
Abstract
The distinctive feature of the adaptive immune system is its ability to generate immunological memory that can provide defense against subsequent infections. In the case of antibody-mediated immune responses, this memory comes in two cellular forms: plasma cells (PCs) and memory B cells (MBCs). PCs protect against reinfection by constitutively producing antibodies. The presence of a diverse pool of MBCs, which can expand and differentiate into PCs in secondary immune responses, is thought to be particularly important for defense against new pathogen variants. Recent studies have shown that the MBC compartment is far more heterogeneous than previously anticipated. This heterogeneity, among other factors, is shaped by their developmental pathway (germinal center (GC) vs non-GC-derived MBCs), the duration and strength of antigenic stimulation, anatomical and microanatomical localization, and the timing of generation in ontogeny. Combinations of these “layers” of MBC identities can define MBCs’ properties and their fate in recall responses. Here, we review the mechanisms underlying MBC differentiation, maintenance, and reactivation and explore how the layered identity of MBCs contributes to the functions of these cells.
Similar content being viewed by others

Introduction
Humoral immune responses to vaccination or infection involve the activation of rare antigen-specific B cells, their clonal expansion and differentiation into antibody-secreting plasma cells (PCs). PCs contribute to immune defense by secreting antibodies that help neutralize and eliminate invading agents. While many of these PCs are short-lived, some are maintained for years or even decades as long-lived plasma cells (LLPCs) that continue to secrete antibodies, serving as a first line of defense upon pathogen reencounter.
Some activated B cells, instead of undergoing terminal differentiation to the PC lineage, exit the cell cycle and persist in a quiescent state as memory B cells (MBCs). Upon subsequent antigen exposure—either in secondary immune responses or after rerecruitment into the primary immune response if the antigen persists—MBCs reinitiate the cascade of B cell activation and differentiation. This expanded pool of antigen-specific B cells—greater in number and often functionally distinct from the naïve precursors present before initial exposure—ensures a faster and more robust response if the pathogen manages to bypass the existing antibody-mediated barrier. Notably, recent evidence suggests that the MBC compartment is more clonally diverse than the PC compartment is [1,2,3,4] and may play a crucial role in mounting effective responses against novel pathogen variants [3,4,5,6,7]. As the maintenance of LLPCs, cells constantly secreting large amounts of antibodies, is energetically costly, archiving a broader repertoire of antigen-specific B cells within the quiescent MBC compartment may represent a more economical strategy for preserving the diversity of antigen-specific B lineage cells over time.
Historically, studies of B cell memory have focused on class-switched B cells (cells that have undergone switching from IgM/IgD to other antibody isotypes during the course of the immune response), which express a few canonical memory markers and recirculate between secondary lymphoid organs. While these studies have provided valuable insights, they represent a somewhat limited view of the true diversity within the MBC pool. It is now evident that the MBC compartment is highly heterogeneous and includes sizable populations of unswitched cells, which are sometimes phenotypically indistinguishable from naïve B cells in terms of the currently used cell-surface markers. Moreover, it becomes increasingly clear that nonrecirculating tissue-resident MBCs can be found in some anatomical locations where they ensure rapid local recall responses. Finally, MBCs can be heterogeneous in terms of their developmental origin (e.g., germinal center (GC) vs non-GC-derived MBCs), the duration and strength of antigenic stimulation they have experienced, and the timing of their generation during ontogeny. These “layers” of MBC identity, discussed in detail in this review, in turn determine the functional properties of MBCs in secondary immune responses.
GC-dependent and independent MBCs
Antigen-dependent B cell differentiation is an unusual developmental process in which both main cellular outputs—PCs and MBCs—can arise through two distinct routes, either directly from early activated B cells or after participation in the GC reaction [8]. To elucidate the mechanisms that regulate the generation of MBCs via these pathways, in the following sections, we discuss how the developmental trajectories of MBCs diverge from those leading to alternative fates. We begin with the better-characterized fate decisions within GCs (“Memory formation in GCs”) and then turn to the less understood events in early B cell activation that lead to the generation of GC-independent MBCs (“Pre-GC memory formation”). Finally, we compare the resulting MBC populations, highlighting both shared and distinguishing features and considering how their developmental origins influence their behavior upon reactivation (“Comparison of GCMBCs and EMBCs”).
Memory formation in GCs
The GC reaction
B cells exhibit the remarkable capacity to refine the binding strength of their B cell receptors (BCRs) to antigens—a process known as affinity maturation. This evolution of antibody affinity occurs within transient microanatomical structures called germinal centers (GCs), which form in the B cell follicles of secondary lymphoid organs [9,10,11,12] as well as in tertiary lymphoid structures (TLSs) [13]. B cells participating in the GC reaction ultimately give rise to GC-derived plasma cells (GCPCs) and GC-derived memory B cells (GCMBCs) (Fig. 1). Although most GCs emerge temporarily in response to infections or immunizations, some lymphoid organs, such as Peyer’s patches (PPs) in the gut, can host persistent GCs [14, 15]. The GC reaction has been predominantly studied in the context of T-dependent responses; however, it is important to note that short-lived GCs can also form in response to T-independent antigens [16,17,18]. These GCs can support the generation of GCMBCs and GCPCs [18], although the mechanisms and functional significance of such GC reactions remain to be further elucidated.

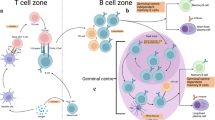
Cell fate decisions upon B cell activation. Upon activation, naïve B cells give rise to tripotent activated precursors (APs). APs can give rise to early plasma cells (EPCs) or early memory B cells (EMBCs) or differentiate into germinal center B cells (GCBCs) by initiating a specialized effector program enabling their participation in the germinal center (GC) reaction. In the GC reaction, through iterative rounds of competition for antigen and T cell help, proliferation and mutation of their antibody genes, GCBCs can improve their affinity for the antigen. Some GCBCs differentiate into GC-derived PCs (GCPCs) or MBCs (GCMBCs). See the text for further details
GCs are formed in B cell follicles by antigen-specific B cells that, after encountering their cognate antigen and receiving T cell help at the T-B border, undergo a burst of proliferation [19] (Fig. 1). Early GCs are polyclonal, often initiated by multiple “founder” clones [20], and remain accessible to additional antigen-specific B cells that can enter the GC reaction later on and join the ongoing response [21,22,23,24,25]. GCs are architecturally organized into two functionally distinct zones [26,27,28]: the dark zone (DZ), where B cells proliferate and introduce mutations into their immunoglobulin (Ig) genes through somatic hypermutation (SHM) [29], and the light zone (LZ), where mutated B cells with varying antigen affinities migrate to and undergo a two-step selection process [27, 28, 30,31,32,33] (Fig. 1). First, they compete for antigen displayed by follicular dendritic cells (FDCs) [34,35,36]. Cells that successfully acquire antigen present its epitopes on MHC class II molecules to GC T follicular helper (GC-TFH) cells—a specialized subset of CD4⁺ T cells that reside within GCs [37,38,39]. These TFH cells, in turn, provide GC B cells (GCBCs) with prosurvival and mitogenic signals through the CD40-CD40L interaction and cytokine secretion, thereby promoting positive selection [28, 40,41,42]. Positively selected GCBCs then continue their diversification within the GC. The GC reaction is an iterative process that can continue for weeks or even months [43,44,45,46], during which surviving B cells undergo many rounds of proliferation, SHM and selection [27, 28, 30,31,32,33]. Throughout this process, GCBCs continuously choose between four distinct fates: cell death [33], continued participation in the GC reaction [27, 28, 30,31,32,33], and differentiation into GCPCs or GCMBCs [47, 48]. In the following two sections, we discuss how the signals received by GCBCs are translated into these cell fate decisions.
Cell fate decisions in GCs
Most GCBCs are thought to undergo apoptosis during the GC reaction, either because of a lack of positive selection signals in the LZ or because of SHM in the DZ that generates nonfunctional BCRs [33]. While it was initially thought that GCBCs with lower-affinity BCRs would be predominantly eliminated by apoptosis due to the lack of survival signals coming from BCR signaling and/or T cell help [12], direct evidence for this is lacking. In fact, comparisons of the ratios of lower-affinity B cells to higher-affinity B cells within the apoptotic and nonapoptotic compartments of the LZ revealed no significant differences [11, 33]. It is therefore conceivable that reduced proliferation rather than increased apoptosis is the main driver of the elimination of lower-affinity clones over time. Indeed, higher-affinity GCBCs capture and present antigen to TFH cells in the LZ more efficiently [28, 49, 50] (Fig. 1), resulting in stronger delivery of T cell-derived signals, increased mTORC1 signaling and greater upregulation of the transcription factor MYC, which in turn leads to a greater burst of proliferation upon their reentry into the DZ [51,52,53]. Thus, even if higher- and lower-affinity clones experienced similar apoptosis rates, higher-affinity B cells are more likely to undergo additional rounds of proliferation and SHM in the DZ, increasing their representation in subsequent cycles of selection [27, 28, 30,31,32,33].
It is important to note that the GC reaction is an inherently “noisy” process, and GC dynamics are not governed solely by affinity-based interclonal competition in a strictly deterministic fashion. Indeed, a recent analysis of hundreds of GC reactions from monoclonal B cells specific for a model antigen revealed that, while an organism-level increase in affinity was reliably achieved, individual GCs exhibited broad variation in both the efficiency and trajectories of affinity maturation [54], highlighting the stochasticity of selection events in GCs. It is conceivable that this “noise” in the selection events could be introduced by constraints in access to antigen and T cell help, as well as by “jackpot” events such as acquisition of antigen aggregates or even antigen-loaded fragments of FDCs. Thus, while relative BCR affinity is a major determinant of cell fate decisions in GCs on average, the fate of an individual GCBC at a given timepoint may be heavily influenced by stochastic factors.
Despite this stochasticity, the GCPC compartment is enriched for higher-affinity clones compared with both contemporaneous GCMBC and GCBC populations [2, 55,56,57], and BCR transgenic B cells with higher affinity BCRs generate more PCs than B cells bearing a lower affinity BCR recognizing the same antigen [58]. These findings suggested that stronger T cell help (and/or stronger BCR signaling) may preferentially instruct higher-affinity clones to differentiate into GCPCs. However, in line with the inherent stochasticity of the GC reaction discussed above, tracing of GCPCs in polyclonal responses has demonstrated that even lower-affinity clones can make some contribution to the GCPC compartment [59,60,61]. These results indicate that GCPC differentiation is compatible with a spectrum of affinities rather than being restricted to clones at the highest end of the affinity range (Fig. 2). In addition to the selection events taking place within GCs, GCPCs remain transiently proliferative after GC egress, with their expansion capacity preprogrammed by the magnitude of TFH help received during LZ selection [56]. This preferential post-GC expansion of higher-affinity GCPCs contributes to the enrichment of high-affinity cells within the GCPC compartment [56]. How much the formation of the high-affinity GCPC compartment depends on GCBC commitment to the PC lineage driven by BCR signaling and T cell help, as opposed to postcommitment expansion of higher-affinity PCs, remains to be fully elucidated.

A possible model of cell fate decisions in GCs. The relative affinity of a GCBC’s BCR for an antigen influences the strength and duration of positively selecting signals (signals from BCR and TFH cells) and may instruct cell fate decisions along the axis: death (absence of positively selecting signals) < GCMBC differentiation < DZ reentry < GCPC differentiation (top). However, the exact boundaries between these outcomes are likely to be strongly blurred by stochastic events in the GC environment (bottom)
GCMBC differentiation
Although several insights have been provided into how affinity maturation progresses in GCs and how the GCPC compartment is formed over time, the mechanisms shaping the GCMBC pool remain much less well defined. A series of studies have demonstrated that the GCMBC compartment is seeded by B cells spanning a broad spectrum of antigen affinities, including both lower- and higher-affinity clones [1, 2, 62, 63], with some MBCs exhibiting extremely low affinity for their antigen [1]. Strikingly, a recent study suggested that the majority of GCMBCs, unlike contemporaneous GCBCs, were unable to bind the antigen used for immunization [1]. However, these MBCs were antigen specific, as antibodies cloned from such cells were able to interact with the antigen when the valency of the interaction was increased by multimerization [1]. In addition to their lower average affinity, GCMBCs also exhibit greater clonal diversity compared to GCPCs [1,2,3,4]. This diversity of MBCs, which includes cells of very low affinity, is thought to be beneficial for responses against pathogen variants. Indeed, the importance of this diversity in recall responses has been demonstrated by studies showing that protective antibodies against flavivirus and influenza variants arise primarily from the rapid differentiation of MBCs into PCs rather than from LLPCs established during primary infection [3, 4, 7].
In contrast to DZ reentry or GCPC fate commitment, which are linked to bursts of proliferation, GCMBC differentiation is associated with a transition to quiescence. Immunization experiments using cell cycle reporter mouse lines have revealed the presence of antigen-experienced, MBC-like cells carrying SHMs in their BCRs among cells with the GCBC surface phenotype and/or among cells physically located in GCs [63, 64]. These cells display features more consistent with MBCs than with typical GCBCs, including quiescent G0 cell cycle status, upregulation of cell-surface markers typical for naïve and memory B cells, and reduced expression of the transcription factor BCL6, a master regulator of the GCBC transcriptional program [63, 64]. Multiple studies have identified similar or overlapping populations exhibiting MBC-like characteristics within the GCBC compartment, which have been used to investigate the mechanisms underlying GCMBC differentiation [62,63,64,65,66]. Notably, across these studies, these putative GCMBC precursors were consistently enriched among GCBCs with LZ-associated surface phenotype [62,63,64,65,66]. These findings, along with evidence from experiments showing enhanced MBC output from CXCR4-deficient GCBCs (which are confined to the LZ) [67], support the notion that the GC LZ may serve as the primary site of GCMBC commitment. Overall, attenuation of the GC program in the LZ, coupled with a transition to quiescence, seems to constitute the key process driving GCMBC differentiation.
