Funct. 2016;7:3176–83.
Veeraperumal S, Qiu HM, Tan CS, Ng ST, Zhang W, Tang S, et al. Restitution of epithelial cells during intestinal mucosal wound healing: The effect of a polysaccharide from the sclerotium of Lignosus rhinocerotis (Cooke) Ryvarden[J/OL]. J Ethnopharmacol. 2021;274:114024.
Bartok B, Hammaker D, Firestein GS. Phosphoinositide 3-kinase δ regulates migration and invasion of synoviocytes in rheumatoid arthritis[J/OL]. J Immunol. 2014;192:2063–70.
Kume H. RhoA/Rho-kinase as a therapeutic target in asthma[J/OL]. Curr Med Chem. 2008;15:2876–85.
Zandvakili I, Lin Y, Morris JC, Zheng Y. Rho GTPases: Anti- or pro-neoplastic targets?[J/OL]. Oncogene. 2017;36:3213–22.
Acknowledgements
This project was supported by grants from the National Natural Science Foundation of China (#82270555, #82070538, #81870374) and Guangdong Science and Technology Department (#2021A1515220107).
Author information
Authors and Affiliations
Contributions
Guarantor of the article: SZ. SZ designed and oversaw the study. XL, MZ and GZ wrote and revised the manuscript. ZX, YW, JH, LL and QW revised the contents of the manuscript. All authors approved the final manuscript and agreed to be responsible for this review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Zhang, M., Zhou, G. et al. Role of Rho GTPases in inflammatory bowel disease. Cell Death Discov. 9, 24 (2023). https://doi.org/10.1038/s41420-023-01329-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-023-01329-w
This article is cited by
-
Hyperglycemia alters the gene and protein expression of CDC42 in small and large intestine of Sprague-Dawley rats
Molecular and Cellular Biochemistry (2026)
-
Unraveling the molecular dynamics of wound healing: integrating spatially resolved lipidomics and temporally resolved proteomics
Analytical and Bioanalytical Chemistry (2025)
