
Abstract
Hypertension induced by hypoxia at high altitude is one of the typical symptoms of high-altitude reactions (HARs). Emerging evidence indicates that endothelial abnormalities, including increases in angiotensin-2 (Ang-2) and endothelin-1 (ET-1), are closely associated with hypertension. Thus, low blood oxygen-induced endothelial dysfunction through acceleration of Ang-2 and ET-1 synthesis may alleviate HARs. In this study, we investigated the effects of hypoxia on rat blood pressure (BP) and endothelial injury. We found that BP increased by 10 mmHg after treatment with 10% O2 (~5500 m above sea level) for 24 h. Consistently, serum Ang-2 and ET-1 levels were increased along with decreases in NO levels. In endothelial cells, angiotensin-1-converting enzyme (ACE) and ET-1 expression levels were upregulated. Interestingly, nuclear respiratory factor 1 (NRF1) levels were also upregulated, consistent with the changes in ACE and ET-1 levels. We further demonstrated that NRF1 transcriptionally activated ACE and ET-1 by directly binding to their promoter regions, suggesting that the endothelial cell dysfunction induced by hypoxia was due to NRF1-dependent upregulation of ACE and ET-1. Surprisingly, testosterone supplementation showed significant protective effects on BP, while castration induced even higher BPs in rats exposed to hypoxia. We further showed that physiological testosterone repressed NRF1 expression in vivo and in vitro and thereby reduced Ang-2 and ET-1 levels, which was dependent on hypoxia. In summary, we have identified that physiological testosterone protects against hypoxia-induced hypertension through inhibition of NRF1, which transcriptionally regulates ACE and ET-1 expression.
Similar content being viewed by others

Introduction
Hypoxia severely affects physiological activities, resulting in memory damage and sensory and motor dysfunction [1]. Systemic hypoxia is associated with conditions such as sleep disorders, asphyxia, lung diseases, anemia, and high-altitude exposure. Physiological responses to acute, chronic, and intermittent normobaric and hypobaric hypoxia have been widely studied in recent years, including blood pressure (BP) regulation, erythropoiesis changes, and cardiopulmonary regulation. Exposure to hypobaric hypoxia is accompanied by increased heart rate and ventilation, decreased oxygen saturation, and activation of angiogenesis and the endothelium [2]. Acute high-altitude exposure causes an increase in BP during the initial days [3,4,5]. The underlying mechanisms include activation of the adrenergic system, increased arterial stiffness, and endothelial dysfunction [6,7,8].
As the first barrier between the vasculature and the blood, vascular endothelial cells respond to blood oxygen concentrations, thus playing a key role in BP regulation [9, 10]. Vascular endothelial cells also form the largest secretory gland and produce bioactive substances through endocrine, paracrine, or autocrine mechanisms, including vasoconstrictor endothelin-1 (ET-1), angiotensin-2 (Ang-2), and nitric oxide (NO), to regulate BP, affect smooth muscle cell proliferation, and repair vascular inflammation [11]. It has been reported that pathological hypoxia induced by inflammation affects tight junctions through cytoskeletal rearrangement and decreased cell adhesion [12]. Acute hypoxia rapidly activates endothelial cells, leading to increased vascular permeability and resulting in endothelium-dependent vasoconstriction via the release of endothelium-derived substances, including ET-1 and Ang-2, and decreases in NO bioavailability [13].
The known mechanism of hypoxia-induced endothelial dysfunction is related to hypoxia-inducible factors (HIFs; HIF-1 and HIF-2), which respond to low oxygen levels and upregulate a number of genes involved in angiogenesis, energy metabolism, and cell survival. Severe hypoxia or prolonged exposure to mild hypoxia induces high-oxidative-stress-related cell damage and eventually leads to apoptosis through strong increases in HIF-1α and accumulation of DNA lesions [14]. NF-κB, a major transcription factor responding to cellular stress, is activated by hypoxia in endothelial cells, inducing the release of proinflammatory factors and leading to the recruitment of neutrophils and the promotion of neutrophil adherence [15].
Nuclear respiratory factor 1 (NRF1), a transcription factor, regulates a series of substances involved in mitochondrial biogenesis and oxidative phosphorylation [16,17,18,19]. In addition to its housekeeping function in mitochondrial homeostasis, NRF1 theoretically binds to 2470 genes that are related not only to mitochondria but also to RNA metabolism, splicing, the cell cycle, DNA damage repair, protein translation initiation, and ubiquitin protein degradation [20]. Furthermore, our past studies have demonstrated that NRF1 acts as a master regulator of the hypoxia response through transcriptional regulation of HIF-1α [21] and affects the endocrine system through steroidogenic acute regulatory protein [22], indicating that NRF1 is involved in the regulation of various cell functions.
A main male sex hormone relaxes blood vessels, inhibits the proliferation of vascular smooth muscle, and reduces BP [23, 24]. Reductions in testosterone reduce the activity of renin–angiotensin, increase vascular resistance, and increase BP [25, 26]. Testosterone also reduces platelet endothelial cell adhesion molecule-1 (PECAM-1) levels in endothelial cells, reduces the aggregation of inflammatory cells in the arterial wall, and promotes the release of NO in endothelial cells [27, 28]. Emerging evidence indicates that decreases in testosterone levels are caused by high-altitude exposure [29].
Here, we present evidence that high-altitude-induced hypotension is related to a reduction in testosterone. Removal of testosterone exacerbated the increase in BP induced by hypoxia, while testosterone supplementation effectively stabilized BP. The protective effect of physiological testosterone was due to inhibition of NRF1, which was upregulated by hypoxia. The increase in NRF1 under hypoxia accelerated the release of ET-1 and Ang-2 and reduced NO activity through transcriptional regulation of ET-1 and angiotensin-1-converting enzyme (ACE).
