
Abstract
Oxygen defects play an important role in many catalytic reactions. Increasing surface oxygen defects can be done through reduction treatment. However, excessive reduction blocks electron channels and deactivates the catalyst surface due to electron-trapped effects by subsurface oxygen defects. How to effectively extract electrons from subsurface oxygen defects which cannot directly interact with reactants is challenging and remains elusive. Here, we report a metallic In-embedded In2O3 nanoflake catalyst over which the turnover frequency of CO2 reduction into CO increases by a factor of 866 (7615 h−1) and 376 (2990 h−1) at the same light intensity and reaction temperature, respectively, compared to In2O3. Under electron-delocalization effect of O-In-(O)Vo-In-In structural units at the interface, the electrons in the subsurface oxygen defects are extracted and gather at surface active sites. This improves the electronic coupling with CO2 and stabilizes intermediate. The study opens up new insights for exquisite electronic manipulation of oxygen defects.
Similar content being viewed by others

Introduction
Oxygen defects play a key role in the adsorption and activation of substrates and have attracted widespread attention in the field of catalysis1,2,3,4. They are made available for most of reactions involving photocatalysis, electrocatalysis, thermocatalysis, and photothermocatalysis. In recent years, applications of oxygen defects have made significant progress in CO2 reduction5,6,7, CO oxidation8,9,10, and NH3 synthesis11,12,13. But these previous studies focused on surface oxygen defects, especially increasing the density of active sites to enhance apparent catalytic activity, while the “quality” (namely, reactivity) of oxygen defects is usually neglected.
To date, the methods of creating oxygen defects include liquid-phase reduction, CO or H2 reduction, thermal annealing in oxygen-deficient environment, flame reduction and electrochemical reduction, and interface engineering4. Among them, H2 reduction treatment is relatively simple and does not introduce other undesired impurities. However, besides surface oxygen defects, subsurface or even deeper counterparts are also produced by this method14,15. The latter is distributed below the surface layer of 5–10 nm and cannot directly interact with substrates. Usually, every oxygen defect retains two electrons when neutral coordinated O atom is removed. One feasible redox reaction at oxygen defects must require effective electron exchange between oxygen defect and substrate to weaken chemical bond of the substrate molecule. Nonetheless, the electrons of oxygen defects are usually fettered by these oxygen defects (Coulomb interaction from adjacent metal ions)8,16, retarding electron delivery to substrates and greatly reducing the catalyst activity.
Scientists have found that the metal loading on a semiconductor surface is beneficial to charge delocalization of the active sites on a semiconductor surface16,17,18,19. Intensively adopted metals were generally transition metals. Non-transition metal, such as In, has been reported to possess superior charge-conducting capability20 and hence it could be considered to delocalize charges. Furthermore, In2O3 is an ideal catalyst for studying oxygen defects because of the richness and controllability of oxygen defects7,21,22,23,24,25. Therefore, we consider a special microstructure design of the catalyst to transfer such electrons bound in the subsurface oxygen defects to the surface oxygen defects via introduction of metallic In, which is expected to promote intrinsic activity. Inspired by this, In2O3 nanoflakes containing embedded metallic In were constructed, wherein In is a native element of In2O3, and thus it possesses better compatibility and affinity with an In2O3 lattice compared to foreign metal elements. The embedded metallic In is competent for constructing subsurface-surface electron channels, which reverses the disadvantage of charge localization by subsurface oxygen defects. Detailed characterizations and performance evaluations demonstrate that the configuration of metallic In embedded in In2O3 lattice can promote the electrons of subsurface oxygen defects to transport to surface oxygen defects, which improves the “quality” of the active sites and thereby boosts the intrinsic activity of CO2 reduction (turnover frequency (TOF)).
Results and discussion
Temperature-dependent surface reconstruction of In2O3 nanoflakes
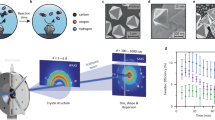
Using indium nitrate and urea as raw materials, amorphous In(OH)3 was prepared via a simple hydrothermal method, followed by calcination dehydration to obtain cubic bixbyite In2O3 (Supplementary Fig. 1). Then In2O3 was annealed in an atmosphere of mixed H2 and Ar (VH2/VAr = 1/9). As displayed in Fig. 1a and Supplementary Fig. 2, only surface reduction occurs before 300 °C whereas surface/subsurface simultaneous reduction to form metallic In appears at the temperature above 450 °C14,26. Accordingly, the catalyst (In-Em In2O3) comprising metallic In was prepared. It is worth noting that the further reduction to In-Em In2O3 does not cause any surface reduction as shown in Supplementary Fig. 2 because the surface reduction degree of In-Em In2O3 has reached the maximum. X-ray diffraction (XRD) pattern (Supplementary Fig. 3) shows characteristic diffraction peaks of metallic In. In X-ray photoelectron spectra (XPS) (Supplementary Fig. 4), In and O elements are observed in In2O3 and In-Em In2O3 and the inclusion of other elements is excluded. Field emission scanning electron microscope (FE-SEM, Supplementary Fig. 5) demonstrates the two-dimensional irregular overall morphologies of In2O3 and In-Em In2O3. Transmission electron microscope (TEM, Fig. 1b) presents the nanoflake morphology of In2O3. In-Em In2O3 displays a compact profile with shrinking size and largely decreased specific surface area relative to In2O3 (Fig. 1c, Supplementary Fig. 6, and Supplementary Table 1) but with a similar strain effect (Supplementary Fig. 7). In the high-resolution TEM image (Fig. 1d), the lattice fringes with an interplanar spacing of 2.92 and 2.72 Å for In2O3 (222) and metallic In (101) facets can be observed, respectively. In2O3 phase of In-Em In2O3 preserves the pristine facets of In2O3 (Fig. 1d and Supplementary Fig. 8). Different from the visually dark oxides, the white color of the dots is due to the absence of lattice O atoms forming a lower-density stacking structure for easier TEM electron transmission.

a Schematic temperature-dependent surface reconstruction. TEM images of (b) In2O3 and (c) In-Em In2O3. d HRTEM image of In-Em In2O3. AFM images of (e) In-Em In2O3 and f In2O3 with metallic In nanoparticles loaded on the surface (Inset: the height along the blue line). g Scheme of three types of oxygen defects.
The metallic In is most likely embedded into In2O3 nanoflakes according to the following three facts. First, based on in-situ reduction characteristics of In2O314, surface oxygen atoms are removed firstly whereupon subsurface counterparts are out to leave some voids on the surface and metallic In fills the voids (Fig. 1a). Second, numerous subsurface oxygen defects were generated and buried by metallic In (referring to temperature-programmed desorption (CO2-TPD) of Fig. 2b). Third, atom force microscopy (AFM) images (Fig. 1e, f and Supplementary Fig. 9) exhibit a noticeable difference in the undulate surface height between the embedded (The highest is ~4 nm.) and supported structures (the highest is ~6 nm, and the height of In nanoparticle in AFM is ~2 nm (Supplementary Fig. 10).), excluding the possibility of a metal-supported structure. Unlike metal nanoparticles loaded on an oxide, the metal-embedded structure can firmly immobilize metallic In, which is similar to Ni nanocatalyst embedded in a hierarchical Al2O3 matrix27 and prevent metal diffusion and agglomerate. Before the performances are discussed, it is worth mentioning that only surface oxygen defects interact with CO2, whereas subsurface counterparts are not accessible to CO2 (Fig. 1g). However, subsurface oxygen defects surrounding metallic In can indirectly interact with CO2 (referring to the statements in the last two parts).

