
Abstract
Genomes capture the adaptive and demographic history of a species, but the choice of sequencing strategy and sample size can impact such inferences. We compared whole genome and reduced representation sequencing approaches to study the population demographic and adaptive signals of the North American mountain goat (Oreamnos americanus). We applied the restriction site-associated DNA sequencing (RADseq) approach to 254 individuals and whole genome resequencing (WGS) approach to 35 individuals across the species range at mid-level coverage (9X) and to 5 individuals at high coverage (30X). We used ANGSD to estimate the genotype likelihoods and estimated the effective population size (Ne), population structure, and explicitly modelled the demographic history with δaδi and MSMC2. The data sets were overall concordant in supporting a glacial induced vicariance and extremely low Ne in mountain goats. We evaluated a set of climatic variables and geographic location as predictors of genetic diversity using redundancy analysis. A moderate proportion of total variance (36% for WGS and 21% for RADseq data sets) was explained by geography and climate variables; both data sets support a large impact of drift and some degree of local adaptation. The empirical similarities of WGS and RADseq presented herein reassuringly suggest that both approaches will recover large demographic and adaptive signals in a population; however, WGS offers several advantages over RADseq, such as inferring adaptive processes and calculating runs-of-homozygosity estimates. Considering the predicted climate-induced changes in alpine environments and the genetically depauperate mountain goat, the long-term adaptive capabilities of this enigmatic species are questionable.
Similar content being viewed by others


Introduction
Genomes contain a detailed evolutionary record that provides a lens into the past. Reconstructing past population processes and demographic events is a key component of understanding species’ biology and predicting future responses to environmental change (Aguirre-Liguori et al. 2021). The development of population genetic analyses, most notably the Kingman coalescent (Kingman 1982), has provided novel insights like timing of migration and contraction events to the study of demography, and has supplemented more traditional observational studies and inferences from the fossil record (Salmona et al. 2019). Likewise, standard ordinations can be used to identify key environmental correlates underlying genetic structure (Briñoccoli et al. 2021) allowing for adaptive inferences to be made. These analytical tools have expanded to include vast amounts of DNA data and the wealth of information stored in genome-scale sequence. Approaches to parse out environmental and geographic correlates to genetic attributes can scale to large SNP panels (Papadopoulou and Knowles 2015), while analyses such as Sequentially Markovian Coalescent (SMC) capitalize on the unique attributes of genome-scale data to reconstruct historical demography (Li and Durbin 2011; MacLeod et al. 2013).
Summarizing population genomic variation can be effectively done in the form of the site frequency spectrum (SFS), also referred to as the allele frequency spectrum. The SFS is a histogram of allele counts across a sample and is amenable to more than one population (i.e., joint SFS); simply visualizing the SFS sheds light on demographic patterns such as contractions that flatten the histogram, and migration, which leads to increased shared allele sharing in the joint SFS. The shape and distribution of variants in the SFS are directly tied to the population(s) demographic history, and many standard population genetic summary statistics can be derived from it (Korneliussen et al. 2013). The increasing number of genomic markers that can be obtained from multiple individuals has allowed for development and testing of complex demographic models via the SFS (Zhao et al. 2013; Gamble et al. 2015; Vijay et al. 2016; Hu et al. 2020). Programs like δaδi rely on diffusion approximation of the SFS to model historical changes in effective population size (Ne), population divergence and migration rates in multiple populations (Gutenkunst et al. 2009). Similarly, the now ubiquitous sequential Markovian coalescent (SMC) approaches aim to reconstruct demographic patterns of a population or species from whole genome data in a single individual (Beichman et al. 2018); here coalescent rates are inferred across segments of the chromosome and scaled to produce a trajectory of Ne over time.
Modern population genomic data can generally be split into two types: reduced representation and whole genome resequencing (WGS). The latter most often involves random fragmentation of genomic DNA from a few to dozens of individuals, with sequencing of those fragments that ultimately encompasses the entirety of the genome. Reduced representation approaches, specifically restriction site-associated DNA sequencing (RADseq), first fragment the genome via restriction enzymes to generate a library, which is followed by high-throughput sequencing, often in hundreds of individuals (Andrews et al. 2016). While debated (Lowry et al. 2017; Catchen et al. 2017), it is assumed that the sparseness of such data makes them less effective at capturing the outlier loci (Andrews et al. 2016). Approaches like distance-based redundancy analysis (dbRDA) are more suitable to infer adaptive divergence with RADseq as the dependent variable is a genetic distance between populations, but a quantitative comparison of WGS as RADseq has not been conducted to date.
Both whole genome and reduced representation approaches are vulnerable to sequencing errors due to the use of the same sequencing technology (Shendure and Ji 2008). Additional sources of error and bias in RADseq data are null alleles and allelic dropout that occurs when a polymorphism alters the restriction enzyme cut-site (Gautier et al. 2013). RADseq is also more susceptible to genotyping errors resulting from PCR duplicates (Davey et al. 2013). The choice of sequencing strategy in study design has an effect on the inferences drawn from the data. Shafer et al. (2015) showed via simulations that while the correct demographic model was identified with RADseq-like data, some demographic parameter estimates could not be recovered with 50,000 variable loci. Similarly, Warmuth and Ellegren (2019) showed that the optimally identified demographic model was generally the same between RADseq and WGS data sets. Furthermore, density of markers, sequencing coverage and number of individuals are the factors that determine which analyses can be performed with genomic data; for example, for Pairwise Sequentially Markovian coalescent a genome coverage of approximately 20X is recommended (Nadachowska-Brzyska et al. 2016), but only one individual is needed. SMC approaches have been applied to simulated RADseq data showing some relevant coalescent information is retained, but this requires a relative high density of loci (Liu and Hansen 2017). Sample size also has a clear effect on diversity statistics tests of neutrality (Subramanian 2016). With sequencing usually comprising the largest portion of the cost of genomic studies, a decision must be made regarding sequencing effort and the number of individuals; specifically, sequencing fewer individuals at a higher coverage, or a greater number of individuals at a lower depth or breadth of coverage. At the same sequencing effort, RADseq offers the possibility of including hundreds of individuals in the study but at the expense of capturing a much lower number of variants. Thus, we set out to quantitively contrast demographic and adaptive inferences from RADseq (large sample number) and WGS at mid and high sequencing depth. All three data sets are commonly applied with a suite of unique and overlapping methods; we took advantage of a well-studied alpine mammal with clear predictions based on microsatellite and mitochondrial data (Shafer et al. 2011) to compare inferences from the different data sets.