Since the GCMBC compartment appears to be enriched for relatively low-affinity clones [1,2,3, 62] and GCMBC commitment is associated with a transition to quiescence rather than a proliferative state, a key question arises: how do these cells avoid apoptosis during this process? Genetic ablation of BCR expression in GCBCs has been shown to trigger apoptosis in candidate GCMBC precursors [63], making it unlikely that cells lacking positively selecting pro-survival signals (or, at the very least, lacking tonic BCR signaling [68,69,70]) in the LZ could successfully develop into GCMBCs. This requirement for positive selection signals, together with the enrichment of relatively low-affinity clones in the GCMBC compartment and their transition to quiescence within GCs, supports the notion that GCBCs receiving signals strong enough to sustain survival but too weak to trigger proliferation are preferentially directed toward the GCMBC fate (Fig. 2). One factor implicated in the “translation” of weak positive signals into GCMBC differentiation is the transcription factor BACH2, which is expressed in naïve and activated B cells but not in PCs [2, 71, 72]. LZ GCBCs with lower-affinity BCRs that receive weaker T cell help have been shown to upregulate BACH2 and decrease mTORC1 signaling [63] (the latter is driven by T cell help that promotes positive selection and proliferation within GCs [73]). BACH2 upregulation appears to be directly linked to the transition to quiescence, as BACH2 ablation in GCBCs leads to hyperactivation of mTORC1 and elevated MYC expression [63], both of which promote proliferation in GCs [52, 53, 73, 74]. In parallel, BACH2 has been shown to actively repress the expression of Prdm1, encoding the transcriptional regulator BLIMP1, which is essential for commitment to the PC lineage [71, 75] (Fig. 3). Notably, these observations, together with the established role of the BACH2-BLIMP1 axis in T cell differentiation, where BACH2 promotes memory cell fate by antagonizing BLIMP1-driven effector differentiation [76,77,78,79], point toward conserved regulatory circuits governing memory versus effector differentiation across the B and T cell branches of the adaptive immune system [76].

Molecular regulation of GCMBC differentiation. GCMBC differentiation includes downregulation of the GCBC transcriptional program, transition to quiescence, and upregulation of prosurvival factors. This figure summarizes the role of the factors discussed in the “GCMBC differentiation” section in each of these three processes. Lines depict both direct and indirect regulation. See the text for further details
The antiapoptotic protein BCL2 may play a central role in supporting the survival of GCBCs differentiating into GCMBCs. In contrast to the majority of GCBCs, which express very low levels of BCL2, the candidate GCMBC precursors have been shown to upregulate this prosurvival factor [63]. Moreover, studies using Bcl2-transgenic mice demonstrated that inhibition of apoptosis within GCs leads to a significant increase in MBC numbers [80, 81]. However, it remains unclear whether this increase was due to enhanced survival of GCBCs in general, increased survival and MBC differentiation of GCBCs that otherwise would undergo apoptosis, increased survival of MBCs after their differentiation, or a combination of these mechanisms [80, 81]. Interestingly, a more recent study showed that constitutive Bcl2 expression in GCBCs can allow cells with nonfunctional BCRs generated in the DZ to become quiescent and begin expressing MBC-associated markers, although these cells fail to transition from the DZ back to the LZ [82]. While the exact mechanisms that induce Bcl2 expression remain unclear, downregulation of the GC program may play a key role, as the transcriptional repressor BCL6, a master regulator of the GCBC gene expression program, has been shown to repress Bcl2 expression in GCBCs [83] (Fig. 3). BCL6 also appears to repress Hhex expression, which encodes the transcription factor HHEX [66]. HHEX was suggested to positively regulate GCMBC generation through the downregulation of BCL6 and release of BCL6-mediated repression of BCL2 [66, 84] (Fig. 3). Overall, these observations suggest an antagonism between the GCBC program maintained by BCL6 and the prosurvival program necessary for GCMBC differentiation.
Recently, the transcriptional repressor ZBTB18, which is induced in GCMBC precursors by IL-9 [64], has been implicated in the regulation of all key aspects of GCMBC differentiation, including the suppression of cell cycle-associated genes, the repression of some GCBC program components, and the promotion of GCMBC precursor survival [85] (Fig. 3). The availability of another TFH-derived cytokine, IL-4, has also been shown to influence GCMBC differentiation, with one study suggesting that IL-4-mediated downregulation of BCL6 may facilitate GC exit [86]. However, assessing the overall effect of IL-4 on GCMBC differentiation has been challenging, as it regulates multiple key processes throughout B cell responses, including GC initiation [86, 87], and has been reported to either promote or suppress GCMBC development, depending on the timing of its experimental manipulation [86, 88].
Collectively, these observations suggest that relatively low levels of selection signals may allow certain GCBCs to exit the GC reaction as GCMBCs. These low-level signals appear to be translated into downregulation of the GCBC transcriptional program, a transition to quiescence, and upregulation of prosurvival factors such as BCL2 (Fig. 3). It is plausible that this quiescence-based, rather than proliferation-driven, mode of generation contributes to the increased clonal diversity observed within the GCMBC compartment [1,2,3,4]. Indeed, mechanisms that translate affinity differences into differential proliferation rates may result in strong overrepresentation of certain clones, thereby reducing diversity. In contrast, the differentiation of GCMBCs is not associated with such clonal outgrowth and therefore may preserve greater clonal diversity. In addition, as the GCMBC fate is compatible with a broad range of affinities—and as affinity-reducing and neutral mutations should be more common than those that increase affinity—this may further promote diversification of the GCMBC repertoire.
Putting the full spectrum of cell fate outcomes in the GC reaction into perspective, the insights discussed above suggest that the four possible fates of GCBCs can be broadly classified into two major trajectories. Stronger positively selecting signals drive proliferative outcomes, including DZ reentry for continued participation in the GC reaction, as well as differentiation into GCPCs. In contrast, weaker—or even absent—positively selecting signals can lead to either cell death or entry into a quiescent state, marking commitment to the GCMBC fate. While the strength of positive selection signals clearly influences the choice between “proliferative” and “quiescent” trajectories, the hierarchy of cell fate decisions within these categories remains less well defined. It is conceivable that all four fates can be positioned along one axis of positively selecting signal strength (e.g., cell death < GCMBC differentiation < DZ reentry < GCPC differentiation) (Fig. 2). If so, the exact boundaries between these outcomes are likely to be strongly blurred by stochastic events in the GC environment [54] (Fig. 2). For example, a high-affinity B cell that fails to engage with a TFH cell by chance may enter the GCMBC pool, explaining the existence of high-affinity MBCs. Conversely, a lower-affinity cell that happens to capture a large antigen aggregate might expand disproportionately within the GC and/or GCPC compartment. Finally, it is also conceivable, although perhaps less likely, that fate decisions within each branch—proliferative or quiescent—are entirely stochastic. Overall, the findings described above illustrate the complexity and dynamic nature of cellular decision-making within the GC. As discussed below, conceptually similar lineage choices also take place at the earlier stages of B cell activation prior to initiation of the GC reaction.
Pre-GC memory formation
Early B cell activation
Although the GC pathway is the most extensively studied route for MBC and PC generation, both cell types can also arise directly from early activated B cells (or from reactivated MBCs) independently of the GC reaction (Fig. 1). After recognizing their cognate antigen in the follicles of secondary lymphoid organs, B cells internalize it, process it for presentation on MHC class II molecules [89, 90], migrate to the outer edge of the follicle—the border between the B and T cell zones—and engage with antigen-specific CD4⁺ T cells [89, 90]. Productive interactions between MHC II-peptide complexes on B cells and T cell receptors on CD4⁺ T cells trigger the delivery of T cell help, including costimulatory prosurvival and mitogenic signals through CD40-CD40L interactions and the secretion of cytokines such as IL-4 and IL-21, which together drive the rapid proliferation of B cells at the follicular perimeter [19, 89,90,91]. These activated B cells acquire a unique cell-surface phenotype characterized by the coexpression of markers associated with both naïve/memory (CD38, CCR6) and activated/GCBC (CD95, GL7) cell states [92,93,94,95]. Like naïve B cells, these cells can initially retain IgD expression [92,93,94,95], although some of them undergo class-switch recombination (CSR) early on and therefore lose IgD and IgM BCRs [92, 96, 97]. These cells express intermediate levels of the transcription factor IRF4 [95, 98, 99], which is required for both PC and GCBC differentiation [99, 100], and exhibit either absent or low expression of BCL6 [95, 98, 99, 101], the key transcriptional regulator of GCBC fate [102, 103]. This population, often referred to as “activated precursors”, is thought to represent common tripotent progenitors of GC-independent early plasma cells (EPCs), early memory B cells (EMBCs) and GCBCs (Fig. 1). In support of this tripotency, limiting dilution cell transfer experiments, which enable tracking of the fate of single naïve B cells in vivo, have demonstrated that an individual naïve B cell is capable of generating all three lineages—GCBCs, EPCs, and EMBCs—although cell death may prevent some clones from contributing to all compartments [104]. These lineage choices take place in the first days after antigen encounter, and at least in some experimental systems, the split between the three lineages is evident by day 4 after immunization [92, 95, 98, 99, 105]. Although these pre-GC events are much less well characterized than those occurring within GCs are, it is evident that, like the lineage choices made in GCs, these fate decisions are largely shaped by the availability of antigen and T cell help.
Cell fate decisions early upon B cell activation: EPC and GCBC differentiation
As discussed in the section “Cell fate decisions in GCs”, increased access to antigen and/or T cell help in GCs favors GCPC generation. Similarly, injection of antigen or anti-CD40 antibody during the early stages of the response has shown that increased availability of antigen and T cell-derived signals shifts the composition of the early activated B cell compartment toward the EPC lineage [92, 98]. Under physiological conditions, where antigen can become limiting very early on [106,107,108], resulting in increased competition, BCR affinity likely plays an important role in determining access to antigen and T cell help. Indeed, experiments comparing the fate of early activated BCR transgenic B cells in response to immunogens of varying affinities have shown that higher-affinity interactions increase the frequency of EPCs relative to that of GCBCs or EMBCs [109,110,111]. At the single-cell level, while the limiting dilution cell transfer experiments described above clearly demonstrated that individual naïve B cells can generate all three lineages, their fate varied with antigen affinity. While in response to both higher-affinity antigens and lower-affinity antigens, naïve B cells showed similar potential to generate GCBCs [104], higher-affinity immunization favored EPC formation, whereas EMBCs were generated more frequently in response to lower-affinity antigen [104]. Overall, these observations suggest that increased antigen availability, the expression of higher-affinity BCRs, and stronger T cell-derived signals favor the generation of EPCs.
The experiments described above indicate that GCBCs can, in principle, be generated by both higher-affinity and lower-affinity B cells; however, in a competitive setting, lower-affinity clones compete poorly for T cell help and may not contribute efficiently to the GCBC compartment [94, 112,113,114,115]. Although the precise quantity or quality of signals that might favor GCBC over PC differentiation remains unclear, it is conceivable that GCBC differentiation is induced by intermediate levels of BCR signaling and/or T cell help. However, unlike PC differentiation, which can be easily induced in vitro by anti-IgM or anti-CD40 stimulation in the presence of cytokines, the GCBC fate, to our knowledge, cannot be induced by a simple reduction in the levels of these stimuli, suggesting that GCBC specification requires a more complex regulatory framework. In line with this notion, an interesting alternative hypothesis suggests that interruptions in CD40-CD40L signaling, rather than signaling strength per se, may bias differentiation toward the GCBC lineage [98]. Indeed, short-term removal of the anti-CD40 antibody after in vitro B cell stimulation was sufficient to induce BCL6 upregulation in a fraction of cultured B cells [98], and the transient decrease in T cell-derived signals in vivo through anti-CD4 or anti-CD40L treatment during the early stages of B cell activation seems to increase the propensity of activated B cells to generate GCBCs [98]. In contrast, increasing and/or prolonging signals associated with T cell help by injecting anti-CD40 antibodies blocked GCBC generation and induced a wave of EPCs [98].
Finally, it is also important to note that, at least in some immunization scenarios, EPCs and GCBCs emerge asynchronously, and EPC generation is restricted to a narrow time window very early in the response before the emergence of the first GCBCs [92]. This suggests that at a given timepoint, an activated precursor may choose between turning on a single possible “effector program” (either EPC or GCBC) or remaining in the precursor state rather than making an actual choice between the induction of the GCBC and EPC programs. A very large fraction of these activated precursors, however, do not induce either the GCBC or EPC molecular programs. Instead, as discussed in the next section, they undergo “differentiation by default” to the EMBC lineage [92].
EMBC differentiation
While most studies of B cell memory have focused on GCMBCs, it has long been recognized that MBCs can also arise from cells that never participate in the GC reaction. Early evidence for such generation of MBCs outside of GCs comes from studies in mice lacking BCL6 in their hematopoietic compartment [116], as well as from patients with CD40L deficiency [117], both of which are unable to form functional GCs but still generate detectable populations of MBCs. Further support for a GC-independent MBC pathway came from studies that showed the early emergence of antigen specific, largely unswitched cells with an MBC phenotype before the appearance of the first GCBCs [93, 118]. Moreover, at least in some immunization scenarios, most early activated B cells start exiting the cell cycle within the first few days of the response, giving rise to a large wave of such EMBCs rather than contributing to the GCBC or EPC compartments [92]. Notably, these EMBCs are also not affected by BCL6 deficiency [93]. Finally, BrdU pulse-chase experiments that allow tracking of B cells exiting the cell cycle in the first days of the response, prior to GC formation, demonstrated that EMBCs can be long-lived [119].
Reminiscent of the MBC differentiation in GCs, which is driven by weaker—or even absent—access to antigen and/or T cell help, the transition to quiescence, which marks the generation of EMBCs during early B cell activation, also appears to be linked to limited or missing signals that would otherwise drive sustained proliferation and differentiation into GCBCs or EPCs. In line with that notion, it was shown that the early cell cycle exit of the majority of activated B cells that marks EMBC differentation is driven by a rapid decline in antigen abundance outside of GCs during the first few days of the response [92]. Indeed, in common immunization scenarios, antigen is readily detectable across B cell follicles in the first hours after immunization, but its presence becomes rapidly restricted to GCs thereafter [106,107,108]. The provision of additional antigen several days after immunization prevents the transition to quiescence associated with EMBC differentiation and instead promotes the differentiation of early activated B cells to the EPC lineage [92]. A similar dominance of quiescent EMBCs early in the response that coincided with rapid loss of antigen outside of GCs has been observed after immunization in nonhuman primates, suggesting that the pathway leading to EMBC differentiation is evolutionarily conserved [92].