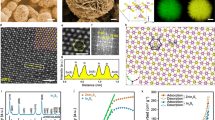
a O1s XPS spectra of In2O3 and In-Em In2O3. b CO2-TPD profiles of In2O3, In-Em In2O3, and In-Em In2O3(HCl) with HCl etching. c ESR spectra of In2O3 and In-Em In2O3 at 100 K in the dark and under light irradiation. d Lifetimes (τ) of positrons in PAS spectra of In2O3 and In-Em In2O3. e Model of distribution and structure of oxygen defects of In-Em In2O3.
Subsurface oxygen defects surrounding metallic In
Surface compositions of the catalysts were tracked via XPS spectra. The peaks of the In3d core level at 444.7 and 452.3 eV are assigned to the characteristic spin-orbit splitting 3d5/2 and 3d3/2, respectively25 (Supplementary Fig. 11). Compared with In2O3, the peaks of In-Em In2O3 shift to lower energy on account of the In0 component28. Through peak deconvolution to In-Em In2O3, two groups of characteristic splitting peaks are attributed to In3+ (444.7 and 452.3 eV, atom percent: 20.3%) and In0 (444.1 and 451.7 eV, atom percent: 79.7%), respectively (Supplementary Fig. 11). The former belongs to In2O3 phase whereas the latter aggregates to form metallic In in the In2O3 lattice. On the other hand, O1s XPS peaks for the catalysts can be deconvoluted into three bands at 529.7, 531.3, and 532.5 eV (Fig. 2a), attributed to lattice oxygen, oxygen in the vicinity of the oxygen defects, and surface −OH, respectively21,22. Although In2O3 was not treated with H2, the phase transformation from In(OH)3 to In2O3 also generated oxygen-vacancy defects29, due to undercoordinated sites. In2O3 and In-Em In2O3 exhibit the same binding energies pertaining to oxygen defects, for electrons in oxygen defects are principally centered on three adjacent In atoms rather than O atoms20,24,25. The binding energy of lattice oxygen of In-Em In2O3 is lower than that of In2O3, implying a weakened binding of In and O in In-Em In2O3 and its electron richness30. The concentrations of the oxygen defects are measured to be virtually equivalent for the two catalysts (Supplementary Fig. 12 and Supplementary Table 2). But their color and light absorption properties are dramatically different because the light absorption characteristics of metallic In endows In-Em In2O3 full-spectrum absorption (referring to the last paragraph in the performance part).
CO2-TPD profiles of the catalysts were measured to verify the number/type of oxygen defects on the surface. In2O3 presents marked chemisorption bands in CO2-TPD pattern (Fig. 2b). During catalysis at T > 300 °C, the sites for chemisorption function as active sites of CO2 reduction7,26. Unexpectedly, CO2 uptake on In-Em In2O3 becomes almost negligible and thus oxygen defects of In-Em In2O3 seem not responsible for CO2 chemisorption. Nevertheless, most of the reports have evidenced that oxygen defects of In2O3 are active sites of CO2 reduction and the O1s XPS peak in Fig. 2a proves the existence of oxygen defects of In-Em In2O3. As is well-known, XPS technology can probe the components of several-nanometer depth31 in line with the size of the metallic In of In-Em In2O3, whereas the TPD profile correlates with the adsorption property of a gas-solid interface. We speculate that oxygen defects of In-Em In2O3 exist at the interface, within a subsurface depth of 1–5 nm31, thereby mitigating the CO2 adsorption amount. Furthermore, the chemisorption appears by using HCl to remove a part of the metallic In, giving more powerful evidence on some subsurface oxygen defects being buried by metallic In. In comparison, with In2O3 etched by HCl, the CO2 adsorption amount does not change, excluding the effect of HCl on the In2O3 phase of In-Em In2O3 for increasing CO2 adsorption (Supplementary Fig. 13). On the other hand, the interface contains rich In-O–In structures as shown in Supplementary Fig. 14, which contributes to the formation of oxygen defects around metallic In32. These findings suggest that subsurface oxygen defects gather surrounding metallic In. The proportions of surface oxygen defects of In2O3 and In-Em In2O3 are estimated to be 0.032% and 0.0017%, respectively.
Then we attach importance to the local structure of the oxygen defects that exerts a substantial impact on the performance of these catalysts. Usually, it is unfavorable in thermodynamics to remove multi-oxygen atoms from the irregular octahedral InO6 unit cell (C2v group) via thermal treatment because of the rather high energy required26,33. This gives rise to the feature of O-penta-coordinated state in In2O3 consisting of one central In, five coordinated O, and one oxygen vacancy with two electrons which are paired most frequently in one general unit, being the most stable conformation of In2O333. The O-penta-coordination mode shackles an electron-transfer event since local electrons are imprisoned by a strong electrical field around oxygen defect8. With more coordinated O atoms removed via reduction, In atom clusters emerge in one general unit around oxygen defects, with more In–O–In attached on metallic In for enhanced electronic interaction between In2O3 and metallic In (Supplementary Fig. 14). Moreover, more than two electrons are required to compensate local non-balance charge around oxygen defect induced by In0 atom cluster (Supplementary Fig. 15). This In0-mediated oxygen defect structure would increase the number of free electrons in In-Em In2O3 compared with In2O3, as verified by electron spin resonance (ESR) spectra (Fig. 2c). Obviously, the ESR signal of In-Em In2O3 demonstrates a 1.6-fold enhancement related to In2O3 under light irradiation. The enhancement of In-Em In2O3 (2.1-fold) is greater in the dark than under light irradiation, suggesting that In-Em In2O3 possesses more unpaired electrons (z > 2) unbound dominantly in defect states. The ESR increase is attributed to In0-induction spin enhancement originating from electric field polarization around oxygen defect.
Positron annihilation spectroscopy (PAS) is a useful tool to unravel the microstructure of the catalysts, where the lifetime and intensity of the positrons relate to the size, relative content, and distribution density of oxygen defects34. The results from this analytical method were fitted best with three-lifetime components and the lifetimes and relative intensities of the positrons for In2O3 and In-Em In2O3 were presented in Fig. 2d and Supplementary Table 3. The small amount of the third component arises from ortho-positron annihilation inside a few large voids (defect clusters or micropores) in the catalysts35. Compared with In2O3, In-Em In2O3 displays a longer third lifetime (τ3), reflecting defect accumulation at the interface. The first component is attributed to free annihilation of the positrons by a bulk state in a crystal36,37. The dissimilarity of the first lifetimes (τ1) between In2O3 and In-Em In2O3 is only 9 picoseconds, attributable to small vacancies38,39 or shallow positron traps40 in the bulk. Such a tiny distinction also indicates their similar isolated vacancy structures (the form of O–In-(O)Vo-In–O) which could be distributed on the surface and in the bulk of In2O3 but predominantly exist in the bulk of In-Em In2O3, on account of the previous catalyst characterization on the marked difference of sub- and surface oxygen defects of In2O3 and In-Em In2O3. The second component originates from the trapping of free positrons by larger-size defects41. In-Em In2O3 presents a much higher second lifetime (τ2) than In2O3, implying that In2O3 includes oxygen-vacancy associates on the surface35 whereas larger-size defect complexes exist in In-Em In2O3, such as a metal-mediated defect complex in the form of O–In-(O)Vo-In–In (Fig. 2e).