Materials and methods
Animals and treatments
Adult Wistar rats weighing 180–200 g were provided by the Experimental Animal Center of Nantong University and were allowed to acclimate for 1 week before experiments with a 12 h light–dark cycle.
BP measurement
Beginning 3 weeks before the experiments, the BPs of the rats were repeatedly tested at the tail artery every day for habituation. Briefly, the rat tails were warmed up at 37 °C for 30 min. Then, the rat tails were held on a rat tail artery BP detector (RM6240bd, Chengdu Instrument Factory, China) for 1 min for BP recording. The average BP of the last 10 days was taken as the baseline BP.
Animal surgery
Rats were anesthetized with 50 mg/kg b.w. ketamine and 10 mg/kg b.w. xylazine by intraperitoneal injection. In the sham group, the skin of the testes was cut and then sutured. In the GD group, the rats were subjected to gonadectomy. In the GD + T group, the rats underwent gonadectomy and were given additional testosterone propionate (1250 μg/kg b.w.) through subcutaneous injection.
Hypoxia treatment
Rats were incubated with 10% O2 in a hypoxic chamber for 24 h. The same number of rats were placed in another chamber with normal air as controls.
Serum hormones and NO assays
Blood samples were collected from rat hearts after the rats were euthanized. Plasma samples were collected and diluted five times. The test procedures were carried out with a Testosterone Parameter Assay Kit (KGE010, R&D, MN, USA), a Corticosterone Parameter Assay Kit (KGE009, R&D), an Aldosterone Parameter Assay Kit (KGE016, R&D), and a Total Nitric Oxide and Nitrate/Nitrite Parameter Assay Kit (KGE001, R&D) following the manufacturer’s instructions.
Aortas were collected and cut into small pieces. All the studies reported here were submitted to the Ethics Committee on Animal Experimentation of Nantong University, and all procedures were approved by the Animal Care and Use Committee of Nantong University (Approval ID: S20200315-011) and the Jiangsu Province Animal Care Ethics Committee (Approval ID: SYXK(SU)2007-0021).
Cell culture and treatments
Human umbilical vein endothelial cell (HUVEC) culture
Cells were purchased from ScienCell Research Laboratories. The cells were cultured in endothelial cell medium (1001, ScienCell, CA, USA) containing 2.5% fetal bovine serum (0025, ScienCell), 0.1% endothelial cell growth supplement (1052, ScienCell), and 0.1% penicillin/streptomycin (0503, ScienCell). HUVECs were identified using an anti-Von Willebrand factor antibody (Ab6994, Abcam, USA) and an anti-PECAM-1 antibody (ab24590, Abcam) by immunofluorescence (Supplementary Fig. 2). The HUVECs were transfected with NRF1 siRNA using Lipofectamine 2000 (Thermo Fisher, USA) for 72 h. For hypoxia treatments, cells were cultured in 1% O2 and 5% CO2 at 37 °C in hypoxia-equilibrated medium for 24 h.
Transfection
HUVECs were seeded in six-well plates in antibiotic-free medium 1 day before transfection. NRF1 siRNA (5′-CACAUUGGCUGAUGCUUCAUU-3′) was transfected into the HUVECs using Lipofectamine 2000 (11668019, Thermo Fisher) according to the provided instructions.
Western blot analysis
Tissue and cells were lysed with RIPA buffer, and the protein concentration was calculated by bicinchoninic acid assay. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. Then, the membranes were blocked with 5% nonfat dry milk for 1 h and incubated with anti-HIF-1α (AF1935, R&D), anti-ACE (MAB4051, Sigma, USA), anti-ET-1 (ab2786, Abcam), anti-NRF1 (ab175932, Abcam), and anti-Histone 3 (ab2786, Abcam) antibodies overnight at 4 °C. Binding of the primary antibodies was visualized with HRP-conjugated donkey anti-rabbit (305-005-003, Jackson, USA), goat anti-rat (31470, Thermo Fisher), and rabbit anti-goat (111-005-003, Jackson) antibodies and an ECL-Plus system. Grayscale analysis was carried out using ImageJ (https://imagej.nih.gov/ij/).
qRT-PCR
Tissue total RNA was isolated by using the Column Animal RNAOUT kit (Tiandz, Beijing, China) according to the manufacturer’s instructions. Cellular total RNA was isolated with TRIzol reagent. The purified total RNA was then reverse-transcribed with a HiScript 1st Strand cDNA Synthesis Kit (Vazyme, Jiangsu, China). qRT-PCR was performed with SYBR premix (Roche, USA) with the following procedure: 98 °C for 4 min followed by 40 three-step cycles of 98 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. The relative amount of gene expression normalized to the reference gene was calculated using the ΔΔCt method, where Ct is the threshold cycle in PCR.
Immunohistofluorescence
HUVECs were fixed with 4% paraformaldehyde for 15 min at room temperature. Tissue was fixed with 4% paraformaldehyde overnight, incubated with 30% sucrose for 3 days, and sectioned at 10 μm. The samples were permeabilized with Triton (0.1% in PBS) for 30 min and blocked with 10% BSA for 1 h. The samples were then incubated with anti-ACE (MAB4051, Sigma, USA), anti-ET-1 (ab2786, Abcam), and anti-NRF1 (ab175932, Abcam) primary antibodies. Binding of the primary antibodies was visualized via incubation with Alexa Fluor 488-conjugated anti-mouse IgG (A21202, Thermo Fisher), Alexa Fluor 488-conjugated anti-rat IgG (A-21208, Thermo Fisher), and Alexa Fluor 555-conjugated anti-rabbit IgG (A-32794, Thermo Fisher) for 2 h. Finally, 5 μg/μL DAPI was used for nuclear counterstaining. Images were acquired using a Leica SP8 confocal microscope.
