
Abstract
T cells are central components of the adaptive immune system and play key roles in antitumor and antiviral responses. The diverse cell fates of T cells enable them to respond to different durations and strengths of antigen stimulation and various cytokine milieus in a context-dependent manner. During acute infection or vaccination, T cells differentiate into effector cells and later develop into memory cells after antigen clearance, which mediate immune protection against the same antigen. In contrast, during cancer and chronic infection, T cells fail to enter the canonical effector or memory cell differentiation path. Instead, antigen-specific T cells enter a dysfunctional, partially responsive state called exhaustion. Exhausted T cells are heterogeneous. A subset of exhausted T cells exhibits stem cell-like properties. These stem-like T cells sustain immunity through self-renewal and repopulation of terminally differentiated progenies. Stem-like properties are critical for T cell immunity induced by immunotherapy. This review summarizes recent advances in understanding the molecular mechanisms controlling the exhaustion and stemness of T cells and explores the potential of rewiring these circuits to increase the efficiency of T-cell-based immunotherapy.
Similar content being viewed by others
Introduction
The differentiation trajectory of CD8 T cells is dictated by the duration of antigen stimulation. During acute infection, naive T cells (TNs) differentiate into either effector T cells (TEFFs) to clear antigens or memory precursor cells (TMPs) [1, 2]. After the antigen is cleared, terminally differentiated short-lived TEFF cells go through a contraction phase to avoid immune pathology, while TMP cells develop into memory T cells (TMEMs) to provide a self-renewing antigen-specific T cell pool for long-term immune protection against potential reinfection [1, 2]. Human TMEM cells contain various subsets, including central memory (TCM), stem-like memory (TSCM), effector memory (TEM), and CD45RA+ effector memory (TEMRA) T cells, that circulate throughout the body and maintain immune memory [3, 4]. Although both TCM and TSCM cells can self-renew, the TSCM population has greater proliferation capacity and multipotency and displays superior antitumor immunity during adoptive cell therapy [4,5,6,7]. In addition to the circulating TMEM subsets, a distinct noncirculating tissue-resident memory T cell population (TRM) that mediates local immune protection has been described [8,9,10,11].
During cancer and chronic infection, antigen-specific CD8 T cells undergo constant TCR stimulation in an immunosuppressive environment, which drives T cells to enter a dysfunctional state called exhaustion [12]. Exhaustion prevents T cells from eradicating infected cells or cancer cells [13,14,15,16,17,18,19,20,21]. T-cell exhaustion has been characterized in landmark studies in a mouse model of chronic lymphocytic choriomeningitis virus (LCMV) clone 13 infection and has been observed in humans with chronic HIV, HBV, and HCV infections and cancers [13,14,15,16,17,18,19,20,21]. Exhausted CD8 T (TEX) cells progressively lose their effector function, upregulate inhibitory receptors (also termed immune checkpoints), fail to persist or form memory, and become metabolically dysregulated [19,20,21]. Immune checkpoints, such as CTLA-4, PD-1, LAG-3, TIM3, and TIGIT, transduce inhibitory signals to suppress T cell responses. By blocking these signals, immune checkpoint blockade (ICB) reinvigorates T cell responses. In addition to constant TCR stimulation, interactions with suppressive cells, including myeloid cells, in the tumor microenvironment promote T-cell exhaustion through immune checkpoints, including TIGIT [22]. The epigenetic program of T cell exhaustion is largely unaffected by checkpoint blockade and drives re-exhaustion after cessation of PD-1 blockade [23, 24]. These “epigenetic scars” are characterized by the maintenance of open chromatin at genes associated with T-cell exhaustion after elimination of chronic antigen stimulation [25, 26], suggesting that at least part of the epigenetic program associated with T-cell exhaustion is irreversible once it is established.
In autoimmune diseases, T-cell exhaustion restrains excessive immune activation and is paradoxically associated with favorable clinical outcomes, in contrast to its detrimental role during chronic infection or cancer. Transcriptomic analyses of CD8 T cells from patients with autoimmune disorders such as antineutrophil cytoplasmic antibody-associated vasculitis, systemic lupus erythematosus, and type 1 diabetes revealed that a gene expression signature resembling that of exhausted CD8 T cells in chronic viral infection correlated with reduced relapse frequency and sustained remission [27, 28]. T-cell exhaustion in autoimmune diseases arises from persistent stimulation by autoantigens combined with insufficient CD4 T-cell help [28, 29]. Exhaustion limits immunopathology by dampening autoreactive CD8 T-cell responses, acting as a form of peripheral tolerance once self-reactivity is established [27, 30]. Indeed, genetic or pharmacologic blockade of PD-1 or LAG-3 signaling in mice promotes autoimmune diseases, underscoring the protective role of exhaustion-associated immune checkpoint pathways [29]. Conversely, therapeutic induction of exhaustion, for example, by enhancing PD-1 signaling, has been proposed as a means to mitigate autoimmunity [30]. In organ transplantation, T-cell exhaustion suppresses alloimmune activation and thereby promotes graft tolerance. Persistent alloantigen exposure induces exhausted CD8 T cells with reduced cytokine production, facilitating long-term transplant acceptance [31]. Conversely, disruption of PD-L1–mediated inhibitory signaling enhances T-cell activation and accelerates cardiac allograft rejection and vasculopathy [32]. Clinically, PD-1/PD-L1 blockade restores antitumor responses but often promotes graft rejection, suggesting that maintaining T-cell exhaustion is crucial for sustaining transplant tolerance [33]. Collectively, T-cell exhaustion limits antiviral and antitumor immunity but is beneficial for preventing chronic autoreactivity or alloreactivity.
Heterogeneity within the TEX lineage
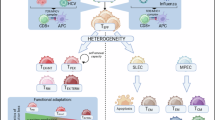
Like the TEFF and TMEM cells, TEX cells are also heterogeneous (Fig. 1). An early study revealed that a subset of TEX cells is more responsive to PD-1 blockade [34]. Transcription factors (TFs), such as T-BET and EOMES, and surface proteins, such as CD39, have been used to identify TEX cells with progenitor-like or terminally exhausted phenotypes [35, 36]. Perhaps one of the most exciting developments in the field of TEX biology is the discovery of a stem cell-like T cell population that expresses the TF TCF1 during chronic infection and cancer [37,38,39,40,41,42,43,44,45,46,47]. Stem-like T cells (TSLs), also termed exhausted progenitor CD8 T cells or TPEX, are critical for long-term cellular immunity. To maintain long-term control over chronic infection and cancer, TSL cells self-renew and replenish other exhausted TCF1− populations [37,38,39,40,41,42,43,44,45,46,47]. Stem-like T cells are maintained by conventional type 1 dendritic cells in their niches in the lymph node and tumor stroma, which serve as reservoirs for antitumor T cells in cancer or antiviral T cells during chronic infection [38,39,40, 48,49,50,51,52,53,54,55]. Compared with their TCF1− counterparts, TSL cells exhibit a superior ability to proliferate in response to immunotherapies, including PD-1 blockade and adoptive cell therapy [37,38,39, 41, 42, 45, 46, 56,57,58]. In addition, TSL cells are endowed with greater mitochondrial fitness, which is critical for tumor control [56, 58,59,60]. The frequency of TCF1+CD8 T cells in cancer patients treated with checkpoint inhibitors is associated with favorable clinical outcomes [42, 61, 62]. In addition, the gene signatures of T-cell stemness and/or T-cell memory in premanufactured T cells and in chimeric antigen receptor (CAR) T-cell infusion products positively correlate with the response to CAR-T-cell therapy in cancer patients [63,64,65]. These studies suggest that the properties of TSL cells are ideal for eliciting optimal T-cell immunity by immunotherapy. Notably, in autoimmune diseases, TSL cells sustain autoreactivity and tissue destruction [66,67,68].

During acute infection, naïve T cells (TNs) differentiate into effector cells that mediate rapid pathogen clearance and memory cells that provide long-term protection. In contrast, persistent antigen stimulation during chronic viral infection or cancer drives an alternative pathway. Early in the immune response, terminal effector-like cells emerge from naïve precursors but decline quickly and show limited persistence. In parallel, stem-like T (TSL) cells, which serve as progenitors of the exhausted lineage, increase and retain self-renewal capacity. TSL cells differentiate into transitory exhausted cells (TEX-Trans), which can transiently expand but ultimately progress irreversibly into terminally exhausted cells (TEX-term) with fixed dysfunction or into effector-like exhausted cells (eff-like TEX)
When and how TEX cells diverge from the differentiation trajectory of TEFF ->TMEM are under active investigation. Rather than being imprinted to become TEFF or TEX during priming, antigen-specific T cells are continuously adapting to the antigenic environment [69]. Recent developments in single-cell omics profiling technologies have enabled us to pinpoint the bifurcation point of the two distinct cell fates. In LCMV infection, single-cell RNA sequencing (scRNA-seq) of CD8 T cells responding to acute infection versus chronic infection diverged during the late stage of initial clonal expansion [44]. Notably, despite the similarities between TSL cells and TMP cells, the TF TOX is expressed only by TSL cells [44]. TOX is essential for the development and persistence of the TEX lineage, including TSL cells, whereas the loss of TOX favors the fate of TEFF but ultimately impairs the persistence of antigen-specific T cells under chronic antigen stimulation [44, 70,71,72,73,74]. Recent discoveries of the common progenitors of TSL cells and TMP cells have revealed the flexibility of early cell fate decisions in both memory formation and exhaustion progression [75, 76].
More recent studies revealed further heterogeneity among TSL or TPEX cells. The expression of CD69 divides these cells into two subsets: a CD69+ lymphoid tissue-resident subset (TEX-Prog1) and a CD69− TEX-Prog2 subset that downregulates TCF1 and enters the blood [77]. Notably, single-cell ATAC+RNA-sequencing analysis of CAR-T cells revealed a TSL subset that shows greater activity of T-box TFs, including EOMES, and may represent a transitory state between the TSL and its progeny [78]. The proliferative potential and multipotency are not evenly distributed among TSL cells. In chronic LCMV infection, a subset of TF-MYB-dependent CD62L+ cells within TSL cells retain the highest level of stemness [79]. Importantly, the capacity for long-term self-renewal and a proliferative burst in response to PD-1 blockade are selectively preserved in this small subset of TSL cells [79]. Similarly, TSL cells exhibit a hierarchical distribution of stemness characteristics in cancer. Compared with their TCF1+TOX+ counterparts, a TCF1+TOX− subset in the draining lymph node of the tumor is protected from the epigenetic scar of exhaustion and demonstrates superior antitumor immunity in adoptive cell therapy and PD-1 blockade [51]. Two TSCM subsets, a functional progenitor subset lacking expression of inhibitory receptors and a PD-1+TIGIT+ exhausted-like subset, are found in human T cells [80].
The progenies of TSL cells are also heterogeneous. TSL cells first differentiate into CD101−TIM3+ transitory TEX cells, which exhibit partial effector function and respond to PD-1 blockade [81]. The transitory subset then differentiates into the terminally exhausted CD101+TIM3+ subset (TEX-Term) [81]. Compared with intermediate TEX cells, TEX-Term cells upregulate CD69 expression [77]. scRNA-seq revealed another potential differentiation pattern in which the TSL subset bifurcates into two distinct progenies, the TEX-Term subset and an IL-21-dependent KLRG1+CX3CR1+ subset that exhibits superior effector function [47, 82]. In chronic LCMV infection, CX3CR1+ eff-like TEX cells are closer to the circulation, whereas CXCR6+CX3CR1− TEX-Term cells reside in tissues [83]. Because of its short-lived nature, the eff-like TEX subset needs to be continuously replenished by TCF1+ TSL cells [47]. In cancer patients, CX3CR1 is expressed in a CD8 T cell population that responds to chemoimmunotherapy [84]. The recent development of single-cell multiomics provides further insight into the diversity within the TEX-Term and eff-like TEX subsets [85, 86]. Notably, CXCR6 is required to position eff-like TEX cells in proximity to CCR7+ conventional DCs that trans-present IL15 to facilitate the survival of T cells [87]. Thus, CXCR6 itself may not drive T cell exhaustion.