TOF activity over In-Em In2O3 is 866 times higher than that over In2O3 under light irradiation
The photothermocatalytic CO2 reduction was conducted under light irradiation. Under the molar ratio of H2/CO2/Ar = 9/3/8 and pressure at 0.18 MPa, CO was the main product with a selectivity of 99.99%. The reaction time of CO2 reduction for calculating TOF and the mass and area-specific activity as follows is 1 h unless a special reaction time is mentioned. For apparent activity, two types of catalyst evaluation indexes were considered: mass-specific activity normalized by the mass of catalyst and area-specific activity by the surface area of the catalyst. The mass and area-specific activity over In-Em In2O3 (380 °C) are 2.7-fold (8.6 vs. 3.2 mmol g−1 h−1) and 40.0-fold (1.2 vs. 0.03 mmol m−2 h−1) higher than that over In2O3 (310 °C), respectively (Supplementary Fig. 16), at the same light intensity (8.2 W cm−2). As shown in Supplementary Fig. 17, In-Em In2O3 exhibited a stronger light-to-heat conversion in relation with In2O3. To eliminate the influence of different temperatures caused by the same light irradiation, the activities were measured at the identical reaction temperature. Their mass-specific activities are almost equivalent while the area-specific activities exhibit a difference of one order of magnitude (17-fold) (Supplementary Fig. 16). However, the apparent activity fails to reflect the exact essence of active sites; hence we evaluated the performance in terms of one active site. The CO2 adsorption site (oxygen defect) of In2O3 definitely acts as active site of CO2 reduction7,21,22,23,26. From CO2-TPD, we derived the adsorbed CO2 amount to estimate the molar quantity of active sites. Accordingly, there exists an incredible distinction between In2O3 and In-Em In2O3 in the TOF activities (mass-specific activity/number of active sites, see methods) under identical light intensity (866-fold) and identical reaction temperature (376-fold) (Fig. 3a). The reason for this obvious difference will be discussed in the following section. Moreover, TOF activity of CO production over the catalyst surpasses that over most of the reported catalysts (Supplementary Table 4).

a TOF activities over In2O3, In-Em In2O3 and In-Sup In2O3. b Cycling experiment over In-Em In2O3 for 10 runs. c Reaction temperature dependence of CO2 conversion, the corresponding curve of ln k (k: rate constant) vs. 1000/(T + 273.15) and corresponding ∆H≠ and ∆S≠ extracted from Eyring plot. d CO2 conversion over In2O3, In-Em In2O3 and In-Sup In2O3 under light irradiation. Note that insufficient temperature was compensated via electric heating to 300 °C. (The error bars represent standard deviation).
The TOF activity over In-Em In2O3 turns out to be higher than that over In2O3. But the active sites for the calculations only correspond to ones before reaction. As we know, oxygen defects could undergo transformations during catalysis: oxygen defects could be replenished by foreign oxygen from COOH intermediate dissociation; the neighboring chemical composition or the structure of oxygen defects could be altered42. To avoid the change in the concentration of oxygen defects and retain the structure of initial oxygen defects in the process of catalysis as far as possible, low-temperature and short-time reactions were conducted, respectively (Supplementary Fig. 18). First, the low-temperature reaction was operated at 250 °C over 30 min and the initial rate over one active site of In-Em In2O3 becomes 557-fold faster than that of In2O3. Then, turnover numbers (TONs) were measured at 350 °C over 10 min and In-Em In2O3 exhibits 327-fold higher vs. In2O3. These results further verify that the active sites of In-Em In2O3 have much stronger catalytic function than that of In2O3 (two orders of magnitude enhancement).
We have measured the time-dependent catalytic activity over In-Em In2O3 to verify the stability of the catalyst. Under the same condition (300 °C, H2/CO2/Ar = 9:3:8), the catalytic reaction over In-Em In2O3 was cycled for ten runs (the reaction time of each run was 1 h). The result shows that the catalytic cycling performance over In-Em In2O3 is stable (Fig. 3b). The content, structure and composition of oxygen defects of the spent catalyst were revealed by the characterization measurement for the nature of performance stability (Supplementary Fig. 19 and Supplementary Table 5).
The improvement of TOF activity is closely related to the microstructure of the catalyst, such as the transition of composition and phase. The formation of metallic In in In2O3 is the most significant change between In2O3 and In-Em In2O3 in their microstructures. Therefore, the correlation between the catalyst performance and metallic In was investigated. We measured a suite of activities over In2O3 at different reaction temperatures and obtained the initial curve (CO2 conversion vs. T, dashed line) and the derived curve (ln k vs. 1000/(T + 273.15), k is rate constant, solid line) in Fig. 3c. Based on the Arrhenius equation which describes the relationship between the reaction rate constant and reaction temperature, the change in the apparent activation energy can be reflected by the slope of the derived curve. If the derived plot is a straight line, its apparent activation energy remains. But in fact, it is a curve, signifying that the active component around the oxygen defects of In2O3 varies with reaction temperatures and a new phase (metallic In) in In2O3 appeared upon elevating reaction temperatures. The high-temperature part of the curve is consistent with the apparent activation energy over In-Em In2O3. Moreover, this suggests that the catalyst underwent distinctly different reaction pathways43. Indeed, after a high-temperature reaction, a part of the In2O3 was reduced to metallic In (Supplementary Fig. 20). The result indicates that metallic In modifies the structure of the oxygen defects and engages in the TOF activity enhancement.
The transition-state theory can describe the reactivity of the catalyst, and the changes in enthalpy (∆H≠) and entropy (∆S≠) of the transition state are given by the Eyring formula (the plot of ln (k/T) vs. 1/T) for the rate-limiting step. ∆H≠ and ∆S≠ on In2O3 at the low and high temperature were calculated to ∆H≠ (18.1 and 78.6 J mol−1) and ∆S≠ (−179.3 and −69.4 J mol−1 K−1), respectively. The transition-state complex is penalized by the negative entropy, but is strongly chemisorbed by oxygen defects. In addition, the reaction underwent different activated complexes. The complex is dominantly controlled by entropy because of H/TS« 1, and the entropy reflects the bound state of the transition state at the active site. The much more favorable entropy change in high-temperature reaction indicates that the transition state is in a more disordered state, driving its transformation to products more readily. Meanwhile, this also gives rise to a lower Gibbs free energy of activation at a constant temperature.
In addition, we adopted mild hydrogen peroxide to oxidize the surface of metallic In of In-Em In2O3 (named as In-Em In2O3(H2O2)), forming a thin layer of In2O3 with more surface oxygen defects. This can create more surface oxygen defects around metallic In. The performance evaluation displays the enhanced activity over In-Em In2O3(H2O2) compared with In-Em In2O3 (Supplementary Fig. 21). The characterization (Supplementary Fig. 22) suggests that the overall structure of In-Em In2O3(H2O2) changes little, but the content of surface oxygen defects around increases. This further verifies that the oxygen defects around metallic In exhibit the stronger reactivity.