ChIP assay
ChIP experiments were performed using a SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) (#9003, CST, USA) according to the manufacturer’s instructions. HUVECs were fixed in culture medium containing 1% formaldehyde at room temperature for 10 min to crosslink proteins and DNA. Then, the cells were lysed and incubated with 0.5 µL of micrococcal nuclease for 30 min at 37 °C to digest DNA into fragments of 200–500 bp. The mixture was then immunoprecipitated with 2.5 μg of anti-NRF1 antibody (ab34682, Abcam) or a negative control IgG at 4 °C overnight. The purified DNA was amplified by qRT-PCR with primers designed to target the following regions: the Ace promoter (forward: 5′-CTGGGAAGATCGAGCCGGAG-3′, reverse: 5′-CGGTGCGCGGTGCTC-3′), the Edn1 promoter (forward: 5′-CTTTTTCTTAGCCCTGCCCC-3′, reverse: 5′-GTGGAGAGCTGGGACTGC-3′), and the Cyt c promoter (forward: 5′- TTCCTGTCCGACTGTGGTGT-3′, reverse: 5′-GGCGGTCTTGTAGTTCTTGATT-3′).
Dual-Luciferase reporter assays
Fragments of the human Ace promoter region (−348/+20) and Edn1 promoter region (−221/+63) were cloned into the pGL3-Basic vector (Promega, Madison, WI, USA) between the KpnI and HindIII restriction sites. HEK293T cells (4 × 104) were cultured in 24-well plates in antibiotic-free culture medium 1 day before transfection. The cells were cotransfected with 250 ng of reporter plasmids, 250 ng of NRF1 overexpression (NRF1 OE) construct or siRNA (siNRF1), and 10 ng of Renilla reporter plasmid (pRL-TK, Promega) as an internal control with Lipofectamine 2000 reagent. The cells were treated with 1% O2 for 24 h after 48 h of transfection. Finally, the transcriptional activity levels were detected with a dual-luciferase reporter assay system. The firefly luminescence signal was normalized to the Renilla luminescence signal.
Statistical analysis
The data were analyzed with GraphPad Prism v. 5 (GraphPad) by Student’s t test, one-way ANOVA, or two-way ANOVA. All the data are presented as the mean ± SEM or the mean ± SD; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Results
Hypoxia-induced upregulation of BP and contractile protein levels along with NRF1 expression in rats
To verify the effects of hypoxia on BP, we treated male Wistar rats with 10% O2 for 24 h. BP was measured at 12 and 24 h from the rat tail artery. The changes in BP were obtained by subtraction of the baseline from the test values. As shown in Fig. 1A, there was no obvious change in BP in the normoxia group at either 12 or 24 h. However, the mean BP rose by 10 and 15 mmHg at 12 and 24 h in hypoxic rats, respectively, showing that hypoxia directly induced BP upregulation. Consistent with the BP results, serum Ang-2 was significantly upregulated, while the NO concentration was reduced after hypoxia treatment (Fig. 1B, C). The serum testosterone level was significantly reduced in the hypoxia group (Fig. 1D), which was consistent with the findings of our previous studies [22, 29, 30]. Serum corticosterone and aldosterone levels were not significantly different between the two groups (Supplementary Fig. 1), indicating that hypoxia-induced high BP was independent on the renin–angiotensin system. Since Ang-2 and NO are secreted mainly by endothelial cells, we hypothesized that the hypoxia-induced high BP was caused by changes in endothelial function. We measured the protein levels of ACE and ET-1, which are the most important contractile proteins in the endothelium. As shown in Fig. 1E, F, the levels of both ACE and ET-1 were significantly increased after hypoxia treatment. Furthermore, the levels of both ET-1 and ACE were also increased in the endothelium in situ (Fig. 1H, I), confirming that hypoxia upregulated contractile protein levels in the endothelium, helping to elevate BP. To further investigate the effect of hypoxia on endothelial function, we also detected the expression of contractile factors along with other factors related to the hypoxia response. The results in Fig. 1G show that Edn1, Ace, and Vegfa were significantly upregulated in the hypoxia group, consistent with their protein changes. Interestingly, we found that the transcription factor NRF1 was also upregulated (Fig. 1E, F) and aggregated in the nucleus (Fig. 1H–J), suggesting that there might be a correlation between NRF1 and the contractile proteins ACE and ET-1.

Hypoxia treatments upregulated BP and contractile protein levels in rats. Wistar rats were treated with 10% O2 for 24 h. A BP was measured from the rat tail artery. The baseline pressure of each animal was the average value from 10 days of repeated determination. The final values were obtained by subtraction of the baseline pressure (n = 8, mean ± SEM). After 24 h of hypoxia treatment, serum Ang-2 (B), NO (C), and testosterone (D) levels were measured by ELISA (n = 8, mean ± SEM, **p ≤ 0.01 by Student’s t test). Protein and total RNA from rat aortic tissue were isolated for Western blot (E, F) and qRT-PCR (G) analyses (n = 8, mean ± SEM, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Student’s t test). Rat aortic tissue was fixed and stained with anti-NRF1/ACE (H) and anti-NRF1/ET-1 (I) antibodies. The protein levels of NRF1 (J), ACE (K), and ET-1 (L) were quantified from the mean intensities by ImageJ (n = 5, mean ± SEM, *p ≤ 0.05, **p ≤ 0.01 by Student’s t test). Nor normoxia, Hyp hypoxia
Hypoxia promoted synchronous changes in NRF1, ACE, and ET-1 in HUVECs
To further confirm the effects of hypoxia on endothelial cells, we cultured HUVECs in 1% O2 for 0, 12, 24 and 48 h. As shown in Fig. 2A, B, the expression of HIF-1 and its target gene VEGFA was significantly increased in hypoxia-treated cells, confirming the hypoxia response of HUVECs. We also found significant upregulation of ACE and ET-1 at both the mRNA and protein levels after hypoxia treatment. Consistent with the in vivo results, NRF1 was also upregulated in hypoxia-treated cells. Furthermore, the immunofluorescence results showed that NRF1 always had a trend similar to those of ACE and ET-1 (Fig. 2C, D), further confirming the correlations among the changes in these three proteins.

