
Abstract
Solar conversion of CO2 into energy-rich products is one of the sustainable solutions to lessen the global energy shortage and environmental crisis. Pitifully, it is still challenging to attain reliable and affordable CO2 conversion. Herein, we demonstrate a facile one-pot approach to design core-triple shell Mn, C-codoped ZnO hollow spheres as efficient photocatalysts for CO2 reduction. The Mn ions, with switchable valence states, function as “ionized cocatalyst” to promote the CO2 adsorption and light harvesting of the system. Besides, they can capture photogenerated electrons from the conduction band of ZnO and provide the electrons for CO2 reduction. This process is continuous due to the switchable valence states of Mn ions. Benefiting from such unique features, the prepared photocatalysts demonstrated fairly good CO2 conversion performance. This work is endeavoured to shed light on the role of ionized cocatalyst towards sustainable energy production.
Similar content being viewed by others

Introduction
Worldwide consumption of fossil fuels is unambiguously accountable for two critical issues, i.e. global warming by the augmented greenhouse gases (especially CO2) emission and depletion of such non-renewable sources1,2,3,4,5,6. Nowadays, it becomes a global appeal to develop sustainable energy grids based on renewable energy sources such as solar, wind, and geothermal energies7,8. Reliable conversion of CO2 by the aid of solar light to energy-rich fuels (i.e. artificial photosynthesis) is an ideal solution for simultaneously relieving both energy and environmental crises9,10,11. Albeit great efforts have been devoted to exploring the potential of artificial photosynthetic materials for CO2 conversion, its efficiency is still unsatisfactory due to difficulties in activating thermodynamically stable CO212,13,14. Therefore, it is critical to construct suitable photocatalysts and coupled with active cocatalysts. Cocatalysts can effectively promote the separation and transfer of photogenerated charge carriers to attain appealing CO2 reduction ability11,15,16.
Transition metal-based materials, such as ions, oxides, sulfides, and phosphides, exhibited outstanding potentials as cocatalysts for both oxidation and reduction reactions17,18,19,20,21. They can not only effectively lower the barrier for CO2 activation but also promote the separation of photogenerated charge carriers22,23. To date, most cocatalysts are in solid state. We wonder whether ionized cocatalysts which are embedded in the lattice of photocatalyst can also serve as a cocatalyst. They can become active sites for the photocatalytic CO2 reduction. Due to their multiple oxidation states, they can swap electrons with photocatalyst as well as CO2. Besides, the introduction of ionized cocatalyst avoids the difficulty of loading a cocatlyst on the photocatalyst. And the difficulty in maintaining intimate contact no longer exists.
Intricate hollow structures with tailored compositions have surpassed their solid counterparts in widespread applications. However, the fabrication of such intricate structures is still an arduous challenge. Multishell hollow structures (MSHSs) are characterized by manifold features such as large specific surface area, low density, as well as reduced mass and charge diffusion lengths24. All of these endow MSHSs with potential in multiple applications, such as photocatalysis, energy storage and conversion, drug delivery and sensors25,26,27. Concerning CO2 photoreduction (PR), MSHSs possess prodigious advantages over their solid counterparts. These advantages particularly spring from esthetic shell layers. For one thing, their large surface area promotes CO2 adsorption and surface redox reactions on both sides of the shells. For another, they provide a reduced path length for charge carriers, favoring charge migration and separation28,29. All these merits make MSHSs ideal support materials for anchoring photocatalysts aiming at designing elaborate catalytic system for CO2 PR. However, the fabrication of such MSHSs is still an arduous challenge.
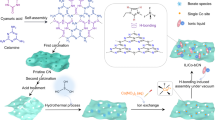
Herein, we develop a facile one-pot coordination polymer (CP) strategy to fabricate Mn, C-codoped ZnO core–triple shell hollow spheres (Mn, C-ZnO CTSHSs). The process involves two steps: (i) formulation of Zn, Mn CP via interaction of salicylate ligand with the corresponding metal ions in the presence of polyvinylpyrrolidone (PVP) and (ii) air calcination of the resultant Zn, Mn CP (Fig. 1). The obtained photocatalyst with fascinating core–multishell morphology and tailored composition affords adequate CO2 reduction activity, i.e. two times higher than commercial ZnO. Such enhanced activity is ascribed to the improved light absorption, favorable transfer of mass and charges across the thin shell layers, and aided CO2 adsorption and activation. Interestingly, Mn species formed in the system as substituent of Zn ions in the ZnO lattice play an indispensable role during CO2 PR. They act as active centers for CO2 adsorption and subsequent favored CO2 reduction by suppressing hydrogen evolution. Most importantly, in situ X-ray photoelectron spectroscopy results reveal that Mn species can restore their primal oxidation state after CO2 PR without external treatment or subsidiary reducing agents. Instead, Mn(n+1)+ species can extract one photogenerated electron from the system to regenerate the original Mnn+ moieties. The switching between Mn(n+1)+/Mnn+ is recognizable by virtue of the multiple oxidation states of Mn and affordability of photogenerated electrons. These results suggest that ionized Mn can act as a durable and efficient ionized cocatalyst toward practical CO2 PR.

a Preparation of Mn, Zn-CPSs from solvothermal treatment of zinc acetate and manganese acetate with salicylic acid in the presence of PVP. b Step-by-step conversion of Mn, Zn-CPSs into Mn, C-codoped ZnO CTSHSs upon calcination at 550 °C for 3 h. c The underlying driving force for such hollow structure formation.
Results and discussions
Formulation and characterization of Mn, C-ZnO CTSHSs
The core–triple shell hollow spheres are obtained via a facile one-pot solvothermal preparation of Mn, Zn CP spheres (Mn, Zn-CPSs) followed by controlled air-annealing as depicted in Fig. 1. The CPSs as precursors were firstly formulated by the coordination interaction between Zn- and Mn-acetate and salicylate ligands (Fig. 1a). The carboxylate groups of salicylic acid play a significant role that they can coordinate to metal ions, enabling a delicate control over the composition. Moreover, this method is universal to most of the transition metals because non-selectivity is witnessed for salicylic acid30.