To eliminate the role of metallic In as a co-catalyst, metallic In supported In2O3 (In-Sup In2O3) was prepared44. XRD patterns show that the catalyst contains In2O3 and metallic In and the amount is similar with that of In-Em In2O3 (Supplementary Fig. 23). The 2θ of In-Sup In2O3 moves to the lower, suggesting a strong interaction between metallic In and In2O3 in In-Sup In2O3. In-Sup In2O3 does not contain other foreign substances with washed by large amount of water/alcohol (Supplementary Fig. 24). The activities over In2O3, In-Sup In2O3 and In-Em In2O3 were compared at the same temperature (Fig. 3d) and the result suggests that there is no obvious change in their activities upon changing the light intensity, consistent with Supplementary Fig. 25, corroborating the slight effect of the photocatalysis and charge transfer on CO2 reduction. If metallic In acts as the photogenerated electron separator, with light irradiation, more electrons are injected into metallic In and the catalyst performance would be improved. However, the activity over In-Sup In2O3 is lower than that over In2O3 (Fig. 3d), thus the effect of electron promoter of metallic In is excluded. Moreover, compared with In-Em In2O3, the activity over In-Sup In2O3 is significantly decreased under light irradiation (Fig. 3d), despite the markedly higher CO2 adsorption for In-Sup In2O3 (Supplementary Fig. 26), indicating that metallic In is not the co-catalyst for favoring the dissociation of C–O of CO2 or COOH intermediate. As expected, TOF activity over In-Sup In2O3 is so much lower than that over In-Em In2O3 (Fig. 3a), suggesting that the simple In-supporting structure cannot improve intrinsic activity.
Photothermal conversion over the system is one of the key factors dictating the photothermocatalytic performance of CO2 reduction. The photothermal conversion capability of In2O3 is rather low (Supplementary Fig. 17a) because of the light absorption of the wavelength below 500 nm and dominant radiative emission. Compared with In2O3, In-Em In2O3 displays a more efficient photothermal conversion due to full-spectral light absorption (to near-infrared light) and a high probability of nonradiative relaxation (Supplementary Fig. 17). The photothermal effect of In-Em In2O3 originates from oxygen defects and light absorption characteristics of metallic In. As reported, oxygen defects can create mid-gap energy state and thus increase light-to-heat conversion due to enhanced light absorption7 and “trap-assisted recombination”45. Compared with oxygen defects, metallic In of In-Em In2O3 plays a dominant role in light-to-heat conversion which heats up the metal lattice by electron–phonon scattering. Due to superior thermal conduction of metallic In, the concentrated energy in metallic In is then rapidly transferred to the active site of In2O3 portion for CO2 reduction via phonon-phonon relaxation46. However, in the future, it is worth investigating which of the light absorption modes from metallic In exhibits the highest efficiency of light-to-heat conversion, including interband-transition absorption, intraband-transition absorption, and plasmon-resonance absorption45,46,47.
Electron delocalization among oxygen defects
The capability to activate CO2 over the catalyst is closely linked with the electron density of the active sites which is correlated with electron transfer. Variable temperature ESR spectroscopy (Fig. 4a and Supplementary Fig. 27) can be used to analyze the kinetic behavior of electron transfer in oxygen defect at the interface between In2O3 and metallic In. The dynamics includes two processes: (a) electron injection from In2O3 to metallic In and (b) electron migration from the subsurface to the surface. At a constant temperature of 100 K, the ESR signal was tested in the dark and under light irradiation, respectively. In the dark, the ESR signal of In-Em In2O3 is twice that of In2O3. At this time, the unpaired electrons in the catalyst are in an equilibrium state, and the population of unpaired electrons of In-Em In2O3 is higher than that of In2O3. The previous characterization demonstrates that the sum of their oxygen defects appears the same, and in general, each oxygen defect contains two electrons4. But as for In-Em In2O3, the average number of electrons per oxygen defect is apparently greater than two, implying the sharing of electrons among oxygen defects. Irradiation to the catalyst produces some non-equilibrium electrons, of which the population in In-Em In2O3 is 2.1 times higher, related to In2O3. Under the interface polarization force, some of the non-equilibrium electrons generated in the In2O3 phase are injected into the metallic In, and the O–In-VO(O)-In–In structure acts as an electron “bridge” (Fig. 4b).

a Temperature-dependent change of unpaired electrons in In2O3 and In-Em In2O3 at 100, 200, and 300 K under light irradiation and numbers of unpaired electrons at 100 K in the dark. b The schematic presence of electron transfer at the interface and its driving forces. c Normalized In K-edge XANES spectra of In2O3 and In-Em In2O3. d LDOS of In–In2O3 near the Fermi level (N: number of energy levels) and orbital contribution in LDOS of In–In2O3. Charge distribution derived from the wave function of the energy levels at a CBM of (e) In2O3 and (f) In–In2O3.
The ESR signal shows temperature-dependent decay, mainly due to the increase in electron conduction resistance with temperature increasing. In-Em In2O3 exhibits a faster ESR signal decaying rate compared with In2O3 (Fig. 4a), implying that unpaired electrons are transported along a different approach in the In-Em In2O3; actually, the unpaired electrons of In-Em In2O3 move along metallic In after electron injection, because the resistance of metallic In increases faster with a temperature rising compared with In2O3, which results in a faster ESR signal decay in In-Em In2O3. Surface oxygen defects are affected by adsorption state and surface state. The number of surface microscopic states turns out to be greater than that of the bulk counterpart, that is, entropy (S) divergence of Ssurface > Sbulk, therefore, the chemical potential of the bulk phase is often higher than that of the surface phase, driving the electrons in subsurface oxygen defects to transport to surface counterparts (Fig. 4b), thereby incrementing the number of unpaired electrons in active sites.
The migration of electrons from the subsurface to the surface is affected by a relatively large resistance in the oxide semiconductor, causing them to be annihilated during the movement through electron-hole recombination, electron–phonon and electron–electron interaction. The mean free path of electrons in oxide semiconductors is less than 5 nm regardless of bound force from metallic ions around, thus only surface electrons would participate in the reaction. The metallic In greatly reduces the resistance to electron migration through the lattice from the subsurface to the surface (Maximum conductivity multiple is 12 orders of magnitude compared with semiconductors48.) and increases the mean free path of electrons. Without feeding with reactants, the electrons stay in equilibrium between the surface and the subsurface. Due to the existence of the surface state, the electron concentration on the surface presents generally higher than that of the subsurface. The surface/subsurface electron-density ratio of In-Em In2O3 is greater than that of In2O3 due to the enrichment of electrons via metallic In. When the active site interacts with the reactant electronically, the electron density on the surface decreases, and the redistribution of the subsurface electrons is required to maintain a dynamic equilibrium state.
Electronic characteristics of oxygen defects were also explored via PAS. The positron trapping rates were calculated, which is proportional to the number of defects and density of negative charges in an individual defect. The positron trapping rate is directed against the second component exclusively pertaining to defects37. As shown in Supplementary Figs. 28 and 29, though In2O3 surpasses In-Em In2O3 in the positron trapping rate, the trapping rate constant over In-Em In2O3 is higher, implying that the defects of In-Em In2O3 have a stronger capability to trap positrons, namely, more negative charges. Besides, the electrons in the defects of In-Em In2O3 display higher delocalization energy (Supplementary Fig. 30). Therefore, different from electrons trapped in the oxygen defects of In2O3, electrons in the oxygen defects of In-Em In2O3 are shared by each other at the interface.