Thus, although the EMBC fate appears to be adopted by a large fraction—if not the majority—of activated B cells from very early stages upon activation [92], this state seems to reflect not an active differentiation process but rather a passive transition to quiescence driven by the loss of activating signals. This transition is marked by minimal transcriptional changes, in sharp contrast to the rapid and extensive transcriptomic divergence observed in cells committing to either the GCBC or EPC fates [92]. These findings suggest that EMBC differentiation may represent a “default” pathway in which cells largely retain the transcriptional identity of the common precursor state. Moreover, these findings could imply that the relative abundance of EMBCs within the MBC pool may be shaped primarily by the extent of initial expansion of early activated B cells in the first days of the response, which in turn can be influenced by the naïve precursor frequency, affinity of their BCRs to antigen, and antigen abundance early in the response. In support of this idea, it has been shown that mice with a greater frequency of naïve B cell precursors bearing germline-encoded, high-affinity BCRs for phycoerythrin (PE) developed MBC compartments dominated by IgM⁺ MBCs [120]—a subset likely enriched for EMBCs (see the “Shared and distinct properties of GCMBCs and EMBCs” section below).
Another layer of regulation of EMBC abundance may involve competition for extrinsic survival factors in the microenvironment, which could promote the long-term persistence of these cells. The cytokine BAFF, for example, has been suggested to be essential for EMBC generation [121] and for the maintenance of both EMBCs and GCMBCs [121, 122]. Additionally, a recent study suggested that Notch2 deficiency preferentially impairs EMBC development [123]. BAFF is essential for the survival of all naïve B cells [124,125,126], whereas Notch signaling is critical for the development of marginal zone (MZ) B cells [127, 128], which make up a substantial fraction of the naïve B cell pool in the spleen. The reliance of EMBCs—and in some instances GCMBCs—on maintenance factors shared with naïve B cells supports the notion that the MBC lineage choice may not entail the activation of a complex, specialized differentiation program but rather represents a partial reversion to a naïve-like quiescent state. Nevertheless, as discussed in the following sections, MBCs acquire functional properties distinct from those of naïve B cells, which shape their fate upon reactivation.
Comparison of GCMBCs and EMBCs
Shared and distinct properties of GCMBCs and EMBCs
Direct comparison of EMBCs and GCMBCs has remained challenging until recently, as the utilization of cell-surface markers and the Ig class switching status of the cells does not allow definitive discrimination between the two subsets. In recent years, the development of GC-specific fate-mapping systems using tamoxifen-inducible Cre recombinase driven by regulatory elements of GC-specific genes such as S1pr2 and Gcsam (Gcet) has enabled irreversible labeling of GC-derived populations [2, 129]. Such fate mapping allows direct discrimination between EMBCs and GCMBC, independent of isotype, surface marker expression, or the degree of SHM, which previously served as indirect indicators of GC or non-GC origin. These systems have now enabled phenotypic and molecular comparisons of EMBCs and GCMBCs as well as assessments of their contributions to the overall memory cell pool. Unexpectedly, such fate-mapping experiments demonstrated that EMBCs, which are often considered a minor memory subset, outnumber their GC-derived counterparts across multiple immunization scenarios ([92] and unpublished results). These studies also revealed that cell-surface markers, Ig class switching status, and SHM levels previously used to classify MBC subsets, while informative, are insufficient to definitively distinguish between EMBCs and GCMBCs [92, 93, 118, 119, 129,130,131,132]. Nevertheless, since much of our current understanding of EMBC and GCMBC biology is based on experiments employing these surrogate markers, we discuss them below.
The Ig class-switching status of MBCs is frequently used as indirect evidence of a history of participation in the GC reaction (or lack thereof). This was initially based on the assumption that CSR occurs predominantly in the GCs. Although recent studies have suggested that CSR may in fact take place primarily prior to GC formation, switched cells gain a competitive advantage in GCs and are therefore enriched in the course of the GC reaction [96, 97, 133]. In line with this notion, multiple studies have shown that MBCs generated early in the response are enriched for unswitched IgM⁺IgD⁻ or IgM⁺IgD⁺ cells, whereas those arising later are predominantly class-switched [92, 93, 118, 119, 132]. However, this distinction is not absolute since class-switched MBCs can be generated at pre-GC stages, albeit as a minority [92, 93, 118, 119]. Moreover, experiments with GC-specific fate mapping have demonstrated that EMBCs include a significant fraction of class-switched cells [129], whereas some GCMBCs can retain an IgM⁺ [129,130,131] or even IgM⁺IgD⁺ [132] phenotype. Taken together, these findings indicate that the utilization of class switching status is insufficient to definitively discriminate EMBC and GCMBC subsets and that even the IgD+ compartment can contain some antigen-experienced cells of both GC and non-GC origin.
Studies from the Shlomchik laboratory demonstrated that several subsets of MBCs can be phenotypically distinguished on the basis of the expression of the surface markers CD80, PD-L2, and CD73 [134]. Their subsequent work refined this classification by focusing on CD80 and PD-L2, identifying three subsets: double-positive (DP: CD80⁺PD-L2⁺), single-positive (SP: CD80⁻PD-L2⁺), and double-negative (DN: CD80⁻PD-L2⁻) MBCs [135]. A recent study using GC-specific fate mapping revealed that while the DP population is enriched for GCMBCs and that DN MBCs consist almost exclusively of EMBCs, this distinction is again not absolute, as a considerable proportion of DP cells appear to originate from the non-GC pathway [129].
In addition to the cell-surface phenotype, the level of hypermutation can also provide an indirect record of the GC history of a cell. Indeed, GCMBCs display higher mean levels of SHM in their immunoglobulin variable region genes than do their non-GC-derived counterparts [129, 132]. However, the mutation load within each population is highly variable [129, 132], and some GCMBCs display SHM levels similar to those of EMBCs and vice versa [129, 132].
Taken together, these results indicate that EMBCs are enriched for unswitched, DN MBCs with relatively low levels of SHM, whereas GCMBCs are more commonly class-switched, DP cells with higher SHM levels (Fig. 1). However, none of these characteristics are fully exclusive to either subset, and the phenotypic properties of the two populations significantly overlap.
GCMBC vs EMBC function upon reactivation
Despite their overlapping phenotypes, EMBCs and GCMBCs clearly exhibit distinct differentiation potentials upon reactivation. Studies analyzing the fate of IgM⁺ and class-switched MBCs—either upon boost immunization or adoptive transfer of MBCs into unimmunized recipients followed by immunization—have suggested that IgG1⁺ MBCs (enriched for GCMBCs) are biased toward PC differentiation, whereas IgM⁺ MBCs (enriched for EMBCs) are more likely to reenter secondary GCs [118, 136]. Similar observations were made using CD80 and PD-L2 expression as MBC subset markers: DP MBCs (enriched for GCMBCs) tend to be more prone to PC differentiation, whereas DN MBCs (enriched for EMBCs) more efficiently reenter secondary GCs [129, 135]. The propensity of GCMBCs to generate PCs rather than enter secondary GC reactions was directly shown by GC-specific fate-mapping experiments, which demonstrated that upon booster immunization, fate-mapped GCMBCs are predominantly biased toward PC differentiation and contribute only minimally to secondary GC formation [137, 138]. Instead, non-fate-mapped cells, a compartment that can include both naïve B cells [137, 138] and EMBCs, dominated the secondary GCBC pool under these conditions. Finally, a subsequent study revealed that these non-fate-mapped cells found in secondary GCs were derived primarily from naïve B cells [137]. Taken together, these findings suggest that EMBCs possess a greater intrinsic capacity to enter the secondary GC reaction than do GCMBCs, but their overall contribution to the secondary GCs remains smaller than that of naïve B cells.
Recent transcriptomic analyses suggest that EMBCs and GCMBCs are closely related at the transcriptional level, with only relatively subtle differences detected between the two subsets ([129, 132] and unpublished observations). The extent to which these differences may explain the functional differences between the two populations remains unclear. Given this limited transcriptional divergence at steady state, it is tempting to speculate that the distinct behaviors of EMBCs and GCMBCs upon reactivation may be preprogrammed at least in part epigenetically. In line with this notion, it has recently been shown that DN and DP MBCs exhibit distinct epigenetic profiles that may reflect their history of participation—or lack thereof—in the GC reaction, suggesting that these epigenetic signatures may predetermine their responsiveness upon secondary antigen encounter [129]. Mechanistically, this programming may take place at least in part through the epigenetic regulation of BACH2 and BLIMP1 expression [139]. Accumulated stimulation has been shown to progressively increase chromatin accessibility at PC-associated gene loci, including Prdm1, thereby increasing baseline BLIMP1 expression and biasing MBCs toward PC differentiation over reentry into the GC response [139] (Fig. 4). These results imply that repeated or prolonged antigen stimulation may gradually shift the MBC potential, contributing to the functional divergence observed between EMBCs and GCMBCs. In such a scenario, the history of participation in the GC reaction may indirectly determine the reactivation fate of MBCs by increasing the cumulative antigen experience of the cell through repeated cycles of stimulation in the GC. It therefore seems plausible, although remaining to be tested, that a B cell that would undergo repeated or prolonged activation without entering a GC could become biased toward PC differentiation (see also section “Atypical MBCs” below). Conversely, a GCMBC generated early in the GC reaction might be less prone to PC differentiation than a GCMBC derived from a GCBC that experienced many cycles of stimulation. Taken together, these findings align with an intriguing hypothesis that MBC fate upon reactivation might be programmed not by a binary GC versus non-GC origin but rather by the cumulative history of stimulation that is recorded epigenetically (Fig. 4).

Epigenetic “recording” of the cumulative activation history may preprogram the fate of MBCs in secondary immune responses. Repeated BCR stimulation and/or signals associated with T cell help gradually enhance chromatin accessibility at genes linked to the PC program, such as Prdm1, biasing MBCs upon reactivation toward PC differentiation rather than entry into secondary GCs. Such a mechanism could be partly responsible for the differential capacity of EMBCs and GCMBCs to generate PCs and GCBCs upon reactivation (see section “GCMBC vs EMBC function upon reactivation” for further details)
MBCs in the tissues
Historically, most studies on B cell memory have focused on MBCs that recirculate between secondary lymphoid organs and are therefore capable of responding to antigenic challenges at distal sites, for example, after intramuscular vaccination followed by respiratory infection. In humans, the study of these MBCs is further facilitated by their accessibility from peripheral blood. Recent studies have shown that certain nonlymphoid tissues harbor MBCs, some of which exhibit long-term tissue residency. Moreover, secondary lymphoid organs can also host nonrecirculating MBC populations. In the following sections, we review some of the anatomical sites where MBCs have been identified, examine the evidence supporting their potential tissue residency, and discuss the shared and distinct phenotypic, developmental, and functional characteristics of these cells.
Lungs and upper respiratory tract
Most of what is currently known about tissue-resident MBCs (BRMs) originates from studies of the lung, a primary site of exposure to respiratory pathogens. The presence of BRMs was first established in influenza-infected mice, where approaches such as parabiosis, intravenous antibody labeling, and pharmacologic blockade of lymphocyte egress via S1PR1 agonists revealed that influenza-specific MBCs could persist within lung tissue and mount rapid responses upon reinfection, strongly supporting their classification as a tissue-resident population [140, 141]. In line with their tissue residency, lung BRMs, unlike their circulating counterparts, exhibit hallmark features of local retention, including the upregulation of CD69 and the concurrent downregulation of S1PR1 [3, 142,143,144,145,146], a receptor that promotes lymphocyte egress from tissues into the circulation [143, 147, 148]. In most of the studies discussed above, lung BRMs have been shown to express the chemokine receptors CCR6 and CXCR3. Although these receptors have been suggested to regulate the migration and retention of BRMs in lung tissue, their exact contributions remain somewhat controversial [142, 149].
Upon respiratory infection, antigen-specific B cells can initiate responses in the mediastinal lymph nodes (medLNs) and within inducible bronchus-associated lymphoid tissue (iBALT)—a TLS that forms locally in the lung parenchyma—highlighting medLNs and iBALT as the most likely sites of BRM origin in the pulmonary environment [140, 145, 149,150,151,152,153,154,155,156,157]. Additionally, clonal sharing analysis in influenza-infected mice suggested that some BRMs might originate from splenic GCs [150]. Regardless of their site of origin, lung BRMs exhibit transcriptional profiles that are distinct from those of their splenic and lymph node counterparts in both mice and humans [145, 150] but that share some features with the residency program of CD8⁺ tissue-resident memory T cells [158].
While both IgG and IgA antibody responses can be generated following respiratory infection, the BRM pool after primary infection is predominantly composed of IgG⁺ MBCs, with a smaller proportion of IgA⁺ MBCs [140, 151, 152]. Notably, while IgG⁺ MBCs are distributed across both the lung and medLNs, IgA⁺ MBCs are primarily localized within the lung parenchyma, which is in line with the specialized role of IgA in mucosal immunity [149, 151, 156]. This compartmentalization is further supported by the results of experiments showing that systemic immunization with influenza virus fails to induce lung IgA⁺ MBCs, in contrast to intranasal immunization, which effectively promotes their formation [149]. The restricted localization of IgA⁺ MBCs, combined with the spatial association of IgA-secreting PCs with iBALT, suggests that IgA production in the lung is largely driven by these inducible lymphoid structures [145, 149, 151, 156, 157, 159]. Although the presence of IgA⁺ BRMs in the lungs is well established, the mechanisms governing their formation, reactivation, and contribution to protective immunity remain considerably less understood than those of their IgG⁺ BRM counterparts.
While IgG-secreting PC recall responses can originate from both circulating and resident IgG⁺ MBCs, a recent study demonstrated that influenza-boosted mice primed intranasally—a route that favors BRM induction—mounted faster and more localized PC responses than those primed intraperitoneally [156]. In particular, circulating MBCs exhibited delayed differentiation into PCs and were less able to generate PCs in close proximity to infected alveoli, in contrast to their lung-resident IgG⁺ BRM counterparts, which mounted a rapid local response. These IgG⁺ BRMs were not associated with iBALT but were instead dispersed throughout the lung parenchyma, patrolling a much larger area than that occupied by iBALT [154].
