
Abstract
Efficient adsorptive separation of acetylene (C2H2) from carbon dioxide (CO2) or ethylene (C2H4) is industrially important but challenging due to the identical dynamic diameter or the trace amount. Here we show an electrostatic potential compatible strategy in a nitroprusside-based Hofmann-type metal-organic framework, Cu(bpy)NP (NP = nitroprusside, bpy = 4,4’-bipyridine), for efficient C2H2 separation. The intruding cyanide and nitrosyl groups in undulating one-dimensional channels induce negative electrostatic potentials for preferential C2H2 recognition instead of open metal sites in traditional Hofmann-type MOFs. As a result, Cu(bpy)NP exhibits a 50/50 C2H2/CO2 selectivity of 47.2, outperforming most rigid MOFs. The dynamic breakthrough experiment demonstrates a 99.9% purity C2H4 productivity of 20.57 mmol g−1 from C2H2/C2H4 (1/99, v/v) gas-mixture. Meanwhile, C2H2 can also be captured and recognized from ternary C2H2/CO2/C2H4 (25/25/50, v/v/v) gas-mixture. Furthermore, computational studies and in-situ infrared spectroscopy reveal that the selective C2H2 binding arises from the compatible pore electro-environment generated by the electron-rich N and O atoms from nitroprusside anions.
Similar content being viewed by others

Introduction
Acetylene (C2H2) capture from gas-mixtures is of great industrial importance1,2. As a vital chemical feedstock and industrial gas3, C2H2 is typically generated by the partial combustion of methane, a considerable amount of carbon dioxide (CO2) inevitably coexists in the crude products. C2H2 and CO2 molecules both possess linear configurations with subtle differences in molecular dimensions (3.3 × 3.3 × 5.7 Å for C2H2 and 3.2 × 3.3 × 5.4 Å for CO2), and show close similarities in physical properties, including quadrupole moments, boiling point, and polarity (Supplementary Table 1)4,5. On the other hand, the highly reactive C2H2 can induce undesirable side effects in various industrial operations. For instance, in ethylene (C2H4) manufacturing by the steam cracking of naphtha or dehydrogenation of ethane6,7, the trace amount of C2H2 (1%) must be removed to an acceptable level of 40 ppm, otherwise the residue C2H2 will deactivate the Ziegler–Natta catalyst and lower the quality of polyethylene commodities8. In industry, trace C2H2 removal is achieved by partial hydrogenation or solvent absorption, which are energy- and cost-intensive9. Therefore, alternative technologies with high capital efficiency have attracted great interest in separating C2H2 from CO2 and C2H4 due to their industrial relevance and scientific challenge, such as adsorptive separations10.
Since the discovery of Prussian blue in 1704, transition metal cyanide complexes, also referred to as cyanometallates, have been extensively synthesized11. The strong basicity of cyanide, as δ-donor and π-acceptor, endows the ability and versatility of coordination with most transition metals12. Particularly, cyanide-based metal-organic frameworks (MOFs) with interior vacancies and open spaces emerge as an intriguing class of solid adsorbents13,14. As a family of Prussian blue analogs, Hofmann-type MOFs have a general formula of M(pyz)[M’(CN)]4, where M and M’ are divalent metal ions and pyz is the pyrazine bidentate organic ligand15. Besides the confined pore spaces, another prevailing feature is the existence of abundant open metal sites (OMSs) incorporated in cyanometallate ligand that can significantly enhance the C2H2 capacity through strong electrostatic interactions. For example, Co(pyz)[Ni(CN)]4, termed as ZJU-74a, exhibited the record C2H2 adsorption capacity of 49 cm3 g−1 at 0.01 bar and 296 K due to the sandwich-like open Ni sites from adjacent [Ni(CN)]42− ligands13. However, the open Ni sites can also interact with the electronegative O atoms of CO2 (Fig. 1a), as evidenced by the close C2H2 and CO2 capacity (3.82 mmol g−1 vs 3.13 mmol g−1) at 1.0 bar and 296 K.

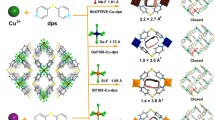
Schematic illustration of a traditional and nitroprusside-based Hofmann-type MOF networks and b the building blocks (Cu2+, [Fe(CN)5(NO)]2−, and bpy) and the 3D sqc-net topology of Cu(bpy)NP; c view of the pore channels by the Connolly surface along the ab-plane, d illustration of the channel size along the bc-plane, and e the ESP mapping for the channel of Cu(bpy)NP (color code: C, gray; H, white; N, blue; O, red; Cu, green; Fe, orange).