Fourier transform infrared spectroscopy (FTIR) spectra of the Mn, Zn-CPSs (Supplementary Fig. 1) evidenced the interaction between deprotonated carboxylate groups of salicylate ligands and metal ions. Such interaction is assured after distinctive intensity drop of the corresponding C=O peak centered at 1657 cm–1 and a slight shift to 1654 cm–130. The deprotonation of the carboxylate groups is very critical to boost such coordination interaction. As a strong conjugated base, acetate anions can deprotonate the carboxylate groups of salicylic acid and thus enable reliable coordination between salicylate ligand and metal cations. To confirm this assumption, we replaced Zn- and Mn-acetates by their sulfates, nitrates, or chlorides and followed the same preparation recipe. All of them fail to fabricate such spherical architecture. We attribute such failure to the inability of the corresponding anions to deprotonate carboxylate groups of salicylic acid. After formation of Mn, Zn-CPSs, the generation of hollow structures derives from the calcination process. In this step, the Mn, Zn-CPS precursor is subjected to air calcination at 550 °C, a step that culminates the unique CTSHS morphology by heterogeneous contraction process (Fig. 1b, c).
Transmittance electron microscopy (TEM) (Fig. 2) images show Mn, Zn-CPSs as high uniform solid spheres (Fig. 2a) with an average diameter of about 1.3 μm and smooth surface texture. After calcination for 3 h, unexpectedly, the solid Mn, Zn-CPSs were converted into unique concentric Mn, C-ZnO CTSHSs, as noticed from the high contrast between the shell edges and centered hollow regions (Fig. 2b). The CTSHSs retain the spherical shape of the CPS precursor but the diameter is dramatically shrunk to 650 nm due to the contraction upon calcination. The surface turns rough, pervaded with crystallites with invasive pores. The size of the crystallites was measured to be 25 nm from high-resolution TEM image. Its porous structure was also confirmed by N2 adsorption–desorption isotherm (Supplementary Fig. 2 and Supplementary Table 1). Additionally, uniform distribution of the constituent elements ca. Zn, O, Mn, and C are observed from energy dispersive X-ray spectroscopy (EDS) elemental mapping of the calcined sample (as taken for sample calcined for 1h, Fig. 2c). The elemental distribution is presented in Supplementary Fig. 3 and the actual compositional percentages of Mn and C in 2% Mn, C-ZnO sample are 1.8 and 8.3%, as derived from elemental analysis and inductively coupled plasma atomic emission spectroscopy measurements, respectively.

TEM images of the Mn, Zn-CPSs a before calcination and b after calcination for 3 h. c HAADF-STEM image of Mn, C-ZnO and EDS mapping of the sample show the constituent elements Zn, O, Mn, and C.
To elucidate the evolution mechanism of the CTSHS morphology, TEM and field emission scanning electron microscopy (FESEM) images of the intermediates at different calcination stages were recorded (Fig. 3 and Supplementary Fig. 4). Before calcination, the solid spheres with distinct boundary are observed (Fig. 3a). At the initial stages of the calcination process (after 0.5 h), a significant temperature gradient (∆T) along the radial direction of the CPSs was generated where the outermost building units are subjected to abrupt heating. Meanwhile, the carbon entities vanish and ZnO nanoparticles were crystallized at the outer surface. Thus, a core–shell structure (the first ZnO shell conjoined to the entire core) was generated (Fig. 3b). Once forming, the rigid ZnO shell plays an indispensable role in preventing further contraction of the outer diameter, even if the entire core suffers from continuous shrinkage upon the disintegration of organic moieties by further annealing, known as heterogeneous contraction31,32,33,34.

a Before calcination and after calcination for b 0.5 h, c 1 h, d 2 h, and e 3 h. f Depicts the HRTEM of Mn, C-codoped ZnO with lattice fringes of 0.248 nm corresponds to the (101) plane of Wurtzite ZnO.
With extended calcination time, both the core and the emerged new shell experienced two opposite forces. Namely, inward cohesion force (Fc) originated from the sharp contraction of the interior core upon continuous disintegration of the skeletal carbon and outward adhesion force (Fa) emanated from the cling of adjacent crystallites. The balance between both forces culminated in varied structures. When the CPSs were calcined for 1 h, the temperature gradient gets smaller. And Fc exceeds Fa, which leads to separation of the entire core from the outer shell, forming a yolk-shell structure (Fig. 3c and Supplementary Fig. 4b). With calcination continued for 2 or 3 h, the entire yolk underwent similar processes described before, forming the unique second (core–double shell, Fig. 3d and Supplementary Fig. 4c) and the third shell (core–triple shell, Fig. 3e and Supplementary Fig. 4d). It is noteworthy that the disintegration of the carbon moieties on both exterior and interior surfaces induces the formation of observable pores within the shell layers (Supplementary Fig. 4e), which facilitates the mass transfer of the reactants/products35,36.
All the calcined samples together with commercial ZnO (for comparison) show diffraction peaks (Supplementary Fig. 5) that are well indexed to Wurtzite hexagonal ZnO (JCPDS PDF Card No. 36-1451), which are in line with the (101) lattice fringes of ZnO (Fig. 3f)37. Adequate purity and good crystallinity of the prepared ZnO CTSHS samples are reflected from the sharp peaks and absence of secondary peaks even with 5% Mn loading. Additionally, compared with commercial ZnO, the lattice of the prepared catalysts is mostly contracted after the doping process (see Supplementary Note 1 for more discussion). These results suggest the successful doping of Mn and/or C within the crystal lattice of ZnO and Mn species are implemented to substitute Zn ions in the lattice.