The North American mountain goat (Oreamnos americanus) species range extends from the Pacific Northwest to central Alaska, Yukon and Northwest Territories (44°N to 63°N), and environmental conditions such as precipitation, temperature and elevation vary greatly. An assessment of genetic diversity indicated the presence of isolation-by-distance (IBD) and two main refugial lineages designated north and south that aligns with the range edges (Shafer et al. 2011); the species appears to have gone through a bottleneck that dramatically reduced Ne at the end of last glacial period approximately 20 kya (Martchenko et al. 2020). Likewise, environmental variation appears to underly current phenotypic differences across the range (Martchenko et al. 2022), suggesting varying selection pressures. These reported patterns lead to clear demographic and environmental predictions when scaling the analysis up to range-wide genome data. Specifically, we hypothesized that northern and southern lineages underwent separate demographic declines during the last glacial maximum (LGM; sensu Shafer et al. 2011). And while drift will be a paramount factor driving population structure, we predict environmental variation underlies some genetic structure (sensu Martchenko et al. 2022); here we predict that RADseq will capture less of the adaptive signal (Lowry et al. 2017).
Materials and methods
Library construction and sequencing
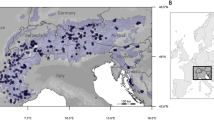
All tissue samples (muscle and skin) were obtained from hunter harvested individuals across the mountain goat range through a research collaboration with government authorities in Canada and United States (Fig. 1). DNA was extracted from all samples using the Qiagen DNeasy Blood & Tissue kit or a phenol chloroform method (Sambrook and Russell 2006) and quantified on a Qubit and TapeStation prior to sequencing. Thirty WGS individuals were sequenced on an Illumina HiSeqX machine to an average coverage of 9X; four individuals were sequenced to 30X. Following the protocol of (Brelsford et al. 2016), we generated three double-digest RADseq libraries with 96 individuals pooled per library (total of 288 individuals, with 14 individuals also sequenced at 9X coverage and included in the previous WGS data set. Briefly, we digested the sample DNA with SbfI and MseI restriction enzymes and size-selected 450–700 bp fragments via a gel extraction. Individual samples that did not amplify during library preparation were not included in the final library. Three RADseq libraries were sequenced on an Illumina 2500 machine with 125 bp paired-end sequencing, totalling three sequencing lanes. All sequencing was conducted at the Centre for Applied Genomics in Toronto, Canada and raw data have been deposited in the SRA (Accession numbers PRJNA510081, PRJNA909333, PRJNA909334).

High coverage samples are labelled as AK – Alaska (West), BC – British Columbia (East), ID – Idaho (South), YT – Yukon (North) and G – sample used for genome assembly.
Bioinformatic processing and SNP calling
Two WGS data sets were compiled from the raw sequencing data: (i) high coverage WGS data set that included 4 individuals sequenced at 30X coverage and the original genome sample at 85X from Martchenko et al. (2020) downsampled to 30X coverage; and (ii) mid-coverage WGS data set that included 35 individuals: 30 individuals sequenced to 9X coverage plus the five individuals sequenced at high coverage from set (i) downsampled to 9X coverage; here. The WGS data was first assessed for quality with FastQC (Andrews 2010); we trimmed both adapters and low quality bases (phred scores under 20) and required a minimum read length of 50 with BBDuk, part of BBTools (Bushnell 2018). We then mapped the reads to the mountain goat genome (GCA_009758055.1; length = 2506 Mbp, N50 = 67 Mbp, # of scaffolds = 3217; (Martchenko et al. 2020)) using the mem algorithm of BWA (Li 2013); we removed sequencing duplicates using the MarkDuplicates module of Picard toolkit (http://broadinstitute.github.io/picard/) and isolated primary alignments with sambamba v. 0.7.1 filtering (Tarasov et al. 2015). We calculated genotype likelihoods on the post-processed bam files with the GATK model (-GL 2) implemented in ANGSD v. 0.918 with the following filters: -SNP_pval 1e-6 -minMapQ 20 -minQ 20 -skipTriallelic 1 (Korneliussen et al. 2014).
For data set (iii) the RADseq libraries were demultiplexed based on adaptor indices. Using the Stacks v. 2.3 (Catchen et al. 2013) reference-based pipeline, we used process_radtags module to demultiplex the reads to individual level based on 24 inline barcodes. We removed individuals with less than 100,000 total reads resulting in 254 individuals in the final data set. We then mapped the reads to Oreamnos americanus reference genome using the mem algorithm of BWA (Li 2013) and used the same ANGSD parameters for SNP calling as for WGS data (-SNP_pval 1e-6 -minMapQ 20 -minQ 20) but we added a -minInd 203 flag to only include sites that have data for at least 80% of individuals. For compatibility with some downstream analyses that do not use GLs, the ANGSD output was converted to VCF using Beagle 5.0 (Browning and Browning 2007) where hard-calls were derived from the highest genotype probability.
Genome-wide statistics and population demographic inference
We calculated a series of genome-wide summary statistics that reflect genetic diversity and attributes of the SFS. For the mid-coverage WGS data set we used VCFtools v. 0.1.14 to calculate Tajima’s D averaged over non-overlapping 50 kbp windows, genome-wide nucleotide diversity (π) using 250 kbp non-overlapping windows, mean transition‐to‐transversion ratio (Ts/Tv) and observed individual heterozygosity (Hetobs). For RADseq data set we used Stacks v. 2.3 to calculate Ts/Tv and Hetobs and VCFtools v. 0.1.14 to calculate Tajima’s D and genome-wide nucleotide diversity (π) averaged over 750 bp non-overlapping windows to reflect the insert size selection during library preparation.