Nitroprusside is a unique type of cyanide ligand in which the Fe(II) atoms are octahedrally coordinated by five carbon atoms from independent cyanide groups and one nitrogen atom from a nitrosyl group (Fig. 1b). The straightforward coordination of Cd2+ and Ni2+ ions with nitroprusside (NP) have yielded Cd-NP and Ni-NP that showed superior CO2/C2H2 and C3H6/C3H8 separation performances11,16, respectively. Nevertheless, to the best of our knowledge, the successful preparation of Hofmann-type MOFs using nitroprusside ligand has never been reported. Unlike the directly exposed OMSs in cyanometallate ligand, the intruding cyanide and nitrosyl groups can create a negative-charged electrostatic potential field for preferred C2H2 capture, because C2H2 possesses a positive charge distribution at both ends (Fig. 1a)14. Note that creating a negative-charged pore environment is challenging due to the limited choice of suitable functional groups.
Herein, we report a Hofmann-type MOFs employing nitroprusside ligand, Cu(bpy)NP (NP = nitroprusside, bpy = 4,4’-bipyridine), for efficient and selective capture of C2H2 from C2H2/CO2 and C2H2/C2H4 gas-mixtures. In particular, Cu(bpy)NP exhibits a three-dimensional sqc topology with a suitable cavity size of 6.0 × 7.0 Å2. The incorporation of cyanide and nitrosyl groups on the pore walls afford a complementary negative electrostatic potential field for C2H2 binding. As a result, Cu(bpy)NP shows a large C2H2 capacity (50.7 cm3 g−1) and superior C2H2/CO2 (47.2) and C2H2/C2H4 (28.5) selectivity at 298 K and 1.0 bar. Dynamic breakthrough experiments confirm its practical C2H2 capture performances from binary C2H2/CO2 and C2H2/C2H4 gas-mixtures and ternary C2H2/CO2/C2H4 gas-mixture. Cu(bpy)NP affords C2H4 productivity of 20.57 mmol g−1 from C2H2/C2H4 (1/99, v/v) gas-mixture. Theoretical modeling and in-situ IR studies reveal that C2H2 molecules are collectively bound by two nitrosyl groups and one cyanide group via multiple strong interactions.
Results
Synthesis and characterization
By dripping the aqueous solution of Cu(NO3)2·3H2O and sodium nitroprusside (NP) into a methanol solution of 4,4’-bipyridine (bpy), the Cu(bpy)NP can be obtained under mild conditions with a yield of 67% (Fig. 1b). Cyan square-block single-crystals of Cu(bpy)NP were also successfully prepared by the slow-diffusion method (Supplementary Fig. 1). Single-crystal X-ray diffraction studies revealed that Cu(bpy)NP crystallizes in an orthorhombic space group Pmma and adopts a 4,6-connected sqc topology (Fig. 1b, CCDC: 2124121). Each Cu moiety is octahedrally coordinated by two ditopic bridging bpy linkers at the axial positions and four independent cyanide groups from [Fe(CN)5(NO)]2− anions at equatorial positions (Supplementary Fig. 2). In the structure of Cu(bpy)NP, the undulating two-dimensional (2D) channels across the ab-plane are different from the regular one-dimensional (1D) channels in previously reported Hofmann-type MOFs (Fig. 1c)12,13. The channels in Cu(bpy)NP showed a channel dimension of 6.0 × 7.0 Å2 along the bc-plane (Fig. 1d). The volume of its solvent-accessible void was calculated to be 34.4% of the total volume. Moreover, the electrostatic potential (ESP) of channel interior in Cu(bpy)NP was mapped by density functional theory (DFT) calculations (Fig. 1e), and strong negative potentials were disclosed near the nitrogen atom in the cyanide group and oxygen atom in the nitrosyl group that are compatible for C2H2 adsorption, however, which are mutually repulsive to CO2 with negatively charged ends (Supplementary Fig. 3).
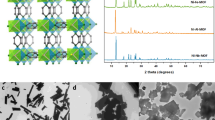
The phase purity of the as-synthesized Cu(bpy)NP was verified by the powder X-ray diffraction (PXRD) pattern, which is consistent with the simulated one, while no noticeable change was observed on the activated sample (Supplementary Fig. 4 Supplementary Table 2). Furthermore, the Rietveld refinement of PXRD revealed that the activated Cu(bpy)NP crystallized in the same crystal system and showed similar cell parameters with the as-synthesized sample, indicating its rigid framework after solvent removal (Supplementary Fig. 5 and Supplementary Table 3). The PXRD patterns of C2H2-loaded samples under different pressures (0–1.0 bar) at 298 K showed inconspicuous changes in position and intensity of diffraction peaks, manifesting structural robustness during adsorption processes (Supplementary Fig. 6). The structural integrity was examined by immersing Cu(bpy)NP in various organic solvents for 1 week and boiling water for 3 h, intact PXRD patterns can be maintained, suggesting its good chemical stability (Supplementary Fig. 7a). Meanwhile, Cu(bpy)NP could survive wide range acid/basic solutions, whereas the crystal structure was destroyed at extreme conditions, i.e., pH = 13 and 1 (Supplementary Fig. 7b). TGA revealed a weight loss of 2.9% at 220 °C, corresponding to the removal of guest molecules (Supplementary Fig. 8). The Brunauer-Emmett-Teller (BET) surface area of Cu(bpy)NP was measured to be 459 m2 g−1 with a total pore volume of 0.45 cm3 g−1 by CO2 adsorption at 195 K (Supplementary Fig. 9). The pore size was determined to center at 6.6 Å, consistent with the channel size derived from the crystal structure. The pore properties were also probed by N2 at 77 K (Supplementary Fig. 10), the decreased BET specific surface area of 121 m2 g−1 can be attributed to the larger size and property inertness of N2 as the probe molecule.