CO2 PR activity of resultant samples
The photocatalytic performance of comm. ZnO, CZ, and MCZ-x (CZ represents C-ZnO CTSHS sample, MCZ-x represents Mn, C-ZnO CTSHS samples with different percentages of Mn (x = 1, 2, and 5)) was evaluated in an online closed gas-circulation system under simulated solar light with a Quartz and Pyrex glass hybrid reaction cell (Supplementary Fig. 6). Ambient conditions were kept, and neither photosensitizer, precious cocatalyst nor sacrificial reagents were used. Carbon monoxide (CO) was the predominant reduction product, whereas oxygen (O2) was the oxidation product (Fig. 4). The 2% Mn, C-ZnO CTSHS photocatalyst (MCZ-2 in Fig. 4a) afforded the highest PR activity with CO yield rate of 0.83 μmol g–1. When Mn loading is too high, i.e. 5%, excessive defect states are introduced into the lattice of ZnO, which act as local recombination centers for photogenerated charge carriers38. H2 gas was hardly detectable, suggesting that the competitive H2 evolution from water is inhibited without the help of noble metal cocatalyst. The initial detected O2 gas came from the input high-purity CO2. As the CO2 PR proceeded, the amount of O2 increased gradually, indicating that water was photooxidized to O2 (Fig. 4b). Taking into account the sluggish kinetics of water oxidation and the need of four hole equivalents to produce one oxygen molecule39, it is reasonable to observe a lower yield of O2 (oxidation product) than CO (reduction product).

Photocatalytic CO2 reduction performance of comm. ZnO and the prepared samples: time course of a CO and b O2 production yields. c Wavelength dependence of the AQE and the UV–vis absorption spectrum of 2% Mn, C-ZnO sample. d Isotopic labeling chromatogram using 13NaHCO3 as a carbon source and e GC-MS data of the peak at retention time (RT) of 11.57 coressponding to 13CO.
The apparent quantum efficiency (AQE) of CO is defined by the ratio of the effective electrons used for CO production to the total input photon flux40,41. It was measured by applying a 300 W xenon arc lamp with monochromatic light band-pass filters (λ = 350, 365, 380, 400, 420, and 450 nm; Beijing Perfectlight, China). The measured light intensity per unit area (E), the CO production yield (MCO), and the calculated CO production rate (RCO), AQE at different monochromatic wavelengths over the 2% Mn, and C-ZnO sample are shown in Table 1. Further, the wavelength dependence of the AQE and the UV–vis absorption spectrum of 2% Mn, C-ZnO sample is shown in Fig. 4c. Clearly, the trends of AQE match well with the light absorption spectrum, indicating the photocatalytic feature of the CO2 reduction reaction.
The recyclability and stability of MCZ-2 for photocatalytic CO2 reduction were also investigated (Supplementary Fig. 7). After four cycles, the decay of CO production yield is hardly perceptible. Meanwhile, the recycled photocatalyst shows identical phase composition as the fresh sample as confirmed from X-ray diffraction (XRD) results (Supplementary Fig. 8). These findings unambiguously disclosed the promising potential of the prepared CTSHS photocatalysts for CO2 PR.
Control experiments showed that no products were detected in the absence of photocatalysts and/or CO2 either in dark or under light irradiation, suggesting that CO is exclusively generated from CO2 PR. We performed isotopic labeling experiment using 13CO2 as isotopic tracer to investigate the carbon source of the obtained products. As shown in Fig. 4d, the total ion chromatographic signal around 11.57 min corresponds to CO, which produces three signals in the mass spectra (Fig. 4e). The main MS signal at m/z = 29 belongs to 13CO and the others at m/z = 13 and m/z = 16 are assigned to the fragments of 13CO, i.e. 13C and 16O, respectively. This result confirms that CO is indeed derived from CO2 reduction over MCZ-2 rather than any other carbon source42,43. In addition, the total ion chromatographic signals around 2.67 and 3.55 min are assigned to O2 and N2, respectively (Supplementary Fig. 9).
Photocatalytic mechanism for enhanced performance
Appreciable enhancement of the visible light absorption was sustained by the fascinating hollow morphology, thanks to the multiple reflections of the incident light inside the interior cavities of the fascinating core–triple shell hollow structure44 (inset of Fig. 5a). Moreover, the codoping process introduces impurity levels within the forbidden gap of ZnO, which indeed contributes to narrowing the bandgap (as depicted from Tauc plots, Supplementary Fig. 10) and enhancing the light-harvesting ability of the doped samples. A stepwise improvement of the light absorptivity of the prepared photocatalysts is observed in line with Mn-content (Fig. 5a). This finding should be correlated to the fact that more impurity states are developed within ZnO bandgap with increasing Mn doping. In addition, the doping with Mn could initiate other electronic transitions such as ligand to metal charge transfer, metal to ligand charge transfer, and d–d transition45,46,47. All these transitions are accounted for the whole system light harvesting, which leads to enlarged absorption response48,49.

a UV–vis light absorption of the prepared photocatalysts and the inset represents the multiple reflection effect of the incident light inside the hollow cavities. b PL spectra of photocatalysts at excitation wavelength of 380 nm. c TRPL spectra of the samples. d CO2 adsorption isotherm of C-ZnO CTSHSs and 2% Mn, C-ZnO-CTSHS samples.
Furthermore, the photoluminescence (PL) spectra of the prepared catalysts are step-wisely quenched after Mn and C doping. This reveals that suppressed charge recombination is achieved by doping. The 2% Mn, C-ZnO sample exhibits the lowest PL behavior (Fig. 5b), benefiting from two factors. One is (i) the defect state. The rest is (ii) the core–shell morphology and porous structure. The former could act as trapping centers, quenching the electron–hole inhalation process. Whereas the latter reduces both diffusion lengths of charges and reactants, thus minimizing charges recombination opportunities and providing accessible channels for mass/charge transfer50. Time-resolved photoluminescence (TRPL) results also demonstrate that 2% Mn, C-ZnO can retard charge carriers recombination and prolong their lifetime (average charge carrier-lifetime ca. 1.1, 0.9, and 0.8 ns for 2% Mn, C-ZnO, C-ZnO, and Comm. ZnO, respectively, Fig. 5c and Supplementary Table 3). These results indeed affirm the potential of Mn doping to delay charge recombination.