Metallic In is responsible to electron delocalization at the interface and more essentially, the O–In-(O)Vo-In–In structure of In-Em In2O3 makes contribution. Therefore, the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra were measured. In the XANES spectra (Fig. 4c), the tiny energy difference (0.44 eV) of the K-edge absorption at 27922 eV between In2O3 and In-Em In2O3 suggests that the crystal network of In-Em In2O3 remains. Moreover, compared with the In2O3 stander, their K-edge absorption is shifted to the lower energy, attributed to a lower average oxidation state of In species which corresponds to a lower coordination around In atoms, namely, oxygen defects7,49,50. Metallic In of In-Em In2O3 does not exhibit obvious electronic feature of In foil, because of the similar In–In bonding lengths of In2O3 and metallic In and low sensitivity to low ordered structure of metallic In51. The intensity of the “white line” of In species in In-Em In2O3 is lower than that in In2O3 (Fig. 4c, inset), attributable to a slower electron transition52 from the 1s orbital to unoccupied N4,5 states. The electron transition needs to reach certain energy. The reduction of the electron-transition probability is because the delocalization of the electron reduces the population of high-energy electrons. The R and K space EXAFS curves and wavelet transform analysis are shown in Supplementary Figs. 31–34. The fitting of the EXAFS result (Supplementary Table 6) gives a lower coordination number and higher Debye-Waller factor for In-Em In2O3, implying a more disordered surface structure. The In–In coordination for In-Em In2O3 could correspond to the dsp2 hybrid at the interface whereas the lattice region in In2O3 predominantly to the d2sp3 hybrid for the octahedral unit. Obviously, In-Em In2O3 possesses more s-orbital and less d-orbital components, and consequently, the overlapping degree of In–In orbitals turns greater due to high dispersion of s-orbital. Therefore, In-Em In2O3 exhibits a stronger electron-delocalization effect at the interface. Moreover, the O–In-(O)Vo-In–In structure partly contributes to the In–In shell and thus facilitates a larger extent of electron delocalization.
Density functional theoretical (DFT) calculations were carried out to verify the electron delocalization. Local density of states (LDOS) of defective In2O3 (Supplementary Fig. 35a) presents a small LDOS defect state dominated by an unoccupied In5s level in the bandgap. LDOS in Supplementary Fig. 35b displays metallic continuity behaviors of metallic In that can match well with defect state and delocalize electrons of defect state53,54. Upon interaction of In2O3 with metallic In of low coverages, the Fermi level of In2O3 shifts to higher energy and a new free-electron-like band appears near Fermi level30. On the In2O3 slab with one oxygen vacancy, some In atom clusters were constructed (donated as In–In2O3) for LDOS calculation. The overall energy band of In–In2O3 remains unchanged (Supplementary Fig. 36), complying with the XANES and EXAFS results. However, only one defect state (0.30 eV) appears in the bandgap of In2O3 while In–In2O3 has continuous defect states (Fig. 4d) which suggests the shareability of electrons of oxygen defects. Integrating LDOS(E) from the band bottom to the Fermi level can get the electron occupied energy levels (N) in the defect state, NIn2O3 = 1.2 and NIn-In2O3 = 6.3, so the corresponding filling electron number is NIn2O3 = 2.4 and NIn-In2O3 = 12.6, respectively. This defect state belongs to the s–p band and exhibits a good electron delocalization between defect states (Fig. 4d). In addition, the charges in the valence band maximum (VBM) and the conduction band minimum (CBM) of In2O3 are restricted in the oxygen defects, while the charges in the CBM of In–In2O3 demonstrate more extensive distribution (Fig. 4e, f and Supplementary Fig. 37). Consequently, the active sites on the surface converge more available electrons from subsurface oxygen defects to ensure successful CO2 activation.
Enhanced CO2 adsorption and activation
To verify the existence of the electron exchange between the active sites of In-Em In2O3 and CO2, Fe3+ salt was selected as an electron scavenger. As expected, the grinding mixture of In-Em In2O3 and Fe3+ salt markedly attenuated its photothermocatalytic and thermocatalytic performances (Supplementary Fig. 38), confirming that the electrons are key active species for CO2 reduction. At room temperature, CO2 adsorption over In2O3 and In-Em In2O3 was measured through Fourier transform infrared (FT-IR) spectroscopy (Supplementary Fig. 39). Enhanced adsorption of CO2 on In-Em In2O3 is observed (Fig. 5a) and CO2 adsorption band shifts from 1304.5 cm−1 for In2O3 to 1286.3 cm−1 for In-Em In2O3 because of more electrons delivered to the anti-bond orbital of CO2. The peaks at 3500–3800 cm−1 in FT-IR spectra were chosen as the standard of the CO2 adsorption amount, which are assigned to a combination mode (ν1 + ν3) of the adsorbed CO255. In Fig. 5b, In-Em In2O3 performs a significantly faster CO2 adsorption rate compared with In2O3 and reached chemical equilibrium within 110–130 s. DFT calculations were carried out to further unravel the electronic interaction between the active sites and the reactants/intermediates. The binding configurations of each adsorbate on the surfaces of In2O3 and In–In2O3 were obtained through theoretical structural optimization (Supplementary Fig. 40, 41), respectively. Adsorption energies were employed to evaluate the strength of the electronic interaction. By contrast with In2O3, all the adsorption energies over In–In2O3 are more negative especially H uptake (Fig. 5c), indicating facile CO2 hydrogenation and COOH dissociation at the active sites via consecutive electron-transfer events. The stronger interaction prolongs the residence times of the adsorbates at the interface, 108-fold for CO2 and 1029-fold for hydrogen higher than that on In2O3 calculated from the equation
where τ is the lifetime of adsorbate, Eads is adsorption energy, R is the molar gas constant, T is temperature and τ0 is the lifetime of a surface vibration (~10−13 s)56. Therefore, the steady-state concentrations of CO2 and H are extremely high at the interface, favoring intermolecular collision. The adsorption energies of CO2 and H on metallic In were calculated to be −0.081 and −1.058 eV, respectively. The adsorption energies of CO2 and H on In–In2O3 (namely at the interface between metallic In and In2O3) are much lower than that on In2O3 and metallic In, which most likely evidence that the active sites are at the interface between metallic In and In2O3. This is consistent with the conclusion above that the oxygen defects at the interface function as the active sites for CO2 reduction. In addition, electron exchange capability can be deduced by the change of the C–O bond length. Over In–In2O3, the bond of CO2 is elongated by 0.099 Å while it is only extended by 0.034 Å over In2O3 (Fig. 5d), indicating the stronger electronic interaction with CO2 over In–In2O3. Noteworthy, only one step is required to vigorously activate the adsorbed CO2 into COOH over In–In2O3, whereas it takes multiple steps of energy conversion to convert CO2 to CO over In2O3. The advantages (Ad.) of electron richness from electron delocalization of subsurface oxygen defects are illustrated in Fig. 5e.

a FT-IR spectra of CO2 adsorption in the range of 1200–1400 cm−1. b Normalized CO2 adsorption amount by the number of active sites. c Adsorption energies on In2O3 and In–In2O3. d Change in C–O bond lengths over In2O3 and In–In2O3. e The schematic picture emphasizes three types of catalytic roles for CO2 reduction over In-Em In2O3.