Adsorption and separation performances
Single-component gas adsorption isotherms of C2H2, CO2, and C2H4 were collected at 273, 298, and 323 K, respectively (Fig. 2a, Supplementary Fig. 11, and Supplementary Tables 6-8). Cu(bpy)NP exhibited a higher C2H2 adsorption capacity (50.7 cm3 g−1) than that of C2H4 (40.8 cm3 g−1) and CO2 (25.1 cm3 g−1) at 298 K and 1.0 bar. Approximately, two C2H2, one CO2, and one and a half C2H4 molecules are adsorbed per unit cell, respectively. Generally, the gas separation performances greatly correlate with the gas adsorption behaviors in low-pressure ranges, in which the adsorbent-adsorbate affinity dominates17. As shown in Fig. 2b, Cu(bpy)NP displayed a C2H2 uptake of 22.4 cm3 g−1 at 0.01 bar and 298 K, notably, even comparable to adsorbents with OMSs, including CuI@UiO-66-(COOH)2 (20.2 cm3 g−1)18, UTSA-74a (19.2 cm3 g−1)19, Zn-MOF-74 (14.8 cm3 g−1)20, and JNU-1 (5.5 cm3 g−1)21 (Fig. 2c and Supplementary Table 9). In sharp contrast, the adsorption uptake of C2H4 (2.1 cm3 g−1) and CO2 (0.6 cm3 g−1) was almost negligible at 0.01 bar. The adsorption isotherms have experimentally confirmed the concept that the negative electrostatic potential can selectively adsorb C2H2 over CO2 and C2H4.

C2H2, CO2, and C2H4 adsorption isotherms a in linear form and b in logarithmic form of Cu(bpy)NP at 298 K; c comparison of C2H2 uptakes with best-performing materials at 0.01 bar and 298 K; d Qst plots of C2H2, CO2, and C2H4 on Cu(bpy)NP; e comparison of C2H2 Qst and 50/50 C2H2/CO2 IAST selectivity with leading adsorbents; f comparison of 50/50 C2H2/CO2 and 1/99 C2H2/C2H4 IAST selectivity of Cu(bpy)NP with reported adsorbents at 298 K and 1.0 bar.
To evaluate the affinity of Cu(bpy)NP toward C2H2, CO2, and C2H4, the isosteric enthalpy of adsorption (Qst) was calculated by Virial fitting from adsorption isotherms at three temperatures (Supplementary Fig. 12 and Supplementary Table 10). The calculated Qst for C2H2 is 40.8 kJ mol−1, higher than that of CO2 (14.6 kJ mol−1) and C2H4 (31.3 kJ mol−1) at near zero coverage (Fig. 2d). The moderate Qst for C2H2 indicates both strong binding and energy-efficient regeneration (Fig. 2e)22. Moreover, the difference in Qst (ΔQst) represents the binding selectivity of adsorbents, the ΔQst for C2H2 and CO2 is calculated to be 26.2 kJ mol−1, which is the most prominent except for two MOFs with rich OMSs (Supplementary Fig. 17), i.e., CuI@UiO-66-(COOH)2 (45.6)18 and ATC-Cu (42.6)23.
The ideal adsorbed solution theory (IAST) was employed to evaluate the adsorptive separation selectivity, and the adsorption isotherm fittings by the dual-site Langmuir-Freundlich model were shown in Supplementary Figs. 13–15 and Supplementary Table 11. First, the adsorption kinetics of C2H2, CO2, and C2H4 were evaluated on Cu(bpy)NP, which depicted that the equilibrium times for all gases were about 6 min at 298 K and 0.4 bar (Supplementary Fig. 16). Benefiting from the preferred C2H2 adsorptions at low pressures, Cu(bpy)NP exhibited a benchmark 50/50 C2H2/CO2 selectivity of 343.8 at 0.01 bar and 298 K, and gradually decreased to 47.2 at 1.0 bar (Supplementary Fig. 18). Notably, such C2H2/CO2 selectivity at ambient conditions, to our knowledge, is only lower than benchmark adsorbents such as CuI@UiO-66-(COOH)2 (185)18, CPL-1-NH2 (119)24, ZNU-1 (56.6)25, and ATC-Cu (53.6)24, and higher than leading materials including ZJU-74a (36.5)13, NKMOF-1-Ni (22.0)17, and HOF-3a (21.5)26 (Fig. 2e). It is worth pointing out that Cu(bpy)NP should be considered as one of the most promising candidates that show favorable C2H2/CO2 selectivity (>40) and suitable C2H2 Qst for regeneration (40~60 kJ mol−1)22. Similarly, the 1/99 C2H2/C2H4 IAST selectivity of 781.2 and 28.5 was obtained at 0.01 bar and 1.0 bar and 298 K, respectively (Supplementary Fig. 19a). Besides the advantageous C2H2/C2H4 IAST selectivity (Supplementary Fig. 19b and Supplementary Table 9), noticeably, the C2H2/CO2 IAST selectivity on Cu(bpy)NP was also superior to many reported adsorbents (Fig. 2f).
