
Abstract
Hereditary diffuse gastric cancer (HDGC) and Lynch syndromes are dominant hereditary diseases caused by pathogenic germline variants in specified genes, and characterised by a broad spectrum of malignancies. Whereas HDGC is associated with CDH1 and CTNNA1 variants and defined by an increased risk of diffuse gastric cancer and lobular breast cancer, Lynch syndrome results from alterations in mismatch repair genes, whose main manifestations include colorectal, endometrial, ovarian, breast, prostate, stomach, and urological tumours. Remarkably, a huge difference remains in the knowledge surrounding the molecular mechanisms that drive these disorders, and in current approaches for patient management. In fact, the HDGC narrative is still in its early stages when compared with Lynch syndrome, which accumulates more than a century of research. Herein, we propose an analogy between HDGC and Lynch syndromes, highlighting intricacies across genetic origin, variant effects, cellular landscapes, and associated clinical outcomes. Further, we postulate that the history of Lynch syndrome may be useful to advance HDGC aetiology, namely strategies for identification of new candidate genes, rules for variant interpretation, sources of phenotypic heterogeneity, and improved surveillance protocols. This collected data will impact clinical perspectives, as well as future research programs addressing HDGC unmet challenges.
Similar content being viewed by others

Introduction
Hereditary cancer syndromes account for 5–10% of all cancers. Although this appears as a modest fraction, an incredible effort has been set to identify at-risk individuals carrying variants in cancer predisposing genes and who might benefit from enhanced surveillance and/or risk reduction measures. More so, uncovering a carrier of a causative variant elicits a cascade of genetic testing among family members to guide clinical management of susceptible individuals [1].
Hereditary diffuse gastric cancer (HDGC), also known as diffuse gastric and lobular breast cancer syndrome (DGLBCS), and Lynch syndromes are both dominant hereditary diseases caused by pathogenic germline variants in “specific genes” and characterised by a panel of malignancies [2, 3].
Originally described in 1998, in a New Zealand Maori family, HDGC is mostly associated with inactivating germline variants in the CDH1 tumour suppressor gene, with pathogenic variants in CTNNA1 also described in a minority of families [4, 5]. This syndrome is characterised by a significant increase in the lifetime risk of diffuse gastric cancer and lobular breast cancer, though other disease entities such as cleft lip/palate (CL/P) have been described in affected families [6, 7]. In the past decade, significant advances in next-generation sequencing technologies have expanded our capacity to identify patients at-risk, while highlighting our lack of knowledge regarding mechanisms that drive HDGC and effective approaches for patient management [8]. Further, increased genetic testing has resulted in the identification of CDH1 variants associated with other clinical conditions unrelated with HDGC, including the Blepharocheilodontic syndrome, of which cleft lip/palate is a main manifestation [9].
Lynch syndrome, also known as hereditary non-polyposis colorectal cancer, was one of the first cancer predisposition syndromes to be described and, as a result of more than a century of multidisciplinary discoveries, our current understanding of this condition represents a role model for studying other hereditary cancer syndromes [10]. In fact, following the first documentation in 1895 of a family pedigree aggregating colorectum, stomach and uterus cancers, the term “Lynch syndrome” was coined in 1984 to describe an autosomal dominant condition characterised by a significantly increased risk of developing a spectrum of cancers that includes colorectal, endometrial, ovarian, small bowel, stomach, hepatobiliary, urinary, brain or central nervous system cancers, as well as sebaceous tumours [3]. It is estimated that Lynch syndrome accounts for approximately 3% of all diagnosed colorectal cancers [10].
The aetiology of Lynch syndrome is attributed to germline alterations in mismatch repair (MMR) genes that lead to microsatellite instability (MSI) [11]. Indeed, the primary diagnostic strategy for Lynch syndrome relies on germline testing for MMR gene variants in patients harbouring tumours with high-level of MSI and/or deficient MMR protein expression [3].
Herein, we provide an overview of the genetic landscape, molecular players and modifiers underlying HDGC and Lynch syndromes. We outline challenges regarding variant classification, as well as management and surveillance protocols that are common to both syndromes. The comparison does not aim to imply any molecular or mechanistic overlap but rather, the analogy is drawn at the conceptual level, emphasising how lessons learned from Lynch syndrome can inform similar approaches in HDGC.
Genetic cause
Over the years, several studies have identified specific genes that, when mutated, promote uncontrolled cell growth and invasion of malignant cells [12]. It is widely accepted that acquired variants in somatic cells cause most of the sporadic cancers, while germline variants explain rare hereditary syndromes [13]. Moreover, cancer-related genes can be categorised into oncogenes and tumour suppressor genes. Oncogenes are phenotypically dominant, with the activation of a single mutated copy (allele) of a proto-oncogene being sufficient to promote cancer [13]. In contrast, tumour suppressor genes are phenotypically recessive, where deletion or inactivation of both alleles is necessary to impair gene function and cause cancer [13]. Inherited cancer syndromes often result from loss of function of tumour suppressor genes rather than from oncogene activation. In particular, HDGC [OMIM #137215] and Lynch syndromes [OMIM #120435] are the most common forms of hereditary gastric and colorectal cancer, respectively, and present an autosomal-dominant inheritance pattern [14]. Since mutations are inherited from one parent, every cell from the progeny carries a functional and a defective copy of the gene. Complete gene inactivation occurs upon somatic loss of the second allele (Knudson’s two-hit hypothesis) through additional mutation, hypermethylation, or other silencing mechanism [15, 16].