Hypoxia exposure upregulated contractile proteins in HUVECs. HUVECs were cultured in 1% O2 pre-equilibrated medium for the indicated duration at 37 °C and 1% O2. A The protein levels of ACE, HIF-1α, NRF1, and ET-1 were measured using western blot (mean ± SD, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by one-way ANOVA). B After treatment with 1% O2 for 24 h, the expression levels of VEGFA, NRF1, ACE, and EDN1 in HUVECs were detected by qRT-PCR (mean ± SD, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Student’s t test). C, D After treatment with 1% O2 for 24 h, HUVECs were fixed and stained with anti-NRF1/ACE or anti-NRF1/ET-1 antibodies. The total intensity of each protein in single cells was quantified using ImageJ. At least 25 cells were used for quantification in each treatment group (mean ± SD, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Student’s t test). Nor normoxia, Hyp hypoxia
Interference with NRF1 downregulated ACE and ET-1 independently of O2
To verify the relationship between NRF1 and ACE/ET-1, we interfered with NRF1 with 70% efficiency under both normoxic and hypoxic conditions (Fig. 3A, B). Then, we determined the mRNA levels of ACE and ET-1 using qRT-PCR. As shown in Fig. 3C, D, the expression of ACE and EDN1 was downregulated when NRF1 was knocked down independent of O2 concentration. We further determined the levels of ACE and ET-1 in situ. The results showed downregulation of both ACE and ET-1 in NRF1-silenced cells (Fig. 3E, F) under both normoxia and hypoxia. All these results indicated that the NRF1-promoted ACE and ET-1 transcription was independent of O2. The upregulation of ACE and ET-1 was due to the upregulation of NRF1 under hypoxic conditions. Furthermore, we have found that NRF1 transcriptionally regulates p65 and thus participates in inflammatory responses in L132 cells [31]. Here, we found a similar regulatory effect of NRF1 on p65 in HUVECs (Supplementary Fig. 3), showing that NRF1 plays a broad role in the regulation of endothelial function.

Interference with NRF1 downregulated ACE and ET-1 under both hypoxia and normoxia. HUVECs were transfected with NRF1 siRNA and cultured for 48 h. After hypoxia (1% O2, 24 h) or normoxia (20% O2, 24 h) treatments, interference efficiency was detected using western blot analysis (A, B). C, D The expression of NRF1, ACE, and ET-1 was measured through qRT-PCR (mean ± SD, *p ≤ 0.05, **p ≤ 0.01 by Student’s t test). E, F Cells were fixed and stained with anti-NRF1/ACE or anti-NRF1/ET-1 antibodies. The total intensity of each protein in single cells was quantified using ImageJ. At least 30 cells were quantified in each treatment group (mean ± SD, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Student’s t test)
NRF1 transcriptionally regulated ACE and EDN1 by binding to their promoter regions
To observe the direct interaction of NRF1 and ACE with the EDN1 promoter region, we performed ChIP experiments. The positive results showed that NRF1 was enriched in the promoter regions of ACE (−348/+20) and EDN1 (−221/+63), with high enrichment under both normoxia and hypoxia (Fig. 4A). To evaluate the transcriptional regulation of NRF1, we used a luciferase assay to detect the transcriptional activity levels of the ACE and EDN1 promoters under NRF1 regulation. The luciferase assay results showed that upregulation of NRF1 significantly increased the relative luciferase activity levels of both ACE (−348/+20) and EDN1 (−221/+63) in HEK293T cells (Fig. 4B), while downregulation of NRF1 significantly decreased the relative luciferase activity levels (Fig. 4C), indicating that NRF1 transcriptionally activates the target genes ACE and EDN1. Furthermore, we determined the transcriptional effects of NRF1 on ACE and EDN1 under hypoxia. The results in Fig. 4D show similar changes in ACE and EDN1 transcriptional activity, indicating that the regulatory effects of NRF1 on ACE and ET-1 were independent of O2.