Transient breakthrough experiments
To further experimentally validate the separation performances of Cu(bpy)NP, dynamic breakthrough experiments were carried out under ambient conditions. As shown in Fig. 3a, Cu(bpy)NP can efficiently separate an equimolar gas-mixture of C2H2/CO2, at a flow rate of 2.0 mL min−1, CO2 was detected first at the outlet of the separation bed at around 20 min, while C2H2 was not eluted from the column until 78 min. The separation selectivity (αAC) for equimolar C2H2/CO2 mixture was calculated to be 3.9 on Cu(bpy)NP based on the breakthrough curve, outperforming SNNU-45 (2.9)27, NKMOF-1-Ni (2.6)17, HOF-3a (2.0)27, and FeNi-M’MOF (1.7)14 (Supplementary Fig. 20). When the flow rate increased to 3.0 and 5.0 mL min−1, the breakthrough time decreased as expected but good separation effects remained. Meanwhile, as illustrated by the desorption curves, CO2 desorbed quickly in 10 min and the complete desorption of C2H2 was achieved at 90 min, indicating a long operation window for C2H2 collection with high C2H2 purity above 99% (Supplementary Fig. 21). As for C2H2/C2H4 (1/99, v/v) gas-mixture, C2H4 immediately broke through the packed column with Cu(bpy)NP at 22 min, while C2H2 was trapped and eluted until 400 min (Fig. 3b). The C2H4 productivity with 99.9 + % purity was calculated to be 20.57 mmol g−1 on Cu(bpy)NP, which is only lower than that of SIFSIX-2-Cu-i (47.4 mmol g−1)9 and UTSA-200a (85.7 mmol g−1)28 (Fig. 3c). Even when the concentration of C2H2 increased to 50%, Cu(bpy)NP also maintained the efficient and clean separation for C2H2/C2H4 (50/50, v/v) gas-mixture (Supplementary Fig. 22).

Dynamic breakthrough curves of Cu(bpy)NP for a C2H2/CO2 (50/50, v/v) and b C2H2/C2H4 (1/99, v/v) gas-mixtures; c comparison of C2H4 productivity with representative MOFs; d dynamic breakthrough curve of Cu(bpy)NP for ternary C2H2/CO2/C2H4 (25/25/50, v/v/v) gas-mixture; e cycling dynamic breakthrough tests for C2H2/C2H4 (1/99, v/v) on Cu(bpy)NP; f continuous five C2H2 adsorption isotherms of Cu(bpy)NP at 298 K.
Furthermore, we also carried out the dynamic breakthrough experiment with ternary C2H2/CO2/C2H4 (25/25/50, v/v/v) gas-mixture. As shown in Fig. 3d, CO2 and C2H4 concurrently broke through the adsorption column at 28 min, whereas C2H2 was not detected at the exit until 50 min. The cycling and regeneration capabilities of Cu(bpy)NP are important parameters for practical industrial applications. Therefore, cycling breakthrough cycle experiments for both C2H2/CO2 (50/50, v/v) and C2H2/C2H4 (1/99, v/v) were conducted (Fig. 3e and Supplementary Fig. 23), during five continuous cycles, no noticeable decay in the residual time was observed. Also, five successive C2H2 adsorption-desorption isotherms showed intact C2H2 uptakes, manifesting the excellent reusability of Cu(bpy)NP (Fig. 3f).
In-situ IR and modeling simulation studies
In-situ infrared (IR) spectroscopic measurements were conducted on gas-loaded samples to illustrate the interactions between three adsorbates and Cu(bpy)NP. It is recognized that acidic C2H2 molecules tend to form strong interactions with basic sites8,29. As shown in Fig. 4a, the νas(C2H2) stretching band of adsorbed C2H2 down-shifted to 3170 cm−1 with reference to the gas-phase value at 3287 cm−1, indicating the existence of guest–host interactions with possible charge transfers30,31,32. The strong perturbation of characteristic bands, i.e., ν(CN)NP, ν(CC)bpy, ν(CN)bpy, δ(CH)bpy, ν(NO)NP, and δ(ring), suggested that C2H2 also interacted with Cu(bpy)NP framework via multiple weak interactions. The adsorbed CO2 within Cu(bpy)NP was confirmed by its asymmetric stretching band νas(CO2) at 2340 cm−1 33. Compared to the C2H2-loaded framework, the perturbations of characteristic bands were obviously decreased in intensity, especially for ν(CN)NP and ν(NO)NP (Fig. 4a inset), verifying that the complementary electrostatic potential created by cyanide and nitrosyl groups were responsible for selective C2H2 capture. As for C2H4, the νas(C2H4) stretching band appeared at 949 cm−1 34,35, and the interaction mode was similar to that of C2H2-loaded samples. It was noted that the ν(NO)NP bond was intensified, indicating the favorable affinity toward nitrosyl groups.