To detect population structure in mid-coverage WGS and RADseq data we used NGSadmix with a K from 1 to 10 (Skotte et al. 2013) and selected the best K as indicated by the largest increase in likelihood for K between each step in K (Bohling et al. 2019). We used PCAngsd to conduct the PCA (Meisner and Albrechtsen 2018) and regressed the first two principal components against each individual’s latitude and longitude as a proxy for IBD using simple least squares regression in R v. 3.6.1. We estimated the folded SFS for mid-coverage WGS and RADseq data sets using ANGSD realSFS EM optimization. To explore adaptive divergence, we conducted a genotype-environment associations analysis, specifically a redundancy analysis (RDA) (Legendre and Legendre 2012) using the rda function in the vegan R package (Oksanen et al. 2020) with the 19 bioclimatic variables with 2.5 min of a degree resolution from the WorldClim 2 data set (Fick and Hijmans 2017), elevation from GTOPO30 USGS data set (Earth Resources Observation And Science (EROS) Center 2017), and latitude and longitude of each sample as predictor variables. We tested the environmental variables for collinearity and removed one variable from each pair with greater than 0.8 Pearson correlation coefficient. We filtered the genotype data for both RADseq and mid-coverage WGS data set to exclude sites with minor allele frequencies below 0.05 and linkage disequilibrium r2 above 0.8. As RDA requires no missing genotype data, we imputed the missing genotypes by using the most common genotype for each SNP using VCFR R package (Knaus and Grünwald 2017).
We subsetted two geographically disparate sampling units in the North and South to avoid confounding effects of population structure (Chikhi et al. 2010): the mid-coverage WGS data sets contained 10 North and 10 South samples and RADseq data sets contained 84 North and 63 South samples (Fig. 1). We estimated contemporary Ne using the linkage-disequilibrium equation based on genotype correlations (r2) and applied the sample size correction of Waples et al. (2016). Genome scale data can inflate r2 due to the higher probability of sequencing many physically linked loci (Waples et al. 2016); to minimize that bias we generated 100 VCF files each containing 10,000 random SNPs, estimated r2 and applied –maf 0.05 filter using VCFtools as per Dedato et al. (2022). We calculated the mean and standard deviation of Ne using the same North and South populations. We ran δaδi v. 2.1.1 (Gutenkunst et al. 2009) on both data sets to test the following population demographic models included in the one- and two-dimensional preset models: (i) snm – standard neutral model, (ii) bottlegrowth – size change followed by exponential growth, (iii) bottlegrowth_split – size change followed by exponential growth then split, (iv) bottlegrowth_split_mig – size change followed by exponential growth then split with migration, (v) split_mig – a split with migration, (vi) split_asym_mig – a split with asymmetric migration and (vii) IM – isolation-with-migration model with exponential growth. To verify convergence, we ran each model 20 times. The best fitting model was determined by comparing log-likelihoods across models. To estimate the historical Ne within the last 200 generations, we used the GONE software that uses the linkage disequilibrium information for markers across the genome (Santiago et al. 2020). We used a maximum of 50,000 SNPs per chromosome, the typical mammalian recombination rate of 1 cM/Mb, the default parameter hc of 0.05, and applied the Haldane correction for genetic distance; we repeated the analysis 40 times.
Lastly, using the four new high-coverage WGS samples we inferred the historical demography by implementing the multiple sequentially Markovian coalescent (MSMC2) model (Schiffels and Wang 2020) that generates estimates of Ne over time. Because we were running MSMC2 for more than one sample, we first generated a 35-mer mappability mask file using SNPable pipeline (http://lh3lh3.users.sourceforge.net/snpable.shtml) which identifies the regions of the genome to which short read data can be uniquely mapped (Schiffels and Wang 2020). We limited analysis to scaffolds >500 KB (Gower et al. 2018) and phased SNPs using WhatsHap 1.4 (Martin et al. 2016) prior to generating the MSMC2 input files. We ran MSMC2 on the input data and 100 generated bootstrapped data sets using the default time segment pattern 1 × 2 + 25 × 1 + 1 × 2 + 1 × 3 of 32 time intervals which describes which segments are joined by having the same coalescence rate (the first 2 segments have the same rate, the following 25 have individual rates etc.). We estimated the relative cross coalescence rate (rCCR) across populations by comparing the first haplotype from each sample in a pairwise fashion using the -P flag and skipping any sites with ambiguous phasing. rCCR ranges from 0 to 1, where 1 indicates that the two populations are one connected population and 0 indicates complete separation. To estimate migration rate (m), we took the MSMC2 output and ran the MSMC-IM model (Wang et al. 2020), and also estimated the cumulative migration probability M(t) which is related to the rCCR and estimates the proportion of ancestry already merged at time t. We assumed a generation time of 6 years and mutation rate of 1.33 × 10−8 mutations/site/generation calculated as the average mammalian mutation rate of 2.22*10−9 mutations/site/year (Kumar and Subramanian 2002) multiplied by the generation time of 6 years as per Martchenko et al. (2020).
Results
We identified 5,509,220 SNPs for the high coverage WGS data, 9,196,294 SNPs for the mid-coverage WGS data and 65,890 SNPs for the RADseq data with the ANGSD pipeline. The average SNP density was 102 SNPs/Mbp per scaffold for RADseq. For the three data sets π ranged from 4.2 × 10−4 to 9.0 × 10−4, Tajima’s D between 0.087 and 0.500, observed heterozygosity between 0.141 and 0.291 and Ts/Tv ratio between 1.89 and 2.06 (Table 1). The proportion of singletons was slightly higher in the mid-coverage WGS data set compared to the RADseq data set, but the overall shape of the SFS was similar (Fig. 2). We observed strong and consistent correlations between PC1 and latitude (R2 = 0.60 for mid-coverage WGS data, and R2 = 0.61 for RADseq data) and longitude (R2 = 0.95 for mid-coverage WGS data, and R2 = 0.91 for RADseq data; Table S1). Climatic variables that were retained for RDA included annual mean temperature (BIO1), maximum temperature of the warmest month (BIO5), mean temperature of the wettest quarter (BIO8), annual precipitation (BIO12), precipitation seasonality (BIO15) and elevation. Full models that included both climatic and geographical variables (latitude and longitude) explained 21% and 36% of total genetic variation for RADseq and WGS data sets respectively (Tables 2 and 3). Partial RDA models revealed that 5% and 18% of the total variation for RADseq and WGS data sets respectively was explained solely by geography, 6% and 9% solely by climate, and 10% and 9% of the total variation was confounded between the two sets of variables (Tables 2 and 3). Analysis of population structure with NGSadmix revealed 3 subpopulation clusters for both WGS and RADseq data (Fig. S1). North and South population representatives were separated by PCA (Fig. 3); FST between them was 0.21 for RADseq data set and 0.25 for the mid-coverage WGS data set, indicating moderate to strong genetic differentiation between regions. Contemporary Ne ranged from 10 ± 0.2 and 11 ± 0.4 individuals for the North subpopulation and 24 ± 1 and 28 ± 2 for the South population representatives for RADseq and WGS data sets respectively. Recent estimates of Ne (within the last 10 generations) inferred with GONE varied between 50 and 80 individuals and are slightly higher than the Waples method of Ne estimates, but the more historical estimate confidence intervals were wide (Fig. S2).