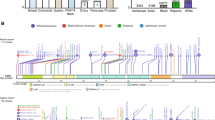
CDH1 (OMIM #192090) and CTNNA1 (OMIM #116805) are the only proven genes implicated in HDGC [17, 18]. CDH1 is located on chromosome 16q22.1 and encodes for epithelial cadherin (E-cadherin). E-cadherin is a 120 kDa glycoprotein that comprises three major portions: an extracellular domain responsible for homophilic binding of E-cadherin molecules on adjacent cells; a transmembrane domain that functions as an anchor and stabiliser; and the intracellular portion that mediates linkage between E-cadherin and the cytoskeleton [19]. The CTNNA1 gene is mapped into chromosome 5q31.2 and codes for α-catenin, one of the cytoplasmic binding partners of E-cadherin [20]. In fact, the cytoplasmic domain of E-cadherin attaches to different proteins, namely α-, β-, and p120-catenins, forming a cadherin-catenin complex. These binding partners regulate E-cadherin trafficking and activity at the plasma membrane, by interacting with signalling moieties, as well as with distinct cytoskeletal components [19]. In particular, α-catenin displays mechanosensory properties and controls a dynamic interplay with filamentous actin (F-actin), improving the recognition of intercellular cadherin bonds [21, 22]. E-cadherin and α-catenin play an indisputable role in cell-cell adhesion, contributing to tissue architecture and integrity (Fig. 1) [19]. More so, E-cadherin is a critical player in the orientation of the mitotic spindle in the epithelium, ensuring planar cell divisions and epithelial polarity [23]. Accordingly, an elegant model involving the misalignment of the mitotic spindle has been proposed as a mechanism underlying the initial steps of diffuse gastric cancer [8, 24].

Schematic representation of the main molecules and corresponding functions involved in the aetiology of both cancer syndromes. E-cadherin and α-catenin are critical players for cell-cell adhesion, contributing to structural and mechanical integrity of normal epithelial tissues. HDGC arises as a consequence of germline variants in CDH1 and CTNNA1, which encode E-cadherin and α-catenin, impacting cell-cell adhesion and invasion capacity. MMR genes MLH1, MSH2, MSH6, and PMS2 code for proteins that assemble to recognise and repair DNA replication errors (wild-type context). In Lynch syndrome, MMR deficiency leads to accumulation of DNA errors and abnormal cell proliferation.
Underlying the aetiology of Lynch syndrome is a set of genes involved in DNA MMR: MLH1 (OMIM #120436), MSH2 (OMIM #609309), MSH6 (OMIM #600678), PMS2 (OMIM #600259), and EPCAM (OMIM #185535) [3]. The MMR system corrects DNA errors occurring during the process of DNA replication, assuring fidelity of the daughter DNA strand and thus tissue homeostasis [25].
MLH1, MSH2, MSH6, and PMS2 genes code for proteins that interact as heterodimers. Specifically, MLH1 pairs with PMS2, PMS1 or MLH3 to form MutLα, MutLβ or MutLγ complexes, whereas MSH2 couples with MSH6 or MSH3, forming MutSα or MutSβ complexes [25]. MutSα or MutSβ complexes recognise single base-pair mismatches and small insertions or deletion loops of DNA that result from replication errors of repetitive sequences – the so-called microsatellite regions [25]. MutLα, MutLβ or MutLγ promote the excision and resynthesis of DNA. It is thus not surprising that defects in MMR machinery cause an accumulation of DNA errors, originating MSI [25]. Importantly, depending on the affected gene, the extent of MMR deficiency is different and results in distinguishable phenotypes. For instance, MLH1 or MSH2 variants are associated with complete inactivation of MMR and high levels of MSI (MSI-H phenotype). In contrast, MSH6 alterations cause partial MMR deficiency and, accordingly, a MSI-low phenotype can be observed [25, 26]. Of note, pathogenic germline variants in MLH1 and MSH2 account for the majority of clinically diagnosed cases, although estimates of the population prevalence pinpoint these alterations as the less prevalent among MMR variants [27, 28]. MSH6 and PMS2 variants, which are in fact more prevalent in the population, are identified as causative of Lynch syndrome in a minority of cases and are often underdiagnosed due to their lower penetrance and later age of onset [27, 28]. Defects in EPCAM, that lead to loss of MSH2 expression by epigenetic inactivation, are even rarer [26, 29, 30].
Overall, it is clear that HDGC and Lynch syndromes are caused by deregulation of two unique cellular mechanisms – cell-cell adhesion and MMR function – which suggests that alterations in other genes from those specific pathways may underlie cases that remain genetically unexplained. Despite comprehensive germline and tumour testing, no pathogenic variant in the currently known genes have been identified in 60-70% of clinically defined HDGC cases [8], and in 10-15% of cases with suspected Lynch syndrome [31, 32].
Variant landscape
The frequencies of CDH1 variant types may vary depending on the compliance with clinical criteria for genetic screening in the HDGC context. A systematic review collected and analysed all published studies describing CDH1 gene variants in gastric cancer patients from both high- and low-prevalence countries to understand how these alterations appear across different populations and study designs [33]. Selected studies were classified into three categories: series studies, which included multiple gastric cancer patients not necessarily tested according to specific criteria; family studies, where patients were tested based on the clinical criteria established by the International Gastric Cancer Linkage Consortium (IGCLC); and unknown studies, which included individuals lacking familial information regarding cancer phenotypes. This comprehensive evaluation revealed that missense variants represent the largest subset, with a frequency of 28.9%, in individuals affected by gastric cancer without compliance with criteria [33]. Deletions were detected in 24.6% of the cases, splice-site in 21.9%, and non-sense alterations in 19.3%. Insertions are less frequently identified, constituting 4.8% of all CDH1 variants detected [33]. In contrast, screening in families fulfilling HDGC criteria shows the highest frequency of deletions (31.6%) and non-sense alterations (27.4%), followed by missense (16.8%), splice-site (15.9%), and insertions (8%) [33]. This shift in frequencies reflects lower penetrance of missense variants and, consequently, limited recognition of HDGC manifestations [34].
The effects of missense variants are often mild, when compared with truncating variants, due to the presence of a mutated protein with potential residual function, thus masking disease penetrance. In the case of truncated mRNA CDH1 transcripts, it is established that nonsense mediated decay mechanisms are activated, leading to their downregulation and disease development [35].
With regard to CTNNA1, a few number of germline alterations have been reported in HDGC families, comprising frameshift, nonsense, and missense variants [5, 17, 18, 36]. While first estimates defined a 49–57% cumulative risk of diffuse gastric cancer in CTNNA1 carriers at 80 years [37], this may represent an overestimation. More recent evidence supports that, in the context of HDGC, CTNNA1 is best characterised as a moderate-penetrance gene, rather than conferring the very high risks initially suggested [38]. A large-scale study including 38112 individuals, ascertained on the basis of personal or family history of cancer, compared outcomes in CTNNA1 and CDH1 variant carriers. Among the 270 individuals carrying CTNNA1 truncating variants, the observed prevalence was 2.6% for unspecified gastric cancer (7/270) and 3.7% for lobular breast cancer (10/270) [38]. These findings provide a more balanced picture of the cancer risk in CTNNA1 carriers, although they also underscore that robust, prospective studies are still needed to accurately quantify lifetime risks.