The reaction pathway in the system is clarified including the roles of heat, photogenerated carrier and H2 in CO2 reduction. For the present photothermocatalysis, the main contribution comes from the thermochemical pathway generated by light irradiation while the photochemical pathway makes minor contribution (Supplementary Fig. 25). Therefore, the catalytic reaction is called light-induced thermocatalysis47. Here, a population of photons are absorbed by metallic In portion and oxygen defects (via “trap-assisted recombination”) and converted into thermal energy, respectively. The thermal chemistry facilitates the transfer of charge carrier, the excited vibration of the related species and the formation of the phonon of the ground state of In2O3, which lower the reaction barrier of CO2 reduction. Simultaneously, some photons are absorbed by In2O3 portion, forming photogenerated carriers. There are two types of evolution directions for the photogenerated carriers. One way is electron-hole recombination generating more heat, which is dominant, and the other is to be transferred to CO2 adsorbed. It is worth noting that most of the photogenerated charges in In2O3 portion are not delivered to CO2 adsorbed specifically at the oxygen defects on the surface because of the very small chemical reaction region in spite of increasing ESR signals upon light irradiation as demonstrated in Supplementary Fig. 27. If the photogenerated electrons can interact with CO2 adsorbed, the energetic electrons would be injected, generating anion species57. Otherwise, the photochemical pathway makes a minor contribution to the catalysis. The H2 is dissociated into two H species in either a heterolytic or a homolytic way with the participation of lattice In and O58,59,60,61, which binds with CO2 to form COOH or binds with OH from the dissociation of COOH to form H2O, respectively7. However, the atmosphere including H2 would not increment the number of surface oxygen defects of In2O3 and the amount of metallic In during the catalytic process, as indicated by H2-TPR pattern (Supplementary Fig. 2) and XRD pattern (Supplementary Fig. 19a).
In summary, a strategy of extracting electrons in subsurface oxygen defects for CO2 reduction was reported via constructing In-Em In2O3 nanoflake with metallic In embedded. The oxygen defects of In–In2O3 are distributed at the interface between In2O3 and metallic In and comprise large-size defect complexes featuring the basic structure of O–In-(O)Vo-In–In. In-Em In2O3 exhibits remarkably higher TOF activities than In2O3 under light irradiation. The O–In-(O)Vo-In–In structure at the interface engenders delocalization of electrons in the subsurface oxygen defects mediated by metallic In that greatly increases the electron density of the active sites, facilitating electron exchange between the active site and CO2 and stabilizing COOH intermediate. The study helps to understand the active sites of In2O3 and paves the way to develop new catalysts and improve catalyst performance involving oxygen defects.
Methods
Preparation of In2O3
In(NO3)3•4H2O (0.013 mol, 5.0 g) and urea (0.039 mol, 2.4 g) were dissolved in deionized water (600 mL), followed by magnetic stirring for 15 min. The obtained solution was transferred into a Teflon-lined autoclave. The autoclave was sealed and heated at 140 °C for 16 h. After cooling down, the suspension was centrifuged and washed with deionized water. The white solid was dried overnight under vacuum at 60 °C and then underwent calcination in a muffle furnace at 300 °C (with the temperature-ramp rate of 7~8 °C min−1) for 3 h to produce a yellow powder.
Preparation of In-Em In2O3
After grinded with a mortar, the virgin In2O3 was placed in a porcelain boat without a lid and underwent calcination at different times or temperatures in H2/Ar (1/9) atmosphere with a temperature-ramp time of 20 min.
Preparation of In-Sup In2O3
According to the literature44, highly dispersed metallic In in H2O was prepared first. InCl3•4H2O (2.5 mmol, 735 mg) and disodium citrate hydrate (1.9 mmol, 500 mg) were dissolved in diethylene glycol (100 mL) in a three-necked flask. Under N2 protection and vigorous stirring, the solution was heated to 100 °C. Subsequently, NaBH4 (25.0 mmol, 945 mg) was dissolved in deionized water (2 mL) and added to the solution which finally turns dark brown. The reaction continued to be stirred for 5 min at 100 °C. After cooling down, the suspension was centrifuged and washed with a large amount of alcohol and deionized water. The obtained precipitate was dispersed in deionized water (30 mL) and In2O3 (300 mg) was added. After stirring for 30 min, the suspension was centrifuged and washed with a large amount of deionized water. The gray solid was dried overnight under vacuum at 60 °C.
Preparation of In-Em In2O3(H2O2)
200 mg of In-Em In2O3 was added into 1% diluted H2O2 solution. After ultrasonic treatment, the dispersion was stirred for 3 h, followed by centrifugation and drying under vacuum at 60 °C.
Catalyst characterization
The morphologies of the catalysts were characterized by FE-SEM (JEOL JEM-6700F) at a working voltage of 8 kV, TEM (HITACHI H-7000FA, 100 kV), high-resolution TEM (JEM 2100 F) and AFM (BRUKER Dimension Icon). XRD patterns were recorded on an X-Pert diffractometer (BRUKER D8 ADVANCE) equipped with graphite monochromatized Cu-Kα radiation. XPS were obtained on a ThermoFisher EscaLab 250Xi using monochromatic Al Kα source (Ephoton = 1486.6 eV) with 10 mA filament current and 14.7 keV filament voltage source energy spectrometer (Correction value of C1s in the XPS spectra was 284.7 eV.). CO2-TPD and H2-TPR curves were carried out on an Auto Chem II2920 chemisorption apparatus with a temperature-ramp rate of 10 °C min−1 after pretreatment at 200 °C for 0.5 h in Ar. Specific surface area and pore size distribution were measured through a high-speed automated surface area and pore size analyzer (TriStar II 3020 V1.03.01) using the multipoint Brunauer-Emmet-Teller (BET) analysis method. Absorption spectra were analyzed through an Agilent Cary60 spectrophotometer and steady/transient fluorescence spectra were measured with a FLS1000 fluorescence spectrometer. Raman spectra were acquired using a Thermo Scientific DXR Raman Microscope at the laser excitation wavelength of 780 nm and an intensity of 20 mW. TG analysis was performed on a Mettler Toledo TGA/DSC 1STARe system (gas flow rate: 15 mL min−1, temperature range: from 50 to 900 °C, temperature-ramp rate: 10 K min−1). ESR data were collected using a Bruker EMXmicro-6 X-band spectrometer. PAS was measured via the apparatus DPLS3000 (the size of the sample film is 12 × 12 × 2 mm3). XANES and EXAFS spectra at In K-edges were recorded at the XAS station (BL14W1) of the Shanghai Synchrotron Radiation Facility using the method given in the literature62.