The spectra were calculated using genotype likelihoods with realSFS (ANGSD) for (A) RADseq data (n = 254) and (B) mid-coverage WGS data (n = 35).

PCAngsd results for (A) RADseq data (n = 254) and (B) mid-coverage WGS data (n = 35). Samples used for population demography inference are shown in colour.
Bottlegrowth split model where there is an instantaneous population size change followed by exponential growth and a split into two populations, and bottlegrowth split with migration model had the highest log-likelihoods in the set of the compared demographic models (Table S2). All models reported a recent split; for the mid-coverage WGS data set the split was dated at 63 generations for bottlegrowth split and 141 generations for the bottlegrowth split with migration model; for the RADseq data set, both models reported a very recent split of 48 and 41 generations ago (Table 4). MSMC2 from four samples representing range extremes and North-South clusters showed remarkably similar Ne trajectories during the last glacial period (Fig. S3; trajectories appeared to diverge approximately ~20–30 KYA, though the northern (YT-AK) and southern (BC-ID) have starting rCCR at >0.5 (Fig. S4) and elevated M(t) ~ 1 suggesting minimal divergence or ongoing gene flow (Fig. 4).

Individuals are abbreviated as AK – Alaska (West), BC – British Columbia (East), ID – Idaho (South), and YT – Yukon (North). A Timing and dynamics of separation process between two groups visualized by the time-dependent-symmetric migration rate m(t). Time is represented as years in the past. The dashed lines represent the median, or the time when 50% of the ancestry between the two groups merged. Shading indicates 1–99% (lighter shade) and 25–75% (darker shade) percentiles of the cumulative migration probabilities. B Cumulative migration probabilities that estimate the proportion of ancestry already merged at time t, and represents proportions of gene flow through time. M(t) values close to 0 denote complete separation between the two groups, while 1 shows a complete mix as one population. Dashed lines represent the relative cross-coalescent rate.
Discussion
Population genomic studies and demographic inference require several decisions to be made regarding sampling and sequencing strategy. In contrast with previously published studies, where whole genome and reduced representation sequencing are compared on the same set of individuals (Heller et al. 2021; Warmuth and Ellegren 2019), we used, arguably, a more realistic scenario of analysing data from a 10-fold larger number of individuals with the reduced representation approach which is more in line with current applications (Kjeldsen et al. 2016; Gervais et al. 2019; Sin et al. 2021). We showed that in this scenario the ecological associations and demographic models, along with broader inferences including IBD and estimates of Ne were largely robust to the sampling and sequencing strategy (Tables 1–4). A notable exception is the discrepancy in genome-wide estimates of Tajima’s D, which is important given the potential link to conservation status (Peart et al. 2020). Coverage differences between data sets will skew the SFS (Benjelloun et al. 2019) including Tajima’s D (Harvey et al. 2016), but sample size is likely the primary cause (Subramanian 2016). WGS showed more rare alleles than RADseq (Fig. 2), resulting also in less of an inferred population reduction (Table 4); thus, inferences from tests of neutrality, especially if informing conservation designations (Peart et al. 2020) must be cognizant of this factor and consider alternatives (e.g. Ramos-Onsins and Rozas 2002).
Importantly, both data sets recovered virtually identical estimates of contemporary Ne, a metric that is often used in conservation and management decisions (Hare et al. 2011). In terms of quantifying diversity and population structure RADseq has been more common to date, especially for samples without a reference genome. In the current sequencing era, however, this study highlights that a subset of WGS individuals is a viable, if not preferable, alternative to hundreds of samples with RADseq. The notable difference in Tajima’s D between data sets did not result in dramatically different model parameters, and qualitative output and demographic inference from SMC-based approaches is in-line with the SFS-based demographic models – all data-sets support a massive bottleneck during the last glacial maximum and no population recovery. The primary consideration should now extend beyond demographic inference, as RADseq will be less effective in identifying adaptive loci and runs of homozygosity due to the lower proportion of the genome variation being captured (Andrews et al. 2016; Lowry et al. 2017); this could explain the lower proportion of variance explained in the RADseq RDA models. Given the high similarities in adaptive and demographic signals here, if given the choice in sequencing strategy, the accumulated weight of evidence, in our opinion, suggests mid-coverage WGS for fewer samples to be preferrable to RADseq in hundreds of individuals for adaptive and demographic inference.
Mountain goat case study
The congruence among data sets allowed us to generate a detailed assessment of mountain goat adaptive differentiation and demography, and revealed a system with low genomic diversity that has been heavily impacted by glacial cycles and genetic drift. Consistent patterns emerged across data sets with respect to mountain goat populations, notably a very low contemporary Ne (Figs. S2; S3) and clear effect of geography on genetic structure (Tables 2 and 3).