Reflecting long term efforts and dedicated research to identify genetic factors associated with Lynch syndrome, the number of germline variants reported affecting MMR genes is currently higher than 3000. Among these alterations, approximately 40% affect MLH1, 34% MSH2, 18% occur in MSH6 and 8% in PMS2, with variable distribution of variant types [26, 29, 30]. For instance, MLH1 variants comprise 40% nonsense or frameshift, 40% missense, 11% splice-site, 7% large rearrangements, and 2% in-frame alterations [26]. MSH2 variants have a similar distribution pattern with 49% of variants classified as nonsense or frameshift, 31% missense, 10% large rearrangements, 8% splice-site, and 2% in-frame [26]. In the case of MSH6 variants, those include missense in 49% of cases, nonsense or frameshift in 43%, splice-site in 3%, in-frame in 3%, and large rearrangements in 2% [26]. Strikingly, PMS2 presents the highest proportion of missense variants - 62%, followed by 24% of nonsense or frameshift, 10% of large rearrangements, 3% of splice-site and 1% of in-frame alterations [26]. Of note, penetrance of MSH6 and PMS2 variants is reduced, as verified by higher average age of onset, when compared with carriers of MLH1 or MSH2 variants [3, 26]. This might be associated with the higher frequency of missense alterations in MSH6 and PMS2 than in MLH1 or MSH2.
Common to both cancer syndromes is the lack of variant hotspots, since alterations span the full coding sequence of all specified genes. Such feature can be attributed to the loss of function of tumour suppressor genes, whose effects can vary in penetrance and residual activity, but often converging on a similar final outcome. In addition, no definite correlations can be established between variant type and functional domains affected with clinical phenotypes. Despite that in a large multicentre study with 854 families no pathogenic/likely pathogenic missense variants could be associated with HDGC phenotypes [39], it has been verified that several other missense variants segregate with disease and are supported by functional data and histopathological findings [40, 41]. Concerning variant location, some studies claim to have defined a correlation with specific clinical presentations, although based in limited information in extent or scope [42].
Variant interpretation
A major challenge in cancer syndromes is the accurate risk prediction for developing the disease, in order to provide patients with adequate surveillance and management schemes [5, 10]. In this setting, efforts have been made to implement variant classification systems discriminating potential pathogenic from benign genetic alterations. Variants that disrupt gene sequence, such as nonsense or frameshift, are more frequently identified as risk-conferring, while most benign variants are intronic, missense and splice site alterations. Remarkably, 28% of variants affecting CDH1, 31% for MLH1, 28% for MSH2, 47% for MSH6, and 26% for PMS2 remain without a definite resolution and are classified as variants of uncertain significance (VUS), of which non-synonymous missense variants represent a large fraction, if not the largest [43, 44].
Currently, classification of variants occurring in MMR genes takes into account clinical and familial data, variant recurrence, allele frequency in control reference groups, in silico prediction algorithms, and studies on variant functional impact (InSiGHT Variant Interpretation Committee: Mismatch Repair Gene Variant Classification Criteria - Version 2.4 June 2018 https://www.insight-group.org/content/uploads/2018/08/2018-06_InSiGHT_VIC_v2.4.pdf). Irrespective of the difficulties that an integrative approach can raise, clinicians and genetic counsellors are committed to achieve a rigorous categorisation for better patient care.
Classification of CDH1 variants is still immature due to a persisting conservative view that neglects high-quality scientific evidence and advances in experimental models [44]. Moreover, specific classification guidelines are not available for CTNNA1 variants given the limited data available and moderate cancer risk verified in the context of HDGC [38]. Indeed, not disregarding that the molecular effects of variants in MMR and adhesion complex genes are distinct, a huge difference remains among the pipelines developed to access the functional impact of variants affecting MMR or CDH1 genes. The effects of CDH1 variants have been investigated through transfected cell lines and a combination of methodologies, which quantitatively evaluate the levels of protein yield, its localization, adhesive and anti-invasive functions, as well as cellular morphology and organisation [34, 40, 45]. These assays are low throughput and time-consuming, and are performed on a research-based standpoint, which is not compatible with current demands upon genetic panel testing. A simpler approach is settled in the case of MMR gene variants, encompassing examination of protein levels and MMR activity [46]. Whereas the deleterious outcome of missense variants leading to lower protein levels, as a result of their structural destabilization, is well-established in Lynch syndrome [47, 48], this effect is not considered in CDH1 missense variants regardless of the vast number of studies showing similar phenotypes [40, 49, 50]. Likewise, a significant decrease in DNA repair efficiency tested in a cell-free system is sufficient to classify an MMR variant as “likely pathogenic” [51], while robust cellular assays showing loss of adhesive and anti-invasive functions of E-cadherin do not change variant classification. As an example, Figueiredo et al. have recently addressed the significance of the CDH1 G212E variant identified in an unusually large pedigree displaying strong aggregation of diffuse gastric cancer [40]. The group has explored clinical observations, histological findings, in silico predictions, in vitro assays and in vivo strategies – all validating the deleterious nature of the genetic alteration [40]. Of note, an innovative Drosophila model was developed, which demonstrated that the G212E variant disrupts epithelial architecture and tissue organisation in vivo [40]. To date no CDH1 missense variants have been recognised as disease-causing by the CDH1 Variant Curation Expert Panel (VCEP) from the Clinical Genome Resource (ClinGen) [44]. In contrast, missense variant effects have been suggested to be mediated through impact on splicing, despite being based on software predictions, often in the absence of experimental validity (using minigene or additional RNA assays) and sequencing of expected alternative transcripts [44].