In addition, the prepared samples (C-ZnO and Mn, C-ZnO CTSHSs) attain very low resistance for charges transfer (Rct) process compared to comm. ZnO sample (as speculated from the electrochemical impedance spectroscopy measurements, Supplementary Fig. 11, where smaller arc diameter represents lesser resistance). Obviously, the fascinating architecture favors charge carrier separation and diffusion from bulk to surface region through few nanometer-thick shells. Especially, the Mn-doped sample shows the least Rct, benefiting from charge trapping action exerted by the doping-based defect states (at optimum content) and the following interfacial charge transfer51,52.
Besides, Mn species could be beneficial for enhancing the adsorption of CO2. To authenticate such a hypothesis, the CO2 adsorption isotherms for C-ZnO and 2% Mn, C-ZnO-CTSHSs are provided in Fig. 5d. Obviously, the 2% Mn-doped sample shows much more adsorption affinity (merely twice) toward CO2 than the undoped sample does. This result coincides well with the photocatalytic activity of both samples. The higher the CO2 adsorption affinity of a photocatalyst is, the better the PR activity will be expected. Therefore, it is reasonable to notice such enhanced activity for Mn-doped photocatalyst. The improvement of CO2 adsoprtion after Mn doping was also reported for Mn-In2S3 tested for CO2 electroreduction53.
Two plausible factors contributing to the high CO2 adsorption affinity are proposed. One is the higher content of surface oxygen-species (see XPS interpretation, Supplementary Note 2 for details) than C-ZnO sample. The other is the potential of Mn species replaced Zn ions in the lattice to activate CO2 molecules.
To further explore the functional role of Mn, in situ irradiated XPS (ISI-XPS) analysis for Mn, C-ZnO CTSHS sample saturated with CO2 was conducted under both dark and irradiated conditions. The high-resolution XPS peaks are demonstrated in Fig. 6 and Supplementary Fig. 12 together with the same peaks from bare sample (without CO2 treatment). The XPS spectra further indicate the successful doping of Mn within the crystal lattice of ZnO, originating from the distinctive Mn 2p and Mn 3s peaks appeared in the XPS profiles and the shift of Zn 2p3/2 peak (see Fig. 6 and Supplementary Fig. 12 for more details about the chemical state of Mn within the ZnO lattice). The presence of Mn is believed to promote catalytic conversions owing to their ability to share electrons with reactant molecules and/or intermediates54,55,56,57. Besides, doping gives rise to defect states and impurity levels. They not only reduce the bandgap of the semiconductor but also improve the light absorptivity of the photocatalyst. The defect states could support the charge separation process through obstructing the electron–hole recombination by electron trapping58,59,60,61. Specifically, introduced transition metal ions could act as distinctive active sites or enrich present active sites, which evidently support reactants’ adsorption, activation, and further chemical conversion62,63,64.

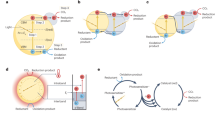
In situ XPS analysis showed Mn 3s profiles for a untreated and b CO2-adsorbed Mn, C-ZnO CTSHS samples. The multiple splitting of Mn 3s (∆S) is used to calculate average oxidation state of Mn ions within the structure (Eq. (1)). c Schematic illustration of CO2 activation and reduction over Mn, C-codoped ZnO sample. The CO2 molecules are firstly activated at the confined Mn active centers to the highly active radical anion. Such intermediate anion is then subjected to successive electron-coupled proton transfer process (electrons from ZnO CB and protons from water) and finally produces CO. The photogenerated holes are consumed for water oxidation (oxygen evolution reaction).
The loading of CO2 molecules onto the surface was assured by emergence of a tiny M-CO3 peak in C 1s XPS (Supplementary Fig. 12d), suggesting the successful bonding of CO2 molecules to metallic sites. To probe how Mn ions could assist activation of CO2 molecules during the PR process, we calculated the average oxidation states (AOS) of Mn under test according to the following equation65:
where ∆S is the multiple splitting of Mn 3s peak (Fig. 6a, b). By substituting ∆S values in Eq. (1), we get a unit increase in the AOS for sample saturated with CO2 and kept in darkness. Then, a decrease in AOS by half a unit is observed for sample saturated with CO2 and irradiated with light. The significant increase in AOS of Mn after CO2 loading and before illumination could be explained in terms of electron transfer from Mn centers to adsorbed CO2 molecules54,55. In our system, we noticed that the primal AOS of Mn in CO2-loaded photocatalyst can be restored again under light irradiation. This is due to photogenerated electrons transfer from ZnO-conduction band (CB) to Mn(n+1)+ centers to regenerate the primal Mnn+ again (Fig. 6c). This light-switchable conversion between Mnn+/Mn(n+1)+ is very critical for restoring the original activity of the Mn-doped catalyst without external treatment or activation steps. In addition, this conversion holds a promising potential for all-in-one CO2 reduction technology. Concisely, Mn(n+1)+ species can capture the photogenerated electrons from the CB of ZnO, yielding Mnn+, which functions as the active centers for CO2 adsorption and activation via one electron-transfer process to yield CO2·– and Mn(n+1)+. At the same time, if more electrons are captured, the generation of CO becomes possible. This finding authenticates the role of Mn species as ionized cocatalyst in the system. Because we measure the change of AOS, further investigations are required to elucidate the actual functional oxidation state involved in CO2 activation.
In fact, the regeneration of the Mn species after the activation step is very critical to maintain the activity. Moreover, the charge trapping action is of great essence for charge carrier’s separation. Thus the optimal doping from Mn results in lowest charges recombination as revealed from PL results.
In this vein, Mn functions as Lewis base center, which supports the adsorption of acidic CO2 molecules and further transfer of one electron to culminate activated CO2·– (Fig. 6c). Such a step is followed by proton-coupled electron-transfer process to produce the desired molecules (Supplementary Eqs. (1)–(7)).