Throughout the range mountain goat populations are not continuous and dispersal is limited (Côté and Festa-Bianchet 2003) which could result in local adaptation across the range. This observation is supported by some variation explained by climatic variables in the GEA analysis for both RADseq and WGS data sets. However a pattern of isolation-by-distance (IBD) where individuals from more distant populations are more genetically distinct (Wright 1943; Shafer et al. 2011) is also observed in the mountain goat as shown by the remarkably strong (R2 > 0.90) correlation between the first principal component axis and longitude for both WGS and RADseq data sets (Table S1). The effect of geography is evident in the RDA, where a large portion of genetic variation that is explained by models is attributable to (or confounded by) latitude and longitude (Tables 2 and 3). This suggests that genetic drift is the preeminent evolutionary force shaping genome-wide diversity and is further corroborated by the extremely low Ne estimates in both the North and South population as small populations that have experienced a bottleneck are more susceptible to genetic drift.
A pattern consistent with a recent bottleneck and separation was revealed by demographic modeling approaches based both on the SFS (δaδi) and the coalescent (MSMC2). Geographic separation into clusters across the range is broadly consistent with previous microsatellite analysis, at least spatially (Shafer et al. 2011); here, the larger SNP data sets reduced the number of clusters from 17 with microsatellites to 3 clusters with WGS data and also indicated a more recent split time (Shafer et al. 2011), perhaps due to the ascertainment bias in selecting microsatellite markers (see Poissant et al. 2009), as they are selected to maximize the potential variation. A less recent split between northern (YT) and southern (ID) samples compared to the western (AK) and eastern (BC) samples is indicated by the cumulative migration probability (Fig. 4); this is consistent with a clear separation of North and South samples in PCA (Fig. 3) and split time estimates from the best fit δaδi models. Previous analysis with PSMC indicated a severe decline in Ne during the last glacial maximum (Martchenko et al. 2020). PSMC can struggle with recent estimates of Ne (Nadachowska-Brzyska et al. 2016), but it is clear from Martchenko et al. (2020) the population massively contracted, likely to a species-wide Ne of less than one thousand. Our estimates of contemporary Ne are low, and account for physical linkage across the genome (Waples et al. 2016); likewise, the new MSMC2 inferences support a massive species-wide decline.
The low mountain goat Ne has likely reduced the efficacy of selection (Fisher 1930), including that of artificial selection potentially imposed by harvest (Martchenko et al. 2022). This low diversity has likely been low for a while (Wootton et al. 2023) and might not be cause for immediate conservation concern if observed genome-wide genetic diversity is not linked to adaptive potential (reviewed in Teixeira & Huber (2021); that however is usually an exception to the general pattern (see Bertola et al. (2021), DeWoody et al. (2021) and Kardos et al. (2021)). While functional genes might not reflect the observed low Ne in mountain goats (Shafer et al. 2012), genome-wide diversity preservation should still form the foundation of any management or conservation plan going forward (Kardos et al. 2021). Historically low mountain goat Ne might have resulted in purging of deleterious alleles (Wootton et al. 2023); however this predates the current anthropogenic pressures, including climate change, faced by mountain goats. Whether there is enough standing and additive genetic variation (Bonnet et al. 2022) in mountain goats to cope with future environmental change remains unknown at the moment. Regardless, the accelerated rate of climate change in the alpine (Grabherr et al. 2010; Verrall and Pickering 2020) combined with the extremely low species Ne, is a clear cause for concern.
Conclusion
In terms of quantifying diversity and modelling population structure RADseq is often touted as a viable alternative to WGS data; we suggest, however, that in the current sequencing era, a subset of WGS individuals is preferrable to RADseq if a reference genome assembly is available; even a closely related reference would suffice (Paris et al. 2017; Shafer et al. 2017). For both methodologies sequencing costs and access to computing resources remain a limitation but are becoming more accessible. The primary consideration should now extend beyond demographic inference, as RADseq will be less effective in identifying adaptive loci and runs-of-homozygosity due to the lower proportion of the genome variation being captured. Differences in cost and library preparation, while a decade ago were considerable, are less of an issue today (Lou et al. 2021). In our case study of the population demography of Oreamnos americanus, the remarkably high similarities between data sets are a positive and reassuring finding for researchers undecided on the sequencing strategy; the results, however, paint a worrisome picture for the long-term persistence of mountain goats in the face of continued and rapid anthropogenetic disturbance.
Data availability
The raw DNA sequence data raw data have been deposited in the SRA under accession numbers PRJNA510081, PRJNA909333, PRJNA909334.
References
Aguirre-Liguori JA, Ramírez-Barahona S, Gaut BS (2021) The evolutionary genomics of species’ responses to climate change. Nat Ecol Evol 5:1350–1360. https://doi.org/10.1038/s41559-021-01526-9
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA (2016) Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 17:81–92. https://doi.org/10.1038/nrg.2015.28
Andrews S (2010) Babraham Bioinformatics - FastQC - a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 25 Apr 2017