The controversy surrounding missense type variants spans a large panel of genes and clinical contexts, including those of other cancer syndromes such as Hereditary Breast and Ovarian Cancer (HBOC) [52], or inherited conditions like Cystic Fibrosis [53] and Familial Partial Lipodystrophy [54]. This lack of consensus is mainly due to incomplete penetrance of associated disease phenotypes, variability on family presentation, and residual protein activity, which impair a straightforward translation of functional defects into evident clinical outcomes. Mounting evidence highlights the need to develop research programs aiming to disclose the paradigm surrounding VUS and the role of other determinants impacting variant effects. In this sense, novel experimental approaches would provide additional indication of variant functional outcomes and improve dedicated classification schemes. For instance, CDH1-specific mutant cell lines could be engineered and cultured in 3D systems for subsequent monitoring of cell division orientation and spheroid organisation. Previous studies using CDH1-knockout organoid models have already demonstrated that complete loss of E-cadherin alters epithelial organisation and mitotic spindle dynamics, providing important mechanistic insight into HDGC pathogenesis [24, 55]. However, knockout systems represent total loss of function, and thus a homogeneous phenotype. Engineering organoids carrying specific CDH1 variants, particularly of the missense type, could be viewed as complementary tools for the functional validation of clinically ambiguous variants, addressing a wider range of biological outputs, including partial protein destabilization or altered adhesive interactions with relevant partners.
Disease phenotypes
HDGC and Lynch syndromes exemplify how germline variants can exert context-dependent effects on tumour development across different tissues. Although the mechanisms underlying tissue susceptibility remain largely unknown, contributing factors may include local gene expression and function, activation of downstream signalling pathways, and features of the tissue microenvironment.
The tumour spectrum associated with Lynch syndrome has evolved over time. Cancers of the colorectum and endometrium were the first manifestations documented, with the publication of family G by Warthin in 1913. Increased frequencies of cancers of the stomach, small bowel, hepatobiliary system, upper urologic tract and ovary were reported 80 years later, in 1994 [56]. A study in 2008 added glioblastomas to the tumour spectrum [57], and only recently cancers of the pancreas, breast, and prostate, as well as the rare adrenocortical tumours, were considered to be phenotypic expressions of the Lynch syndrome [58,59,60]. Reliable associations between affected genes and clinical presentations have been observed for the broad spectrum of cancer types in Lynch syndrome (Fig. 2). Carriers of MLH1 alterations have increased lifetime risk of developing colorectal (46%) and endometrium (43%) cancers, followed by prostate (17%), breast (12%), and ovarian (10%) neoplasms [61]. Cancers affecting urological tract (bladder, ureter or kidney, 8%), stomach (7%), and pancreas (6%) can also occur although at lower frequencies [61]. In the case of MSH2 variants, endometrial (57%), colorectal (43%), prostate (32%), and urological tract (25%) cancers constitute the most frequent presentations, whereas MSH6 loss of function confers higher risk for endometrium (47%) and lower risk for prostate (18%), colorectal (15%), ovarian (13%), breast (13%), urinary tract (11%), gastric (5%), and pancreatic cancers (1%) [61]. Concerning PMS2 variants, major risks were reported for breast (56%), prostate (38%) and endometrium (26%) cancers [61], but according to recent studies these were not confirmed and only a marginal increase is detected, when compared with risks for the general population [28]. In addition, Lynch syndrome-associated tumours include those of the brain (usually glioblastomas), small bowel, hepatobiliary tract and skin (sebaceous tumours) [10, 62].

Disorders described in HDGC context comprise diffuse gastric cancer, lobular breast cancer and orofacial clefts (cleft lip and palate). Lynch syndrome is associated with a large panel of cancer types. These include colorectal, endometrium, prostate, breast, ovarian, urological tract (bladder, ureter or kidney), stomach, pancreas, brain (usually glioblastomas), small bowel, hepatobiliary tract, and skin tumours.
So far, a restricted panel of disorders has been described in HDGC patients. These comprise diffuse gastric cancer, which occurs in up to 70–80% of HDGC cases, with lifetime risks varying from 25% to 33% for women and 37% to 42% for men, and a mean age at onset of 45 years [63, 64]. This contrasts with Lynch syndrome, where gastric cancer represents only a minor fraction of the clinical presentations, underscoring the substantial differences in tissue-specific cancer risk between the two syndromes. Likewise, lobular breast cancer is highly prevalent in HDGC, accounting for the second most frequent neoplasia in 39% to 55% of female variant carriers, with a mean age at onset of 48–53 years [63, 64]. Importantly, among families from North America with germline CDH1 pathogenic/likely pathogenic variants, the cumulative risk of gastric cancer was recently conveyed as only 7% to 10%, whereas the cumulative risk of breast cancer in female carriers remained similar to prior estimates [65].
HDGC kindreds have also been reported to display cases of colorectal cancer. In fact, colorectal cancer with signet ring cell features appears to be enriched in patients harbouring pathogenic or likely pathogenic CDH1 variants, but the number of cases identified in HDGC families are scarce [4, 66, 67]. Moreover, data suggest that the lifetime risk of invasive colorectal cancer in HDGC may be similar to that of the general population, hampering its inclusion within the HDGC disease spectrum [66, 68]. Congenital malformations, like orofacial clefts, have also been associated with HDGC [6, 7], and in accordance, occurrence of cleft lip, cleft lip and palate, or cleft palate alone now constitutes a criterion for CDH1 genetic screening [5]. Of note, phenotype presentation varies largely among families as these may display diffuse gastric cancer only or combined with the abovementioned manifestations.
CDH1 germline variants are not restricted to the classical HDGC spectrum since individuals who do not meet the clinical criteria for HDGC may still be at risk of hereditary lobular breast cancer (HLBC). The study by García-Peláez et al. demonstrated that 7% of pathogenic/likely pathogenic variants were detected in families presenting only lobular breast cancer, not fulfilling the 2020 HDGC criteria [39]. Similarly, Corso et al. conducted a large longitudinal study with 5429 lobular breast cancer patients, revealing that HLBC represents 34.4% of cases. Multigene panel testing of 394 cases identified CDH1 pathogenic/likely pathogenic variants in 1.5% of patients with early-onset disease and a family history of breast cancer [69].
It is clear that clinical presentations in both hereditary conditions differ even in individuals with similar genotypes, suggesting that other genetic and non-genetic factors modulate disease phenotypes. Considering the undisputable role of E-cadherin during embryonic development and in adult epithelia, it is not unexpected that the range of cancers, as well as congenital malformations, arising in HDGC context may expand in the upcoming years. Likewise, we postulate that a plausible profile of disorders may emerge depending on the disturbed gene.