Performance evaluation
Photothermocatalytic CO2 reduction in the presence of H2 was conducted in a sealed batch-type reaction system. After air evacuation of the reaction vessel, the mixed feed gases containing H2, CO2, and Ar with the molar ratio of H2/CO2/Ar = 9/3/8 were introduced. The photothermocatalytic CO2 reduction proceeded under light irradiation equipped with a 300 W Xe lamp (Beijing Perfectlight Technology Co., Ltd. PLS-SXE-300DUV) over 1 h. For all of these experiments, 100 mg of samples were weighed and spread onto a round shape air-permeable quartz fiber filter. The quartz fiber filter film was fixed on the stage of the reactor. The tip of the thermometer was maintained an intimate contact with the sample. The initial pressure in the reactor was kept at 0.18 MPa. The reaction temperature can be adjusted by changing the magnitude of the current of Xe lamp: first, adjust the light-irradiation current to 16 A to increase the reaction temperature; second, finely tune the position of the reactor to make the stable temperature reach the maximum; third, slowly increase the current to make the reaction temperature reach 300 °C. The adjustment time was controlled within 5 min. The reaction gas in the reaction system was collected and measured through a gas chromatograph (Agilent 7890B) equipped with a combination of Porapak Q, Molsieve 5 Å columns, and a thermal conductivity detector which can detect CO2, O2, N2, CH4, and CO.
Where the CO production refers to one over 1 h.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable requests.
References
Campbell, C. T. & Peden, C. H. F. Oxygen vacancies and catalysis on ceria surfaces. Science 309, 713–714 (2005).
Jia, J. et al. Heterogeneous catalytic hydrogenation of CO2 by metal oxides: defect engineering-perfecting imperfection. Chem. Soc. Rev. 46, 4631–4644 (2017).
Wang, G., Yang, Y., Han, D. & Li, Y. Oxygen defective metal oxides for energy conversion and storage. Nano Today 13, 23–39 (2017).
Zhuang, G., Chen, Y., Zhuang, Z., Yu, Y. & Yu, J. Oxygen vacancies in metal oxides: recent progress towards advanced catalyst design. Sci. China Mater. 63, 2089–2118 (2020).
Li, Y. et al. Plasmonic hot electrons from oxygen vacancies for infrared light-driven catalytic CO2 reduction on Bi2O3-x. Angew. Chem. Int. Ed. 60, 910–916 (2021).
Qi, Y. et al. Fabrication of black In2O3 with dense oxygen vacancy through dual functional carbon doping for enhancing photothermal CO2 hydrogenation. Adv. Funct. Mater. 31, 2100908 (2021).
Qi, Y. et al. Photoinduced defect engineering: enhanced photothermal catalytic performance of 2D black In2O3-x nanosheets with bifunctional oxygen vacancies. Adv. Mater. 32, 1903915 (2020).
Huang, J. et al. Manipulating atomic structures at the Au/TiO2 interface for O2 activation. J. Am. Chem. Soc. 142, 6456–6460 (2020).
Jiang, D. et al. Elucidation of the active sites in single-atom Pd1/CeO2 catalysts for low-temperature CO oxidation. ACS Catal. 10, 11356–11364 (2020).
Wang, Y., Widmann, D. & Behm, R. J. Influence of TiO2 bulk defects on CO adsorption and CO oxidation on Au/TiO2: electronic metal-support interactions (EMSIs) in supported Au catalysts. ACS Catal. 7, 2339–2345 (2017).
Sun, Z. et al. Oxygen vacancy enables electrochemical N2 fixation over WO3 with tailored structure. Nano Energy 62, 869–875 (2019).
Zhang, N. et al. Refining defect states in W18O49 by Mo doping: a strategy for tuning N2 activation towards solar-driven nitrogen fixation. J. Am. Chem. Soc. 140, 9434–9443 (2018).
Zhao, Y. et al. Tuning oxygen vacancies in ultrathin TiO2 nanosheets to boost photocatalytic nitrogen fixation up to 700 nm. Adv. Mater. 31, 1806482 (2019).
Bielz, T. et al. Hydrogen on In2O3: reducibility, bonding, defect formation, and reactivity. J. Phys. Chem. C. 114, 9022–9029 (2010).
Esch, F. et al. Electron localization determines defect formation on ceria substrates. Science 309, 752–755 (2005).
Wei, Z. et al. Steering electron-hole migration pathways using oxygen vacancies in tungsten oxides to enhance their photocatalytic oxygen evolution performance. Angew. Chem. Int. Ed. 60, 8236–8242 (2021).
Bi, Y. et al. Selective growth of Ag3PO4 submicro-cubes on Ag nanowires to fabricate necklace-like heterostructures for photocatalytic applications. J. Mater. Chem. 22, 14847–14850 (2012).
Xue, J., Fujitsuka, M. & Majima, T. Near bandgap excitation inhibits the interfacial electron transfer of semiconductor/cocatalyst. ACS Appl. Mater. Interfaces 12, 5920–5924 (2020).
Huang, H. et al. Heteroatomic Ni, Sn clusters-grafted anatase TiO2 photocatalysts: structure, electron delocalization, and synergy for solar hydrogen production. J. Phys. Chem. C. 119, 10478–10492 (2015).
Yang, W. et al. Defective indium/indium oxide heterostructures for highly selective carbon dioxide electrocatalysis. Inorg. Chem. 59, 12437–12444 (2020).
Hoch, L. B. et al. The rational design of a single-component photocatalyst for gas-phase CO2 reduction using both UV and visible light. Adv. Sci. 1, 1400013 (2014).
Martin, O. et al. Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation. Angew. Chem. Int. Ed. 55, 6261–6265 (2016).
Wang, J. et al. CO2 hydrogenation to methanol over In2O3-based catalysts: from mechanism to catalyst development. ACS Catal. 11, 1406–1423 (2021).
Frei, M. S. et al. Nanostructure of nickel-promoted indium oxide catalysts drives selectivity in CO2 hydrogenation. Nat. Commun. 12, 1960 (2021).
Lei, F. et al. Oxygen vacancies confined in ultrathin indium oxide porous sheets for promoted visible-light water splitting. J. Am. Chem. Soc. 136, 6826–6829 (2014).
Gao, P. et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 9, 1019–1024 (2017).
He, S. et al. A surface defect-promoted Ni nanocatalyst with simultaneously enhanced activity and stability. Chem. Mater. 25, 1040–1046 (2013).
Heilmann, A., Werner, J., Kelly, M., Holloway, B. & Kay, E. XPS depth profiles and optical properties of plasma polymer multilayers with embedded metal particles. Appl. Surf. Sci. 115, 365–376 (1997).
Gan, J. et al. Oxygen vacancies promoting photoelectrochemical performance of In2O3 nanocubes. Sci. Rep. 3, 1021 (2013).
Nazarzahdemoafi, M. et al. Comparative study of the electronic structures of the In and Sn/In2O3(111) interfaces. Phys. Rev. B 93, 081303 (2016).
Cored, J. et al. Hydrothermal synthesis of ruthenium nanoparticles with a metallic core and a ruthenium carbide shell for low-temperature activation of CO2 to methane. J. Am. Chem. Soc. 141, 19304–19311 (2019).
Yang, C. et al. Strong electronic oxide-support interaction over In2O3/ZrO2 for highly selective CO2 hydrogenation to methanol. J. Am. Chem. Soc. 142, 19523–19531 (2020).
Albani, D. et al. Semihydrogenation of acetylene on indium oxide: proposed single-ensemble catalysis. Angew. Chem. Int. Ed. 56, 10755–10760 (2017).
Tuomisto, F. & Makkonen, I. Defect identification in semiconductors with positron annihilation: experiment and theory. Rev. Mod. Phys. 85, 1583–1631 (2013).
Dutta, S. et al. Annealing effect on nano-ZnO powder studied from positron lifetime and optical absorption spectroscopy. J. Appl. Phys. 100, 114328 (2006).