Boosting experiments with influenza virus or virus-like particles that lack antigenic similarity to influenza virions have demonstrated that BRMs of diverse specificities—including those unrelated to the invading threat—are rapidly recruited to the site of infection, presumably to “screen“ the local environment for the presence of their cognate antigen [154]. This rapid migratory process is orchestrated by alveolar macrophages, in part through the initiation of a cytokine cascade that involves IFN-γ production by NK cells, which in turn drives the induction of CXCL9 and CXCL10, thereby attracting CXCR3-expressing BRMs to the site of infection [154, 156]. Even more intriguingly, some of these BRMs can differentiate into PCs in response to innate signals alone, producing antibodies despite a lack of specificity for the current threat [156]. This phenomenon suggests that the immune system may, in certain contexts, prioritize speed over precision, particularly in vital tissues such as the lung, where delays in immune responses could be life-threatening. Whether this form of “bystander“ PC differentiation is unique to the lung or also occurs in other tissues remains an open question. It is also unclear whether this capacity is an intrinsic feature of BRMs established during their differentiation in the lung or instead instructed by external cues, potentially specific to highly vulnerable microenvironments such as lung tissue, where rapid local antibody production may be critical for host survival.
Overall, lung BRMs are the best-characterized population of resident MBCs in nonlymphoid tissues, yet their biology remains an area of active investigation, with many questions still unanswered regarding their heterogeneity and functional specialization. For example, the relative contribution and role of GC-derived versus GC-independent MBCs to the lung-resident MBC pool remains unclear. In many of the studies discussed above, BRMs have been identified and visualized via fate-mapping systems driven by the Aicda gene, which encodes AID, a key enzyme required for CSR and SHM [142, 154, 156]. However, because both AID expression and CSR can occur outside the GC reaction [92, 96, 97, 160,161,162], these approaches do not definitively distinguish the developmental origin of BRMs, leaving the precise composition of this population unresolved. Influenza-specific BRMs have frequently been shown to express high levels of CD80, PD-L2, and CD73 [140, 152, 163], which are surface markers associated with GCMBCs [129, 135]. However, most studies focusing on BRMs defined by these markers excluded from their analysis IgM⁺ and/or IgM⁺IgD⁺ populations, which are often enriched for EMBCs [92,93,94, 118].
Local immune responses in the respiratory tract are not restricted to the lower airways. Indeed, recent studies have identified antigen-specific GCs and populations of MBCs and PCs within the nasal-associated lymphoid tissue (NALT) of mice following intranasal immunization or infection [164, 165]. In line with these observations, CD69⁺ BRM-like cells have been detected in nasopharyngeal swabs from healthy individuals [166]. Together, these findings suggest that local humoral memory in response to inhaled threats could also be established in the upper respiratory tract, although the candidate BRM compartment at this site requires further characterization.
Skin
The skin represents another major body surface that is continuously exposed to environmental antigens and microbial challenges, where local immune memory could play an important role. While direct evidence for the presence of MBCs in the skin is lacking, B cells have been identified in the dermis under both homeostatic and inflammatory conditions [167,168,169], and although they are present in very low numbers, they can include expanded clones [167,168,169] and class-switched cells [169]. A recent study has shown that exposure to Staphylococcus epidermidis induces the formation of dermal TLSs and triggers antigen-specific antibody responses, including the generation of LPPCs that persist for months after exposure [170]. Within these TLSs, skin-associated B cells were shown to undergo class switching to produce IgG2b and IgG2c antibodies, supporting localized humoral responses independent of secondary lymphoid organs [170]. However, whether this cutaneous B cell response can also generate skin-resident B cell memory remains to be tested.
Gut
Like the respiratory tract and the skin, the gastrointestinal tract represents a highly immunologically active site owing to its constant exposure to dietary antigens, commensal microbes, and potential pathogens. PCs in the gut, which predominantly secrete IgA, arise through both T-dependent and T-independent mechanisms [171,172,173,174] and not only protect against pathogens but also shape the composition of the commensal microbiota [171, 173,174,175,176,177,178]. Studies using oral immunization have demonstrated rapid and enhanced mucosal antibody responses upon secondary exposure, suggesting the presence of MBC populations in the gut [179,180,181,182,183]. Recently, such a population of antigen-specific IgA⁺ MBCs was detected among adoptively transferred model antigen-specific B cells that were activated via oral immunization with an antigen conjugated to cholera toxin [184]. These MBCs were found in PPs and mesenteric lymph nodes (mesLNs) and could be detected even one year after priming [184]. Notably, a recent study employing a B cell specific, tamoxifen-inducible Cre recombinase to label B cells generated during a defined developmental window demonstrated that many labeled IgA⁺ MBCs in PPs and PCs in the lamina propria (LP) of the gut are clonally related [185]. Moreover, these MBCs likely contributed to rapid LP PC reconstitution upon depletion of the latter cells with bortezomib [185]. Taken together, these results suggest that these cells may be part of a gut-resident MBC compartment that contributes to the long-term replenishment of the PC pool in the intestinal LP.
However, whether the intestinal MBC compartment contains bona fide BRM cells remains to be formally tested. It also remains to be determined whether these cells are strictly confined to gut-associated lymphoid tissues (i.e., PPs and isolated lymphoid follicles) or whether they can also populate the LP. Serving as indirect evidence for the latter, in human intestinal samples, excluding epithelial tissues and PPs, MBC-like cells have been identified, with nearly half expressing CD69 [186], a marker widely associated with tissue residency in both B and T cells. Moreover, although evidence supports the presence of GCMBCs in the gut [184, 187], the existence and potential roles of non-GC-derived MBCs, whether arising through T-dependent or T-independent pathways, remain to be investigated. In addition, the relative contribution of pathogen-specific and microbiota-specific cells to the MBC compartment of the gut as well as their role in maintaining antibody responses remain to be elucidated.
Liver
Beyond barrier tissues, the liver represents an organ in which local infection may give rise to MBCs with features of tissue residency, with the most compelling evidence coming from Ehrlichia muris infection in mice. In this experimental model, somatically hypermutated IgM⁺ T-bet⁺ MBC-like cells can be found in both the liver and the spleen [188]. In the liver, these cells reside within the tissue, as evidenced by their lack of labeling following intravenous antibody injection [188]. These MBCs are likely generated through a GC-independent pathway (as GCs are absent in the spleen in this infection model) and persist after infection clearance, suggesting their potential role in long-term protection against reinfection [188]. Repertoire analysis of spleen and liver MBCs, although predominantly identifying spleen-specific clones, suggested that approximately half of the liver-derived clones were exclusive to the liver [188]. Additionally, RNA-seq analysis revealed that liver but not splenic MBCs expressed Cd69 [188]. Moreover, in humans, some studies suggest the presence of atypical MBCs (see the “Atypical MBCs” section below) in the livers of chronic hepatitis B patients [189, 190]. Taken together, these findings indicate the presence of potential tissue-resident MBCs in the liver, but further experiments are needed to assess their migratory properties, the mechanisms underlying their generation, and their possible functional role.
Evidence for lymph node-resident B cell memory
In addition to the presence of MBCs in nonlymphoid tissues, substantial evidence suggests the existence of resident MBCs at strategic locations within secondary lymphoid organs, enabling them to mount rapid recall responses upon antigen re-exposure. Studies using parabiosis, intravenous antibody labeling, and live imaging of MBCs after immunization or infection have demonstrated that a large fraction of antigen-specific MBCs can persist in draining lymph nodes (dLNs) [140, 191]. Although these MBCs display migratory behavior within lymph nodes similar to that of naïve B cells, which frequently scan the subcapsular sinus (SCS) [191], where the lymph first enters and lymph-borne antigens are captured and displayed by SCS macrophages [192, 193], their overall localization is distinct. Unlike naïve B cells, which show no apparent bias between the inner and outer regions of B cell follicles, MBCs in dLNs are strongly biased toward the outer follicular area close to the SCS [191]. Their positioning in this strategic niche, which is enriched for antigen and memory TFH cells [194], ensures rapid recall responses [191]. Although this localization is dependent on signals from SCS macrophages [195], the precise cues that guide it remain to be identified.
Interestingly, antigen-specific MBCs found in distant nondraining lymph nodes (ndLNs) upon immunization do not show a comparable residency pattern, indicating a unique resident subset in dLNs [195]. Consistent with this, transcriptional differences in dLN and ndLN MBCs have been observed, some of which may underpin their distinct localization patterns within LNs [195]. These observations may have implications for vaccination and boosting strategies, as the two MBC populations respond differently upon reactivation, with MBCs in dLNs more efficiently re-entering secondary GCs following boosting than those in ndLNs do [138, 195, 196]. These findings indicate that the site of booster administration, for example, whether booster vaccinations in humans are given in the same arm or the opposite arm as the primary injection, can influence the quality of the resulting immune response.
MBCs in the marginal zone of the spleen
While MBCs in the SCS of LNs represent a key population poised to mount rapid recall responses to lymph-borne antigens, substantial evidence also supports the presence of a distinct MBC subset in the marginal zone (MZ) of the spleen. Like the SCS in LNs, the MZ is strategically positioned to intercept circulating antigens since it lies at the interface of red and white pulp and is directly exposed to blood circulating through the splenic sinuses [197]. Its direct exposure to circulating blood makes the MZ an ideal location for rapid immune surveillance of bloodborne pathogens and for mounting immediate responses to systemic threats. The MZ is populated by marginal zone B (MZ B) cells, a specialized subset of B-2 cells that are thought to diverge from the conventional follicular B (FO B) lineage during peripheral maturation in the spleen [197, 198]. While the human spleen has a different anatomy than that of mice [197], B cells with an MZ B cell phenotype can be found within B cell follicles near the red–white pulp border, distal from the T cell zone, which is an area with increased accessibility to bloodborne antigens [197, 199, 200]. The development of naïve MZ B cells critically depends on Notch signaling, [127, 128] which imprints a unique transcriptional identity on this population [201,202,203,204]. BCR signaling has also been suggested to play a role in the FO vs MZ B cell fate decision; however, the exact mechanism has been a matter of debate [199, 205,206,207,208].
Although the MZ B cell compartment in rodents is thought to be mostly composed of naïve innate-like B cells that, upon activation, rapidly generate PCs producing low-affinity IgM antibodies as a first line of defense against bloodborne pathogens [199, 209,210,211], the situation appears to be different in humans. Indeed, a large fraction of human MZ B cells are considered to represent MBCs, as judged by the expression of the memory marker CD27 [212, 213] and, more importantly, the fact that many of those cells carry BCRs that have undergone SHM [214,215,216,217]. This apparent discrepancy between mice and humans might be explained, at least in part, by the use of mice raised under relatively “clean” laboratory conditions with limited microbial exposure. In support of this notion, immunization studies in mice have shown that MBCs with MZ B cell characteristics (as evidenced by cell-surface marker expression, transcriptional profiles, and localization) can emerge following antigenic challenge [123, 142, 218,219,220,221,222]. These observations suggest that the splenic MZ can serve as a niche for MBCs both in mice and in humans. Notably, despite the reported shuttling of B cells between the MZ and B cell follicles [223], parabiosis experiments have shown that naïve MZ B cells represent largely a spleen-resident nonrecirculating population—at least within the tested 3-week time frame—in contrast to FO B cells, which are distributed equally between parabiotic partners [224]. The retention of these cells within the MZ is mediated by several G protein-coupled receptors, including S1PR1 [225], CNR2 [226] and CD97 [227]. Of note, these molecules are expressed at higher levels in MZ B cells than in their FO counterparts, suggesting a specialized tissue residency program. It is intriguing to speculate that MZ-MBCs could likewise induce this residency program, although this remains to be formally demonstrated.
The developmental origin and functions of MZ-MBCs remain largely unknown. It remains unclear whether these cells arise from early activated B cells, through the GC reaction, or via both pathways and whether they preferentially originate from responses initiated by naïve MZ B cells. Similarly, it is not known whether MZ-MBCs can be generated in response to a broad range of immunological challenges in various locations or whether they are primarily induced by antigenic encounters in the spleen. Functionally, it is tempting to speculate that their strategic localization facilitates rapid antibody production upon reencounter with systemic threats; however, this hypothesis remains untested. Pulse-chase labeling experiments have suggested that naïve MZ B cells persist longer than their FO counterparts in the steady state [185, 228, 229]. Given that longevity is a hallmark of immunological memory, it is tempting to speculate that the splenic MZ could serve as an MBC survival niche. Indeed, a recent study demonstrated that long-lived anti-smallpox MBCs with an MZ B cell phenotype and transcriptional features dominate the switched MBC compartment in the spleens of individuals immunized decades earlier with the smallpox vaccine [230], although it remains unclear whether this dominance reflects preferential generation of such MBCs in response to this vaccine or their preferential long-term survival. Taken together, these findings suggest the intriguing – albeit still speculative – possibility that MZ-MBCs represent a specialized, long-lived, strategically positioned BRM subset that has evolved to mediate rapid recall responses to systemic threats.
Bone marrow
While the BM has long been recognized as a primary niche for antibody-secreting LLPCs [231,232,233,234], some evidence suggests that it also serves as a site for MBC residence. In humans, IgM⁺ mature B cells bearing somatically hypermutated BCRs were reported in BM samples from healthy individuals [235]. Moreover, a recent study revealed the accumulation of antigen-specific MBCs in the BM of immunized mice [222]. These cells are detectable more than one year post-immunization [222]. Most strikingly, BM MBCs had a transcriptional signature and BCR repertoire distinct from those of splenic MBCs [222]. The latter result indicates that there is no complete exchange between the MBC compartments of the two organs and suggests that at least some of the MBCs in the BM may represent resident cells. In line with this possibility, MBCs in the BM expressed higher levels of adhesion molecules, such as integrins α4β1 and α6β1, and were found to be in close contact with stromal cells expressing VCAM-1, the ligand for α4β1 [222].
From a teleological perspective, the reasons for the accumulation of MBCs in the BM remain unclear. It is possible that, similar to PCs, they may benefit from survival signals provided by specialized stromal niches in this tissue. However, unlike PCs, which confer protection at a distance through continuous antibody secretion, the functional advantage of MBCs in the BM is less obvious. One possibility is that these cells can still recirculate and/or be mobilized to peripheral lymphoid organs in the course of secondary immune responses. Alternatively, although the extent to which the antigen can reach the BM via the circulation remains unknown, it is tempting to speculate that this site may support the local activation and differentiation of MBCs into PCs, potentially providing a strategic location for the generation of antibody-secreting cells directly within a niche that also supports their long-term maintenance.
Additional “layers” of MBC identity
While much of our current understanding of B cell memory stems from studies of MBCs generated in adulthood following vaccination or acute infection, recent studies are beginning to shed light on the unique properties of MBCs that arise under less studied conditions. This, for example, includes B cell memory that emerges early in ontogeny as well as atypical MBCs that are generated and persist under the conditions of chronic antigenic stimulation. In the following sections, we highlight recent advances—as well as remaining open questions—in our understanding of these MBC populations.