Beichman AC, Huerta-Sanchez E, Lohmueller KE (2018) Using genomic data to infer historic population dynamics of nonmodel organisms. Annu Rev Ecol Evol Syst 49:433–456. https://doi.org/10.1146/annurev-ecolsys-110617-062431
Benjelloun B, Boyer F, Streeter I, Zamani W, Engelen S, Alberti A, Alberto FJ, BenBati M, Ibnelbachyr M, Chentouf M, Bechchari A, Rezaei HR, Naderi S, Stella A, Chikhi A, Clarke L, Kijas J, Flicek P, Taberlet P, Pompanon F (2019) An evaluation of sequencing coverage and genotyping strategies to assess neutral and adaptive diversity. Mol Ecol Resour 19:1497–1515. https://doi.org/10.1111/1755-0998.13070
Bertola LD, Sunnucks P, Bruford (SANBI) M, Brüniche-Olsen A, Cadena CD, Frankham D, Guayasamin JM, Grueber CE, Hoareau TB, Hoban S, Hohenlohe P, Hunter ME, Kershaw F, Kotze (SANBI) A, Lacy RC, Laikre L, MacDonald AJ, Meek MH, Mergeay J, Mittan C, O´Brien D, Orozco-terWengel P, Ogden R, Páez-Vacas M, Ralls K, Ramakrishnan U, Russo I-RM, Shaw RE, Stow A, Steeves T, Vernesi C, Waits L, Segelbacher G (2021) Genome-wide diversity is informative of extinction risk and critical for conservation: a response to Teixeira and Huber (2021)
Bohling J, Small M, Von Bargen J, Louden A, DeHaan P (2019) Comparing inferences derived from microsatellite and RADseq datasets: a case study involving threatened bull trout. Conserv Genet 20:329–342. https://doi.org/10.1007/s10592-018-1134-z
Bonnet T, Morrissey MB, de Villemereuil P, Alberts SC, Arcese P, Bailey LD, Boutin S, Brekke P, Brent LJN, Camenisch G, Charmantier A, Clutton-Brock TH, Cockburn A, Coltman DW, Courtiol A, Davidian E, Evans SR, Ewen JG, Festa-Bianchet M, de Franceschi C, Gustafsson L, Höner OP, Houslay TM, Keller LF, Manser M, McAdam AG, McLean E, Nietlisbach P, Osmond HL, Pemberton JM, Postma E, Reid JM, Rutschmann A, Santure AW, Sheldon BC, Slate J, Teplitsky C, Visser ME, Wachter B, Kruuk LEB (2022) Genetic variance in fitness indicates rapid contemporary adaptive evolution in wild animals. Science 376:1012–1016. https://doi.org/10.1126/science.abk0853
Brelsford A, Dufresnes C, Perrin N (2016) High-density sex-specific linkage maps of a European tree frog (Hyla arborea) identify the sex chromosome without information on offspring sex. Heredity 116:177–181. https://doi.org/10.1038/hdy.2015.83
Briñoccoli YF, Jardim de Queiroz L, Bogan S, Paracampo A, Posadas PE, Somoza GM, Montoya‐Burgos JI, Cardoso YP (2021) Processes that drive the population structuring of Jenynsia lineata (Cyprinidontiformes, Anablepidae) in the La Plata Basin. Ecol Evol 11:6119–6132. https://doi.org/10.1002/ece3.7427
Browning SR, Browning BL (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81:1084–1097. https://doi.org/10.1086/521987
Bushnell B (2018) BBTools. Available online at: https://jgi.doe.gov/data-and-tools/bbtools/
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124. https://doi.org/10.1111/mec.12354
Catchen JM, Hohenlohe PA, Bernatchez L, Funk WC, Andrews KR, Allendorf FW (2017) Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Mol Ecol Resour 17:362–365. https://doi.org/10.1111/1755-0998.12669
Chikhi L, Sousa VC, Luisi P, Goossens B, Beaumont MA (2010) The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes. Genetics 186:983–995. https://doi.org/10.1534/genetics.110.118661
Côté SD, Festa-Bianchet M (2003) Mountain goat. In: Feldhamer GA, Thompson B, Chapman J (eds) Wild Mammals of North America: Biology, Management, Conservation. The John Hopkins University Press, Baltimore, Maryland
Davey JW, Cezard T, Fuentes‐Utrilla P, Eland C, Gharbi K, Blaxter ML (2013) Special features of RAD Sequencing data: implications for genotyping. Mol Ecol 22:3151–3164. https://doi.org/10.1111/mec.12084
Dedato M, Robert C, Taillon J, Shafer A, Cote S (2022) Demographic history and conservation genomics of caribou (Rangifer tarandus) in Québec. Evol Appl https://doi.org/10.1111/EVA.13495
DeWoody JA, Harder AM, Mathur S, Willoughby JR (2021) The long-standing significance of genetic diversity in conservation. Mol Ecol 30:4147–4154. https://doi.org/10.1111/mec.16051
Earth Resources Observation And Science (EROS) Center (2017) Global 30 Arc-Second Elevation (GTOPO30) data. https://doi.org/10.5066/F7DF6PQS
Fick SE, Hijmans RJ (2017) WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int J Climatol 37:4302–4315. https://doi.org/10.1002/joc.5086
Fisher RA (1930) The genetical theory of natural selection. Clarendon Press, Oxford, England
Gamble T, Coryell J, Ezaz T, Lynch J, Scantlebury DP, Zarkower D (2015) Restriction site-associated DNA sequencing (RAD-seq) reveals an extraordinary number of transitions among gecko sex-determining systems. Mol Biol Evol 32:1296–1309. https://doi.org/10.1093/molbev/msv023
Gautier M, Gharbi K, Cezard T, Foucaud J, Kerdelhué C, Pudlo P, Cornuet J-M, Estoup A (2013) The effect of RAD allele dropout on the estimation of genetic variation within and between populations. Mol Ecol 22:3165–3178. https://doi.org/10.1111/mec.12089
Gervais L, Perrier C, Bernard M, Merlet J, Pemberton JM, Pujol B, Quéméré E (2019) RAD-sequencing for estimating genomic relatedness matrix-based heritability in the wild: a case study in roe deer. Mol Ecol Resour 19:1205–1217. https://doi.org/10.1111/1755-0998.13031
Gower G, Tuke J, Rohrlach AB, Soubrier J, Llamas B, Bean N, Cooper A (2018) Population size history from short genomic scaffolds: how short is too short? 382036
Grabherr G, Gottfried M, Pauli H (2010) Climate change impacts in alpine environments. Geogr Compass 4:1133–1153. https://doi.org/10.1111/j.1749-8198.2010.00356.x
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD (2009) Inferring the joint demographic History of multiple populations from multidimensional SNP frequency data. PLOS Genet 5:e1000695. https://doi.org/10.1371/journal.pgen.1000695