The molecular circuit regulating the exhaustion and stemness of T cells
The transcriptional program of T cell exhaustion
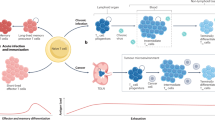
While many transcriptional regulatory circuits are shared between acute and chronic antigen exposure, some transcriptional signaling cascades are specific to the adaptation of TEX subsets to chronic antigen stimulation (Fig. 2). The typical “exhaustion-specific” TF is TOX, which defines the TEX lineage and plays key roles in all exhausted T cell subset differentiation processes [44, 70,71,72,73,74]. High expression of TOX is a direct consequence of strong and constant TCR stimulation, and it can be a potential adaptive mechanism to maintain CD8 T cell internal homeostasis at the transcriptional and epigenetic level [44, 70,71,72,73,74]. Indeed, overexpression of TOX improves the persistence of virus-specific CD8 T cells during chronic infection [44], suggesting that TOX plays a beneficial role in the adaptation of T cells to chronic stimulation. Removing TOX drives the differentiation of antigen-specific CD8 T cells to short-lived TEFF cells while impairing the differentiation of the TSL population [44, 70,71,72,73,74]. Consequently, in the long term, all TEX cell subsets fail to persist [44, 70,71,72]. In addition to TCR signaling, LAG-3 also sustains TOX to facilitate the development of TEX lineages [88]. TSL cells are maintained by sustained TCR stimulation [55]. Cessation of TCR signaling prompts TSL cells to diverge to a TMEM fate, which is accompanied by downregulation of TOX [75, 76, 89]. These findings are consistent with the notion that TOX is a key feature that distinguishes TSL cells from TMP cells [44]. Notably, the effect of TOX may be dosage dependent. A partial reduction in TOX levels results in effective tumor control without compromising long-term T cell immunity [70]. Other TFs critical for the development of T cell exhaustion, such as NR4A family members [73, 90,91,92], NFAT-AP-1 signaling [93,94,95,96], and BATF/IRF4 [97,98,99,100], are also associated with TCR signaling.

Development and/or maintenance of TSL is driven by transcription factors such as TCF1, BACH2, MYB, FOXP1, FOXO1, ID3, BCL6, c-JUN, and SATB1. The transcription factors BATF, IRF4, ID2, and RUNX3 promote the terminal differentiation of CD8 T cells and suppress stem-like cell fate. Among the terminally differentiated subsets, the eff-like TEX subset is positively regulated by KLF2, ZEB2, T-BET, and BHLHE40, whereas NR4A and BLIMP1 promote terminal exhaustion. Both the TSL and TEX-Term lineages require TOX
TCF1– the “identity” of stemness
TCF1, the most broadly reported pro-stem TF, has two major isoforms (short p33 and long p45) with distinct functions in different biological processes [101]. TCF1 was first reported as a master regulator during T cell development in the thymus via Notch signaling [102, 103]. As a pioneer factor, TCF1 has the capacity to shape and reprogram the epigenetic landscape toward a functional mature T cell stage to initiate T cell identity [104]. Together with its homolog LEF1, TCF1 establishes the epigenetic landscape of T cells by controlling both histone acetylation and chromatin architecture via the intrinsic HDAC activity of its short isoform p33 [105] and by coordinating with CTCF [106]. Thus, from a developmental perspective, TCF1 has the capacity to modulate T cell identity toward a naive mature T cell stage and maintain T cell stemness with strong epigenetic footprints.
During acute antigen exposure, TCF1 restrains hyper-effector proliferation and modulates the memory T cell pool to maintain its capacity for secondary responses [6, 107,108,109]. A lack of TCF1 has a limited effect on the initial immune response; however, it has a significantly strong effect on recall toward the same antigen, suggesting that it plays a key role in maintaining the stemness of memory T cells [107,108,109]. In particular, the TCF1 p45 isoform contributes to optimal memory formation [110]. TCF1 has been shown to regulate multiple downstream pathways contributing to memory formation, including EOMES and BCL-2 [107, 108]. Interestingly, moderate levels of coinhibitory signaling molecules such as PD-1 and LAG-3 during acute infection help T cells maintain memory capacity and high expression of TCF1. In fact, intermediate levels of PD-1 expression marked a TSCM population with high CD62L expression, high self-renewal capacity with secondary transfers, and, most importantly, better genomic protection [5].
During chronic infection, TCF1 acts as a master regulator and maintains a pool of TSL cells that renew themselves even in the presence of constant antigen stimulation and mount a proliferative burst to ICB. In the LCMV chronic infection model, TCF1 was first defined as the core TF that regulates a follicular-like CD8 T cell population with a major capacity for self-renewal during chronic infection and response to PD-1 blockade [37,38,39,40,41], similar to the regulatory circuitry of follicular helper T cells (TFHs) [111]. This TCF1+ population is defined as progenitors for exhausted T cells during chronic infection and is negatively associated with IFN signaling sensing [37, 112]. The major follicular features of TSL cells are that they are CXCR5+ and Ly108+ and are exclusive to terminal differentiation markers such as TIM3 and CD39 [37,38,39, 47, 82]. In the early stages of chronic infection, TCF1 restrains terminal differentiation and promotes TSL generation [47]. At the late stage of chronic infection, TCF1 is required for the persistence of the antigen-specific CD8 T cell population, including the TEX-Term and eff-like TEX subsets, which can be either binarily differentiated from TSL cells [81, 82] or from a tissue-circulation-tissue-migration manner for peripheral proliferation of antigen-specific T cells [77, 113]. Notably, similar to the acute setting, the TCF1 isoform p45 plays a strong role in maintaining TSL cell fate [47], suggesting a potential regulatory role of Wnt-β-catenin signaling in regulating TSL cell identity maintenance [6].
Master regulators of stem-like T cells and their progenies
In addition to TCF1, multiple TFs are involved in maintaining T cell stemness during either acute or chronic infection and targeting different signaling cascades, such as those involved in T cell quiescence, survival, metabolism, and transcription.
BACH2 maintains the naïve differentiation state of mature T cells and is critical for the development of TCM and regulatory T cells [114,115,116,117,118]. This effect of BACH2 further extends as a major factor in repressing the terminal exhaustion program and maintaining the transcriptional and epigenetic landscape of TSL cells during both acute and chronic infection [119, 120]. Transcriptionally, BACH2 has a shared motif comparable to that of AP-1, and the expression/motif usage of BACH2 and AP-1 strongly antagonizes both mouse and human T cell differentiation trajectories [116, 119, 120]. This potential transcriptional competition between BACH2 and AP-1 occurs on the enhancer of TCR-induced activation genes, and BACH2 reshapes the whole epigenetic landscape of CD8 T cells toward a more stemness-restrained pattern [116, 119, 120]. Notably, the expression level of BACH2 is correlated with the degree of stemness in CAR-T cells and can be harnessed to optimize the antitumor immunity of CAR-T cells [121]. In addition to AP-1 TFs, BACH2 also antagonizes RUNX3 and BLIMP1, both of which are important players in the terminal differentiation and exhaustion of T cells [119].
MYB has been reported to be among the major regulators of hematopoietic stem cell maintenance and lymphocyte lineage commitment [122,123,124]. It is also directly involved in regulating T cell development at different stages [125, 126], suggesting that it may have a role similar to that of TCF1 in reshaping the epigenetic landscape of “naive-like” T cells. During acute infection, MYB is strongly enriched in memory-associated populations, particularly in the CD62L+ TSCM population, and is the central factor that maintains memory cell homeostasis [127, 128]. Manipulating MYB strongly affects memory recall responses and polyfunctionality. For example, enhancing MYB expression results in better memory T cell survival via increased BCL-2/BCL-XL expression and durable polyfunctional responses of multiple cytokines via potential oxidative metabolic processes [127, 128]. This MYB-driven enhanced stemness signaling cascade is also associated with an increased CD62L+ TPEX population during chronic infection, which further strongly contributes to TPEX population survival as well as the response to PD-1 blockade [47, 79]. MYB overexpression in therapeutic T cells enhances tumor control during adoptive cell therapy [128].
FOXO1, a major TF that is involved in multiple metabolic processes to increase gluconeogenesis [129], also plays a key role in stemness maintenance in TN and TCM cell populations [130, 131]. FOXO1 generally enhances memory T cell formation via the inhibition of effector-associated genes such as T-bet [132] and promotes the expression of memory-associated receptors such as IL7RA and CCR7, even at the early stage of activation [133, 134]. During chronic infection, FOXO1 is a key regulator that maintains PD-1 expression and ensures the survival of the TSL population [135, 136]. This FOXO1-PD-1 axis further influences metabolic regulation, where the FOXO1-PGC1a pathway counterbalances PD-1-driven exhaustion and maintains partial functionality of antigen-responsive T cells [137].
FOXP1 is a member of the Forkhead box (FOX) TF family. In T cells, FOXP1 was first described as a TF that enforces T cell quiescence and suppresses FOXO1 and the MEK/ERK pathway [138]. Deletion of FOXP1 in naïve T cells induces an effector-like phenotype in lymphopenic mice [138]. FOXP1 is also required for the homeostasis and suppressive function of regulatory T cells [139, 140]. CD8 T cells from mice in which FOXP1 is deleted from T cells during development exhibit increased effector function and antitumor immunity [141]. Surprisingly, acutely disrupting FOXP1 in CD8+ CAR-T cells impaired expansion and tumor control by CAR-T cells [78]. In addition, FOXP1 deficiency compromises the differentiation of TSL cells and promotes the premature transition from TSL to TEFF CAR-T cells [78]. Mechanistically, FOXP1 deficiency increases chromatin accessibility to TCR downstream TFs, including AP-1 and NR4A family TFs. Thus, FOXP1 may play context-dependent roles at different stages of T cell differentiation. Future studies are warranted to determine how to optimize T cell immunity during immunotherapy by harnessing the activity of FOXP1.
The zinc finger TF KLF2 is best known as a master regulator that promotes T cell egress from lymphoid tissues and regulates the expression of S1PR1, CD62L, and integrin β7 [142]. KLF2, which is highly expressed in naïve T cells, is a gatekeeper for T cell activation and restrains cytokine production [143]. The downregulation of KLF2 and S1PR1 is required for the formation of TRM cells [144]. TSL and TEFF cells are predominantly found in CAR-T cells recovered from hosts that have cleared tumors [78]. Simultaneous profiling of the single-cell transcriptome and epigenome established the gene regulatory network of TEFF-like CAR-T cells and revealed that KLF2 is a hub TF [78]. KLF2-deficient CAR-T cells exhibit profound defects in the generation of the TEFF-like subset [78]. Instead, KLF2-deficient CAR-T cells display an exhaustion-like phenotype and upregulate the expression of inhibitory receptors and TOX [78]. KLF2 deficiency also downregulates effector molecules and impairs in vitro killing by T cells [78]. In mice with solid tumors, KLF2 deficiency reduces tumor infiltration by CAR-T cells [78]. Single-cell epigenetic analysis revealed that KLF2 deficiency increases chromatin accessibility at binding sites of AP-1 and NFAT TFs while decreasing chromatin accessibility at binding sites of KLF and T-box TFs [78]. Like CAR-T cells, virus-specific CD8 T cells that respond to acute LCMV or MHV infection also exhibit marked defects in TEFF differentiation and upregulation of the exhaustion signature [145, 146]. Thus, KLF2 may represent a master switch controlling the cell fate decision between TEFF and TEX lineages. It is worth further investigating whether the effect of KLF2 on TEFF versus TEX differentiation is connected to its role in regulating T cell migration. In addition, whether targeting KLF2 activity in therapeutic T cells improves their synergy with ICB awaits further investigation.