The adsorption, activation and subsequent reduction of CO2 molecules over Mn-doped ZnO photocatalyst are further confirmed by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) results (Supplementary Fig. 13 and Supplementary Note 3), which assure the adsorption, activation, and successful conversion of activated CO2 molecules onto CO at the photocatalyst surface (Fig. 6c).
All these results unambiguously disclosed the potential of the prepared and light-switchable ionized cocatalyst (Mnn+/Mn(n+1)+) integrated C-ZnO CTSHSs as an efficient photocatalyst for CO2 PR under ambient conditions without sacrificial reagents, precious cocatalysts, or external treatment set up for activity restoration.
In summary, core–triple shell Mn, C-codoped ZnO hollow spheres were rationally fabricated via a one-pot synthetic protocol. In addition, they were tested as a highly active and selective catalyst for CO2 PR under simulated sunlight and ambient conditions. The unique core–triple shell hollow structures codoped with Mn and C hold manifold structural and functional features. These unique features culminated improved light absorption capability, enhanced charge separation and migration efficiency, suppressed e−/h+ pairs undesired recombination probability and abundant adsorption and activation active sites for effective photoredox catalysis. All of these accounted for the superior activity of the prepared photocatalyst. The Mn species played a significant role to activate CO2 molecules through an electron-transfer mechanism. We have observed that the ionized Mn species can restore their primal oxidation state and hence their activity by virtue of the photogenerated electrons at the ZnO CB. Therefore, it can be operated as a light-switchable ionized cocatalytic system. These findings will support the design of efficient photocatalytic materials for sustainable clean energy production and beyond. The present study sheds light on the significance of hollow structure materials as an active photocatalyst for CO2 reduction as well as the importance of light-switchable ionized cocatalysts during this process.
Methods
Materials
All chemicals are of analytical grade and used without additional purification. Zinc acetate dihydrate (Zn(CH3COO)2·2H2O) and absolute ethanol (EtOH) were purchased from Beijing Sinopham Chemical Reagent Co., Ltd. Manganese acetate tetrahydrate (Mn(CH3COO)2·4H2O)) and salicylic acid (C7H6O3) were purchased from Tianjin BODI Chemical Co., Ltd. PVP (C6H9NO)n (K23-27, MWt ~24,000) was supplied by Aladdin Industrial cooperation, Shanghai, China.
Synthesis of Mn, C-ZnO CTSHSs
The Mn, Zn-CPSs were firstly prepared by a solvothermal step. Typically, 1 mmol of Zn(CH3COO)2·2H2O, x mmol of Mn(CH3COO)2·4H2O corresponding to the desired Mn2+/Zn2+ mole ratio (0, 0.01, 0.02 or 0.05), and (2 + x) mmol of salicylic acid were magnetically dissolved in 50 mL of EtOH. Afterward, a calculated amount of PVP (to keep the concentration ratio MPVP/MM2+ = 5, M of PVP was calculated based on the molecular weight of its monomer) was added to the above solution and further stirred for 30 min. The mixture was transferred into a 100 mL of Teflon-lined stainless steel autoclave, heated at 160 °C for 12 h, and then air-cooled to room temperature. The precipitate was separated by centrifugation, washed three times with EtOH, and dried under vacuum at 60 °C overnight. The as-obtained Mn, Zn-CPSs were calcined at 550 °C for 3 h by 5 °C/min rate under air atmosphere. The samples with 0, 1, 2, and 5% Mn were denoted as C-ZnO (CZ), 1% Mn, C-ZnO (MCZ-1), 2% Mn, C-ZnO (MCZ-2), and 5% Mn, C-ZnO (MCZ-5), respectively.
Characterization
TEM, HRTEM, and elemental mapping were obtained by Titan G2 60–300 operated at 300 kV. FESEM images were taken by JSM-7500F, JEOL, Japan, equipped with X-Max 50 EDS unit (Oxford Instruments, UK). XRD patterns of the prepared samples were recorded using D/Max-RB X-ray diffractometer (Rigaku, Japan) with Cu Kα radiation (λ = 1.5406 Å) and scan rate = 0.05 ° S–1. The surface composition and chemical states of C-ZnO and 2% Mn, C-ZnO CTSHS samples were analyzed by XPS analysis (Thermo ESCALAB 250). ISI-XPS analysis for 2% Mn, C-ZnO CTSHS sample saturated with CO2 was conducted in darkness as well as under light irradiation to probe the oxidation state change of Mn during CO2 PR. The 2% Mn, C-ZnO CTSHS sample was firstly degassed at 150 °C for 4 h to remove pre-adsorbed species. Afterwards, it was saturated with CO2 by continuously purging with pure CO2 for 4 h prior to the XPS test. A 3 W 365 nm LED was used as the light source during ISI-XPS test. The UV–visible light absorption spectra of the samples were recorded by UV-2600, Shimadzu spectrophotometer (Japan) where BaSO4 was used as the reflectance standard. The Brunauer–Emmett–Teller specific surface area and porosity of the prepared samples, nitrogen adsorption–desorption isotherms were measured on a Micromeritics ASAP 2020 apparatus (USA) and the adsorption data at relative pressure (p/p0) range of 0.05–0.3 were used. The average pore size and pore volume of the samples were calculated through the Barret–Joyner–Halender method using the adsorbed nitrogen volume at the relative pressure of 0.994. PL spectra were recorded on a Hitachi F-7000 (Japan) fluorescence spectrophotometer. FLS1000 fluorescence lifetime spectrophotometer (Edinburgh, Instruments, UK) was employed to record TRPL spectra of the prepared samples.
FTIR and DRIFTS analyses were obtained by an FTIR spectrometer (Thermo Scientific, Nicolet iS50, USA). For DRIFT test, the prepared sample was placed into the reaction chamber, then the chamber was sealed and purged with the mixed CO2 and H2O gases. The DRIFTS analysis was conducted under both dark and light irradiation (3 W 365 nm LED) conditions.