a In-situ IR spectra of gas-loaded Cu(bpy)NP samples; b Charge density difference plots showing the interaction between C2H2; DFT-D calculated binding sites of c C2H2, d CO2, and e C2H4 in Cu(bpy)NP. The distances include the Van der Waals radius of atoms (Color code: C, gray; H, white; N, blue; O, red; Cu, green; Fe, orange).
To gain insight into the adsorption mechanisms of C2H2, CO2, and C2H4 in Cu(bpy)NP, theoretical molecular simulations using the grand canonical Monte Carlo (GCMC) and first-principles dispersion-corrected density functional theory (DFT-D) were carried out. The GCMC-simulated C2H2, CO2, and C2H4 adsorption isotherms agreed well with the experimental isotherms (Supplementary Fig. 24). The distribution densities were firstly investigated at 1 kPa, C2H2 molecules are mainly distributed in two locations near the cyanide and nitrosyl groups. In sharp contrast, the CO2 molecules are distributed in the middle of the channels that are apart from nitroprusside anions. Whereas, the C2H4 molecules occupy almost identical locations in the channels of Cu(bpy)NP but with less distribution density compared to C2H2 (Supplementary Fig. 25a–c). As the loading pressure increased to 100 kPa, the adsorption locations for each adsorbent remained, while the contribution density is in the order of C2H2 > C2H4 > CO2, which is consistent with the experimental adsorption capacities (Supplementary Fig. 25d–f).
We firstly carried out the charge transfer analysis on the gas-loaded structures by DFT calculations. As shown in Fig. 4b, the blue and yellow surfaces indicate charge accumulation and charge depletion, respectively. The close end-on adsorption configuration of H atom of C2H2 and N atom of cyanide groups implied the strong electro-field induced C2H2 capture (Fig. 4b). Moreover, the negatively charged H atom of C2H2, opposite to its original positive-charged state, revealed the charge transfer from Cu(bpy)NP framework to adsorbed C2H2. In contrast, CO2 and C2H4 molecules displayed the side-on adsorption configurations in Cu(bpy)NP skeleton, and no noticeable charge transfer was observed (Supplementary Fig. 26). These results highlighted the effect of negatively charged pore environment in enhancing the adsorption and recognition of C2H2.
The lowest-energy gas binding configurations of C2H2, CO2, and C2H4 in Cu(bpy)NP were also calculated. As shown in Fig. 4c, C2H2 was roughly oriented parallel to the bpy ligand along the a-axis, where it was captured by three independent [Fe(CN)5(NO)]2− anions via strong interactions of two C-H•••O bonds (2.78–2.93 Å) and one C-H•••N bond (2.82 Å). The binding energy of C2H2 in Cu(bpy)NP was calculated to be 38.4 kJ mol−1, slightly lower than the experimental C2H2 Qst (40.8 kJ mol−1). In contrast, CO2 was adsorbed by two opposite bpy ligands through weak interactions of N•••O(CO2) (3.06 Å) and C•••C(CO2) (3.18 Å) with a calculated CO2 binding energy of 13.4 kJ mol−1 (Fig. 4d). Clearly, the negative electrostatic potential generated by [Fe(CN)5(NO)]2− anions induced the preferential binding with C2H2 over CO2. Similarly, Cu(bpy)NP displayed relatively weak interactions with C2H4 via the C-H•••N bond (3.16 Å) and C-H•••O bond (3.41 Å), the C2H4 binding energy was calculated to be 28.5 kJ mol−1 (Fig. 4e).
Discussion
In summary, we synthesized and reported a nitroprusside-based Hofmann-type MOF adsorbent, Cu(bpy)NP, for simultaneous efficient separation of C2H2 from C2H2/CO2 and C2H2/C2H4 gas-mixtures. Compared to the traditional Hofmann-type MOFs with rich OMSs, the cyanide and nitrosyl groups of nitroprusside anions generated a negative electro-environment in the undulating 1D channels for the preferential discrimination of C2H2 over CO2. Consequently, Cu(bpy)NP exhibited a high 50/50 C2H2/CO2 selectivity and 1/99 C2H2/C2H4 selectivity. A considerable balance of separation selectivity and adsorption enthalpy was achieved on Cu(bpy)NP. Dynamic breakthrough experiments revealed the advantageous C2H4 productivity with 99.9% purity. Modeling studies and in-situ IR measurements indicated the compatible pore environment and strong guest-host interactions for efficient C2H2 separation. This work encourages the community to design and synthesize Hofmann-type adsorbents for challenging separation tasks.