Hare MP, Nunney L, Schwartz MK, Ruzzante DE, Burford M, Waples RS, Ruegg K, Palstra F (2011) Understanding and estimating effective population size for practical application in marine species management. Conserv Biol J Soc 25:438–449. https://doi.org/10.1111/j.1523-1739.2010.01637.x
Harvey MG, Smith BT, Glenn TC, Faircloth BC, Brumfield RT (2016) Sequence capture versus restriction site associated DNA sequencing for shallow systematics. Syst Biol 65:910–924. https://doi.org/10.1093/sysbio/syw036
Heller R, Nursyifa C, Garcia-Erill G, Salmona J, Chikhi L, Meisner J, Korneliussen TS, Albrechtsen A (2021) A reference-free approach to analyse RADseq data using standard next generation sequencing toolkits. Mol Ecol Resour 21(4):1085–1097. https://doi.org/10.1111/1755-0998.13324
Hu J-Y, Hao Z-Q, Frantz L, Wu S-F, Chen W, Jiang Y-F, Wu H, Kuang W-M, Li H, Zhang Y-P, Yu L (2020) Genomic consequences of population decline in critically endangered pangolins and their demographic histories. Natl Sci Rev 7:798–814. https://doi.org/10.1093/nsr/nwaa031
Kardos M, Armstrong EE, Fitzpatrick SW, Hauser S, Hedrick PW, Miller JM, Tallmon DA, Funk WC (2021) The crucial role of genome-wide genetic variation in conservation. Proc Natl Acad Sci. 118. https://doi.org/10.1073/pnas.2104642118
Kingman JFC (1982) The coalescent. Stoch Process Their Appl 13:235–248. https://doi.org/10.1016/0304-4149(82)90011-4
Kjeldsen SR, Zenger KR, Leigh K, Ellis W, Tobey J, Phalen D, Melzer A, FitzGibbon S, Raadsma HW (2016) Genome-wide SNP loci reveal novel insights into koala (Phascolarctos cinereus) population variability across its range. Conserv Genet 17:337–353. https://doi.org/10.1007/s10592-015-0784-3
Knaus BJ, Grünwald NJ (2017) vcfr: a package to manipulate and visualize variant call format data in R. Mol Ecol Resour 17:44–53. https://doi.org/10.1111/1755-0998.12549
Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD: analysis of next generation sequencing data. BMC Bioinforma 15:356. https://doi.org/10.1186/s12859-014-0356-4
Korneliussen TS, Moltke I, Albrechtsen A, Nielsen R (2013) Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinforma 14:289. https://doi.org/10.1186/1471-2105-14-289
Kumar S, Subramanian S (2002) Mutation rates in mammalian genomes. Proc Natl Acad Sci 99:803–808. https://doi.org/10.1073/pnas.022629899
Legendre P, Legendre L (2012) Chapter 11 - Canonical analysis. In: Legendre P, Legendre L (eds) Developments in Environmental Modelling. Elsevier, pp 625–710
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv E-Prints 1303:arXiv:1303.3997
Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature 475:493–496. https://doi.org/10.1038/nature10231
Liu S, Hansen MM (2017) PSMC (pairwise sequentially Markovian coalescent) analysis of RAD (restriction site associated DNA) sequencing data. Mol Ecol Resour 17:631–641. https://doi.org/10.1111/1755-0998.12606
Lou RN, Jacobs A, Wilder AP, Therkildsen NO (2021) A beginner’s guide to low-coverage whole genome sequencing for population genomics. Mol Ecol 30:5966–5993. https://doi.org/10.1111/mec.16077
Lowry DB, Hoban S, Kelley JL, Lotterhos KE, Reed LK, Antolin MF, Storfer A (2017) Breaking RAD: an evaluation of the utility of restriction site-associated DNA sequencing for genome scans of adaptation. Mol Ecol Resour 17:142–152. https://doi.org/10.1111/1755-0998.12635
MacLeod IM, Larkin DM, Lewin HA, Hayes BJ, Goddard ME (2013) Inferring demography from runs of homozygosity in whole-genome sequence, with correction for sequence errors. Mol Biol Evol 30:2209–2223. https://doi.org/10.1093/molbev/mst125
Martchenko D, Chikhi R, Shafer ABA (2020) Genome assembly and analysis of the North American mountain goat (Oreamnos americanus) reveals species-level responses to extreme environments. G3 Genes Genomes Genet 10:437–442. https://doi.org/10.1534/g3.119.400747
Martchenko D, White KS, Shafer ABA (2022) Long-term data reveal effects of climate, road access, and latitude on mountain goat horn size. J Wildl Manag 86:e22195. https://doi.org/10.1002/jwmg.22195
Martin M, Patterson M, Garg S, Fischer OS, Pisanti N, Klau GW, Schöenhuth A, Marschall T (2016) WhatsHap: fast and accurate read-based phasing. bioRxiv 085050. https://doi.org/10.1101/085050
Meisner J, Albrechtsen A (2018) Inferring population structure and admixture proportions in low-depth NGS data. Genetics 210:719–731. https://doi.org/10.1534/genetics.118.301336
Nadachowska-Brzyska K, Burri R, Smeds L, Ellegren H (2016) PSMC analysis of effective population sizes in molecular ecology and its application to black-and-white Ficedula flycatchers. Mol Ecol 25:1058–1072. https://doi.org/10.1111/mec.13540
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens HH, Szoecs E, Wagner HH (2020) vegan: Community Ecology Package. R package version 2.5-7
Papadopoulou A, Knowles LL (2015) Genomic tests of the species-pump hypothesis: Recent island connectivity cycles drive population divergence but not speciation in Caribbean crickets across the Virgin Islands. Evolution 69:1501–1517. https://doi.org/10.1111/evo.12667
Paris JR, Stevens JR, Catchen JM (2017) Lost in parameter space: a road map for stacks. Methods Ecol Evol 8:1360–1373. https://doi.org/10.1111/2041-210X.12775
Peart CR, Tusso S, Pophaly SD et al. (2020) Determinants of genetic variation across eco-evolutionary scales in pinnipeds. Nat Ecol Evol 4:1095–1104. https://doi.org/10.1038/s41559-020-1215-5
Poissant J, Shafer ABA, Davis CS, Mainguy J, Hogg JT, Côté S, Coltman DW (2009) Genome-wide cross-amplification of domestic sheep microsatellites in bighorn sheep and mountain goats. Mol Ecol Resour 9:1121–1126. https://doi.org/10.1111/j.1755-0998.2009.02575.x
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100. https://doi.org/10.1093/oxfordjournals.molbev.a004034
Salmona J, Heller R, Lascoux M, Shafer A (2019) Inferring Demographic History Using Genomic Data. In: Rajora OP (ed) Population Genomics: Concepts, Approaches and Applications. Springer International Publishing, Cham, pp 511–537