Kong, M. et al. Tuning the relative concentration ratio of bulk defects to surface defects in TiO2 nanocrystals leads to high photocatalytic efficiency. J. Am. Chem. Soc. 133, 16414–16417 (2011).
Fujimori, K. et al. Visualizing cation vacancies in Ce:Gd3Al2Ga3O12 scintillators by gamma-ray-induced positron annihilation lifetime spectroscopy. Appl. Phys. Express 13, 085505 (2020).
Chen, Z. Q. et al. Evolution of voids in Al+-implanted ZnO probed by a slow positron beam. Phys. Rev. B 69, 035210 (2004).
Nambissan, P. M. G., Upadhyay, C. & Verma, H. C. Positron lifetime spectroscopic studies of nanocrystalline ZnFe2O4. J. Appl. Phys. 93, 6320–6326 (2003).
Tuomisto, F., Saarinen, K., Look, D. C. & Farlow, G. C. Introduction and recovery of point defects in electron-irradiated ZnO. Phys. Rev. B 72, 085206 (2005).
Liu, X. W., Zhou, K. B., Wang, L., Wang, B. Y. & Li, Y. D. Oxygen vacancy clusters promoting reducibility and activity of ceria nanorods. J. Am. Chem. Soc. 131, 3140–3141 (2009).
Tsoukalou, A. et al. Structural evolution and dynamics of an In2O3 catalyst for CO2 hydrogenation to methanol: an operando XAS-XRD and in situ TEM study. J. Am. Chem. Soc. 141, 13497–13505 (2019).
Su, Y. et al. Unexpected catalytic performance in silent tantalum oxide through nitridation and defect chemistry. J. Am. Chem. Soc. 135, 11433–11436 (2013).
Kind, C. & Feldmann, C. One-pot synthesis of In0 nanoparticles with tuned particle size and high oxidation stability. Chem. Mater. 23, 4982–4987 (2011).
Mateo, D., Cerrillo, J. L., Durini, S. & Gascon, J. Fundamentals and applications of photo-thermal catalysis. Chem. Soc. Rev. 50, 2173–2210 (2020).
Fang, M., Tan, X., Liu, Z., Hu, B. & Wang, X. Recent progress on metal-enhanced photocatalysis: a review on the mechanism. Research 80, 9794329 (2021).
Song, C., Wang, Z., Yin, Z., Xiao, D. & Ma, D. Principles and applications of photothermal catalysis. Chem. Catal. 2, 52–83 (2022).
Saito, G. & Yoshida, Y. Development of conductive organic molecular assemblies: organic metals, superconductors, and exotic functional materials. Bull. Chem. Soc. Jpn. 80, 1–137 (2007).
Haneda, M., Kintaichi, Y., Bion, N. & Hamada, H. Mechanistic study of the effect of coexisting H2O on the selective reduction of NO with propene over sol-gel prepared In2O3-Al2O3 catalyst. Appl. Catal. B: Environ. 42, 57–68 (2003).
Zhao, Y. et al. Defect-rich ultrathin ZnAl-layered double hydroxide nanosheets for efficient photoreduction of CO2 to CO with water. Adv. Mater. 27, 7824–7831 (2015).
Liu, L. & Corma, A. Identification of the active sites in supported subnanometric metal catalysts. Nat. Catal. 4, 453–456 (2021).
Pacile, D. et al. Near-edge X-ray absorption fine-structure investigation of graphene. Phys. Rev. Lett. 101, 066806 (2008).
Duan, Z. & Henkelman, G. CO oxidation at the Au/TiO2 boundary: the role of the Au/Ti5c site. ACS Catal. 5, 1589–1595 (2015).
Saqlain, M. A., Hussain, A., Siddiq, M., Ferreira, A. R. & Leitao, A. A. Thermally activated surface oxygen defects at the perimeter of Au/TiO2: a DFT+U study. Phys. Chem. Chem. Phys. 17, 25403–25410 (2015).
Pulido, A. et al. Combined DFT/CC and IR spectroscopic studies on carbon dioxide adsorption on the zeolite H-FER. Energy Environ. Sci. 2, 1187–1195 (2009).
Walenta, C. A. et al. Surface species in photocatalytic methanol reforming on Pt/TiO2(110): learning from surface science experiments for catalytically relevant conditions. ACS Catal. 10, 4080–4091 (2020).
Lee, J., Sorescu, D. C. & Deng, X. Electron-induced dissociation of CO2 on TiO2(110). J. Am. Chem. Soc. 133, 10066–10069 (2011).
Ye, J., Liu, C. & Ge, Q. DFT study of CO2 adsorption and hydrogenation on the In2O3 surface. J. Phys. Chem. C. 116, 7817–7825 (2012).
Ye, J., Liu, C., Mei, D. & Ge, Q. Active oxygen vacancy site for methanol synthesis from CO2 hydrogenation on In2O3(110): a DFT study. ACS Catal. 3, 1296–1306 (2013).
Serykh, A. I. Low-dimensional indium oxo-species on the surface of In2O3/Al2O3 catalytic material: the sites of dissociative adsorption of hydrogen. J. Phys. Chem. C. 120, 21436–21440 (2016).
Qin, B. & Li, S. First principles investigation of dissociative adsorption of H2 during CO2 hydrogenation over cubic and hexagonal In2O3 catalysts. Phys. Chem. Chem. Phys. 22, 3390–3399 (2020).
Zhao, Y., Zhou, H., Zhu, X., Qu, Y. & Wu, Y. Simultaneous oxidative and reductive reactions in one system by atomic design. Nat. Catal. 4, 134–143 (2021).
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 21972052). We acknowledge Prof. Tierui Zhang for precious advice of the original idea. We acknowledge Yanbiao Shi for in-situ FT-IR measurement and Jianwei Wang for theoretical calculations. We thank Prof. Jian Wang for comments and manuscript polishing. S.O. thanks for the financial support from the “Guizi Scholar” Program of Central China Normal University.
Author information
Authors and Affiliations
Contributions
W.W. conducted all the experiments. W.W. and S.O. developed the idea and wrote the manuscript. S.O. directed the project and provided guidance for the experimental and theoretical work. Z.W., R.L., Z.L., R.S., Y.Q., D.P., and H.Y. conceived and performed some of the experiments and provided advice for the experiments. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Emma Lovell, Dequan Xiao and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, W., Wei, Z., Li, R. et al. Subsurface oxygen defects electronically interacting with active sites on In2O3 for enhanced photothermocatalytic CO2 reduction. Nat Commun 13, 3199 (2022). https://doi.org/10.1038/s41467-022-30958-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-022-30958-5
This article is cited by
-
Subsurface engineering for directional-selective CO₂-to-ethanol electrocatalysis at industrial-level
Nature Communications (2025)
-
InNi alloy-induced interface effect promoting the parallel electron transfer: improved photothermal catalytic CO2 hydrogenation
Science China Chemistry (2025)
-
Dynamics of bulk and surface oxide evolution in copper foams for electrochemical CO2 reduction
Communications Chemistry (2024)
-
Progress of Main-Group Metal-Based Single-Atom Catalysts
Electrochemical Energy Reviews (2024)
-
Three-dimensional porous In2O3 arrays for self-powered transparent solar-blind photodetectors with high responsivity and excellent spectral selectivity
Nano Research (2024)