Transcriptional circuits determine T cell effectiveness versus persistence
While these major TFs individually contribute to T cell stemness, they also form a transcriptional network core in which these TFs maintain the expression level of one another. For example, TCF1 is critical for maintaining the expression of MYB; however, alteration of the expression level of TCF1 is among the major phenotypes of MYB genetic perturbation [47, 79]. While FOXO1 may coordinate with TCF1 and BACH2 to promote stemness at the epigenetic level [119, 147, 148], both TCF1 and BACH2 can be potential direct targets of FOXO1 [134]. These TFs inside the stemness core self-enhance the expression and function of each other to restrain T cells in a high-proliferative capacity but low-activation stage. Removal of these TF core candidates usually results in a limited or no reduction in TEFF populations but has a significant effect on the development of TSL cells during chronic antigen stimulation.
Another important feature of the function of this stemness TF core is that multiple candidates can respond to the same upstream signals. For example, an intermediate level of PD-1 expression is important for maintaining the expression of both TCF1 and FOXO1 [5, 47, 135, 137], whereas type 1 interferon signaling inhibits both of them [37, 149]. MYB and FOXO1 share the same upstream miRNA, miR-150, to inhibit their expression [127, 150]. These coordinated upstream signals can upregulate or downregulate TFs in this stemness transcriptional core to make the cell fate decide whether to maintain commitment to a stem-like state or terminally differentiate in response to an antigen.
In addition to the core module, multiple polarized TF pairs affect “effectiveness” versus “persistence” during CD8 T cell responses. These TF pairs include T-BET versus EOMES [35, 151], ID2 versus ID3 [152, 153], BLIMP1 versus BCL6 [154, 155], STAT4 versus STAT3 [156,157,158], and ZEB2 versus ZEB1 [159, 160]. Most of these TF pairs have the feature of tuning the same functional module with different activation intensities. For example, both T-bet and Eomes are T-box family members that can drive the expression of Ifng and other effector genes. T-bet, however, has much stronger functionality in pushing cells into an “effective” module and overactivation via T-bet-triggered terminal Teff differentiation [2], whereas Eomes maintains a partial response capacity to the antigens in both post-Teff contraction during acute infection and cellular persistence during chronic infection and cancer progression [161,162,163]. Similar rationales apply to Blimp1 versus Bcl6 [154], although Blimp1+ cells are considered to be more terminal CD8 T cells with high cytotoxicity but limited cytokine-secreting capacity [164,165,166,167].
TFs involved in stem cell maintenance primarily function by promoting “persistence-biased” TFs to retain the stem-like or progenitor identity of CD8 T cells or by directly inhibiting “effectiveness-biased” TFs. TCF1, FOXO1, and MYB are known to promote Eomes expression and mediate the T-bet-to-Eomes transition after the Teff boost phase, both in acute and chronic infections [47, 168]. TCF1 also enhances and maintains the expression of Bcl6 and Id3 to promote TMP or TSL cell fate, and the latter TFs drive a function-specific molecular module to ensure T cell persistence [37, 39, 40, 169, 170]. The chromatin organizer SATB1 maintains the quiescent and stem-like state of TSL cells and inhibits expansion and effector differentiation during chronic infection and cancer by regulating transcriptional programs, chromatin accessibility, and genome architecture at key stemness-associated loci such as Tcf7, Bach2, and Myb [171, 172].
Recently, researchers have discovered the functions of more transcriptional circuits that are related to a “persistence-to-terminal effectiveness” transition rationale. For example, a study revealed that the ETS family member Fli1 is a transcriptional immune checkpoint that inhibits hyper-Teff responses in both multiple infection and cancer models. Fli1 directly inhibits the cis-regulatory elements on effector-associated genes by competing with Runx3 [173], which drives TEFF responses, particularly in pathological tissue [174,175,176,177]. Furthermore, in addition to these transcriptional checkpoints, several TFs previously known to regulate TH2 versus TH1 responses, including GATA3 and EGR2, also promote “naiveness” or “stemness” modules during CD8 T cell responses. GATA3 inhibits Teff differentiation, potentially by suppressing the expression of the terminal Teff TF BHLHE40 [178,179,180], whereas EGR2 contributes to the expression of multiple persistence module TFs, including FOXO1 and Eomes [181].
Metabolic adaptation of TEX cells to chronic antigen stimulation
Metabolism is a critical determinant of T-cell function. The exchange of metabolites between T cells and their surrounding environment profoundly influences T cell fate. Dysregulation of cellular energy metabolism in exhausted T cells not only limits their bioenergetic capacity but also reshapes their epigenetic program.
Exhausted CD8 T cells in chronic infections and tumors exhibit marked impairment in core bioenergetic pathways, with both glycolysis and oxidative phosphorylation (OXPHOS) substantially reduced [137, 182,183,184,185]. Mitochondria in exhausted T cells display reduced mass, lower membrane potential, and impaired respiratory reserve, changes that are closely associated with decreased expression of PGC1α, a central regulator of mitochondrial biogenesis and antioxidant defense [137, 182,183,184,185]. Depolarized mitochondria in CD8⁺ tumor-infiltrating lymphocytes (TILs), resulting from impaired mitophagy, drive terminal exhaustion through epigenetic reprogramming. Enhancing mitochondrial fitness with nicotinamide riboside alleviated dysfunction and improved the response to PD-1 blockade [186]. In parallel, glycolytic flux is suppressed through both extrinsic and intrinsic mechanisms [137, 185, 187]. Nutrient limitation in the tumor microenvironment restricts glucose uptake, while persistent signaling through the PD-1 pathway inhibits aerobic glycolysis, further exacerbating metabolic insufficiency [137]. Metabolic reprogramming of T cells by increasing phosphoenolpyruvate production via PCK1 overexpression enhances effector function and tumor control by T cells [187]. Together, mitochondrial and glycolytic defects create an energy-deficient state that reinforces functional decline in exhausted T cells.
The metabolic state of exhausted T cells directly shapes their epigenetic landscape. Key metabolites such as acetyl-CoA, α-ketoglutarate, and S-adenosylmethionine (SAM) act as substrates or cofactors for histone acetylation and methylation, thereby modulating gene expression profiles that are central to T-cell fate [188,189,190,191,192]. Perturbations in amino acid metabolism can also have lasting epigenetic consequences. Methionine availability regulates methyl group donation for histone and DNA methylation, whereas tryptophan catabolism alters chromatin states [188, 193,194,195]. Metabolic–epigenetic coupling stabilizes exhaustion-associated programs, making T cells resistant to functional reprogramming even when inhibitory receptor signaling is blocked.
Beyond mitochondrial bioenergetics and metabolic–epigenetic coupling, additional metabolites and nutrient pathways critically influence the establishment and persistence of T-cell exhaustion. In the tumor microenvironment, the depletion of amino acids such as arginine and serine impairs proliferation, cytokine secretion, and receptor expression [196, 197]. Dysregulated lipid metabolism is a common feature of PD-1hi TILs and is characterized by the accumulation of cholesterol and fatty acids [198, 199]. These lipid deposits induce endoplasmic reticulum stress, thereby impairing effector T-cell function. However, cholesterol deficiency also impairs the effector function of tumor-infiltrating T cells [200]. Conjugated bile acids accumulate in liver cancer, whereas inhibiting their synthesis improves T cell function and sensitivity to ICB [201]. Hypoxia has dual effects on T cells. While HIF signaling promotes glycolytic metabolism and augments effector activity in certain contexts [202,203,204], it may simultaneously induce inhibitory receptor expression and dampen cytotoxic function [205]. Importantly, under persistent antigenic stimulation, hypoxic stress accelerates this dysfunction by enforcing Blimp1–mediated repression of PGC1α-dependent mitochondrial reprogramming [206, 207]. Other metabolic by-products regulate T-cell exhaustion and differentiation. Succinate, a TCA cycle metabolite that accumulates in SDH-deficient tumors, enhances CD8 T cell stemness and persistence through mitochondrial and epigenetic remodeling and thereby improves the response to CAR-T and checkpoint blockade therapies [208]. Acidic metabolic waste accumulated in the tumor microenvironment paradoxically preserved T cell stemness and enhanced persistent antitumor T-cell immunity [209]. Clearance of ammonia, a byproduct of amino acid metabolism, is required for the development of T cell memory and can be targeted to improve adoptive cell therapy [210]. Additional by-products, such as tumor-derived lactate and excess extracellular potassium, also regulate exhaustion by directly impairing effector function or skewing differentiation toward stem-like states [211,212,213]. Notably, stiffness of the extracellular matrix is a hallmark of cancer and promotes exhaustion through the PIEZO1-OSR2 axis [214]. Thus, T-cell exhaustion is not caused by a single metabolic defect but by a complex interplay of nutrient availability, metabolic activity, and environmental stressors.
TCR activation triggers calcium release from the endoplasmic reticulum [215]. Increased cytosolic calcium is subsequently taken up by mitochondria, which are the primary sites of oxidative phosphorylation, and increases the activity of multiple TCA cycle enzymes. This increase in enzymatic activity promotes the generation of redox cofactors and increases reactive oxygen species (ROS) production. In both tumor and chronic infection models, persistent antigenic stimulation drives mitochondrial dysfunction in T cells, leading to impaired oxidative phosphorylation, ATP depletion, and ROS accumulation [216]. These redox-driven defects enforce terminal exhaustion by suppressing self-renewal programs and activating exhaustion-associated TFs, whereas antioxidant treatment restores proliferation, effector function, and progenitor-like features, thereby enhancing antitumor immunity [216]. While excessive ROS are detrimental to the T cell response, ROS also play an important role in T cell activation [217]. KEAP1 is a key sensor of oxidative stress. Under basal conditions, it targets the TF NRF2 for proteasomal degradation. Upon oxidation of reactive cysteine residues, NRF2 is released from KEAP1, which is subsequently translocated to the nucleus, where it activates the expression of antioxidant genes. KEAP1 expression is essential for CD8 T cells to adapt to chronic antigens because it prevents NRF2-driven hyperactivation of TCR signaling, cell death, and metabolic dysregulation [218]. KEAP1 deficiency and NRF2 hyperactivation reduce the TSL subset and lead to the accumulation of TEX with a terminal exhaustion phenotype [218]. NRF2 promotes exhaustion by upregulating the expression of the immune checkpoint PTGIR, which impairs metabolism and cytokine production by T cells [219]. In the context of asparagine restriction, however, NRF2 plays a positive role in the metabolic fitness and antitumor response of T cells [220]. The precise impact of the KEAP1-NRF2 axis on the T cell response may be context dependent.
Targeting the molecular program of T cell exhaustion and stemness to improve immunotherapy efficacy
Successful T-cell-based cancer immunotherapy depends on the balanced differentiation of T-cell effectiveness and persistence. It has been shown that the TCF1+ TSL population in the tumor microenvironment is the major population that responds to ICB, and these cells differentiate into further reinvigorated eff-like TEX cells to eliminate tumor growth [42, 45, 221, 222]. According to multiple scRNA-seq studies of tumor-infiltrated immune cells, the abundance of TSL cells is a prognostic marker for ICB treatment in different cancer types, including melanoma [223], breast cancer [224], and renal cell carcinoma [225]. While this persistent transcriptional module is important for maintaining the antigen-specific cellular response pool to ICBs, the major reinvigoration feature of ICB-treated antigen-specific CD8 T cells is eff-like TEX reactivation [23]. In clinical studies, enhanced T cell response features, such as stronger cell cycling and effector-associated molecule expression, have also been reported to be associated with better outcomes [226, 227], although these effector-like cells share TEX receptor profiles [228,229,230,231,232]. Thus, modulating transcriptional circuits to reinforce TEFF-associated responses has also been a working hypothesis in several studies that involved targeting TOX [44, 70,71,72,73,74], Fli1 [173], and Blimp1/NR4A3 [233] or enhancing STAT5 signaling [113].