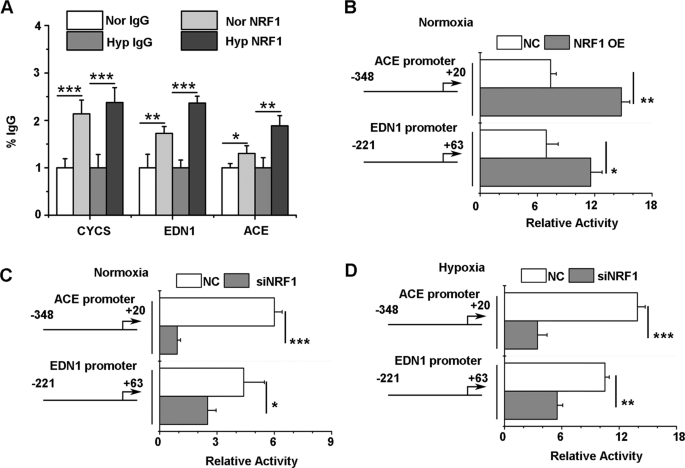
NRF1 bound to the Ace and Edn1 promoter regions and activated gene expression. A HUVECs were cultured in 1% O2 pre-equilibrated medium for the indicated duration at 37 °C under 1% O2. To identify the interaction between NRF1 and the Edn1 or Ace promoter, cells were fixed and lysed for ChIP experiments. Enriched DNA fragments were amplified by real-time PCR (mean ± SD, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Student’s t test). B–D A luciferase reporter gene assay was carried out after cotransfection of an ACE (−348/+20 Luc) or EDN1 (−221/+63 Luc) plasmid and the pRL-TK vector to normalize the transfection efficiencies. To interfere with NRF1 levels, HEK 293T cells were cotransfected with NRF1 overexpression plasmids (NRF1 OE, B) under normoxia or with NRF1 siRNA (siNRF1) under normoxia (C) or hypoxia (D). The results were normalized with the luciferase/Renilla ratio (mean ± SD, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Student’s t test). Nor normoxia, Hyp hypoxia
Testosterone suppressed hypoxia-induced hypertension through inhibition of NRF1
To investigate the function of testosterone in BP regulation under hypoxic conditions, gonadectomy was performed in rats on day 1. Then, at day 5 and day 12, the castrated rats were subcutaneously injected with testosterone propionate (1250 μg/kg b.w.). Finally, the rats were treated with 8% O2 for 24 h (Fig. 5A). As shown in Fig. 5B, serum testosterone in the GD group was significantly lower than that in the sham group, while testosterone levels were raised to physiological levels in the GD + T group. Interestingly, the increase in BP was related to the level of testosterone, as shown in Fig. 5C. BP increased by 10 mmHg in the GD group but only by 5 mmHg in the sham and GD + T groups. These results indicated that the level of testosterone was negatively correlated with the increase in BP; thus, physiological testosterone might have a protective effect against increase in BP. We further stained NRF1 and ACE in vascular endothelial tissue. The results in Fig. 5D, E show that both NRF1 and ACE were upregulated in the GD group compared to the sham group. However, in the GD + T group, these two proteins returned to the sham group level, suggesting that physiological testosterone prevented the increase in BP through inhibition of NRF1. We also tested the expression levels of Ace, Edn1, and Nrf1 through qRT-PCR. Consistent with the immunofluorescence results, these three genes were upregulated in the GD group and returned to sham levels in the GD + T group (Fig. 5F).

Testosterone replacement therapy attenuated the high BP induced by hypoxia. A Male rats were castrated on day 1. Gonadectomized rats were injected subcutaneously with testosterone propionate (20 mg/kg b.w.) on day 7 and day 12. On day 15, the rats were treated with 10% O2 for 24 h. B Serum testosterone levels were measured by ELISA (n = 7, mean ± SEM, *p ≤ 0.05 by two-way ANOVA). C BP was measured from the rat tail artery. The baseline BP was the average value from 10 days of repeated testing. The final values were obtained by subtraction of the baseline from the test values (n = 7, mean ± SEM). D Rat aortic tissue was fixed and stained with anti-NRF1 and anti-ACE antibodies. E The intensity of NRF1 and ACE was quantified with ImageJ (n = 7, mean ± SEM, **p ≤ 0.01, ***p ≤ 0.001 by two-way ANOVA). F The expression of Ace, Edn1, and Nrf1 was detected by qRT-PCR (n = 7, mean ± SEM, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.0001 by two-way ANOVA). G–J HUVECs were incubated with the indicated concentration of testosterone for 24 h under 1% O2. Ang-2 in the supernatant was measured by ELISA (G, mean ± SD, **p ≤ 0.01, ***p ≤ 0.001 by two-way ANOVA). The expression of ACE, EDN1, NRF1, and VEGFA was measured by qRT-PCR (H–J, mean ± SD, *p ≤ 0.05, **p ≤ 0.01 by two-way ANOVA). Nor normoxia, Hyp hypoxia. K–M L132 cells were cotreated with 200 μM CoCl2 and testosterone at the indicated concentrations. The protein levels of HIF-1α and NRF1 were measured by western blot analysis (K), and the mRNA levels of NRF1 and ACE were detected by qRT-PCR (L, M, mean ± SD, two-way ANOVA). N L132 cells were transfected with NRF1 siRNA for 48 h and then subjected to hypoxia treatments. The mRNA level of ACE was detected by qRT-PCR (L, mean ± SD, ***p ≤ 0.001 by two-way ANOVA)
To confirm the protective effect of testosterone on endothelial function, we also measured the Ang-2 concentrations in the supernatants of testosterone-treated HUVECs. As shown in Fig. 5G, Ang-2 concentrations significantly decreased in a testosterone dose-dependent manner under hypoxic conditions. To further verify that testosterone directly repressed NRF1 and ACE expression in endothelial cells, we treated hypoxic HUVECs with testosterone. As shown in Fig. 5H, I, both NRF1 and ACE mRNA levels were decreased in a testosterone dose-dependent manner under hypoxic conditions, while there was no obvious change after testosterone treatment in the normoxia group, indicating that the inhibition of NRF1 and ACE by testosterone was dependent on low oxygen. However, Vegfa, a HIF-1 target gene, showed no obvious change in either the normoxia group or the hypoxia group (Fig. 5I), suggesting that testosterone did not affect the HIF-1 signaling pathway. To further confirm that testosterone protected endothelial function independent of HIF-1, we accumulated HIF-1 by incubating cells with 200 μM CoCl2 under normoxic conditions. The results in Fig. 5K–M show that there were no significant changes in NRF1 and ACE in the CoCl2-treated groups. Furthermore, L132 cells were transfected with Hif1a shRNA to inhibit HIF-1α and then subjected to hypoxia. The evidence in Fig. 5N shows that similar trends were observed in the hypoxia and shHIF1a groups. In summary, testosterone inhibited NRF1 in a hypoxia-dependent manner but did not inhibit HIF-1 signaling and further suppressed hypoxia-induced hypertension.