Management and surveillance protocols
As described above, individuals with inherited cancer syndromes have increased susceptibility to develop tissue-specific cancers at an early age. Therefore, a successful outcome and improved overall survival is dependent on long term and adequate management protocols at specialised centres. While proper surveillance guidelines are already at place for some hereditary cancer syndromes, comprehensive recommendation protocols are still underdeveloped for others.
Lynch syndrome is a valuable example of integrative patient management, in view of its association with an increased lifetime risk of developing many cancer types. Improvements in patient follow-up procedures have indeed accompanied the emergence of new data, mostly due to technological advances and accurate identification of susceptible individuals. Patients diagnosed with Lynch syndrome are being offered coordinated multidisciplinary support, strongly reducing the risk of developing cancer and impacting their quality of life. Clinical surveillance and intervention guidelines differ in screening starting age and intervals, depending on the affected gene (MLH1, MSH2, MSH6 or PMS2) and the proposing entity [3, 70, 71]. In particular, for patients with MLH1 or MSH2 variants, the National Comprehensive Cancer Network (NCCN, United States) suggests screening should start at 20-25 years for colorectal, 30-35 years for gynaecological and urological, and 40 years for gastroduodenal cancer if associated with risk factors [3]. In these cases, screening intervals range from 1-2 years for colorectal and gynaecological, and 3-5 years in gastroduodenal surveillance. There are no recommendations for urological screening interval. According to the observed lower risk for colorectal cancer associated with MSH6 or PMS2 variants, screening should start at a later age (30-35 years) and be maintained at intervals of 1-3 years [3]. In contrast, European guidelines are less strict and suggest colorectal surveillance should initiate at the age of 25 years for MLH1 or MSH2 variant carriers, or 35 years for MSH6 or PMS2 variants. Screening intervals range from 2-3 years for MHL1, MSH2, and MSH6 variants or 3-5 for PMS2 [3, 70]. Likewise, urological surveillance should start at 40-50 years and should comprise urinalysis, urine cytology, and abdominal ultrasound every 2 years for MLH1, MSH6, and PMS2 variants, or yearly for MSH2 variants [72]. Notably, no surveillance is recommended for gastroduodenal lesions, whereas for gynaecological it is offered an optional annual review starting at the age of 25 years [70, 73]. In addition to these, risk-reducing colon and gynaecological surgeries, as well as aspirin intake can be considered as part of the NCCN and European guidelines [70, 71]. Clinical management encompassing endoscopy, screening for H. pylori infection, and assessment of physical/neurological capabilities have also been suggested [70]. Nonetheless, the potential benefits of the latest remain unclear and are not performed on a regular basis [74]. To ameliorate the identification of individuals with Lynch syndrome, genetic testing has been expanded and is now proposed for all patients diagnosed with colorectal cancer before the age of 50 years [11, 70].
Multidisciplinary surveillance in the context of HDGC is still in its infancy. Diffuse gastric cancer and lobular breast cancer are undoubtedly the main cancer presentations in HDGC, although with variable penetrance. According to current guidelines, genetic testing for HDGC should be offered from the legal age of consent (generally 16-18 years) in individuals meeting testing criteria, while testing of younger family members may be considered when warranted by family history. Clinical management of CDH1 carriers involves intense endoscopic surveillance and/or prophylactic gastrectomy, as well as mammography and magnetic resonance imaging screening [5]. Specifically, for carriers of pathogenic/likely pathogenic CDH1 variants with confirmed cases of diffuse gastric cancer in first- or second-degree relatives, prophylactic total gastrectomy is advised [5]. Alternatively, carriers who have decided to undergo surveillance, should perform annual endoscopy at specialised centres, with IGCLC guidelines recommending extensive analysis of 30 random biopsies along the cardia, fundus, body, transition zone, and antrum, as well as targeted biopsies of visible abnormalities [5]. Asif et al. have provided evidence highlighting the critical role of targeted biopsies in HDGC surveillance [75]. In a prospective study, the authors demonstrated that targeted sampling of subtle mucosal changes substantially improved the detection of early signet-ring cell carcinoma, emphasising the need for combining both approaches in endoscopic protocols [75]. Indeed, the presence of latent or transient intramucosal signet-ring cell carcinoma foci are pathognomonic of HDGC with no equivalent in Lynch syndrome. Early immune changes within the gastric mucosa of CDH1 variant carriers may promote signet-ring cell dormancy, justifying the persistence of latent foci and their variable progression to invasive cancer [76].
As for individuals with CDH1 VUS, the IGCLC recommends annual endoscopic surveillance for at least 2 years. After this period, the interval may be increased [5]. It is important to note that counselling of these patients should balance the burden of gastric cancer risk and the psychological impact of prophylactic surgery and intense surveillance [5]. NCCN guidelines propose a more balanced approach, placing endoscopic surveillance on equal footing with prophylactic gastrectomy. This reflects emerging data on the potential safety of surveillance strategies, and the recognised risks and quality-of-life impact associated with prophylactic gastrectomy [77].
Considering breast cancer, it is recommended that women with a pathogenic/likely pathogenic CDH1 variant undergo annual surveillance from 30 years of age, which includes annual magnetic resonance imaging possibly supplemented with mammography, starting at 35-40 years of age [5]. Bilateral risk-reducing mastectomy can also be considered in a case-by-case scenario [5]. In addition to diffuse gastric cancer and lobular breast cancer, other cancer types and congenital disorders have been observed in HDGC kindred, although at a much lower incidence [6, 7, 66]. In contrast to Lynch syndrome, these less frequent manifestations within the HDGC tumour spectrum are not yet considered for surveillance protocols in the guidelines since their incidence has been shown to be comparable to that of the general population [5]. We envisage that future research will provide robust data concerning risk estimates of other cancers and congenital manifestations within HDGC kindreds.
The evolution of genetic testing criteria and the trend towards a more liberal testing approach represent an emergent aspect of both Lynch syndrome and HDGC management. While initial testing strategies were highly restrictive, relying on stringent clinical and familial criteria, broader tumour screening and germline testing criteria have been adopted in Lynch syndrome, resulting in increased detection rates. A similar movement is now seen in HDGC, with recent NCCN guidelines recommending more inclusive criteria for genetic testing of CDH1 and, more cautiously, of CTNNA1, overcoming potential dismissal of at-risk individuals [77].