Methods
All reagents were purchased from commercial companies and used without further purification. Sodium nitroprusside dihydrate (C5FeN6Na2O·2H2O, 99.98%, Aladdin), copper nitrate trihydrate (Cu(NO3)2·3H2O, 99.99%, Aldrich), 4,4’-bipyridine (C10H8N2, 98%, Aladdin), and methanol (CH4O, anhydrous, 99.9%, Aladdin) were commercially available and used as supplied without further purification. N2 (99.999%), acetylene (C2H2, 99.99%), CO2 (99.99%), ethylene (C2H4, 99.99%), He (99.999%), and mixed gas-mixtures of C2H2/CO2 (50/50, v/v), C2H2/C2H4 (1/99, v/v), C2H2/C2H4 (50/50, v/v), and C2H2/CO2/C2H4 (25/25/50, v/v/v) were purchased from Nanchang Guoteng Gas Co., Ltd (China).
Synthesis of Cu(bpy)NP
Typically, Cu(NO3)2·3H2O (0.25 mmol, 60 mg) and sodium nitroprusside (NP, 0.25 mmol, 75 mg) were dissolved in 3 mL H2O and then dripped into a methanol solution (30 mL) of 4,4’-bipyridine (bpy, 0.2 mmol, 35 mg) under vigorous stirring. After reacting at room temperature in the dark for 48 h, cyan precipitates were collected by centrifugation, washed thrice with methanol, and dried overnight in a vacuum oven at 333 K (yield: ca. 67% based on NP).
Single-crystals of Cu(bpy)NP were synthesized by slow diffusion of a methanol solution (0.4 mL) of NP (0.003 mmol, 0.89 mg) and bpy (0.002 mmol, 0.35 mg) into an aqueous solution (0.4 mL) of Cu(NO3)2·3H2O (0.003 mmol, 0.72 mg) at room temperature in the dark in a watch glass and kept undisturbed, and 1.2 mL of methanol/H2O (1:1) was layered between the top and bottom solutions to slow the reaction rate. Light cyan and square prismatic crystals were formed after 1 week.
Structure simulations
The unit cell structures (e.g., cell parameters and atomic positions) of Cu(bpy)NP were calculated using the Forcite and Castep modules. The Rietveld refinement, a software package for crystal determination from the XRD pattern, was performed to optimize the lattice parameters iteratively until the wRp value converges. The pseudo-Voigt profile function was used for whole profile fitting, and the Berrar-Baldinozzi function was used for asymmetry correction during the refinement processes. Line broadening from crystallite size and lattice strain were both considered.
Gas adsorption measurements
Equilibrium and kinetic adsorptions of C2H2, CO2, and C2H4 at 273, 298, and 323 K were measured on Micromeritics ASAP 2460 adsorption apparatus (Micromeritics Instruments, USA). The kinetic adsorptions of C2H2, CO2, and C2H4 were obtained on Intelligent Gravimetric Analyzer (IGA-100, HIDEN). To remove all the guest solvents in the framework, the fresh powder samples were evacuated under a high vacuum at 373 K for 12 h. The BET surface area was calculated using the adsorption branch with the relative pressure P/P0 range of 0.005–0.3. The total pore volume (Vtot) was calculated based on the adsorbed amount of nitrogen at the P/P0 of 0.99. The pore size distribution (PSD) was calculated based on CO2 and N2 adsorption isotherms at 195 K and 77 K, respectively. The helium gas was used to determine the free space of the system. The sample was degassed for 24 h between each measurement.
In-situ infrared (IR) spectroscopic measurements
All the IR spectroscopic data are recorded in a Tensor 27 FTIR spectrometer (Bruker, GER) equipped with a liquid N2-cooled mercury cadmium telluride MCT-A detector. A vacuum cell, purchased from Specac Ltd., UK (product number P/N 5850c), is placed in the sample compartment of the infrared spectrometer with the sample at the beam’s focal point. The cell is connected to different gas lines (C2H2, CO2, and C2H4) and a vacuum line for evacuation. The Cu(bpy)NP (powder, ~30 mg) was placed in the cell, and firstly annealed at 100 °C under vacuum for activation and then cooled to RT for recording the reference spectrum. C2H2 was introduced into the cell, and the spectra were recorded during the gas exposure till 60 min. After fully evacuating the same sample by pumping the cell, the reference spectrum was retaken. Loading of CO2 and C2H4 was performed separately, and the infrared data were recorded in the same manner.
Breakthrough experiments
The breakthrough experiments were performed on a self-assembly device. Typically, the activated Cu(bpy)NP (about 1.65 g) was packed into a stainless-steel column (4.6 mm inner diameter × 100 mm). The column was first purged with a He flow (10 mL min−1) at room temperature for 8 h before breakthrough measurements. The binary C2H2/CO2 (50/50, v/v), C2H2/C2H4 (1/99, v/v), and ternary C2H2/CO2/C2H4 (25/25/50, v/v/v) gas-mixtures were then introduced at a flow rate of 2.0, 3.0, and 5.0 mL min−1, respectively. The outlet gas from the column was monitored using gas chromatography (GC-490 plus) with a flame ionization detector. After the breakthrough measurement, the columns packed with samples were regenerated by purging dry He gas (10 mL min−1) at 100 °C for 24 h.