The electrochemical measurements were performed in a standard three-electrode configuration analyzer (CHI660C instruments, CHI, China) system with Pt wire and Ag/AgCl (saturated KCl) utilized as counter and reference electrodes, respectively, while the working electrode was prepared using the as-synthesized samples deposited on FTO glass, and the three electrodes were immersed in 0.5 M Na2SO4 aqueous solution as electrolyte.
Photocatalytic CO2 reduction and isotope-labeling measurement
The photocatalytic reduction of CO2 was conducted in an online gas-closed system with a gas-circulated pump. Typically, 50 mg of photocatalysts and 1 mL of H2O were added in a Quartz and Pyrex glass hybrid reactor connecting to the CO2 PR system (Supplementary Fig. 6). After complete evacuation of the reaction system, ~80 kPa of high-purity CO2 (99.999%) gas was injected into the air-tight system. After adsorption equilibrium, a 300 W Xe arc lamp (PLS-SXE300D, Beijing Perfectlight, China) was used as the light source. The photocatalytic CO2 reduction products were analyzed by a gas chromatograph (GC-2030, Shimadzu Corp., Japan) equipped with barrier discharge ionization detector (BID) and a capillary column (Carboxen 1010 PLOT Capillary, 60 m × 0.53 mm). The temperatures of the injector and BID were set to be 150 and 280 °C, respectively. The AQE of CO over the 2% Mn, C-ZnO sample was calculated as follows:
where RCO is the CO production rate (mol h–1); t1 is the irradiation time (1 h); NA is Avogadro constant (6.02 × 1023 mol–1); P is the total incident light flux (W, J s–1) and equals light intensity per unit area (E, W cm–2) times effective irradiation area (S, cm2), where E can be measured by the radiant power energy meter (UV-A and FZ-A, Photoelectric Instrument Factory of Beijing Normal University), S is 4.5 cm2 in this experiment; t2 equals 3600 s; λ is the monochromatic light wavelength (m); h is the Planck constant (6.626 × 10–34 J s) and c is the light speed in vacuum (3 × 108 m s–1).
13CO2 isotope tracer experiment was conducted to verify the carbon source of the products by using 13C isotope-labeled sodium bicarbonate (NaH13CO3, Cambridge Isotope Laboratories Inc., USA) and H2SO4 aqueous solution for the photocatalysis examinations. Typically, 20 mg of photocatalysts, 2 mM of [RuII(bpy)3]Cl2·6H2O, 5 mL of triethanolamine, 30 mL of acetonitrile, and 1 mL of water were added in a Pyrex glass reaction cell. After 1 h of photocatalytic reaction, 1 mL of the mixed gas was taken out from the reactor and analyzed by gas chromatography-mass spectrometry (TRACE 1300 and ISQ 7000, Thermo scientific, USA) equipped with the column for detecting the product of 13CO (TG-BOND Msieve 5 A, 30 m × 0.32 mm × 30 μm, Thermo Scientific, USA). The column was maintained at 45 °C for 15 min and then heated to 180 °C at 30 °C min–1 and maintained for 1 min. The temperatures of the injector, ion source, and MS transfer line were set to be 200, 230, and 250 °C, respectively.
References
Yu, J., Jaroniec, M. & Jiang, C. Surface Science of Photocatalysis (Elsevier, London, 2020).
Diercks, C. S., Liu, Y., Cordova, K. E. & Yaghi, O. M. The role of reticular chemistry in the design of CO2 reduction catalysts. Nat. Mater. 17, 301–307 (2018).
Bie, C., Zhu, B., Xu, F., Zhang, L. & Yu, J. In situ grown monolayer N-doped graphene on CdS hollow spheres with seamless contact for photocatalytic CO2 reduction. Adv. Mater. 31, 1902868 (2019).
Zhang, W., Mohamed, A. R. & Ong, W.-J. Z-scheme photocatalytic systems for carbon dioxide reduction: where are we now? Angew. Chem. Int. Ed. 59, 22894 (2020).
Boston, D. J., Xu, C., Armstrong, D. W. & MacDonnell, F. M. Photochemical reduction of carbon dioxide to methanol and formate in a homogeneous system with pyridinium catalysts. J. Am. Chem. Soc. 135, 16252–16255 (2013).
Boston, D. J., Pachón, Y. M. F., Lezna, R. O., de Tacconi, N. R. & MacDonnell, F. M. Electrocatalytic and photocatalytic conversion of CO2 to methanol using ruthenium complexes with Internal pyridyl cocatalysts. Inorg. Chem. 53, 6544–6553 (2014).
Li, W. Advances in CO2 Conversion and Utilization 5–55 (ed. Hu, Y. H.) (American Chemical Society, 2010).
Tiwari, J. N. et al. Multi-heteroatom-doped carbon from waste-yeast biomass for sustained water splitting. Nat. Sustain. 3, 556–563 (2020).
Habisreutinger, S. N., Schmidt-Mende, L. & Stolarczyk, J. K. Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 52, 7372–7408 (2013).
Li, A. et al. Three-phase photocatalysis for the enhanced selectivity and activity of CO2 reduction on a hydrophobic surface. Angew. Chem. Int. Ed. 58, 14549–14555 (2019).
Xia, Y. et al. Highly selective CO2 capture and its direct photochemical conversion on ordered 2D/1D heterojunctions. Joule 3, 2792–2805 (2019).
White, J. L. et al. Light-driven heterogeneous reduction of carbon dioxide: photocatalysts and photoelectrodes. Chem. Rev. 115, 12888–12935 (2015).
Xu, F. et al. Graphdiyne: a new photocatalytic CO2 reduction cocatalyst. Adv. Funct. Mater. 29, 1904256 (2019).
Xu, F. et al. Unique S-scheme heterojunctions in self-assembled TiO2/CsPbBr3 hybrids for CO2 photoreduction. Nat. Commun. 11, 4613 (2020).
Li, K., Peng, B. & Peng, T. Recent advances in heterogeneous photocatalytic CO2 conversion to solar fuels. ACS Catal. 6, 7485–7527 (2016).
Li, X., Yu, J., Jaroniec, M. & Chen, X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 119, 3962–4179 (2019).