Conclusions
Compelling evidence demonstrates that recognition of clinical, pathological and molecular features of Lynch syndrome has evolved drastically since the first reports. Interestingly, it is noticeable that HDGC and Lynch syndromes are attributed to dysfunction of genes involved in two distinct but well-defined cellular processes, namely cell-cell adhesion and MMR system. Germline alterations are evenly distributed across the entire genes, without categorical correlations established between variant types or protein domains affected. Additional complexity incurs in light of missense variants due to residual protein activity and consequent lower penetrance of disease phenotypes.
We foresee that the intricacies and history of Lynch syndrome may serve as a paradigm to progress in HDGC identity. Indeed, the extended knowledge acquired within the scope of Lynch syndrome may unlock new avenues of research to elucidate novel causative genes, the significance of missense variants, determinants of tissue-specific effects, and environmental modulators of clinical presentations in HDGC. Single-cell multi-omics technologies providing information on cell functional states and spatial dynamics will be critical to advance the molecular profile of E-cadherin deficient cells and signalling programs underlying HDGC.
References
Okur V, Chung WK: The impact of hereditary cancer gene panels on clinical care and lessons learned. Cold Spring Harbor Molecular Case Stud 2017;3:a002154.
Figueiredo J, Melo S, Carneiro P, Moreira AM, Fernandes MS, Ribeiro AS, et al. Clinical spectrum and pleiotropic nature of CDH1 germline mutations. J Med Genet. 2019;56:199–208.
Peltomaki P, Nystrom M, Mecklin JP, Seppala TT. Lynch Syndrome Genetics and Clinical Implications. Gastroenterology. 2023;164:783–99.
Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–5.
Blair VR, McLeod M, Carneiro F, Coit DG, D’Addario JL, van Dieren JM, et al. Hereditary diffuse gastric cancer: updated clinical practice guidelines. Lancet Oncol. 2020;21:e386–e397.
Frebourg T, Oliveira C, Hochain P, Karam R, Manouvrier S, Graziadio C, et al. Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J Med Genet. 2006;43:138–42.
Kluijt I, Siemerink EJ, Ausems MG, van Os TA, de Jong D, Simoes-Correia J, et al. CDH1-related hereditary diffuse gastric cancer syndrome: clinical variations and implications for counseling. Int J Cancer. 2012;131:367–76.
Decourtye-Espiard L, Guilford P. Hereditary Diffuse Gastric Cancer. Gastroenterology. 2023;164:719–35.
Ghoumid J, Stichelbout M, Jourdain AS, Frenois F, Lejeune-Dumoulin S, Alex-Cordier MP, et al. Blepharocheilodontic syndrome is a CDH1 pathway-related disorder due to mutations in CDH1 and CTNND1. Genet Med. 2017;19:1013–21.
Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer. 2015;15:181–94.
Rubenstein JH, Enns R, Heidelbaugh J, Barkun A. Clinical Guidelines C: American Gastroenterological Association Institute Guideline on the Diagnosis and Management of Lynch Syndrome. Gastroenterology. 2015;149:777–82. quiz e716-777.
Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–9.
Nenclares P, Harrington KJ. The biology of cancer. Medicine. 2020;48:67–72.
Lerner BA, Llor X. Genetic Gastric Cancer Risk Syndromes. Curr Treat Opt Gastroenterol. 2020;18:604–15.
Oliveira C, Sousa S, Pinheiro H, Karam R, Bordeira-Carrico R, Senz J, et al. Quantification of epigenetic and genetic 2nd hits in CDH1 during hereditary diffuse gastric cancer syndrome progression. Gastroenterology. 2009;136:2137–48.
Peltomaki P. Epigenetic mechanisms in the pathogenesis of Lynch syndrome. Clin Genet. 2014;85:403–12.
Hansford S, Kaurah P, Li-Chang H, Woo M, Senz J, Pinheiro H, et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015;1:23–32.
Majewski IJ, Kluijt I, Cats A, Scerri TS, de Jong D, Kluin RJ, et al. An alpha-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J Pathol. 2013;229:621–9.
van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65:3756–88.
Takeichi M. Multiple functions of alpha-catenin beyond cell adhesion regulation. Curr Opin Cell Biol. 2018;54:24–29.
Bajpai S, Correia J, Feng Y, Figueiredo J, Sun SX, Longmore GD, et al. {alpha}-Catenin mediates initial E-cadherin-dependent cell-cell recognition and subsequent bond strengthening. Proc Natl Acad Sci USA. 2008;105:18331–6.
Wang A, Dunn AR, Weis WI: Mechanism of the cadherin-catenin F-actin catch bond interaction. eLife 2022;11:e80130.
den Elzen N, Buttery CV, Maddugoda MP, Ren G, Yap AS. Cadherin adhesion receptors orient the mitotic spindle during symmetric cell division in mammalian epithelia. Mol Biol Cell. 2009;20:3740–50.
Monster JL, Kemp LJ, Busslinger GA, Vliem MJ, Derks LL, Staes AA, et al. Cell division-dependent dissemination following E-cadherin loss underlies initiation of diffuse-type gastric cancer. J Pathol. 2024;263:226–41.
Pecina-Slaus N, Kafka A, Salamon I, Bukovac A. Mismatch Repair Pathway, Genome Stability and Cancer. Front Mol Biosci. 2020;7:122.
Peltomaki P. Update on Lynch syndrome genomics. Fam Cancer. 2016;15:385–93.
Win AK, Jenkins MA, Dowty JG, Antoniou AC, Lee A, Giles GG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer epidemiology. Biomark Prev. 2017;26:404–12.
Dominguez-Valentin M, Sampson JR, Seppala TT, Ten Broeke SW, Plazzer JP, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020;22:15–25.
Liccardo R, De Rosa M, Rossi GB, Carlomagno N, Izzo P, Duraturo F. Incomplete Segregation of MSH6 Frameshift Variants with Phenotype of Lynch Syndrome. Int J Mol Sci 2017;152:S1–S113.