Early-life-origin MBCs
During the neonatal period, the naïve immune system is rapidly exposed to a wave of novel antigens, which can drive the development of LLPCs and MBCs. Moreover, neonatal B cells possess a distinct BCR repertoire as they develop in the absence of terminal deoxynucleotidyl transferase (TdT) activity [236], and they have been suggested to preferentially utilize proximal (i.e., located close to D and J segments) V segments in their Ig heavy chain rearrangements [237,238,239]. A recent study using a B cell specific, tamoxifen-inducible Cre recombinase for pulse-chase labeling demonstrated that B cell clones generated within the first two weeks of life in mice contribute substantially to antigen-experienced B cell compartments across multiple organs in adulthood [185]. This contribution was particularly pronounced in mucosal tissues, likely reflecting exposure to microbial antigens [185]. The majority of IgA⁺ PCs in both the small intestinal LP and BM, as well as a significant proportion of GCBCs in PPs, were derived from these early-life B cell precursors [185]. Notably, a substantial fraction of both IgA⁺ and IgA⁻ MBCs in PPs (possibly GCMBCs, as evidenced by their expression of CD73 and PD-L2 [129, 135]) originated from this early-life-origin (ELO) B cell wave; these cells carried SHMs in their B cell receptors and were clonally related to the PCs in the LP [185].
These observations demonstrate that a substantial fraction of the adult MBC compartment throughout life can be composed of MBCs generated early in ontogeny. It remains to be investigated whether the ontogenetic timing of MBC generation can influence the functions of these cells, both in terms of the extent of their contribution to secondary B cell responses and in terms of the quality of these responses. In addition, the developmental pathways underlying ELO MBC formation remain to be defined, including whether these cells are driven primarily by T-dependent or T-independent stimuli and whether they are generated exclusively by the GC pathway or can also arise independently of the GC reaction. Interestingly, in addition to being enriched in mucosal tissues, ELO B cells are enriched among splenic MZ B cells [185]. As the spleen MZ may serve as a hub for MBCs (see the “MBCs in the tissues” section above), it would be interesting to test whether ELO B cells contribute to the pool of MZ-MBCs in the spleen.
Atypical MBCs
Atypical B cells (ABCs) are a distinct subset of B cells identified across various immune contexts, including chronic infections, vaccination, and autoimmune diseases [240,241,242,243]. In humans, ABCs typically lack classical MBC markers such as CD27 and CD21 and often express the transcription factor T-bet along with distinct surface markers, including CD11c, CXCR3, FCRL4, and FCRL5 [243,244,245,246,247,248]. A functionally and phenotypically related population has been identified in mice as CD11c⁺CD21lo “age-associated” B cells, as these cells expand with age as well as in the context of chronic infection and autoimmunity [242, 249,250,251,252,253,254,255]. Although primarily studied in chronic settings, ABCs can also expand during acute infections, both in mice [221] and humans [247, 256,257,258,259], as well as upon vaccination in humans [247, 260, 261]. These expanded ABCs then contract over a period of weeks to months in the absence of restimulation [258, 262, 263].
Despite their phenotypic heterogeneity, accumulating evidence points to a conserved transcriptional program. A recent study revealed that ABCs from individuals with malaria, HIV, and autoimmune diseases exhibit similar transcriptional profiles, suggesting a potentially shared differentiation trajectory across a variety of immune contexts [264]. While T-bet was believed to be essential for ABC development [248, 251, 265], recent findings indicate that T-bet is not strictly needed, as CD11c⁺ ABCs can still arise in infection and autoimmune models even in the absence of this transcription factor [266, 267]. In addition, Zeb2 has recently been identified as a key transcription factor involved in ABC development both in mice and humans [268, 269], suggesting that it may play a role in imposing a shared transcriptional program of ABCs across different contexts.
Mechanistically, ABCs are thought to arise following BCR stimulation in an inflammatory cytokine milieu, with their differentiation requiring additional signals from endosomal TLRs, as well as from IFN-γ and IL-21 [242, 249, 266, 270,271,272,273,274,275,276]. Growing evidence strongly supports the view that ABCs are not merely byproducts of inflammation but also, in most cases, antigen-experienced cells. It has been suggested that in autoimmune settings, ABCs originate from B cells that have encountered self-antigen [255, 269, 274, 275, 277]. In support of this, a recent study has shown that anergic B cells, which emerge following chronic self-antigen engagement, can be converted into ABCs and are more prone than naïve B cells to acquire an ABC phenotype under in vitro conditions that promote ABC differentiation [278]. Furthermore, ABCs have been found within the antigen-specific B cell pool following immunization in both mice and humans [221, 268, 279]. Some ABCs can express classical MBC markers [221, 255, 277, 280], carry somatically hypermutated BCRs [255, 277, 281], and respond to antigen restimulation [221, 280]. Moreover, ABC frequencies decrease significantly in patients with mutations affecting CD40/CD40L [276], and ABCs do not develop in mice with fixed BCR specificity under steady-state conditions [278], further supporting the notion that antigen encounter and, at least in some cases, T cell help is required for their formation.
Despite extensive investigations in recent years, many questions regarding ABC differentiation, homeostasis, and functions remain to be answered. For example, the extent to which the ABC compartment is maintained by self-renewal versus replenishment by newly activated B cells remains unclear. Moreover, the relative contributions of GC-dependent and GC-independent pathways to the ABC pool remain unclear. A recent study using GC-specific fate mapping demonstrated that, following acute viral infection in mice, the majority of antigen-specific ABCs are generated via a GC-independent pathway [221]. However, whether this holds true across other immune contexts that give rise to ABCs remains to be elucidated.
The functions of ABCs across different immunological contexts remain an active area of investigation. A recent study demonstrated that a reduction in ABC numbers through B cell-specific deletion of Zeb2 significantly improved disease outcomes in a lupus mouse model [269]. Similarly, depletion of CD11c⁺ or T-bet⁺ B cells decreased autoantibody levels and ameliorated disease in other autoimmune models [254, 255, 274]. While the latter depletion strategy may affect other activated B cell subsets, the findings suggest that ABCs can play an active, pathogenic role in autoimmunity. In the context of infection, B cell-specific deletion of Zeb2 resulted in decreased numbers of GCBCs during persistent Plasmodium infection, indicating a potentially beneficial role for ABCs in sustaining humoral immune responses [268]. Taken together, these findings support the idea that ABCs are functionally involved in shaping humoral immune responses, contributing to protective immunity during infection and sustaining autoreactive responses in autoimmunity. These observations reinforce the notion that ABCs are not merely byproducts of chronic immune activation but represent active players in B cell-mediated immunity. It remains to be investigated whether these cells play a role in autoimmunity and the response to pathogens predominantly through differentiation to antibody-producing PCs, antigen presentation to T cells, or both.
The precise function of ABCs upon reactivation remains incompletely understood. Early in vitro studies indicated that ABCs exhibit limited PC differentiation capacity [245, 246], raising questions about their effector potential. However, later work suggested that the lower PC differentiation observed in earlier studies may have been influenced by the mode of stimulation. Indeed, in contrast to soluble anti-Ig antibodies, membrane-bound anti-Ig effectively drove PC differentiation from ABCs, and it was suggested that soluble antibodies may dampen BCR signaling in these cells through inhibitory receptors such as FcRL5 and FcγRIIB [282]. Moreover, some studies suggest that ABCs are in fact transcriptionally and functionally primed for PC differentiation. This notion is supported by the expression of PC-associated genes by ABCs [221] and the increased propensity of these cells to differentiate into PCs in T cell coculture experiments [272, 283]. Given that ABCs are mostly studied in models involving chronic BCR stimulation, such a propensity would fit with the idea that they may be epigenetically programmed by sustained antigen engagement to favor terminal differentiation into PCs, similar to what has been proposed for conventional MBCs [139]. Nevertheless, a recent study involving adoptive transfer of ABCs between mice of the same autoimmune strain has shown that in addition to PC differentiation, ABCs are capable of both differentiating into GCBCs and self-renewing [255]. Whether this apparent functional plasticity is shaped by the degree of antigenic stimulation experienced by different ABCs prior to restimulation remains unclear. Overall, although the development and function of ABCs remain to be fully elucidated, gaining further insight into their behavior may help clarify how MBCs can develop, persist and function under conditions of chronic antigen exposure.
Concluding remarks and open questions
In this review, we discuss advances in our understanding of MBC development and how key parameters, such as developmental pathways, tissue localization, and the timing of generation, influence MBC properties. Despite substantial recent progress, many questions concerning MBC differentiation, maintenance, and function remain to be answered.
Until recently, most studies on B cell memory have focused on circulating GC-derived MBCs. Therefore, our understanding of the biology of other MBC subsets, such as tissue-resident MBCs (especially those outside of the lungs) and non-GC-derived EMBCs, remains limited. For example, although it is now clear that EMBCs can make a very significant contribution to the MBC pool after immunization [92], their involvement in response to chronic infections, the microbiota or participation in autoimmune diseases remains uninvestigated. Moreover, the lack of suitable tools largely precludes investigation of the participation of these cells in secondary immune responses, as existing fate-mapping approaches cannot simultaneously discriminate the progeny of naïve B cells, EMBCs, and GCMBCs in recall responses. The development of such tools would enable functional characterization of this understudied MBC population.
A hallmark of immunological memory is longevity. While the lifespan of MBCs can range from weeks to decades [230, 284, 285], the determinants underlying this remarkable variability remain unknown and arguably represent one of the most significant unresolved questions in the biology of B cell memory. Many of the parameters discussed in this review, such as developmental origin, activation history, and localization within specific tissue niches, may contribute to the long-term maintenance of MBCs, but this remains to be systematically investigated.
References
Viant C, Weymar GHJ, Escolano A, Chen S, Hartweger H, Cipolla M, et al. Antibody affinity shapes the choice between memory and germinal center B cell fates. Cell. 2020;183:1298–1311.e11.
Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol. 2016;17:861–9.
Wong R, Belk JA, Govero J, Uhrlaub JL, Reinartz D, Zhao H, et al. Affinity-restricted memory B cells dominate recall responses to heterologous flaviviruses. Immunity. 2020;53:1078–1094.e7.
Purtha WE, Tedder TF, Johnson S, Bhattacharya D, Diamond MS. Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J Exp Med. 2011;208:2599–606.
Rodda LB, Morawski PA, Pruner KB, Fahning ML, Howard CA, Franko N, et al. Imprinted SARS-CoV-2-specific memory lymphocytes define hybrid immunity. Cell. 2022;185:1588–1601.e14.
Goel RR, Painter MM, Apostolidis SA, Mathew D, Meng W, Rosenfeld AM, et al. mRNA vaccines induce durable immune memory to SARS-CoV-2 and variants of concern. Science. 2021;374:eabm0829.
Leach S, Shinnakasu R, Adachi Y, Momota M, Makino-Okamura C, Yamamoto T, et al. Requirement for memory B-cell activation in protection from heterologous influenza virus reinfection. Int Immunol. 2019;31:771–9.
Inoue T, Kurosaki T. Memory B cells. Nat Rev Immunol. 2024;24:5–17.
Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–9.
Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centers. Nature. 1991;354:389–92.
Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2022;40:413–42.
Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–57.
Chen Y, Wu Y, Yan G, Zhang G. Tertiary lymphoid structures in cancer: maturation and induction. Front Immunol. 2024;15:1369626.
Chen H, Zhang Y, Ye AY, Du Z, Xu M, Lee CS, et al. BCR selection and affinity maturation in Peyer’s patch germinal centers. Nature. 2020;582:421–5.
Nowosad CR, Mesin L, Castro TBR, Wichmann C, Donaldson GP, Araki T, et al. Tunable dynamics of B cell selection in gut germinal centers. Nature. 2020;588:321–6.
García De Vinuesa C, Cook MC, Ball J, Drew M, Sunners Y, Cascalho M, et al. Germinal centers without T cells. J Exp Med. 2000;191:485–93.
Lentz VM, Manser T. Cutting edge: germinal centers can be induced in the absence of T cells. J Immunol. 2001;167:15–20.
Liu X, Zhao Y, Qi H. T-independent antigen induces humoral memory through germinal centers. J Exp Med. 2022;219:e20210527.
Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and t follicular helper cell development initiates in the interfollicular zone. Immunity. 2011;34:947–60.
Tas JMJ, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, et al. Visualizing antibody affinity maturation in germinal centers. Science. 2016;351:1048–54.
Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, et al. In vivo imaging of germinal centers reveals a dynamic open structure. Nature. 2007;446:83–87.
Turner JS, Benet ZL, Grigorova IL. Antigen acquisition enables newly arriving B cells to enter ongoing immunization-induced germinal centers. J Immunol. 2017;199:1301–7.
Hägglöf T, Cipolla M, Loewe M, Chen ST, Mesin L, Hartweger H, et al. Continuous germinal center invasion contributes to the diversity of the immune response. Cell. 2023;186:147–161.e15.
de Carvalho RVH, Ersching J, Barbulescu A, Hobbs A, Castro TBR, Mesin L, et al. Clonal replacement sustains long-lived germinal centers primed by respiratory viruses. Cell. 2023;186:131–146.e13.
van den Broek T, Oleinika K, Rahmayanti S, Castrillon C, van der Poel CE, Carroll MC. Invasion of spontaneous germinal centers by naïve B cells is rapid and persistent. Sci Immunol. 2024;9:eadi8150.
Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–52.
Allen CDC, Okada T, Cyster JG. GerminAl-center Organization And Cellular Dynamics. Immunity. 2007;27:190–202.
Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605.
McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc Natl Acad Sci USA. 1984;81:3180–4.
Klein U, Dalla-Favera R. Germinal centers: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8:22–33.
Tarlinton DM. Evolution in miniature: selection, survival and distribution of antigen reactive cells in the germinal center. Immunol Cell Biol. 2008;86:133–8.
Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SEE, et al. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity. 2007;26:655–67.
Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, et al. The microanatomic segregation of selection by apoptosis in the germinal center. Science. 2017;358:eaao2602.
Qin D, Wu J, Vora KA, Ravetch JV, Szakal AK, Manser T, et al. Fc gamma receptor IIB on follicular dendritic cells regulates the B cell recall response. J Immunol. 2000;164:6268–75.
Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol. 1998;16:545–68.
Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, et al. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity. 2013;38:1164–75.
Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–63.
Mintz MA, Cyster JG. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol Rev. 2020;296:48–61.
Vinuesa CG, Linterman MA, Yu D, Maclennan ICM. Follicular helper T cells. Annu Rev Immunol. 2016;34:335–68.
Ramiscal RR, Vinuesa CG. T-cell subsets in the germinal center. Immunol Rev. 2013;252:146–55.
Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol. 2015;15:185–9.
Bélanger S, Crotty S. Dances with cytokines, featuring TFH cells, IL-21, IL-4 and B cells. Nat Immunol. 2016;17:1135–6.
Bachmann MF, Odermatt B, Hengartner H, Zinkernagel RM. Induction of long-lived germinal centers associated with persisting antigen after viral infection. J Exp Med. 1996;183:2259–69.
Yewdell WT, Smolkin RM, Belcheva KT, Mendoza A, Michaels AJ, Cols M, et al. Temporal dynamics of persistent germinal centers and memory B cell differentiation following respiratory virus infection. Cell Rep. 2021;37:109961.
Turner JS, O’Halloran JA, Kalaidina E, Kim W, Schmitz AJ, Zhou JQ, et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal center responses. Nature. 2021;596:109–13.
Lee JH, Sutton HJ, Cottrell CA, Phung I, Ozorowski G, Sewall LM, et al. Long-primed germinal centers with enduring affinity maturation and clonal migration. Nature. 2022;609:998–1004.
Suan D, Sundling C, Brink R. Plasma cell and memory B cell differentiation from the germinal center. Curr Opin Immunol. 2017;45:97–102.
Shinnakasu R, Kurosaki T. Regulation of memory B and plasma cell differentiation. Curr Opin Immunol. 2017;45:126–31.
Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal center by regulated proliferation and hypermutation. Nature. 2014;509:637–40.
Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJK, Koren A, et al. T cell help controls the speed of the cell cycle in germinal center B cells. Science. 2015;349:643–6.
Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, et al. Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity. 2017;46:1045–1058.e6.
Finkin S, Hartweger H, Oliveira TY, Kara EE, Nussenzweig MC. Protein amounts of the MYC transcription factor determine germinal center B cell division capacity. Immunity. 2019;51:324–336.e5.
Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol. 2012;13:1083–91.
DeWitt WSS, Vora AA, Araki T, Galloway JG, Alkutkar T, Bortolatto J, et al. Replaying germinal center evolution on a quantified affinity landscape. bioRxiv 2025; preprint: https://doi.org/10.1101/2025.06.02.656870.
Smith KGC, Light A, Nossal GJV, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 1997;16:2996.
MacLean AJ, Deimel LP, Zhou P, ElTanbouly MA, Merkenschlager J, Ramos V, et al. Affinity maturation of antibody responses is mediated by differential plasma cell proliferation. Science. 2025;387:413–20.
Robinson MJ, Dowling MR, Pitt C, O’Donnell K, Webster RH, Hill DL, et al. Long-lived plasma cells accumulate in the bone marrow at a constant rate from early in an immune response. Sci Immunol. 2022;7:eabm8389.
Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med. 2006;203:2419–24.
Sprumont A, Rodrigues A, McGowan SJ, Bannard C, Bannard O. Germinal centers output clonally diverse plasma cell populations expressing high- and low-affinity antibodies. Cell. 2023;186:5486–5499.e13.
Eltanbouly MA, Ramos V, Maclean AJ, Chen ST, Loewe M, Steinbach S, et al. Role of affinity in plasma cell development in the germinal center light zone. J Exp Med. 2024;221:e20231838.
Sutton HJ, Gao X, Kelly HG, Parker BJ, Lofgren M, Dacon C, et al. Lack of affinity signature for germinal center cells that have initiated plasma cell differentiation. Immunity. 2024;57:245–255.e5.
Suan D, Kräutler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, et al. CCR6 defines memory B cell precursors in mouse and human germinal centers, revealing light-zone location and predominant low antigen affinity. Immunity. 2017;47:1142–1153.e4.
Inoue T, Shinnakasu R, Kawai C, Ise W, Kawakami E, Sax N, et al. Exit from germinal center to become quiescent memory B cells depends on metabolic reprograming and provision of a survival signal. J Exp Med. 2021;218:e20200866.
Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, et al. Germinal-center development of memory B cells driven by IL-9 from follicular helper T cells. Nat Immunol. 2017;18:921–30.
Laidlaw BJ, Schmidt TH, Green JA, Allen CDC, Okada T, Cyster JG. The Eph-related tyrosine kinase ligand Ephrin-B1 marks germinal center and memory precursor B cells. J Exp Med. 2017;214:639–49.
Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG. The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat Immunol. 2020;21:1082–93.
Bannard O, Horton RM, Allen CDC, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity. 2013;39:912–24.
Lam KP, Rajewsky K. B cell antigen receptor specificity and surface density together determine B-1 versus B-2 cell development. J Exp Med. 1999;190:471–7.
Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igα/β heterodimer. Cell. 2004;117:787–800.
Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–86.
Ochiai K, Katoh Y, Ikura T, Hoshikawa Y, Noda T, Karasuyama H, et al. Plasmacytic transcription factor Blimp-1 is repressed by Bach2 in B cells. J Biol Chem. 2006;281:38226–34.
Inoue T, Shinnakasu R, Kurosaki T. Generation of high quality memory B cells. Front Immunol. 2022;12:825813.
Keating R, Hertz T, Wehenkel M, Harris TL, Edwards BA, McClaren JL, et al. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat Immunol. 2013;14:1266–76.
Calado DP, Sasaki Y, Godinho SA, Pellerin A, Köchert K, Sleckman BP, et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol. 2012;13:1092–1100.
Muto A, Ochiai K, Kimura Y, Itoh-Nakadai A, Calame KL, Ikebe D, et al. Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J. 2010;29:4048–61.
Sidwell T, Kallies A. Bach2 is required for B cell and T cell memory differentiation. Nat Immunol. 2016;17:744–5.
Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8+ T cells and memory responses. Immunity. 2009;31:283–95.
Roychoudhuri R, Clever D, Li P, Wakabayashi Y, Quinn KM, Klebanoff CA, et al. BACH2 regulates CD8 + T cell differentiation by controlling access of AP-1 factors to enhancers. Nat Immunol. 2016;17:851–60.
Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, et al. BACH2 represses effector programs to stabilize T reg-mediated immune homeostasis. Nature. 2013;498:506–10.
Smith KGC, Weiss U, Rajewsky K, Nossal GJV, Tarlinton DM. Bcl-2 increases memory B cell recruitment but does not perturb selection in germinal centers. Immunity. 1994;1:803–13.
Smith KGC, Light A, O’Reilly LA, Ang SM, Strasser A, Tarlinton D. bcl-2 Transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming. J Exp Med. 2000;191:475–84.
Stewart I, Radtke D, Phillips B, McGowan SJ, Bannard O. Germinal center B cells replace their antigen receptors in dark zones and fail light zone entry when immunoglobulin gene mutations are damaging. Immunity. 2018;49:477–489.e7.
Saito M, Novak U, Piovan E, Basso K, Sumazin P, Schneider C, et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2009;106:11294–9.
Laidlaw BJ, Cyster JG. Transcriptional regulation of memory B cell differentiation. Nat Rev Immunol. 2021;21:209–20.
Luo X, Hou X, Wang Y, Li Y, Yu S, Qi H. An interleukin-9-ZBTB18 axis promotes germinal center development of memory B cells. Immunity. 2025;58:861–874.e6.
Shehata L, Thouvenel CD, Hondowicz BD, Pew LA, Pritchard GH, Rawlings DJ, et al. Interleukin-4 downregulates transcription factor BCL6 to promote memory B cell selection in germinal centers. Immunity. 2024;57:843–858.e5.
Chakma CR, Good-Jacobson KL. Requirements of IL-4 during the Generation of B Cell Memory. J Immunol. 2023;210:1853–60.
Duan L, Liu D, Chen H, Mintz MA, Chou MY, Kotov DI, et al. Follicular dendritic cells restrict interleukin-4 availability in germinal centers and foster memory B cell generation. Immunity. 2021;54:2256–2272.e6.
Harwood NE, Batista FD. Early events in B cell activation. Annu Rev Immunol. 2010;28:185–210.
Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int Immunol. 2010;22:413–9.
Coffey F, Alabyev B, Manser T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity. 2009;30:599–609.
Glaros V, Rauschmeier R, Artemov AV, Reinhardt A, Ols S, Emmanouilidi A, et al. Limited access to antigen drives generation of early B cell memory while restraining the plasmablast response. Immunity. 2021;54:2005–2023.e10.
Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med. 2012;209:597–606.
Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, et al. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J Exp Med. 2011;208:1243–52.
Robinson MJ, Ding Z, Pitt C, Brodie EJ, Quast I, Tarlinton DM. The amount of BCL6 in B cells shortly after antigen engagement determines their representation in subsequent germinal centers. Cell Rep. 2020;30:1530–1541.e4.
Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-switch recombination occurs infrequently in germinal centers. Immunity. 2019;51:337–350.e7.
Sundling C, Lau AWY, Bourne K, Young C, Laurianto C, Hermes JR, et al. Positive selection of IgG+ over IgM+ B cells in the germinal center reaction. Immunity. 2021;54:988–1001.e5.
Zhang TT, Gonzalez DG, Cote CM, Kerfoot SM, Deng S, Cheng Y, et al. Germinal center B cell development has distinctly regulated stages completed by disengagement from T cell help. eLife. 2017;6:e19552.
Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity. 2013;38:918–29.
Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7:773–82.
Taylor JJ, Jenkins MK, Pape KA. Heterogeneity in the differentiation and function of memory B cells. Trends Immunol. 2012;33:590–7.
Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–92.
Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, De Waard R, et al. The BCL-6 proto-oncogene controls germinal-center formation and Th2- type inflammation. Nat Genet. 1997;16:161–70.
Taylor JJ, Pape KA, Steach HR, Jenkins MK. Apoptosis and antigen affinity limit effector cell differentiation of a single naïve B cell. Science. 2015;347:784–7.
Grenov A, Hezroni H, Lasman L, Hanna JH, Shulman Z. YTHDF2 suppresses the plasmablast genetic program and promotes germinal center formation. Cell Rep. 2022;39:110778.
Ols S, Yang L, Thompson EA, Pushparaj P, Tran K, Liang F, et al. Route of vaccine administration alters antigen trafficking but not innate or adaptive immunity. Cell Rep. 2020;30:3964–3971.e7.
Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc Natl Acad Sci USA. 2016;113:E6639–48.
Tokatlian T, Read BJ, Jones CA, Kulp DW, Menis S, Chang JYH, et al. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science. 2019;363:649–54.
Paus D, Chan TriGP, Gardam TD, Basten S, Brink A. R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med. 2006;203:1081–91.
O’Connor BP, Vogel LA, Zhang W, Loo W, Shnider D, Lind EF, et al. Imprinting the fate of antigen-reactive B cells through the affinity of the B cell receptor. J Immunol. 2006;177:7723–32.
Chan TD, Gatto D, Wood K, Camidge T, Basten A, Brink R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J Immunol. 2009;183:3139–49.
Abbott RK, Lee JH, Menis S, Skog P, Rossi M, Ota T, et al. Precursor frequency and affinity determine B cell competitive fitness in germinal centers, tested with germline-targeting HIV vaccine immunogens. Immunity. 2018;48:133–146.e6.
Dal Porto JM, Haberman AM, Kelsoe G, Shlomchik MJ. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J Exp Med. 2002;195:1215–21.
Dosenovic P, Kara EE, Pettersson AK, McGuire AT, Gray M, Hartweger H, et al. Anti–HIV-1 B cell responses are dependent on B cell precursor frequency and antigen-binding affinity. Proc Natl Acad Sci USA. 2018;115:4743–8.
Shih TAY, Meffre E, Roederer M, Nussenzweig MC. Role of BCR affinity in T cell–dependent antibody responses in vivo. Nat Immunol. 2002;3:570–5.
Toyama H, Okada S, Hatano M, Takahashi Y, Takeda N, Ichii H, et al. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity. 2002;17:329–39.
Weller S, Faili A, Garcia C, Braun MC, Le Deist F, De Saint Basile G, et al. CD40-CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci USA. 2001;98:1166–70.
Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–7.
Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity. 2016;44:116–30.
Pape KA, Maul RW, Dileepan T, Paustian AS, Gearhart PJ, Jenkins MK. Naïve B cells with high-avidity germline-encoded antigen receptors produce persistent IgM+ and transient IgG+ memory B cells. Immunity. 2018;48:1135–1143.e4.
Lau AWY, Turner VM, Bourne K, Hermes JR, Chan TD, Brink R. BAFFR controls early memory B cell responses but is dispensable for germinal center function. J Exp Med. 2021;218:e20191167.
Müller-Winkler J, Mitter R, Rappe JCF, Vanes L, Schweighoffer E, Mohammadi H, et al. Critical requirement for BCR, BAFF, and BAFFR in memory B cell survival. J Exp Med. 2021;218:e20191393.
Xu T, Zhang T, Xu C, Yang F, Zhang W, Huang C. Notch2 signaling governs activated B cells to form memory B cells. Cell Rep. 2024;43:114454.
Gross JA, Dillon SR, Mudri S, Johnston J, Littau A, Roque R, et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease: impaired B cell maturation in mice lacking BLyS. Immunity. 2001;15:289–302.
Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, et al. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 2001;293:2108–11.
Schiemann B, Gommerman JL, Vora K, Cachero TG, Shutga-Morskaya S, Dobles M, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–4.
Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18:675–85.
Tanigaki K, Han H, Yamamoto N, Tashiro K, Ikegawa M, Kuroda K, et al. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat Immunol. 2002;3:443–50.
Callahan D, Smita S, Joachim S, Hoehn K, Kleinstein S, Weisel F, et al. Memory B cell subsets have divergent developmental origins that are coupled to distinct imprinted epigenetic states. Nat Immunol. 2024;25:562–75.
Termote M, Marques RC, Hyllner E, Guryleva MV, Henskens M, Brutscher A, et al. Antigen affinity and site of immunization dictate B cell recall responses. Cell Rep. 2025;44:115221.