Sambrook J, Russell DW (2006) Purification of nucleic acids by extraction with Phenol:chloroform. Cold Spring Harb Protoc https://doi.org/10.1101/pdb.prot4455
Santiago E, Novo I, Pardiñas AF, Saura M, Wang J, Caballero A (2020) Recent Demographic History inferred by high-resolution analysis of linkage disequilibrium. Mol Biol Evol 37:3642–3653. https://doi.org/10.1093/molbev/msaa169
Schiffels S, Wang K (2020) MSMC and MSMC2: The Multiple Sequentially Markovian Coalescent. In: Dutheil JY (ed) Statistical Population Genomics. Springer US, New York, NY, pp 147–166
Shafer ABA, Côté SD, Coltman DW (2011) Hot spots of genetic diversity descended from multiple Pleistocene refugia in an alpine ungulate. Evol Int J Org Evol 65:125–138. https://doi.org/10.1111/j.1558-5646.2010.01109.x
Shafer ABA, Fan CW, Côté SD, Coltman DW (2012) (Lack of) genetic diversity in immune genes predates glacial isolation in the North American mountain goat (Oreamnos americanus). J Hered 103:371–379. https://doi.org/10.1093/jhered/esr138
Shafer ABA, Gattepaille LM, Stewart REA, Wolf JBW (2015) Demographic inferences using short-read genomic data in an approximate Bayesian computation framework: in silico evaluation of power, biases and proof of concept in Atlantic walrus. Mol Ecol 24:328–345. https://doi.org/10.1111/mec.13034
Shafer ABA, Peart CR, Tusso S, Maayan I, Brelsford A, Wheat CW, Wolf JBW (2017) Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol Evol 8:907–917. https://doi.org/10.1111/2041-210X.12700
Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26:1135–1145. https://doi.org/10.1038/nbt1486
Sin SYW, Hoover B, Nevitt G, Edwards SV (2021) Demographic history, not mating system, explains signatures of inbreeding and inbreeding depression in a large outbred population. Am Nat. https://doi.org/10.1086/714079
Skotte L, Korneliussen TS, Albrechtsen A (2013) Estimating individual admixture proportions from next generation sequencing data. Genetics 195:693–702. https://doi.org/10.1534/genetics.113.154138
Subramanian S (2016) The effects of sample size on population genomic analyses – implications for the tests of neutrality. BMC Genom 17:123. https://doi.org/10.1186/s12864-016-2441-8
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics 31:2032–2034. https://doi.org/10.1093/bioinformatics/btv098
Teixeira JC, Huber CD (2021) The inflated significance of neutral genetic diversity in conservation genetics. Proc Natl Acad Sci 118. https://doi.org/10.1073/pnas.2015096118
Verrall B, Pickering CM (2020) Alpine vegetation in the context of climate change: a global review of past research and future directions. Sci Total Environ 748:141344. https://doi.org/10.1016/j.scitotenv.2020.141344
Vijay N, Bossu CM, Poelstra JW, Weissensteiner MH, Suh A, Kryukov AP, Wolf JBW (2016) Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat Commun 7:13195. https://doi.org/10.1038/ncomms13195
Wang K, Mathieson I, O’Connell J, Schiffels S (2020) Tracking human population structure through time from whole genome sequences. PLOS Genet 16:e1008552. https://doi.org/10.1371/journal.pgen.1008552
Waples RK, Larson WA, Waples RS (2016) Estimating contemporary effective population size in non-model species using linkage disequilibrium across thousands of loci. Heredity 117:233–240. https://doi.org/10.1038/hdy.2016.60
Warmuth VM, Ellegren H (2019) Genotype-free estimation of allele frequencies reduces bias and improves demographic inference from RADSeq data. Mol Ecol Resour 19:586–596. https://doi.org/10.1111/1755-0998.12990
Wootton E, Robert C, Taillon J, Côté S, Shafer ABA (2023) Genomic health is dependent on long-term population demographic history. Mol Ecol 32:1943–1954. https://doi.org/10.1111/mec.16863
Wright S (1943) Isolation by distance. Genetics 28:114–138
Zhao S, Zheng P, Dong S, Zhan X, Wu Q, Guo X, Hu Y, He W, Zhang S, Fan W, Zhu L, Li D, Zhang X, Chen Q, Zhang H, Zhang Z, Jin X, Zhang J, Yang H, Wang J, Wang J, Wei F (2013) Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet 45:67–71. https://doi.org/10.1038/ng.2494
Acknowledgements
We thank Marty Kardos, Joanna R. Freeland, and Paul J. Wilson for comments on earlier versions of this manuscript.
Funding
This work was supported by a Natural Sciences and Engineering Research Council Discovery Grant, Canada Foundation for Innovation: John R. Evans Leaders Fund, and Compute Canada Awards to ABAS; Earth Rangers Bring Back the Wild grant and WCS Canada W. Garfield Weston Foundation 2017 and 2018 Fellowships for Northern Conservation to DM. DM was supported by the Ontario Graduate Scholarship. The samples were provided by Alaska Department of Fish and Game, BC Ministry of Forests, Lands, and Natural Resource Operations, BC Mountain Goat Conservation Society, Environment Yukon, Montana Fish, Wildlife and Parks, Alberta Fish and Wildlife and Washington Department of Fish and Wildlife.
Author information
Authors and Affiliations
Contributions
ABAS conceived the study. DM and ABAS analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Giorgio Bertorelle.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Martchenko, D., Shafer, A.B.A. Contrasting whole-genome and reduced representation sequencing for population demographic and adaptive inference: an alpine mammal case study. Heredity 131, 273–281 (2023). https://doi.org/10.1038/s41437-023-00643-4
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41437-023-00643-4
This article is cited by
-
Sharing approaches in predictive genomics across animals, plants and humans
Nature Genetics (2026)
-
Genomic insights into the population structure and adaptive variation of Mullus barbatus in the Mediterranean Sea
BMC Ecology and Evolution (2025)
-
Island demographics and trait associations in white-tailed deer
Heredity (2024)
-
Next-generation data filtering in the genomics era
Nature Reviews Genetics (2024)