Liccardo R, Della Ragione C, Mitilini N, De Rosa M, Izzo P, Duraturo F. Novel variants of unknown significance in the PMS2 gene identified in patients with hereditary colon cancer. Cancer Manag Res. 2019;11:6719–25.
Eikenboom EL, van der Werf-‘t Lam AS, Rodriguez-Girondo M, Van Asperen CJ, Dinjens WNM, Hofstra RMW, et al. Universal Immunohistochemistry for Lynch Syndrome: A Systematic Review and Meta-analysis of 58,580 Colorectal Carcinomas. Clin Gastroenterol Hepatol. 2022;20:e496–e507.
Walker R, Mahmood K, Joo JE, Clendenning M, Georgeson P, Como J, et al. A tumor focused approach to resolving the etiology of DNA mismatch repair deficient tumors classified as suspected Lynch syndrome. J Transl Med. 2023;21:282.
Corso G, Corso F, Bellerba F, Carneiro P, Seixas S, Cioffi A, et al. Geographical Distribution of E-cadherin Germline Mutations in the Context of Diffuse Gastric Cancer: A Systematic Review. Cancers (Basel) 2021;13:1269.
Melo S, Figueiredo J, Fernandes MS, Goncalves M, Morais-de-Sa E, Sanches JM, et al. Predicting the Functional Impact of CDH1 Missense Mutations in Hereditary Diffuse Gastric Cancer. Int J Mol Sci 2017;18:2687.
Karam R, Carvalho J, Bruno I, Graziadio C, Senz J, Huntsman D, et al. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene. 2008;27:4255–60.
Benusiglio PR, Colas C, Guillerm E, Canard A, Delhomelle H, Warcoin M, et al. Clinical implications of CTNNA1 germline mutations in asymptomatic carriers. Gastric Cancer. 2019;22:899–903.
Coudert M, Drouet Y, Delhomelle H, Svrcek M, Benusiglio PR, Coulet F, et al. First estimates of diffuse gastric cancer risks for carriers of CTNNA1 germline pathogenic variants. J Med Genet. 2022;59:1189–95.
Lobo S, Dias A, Pedro AM, Ferreira M, Pinto-Oliveira A, Sao Jose C, et al: Hereditary diffuse gastric cancer spectrum associated with germline CTNNA1 loss of function revealed by clinical and molecular data from 351 carrier families and over 37000 non-carrier controls. Gut 2025. https://doi.org/10.1136/gutjnl-2024-334601.
Garcia-Pelaez J, Barbosa-Matos R, Lobo S, Dias A, Garrido L, Castedo S, et al. Genotype-first approach to identify associations between CDH1 germline variants and cancer phenotypes: a multicentre study by the European Reference Network on Genetic Tumour Risk Syndromes. Lancet Oncol. 2023;24:91–106.
Figueiredo J, Mercadillo F, Melo S, Barroso A, Goncalves M, Diaz-Tasende J, et al. Germline CDH1 G212E Missense Variant: Combining Clinical, In Vitro and In Vivo Strategies to Unravel Disease Burden. Cancers 2021;13:4359.
Betes M, Alonso-Sierra M, Valenti V, Patino A. A multidisciplinary approach allows identification of a new pathogenic CDH1 germline missense mutation in a hereditary diffuse gastric cancer family. Digestive Liver Dis. 2017;49:825–6.
Lo W, Zhu B, Sabesan A, Wu HH, Powers A, Sorber RA, et al. Associations of CDH1 germline variant location and cancer phenotype in families with hereditary diffuse gastric cancer (HDGC). J Med Genet. 2019;56:370–9.
Nolano A, Medugno A, Trombetti S, Liccardo R, De Rosa M, Izzo P, et al. Hereditary colorectal cancer: state of the art in lynch syndrome. Cancers 2022;15:75.
Luo X, Maciaszek JL, Thompson BA, Leong HS, Dixon K, Sousa S, et al: Optimising clinical care through CDH1-specific germline variant curation: improvement of clinical assertions and updated curation guidelines. J Med Genet 2023;60:568–75.
Figueiredo J, Melo S, Gamet K, Godwin T, Seixas S, Sanches JM, et al. E-cadherin signal sequence disruption: a novel mechanism underlying hereditary cancer. Mol Cancer. 2018;17:112.
Koger N, Paulsen L, Lopez-Kostner F, Della Valle A, Vaccaro CA, Palmero EI, et al. Evaluation of MLH1 variants of unclear significance. Genes, Chromosomes Cancer. 2018;57:350–8.
Abildgaard AB, Stein A, Nielsen SV, Schultz-Knudsen K, Papaleo E, Shrikhande A, et al. Computational and cellular studies reveal structural destabilization and degradation of MLH1 variants in Lynch syndrome. eLife 2019;8:e49138.
Arlow T, Scott K, Wagenseller A, Gammie A. Proteasome inhibition rescues clinically significant unstable variants of the mismatch repair protein Msh2. Proc Natl Acad Sci USA. 2013;110:246–51.
Simoes-Correia J, Figueiredo J, Lopes R, Stricher F, Oliveira C, Serrano L, et al. E-cadherin destabilization accounts for the pathogenicity of missense mutations in hereditary diffuse gastric cancer. PLoS ONE. 2012;7:e33783.
Simoes-Correia J, Figueiredo J, Oliveira C, van Hengel J, Seruca R, van Roy F, et al. Endoplasmic reticulum quality control: a new mechanism of E-cadherin regulation and its implication in cancer. Hum Mol Genet. 2008;17:3566–76.
Mahdouani M, Ben Ahmed S, Hmila F, Rais H, Ben Sghaier R, Saad H, et al. Functional characterization of MLH1 missense variants unveils mechanisms of pathogenicity and clarifies role in cancer. PLoS ONE. 2022;17:e0278283.
Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–22.
Raraigh KS, Han ST, Davis E, Evans TA, Pellicore MJ, McCague AF, et al. Functional Assays Are Essential for Interpretation of Missense Variants Associated with Variable Expressivity. Am J Hum Genet. 2018;102:1062–77.
Majithia AR, Tsuda B, Agostini M, Gnanapradeepan K, Rice R, Peloso G, et al. Prospective functional classification of all possible missense variants in PPARG. Nat Genet. 2016;48:1570–5.