Discussion
Studies have proven that testosterone benefits vasodilation, inhibits vascular proliferation, and promotes BP homeostasis [23, 24]. Testosterone deficiency induces endothelial dysfunction, which can lead to vascular dysfunction [32]. In some basic and clinical reports, testosterone has been shown to regulate NO synthesis and thereby influence endothelial function [33, 34]. Testosterone deficiency is also related to increased plasma ET-1 levels [35]. Administration of testosterone leads to a notable reduction in the expression of Ang-2 [36]. In this study, we found the same phenomenon: the reduction in testosterone level caused by hypoxia was associated with upregulation of ET-1 and ACE, enzymes that yield active Ang-2. We have proven, for the first time, the mechanism by which testosterone protects endothelial function through transcriptional regulation of NRF1.
Acute or chronic hypoxia exacerbates hypertension in contexts including high-altitude exposure [37], sleep apnea [38], and chronic obstructive pulmonary disease [39]. The mechanisms involve the renin–angiotensin–aldosterone system, the sympathetic nervous system, and HIF-1 signaling in the endothelium [8]. In this experiment, we found no significant change in serum corticosterone or aldosterone after 8% O2 treatment for 24 h, suggesting that the aldosterone system might not be the cause of acute hypoxia-induced hypertension. The downregulation of NO and upregulation of Ang-2 and ET-1 contributed to the increase in BP. There is evidence that hypoxia upregulates ACE and ET-1 through overexpression of HIF-1α [40, 41], but we further found that another mechanism was involved in hypoxia-induced endothelial dysfunction. Evidence shows that hypoxia stimulates NF-κB signaling and induces endothelial cell injury [42, 43]. It has also been reported that NF-κB transcriptionally regulates NRF1 [44], which is consistent with our finding that NRF1 was upregulated in the hypoxia-treated rat vascular endothelium and HUVECs. We further confirmed that NRF1 transcriptionally regulated ET-1 and ACE. It is a novel finding indicating that NRF1 participates in endothelial injury. In addition, due to the widely known mitochondria-regulating function of NRF1, NRF1 upregulation is also associated with oxidative damage and apoptosis. In summary, NRF1 widely affected endothelial function by affecting the inflammatory response, mitochondrial regulation, and contractile proteins. In our study, we also found a decrease in endothelial NO synthase in hypoxia-treated HUVECs (data not shown) that was not affected by NRF1, indicating that additional mechanisms are involved in the hypoxic responses of endothelial cells.
Testosterone levels declined after acute hypoxia treatments, consistent with the findings in our previous publication [30]. Testosterone supplementation effectively inhibited the upregulation of NRF1. A similar result was found in pulmonary epithelia in our previous study [31], suggesting that the decline in testosterone induced by hypoxia contributed to the increase in BP. However, the safety of testosterone supplements in men remains unclear. Both clinical and basic reports have shown variable effects of testosterone on hypertension. Dalmasso et al. found that short-term testosterone supplements benefitted older males, while long-term supplements significantly worsened hypertension in young males [45]. Velho et al. summarized 455 potentially relevant articles and concluded that testosterone supplementation does not cause obvious changes in BP [46]. Kienitz et al. concluded that acute administration of testosterone seems to decrease vascular tone, while the long-term net effect of androgens appears to be vasoconstriction [47]. Thus, we have demonstrated that testosterone supplementation may be a way to alleviate the BP increase induced by acute hypoxia exposure, but long-term testosterone intake needs careful consideration.
Testosterone supplementation inhibited ACE, ET-1, and NRF1 only under hypoxic conditions, suggesting that the protective effect of testosterone on BP was dependent on hypoxia. It is worth studying the mechanism by which testosterone inhibits NRF1 under hypoxia, which may provide a deeper understanding of cell hypoxia responses and hypoxia-induced endothelial dysfunction. Traish et al. found that testosterone represses Toll-like receptor 4 in macrophages, inhibits TNF-α-induced mastoparan-1 and IL-6 production in monocytes and mast cells, and affects T-cell and lymphocyte proliferation [48]. Toll-like receptor 4 and TNF-α signaling activates NF-κB, which is reported to transcriptionally regulate NRF1 [44], suggesting a possible mechanism by which testosterone inhibits hypoxia-stimulated NRF1 through its anti-inflammatory function.
We found that testosterone may have a protective effect against hypertension under acute hypoxia. However, the cellular response under chronic hypoxia might be different. A report that hypertension is a complication of topical testosterone therapy has suggested that the side effects of testosterone replacement therapy should not be neglected. Testosterone replacement therapy has been suggested to increase the risks of issues including venous thromboembolism, myocardial infarctions, stroke, and death [49]. In 2015, the US Food and Drug Administration issued a warning cautioning that testosterone might increase the risk of heart attack and stroke [50]. It has also been reported that testosterone replacement therapy is related to the risk of erythrocytosis [51]. Thus, the potential disease risks of long-term testosterone replacement therapy still need to be considered. In addition, the current guidelines suggest that a BP above 130 mmHg indicates hypertension. The BP observed in our model was prehypertensive. It is possible that chronic hypertension or hypertension with higher BP will not respond to testosterone due to distinct mechanisms linked to inflammation and oxidative stress.
In summary, acute hypoxia exposure induced an increase in BP through upregulation of ET-1 and Ang-2 along with downregulation of NO. The mechanism involved upregulation of NRF1 induced by low oxygen and subsequent transcriptional stimulation of ACE and EDN1 by direct binding of NRF1 to their promoter regions. Testosterone supplementation at the physiological level inhibited NRF1-derived changes in ACE and ET-1 to attenuate BP in a hypoxia-dependent manner.
References
Disease EC, Tomelleri A, Cavalli G, Berti A, Dagna L. Erdheim-Chester disease. Eur J Intern Med. 2015;26:223–9.