Pritchard GH, Krishnamurty AT, Rodda L, McDougal C, Shehata L, Pepper M. A continuum of CD4+ T cell “help” defines Memory B cell fate. bioRxiv.2024; preprint: https://doi.org/10.1101/564351.
Viant C, Wirthmiller T, ElTanbouly MA, Chen ST, Cipolla M, Ramos V, et al. Germinal center-dependent and -independent memory B cells produced throughout the immune response. J Exp Med. 2021;218:e20202489.
Young C, Brink R. The unique biology of germinal center B cells. Immunity. 2021;54:1652–64.
Tomayko MM, Steinel NC, Anderson SM, Shlomchik MJ. Cutting edge: Hierarchy of maturity of murine memory B cell subsets. J Immunol. 2010;185:7146–50.
Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. 2014;15:631–7.
Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol. 2009;10:1292–9.
Schiepers A, Van’t Wout MFL, Hobbs A, Mesin L, Victora GD. Opposing effects of preexisting antibody and memory T cell help on the dynamics of recall germinal centers. Immunity. 2024;57:1618–1628.e4.
Mesin L, Schiepers A, Ersching J, Barbulescu A, Cavazzoni CB, Angelini A, et al. Restricted clonality and limited germinal center reentry characterize memory B cell reactivation by boosting. Cell. 2020;180:92–106.e11.
Shao W, Wang Y, Fang Q, Shi W, Qi H. Epigenetic recording of stimulation history reveals BLIMP1–BACH2 balance in determining memory B cell fate upon recall challenge. Nat Immunol. 2024;25:1432–44.
Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, et al. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol. 2019;20:97–108.
Allie SR, Randall TD. Resident memory B cells. Viral Immunol. 2020;33:282–93.
Gregoire C, Spinelli L, Villazala-Merino S, Gil L, Holgado MP, Moussa M, et al. Viral infection engenders bona fide and bystander subsets of lung-resident memory B cells through a permissive mechanism. Immunity. 2022;55:1216–1233.e9.
Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–97.
Thome JJC, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial map of human t cell compartmentalization and maintenance over decades of life. Cell. 2014;159:814–28.
Tan HX, Juno JA, Esterbauer R, Kelly HG, Wragg KM, Konstandopoulos P, et al. Lung-resident memory B cells established after pulmonary influenza infection display distinct transcriptional and phenotypic profiles. Sci Immunol. 2022;7:eabf5314.
Lam N, Lee YS, Farber DL. A guide to adaptive immune memory. Nat Rev Immunol. 2024;24:810–29.
Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94.
Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14:1285–93.
Oh JE, Song E, Moriyama M, Wong P, Zhang S, Jiang R, et al. Intranasal priming induces local lung-resident B cell populations that secrete protective mucosal antiviral IgA. Sci Immunol. 2021;6:eabj5129.
Mathew NR, Jayanthan JK, Smirnov IV, Robinson JL, Axelsson H, Nakka SS, et al. Single-cell BCR and transcriptome analysis after influenza infection reveals spatiotemporal dynamics of antigen-specific B cells. Cell Rep. 2021;35:109286.
He HyeMJ, Sangster Y. MY. Broad dispersion and lung localization of virus-specific memory B cells induced by influenza pneumonia. Proc Natl Acad Sci USA. 2008;105:3485–90.
Onodera T, Takahashi Y, Yokoi Y, Ato M, Kodama Y, Hachimura S, et al. Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc Natl Acad Sci USA. 2012;109:2485–90.
Adachi Y, Onodera T, Yamada Y, Daio R, Tsuiji M, Inoue T, et al. Distinct germinal center selection at local sites shapes memory B cell response to viral escape. J Exp Med. 2015;212:1709–23.
MacLean AJ, Richmond N, Koneva L, Attar M, Medina CAP, Thornton EE, et al. Secondary influenza challenge triggers resident memory B cell migration and rapid relocation to boost antibody secretion at infected sites. Immunity. 2022;55:718–733.e8.
Arroyo-Díaz NM, Bachus H, Papillion A, Randall TD, Akther J, Rosenberg AF, et al. Interferon-γ production by Tfh cells is required for CXCR3+ prememory B cell differentiation and subsequent lung-resident memory B cell responses. Immunity. 2023;56:2358–2372.e5.
Maclean AJ, Bonifacio JPPL, Oram SL, Mohsen MO, Bachmann MF, Arnon I. T. Regulation of pulmonary plasma cell responses during secondary infection with influenza virus. J Exp Med. 2024;221:e20232014.
Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, et al. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med. 2004;10:927–34.
MacKay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103+ CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14:1294–301.
Tan HX, Esterbauer R, Vanderven HA, Juno JA, Kent SJ, Wheatley AK. Inducible bronchus-associated lymphoid tissues (IBALT) serve as sites of B cell selection and maturation following influenza infection in mice. Front Immunol. 2019;10:611.
Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science. 2016;352:aaf4822.
Albright AR, Kabat J, Li M, Raso F, Reboldi A, Muppidi JR. TGFβ signaling in germinal center B cells promotes the transition from light zone to dark zone. J Exp Med. 2019;216:2531–45.
Raso F, Liu S, Simpson MJ, Barton GM, Mayer CT, Acharya M, et al. Antigen receptor signaling and cell death resistance controls intestinal humoral response zonation. Immunity. 2023;56:2373–2387.e8.
Barker KA, Etesami NS, Shenoy AT, Arafa EI, Lyon de Ana C, Smith NMS, et al. Lung-resident memory B cells protect against bacterial pneumonia. J Clin Invest. 2021;131:e141810.
Liu J, Stoler-Barak L, Hezroni-Bravyi H, Biram A, Lebon S, Davidzohn N, et al. Turbinate-homing IgA-secreting cells originate in the nasal lymphoid tissues. Nature. 2024;632:637–46.
Gailleton R, Mathew NR, Reusch L, Schön K, Scharf L, Strömberg A, et al. Ectopic germinal centers in the nasal turbinates contribute to B cell immunity to intranasal viral infection and vaccination. Proc Natl Acad Sci USA. 2025;122:e2421724122.
Ramirez SI, Faraji F, Hills LB, Lopez PG, Goodwin B, Stacey HD, et al. Immunological memory diversity in the human upper airway. Nature. 2024;632:630–6.
Geherin SA, Gómez D, Glabman RA, Ruthel G, Hamann A, Debes GF. IL-10+ innate-like B cells are part of the skin immune system and require α4β1 integrin to migrate between the peritoneum and inflamed skin. J Immunol. 2016;196:2514–25.
Nihal M, Mikkola D, Wood GS. Detection of clonally restricted immunoglobulin heavy chain gene rearrangements in normal and lesional skin: Analysis of the B cell component of the skin-associated lymphoid tissue and implications for the molecular diagnosis of cutaneous B cell lymphomas. J Mol Diagn. 2000;2:5–10.
Saul L, Ilieva KM, Bax HJ, Karagiannis P, Correa I, Rodriguez-Hernandez I, et al. IgG subclass switching and clonal expansion in cutaneous melanoma and normal skin. Sci Rep. 2016;6:29736.
Gribonika I, Band VI, Chi L, Perez-Chaparro PJ, Link VM, Ansaldo E, et al. Skin autonomous antibody production regulates host-microbiota interactions. Nature. 2024;638:1043–53.
Bemark M, Boysen P, Lycke NY. Induction of gut IgA production through T cell-dependent and T cell-independent pathways. Ann N Y Acad Sci. 2012;1247:97–116.
Lycke NY, Bemark M. The regulation of gut mucosal IgA B-cell responses: Recent developments. Mucosal Immunol. 2017;10:1361–74.
MacPherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. The immune geography of IgA induction and function. Mucosal Immunol. 2008;1:11–22.
Huus KE, Petersen C, Finlay BB. Diversity and dynamism of IgA−microbiota interactions. Nat Rev Immunol. 2021;21:514–25.
Neutra M, Weltzin R, Winner L, Mack J, Michetti P, Morrison L, et al. Identification and use of protective monoclonal IgA antibodies against viral and bacterial pathogens. Adv Exp Med Biol. 1991;310:179–82.
Michetti P, Mahan MJ, Slauch JM, Mekalanos JJ, Neutra MR. Monoclonal secretory immunoglobulin A protects mice against oral challenge with the invasive pathogen Salmonella typhimurium. Infect Immun. 1992;60:1786–92.
Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci USA. 2004;101:1981–6.
Shroff KE, Meslin K, Cebra JJ. Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect Immun. 1995;63:3904–13.
Burns JW, Krishnaney AA, Vo PT, Rouse RV, Anderson LJ, Greenberg HB. Analyses of homologous rotavirus infection in the mouse model. Virology. 1995;207:143–53.
McNeal MM, Ward RL. Long-term production of rotavirus antibody and protection against reinfection following a single infection of neonatal mice with murine rotavirus. Virology. 1995;211:474–80.
Lycke N, Holmgren J. Intestinal mucosal memory and presence of memory cells in lamina Propria and Peyer’s patches in mice 2 years after oral immunization with Cholera Toxin. Scand J Immunol. 1986;23:611–6.
Herremans TMPT, Reimerink JHJ, Buisman AM, Kimman TG, Koopmans MPG. Induction of mucosal immunity by inactivated poliovirus vaccine is dependent on previous mucosal contact with live virus. J Immunol. 1999;162:5011–8.
Lycke N, Holmgren J. Adoptive transfer of gut mucosal antitoxin memory by isolated B cells 1 year after oral immunization with cholera toxin. Infect Immun. 1989;57:1137–41.
Bemark M, Hazanov H, Strömberg A, Komban R, Holmqvist J, Köster S, et al. Limited clonal relatedness between gut IgA plasma cells and memory B cells after oral immunization. Nat Commun. 2016;7:12698.
Vergani S, Muleta KG, Da Silva C, Doyle A, Kristiansen TA, Sodini S, et al. A self-sustaining layer of early-life-origin B cells drives steady-state IgA responses in the adult gut. Immunity. 2022;55:1829–1842.e6.
Weisel NM, Weisel FJ, Farber DL, Borghesi LA, Shen Y, Ma W, et al. Comprehensive analyses of B-cell compartments across the human body reveal novel subsets and a gut-resident memory phenotype. Blood. 2020;136:2774–85.
Lindner C, Thomsen I, Wahl B, Ugur M, Sethi MK, Friedrichsen M, et al. Diversification of memory B cells drives the continuous adaptation of secretory antibodies to gut microbiota. Nat Immunol. 2015;16:880–8.
Trivedi N, Weisel F, Smita S, Joachim S, Kader M, Radhakrishnan A, et al. Liver is a generative site for the B cell response to Ehrlichia muris. Immunity. 2019;51:1088–1101.e5.
Burton AR, Pallett LJ, McCoy LE, Suveizdyte K, Amin OE, Swadling L, et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J Clin Invest. 2018;128:4588–603.
Osmani Z, Villanueva MA, Joseph-Chazan J, Beudeker BJ, de Knegt RJ, Chung RT, et al. Intrahepatic plasma cells, but not atypical memory B cells, associate with clinical phases of chronic hepatitis B. Eur J Immunol. 2024;54:e2451085.
Moran I, Nguyen A, Khoo WH, Butt D, Bourne K, Young C, et al. Memory B cells are reactivated in subcapsular proliferative foci of lymph nodes. Nat Commun. 2018;9:1–14.
Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450:110–4.
Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol. 2007;8:992–1000.
Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naïve and memory immune responses. Immunity. 2015;42:704–18.
Dhenni R, Hoppé AC, Reynaldi A, Kyaw W, Handoko NT, Grootveld AK, et al. Macrophages direct location-dependent recall of B cell memory to vaccination. Cell. 2025;188:3477–3496.e22.
Kuraoka M, Yeh CH, Bajic G, Kotaki R, Song S, Windsor I, et al. Recall of B cell memory depends on relative locations of prime and boost immunization. Sci Immunol. 2022;7:eabn5311.
Lewis SM, Williams A, Eisenbarth SC. Structure and function of the immune system in the spleen. Sci Immunol. 2019;4:6085.
Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–29.
Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol. 2009;9:767–77.
Steiniger B, Timphus EM, Barth PJ. The splenic marginal zone in humans and rodents: an enigmatic compartment and its inhabitants. Histochem Cell Biol. 2006;126:641–8.
Lechner M, Engleitner T, Babushku T, Schmidt-Supprian M, Rad R, Strobl LJ, et al. Notch2-mediated plasticity between marginal zone and follicular B cells. Nat Commun. 2021;12:1–13.
Gaudette BT, Roman CJ, Ochoa TA, Atria DG, Jones DD, Siebel CW, et al. Resting innate-like B cells leverage sustained Notch2/mTORC1 signaling to achieve rapid and mitosisindependent plasma cell differentiation. J Clin Invest. 2021;131:e151975.
Descatoire M, Weller S, Irtan S, Feuillard J, Storck S, Guiochon-Mantel A, et al. Identification of a human splenic marginal zone B cell precursor with NOTCH2-dependent differentiation properties. J Exp Med. 2014;211:987–1000.
Allman A, Gaudette BT, Kelly S, Alouche N, Carrington LJ, Perkey E, et al. Splenic fibroblasts control marginal zone B cell movement and function via two distinct Notch2-dependent regulatory programs. Immunity. 2025;58:143–161.e8.
Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, et al. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med. 1999;190:75–89.
Cariappa A, Tang M, Parng C, Nebelitskiy E, Carroll M, Georgopoulos K, et al. The follicular versus marginal zone B lymphocyte cell fate decision is regulated by aiolos, Btk, and CD21. Immunity. 2001;14:603–15.
Wen L, Brill-Dashoff J, Shinton SA, Asano M, Hardy RR, Hayakawa K. Evidence of marginal-zone B cell-positive selection in spleen. Immunity. 2005;23:297–308.
Samardzic T, Marinkovic D, Danzer CP, Gerlach J, Nitschke L, Wirth T. Reduction of marginal zone B cells in CD22-deficient mice. Eur J Immunol. 2002;32:561–7.
Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol. 2002;2:323–35.
Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. 2013;13:118–32.
Palm AKE, Kleinau S. Marginal zone B cells: from housekeeping function to autoimmunity?. J Autoimmun. 2021;119:102627.
Agematsu K, Hokibara S, Nagumo H, Komiyama A. CD27: a memory B-cell marker. Immunol Today. 2000;21:204–6.
Grimsholm O. CD27 on human memory B cells–more than just a surface marker. Clin Exp Immunol. 2023;213:164.