Meng, A., Zhang, L., Cheng, B. & Yu, J. Dual cocatalysts in TiO2 photocatalysis. Adv. Mater. 31, 1807660 (2019).
Meng, A., Wu, S., Cheng, B., Yu, J. & Xu, J. Hierarchical TiO2/Ni(OH)2 composite fibers with enhanced photocatalytic CO2 reduction performance. J. Mater. Chem. A 6, 4729–4736 (2018).
Zhu, D. D., Liu, J. L. & Qiao, S. Z. Recent advances in inorganic heterogeneous electrocatalysts for reduction of carbon dioxide. Adv. Mater. 28, 3423–3452 (2016).
Aguirre, M. E., Zhou, R., Eugene, A. J., Guzman, M. I. & Grela, M. A. Cu2O/TiO2 heterostructures for CO2 reduction through a direct Z-scheme: protecting Cu2O from photocorrosion. Appl. Catal. B Environ. 217, 485–493 (2017).
Zhou, R. & Guzman, M. I. CO2 reduction under periodic illumination of ZnS. J. Phys. Chem. C 118, 11649–11656 (2014).
Das, S. & Wan Daud, W. M. A. A review on advances in photocatalysts towards CO2 conversion. RSC Adv. 4, 20856 (2014).
Sajan, C. P., Wageh, S., Al-Ghamdi, A. A., Yu, J. & Cao, S. TiO2 nanosheets with exposed {001} facets for photocatalytic applications. Nano Res. 9, 3–27 (2016).
Zhou, L. et al. Intricate hollow structures: controlled synthesis and applications in energy storage and conversion. Adv. Mater. 29, 1602914 (2017).
Wang, X. et al. A unique disintegration–reassembly route to mesoporous titania nanocrystalline hollow spheres with enhanced photocatalytic activity. Adv. Funct. Mater. 28, 1704208 (2018).
Ye, C. et al. A 3D hybrid of chemically coupled nickel sulfide and hollow carbon spheres for high performance lithium–sulfur batteries. Adv. Funct. Mater. 27, 1702524 (2017).
Potroz, M. G. et al. Plant-based hollow microcapsules for oral delivery applications: toward optimized loading and controlled release. Adv. Funct. Mater. 27, 1700270 (2017).
Fang, B., Xing, Y., Bonakdarpour, A., Zhang, S. & Wilkinson, D. P. Hierarchical CuO–TiO2 hollow microspheres for highly efficient photodriven reduction of CO2 to CH4. ACS Sustain. Chem. Eng. 3, 2381–2388 (2015).
You, F. et al. Lattice distortion in hollow multi-shelled structures for efficient visible-light CO2 reduction with a SnS2/SnO2 junction. Angew. Chem. Int. Ed. 59, 721–724 (2020).
Chooset, S., Kantacha, A., Chainok, K. & Wongnawa, S. Synthesis, crystal structure, luminescent properties and antibacterial activities of zinc complexes with bipyridyl and salicylate ligands. Inorg. Chim. Acta 471, 493–501 (2018).
Guan, J., Mou, F., Sun, Z. & Shi, W. Preparation of hollow spheres with controllable interior structures by heterogeneous contraction. Chem. Commun. 46, 6605–6607 (2010).
Hwang, H., Shin, H. & Lee, W.-J. Effects of calcination temperature for rate capability of triple-shelled ZnFe2O4 hollow microspheres for lithium ion battery anodes. Sci. Rep. 7, 46378 (2017).
Song, X. et al. Triple-shelled ZnO/ZnFe2O4 heterojunctional hollow microspheres derived from Prussian Blue analogue as high-performance acetone sensors. Sens. Actuators B Chem. 256, 374–382 (2018).
Park, G. D., Lee, J. H., Lee, J. K. & Kang, Y. C. Effect of esterification reaction of citric acid and ethylene glycol on the formation of multi-shelled cobalt oxide powders with superior electrochemical properties. Nano Res. 7, 1738–1748 (2014).
Liu, J., Qiao, S. Z., Budi Hartono, S. & Max Lu, G. Q. Monodisperse yolk–shell nanoparticles with a hierarchical porous structure for delivery vehicles and nanoreactors. Angew. Chem. Int. Ed. 49, 4981–4985 (2010).
Jiang, Z. et al. Nature-inspired catalyst for visible-light-driven photocatalytic CO2 reduction. Energy Environ. Sci. 11, 2382–2389 (2018).
Nie, N., Zhang, L., Fu, J., Cheng, B. & Yu, J. Self-assembled hierarchical direct Z-scheme g-C3N4/ZnO microspheres with enhanced photocatalytic CO2 reduction performance. Appl. Surf. Sci. 441, 12–22 (2018).
Li, X. et al. Engineering heterogeneous semiconductors for solar water splitting. J. Mater. Chem. A 3, 2485–2534 (2015).
Waqas, M. et al. Multi-shelled TiO2/Fe2TiO5 heterostructured hollow microspheres for enhanced solar water oxidation. Nano Res. 10, 3920–3928 (2017).
Chen, Z. et al. Accelerating materials development for photoelectrochemical hydrogen production: standards for methods, definitions, and reporting protocols. J. Mater. Res. 25, 3–16 (2010).
Buriak, J. M. et al. Virtual issue on best practices for reporting the properties of materials and devices. Chem. Mater. 28, 3525–3526 (2016).
Sayed, M., Zhang, L. & Yu, J. Plasmon-induced interfacial charge-transfer transition prompts enhanced CO2 photoreduction over Cu/Cu2O octahedrons. Chem. Eng. J. 397, 125390 (2020).
Deng, H., Xu, F., Cheng, B., Yu, J. & Ho, W. Photocatalytic CO2 reduction of C/ZnO nanofibers enhanced by an Ni-NiS cocatalyst. Nanoscale 12, 7206–7213 (2020).
Chen, X., Zhang, H., Zhang, D., Miao, Y. & Li, G. Controllable synthesis of mesoporous multi-shelled ZnO microspheres as efficient photocatalysts for NO oxidation. Appl. Surf. Sci. 435, 468–475 (2018).