DFT calculations
First-principles DFT calculations were performed using Materials Studio’s CASTEP code. All calculations were conducted under the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof (PBE). The optimized structures are in great consistency with the experimentally determined crystal structures. The energy, force, and displacement convergence criteria were set as 1 × 10−5 Ha, 2 × 10−3 Ha, and 5 × 10−3 Å, respectively. Single point energy calculations with the same parameters using Dmol3 were performed on optimized Cu(bpy)NP. The electron density data obtained from these calculations were used to construct the 0.015 e− Å−3 electron density isosurfaces of the C2H2 and CO2 molecules, while the electron density data of both frameworks were used to construct the 0.15 e− Å−3 electron density isosurfaces, with a grid interval of 0.1 Å. The calculated electrostatic potential for Cu(bpy)NP and C2H2 and CO2 molecules were then mapped onto their electron density isosurfaces. A semiempirical addition of dispersive forces to conventional DFT was included in the calculation to account for van der Waals interactions. Cutoff energy of 544 eV and a 2 × 2 × 2 k-point mesh were enough for the total energy coverage within 0.01 meV atom−1. The structures of the synthesized materials were first optimized from the reported crystal structures. The pristine structure and an isolated gas molecule placed in a supercell (with the same cell dimensions as the pristine crystal structure) were optimized and relaxed as references to obtain the binding energy. C2H2, CO2, and C2H4 gas molecules were then introduced to different locations of the channel pore, followed by a full structural relaxation. The static binding energy was calculated by the equation EB = E(gas) + E(adsorbent) − E(adsorbent + gas).
GCMC calculations
All the GCMC simulations were performed in MS 2017R2 package. The crystal structure of the Cu(bpy)NP was chosen after the DFT geometry optimization. The framework and the individual C2H2, CO2, and C2H4 were considered rigid during the simulation. The charges for atoms of the Cu(bpy)NP and gas components were derived from the Muliken method. The simulations adopted the fixed pressure task, the Metropolis method in the sorption module, and the universal force field (UFF). The interaction energy between the adsorbed molecules and the framework was computed through the Coulomb and Lennard–Jones 6-12 (LJ) potentials. The cutoff radius was chosen 18.5 Å for LJ potential and the electrostatic interactions were handled using the Ewald summation method. The loading and equilibration steps were 1 × 107, and the production steps were 1 × 107.
Sample characterizations
PXRD was collected on a PANalytical Empyrean Series 2 diffractometer with Cu Kα radiation (λ = 1.540598 Å), which operated at 40 kV, 40 mA, and a scan speed of 0.0167°, a scan time of 15 s per step, and 2θ ranging from 5 to 60° at room temperature. The thermogravimetric analysis (TGA) data were performed on a NETZSCH Thermogravimetric Analyzer (STA2500) from 25 to 800 °C with a heating rate of 10 °C/min under an N2 atmosphere. The single-component adsorption isotherms of C2H2, CO2, and C2H4 at 273, 298, and 323 K were measured on Micromeritics ASAP 2460 adsorption apparatus (Micromeritics Instruments, USA). The degassing procedure for all samples was carried out at 373 K under vacuum for 12 h before each adsorption measurement. The specific BET surface area was calculated based on the CO2 adsorption isotherm data (0.05 and 0.15 relative pressure) at 195 K. The PSD was derived from the adsorption branch of CO2 isotherms using the non-local density functional theory method and assuming a slit pore model.
Single-crystal X-ray diffraction
Single-crystal X-ray diffraction data for Cu(bpy)NP were collected at 193(2) K on a Bruker-AXS D8 VENTURE diffractometer equipped with a PHOTON-100/CMOS detector (GaKα, λ = 1.3414 Å). Indexing was performed using APEX2. SaintPlus 6.01 was used to complete data integration and reduction. The multi-scan method implemented in SADABS was used to conduct absorption correction. XPREP implemented in APEX2.1 was used to determine the space group. The structures were solved by direct methods and refined by nonlinear least-squares on the F2 method with SHELXL-97 contained in APEX2, OLEX2 v1.1.5, and WinGX v1.70.01 program packages. The Squeeze routine implemented in Platon was used to treat the contribution of disordered solvent molecules as diffuse. Crystallographic data are available free of charge from the Cambridge Crystallographic Data Center (CCDC). The CCDC number for the Cu(bpy)NP crystal is 2124121.
Structural stability tests
Solvent stability tests were performed by placing 100 mg samples in 20 mL vials containing 15 mL of different organic solvents for 1 week, boiling water for 3 h, and acid/basic solutions with different pH for 1 week. Then, the solid was separated by filtration and subsequently activated at 100 °C for 12 h, and PXRD tests characterized the structure of the materials.
Fitting of isotherms
The single-component isotherms for C2H2, CO2, and C2H4 on Cu(bpy)NP were fitted with the dual-site Langmuir-Freundlich isotherm model:
with T-dependent parameters b
Where q is the adsorbed amount for an adsorbent in mmol g−1, q1, q2 is the saturated adsorption capacities in mmol g−1, b is the Langmuir parameters in kPa−1, p is pressure in kPa−1, ν is the Freundlich parameters for single-sites, R is the gas constant, and T (K) is the temperature. The model fits the pure component isotherms well, and the R2 values are more significant than 0.9994 (see Table S6).