Kropfl JM, Kammerer T, Faihs V, Gruber HJ, Stutz J, Rehm M, et al. Acute exercise in hypobaric hypoxia attenuates endothelial shedding in subjects unacclimatized to high altitudes. Front Physiol. 2019;10:1632.
Wolfel EE, Selland MA, Mazzeo RS, Reeves JT. Systemic hypertension at 4,300 m is related to sympathoadrenal activity. J Appl Physiol. 1994;76:1643–50.
Mazzeo RS, Carroll JD, Butterfield GE, Braun B, Rock PB, Wolfel EE, et al. Catecholamine responses to alpha-adrenergic blockade during exercise in women acutely exposed to altitude. J Appl Physiol. 2001;90:121–6.
Bilo G, Caldara G, Styczkiewicz K, Revera M, Lombardi C, Giglio A, et al. Effects of selective and nonselective beta-blockade on 24-h ambulatory blood pressure under hypobaric hypoxia at altitude. J Hypertens. 2011;29:380–7.
Grocott M. Commentary. J Intensive Care Soc. 2012;13:204–5.
Santos RA, Ferreira AJ, Ac SES. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp Physiol. 2008;93:519–27.
Narvaez-Guerra O, Herrera-Enriquez K, Medina-Lezama J, Chirinos JA. Systemic hypertension at high altitude. Hypertension. 2018;72:567–78.
Wood KC, Liu VB, Noguchi A, Wang XD, Raghavachari N, Gladwin MT. Blood cell versus vascular endothelial cell contributions to nitrite homeostasis and blood pressure regulation. Free Radic Biol Med. 2008;45:S118–9.
Rickard AJ, Morgan J, Chrissobolis SB, Miller AA, Sobey CG, Young MJ. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity, but not blood pressure. Hypertension. 2014;63:E160
Amiri F, Paradis P, Reudelhuber TL, Schiffrin EL. Vascular inflammation in absence of blood pressure elevation in transgenic murine model overexpressing endothelin-1 in endothelial cells. J Hypertens. 2008;26:1102–9.
Chuaiphichai S, Rashbrook VS, Hale AB, Trelfa L, Mcneill E, Lygate CA, et al. Deficiency in endothelial cell tetrahydrobiopterin increases resistance vascular remodelling, blood pressure, and susceptibility to aortic abdominal aneurysm in response to angiotensin II. Cardiovasc Res. 2018;114:S90.
Ten VS, Pinsky DJ. Endothelial response to hypoxia: physiologic adaptation and pathologic dysfunction. Curr Opin Crit Care. 2002;8:242–50.
Baldea I, Teacoe I, Olteanu DE, Vaida-Voievod C, Clichici A, Sirbu A, et al. Effects of different hypoxia degrees on endothelial cell cultures—time course study. Mech Ageing Dev. 2018;172:45–50.
Michiels C, Arnould T, Remacle J. Endothelial cell responses to hypoxia: initiation of a cascade of cellular interactions. Biochim Biophys Acta. 2000;1497:1–10.
Adam T, Makaula S, Essop MF. Nuclear respiratory factor 1: a novel inhibitor of the human gene promoter of the cardiac isoform of acetyl-coa carboxylase. Cardiovasc Drug Ther. 2006;20:396.
Scarpulla R. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–38.
Tiranti V, Rossi E, Rocchi M, Didonato S, Zuffardi O, Zeviani M. The gene (NFE2L1) for human NRF-1, an activator involved in nuclear-mitochondrial interactions, maps to 7q32. Genomics. 1995;27:555–7.
Scarpulla RC. Nuclear respiratory factors and the pathways of nuclear-mitochondrial interaction. Trends Cardiovas Med. 1996;6:39–45.
Jun-Ichi S, Natsuki K, Yoji Y. Pathway analysis of ChIP-seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul Syst Biol. 2013;7:139–52.
Wang D, Zhang J, Lu Y, Luo Q, Zhu L. Nuclear respiratory factor-1 (NRF-1) regulated hypoxia-inducible factor-1alpha (HIF-1alpha) under hypoxia in HEK293T. IUBMB Life. 2016;68:748–55.
Wang X, Jin L, Jiang S, Wang D, Lu Y, Zhu L. Transcription regulation of NRF1 on StAR reduces testosterone synthesis in hypoxemic murine. J Steroid Biochem Mol Biol. 2019;191:105370.
Webb CM, Mcneill JG, Hayward CS, Zeigler D, De, Collins P. Effects of testosterone on coronary vasomotor regulation in men with coronary heart disease. Circulation. 1999;100:1690–6.
Lauro FV, Hector L, Carlos CH, Olga MOG, Tomas MC, Guillermo CR. Synthesis and evaluation of the cardiovascular effects of two, membrane impermeant, macromolecular complexes of dextran-testosterone. Steroids. 2002;67:611–9.
Villar IC, Francis S, Webb A, Hobbs AJ, Ahluwalia A. Novel aspects of endothelium-dependent regulation of vascular tone. Kidney Int. 2006;70:840–53.
Montalcini T, Gorgone G, Gazzaruso C, Sesti G, Perticone F, Pujia A. Endogenous testosterone and endothelial function in postmenopausal women. Coron Artery Dis. 2007;18:9.
Suzuki T, Nakamura Y, Moriya T, Sasano H. Effects of steroid hormones on vascular functions. Microsc Res Tech. 2010;60:76.
Yildiz O, Seyrek M. Vasodilating mechanisms of testosterone. Exp Clin Endocrinol Diabetes. 2007;115:1–6.
Wang X, Pan L, Zou Z, Wang D, Lu Y, Dong Z, et al. Hypoxia reduces testosterone synthesis in mouse Leydig cells by inhibiting NRF1-activated StAR expression. Oncotarget. 2017;8:16401–13.