Ma, Q., Lv, X., Wang, Y. & Chen, J. Optical and photocatalytic properties of Mn doped flower-like ZnO hierarchical structures. Opt. Mater. 60, 86–93 (2016).
Qi, K., Cheng, B., Yu, J. & Ho, W. Review on the improvement of the photocatalytic and antibacterial activities of ZnO. J. Alloys Compd. 727, 792–820 (2017).
Strelchuk, V. V. et al. Optical and structural properties of Mn-doped ZnO nanorods grown by aqueous chemical growth for spintronic applications. Thin Solid Films 601, 22–27 (2016).
Dai, B. et al. Construction of infrared-light-responsive photoinduced carriers driver for enhanced photocatalytic hydrogen evolution. Adv. Mater. 32, 1906361 (2020).
Zhu, Y.-P. et al. Highly stable phosphonate‐based MOFs with engineered bandgaps for efficient photocatalytic hydrogen production. Adv. Mater. 32, 1906368 (2020).
Kuang, P., Sayed, M., Fan, J., Cheng, B. & Yu, J. 3D graphene-based H2-production photocatalyst and electrocatalyst. Adv. Energy Mater. 10, 1903802 (2020).
Yu, J., Liu, S. & Zhou, M. Enhanced photocalytic activity of hollow anatase microspheres by Sn4+ incorporation. J. Phys. Chem. C 112, 2050–2057 (2008).
Abe, H. et al. Mesoporous metallic rhodium nanoparticles. Nat. Commun. 8, 15581 (2017).
Zhang, A. et al. Harmonizing the electronic structures of the adsorbate and catalysts for efficient CO2 reduction. Nano Lett. 19, 6547–6553 (2019).
Stanbury, M., Compain, J.-D. & Chardon-Noblat, S. Electro and photoreduction of CO2 driven by manganese-carbonyl molecular catalysts. Coord. Chem. Rev. 361, 120–137 (2018).
Najafpour, M. M. et al. Manganese compounds as water-oxidizing catalysts: from the natural water-oxidizing complex to nanosized manganese oxide structures. Chem. Rev. 116, 2886–2936 (2016).
Huang, J., Zhong, S., Dai, Y., Liu, C. & Zhang, H. J. Effect of MnO2 phase structure on the oxidative reactivity toward bisphenol A degradation. Environ. Sci. Technol. 52, 11309–11318 (2018).
Huang, J. et al. Effects of MnO2 of different structures on activation of peroxymonosulfate for bisphenol A degradation under acidic conditions. Chem. Eng. J. 370, 906–915 (2019).
Sambandam, B., Michael, R. J. V. & Manoharan, P. T. Oxygen vacancies and intense luminescence in manganese loaded ZnO microflowers for visible light water splitting. Nanoscale 7, 13935–13942 (2015).
Guo, Y. et al. Solution-based doping of manganese into colloidal ZnO nanorods. J. Phys. Chem. C 112, 8832–8838 (2008).
Yu, W., Zhang, J. & Peng, T. New insight into the enhanced photocatalytic activity of N-, C- and S-doped ZnO photocatalysts. Appl. Catal. B Environ. 181, 220–227 (2016).
Wang, W., Xu, D., Cheng, B., Yu, J. & Jiang, C. Hybrid carbon@TiO2 hollow spheres with enhanced photocatalytic CO2 reduction activity. J. Mater. Chem. A 5, 5020–5029 (2017).
Tedstone, A. A., Lewis, D. J. & Brien, P. O. Synthesis, properties, and applications of transition metal-doped layered transition metal dichalcogenides. Chem. Mater. 28, 1965–1974 (2016).
Huang, Y. et al. Atomically engineering activation sites onto metallic 1T-MoS2 catalysts for enhanced electrochemical hydrogen evolution. Nat. Commun. 10, 982 (2019).
Xia, P. et al. Designing defective crystalline carbon nitride to enable selective CO2 photoreduction in the gas phase. Adv. Funct. Mater. 29, 1900093 (2019).
Santos, V. P. et al. Structural and chemical disorder of cryptomelane promoted by alkali doping: influence on catalytic properties. J. Catal. 293, 165–174 (2012).
Acknowledgements
This work was supported by NSFC (51872220, 51961135303, 51932007, 21871217, 52073223, U1905215, and 52003213) and the Fundamental Research Funds for the Central Universities (WUT: 2020III027).
Author information
Authors and Affiliations
Contributions
M.S., F.X., P.K., J.L., L.Z., and J.Y. conceived and designed the experiments. M.S., F.X., and P.K. carried out the synthesis of the materials, the photocatalytic test, and the characterizations of the materials. M.S., F.X., J.L., S.W., L.Z., and J.Y. conducted data analysis. J.Y. supervised the project. M.S. and F.X. wrote the paper. L.Z. and J.Y. revised and reviewed the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Jeffrey Chi-Sheng Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sayed, M., Xu, F., Kuang, P. et al. Sustained CO2-photoreduction activity and high selectivity over Mn, C-codoped ZnO core-triple shell hollow spheres. Nat Commun 12, 4936 (2021). https://doi.org/10.1038/s41467-021-25007-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-021-25007-6
This article is cited by
-
Ohmic contact Metal/ZnO heterostructure for CO2 photoreduction to CO
Rare Metals (2025)
-
NiB as a Non-noble Metal Cocatalyst Electronic Bridge to Enhance the Photocatalytic Hydrogen Production of Cd3(C3N3S3)2
Chemical Research in Chinese Universities (2025)
-
Boosting photogenerated charge accumulation of oxidated carbon nitride nanotubes for efficient CO2 photoreduction
Science China Chemistry (2025)
-
Ultrafast electron transfer at the In2O3/Nb2O5 S-scheme interface for CO2 photoreduction
Nature Communications (2024)
-
g-C3N4-based S-scheme heterojunction photocatalysts
Science China Materials (2024)