The transcriptional features of CAR-T cells in the tumor microenvironment are similar to those of infection-model-defined TEX cells [78, 88, 234], with an increase in effector features over time in non-Hodgkin lymphoma patients but an increased AP-1/NR4A/BLIMP1 TF profile in the TIGIT+ CAR-T cell population in the nonresponsive group [234]. Similar dysfunctional CAR-T cell features with increased Blimp1/NR4A3 expression were also observed in metastatic prostate cancer treatment, in which targeting these two TFs increased the therapeutic effect of CAR-T cells in murine models [233]. Deletion of the pro-exhaustion TF ETV7 also enhances the antitumor efficacy of CD8 T cells [235]. Furthermore, in vitro CRISPR screening revealed TLE4 and IKZF2 as negative regulators that restrict the effects of effector-like CAR-T cells against glioblastomas [236]. In addition to tuning the effector and terminal exhaustion balance, several other studies have focused on enhancing stem-like differentiation during CAR-T-cell responses and have highlighted the importance of the stemness module during cancer treatment. An earlier study of Listeria monocytogenes infection revealed that deleting the histone H3 lysine 9 methyltransferase Suv39h1 promotes the stemness of T cells [237]. Consistently, disruption of Suv39h1 in CAR-T cells improves stemness, expansion, persistence, and tumor control [238]. In addition to Suv39h1, disrupting other epigenetic regulators, such as DNMT3a, TET2, and ASXL1, also enhances T cell stemness and antitumor T cell immunity in adoptive cell therapy [239, 240] and ICB [241]. The AP-1 TF c-JUN promotes a TSL-like phenotype in CAR-T cells [242]. Another AP-1 member, BATF, is the key TF downstream of PD-1 [98] and is involved in early T cell activation [243]. BATF can amplify effector-like T cell features during chronicity [244], and targeting BATF in CAR-T cells enhances the stemness module for a long-term robust response [245]. Notably, the deletion of REGNASE-1, which targets BATF, in T cells programs the long-term antitumor efficacy of adoptive cell therapy [99]. Combined deletion of REGNASE-1 and BCOR synergistically induces an immortal stem-like state and enhances the function of CAR-T cells [246]. Furthermore, while activating more IL2-STAT5 transcriptional circuits may be an important strategy for promoting stronger eff-like TEX signatures [77, 113, 247,248,249], activating more stemness-related modules via IL10-STAT3 signaling may also be a viable strategy for achieving better therapeutic outcomes [250, 251]. These studies indicate several potential general mechanisms involved in the transcriptional regulation of CAR-T-cell responses: (1) Effector and exhausted T-cells are defined on the basis of the functional capacity per cell, and both of them can be driven into terminal stages via transcriptional circuits involving TFs such as Blimp1; (2) AP-1 activation is a major feature of CAR-T-cell activation; however, different AP-1 family members may trigger different downstream effects in tuning the effector versus stemness modules, potentially by involving different TF co-binders such as IRF4 or NFAT; and (3) considering that BACH2 inhibits broad AP-1 function and locks the cell into a stemness stage, it is important to orchestrate BACH2 and AP-1 levels to maintain the balance in the differentiation of CAR-T-cells in vivo for the best potential outcomes. (4) STAT signaling activation in the tumor microenvironment is among the key factors involved in the differentiation of the T cell response, with STAT5 signaling being more biased toward effector differentiation and STAT3 signaling being more biased toward stemness. Both strategies may benefit clinical outcomes, but in different scenarios.
How should we choose to enhance short-term effector function or stem-like differentiation and the persistence of CD8 T cell responses during disease treatment, particularly in cancer immunotherapies? One major potential prediagnostic identifier is the “effective immune–tumor intensity ratio.” Previous studies have shown that the “responsive T cell-to-tumor” ratio can be a key marker for predicting the clinical outcome of anti-PD-1 responses [252, 253]. Indeed, combining immunotherapy with chemotherapy or radiotherapy enhances the clinical response and has demonstrated potential benefits across a variety of clinical scenarios [254, 255]. In addition to the effects of extraantigen exposure as well as local inflammatory immune microenvironment reorganization [255], one of the potential reasons for better outcomes in some of these situations is increasing the “effective immune–tumor intensity ratio”. These findings indicate that there are two potential outcomes after treatment: (1) Antigen-specific T cells respond strongly to a limited tumor volume. In this context, a stronger effector T cell response is more likely to trigger a favorable clinical outcome, with the capacity to achieve tumor clearance at least at the given lesion level. (2) The antigen-specific T cell population has some response but is not able to clear tumor cells in the lesion in the short term. Under these conditions, the persistence of exhausted T cells is important for maintaining immune–tumor equilibrium and a partial response or stable disease. This notion is supported by a recent study on the ICB-induced T cell response in murine tumors with different levels of immunogenicity [256].
A topic that has recently garnered increased research efforts is whether we can rewire the molecular circuits of T cells to achieve a context-specific T cell response, particularly in the tumor microenvironment. In the past decade, the syn-Notch system has been developed to sequentially arrange tumor microenvironmental signaling activation toward local CAR expression in T cells, thus triggering only an intratumoral CAR-T cell response at target sites [257,258,259]. Additionally, the development of an orthogonal cytokine system offers the ability to improve cytokine treatment by supporting the persistence of infiltrating T cells and promoting lesion-dependent, local antigen-specific T cell proliferation [113, 260, 261]. Furthermore, the recent development of integrating CAR constructs, particularly into PD-1 loci, suggests the possibility of using exhaustion-specific DNA regulatory elements to achieve tumor microenvironment-specific functional molecular responses [262, 263]. These strategies aim to initiate strong antigen-specific T cell responses in the lesion area while eliminating off-site T cell activation to reduce potential immune-related adverse events in patients. Thus, future efforts may dive deeper into rewiring molecular circuits of the T cell response at the lesion microenvironmental level to achieve precision microenvironmental medicine.
Druggable targets and interventional modalities
Recent studies have revealed diverse regulatory checkpoints that control T cell exhaustion, many of which represent potential druggable targets for immune modulation. At the epigenetic level, inhibition of the histone demethylase LSD1 by a small-molecule drug preserves the progenitor-exhausted T cell pool and sustains durable responses to PD-1 blockade by counteracting TCF1 repression and terminal differentiation [264]. Despite their critical role in controlling T cell fates, most master transcriptional regulators are challenging to target using traditional small-molecule therapeutics. Emerging strategies such as PROTACs and molecular glues offer promising alternatives for modulating these TFs. Degradation of the nuclear receptor NR4A1, which represses the effector program in exhausted T cells, by the PROTAC NR-V04 reprograms the tumor microenvironment by enhancing the response of effector-memory CD8 T cells and reducing suppressive myeloid populations [265]. A molecular glue targeting IKZF2 rescues exhausted T cells and potentiates immune control of tumors [266]. In addition, chemical switches can be fused to TFs such as BACH2 to exert temporal and tunable control of T cell differentiation and improve the efficacy of CAR-T-cell therapy [121]. A screen of chromatin-modifying drugs revealed HDAC inhibitors that increase the persistence and repress the exhaustion of CAR-T cells to promote their antitumor immunity [267]. HDAC inhibitors also synergize with PD-1 blockade to enhance antitumor T cell responses [268]. Inhibitors of the EZH2 protein, a core component of the polycomb repressive complex 2, increase CAR-T-cell efficacy by directly repressing exhaustion [269]. JQ1, a small-molecule inhibitor of BRD4, enhances the persistence and antitumor immunity of T cells in adoptive cell therapy while preventing terminal differentiation [270, 271]. The in vitro manufacturing of therapeutic T cells, including CAR-T cells, offers a unique opportunity to rewire their cellular programs with chemical treatments while avoiding direct drug exposure in patients. Treatment with ibrutinib, which inhibits ITK and BTK, during manufacturing promotes survival and stemness and represses the exhaustion of CAR-T cells [218, 272]. Pretreatment of CAR-T cells with inhibitors targeting AKT or MEK attenuated exhaustion and terminal differentiation and potentiated the antitumor efficacy of CAR-T cells in vivo [273, 274]. Interestingly, lithium carbonate treatment enhances antitumor immunity in T cells through directing lactate to mitochondria and could improve T cell-based immunotherapy [275]. Therefore, targeting key regulators of T cell exhaustion and stemness, either in vivo or in vitro, constitutes a viable strategy to improve the antitumor efficacy of T cells.
In this article, we summarize the major molecular circuits that regulate the stemness and exhaustion of T cells and the differentiation of TSL cells into different TEX progenies. We further discuss how the balance between the short-term response to TEFF cells and long-term persistence sustained by TSL cells affects the outcome of T cell-mediated immune responses in cancer and chronic infection. Finally, we discuss the current status and future directions for harnessing molecular circuits to control T cell differentiation in T-cell-based immunotherapy.
References
Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–8. https://doi.org/10.1038/ni1009.
Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–95. https://doi.org/10.1016/j.immuni.2007.07.010.
Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–34. https://doi.org/10.1038/ni889.
Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–7. https://doi.org/10.1038/nm.2446.
Johnnidis JB, Muroyama Y, Ngiow SF, Chen Z, Manne S, Cai Z, et al. Inhibitory signaling sustains a distinct early memory CD8(+) T cell precursor that is resistant to DNA damage. Sci Immunol. 2021;6. https://doi.org/10.1126/sciimmunol.abe3702
Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–13. https://doi.org/10.1038/nm.1982.
Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med. 2005;11:1299–305. https://doi.org/10.1038/nm1326.
Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10:524–30. https://doi.org/10.1038/ni.1718.
Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. 2010;207:553–64. https://doi.org/10.1084/jem.20090858.
Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci USA. 2010;107:17872–9. https://doi.org/10.1073/pnas.1010201107.
Liu L, Zhong Q, Tian T, Dubin K, Athale SK, Kupper TS. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat Med. 2010;16:224–7. https://doi.org/10.1038/nm.2078.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99. https://doi.org/10.1038/nri3862.
Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–13. https://doi.org/10.1084/jem.188.12.2205.
Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–61. https://doi.org/10.1038/362758a0.
Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. https://doi.org/10.1038/nature05115.
Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–25. https://doi.org/10.1128/JVI.02844-06.
Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, et al. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–403. https://doi.org/10.1128/JVI.01177-06.
Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–202. https://doi.org/10.1038/nm1482.
McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019;37:457–95. https://doi.org/10.1146/annurev-immunol-041015-055318.
Zuniga EI, Macal M, Lewis GM, Harker JA. Innate and adaptive immune regulation during chronic viral infections. Annu Rev Virol. 2015;2:573–97. https://doi.org/10.1146/annurev-virology-100114-055226.
Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu Rev Med. 2018;69:301–18. https://doi.org/10.1146/annurev-med-012017-043208.
Launonen IM, Niemiec I, Hincapié-Otero M, Erkan EP, Junquera A, Afenteva D, et al. Chemotherapy induces myeloid-driven spatially confined T cell exhaustion in ovarian cancer. Cancer Cell. 2024;42:2045–63.e10. https://doi.org/10.1016/j.ccell.2024.11.005.
Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–5. https://doi.org/10.1126/science.aaf2807.
Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354:1165–9. https://doi.org/10.1126/science.aae0491.
Abdel-Hakeem MS, Manne S, Beltra JC, Stelekati E, Chen Z, Nzingha K, et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat Immunol. 2021;22:1008–19. https://doi.org/10.1038/s41590-021-00975-5.
Yates KB, Tonnerre P, Martin GE, Gerdemann U, Al Abosy R, Comstock DE, et al. Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nat Immunol. 2021;22:1020–9. https://doi.org/10.1038/s41590-021-00979-1.
McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–6. https://doi.org/10.1038/nature14468.
McKinney EF, Smith KG. T-cell exhaustion: understanding the interface of chronic viral and autoinflammatory diseases. Immunol Cell Biol. 2016;94:935–42. https://doi.org/10.1038/icb.2016.81.
Okazaki T, Okazaki IM, Wang J, Sugiura D, Nakaki F, Yoshida T, et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. 2011;208:395–407. https://doi.org/10.1084/jem.20100466.
McKinney EF, Smith KG. T cell exhaustion and immune-mediated disease-the potential for therapeutic exhaustion. Curr Opin Immunol. 2016;43:74–80. https://doi.org/10.1016/j.coi.2016.09.005.
Zou D, Dai Y, Zhang X, Wang G, Xiao X, Jia P, et al. T cell exhaustion is associated with antigen abundance and promotes transplant acceptance. Am J Transplant. 2020;20:2540–50. https://doi.org/10.1111/ajt.15870.