Weill JC, Weller S, Reynaud CA. Human marginal zone B cells. Annu Rev Immunol. 2009;27:267–85.
Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, et al. Human blood IgM “memory” B cells are circulating splenic marginal Zone B cells harboring a prediversified immunoglobulin repertoire. Blood. 2004;104:3647–54.
Dunn-Walters DK, Isaacson PG, Spencer J. Sequence analysis of rearranged IgVH genes from microdissected human Peyer’s° patch marginal zone B cells. Immunology. 1996;88:618–24.
Dunn-Walters DK, Isaacson PG, Spencer J. Analysis of mutations in immunoglobulin heavy chain variable region genes of microdissected marginal zone (MGZ) B cells suggests that the MGZ of human spleen is a reservoir of memory B cells. J Exp Med. 1995;182:559–66.
Liu Y-J, Oldfield S, Maclennan ICM. Memory B cells in T cell-dependent antibody responses colonize the splenic marginal zones. Eur J Immunol. 1988;18:355–62.
Anderson SM, Tomayko MM, Ahuja A, Haberman AM, Shlomchik MJ. New markers for murine memory B cells that define mutated and unmutated subsets. J Exp Med. 2007;204:2103.
Babushku T, Lechner M, Ehrenberg S, Rambold U, Schmidt-Supprian M, Yates AJ, et al. Notch2 controls developmental fate choices between germinal center and marginal zone B cells upon immunization. Nat Commun. 2024;15:1–20.
Song W, Antao OQ, Condiff E, Sanchez GM, Chernova I, Zembrzuski K, et al. Development of Tbet- and CD11c-expressing B cells in a viral infection requires T follicular helper cells outside of germinal centers. Immunity. 2022;55:290–307.e5.
Riedel R, Addo R, Ferreira-Gomes M, Heinz GA, Heinrich F, Kummer J, et al. Discrete populations of isotype-switched memory B lymphocytes are maintained in murine spleen and bone marrow. Nat Commun. 2020;11:1–14.
Arnon TI, Horton RM, Grigorova IL, Cyster JG. Visualization of splenic marginal zone B-cell shuttling and follicular B-cell egress. Nature. 2012;493:684–8.
Turchinovich G, Thanh VT, Frommer F, Kranich J, Schmid S, Alles M, et al. Programming of marginal zone B-cell fate by basic Krüppel-like factor(BKLF/KLF3). Blood. 2011;117:3780–92.
Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, et al. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol. 2004;5:713–20.
Muppidi JR, Arnon TI, Bronevetsky Y, Veerapen N, Tanaka M, Besra GS, et al. Cannabinoid receptor 2 positions and retains marginal zone B cells within the splenic marginal zone. J Exp Med. 2011;208:1941–8.
Liu D, Winer BY, Chou MY, Tam H, Xu Y, An J, et al. Dynamic encounters with red blood cells trigger splenic marginal zone B cell retention and function. Nat Immunol. 2024;25:142–54.
Jones DD, Wilmore JR, Allman D. Cellular dynamics of memory B cell populations: IgM+ and IgG+ memory B cells persist indefinitely as quiescent cells. J Immunol. 2015;195:4753–9.
Hao Z, Rajewsky K. Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J Exp Med. 2001;194:1151–63.
Chappert P, Huetz F, Espinasse M-AA, Chatonnet F, Pannetier L, Da Silva L, et al. Human anti-smallpox long-lived memory B cells are defined by dynamic interactions in the splenic niche and long-lasting germinal center imprinting. Immunity. 2022;55:1872–1890.e9.
Slifka MK, Ahmed R. Long-term antibody production is sustained by antibody-secreting cells in the bone marrow following acute viral infection. Ann. N Y Acad. Sci. 1996;797:166–76.
Manz RA, Löhning M, Cassese G, Thiel A, Radbruch A. Survival of long-lived plasma cells is independent of antigen. Int Immunol. 1998;10:1703–11.
Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KGC, Dörner T, et al. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–50.
Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, et al. Long-lived plasma cells are contained within the CD19-CD38hiCD138+ subset in human bone marrow. Immunity. 2015;43:132–45.
Paramithiotis E, Cooper MD. Memory B lymphocytes migrate to bone marrow in humans. Proc Natl Acad Sci USA. 1997;94:208–12.
Benedict CL, Gilfillan S, Thai TH, Kearney JF. Terminal deoxynucleotidyl transferase and repertoire development. Immunol Rev. 2000;175:150–7.
Perlmutter RM, Kearney JF, Chang SP, Hood LE. Developmentally controlled expression of immunoglobulin VH genes. Science. 1985;227:1597–601.
Yancopoulos GD, Desiderio SV, Paskind M, Kearney JF, Baltimore D, Alt FW. Preferential utilization of the most JH-proximal VH gene segments in pre-B-cell lines. Nature. 1984;311:727–33.
Schroeder HW, Hillson JL, Perlmutter RM. Early restriction of the human antibody repertoire. Science. 1987;238:791–3.
Portugal S, Obeng-Adjei N, Moir S, Crompton PD, Pierce SK. Atypical memory B cells in human chronic infectious diseases: an interim report. Cell Immunol. 2017;321:18–25.
Mouat IC, Goldberg E, Horwitz MS. Age-associated B cells in autoimmune diseases. Cell Mol Life Sci. 2022;79:402.
Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118:1294–304.
Cancro MP. Age-associated B cells. Annu Rev Immunol. 2020;38:315–40.
Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O’Shea MA, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med. 2008;205:1797–805.
Portugal S, Tipton CM, Sohn H, Kone Y, Wang J, Li S, et al. Malaria-associated atypical memory B cells exhibit markedly reduced B cell receptor signaling and effector function. eLife. 2015;4:e07218.
Sullivan RT, Kim CC, Fontana MF, Feeney ME, Jagannathan P, Boyle MJ, et al. FCRL5 delineates functionally impaired memory B cells associated with plasmodium falciparum exposure. PLoS Pathog. 2015;11:e1004894.
Sutton HJ, Aye R, Idris AH, Vistein R, Nduati E, Kai O, et al. Atypical B cells are part of an alternative lineage of B cells that participates in responses to vaccination and infection in humans. Cell Rep. 2021;34:108684.
Knox JJ, Myles A, Cancro MP. T-bet+ memory B cells: Generation, function, and fate. Immunol Rev. 2019;288:149–60.
Rubtsova K, Rubtsov AV, Van Dyk LF, Kappler JW, Marrack P. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc Natl Acad Sci USA. 2013;110:E3216–24.
Ehrhardt GRA, Hsu JT, Gartland L, Leu CM, Zhang S, Davis RS, et al. Expression of the immunoregulatory molecule FcRH4 defines a distinctive tissue-based population of memory B cells. J Exp Med. 2005;202:783–91.
Rubtsova K, Rubtsov AV, Cancro MP, Marrack P. Age-associated B cells: a T-bet-dependent effector with roles in protective and pathogenic immunity. J Immunol. 2015;195:1933–7.
Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS One. 2013;8:e67003.
Kleberg L, Courey-Ghaouzi A-D, Lautenbach MJ, Färnert A, Sundling C. Regulation of B cell function and expression of CD11c, T-bet, and FcRL5 in response to different activation signals. Eur J Immunol. 2023;54:2350736.
Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J Clin Invest. 2017;127:1392–404.
Nickerson KM, Smita S, Hoehn KB, Marinov AD, Thomas KB, Kos JT, et al. Age-associated B cells are heterogeneous and dynamic drivers of autoimmunity in mice. J Exp Med. 2023;220:e20221346.
Oliviero B, Varchetta S, Mele D, Mantovani S, Cerino A, Perotti CG, et al. Expansion of atypical memory B cells is a prominent feature of COVID-19. Cell Mol Immunol. 2020;17:1101–3.
Weiss GE, Crompton PD, Li S, Walsh LA, Moir S, Traore B, et al. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J Immunol. 2009;183:2176–82.
Sundling C, Rönnberg C, Yman V, Asghar M, Jahnmatz P, Lakshmikanth T, et al. B cell profiling in malaria reveals expansion and remodeling of CD11c+ B cell subsets. JCI Insight. 2019;4:e126492.
Pape KA, Dileepan T, Kabage AJ, Kozysa D, Batres R, Evert C, et al. High-affinity memory B cells induced by SARS-CoV-2 infection produce more plasmablasts and atypical memory B cells than those primed by mRNA vaccines. Cell Rep. 2021;37:109823.
McGrath JJC, Li L, Wilson PC. Memory B cell diversity: insights for optimized vaccine design. Trends Immunol. 2022;43:343–54.
Steuten J, Bos AV, Kuijper LH, Claireaux M, Olijhoek W, Elias G, et al. Distinct dynamics of antigen-specific induction and differentiation of different CD11c+Tbet+ B-cell subsets. J Allergy Clin Immunol. 2023;152:689–699.e6.
Lopes de Assis F, Hoehn KB, Zhang X, Kardava L, Smith CD, El Merhebi O, et al. Tracking B cell responses to the SARS-CoV-2 mRNA-1273 vaccine. Cell Rep. 2023;42:112780.
Knox JJ, Buggert M, Kardava L, Seaton KE, Eller MA, Canaday DH, et al. T-bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight. 2017;2:e92943.
Holla P, Dizon B, Ambegaonkar AA, Rogel N, Goldschmidt E, Boddapati AK, et al. Shared transcriptional profiles of atypical B cells suggest common drivers of expansion and function in malaria, HIV, and autoimmunity. Sci Adv. 2021;7:eabg8384.
Yang R, Avery DT, Jackson KJL, Ogishi M, Benhsaien I, Du L, et al. Human T-bet governs the generation of a distinct subset of CD11chighCD21low B cells. Sci Immunol. 2022;7:eabq3277.
Levack RC, Newell KL, Popescu M, Cabrera-Martinez B, Winslow GM. CD11c+ T-bet+ B cells require IL-21 and IFN-γ from Type 1 T follicular helper cells and intrinsic Bcl-6 expression but develop normally in the absence of T-bet. J Immunol. 2020;205:1050–8.
Du SW, Arkatkar T, Jacobs HM, Rawlings DJ, Jackson SW. Generation of functional murine CD11c + age-associated B cells in the absence of B cell T-bet expression. Eur J Immunol. 2019;49:170–8.
Gao X, Shen Q, Roco JA, Dalton B, Frith K, Mee Ling Munier C, et al. Zeb2 drives the formation of CD11c+ atypical B cells to sustain germinal centers that control persistent infection. Sci Immunol. 2024;9:4748.
Dai D, Gu S, Han X, Ding H, Jiang Y, Zhang X, et al. The transcription factor ZEB2 drives the formation of age-associated B cells. Science. 2024;383:413–21.
Naradikian MS, Myles A, Beiting DP, Roberts KJ, Dawson L, Herati RS, et al. Cutting edge: IL-4, IL-21, and IFN-γ interact to govern T-bet and CD11c expression in TLR-activated B cells. J Immunol. 2016;197:1023–8.
Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. TLR7 drives accumulation of ABCs and autoantibody production in autoimmune-prone mice. Immunol Res. 2013;55:210–6.
Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat Commun. 2018;9:1758.
Ricker E, Manni M, Flores-Castro D, Jenkins D, Gupta S, Rivera-Correa J, et al. Altered function and differentiation of age-associated B cells contribute to the female bias in lupus mice. Nat Commun. 2021;12:4813.
Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. 2011;118:1305–15.
Manni M, Gupta S, Ricker E, Chinenov Y, Park SH, Shi M, et al. Regulation of age-associated B cells by IRF5 in systemic autoimmunity. Nat Immunol. 2018;19:407–19.
Keller B, Strohmeier V, Harder I, Unger S, Payne KJ, Andrieux G, et al. The expansion of human T-bethighCD21low B cells is T cell dependent. Sci Immunol. 2021;6:eabh0891.
Aranburu A, Höök N, Gerasimcik N, Corleis B, Ren W, Camponeschi A, et al. Age-associated B cells expanded in autoimmune mice are memory cells sharing H-CDR3-selected repertoires. Eur J Immunol. 2018;48:509–21.
Imabayashi K, Yada Y, Kawata K, Yoshimura M, Iwasaki T, Baba A, et al. Critical roles of chronic BCR signaling in the differentiation of anergic B cells into age-associated B cells in aging and autoimmunity. Sci Adv. 2025;11:eadt8199.
Johnson JL, Rosenthal RL, Knox JJ, Myles A, Naradikian MS, Madej J, et al. The transcription factor T-bet resolves memory B cell subsets with distinct tissue distributions and antibody specificities in mice and humans. Immunity. 2020;52:842–855.e6.
Du SW, Arkatkar T, Al Qureshah F, Jacobs HM, Thouvenel CD, Chiang K, et al. Functional characterization of CD11c+ age-associated B cells as memory B cells. J Immunol. 2019;203:2817–26.
William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–70.
Ambegaonkar AA, Kwak K, Sohn H, Manzella-Lapeira J, Brzostowski J, Pierce SK. Expression of inhibitory receptors by B cells in chronic human infectious diseases restricts responses to membrane-associated antigens. Sci Adv. 2020;6:eaba6493.
Louis K, Bailly E, Macedo C, Lau L, Ramaswami B, Chang A, et al. T-bet+CD27+CD21- B cells poised for plasma cell differentiation during antibody-mediated rejection of kidney transplants. JCI Insight. 2021;6:e148881.
Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. Cutting edge: long-term B cell memory in humans after smallpox vaccination. J Immunol. 2003;171:4969–73.
Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357:1903–15.
Acknowledgements
We thank Christopher Sundling for his expert review and valuable feedback on the “Atypical MBCs” section. We are also grateful to Manon Termote and Valentin Zollner for critical reading of the manuscript. The figures for this review were prepared with BioRender.com. Our laboratory is supported by grants from the Swedish Research Council (2021-01468), Cancerfonden (21 1602 Pj and 24 3516 Pj), Barncancerfonden (PR2023-0091), Radiumhemmets Forskningsfonder (231233) and the Knut and Alice Wallenberg Foundation (KAW 2024.0097).
Funding
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
VG wrote the manuscript; TK and NF edited the manuscript; NF made most of the figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Glaros, V., Francis, N. & Kreslavsky, T. The multilayered identity of B cell memory. Cell Mol Immunol 23, 150–167 (2026). https://doi.org/10.1038/s41423-025-01377-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41423-025-01377-5