Decourtye-Espiard L, Bougen-Zhukov N, Godwin T, Brew T, Schulpen E, Black MA, et al: E-Cadherin-Deficient Epithelial Cells Are Sensitive to HDAC Inhibitors. Cancers 2021;14:175.
Watson P, Lynch HT. The tumor spectrum in HNPCC. Anticancer Res. 1994;14:1635–9.
Watson P, Vasen HFA, Mecklin JP, Bernstein I, Aarnio M, Jarvinen HJ, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444–9.
Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790–5.
Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30:958–64.
Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, et al. Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol. 2013;31:3012–8.
Moller P, Seppala TT, Bernstein I, Holinski-Feder E, Sala P, Gareth Evans D, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut. 2018;67:1306–16.
Koornstra JJ, Mourits MJ, Sijmons RH, Leliveld AM, Hollema H, Kleibeuker JH. Management of extracolonic tumours in patients with Lynch syndrome. Lancet Oncol. 2009;10:400–8.
Roberts ME, Ranola JMO, Marshall ML, Susswein LR, Graceffo S, Bohnert K, et al. Comparison of CDH1 Penetrance Estimates in Clinically Ascertained Families vs Families Ascertained for Multiple Gastric Cancers. JAMA Oncol. 2019;5:1325–31.
Xicola RM, Li S, Rodriguez N, Reinecke P, Karam R, Speare V, et al. Clinical features and cancer risk in families with pathogenic CDH1 variants irrespective of clinical criteria. J Med Genet. 2019;56:838–43.
Ryan CE, Fasaye GA, Gallanis AF, Gamble LA, McClelland PH, Duemler A, et al. Germline CDH1 Variants and Lifetime Cancer Risk. JAMA. 2024;332:722–9.
Richards FM, McKee SA, Rajpar MH, Cole TR, Evans DG, Jankowski JA, et al. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 1999;8:607–10.
Benesch MGK, Bursey SR, O’Connell AC, Ryan MG, Howard CL, Stockley CC, et al: CDH1 Gene Mutation Hereditary Diffuse Gastric Cancer Outcomes: Analysis of a Large Cohort, Systematic Review of Endoscopic Surveillance, and Secondary Cancer Risk Postulation. Cancers 2021;13:2622.
Hamilton LE, Jones K, Church N, Medlicott S. Synchronous appendiceal and intramucosal gastric signet ring cell carcinomas in an individual with CDH1-associated hereditary diffuse gastric carcinoma: a case report of a novel association and review of the literature. BMC Gastroenterol. 2013;13:114.
Corso G, Marino E, Zanzottera C, Oliveira C, Bernard L, Macis D, et al. CDH1 Genotype Exploration in Women With Hereditary Lobular Breast Cancer Phenotype. JAMA Netw Open. 2024;7:e247862.
Seppala TT, Latchford A, Negoi I, Sampaio Soares A, Jimenez-Rodriguez R, Sanchez-Guillen L, et al. European guidelines from the EHTG and ESCP for Lynch syndrome: an updated third edition of the Mallorca guidelines based on gene and gender. Br J Surg. 2021;108:484–98.
Monahan KJ, Bradshaw N, Dolwani S, Desouza B, Dunlop MG, East JE, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020;69:411–44.
Lonati C, Necchi A, Gomez Rivas J, Afferi L, Laukhtina E, Martini A, et al. Upper Tract Urothelial Carcinoma in the Lynch Syndrome Tumour Spectrum: A Comprehensive Overview from the European Association of Urology - Young Academic Urologists and the Global Society of Rare Genitourinary Tumors. Eur Urol Oncol. 2022;5:30–41.
Crosbie EJ, Ryan NAJ, Arends MJ, Bosse T, Burn J, Cornes JM, et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet Med. 2019;21:2390–2400.
Vasen HF, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62:812–23.
Asif B, Sarvestani AL, Gamble LA, Samaranayake SG, Famiglietti AL, Fasaye GA, et al. Cancer surveillance as an alternative to prophylactic total gastrectomy in hereditary diffuse gastric cancer: a prospective cohort study. Lancet Oncol. 2023;24:383–91.
Green BL, Gamble LA, Diggs LP, Nousome D, Patterson JC, Joughin BA, et al. Early Immune Changes Support Signet Ring Cell Dormancy in CDH1-Driven Hereditary Diffuse Gastric Carcinogenesis. Mol Cancer Res MCR. 2023;21:1356–65.
Lerner BA, Gupta S, Burke CA, Kupfer S, Katona BW, Grady WM, et al: Advancing the Evaluation and Management of CDH1-Associated Gastric Cancer. J Natil Comprehens Cancer Netw JNCCN 2025;23:e257006.
Acknowledgements
We would like to pay a tribute to Raquel Seruca, expressing our endless love and admiration. To infinity and beyond.
Funding
This work was financed by FEDER funds through the Operational Programme for Competitiveness Factors (COMPETE 2020), Programa Operacional de Competitividade e Internacionalização (POCI) and Programa Operacional Regional do Norte (Norte 2020); and by National Funds through the Portuguese Foundation for Science and Technology (FCT projects EXPL/MED-ONC/0386/2021, 2022.02665.PTDC, 2023.17974.ICDT https://doi.org/10.54499/2023.17974.ICDT, and doctoral grants SFRH/BD/143533/2019-JP and 2023.01033.BD-LC). JF is funded by the “FCT Scientific Employment Stimulus – Institutional Call” (CEECINST/00056/2021). We acknowledge the American Association of Patients with Hereditary Gastric Cancer “No Stomach for Cancer” for funding Seruca and Figueiredo’s research, and the project “P.CCC: Centro Compreensivo de Cancro do Porto” - NORTE-01-0145-FEDER-072678, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF).
Author information
Authors and Affiliations
Contributions
JF and RS were responsible for study concept and design. JP, LC, SM, PC, MSF and JF have draughted the article. JF and PC critically reviewed the manuscript for important intellectual content. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pereira, J., Carvalho, L., Melo, S. et al. Hereditary diffuse gastric cancer in progress: Comparative lessons from Lynch syndrome. Eur J Hum Genet (2025). https://doi.org/10.1038/s41431-025-01992-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41431-025-01992-w