Yang J, Popoola J, Khandwala S, Vadivel N, Vanguri V, Yuan X, et al. Critical role of donor tissue expression of programmed death ligand-1 in regulating cardiac allograft rejection and vasculopathy. Circulation. 2008;117:660–9. https://doi.org/10.1161/CIRCULATIONAHA.107.741025.
Alzahrani N, Al Jurdi A, Riella LV. Immune checkpoint inhibitors in kidney transplantation. Curr Opin Organ Transplant. 2023;28:46–54. https://doi.org/10.1097/MOT.0000000000001036.
Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci USA. 2008;105:15016–21. https://doi.org/10.1073/pnas.0801497105.
Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338:1220–5. https://doi.org/10.1126/science.1229620.
Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog. 2015;11:e1005177. https://doi.org/10.1371/journal.ppat.1005177.
Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. 2016;1. https://doi.org/10.1126/sciimmunol.aai8593
Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–21. https://doi.org/10.1038/nature19330.
He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5-expressing CD8(+) T cells curtail chronic viral infection. Nature. 2016;537:412–28. https://doi.org/10.1038/nature19317.
Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol. 2016;17:1187–96. https://doi.org/10.1038/ni.3543.
Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity. 2016;45:415–27. https://doi.org/10.1016/j.immuni.2016.07.021.
Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20:326–36. https://doi.org/10.1038/s41590-019-0312-6.
Ferrando-Martinez S, Moysi E, Pegu A, Andrews S, Nganou Makamdop K, Ambrozak D, et al. Accumulation of follicular CD8+ T cells in pathogenic SIV infection. J Clin Investig. 2018;128:2089–103. https://doi.org/10.1172/JCI96207.
Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol. 2019;20:890–901. https://doi.org/10.1038/s41590-019-0403-4.
Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50:195–211.e10. https://doi.org/10.1016/j.immuni.2018.12.021.
Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(-)CD8(+) tumor-infiltrating T cells. Immunity. 2019;50:181–94.e6. https://doi.org/10.1016/j.immuni.2018.11.014.
Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, et al. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity. 2019;51:840–55 e845. https://doi.org/10.1016/j.immuni.2019.09.013.
Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan SL, et al. Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1(+) CD8(+) T cells in tumor-draining lymph nodes. Immunity. 2021;54:2338–53.e6. https://doi.org/10.1016/j.immuni.2021.08.026.
Meiser P, Knolle MA, Hirschberger A, de Almeida GP, Bayerl F, Lacher S, et al. A distinct stimulatory cDC1 subpopulation amplifies CD8(+) T cell responses in tumors for protective anti-cancer immunity. Cancer Cell. 2023;41:1498–515.e10. https://doi.org/10.1016/j.ccell.2023.06.008.
Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, et al. A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci Immunol. 2021;6:eabg7836. https://doi.org/10.1126/sciimmunol.abg7836.
Huang Q, Wu X, Wang Z, Chen X, Wang L, Lu Y, et al. The primordial differentiation of tumor-specific memory CD8(+) T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell. 2022;185:4049–66.e25. https://doi.org/10.1016/j.cell.2022.09.020.
Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576:465–70. https://doi.org/10.1038/s41586-019-1836-5.
Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The PD-1/PD-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell. 2020;38:685–700.e8. https://doi.org/10.1016/j.ccell.2020.09.001.
Rahim MK, Okholm T, Jones KB, McCarthy EE, Liu CC, Yee JL, et al. Dynamic CD8(+) T cell responses to cancer immunotherapy in human regional lymph nodes are disrupted in metastatic lymph nodes. Cell. 2023;186:1127–1143.e18. https://doi.org/10.1016/j.cell.2023.02.021.
Lan X, Mi T, Alli S, Guy C, Djekidel MN, Liu X, et al. Antitumor progenitor exhausted CD8(+) T cells are sustained by TCR engagement. Nat Immunol. 2024;25:1046–58. https://doi.org/10.1038/s41590-024-01843-8.
Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128:519–28. https://doi.org/10.1182/blood-2015-11-683847.
Zheng W, Wei J, Zebley CC, Jones LL, Dhungana Y, Wang YD, et al. Regnase-1 suppresses TCF-1+ precursor exhausted T-cell formation to limit CAR-T-cell responses against ALL. Blood. 2021;138:122–35. https://doi.org/10.1182/blood.2020009309.
van Bruggen J, Martens A, Fraietta JA, Hofland T, Tonino SH, Eldering E, et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8(+) T cells and impede CAR T-cell efficacy. Blood. 2019;134:44–58. https://doi.org/10.1182/blood.2018885863.
Gabriel SS, Tsui C, Chisanga D, Weber F, Llano-León M, Gubser PM, et al. Transforming growth factor-beta-regulated mTOR activity preserves cellular metabolism to maintain long-term T cell responses in chronic infection. Immunity. 2021;54:1698–714 e5. https://doi.org/10.1016/j.immuni.2021.06.007.
Wu H, Zhao X, Hochrein SM, Eckstein M, Gubert GF, Knöpper K, et al. Mitochondrial dysfunction promotes the transition of precursor to terminally exhausted T cells through HIF-1alpha-mediated glycolytic reprogramming. Nat Commun. 2023;14:6858. https://doi.org/10.1038/s41467-023-42634-3.
Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175:998–1013.e20. https://doi.org/10.1016/j.cell.2018.10.038.
Liu Z, Yang Z, Wu J, Zhang W, Sun Y, Zhang C, et al. A single-cell atlas reveals immune heterogeneity in anti-PD-1-treated non-small cell lung cancer. Cell. 2025;188:3081–96.e19. https://doi.org/10.1016/j.cell.2025.03.018.
Chen GM, Chen C, Das RK, Gao P, Chen CH, Bandyopadhyay S, et al. Integrative bulk and single-cell profiling of premanufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell Therapy. Cancer Discov. 2021;11:2186–99. https://doi.org/10.1158/2159-8290.CD-20-1677.
Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563–71. https://doi.org/10.1038/s41591-018-0010-1.
Deng Q, Han G, Puebla-Osorio N, Ma M, Strati P, Chasen B, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med. 2020;26:1878–87. https://doi.org/10.1038/s41591-020-1061-7.
Gearty SV, Dündar F, Zumbo P, Espinosa-Carrasco G, Shakiba M, Sanchez-Rivera FJ, et al. An autoimmune stem-like CD8 T cell population drives type 1 diabetes. Nature. 2022;602:156–61. https://doi.org/10.1038/s41586-021-04248-x.
Abdelsamed HA, Zebley CC, Nguyen H, Rutishauser RL, Fan Y, Ghoneim HE, et al. Beta cell-specific CD8(+) T cells maintain stem cell memory-associated epigenetic programs during type 1 diabetes. Nat Immunol. 2020;21:578–87. https://doi.org/10.1038/s41590-020-0633-5.
Shin B, Kress RL, Kramer PA, Darley-Usmar VM, Bellis SL, Harrington LE. Effector CD4 T cells with progenitor potential mediate chronic intestinal inflammation. J Exp Med. 2018;215:1803–12. https://doi.org/10.1084/jem.20172335.
Brooks DG, McGavern DB, Oldstone MB. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J Clin Investig. 2006;116:1675–85. https://doi.org/10.1172/JCI26856.
Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571:265–9. https://doi.org/10.1038/s41586-019-1326-9.
Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571:211–8. https://doi.org/10.1038/s41586-019-1325-x.
Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–4. https://doi.org/10.1038/s41586-019-1324-y.
Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci USA. 2019;116:12410–5. https://doi.org/10.1073/pnas.1905675116.
Wang X, He Q, Shen H, Xia A, Tian W, Yu W, et al. TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol. 2019;71:731–41. https://doi.org/10.1016/j.jhep.2019.05.015.
McManus DT, Valanparambil RM, Medina CB, Scharer CD, McGuire DJ, Sobierajska E, et al. An early precursor CD8(+) T cell that adapts to acute or chronic viral infection. Nature. 2025;640:772–81. https://doi.org/10.1038/s41586-024-08562-y.
Chu T, Wu M, Hoellbacher B, de Almeida GP, Wurmser C, Berner J, et al. Precursors of exhausted T cells are pre-emptively formed in acute infection. Nature. 2025;640:782–92. https://doi.org/10.1038/s41586-024-08451-4.
Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity. 2020;52:825–41.e8. https://doi.org/10.1016/j.immuni.2020.04.014.
Zhu Z, Lou G, Teng XL, Wang H, Luo Y, Shi W, et al. FOXP1 and KLF2 reciprocally regulate checkpoints of stem-like to effector transition in CAR T cells. Nat Immunol. 2024;25:117–28. https://doi.org/10.1038/s41590-023-01685-w.
Tsui C, Kretschmer L, Rapelius S, Gabriel SS, Chisanga D, Knöpper K, et al. MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature. 2022;609:354–60. https://doi.org/10.1038/s41586-022-05105-1.
Galletti G, De Simone G, Mazza E, Puccio S, Mezzanotte C, Bi TM, et al. Two subsets of stem-like CD8(+) memory T cell progenitors with distinct fate commitments in humans. Nat Immunol. 2020;21:1552–62. https://doi.org/10.1038/s41590-020-0791-5.
Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, et al. Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1(+) stem-like CD8(+) T cells during chronic infection. Immunity. 2019;51:1043–58.e4. https://doi.org/10.1016/j.immuni.2019.11.002.
Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. CD4(+) T cell help is required for the formation of a cytolytic CD8(+) T cell subset that protects against chronic infection and cancer. Immunity. 2019;51:1028–42.e4. https://doi.org/10.1016/j.immuni.2019.10.009.
Sandu I, Cerletti D, Oetiker N, Borsa M, Wagen F, Spadafora I, et al. Landscape of exhausted virus-specific CD8 T cells in chronic LCMV infection. Cell Rep. 2020;32:108078. https://doi.org/10.1016/j.celrep.2020.108078.
Yan Y, Cao S, Liu X, Harrington SM, Bindeman WE, Adjei AA, et al. CX3CR1 identifies PD-1 therapy-responsive CD8+ T cells that withstand chemotherapy during cancer chemoimmunotherapy. JCI Insight. 2018;3. https://doi.org/10.1172/jci.insight.97828
Daniel B, Yost KE, Hsiung S, Sandor K, Xia Y, Qi Y, et al. Divergent clonal differentiation trajectories of T cell exhaustion. Nat Immunol. 2022;23:1614–27. https://doi.org/10.1038/s41590-022-01337-5.
Giles JR, Ngiow SF, Manne S, Baxter AE, Khan O, Wang P, et al. Shared and distinct biological circuits in effector, memory and exhausted CD8(+) T cells revealed by temporal single-cell transcriptomics and epigenetics. Nat Immunol. 2022;23:1600–13. https://doi.org/10.1038/s41590-022-01338-4.
Di Pilato M, Kfuri-Rubens R, Pruessmann JN, Ozga AJ, Messemaker M, Cadilha BL, et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell. 2021;184:4512–30.e22. https://doi.org/10.1016/j.cell.2021.07.015.
Ngiow SF, Manne S, Huang YJ, Azar T, Chen Z, Mathew D, et al. LAG-3 sustains TOX expression and regulates the CD94/NKG2-Qa-1b axis to govern exhausted CD8 T cell NK receptor expression and cytotoxicity. Cell. 2024;187:4336–54.e19. https://doi.org/10.1016/j.cell.2024.07.018.
Broomfield BJ, Tan CW, Qin RZ, Abberger H, Duckworth BC, Alvarado C, et al. Transient inhibition of type I interferon enhances CD8+ T cell stemness and vaccine protection. J Exp Med. 2025;222. https://doi.org/10.1084/jem.20241148
Chen J, López-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567:530–4. https://doi.org/10.1038/s41586-019-0985-x.
Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019;567:525–9. https://doi.org/10.1038/s41586-019-0979-8.
Hao J, Li R, Zhao X, Liu X, Chen X, Xie T, et al. NR4A1 transcriptionally regulates the differentiation of stem-like CD8(+) T cells in the tumor microenvironment. Cell Rep. 2024;43:114301. https://doi.org/10.1016/j.celrep.2024.114301.
Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. 2015;42:265–78. https://doi.org/10.1016/j.immuni.2015.01.006.
Tillé L, Cropp D, Charmoy M, Reichenbach P, Andreatta M, Wyss T, et al. Activation of the transcription factor NFAT5 in the tumor microenvironment enforces CD8(+) T cell exhaustion. Nat Immunol. 2023;24:1645–53. https://doi.org/10.1038/s41590-023-01614-x.
Klein-Hessling S, Muhammad K, Klein M, Pusch T, Rudolf R, Flöter J, et al. NFATc1 controls the cytotoxicity of CD8(+) T cells. Nat Commun. 2017;8:511. https://doi.org/10.1038/s41467-017-00612-6.
Agnellini P, Wolint P, Rehr M, Cahenzli J, Karrer U, Oxenius A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc Natl Acad Sci USA. 2007;104:4565–70. https://doi.org/10.1073/pnas.0610335104.
Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity. 2017;47:1129–41.e5. https://doi.org/10.1016/j.immuni.2017.11.021.
Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16:1147–51. https://doi.org/10.1038/nm.2232.
Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576:471–6. https://doi.org/10.1038/s41586-019-1821-z.
Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio CJ, et al. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol. 2021;22:983–95. https://doi.org/10.1038/s41590-021-00964-8.
Escobar G, Mangani D, Anderson AC. T cell factor 1: a master regulator of the T cell response in disease. Sci Immunol. 2020;5. https://doi.org/10.1126/sciimmunol.abb9726
Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, et al. A critical role for TCF-1 in T-lineage specification and differentiation. Nature. 2011;476:63–8. https://doi.org/10.1038/nature10279.
Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, et al. T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc Natl Acad Sci USA. 2011;108:20060–5. https://doi.org/10.1073/pnas.1110230108.
Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, et al. Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity. 2018;48:243–57.e10. https://doi.org/10.1016/j.immuni.2018.01.012.
Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol. 2016;17:695–703. https://doi.org/10.1038/ni.3456.
Shan Q, Zhu S, Chen X, Liu J, Yuan S, Li X, et al. Tcf1-CTCF cooperativity shapes genomic architecture to promote CD8(+) T cell homeostasis. Nat Immunol. 2022;23:1222–35. https://doi.org/10.1038/s41590-022-01263-6.
Jeannet G, Boudousquié C, Gardiol N, Kang J, Huelsken J, Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci USA. 2010;107:9777–82. https://doi.org/10.1073/pnas.0914127107.
Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33:229–40. https://doi.org/10.1016/j.immuni.2010.08.002.
Pais Ferreira D, Silva JG, Wyss T, Fuertes Marraco SA, Scarpellino L, Charmoy M, et al. Central memory CD8(+) T cells derive from stem-like Tcf7(hi) effector cells in the absence of cytotoxic differentiation. Immunity. 2020;53:985–1000.e11. https://doi.org/10.1016/j.immuni.2020.09.005.
Gullicksrud JA, Li F, Xing S, Zeng Z, Peng W, Badovinac VP, et al. Differential Requirements for Tcf1 Long Isoforms in CD8(+) and CD4(+) T Cell Responses to Acute Viral Infection. J Immunol. 2017;199:911–9. https://doi.org/10.4049/jimmunol.1700595.
Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, et al. TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep. 2015;12:2099–110. https://doi.org/10.1016/j.celrep.2015.08.049.
Huang Z, Zak J, Pratumchai I, Shaabani N, Vartabedian VF, Nguyen N, et al. IL-27 promotes the expansion of self-renewing CD8(+) T cells in persistent viral infection. J Exp Med. 2019;216:1791–808. https://doi.org/10.1084/jem.20190173.
Beltra JC, Abdel-Hakeem MS, Manne S, Zhang Z, Huang H, Kurachi M, et al. Stat5 opposes the transcription factor Tox and rewires exhausted CD8(+) T cells toward durable effector-like states during chronic antigen exposure. Immunity. 2023;56:2699–718.e11. https://doi.org/10.1016/j.immuni.2023.11.005.
Tsukumo S, Unno M, Muto A, Takeuchi A, Kometani K, Kurosaki T, et al. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proc Natl Acad Sci USA. 2013;110:10735–40. https://doi.org/10.1073/pnas.1306691110.
Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature. 2013;498:506–10. https://doi.org/10.1038/nature12199.
Roychoudhuri R, Clever D, Li P, Wakabayashi Y, Quinn KM, Klebanoff CA, et al. BACH2 regulates CD8(+) T cell differentiation by controlling access of AP-1 factors to enhancers. Nat Immunol. 2016;17:851–60. https://doi.org/10.1038/ni.3441.
Grant FM, Yang J, Nasrallah R, Clarke J, Sadiyah F, Whiteside SK, et al. BACH2 drives quiescence and maintenance of resting Treg cells to promote homeostasis and cancer immunosuppression. J Exp Med. 2020;217. https://doi.org/10.1084/jem.20190711
Sidwell T, Liao Y, Garnham AL, Vasanthakumar A, Gloury R, Blume J, et al. Attenuation of TCR-induced transcription by Bach2 controls regulatory T cell differentiation and homeostasis. Nat Commun. 2020;11:252. https://doi.org/10.1038/s41467-019-14112-2.
Yao C, Lou G, Sun HW, Zhu Z, Sun Y, Chen Z, et al. BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8(+) T cells. Nat Immunol. 2021;22:370–80. https://doi.org/10.1038/s41590-021-00868-7.
Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, et al. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol. 2020;21:1256–66. https://doi.org/10.1038/s41590-020-0760-z.
Hu T, Zhu Z, Luo Y, Wizzard S, Hoar J, Shinde SS, et al. BACH2 dosage establishes the hierarchy of stemness and finetunes antitumor immunity in CAR T cells. bioRxiv [Preprint]. 2025. https://doi.org/10.1101/2025.08.18.670909
Sandberg ML, Sutton SE, Pletcher MT, Wiltshire T, Tarantino LM, Hogenesch JB, et al. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev Cell. 2005;8:153–66. https://doi.org/10.1016/j.devcel.2004.12.015.
Soza-Ried C, Hess I, Netuschil N, Schorpp M, Boehm T. Essential role of c-myb in definitive hematopoiesis is evolutionarily conserved. Proc Natl Acad Sci USA. 2010;107:17304–8. https://doi.org/10.1073/pnas.1004640107.
Bender TP, Kremer CS, Kraus M, Buch T, Rajewsky K. Critical functions for c-Myb at three checkpoints during thymocyte development. Nat Immunol. 2004;5:721–9. https://doi.org/10.1038/ni1085.
Lieu YK, Kumar A, Pajerowski AG, Rogers TJ, Reddy EP. Requirement of c-myb in T cell development and in mature T cell function. Proc Natl Acad Sci USA. 2004;101:14853–8. https://doi.org/10.1073/pnas.0405338101.
Badiani P, Corbella P, Kioussis D, Marvel J, Weston K. Dominant interfering alleles define a role for c-Myb in T-cell development. Genes Dev. 1994;8:770–82. https://doi.org/10.1101/gad.8.7.770.
Chen Z, Stelekati E, Kurachi M, Yu S, Cai Z, Manne S, et al. miR-150 regulates memory CD8 T cell differentiation via c-Myb. Cell Rep. 2017;20:2584–97. https://doi.org/10.1016/j.celrep.2017.08.060.
Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, et al. The transcription factor c-Myb regulates CD8(+) T cell stemness and antitumor immunity. Nat Immunol. 2019;20:337–49. https://doi.org/10.1038/s41590-018-0311-z.
Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–5. https://doi.org/10.1038/nature01667.
Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–84. https://doi.org/10.1038/ni.1689.
Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity. 2013;39:286–97. https://doi.org/10.1016/j.immuni.2013.07.013.
Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity. 2012;36:374–87. https://doi.org/10.1016/j.immuni.2012.01.015.
Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. 2013;210:1189–200. https://doi.org/10.1084/jem.20130392.
Delpoux A, Lai CY, Hedrick SM, Doedens AL. FOXO1 opposition of CD8(+) T cell effector programming confers early memory properties and phenotypic diversity. Proc Natl Acad Sci USA. 2017;114:E8865–74. https://doi.org/10.1073/pnas.1618916114.
Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41:802–14. https://doi.org/10.1016/j.immuni.2014.10.013.
Utzschneider DT, Delpoux A, Wieland D, Huang X, Lai CY, Hofmann M, et al. Active maintenance of T cell memory in acute and chronic viral infection depends on continuous expression of FOXO1. Cell Rep. 2018;22:3454–67. https://doi.org/10.1016/j.celrep.2018.03.020.
Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity. 2016;45:358–73. https://doi.org/10.1016/j.immuni.2016.07.008.
Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol. 2011;12:544–50. https://doi.org/10.1038/ni.2034.
Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS, Rudensky AY. Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat Immunol. 2019;20:232–42. https://doi.org/10.1038/s41590-018-0291-z.
Ren J, Han L, Tang J, Liu Y, Deng X, Liu Q, et al. Foxp1 is critical for the maintenance of regulatory T-cell homeostasis and suppressive function. PLoS Biol. 2019;17:e3000270. https://doi.org/10.1371/journal.pbio.3000270.
Stephen TL, Rutkowski MR, Allegrezza MJ, Perales-Puchalt A, Tesone AJ, Svoronos N, et al. Transforming growth factor beta-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression. Immunity. 2014;41:427–39. https://doi.org/10.1016/j.immuni.2014.08.012.
Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442:299–302. https://doi.org/10.1038/nature04882.
Weinreich MA, Takada K, Skon C, Reiner SL, Jameson SC, Hogquist KA. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity. 2009;31:122–30. https://doi.org/10.1016/j.immuni.2009.05.011.
Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14:1285–93. https://doi.org/10.1038/ni.2745.
Zhu Z, Lou G, Luo Y, Yihunie K, Hoar J, Daniel JA, et al. Aging compromises terminal differentiation program of cytotoxic effector lineage and promotes exhaustion in CD8(+) T cells responding to coronavirus infection. Aging Cell. 2025;24:e70109. https://doi.org/10.1111/acel.70109.
Fagerberg E, Attanasio J, Dien C, Singh J, Kessler EA, Abdullah L, et al. KLF2 maintains lineage fidelity and suppresses CD8 T cell exhaustion during acute LCMV infection. Science. 2025;387:eadn2337. https://doi.org/10.1126/science.adn2337.
Doan AE, Mueller KP, Chen AY, Rouin GT, Chen Y, Daniel B, et al. FOXO1 is a master regulator of memory programming in CAR T cells. Nature. 2024;629:211–8. https://doi.org/10.1038/s41586-024-07300-8.
Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021;372. https://doi.org/10.1126/science.aba1786
Durand A, Bonilla N, Level T, Ginestet Z, Lombès A, Guichard V, et al. Type 1 interferons and Foxo1 down-regulation play a key role in age-related T-cell exhaustion in mice. Nat Commun. 2024;15:1718. https://doi.org/10.1038/s41467-024-45984-8.
Ban YH, Oh SC, Seo SH, Kim SM, Choi IP, Greenberg PD, et al. miR-150-mediated Foxo1 regulation programs CD8(+) T cell differentiation. Cell Rep. 2017;20:2598–611. https://doi.org/10.1016/j.celrep.2017.08.065.
Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–44. https://doi.org/10.1038/ni1268.
Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12:1221–9. https://doi.org/10.1038/ni.2158.
Milner JJ, Toma C, He Z, Kurd NS, Nguyen QP, McDonald B, et al. Heterogenous populations of tissue-resident CD8(+) T cells are generated in response to infection and malignancy. Immunity. 2020;52:808–24.e7. https://doi.org/10.1016/j.immuni.2020.04.007.
Sun Q, Cai D, Liu D, Zhao X, Li R, Xu W, et al. BCL6 promotes a stem-like CD8(+) T cell program in cancer via antagonizing BLIMP1. Sci Immunol. 2023;8:eadh1306. https://doi.org/10.1126/sciimmunol.adh1306.
Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–20. https://doi.org/10.1038/ni.1837.
Sun Q, Zhao X, Li R, Liu D, Pan B, Xie B, et al. STAT3 regulates CD8+ T cell differentiation and functions in cancer and acute infection. J Exp Med. 2023;220. https://doi.org/10.1084/jem.20220686
Gil MP, Ploquin MJ, Watford WT, Lee SH, Kim K, Wang X, et al. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood. 2012;120:3718–28. https://doi.org/10.1182/blood-2012-05-428672.
Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–61. https://doi.org/10.1038/nri3307.
Guan T, Dominguez CX, Amezquita RA, Laidlaw BJ, Cheng J, Henao-Mejia J, et al. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8(+) T cell fates. J Exp Med. 2018;215:1153–68. https://doi.org/10.1084/jem.20171352.
Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, Nguyen JV, et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J Exp Med. 2015;212:2027–39. https://doi.org/10.1084/jem.20150194.
Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–3. https://doi.org/10.1126/science.1090148.
Li J, He Y, Hao J, Ni L, Dong C. High levels of eomes promote exhaustion of anti-tumor CD8(+) T cells. Front Immunol. 2018;9:2981. https://doi.org/10.3389/fimmu.2018.02981.
Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaëlsson J, et al. T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog. 2014;10:e1004251. https://doi.org/10.1371/journal.ppat.1004251.
Lu P, Youngblood BA, Austin JW, Mohammed AU, Butler R, Ahmed R, et al. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med. 2014;211:515–27. https://doi.org/10.1084/jem.20130208.
Xin A, Masson F, Liao Y, Preston S, Guan T, Gloury R, et al. A molecular threshold for effector CD8(+) T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nat Immunol. 2016;17:422–32. https://doi.org/10.1038/ni.3410.
Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. 2009;31:283–95. https://doi.org/10.1016/j.immuni.2009.06.021.
Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. https://doi.org/10.1016/j.immuni.2009.05.014.
McLane LM, Ngiow SF, Chen Z, Attanasio J, Manne S, Ruthel G, et al. Role of nuclear localization in the regulation and function of T-bet and Eomes in exhausted CD8 T cells. Cell Rep. 2021;35:109120. https://doi.org/10.1016/j.celrep.2021.109120.
Ran L, Yue Z, Ran M, Liu Q, Su X, Wang L, et al. The transcription regulator ID3 maintains tumor-specific memory CD8(+) T cells in draining lymph nodes during tumorigenesis. Cell Rep. 2024;43:114690. https://doi.org/10.1016/j.celrep.2024.114690.
Gago da Graça C, Sheikh AA, Newman DM, Wen L, Li S, Shen J, et al. Stem-like memory and precursors of exhausted T cells share a common progenitor defined by ID3 expression. Sci Immunol. 2025;10:eadn1945. https://doi.org/10.1126/sciimmunol.adn1945.
Lin S, Niu H, Zhang Y, Gai K, Brown R, Brown A, et al. SATB1 is a key regulator of quiescence in stem-like CD8(+) T cells. Nat Immunol. 2025;26:1737–51. https://doi.org/10.1038/s41590-025-02257-w.
Heyden L, Rausch L, Shannon MH, Dryburgh L, Moreira ML, Frolov A, et al. Transcriptional regulator SATB1 limits CD8(+) T cell population expansion and effector differentiation in chronic infection and cancer. Nat Immunol. 2025;26:2312–27. https://doi.org/10.1038/s41590-025-02316-2.
Chen Z, Arai E, Khan O, Zhang Z, Ngiow SF, He Y, et al. In vivo CD8(+) T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell. 2021;184:1262–80.e22. https://doi.org/10.1016/j.cell.2021.02.019.
Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, et al. The transcription factor Runx3 guards cytotoxic CD8(+) effector T cells against deviation towards follicular helper T cell lineage. Nat Immunol. 2017;18:931–9. https://doi.org/10.1038/ni.3773.
Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. 2009;206:51–9. https://doi.org/10.1084/jem.20081242.
Wang D, Diao H, Getzler AJ, Rogal W, Frederick MA, Milner J, et al. The transcription factor Runx3 establishes chromatin accessibility of cis-regulatory landscapes that drive memory cytotoxic T lymphocyte formation. Immunity. 2018;48:659–74.e6. https://doi.org/10.1016/j.immuni.2018.03.028.
Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Erratum: Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. 2018;554:392. https://doi.org/10.1038/nature25445.
Wu JE, Manne S, Ngiow SF, Baxter AE, Huang H, Freilich E, et al. In vitro modeling of CD8(+) T cell exhaustion enables CRISPR screening to reveal a role for BHLHE40. Sci Immunol. 2023;8:eade3369. https://doi.org/10.1126/sciimmunol.ade3369.
Chen Z, Javed N, Moore M, Wu J, Sun G, Vinyard M, et al. Integrative dissection of gene regulatory elements at base resolution. Cell Genom. 2023;3:100318. https://doi.org/10.1016/j.xgen.2023.100318.
Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, et al. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell. 2017;171:1221–3. https://doi.org/10.1016/j.cell.2017.11.006.
Wagle MV, Vervoort SJ, Kelly MJ, Van Der Byl W, Peters TJ, Martin BP, et al. Antigen-driven EGR2 expression is required for exhausted CD8(+) T cell stability and maintenance. Nat Commun. 2021;12:2782. https://doi.org/10.1038/s41467-021-23044-9.
Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45:374–88. https://doi.org/10.1016/j.immuni.2016.07.009.
Schurich A, Pallett LJ, Jajbhay D, Wijngaarden J, Otano I, Gill US, et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep. 2016;16:1243–52. https://doi.org/10.1016/j.celrep.2016.06.078.
Fisicaro P, Barili V, Montanini B, Acerbi G, Ferracin M, Guerrieri F, et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med. 2017;23:327–36. https://doi.org/10.1038/nm.4275.
Sugiura A, Rathmell JC. Metabolic barriers to T cell function in tumors. J Immunol. 2018;200:400–7. https://doi.org/10.4049/jimmunol.1701041.
Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol. 2020;21:1540–51. https://doi.org/10.1038/s41590-020-0793-3.
Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–28. https://doi.org/10.1016/j.cell.2015.08.012.
Sinclair LV, Howden AJ, Brenes A, Spinelli L, Hukelmann JL, Macintyre AN, et al. Antigen receptor control of methionine metabolism in T cells. eLife. 2019;8. https://doi.org/10.7554/eLife.44210
Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354:481–4. https://doi.org/10.1126/science.aaf6284.
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–80. https://doi.org/10.1126/science.1164097.
Ma S, Dahabieh MS, Mann TH, Zhao S, McDonald B, Song WS, et al. Nutrient-driven histone code determines exhausted CD8(+) T cell fates. Science. 2025;387:eadj3020. https://doi.org/10.1126/science.adj3020.
Kaymak I, Watson MJ, Oswald BM, Ma S, Johnson BK, DeCamp LM, et al. ACLY and ACSS2 link nutrient-dependent chromatin accessibility to CD8 T cell effector responses. J Exp Med. 2024;221. https://doi.org/10.1084/jem.20231820
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–42. https://doi.org/10.1016/j.immuni.2005.03.013.
Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1:1460–8. https://doi.org/10.4161/onci.21716.
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. https://doi.org/10.1038/nature10491.
Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553–60. https://doi.org/10.1158/0008-5472.CAN-08-1921.
Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 2017;25:345–57. https://doi.org/10.1016/j.cmet.2016.12.011.
Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30:143–56.e5. https://doi.org/10.1016/j.cmet.2019.04.002.
Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity. 2021;54:1561–77.e7. https://doi.org/10.1016/j.immuni.2021.05.003.
Yan C, Zheng L, Jiang S, Yang H, Guo J, Jiang LY, et al. Exhaustion-associated cholesterol deficiency dampens the cytotoxic arm of antitumor immunity. Cancer Cell. 2023;41:1276–93.e11. https://doi.org/10.1016/j.ccell.2023.04.016.
Varanasi SK, Chen D, Liu Y, Johnson MA, Miller CM, Ganguly S, et al. Bile acid synthesis impedes tumor-specific T cell responses during liver cancer. Science. 2025;387:192–201. https://doi.org/10.1126/science.adl4100.
Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14:1173–82. https://doi.org/10.1038/ni.2714.
Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An HIF-1alpha/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. 2017;32:669–83.e5. https://doi.org/10.1016/j.ccell.2017.10.003.
Phan AT, Doedens AL, Palazon A, Tyrakis PA, Cheung KP, Johnson RS, et al. Constitutive glycolytic metabolism supports CD8(+) T cell effector memory differentiation during viral infection. Immunity. 2016;45:1024–37. https://doi.org/10.1016/j.immuni.2016.10.017.
Vuillefroy de Silly R, Ducimetière L, Yacoub Maroun C, Dietrich PY, Derouazi M, Walker PR. Phenotypic switch of CD8(+) T cells reactivated under hypoxia toward IL-10 secreting, poorly proliferative effector cells. Eur J Immunol. 2015;45:2263–75. https://doi.org/10.1002/eji.201445284.
Scharping NE, Rivadeneira DB, Menk AV, Vignali P, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. 2021;22:205–15. https://doi.org/10.1038/s41590-020-00834-9.
Vignali P, DePeaux K, Watson MJ, Ye C, Ford BR, Lontos K, et al. Hypoxia drives CD39-dependent suppressor function in exhausted T cells to limit antitumor immunity. Nat Immunol. 2023;24:267–79. https://doi.org/10.1038/s41590-022-01379-9.
Ma K, Cheng H, Wang L, Xiao H, Liu F, Yang Y, et al. Succinate preserves CD8(+) T cell fitness to augment antitumor immunity. Immunity. 2025;58:2505–23. https://doi.org/10.1016/j.immuni.2025.07.017.
Cheng H, Qiu Y, Xu Y, Chen L, Ma K, Tao M, et al. Extracellular acidosis restricts one-carbon metabolism and preserves T cell stemness. Nat Metab. 2023;5:314–30. https://doi.org/10.1038/s42255-022-00730-6.
Tang K, Zhang H, Deng J, Wang D, Liu S, Lu S, et al. Ammonia detoxification promotes CD8(+) T cell memory development by urea and citrulline cycles. Nat Immunol. 2023;24:162–73. https://doi.org/10.1038/s41590-022-01365-1.
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–9. https://doi.org/10.1182/blood-2006-07-035972.
Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537:539–43. https://doi.org/10.1038/nature19364.
Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019;363. https://doi.org/10.1126/science.aau0135
Zhang J, Li J, Hou Y, Lin Y, Zhao H, Shi Y, et al. Osr2 functions as a biomechanical checkpoint to aggravate CD8(+) T cell exhaustion in tumor. Cell. 2024;187:3409–26.e24. https://doi.org/10.1016/j.cell.2024.04.023.
Imboden JB, Stobo JD. Transmembrane signalling by the T cell antigen receptor. Perturbation of the T3-antigen receptor complex generates inositol phosphates and releases calcium ions from intracellular stores. J Exp Med. 1985;161:446–56. https://doi.org/10.1084/jem.161.3.446.
Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol. 2020;21:1022–33. https://doi.org/10.1038/s41590-020-0725-2.
Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–36. https://doi.org/10.1016/j.immuni.2012.10.020.
Zhu Z, Luo Y, Lou G, Yihunie K, Wizzard S, DeVilbiss AW, et al. The redox sensor KEAP1 facilitates adaptation of T cells to chronic antigen stimulation by preventing hyperactivation. Sci Immunol. 2024;9:eadk2954. https://doi.org/10.1126/sciimmunol.adk2954.
Dahabieh MS, DeCamp LM, Oswald BM, Kitchen-Goosen SM, Fu Z, Vos M, et al. The prostacyclin receptor PTGIR is a NRF2-dependent regulator of CD8(+) T cell exhaustion. Nat Immunol. 2025;26:1139–51. https://doi.org/10.1038/s41590-025-02185-9.