Wang X, Zou Z, Yang Z, Jiang S, Lu Y, Wang D, et al. HIF 1 inhibits StAR transcription and testosterone synthesis in murine Leydig cells. J Mol Endocrinol. 2018;62:1–12.
Wang X, Huang L, Jiang S, Cheng K, Wang D, Luo Q, et al. Testosterone attenuates pulmonary epithelial inflammation in male rats of COPD model through preventing NRF1-derived NF-kappaB signaling. J Mol Cell Biol. 2021;13:128–140.
Hotta Y, Kataoka T, Kimura K. Testosterone deficiency and endothelial dysfunction: nitric oxide, asymmetric dimethylarginine, and endothelial progenitor cells. Sex Med Rev. 2019;7:661–8.
Marin R, Escrig A, Abreu P, Mas M. Androgen-dependent nitric oxide release in rat penis correlates with levels of constitutive nitric oxide synthase isoenzymes. Biol Reprod. 1999;61:1012–6.
Elesber AA, Solomon H, Lennon RJ, Mathew V, Prasad A, Pumper G, et al. Coronary endothelial dysfunction is associated with erectile dysfunction and elevated asymmetric dimethylarginine in patients with early atherosclerosis. Eur Heart J. 2006;27:824–31.
Kumanov P, Tomova A, Kirilov G, Dakovska L, Schinkov A. Increased plasma endothelin levels in patients with male hypogonadism. Andrologia. 2002;34:29–33.
Zhang HL, Yue ZP, Zhang L, Yang ZQ, Geng S, Wang K, et al. Expression and regulation of angiopoietins and their receptor Tie-2 in sika deer antler. Anim Cells Syst. 2017;21:177–84.
Gassmann M, Cowburn A, Gu H, Li J, Rodriguez M, Babicheva A, et al. Hypoxia-induced pulmonary hypertension—utilizing experiments of nature. Br J Pharm. 2021;178:121–31.
Nanduri J, Peng YJ, Yuan G, Kumar GK, Prabhakar NR. Hypoxia-inducible factors and hypertension: lessons from sleep apnea syndrome. J Mol Med. 2015;93:473–80.
Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J. 2008;32:1371–85.
Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem. 2001;276:12645–53.
Zhang R, Wu Y, Zhao M, Liu C, Zhou L, Shen S, et al. Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L631–40.
Xiao F, Li X, Wang J, Cao J. Mechanisms of vascular endothelial cell injury in response to intermittent and/or continuous hypoxia exposure and protective effects of anti-inflammatory and anti-oxidant agents. Sleep Breath. 2019;23:515–22.
Flamant L, Toffoli S, Raes M, Michiels C. Hypoxia regulates inflammatory gene expression in endothelial cells. Exp Cell Res. 2009;315:733–47.
Suliman HB, Sweeney TE, Withers CM, Piantadosi CA. Co-regulation of nuclear respiratory factor-1 by NFkappaB and CREB links LPS-induced inflammation to mitochondrial biogenesis. J Cell Sci. 2010;123:2565–75.
Dalmasso C, Patil CN, Yanes Cardozo LL, Romero DG, Maranon RO. Cardiovascular and metabolic consequences of testosterone supplements in young and old male spontaneously hypertensive rats: implications for testosterone supplements in men. J Am Heart Assoc. 2017;6:10.
Velho I, Fighera TM, Ziegelmann PK, Spritzer PM. Effects of testosterone therapy on BMI blood pressure, laboratory profile of transgender men: a systematic review. Andrology. 2017;5:881–8.
Kienitz T, Quinkler M. Testosterone and blood pressure regulation. Kidney Blood Press Res. 2008;31:71–9.
Traish A, Bolanos J, Nair S, Saad F, Morgentaler A. Do androgens modulate the pathophysiological pathways of inflammation? Appraising the contemporary evidence. J Clin Med. 2018;7:12.
Lee OD, Tillman K. An overview of testosterone therapy. Am J Mens Health. 2016;10:68–72.
Lo EM, Rodriguez KM, Pastuszak AW, Khera M. Alternatives to testosterone therapy: a review. Sex Med Rev. 2018;6:106–13.
Ohlander SJ, Varghese B, Pastuszak AW. Erythrocytosis following testosterone therapy. Sex Med Rev. 2018;6:77–85.
Acknowledgements
The authors thank Professor Xia Li at Nantong University, who kindly provided the HUVEC line as a gift. In addition, the authors thank Dr. Zhangji Dong at Nantong University for polishing the manuscript.
Funding
The study was supported by the National Natural Science Foundation of China (31671206, 81702874).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jiang, S., Chen, G., Yang, Z. et al. Testosterone attenuates hypoxia-induced hypertension by affecting NRF1-mediated transcriptional regulation of ET-1 and ACE. Hypertens Res 44, 1395–1405 (2021). https://doi.org/10.1038/s41440-021-00703-4
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41440-021-00703-4
Keywords
This article is cited by
-
The Role of Testosterone in Atherosclerosis: View From Cell Cultures and Animal Models
Journal of Cardiovascular Translational Research (2026)
-
Sex Difference of Alcoholic Hypertension: Mechanism and Targeted Therapy
Current Hypertension Reports (2025)
-
Association between Low Serum Testosterone Levels and All-cause Mortality in Patients With Cardiovascular Disease: A Study Based on the NHANES Database
Cardiovascular Toxicology (2025)
-
Canagliflozin alleviates pulmonary hypertension by activating PPARγ and inhibiting its S225 phosphorylation
Acta Pharmacologica Sinica (2024)
-
Ciprofloxacin Accelerates Angiotensin-II-Induced Vascular Smooth Muscle Cells Senescence Through Modulating AMPK/ROS pathway in Aortic Aneurysm and Dissection
Cardiovascular Toxicology (2024)