Gnanaprakasam J, Kushwaha B, Liu L, Chen X, Kang S, Wang T, et al. Asparagine restriction enhances CD8(+) T cell metabolic fitness and antitumoral functionality through an NRF2-dependent stress response. Nat Metab. 2023;5:1423–39. https://doi.org/10.1038/s42255-023-00856-1.
Kallies A, Zehn D, Utzschneider DT. Precursor exhausted T cells: key to successful immunotherapy?. Nat Rev Immunol. 2020;20:128–36. https://doi.org/10.1038/s41577-019-0223-7.
Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. 2018;24:994–1004. https://doi.org/10.1038/s41591-018-0057-z.
Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2019;176:404. https://doi.org/10.1016/j.cell.2018.12.034.
Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell. 2021;39:1578–93.e8. https://doi.org/10.1016/j.ccell.2021.09.010.
Krishna C, DiNatale RG, Kuo F, Srivastava RM, Vuong L, Chowell D, et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell. 2021;39:662–77 e666. https://doi.org/10.1016/j.ccell.2021.03.007.
Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi A, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2020;181:747 https://doi.org/10.1016/j.cell.2020.04.017.
van der Leun AM, Traets J, Vos JL, Elbers J, Patiwael S, Qiao X, et al. Dual immune checkpoint blockade induces analogous alterations in the dysfunctional CD8+ T-cell and activated treg compartment. Cancer Discov. 2023;13:2212–27. https://doi.org/10.1158/2159-8290.CD-22-0851.
Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. 2018;9:2724. https://doi.org/10.1038/s41467-018-05072-0.
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342–56.e16. https://doi.org/10.1016/j.cell.2017.05.035.
Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–72. https://doi.org/10.1038/s41586-018-0694-x.
Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24:978–85. https://doi.org/10.1038/s41591-018-0045-3.
Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374:abe6474. https://doi.org/10.1126/science.abe6474.
Jung IY, Narayan V, McDonald S, Rech AJ, Bartoszek R, Hong G, et al. BLIMP1 and NR4A3 transcription factors reciprocally regulate antitumor CAR T cell stemness and exhaustion. Sci Transl Med. 2022;14:eabn7336. https://doi.org/10.1126/scitranslmed.abn7336.
Jackson Z, Hong C, Schauner R, Dropulic B, Caimi PF, de Lima M, et al. Sequential single-cell transcriptional and protein marker profiling reveals TIGIT as a marker of CD19 CAR-T cell dysfunction in patients with non-Hodgkin lymphoma. Cancer Discov. 2022;12:1886–903. https://doi.org/10.1158/2159-8290.CD-21-1586.
Cheng J, Xiao Y, Peng T, Zhang Z, Qin Y, Wang Y, et al. ETV7 limits the antiviral and antitumor efficacy of CD8(+) T cells by diverting their fate toward exhaustion. Nat Cancer. 2025;6:338–56. https://doi.org/10.1038/s43018-024-00892-0.
Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, et al. CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discov. 2021;11:1192–211. https://doi.org/10.1158/2159-8290.CD-20-1243.
Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, et al. The epigenetic control of stemness in CD8(+) T cell fate commitment. Science. 2018;359:177–86. https://doi.org/10.1126/science.aah6499.
Jain N, Zhao Z, Koche RP, Antelope C, Gozlan Y, Montalbano A, et al. Disruption of SUV39H1-mediated H3K9 methylation sustains CAR T-cell function. Cancer Discov. 2024;14:142–57. https://doi.org/10.1158/2159-8290.CD-22-1319.
Kang TG, Lan X, Mi T, Chen H, Alli S, Lim SE, et al. Epigenetic regulators of clonal hematopoiesis control CD8 T cell stemness during immunotherapy. Science. 2024;386:eadl4492. https://doi.org/10.1126/science.adl4492.
Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–12. https://doi.org/10.1038/s41586-018-0178-z.
Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170:142–57.e19. https://doi.org/10.1016/j.cell.2017.06.007.
Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300. https://doi.org/10.1038/s41586-019-1805-z.
Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol. 2014;15:373–83. https://doi.org/10.1038/ni.2834.
Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, Shen J, et al. BATF regulates progenitor to cytolytic effector CD8(+) T cell transition during chronic viral infection. Nat Immunol. 2021;22:996–1007. https://doi.org/10.1038/s41590-021-00965-7.
Zhang X, Zhang C, Qiao M, Cheng C, Tang N, Lu S, et al. Depletion of BATF in CAR-T cells enhances antitumor activity by inducing resistance against exhaustion and formation of central memory cells. Cancer Cell. 2022;40:1407–22.e7. https://doi.org/10.1016/j.ccell.2022.09.013.
Wang L, Jin G, Zhou Q, Liu Y, Zhao X, Li Z, et al. Induction of immortal-like and functional CAR T cells by defined factors. J Exp Med. 2024;221. https://doi.org/10.1084/jem.20232368
Grange M, Verdeil G, Arnoux F, Griffon A, Spicuglia S, Maurizio J, et al. Active STAT5 regulates T-bet and eomesodermin expression in CD8 T cells and imprints a T-bet-dependent Tc1 program with repressed IL-6/TGF-beta1 signaling. J Immunol. 2013;191:3712–24. https://doi.org/10.4049/jimmunol.1300319.
Hashimoto M, Araki K, Cardenas MA, Li P, Jadhav RR, Kissick HT, et al. PD-1 combination therapy with IL-2 modifies CD8(+) T cell exhaustion program. Nature. 2022;610:173–81. https://doi.org/10.1038/s41586-022-05257-0.
Mo F, Yu Z, Li P, Oh J, Spolski R, Zhao L, et al. An engineered IL-2 partial agonist promotes CD8(+) T cell stemness. Nature. 2021;597:544–8. https://doi.org/10.1038/s41586-021-03861-0.
Zhao Y, Chen J, Andreatta M, Feng B, Xie YQ, Wenes M, et al. IL-10-expressing CAR T cells resist dysfunction and mediate durable clearance of solid tumors and metastases. Nat Biotechnol. 2024;42:1693–704. https://doi.org/10.1038/s41587-023-02060-8.
Qiao J, Liu Z, Dong C, Luan Y, Zhang A, Moore C, et al. Targeting tumors with IL-10 prevents dendritic cell-mediated CD8(+) T cell apoptosis. Cancer Cell. 2019;35:901–15.e4. https://doi.org/10.1016/j.ccell.2019.05.005.
Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med. 2019;25:454–61. https://doi.org/10.1038/s41591-019-0357-y.
Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–5. https://doi.org/10.1038/nature22079.
Salas-Benito D, Pérez-Gracia JL, Ponz-Sarvisé M, Rodriguez-Ruiz ME, Martínez-Forero I, Castañón E, et al. Paradigms on Immunotherapy Combinations with Chemotherapy. Cancer Discov. 2021;11:1353–67. https://doi.org/10.1158/2159-8290.CD-20-1312.
Chen Y, Wang D, Li Y, Qi L, Si W, Bo Y, et al. Spatiotemporal single-cell analysis decodes cellular dynamics underlying different responses to immunotherapy in colorectal cancer. Cancer Cell. 2024;42:1268–85.e7. https://doi.org/10.1016/j.ccell.2024.06.009.
Escobar G, Tooley K, Oliveras JP, Huang L, Cheng H, Bookstaver ML, et al. Tumor immunogenicity dictates reliance on TCF1 in CD8(+) T cells for response to immunotherapy. Cancer Cell. 2023;41:1662–79.e7. https://doi.org/10.1016/j.ccell.2023.08.001.
Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell. 2016;167:419–432 e416. https://doi.org/10.1016/j.cell.2016.09.011.
Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13. https://doi.org/10.1126/scitranslmed.abe7378
Hernandez-Lopez RA, Yu W, Cabral KA, Creasey OA, Lopez Pazmino M, Tonai Y, et al. T cell circuits that sense antigen density with an ultrasensitive threshold. Science. 2021;371:1166–71. https://doi.org/10.1126/science.abc1855.
Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. 2018;359:1037–42. https://doi.org/10.1126/science.aar3246.
Zhang Q, Hresko ME, Picton LK, Su L, Hollander MJ, Nunez-Cruz S, et al. A human orthogonal IL-2 and IL-2Rbeta system enhances CAR T cell expansion and antitumor activity in a murine model of leukemia. Sci Transl Med. 2021;13:eabg6986. https://doi.org/10.1126/scitranslmed.abg6986.
Zhang J, Hu Y, Yang J, Li W, Zhang M, Wang Q, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature. 2022;609:369–74. https://doi.org/10.1038/s41586-022-05140-y.
Weiss SA, Huang AY, Fung ME, Martinez D, Chen A, LaSalle TJ, et al. Epigenetic tuning of PD-1 expression improves exhausted T cell function and viral control. Nat Immunol. 2024;25:1871–83. https://doi.org/10.1038/s41590-024-01961-3.
Liu Y, Debo B, Li M, Shi Z, Sheng W, Shi Y. LSD1 inhibition sustains T cell invigoration with a durable response to PD-1 blockade. Nat Commun. 2021;12:6831. https://doi.org/10.1038/s41467-021-27179-7.
Wang L, Xiao Y, Luo Y, Master RP, Mo J, Kim MC, et al. PROTAC-mediated NR4A1 degradation as a novel strategy for cancer immunotherapy. J Exp Med. 2024;221. https://doi.org/10.1084/jem.20231519
Bonazzi S, d'Hennezel E, Beckwith R, Xu L, Fazal A, Magracheva A, et al. Discovery and characterization of a selective IKZF2 glue degrader for cancer immunotherapy. Cell Chem Biol. 2023;30:235–47.e12. https://doi.org/10.1016/j.chembiol.2023.02.005.
Zhu M, Han Y, Gu T, Wang R, Si X, Kong D, et al. Class I HDAC inhibitors enhance antitumor efficacy and persistence of CAR-T cells by activation of the Wnt pathway. Cell Rep. 2024;43:114065. https://doi.org/10.1016/j.celrep.2024.114065.
Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2016;22:4119–32. https://doi.org/10.1158/1078-0432.CCR-15-2584.
Isshiki Y, Chen X, Teater M, Karagiannidis I, Nam H, Cai W, et al. EZH2 inhibition enhances T cell immunotherapies by inducing lymphoma immunogenicity and improving T cell function. Cancer Cell. 2025;43:49–68.e9. https://doi.org/10.1016/j.ccell.2024.11.006.
Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest. 2016;126:3479–94. https://doi.org/10.1172/JCI86437.
Milner JJ, Toma C, Quon S, Omilusik K, Scharping NE, Dey A, et al. Bromodomain protein BRD4 directs and sustains CD8 T cell differentiation during infection. J Exp Med. 2021;218. https://doi.org/10.1084/jem.20202512
Fan F, Yoo HJ, Stock S, Wang L, Liu Y, Schubert ML, et al. Ibrutinib for improved chimeric antigen receptor T-cell production for chronic lymphocytic leukemia patients. Int J Cancer. 2021;148:419–28. https://doi.org/10.1002/ijc.33212.
Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight. 2017;2. https://doi.org/10.1172/jci.insight.95103
Wang X, Tao X, Chen P, Jiang P, Li W, Chang H, et al. MEK inhibition prevents CAR-T cell exhaustion and differentiation via downregulation of c-Fos and JunB. Signal Transduct Target Ther. 2024;9:293. https://doi.org/10.1038/s41392-024-01986-y.
Ma J, Tang L, Tan Y, Xiao J, Wei K, Zhang X, et al. Lithium carbonate revitalizes tumor-reactive CD8(+) T cells by shunting lactic acid into mitochondria. Nat Immunol. 2024;25:552–61. https://doi.org/10.1038/s41590-023-01738-0.
Acknowledgements
The authors thank M. Iyer for her valuable assistance with manuscript editing. The authors received no financial support for this article.
Author information
Authors and Affiliations
Contributions
ZC, ZZ, TH, CY, and TW wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Z., Zhu, Z., Hu, T. et al. Regulation of T cell exhaustion and stemness: molecular mechanisms and implications for cancer immunotherapy. Cell Mol Immunol 23, 1–14 (2026). https://doi.org/10.1038/s41423-025-01378-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41423-025-01378-4