IAST calculations of adsorption selectivity
IAST calculations of adsorption selectivity for C2H2/CO2 and C2H2/C2H4 separation was defined by
q1 and q2 are the molar loadings in the adsorbed phase in equilibrium with the bulk gas phase, p1 and p2 are partial pressures.
Separation factor/separation selectivity calculations
The amount of adsorbed gas i (qi) is calculated from the breakthrough curve as follows:
Here, Vi is the influent flow rate of gas (cm3 min−1), Ve is the effluent flow rate of gas (cm3 min−1), Vdead is the dead volume of the system (cm3), t is the adsorption time (min) and m is the mass of the sorbent (g)1.
On approximation, this simplifies to:
VT is the total flow rate of gas (cm3 min−1), Pi is the partial pressure of gas i (bar) and ΔT is the time for the initial breakthrough of gas i to occur (min)2. The separation factor, also known as separation selectivity (αAC) for the breakthrough experiment, i.e., breakthrough-derived selectivity is determined as follows:
yi is the partial pressure of gas i in the gas-mixture. If one gas component has negligible adsorption, the amount of gas adsorbed is treated as ≤1 cm3 for calculations.
Isosteric heat of adsorption
The binding energies of C2H2, CO2, and C2H4 are reflected indirectly in the isosteric heat of adsorption, Qst, which are calculated by the Clausius-Clapeyron equation, defined as
where Qst represents the adsorption heat of C2H2, C2H4, and CO2, P, and T represent the pressure and temperature under adsorption measurement conditions, and R is the universal gas constant. Here, the adsorption heat of each component was determined precisely according to the virial fitting parameters of single-component adsorption isotherms measured at 273, 298, and 323 K up to 1.0 bar, which was defined as follows:
where the N is the adsorption amount, and m and n determine the number of items required to precisely fit the adsorption isotherms.
Data availability
All data supporting the finding of this study are available within this article and its Supplementary Information. Crystallographic data for the structures in this article have been deposited at the Cambridge Crystallographic Data Center under deposition nos. CCDC 2124121 (Cu(bpy)NP). Copies of the data can be obtained free of charge from www.ccdc.cam.ac.uk/data_request/cif. Source data supporting this study’s findings are available upon request to J.W.
References
Bao, Z. et al. Fine tuning and specific binding sites with a porous hydrogen-bonded metal-complex framework for gas selective separations. J. Am. Chem. Soc. 13, 4596–4603 (2018).
Fan, W. et al. Optimizing multivariate metal-organic frameworks for efficient C2H2/CO2 separation. J. Am. Chem. Soc. 19, 8728–8737 (2020).
Wang, J. et al. Optimizing pore space for flexible-robust metal-organic framework to boost trace acetylene removal. J. Am. Chem. Soc. 21, 9744–9751 (2020).
Adil, K. et al. Gas/vapour separation using ultra-microporous metal-organic frameworks: insights into the structure/separation relationship. Chem. Soc. Rev. 11, 3402–3430 (2017).
Gu, Y. et al. Host-guest interaction modulation in porous coordination polymers for inverse selective CO2/C2H2 separation. Angew. Chem. Int. Ed. 21, 11688–11694 (2021).
Lin, R.-B. et al. Optimized separation of acetylene from carbon dioxide and ethylene in a microporous material. J. Am. Chem. Soc. 23, 8022–8028 (2017).
Zhang, P. et al. Synergistic binding sites in a hybrid ultramicroporous material for one-step ethylene purification from ternary C2 hydrocarbon mixtures. Sci. Adv. 23, eabn9231 (2022).
Chen, K.-J. et al. Synergistic sorbent separation for one-step ethylene purification from a four-component mixture. Science 6462, 241–246 (2019).
Cui, X. et al. Pore chemistry and size control in hybrid porous materials for acetylene capture from ethylene. Science 6295, 141–144 (2016).
Fan, W. et al. Tetrazole-functionalized zirconium metal-organic cages for efficient C2H2/C2H4 and C2H2/CO2 separations. Angew. Chem. Int. Ed. 32, 17338–17343 (2021).
Xie, Y. et al. Efficient separation of propylene from propane in an ultramicroporous cyanide-based compound with open metal sites. Small Struct. 5, 2100125 (2022).
Xie, Y. et al. Old materials for new functions: recent progress on metal cyanide based porous materials. Adv. Sci. 1, 2104234 (2022).
Pei, J. et al. A chemically stable hofmann-type metal-organic framework with sandwich-like binding sites for benchmark acetylene capture. Adv. Mater. 24, 1908275 (2020).
Gao, J. et al. Mixed metal-organic framework with multiple binding sites for efficient C2H2/CO2 separation. Angew. Chem. Int. Ed. 11, 4396–4400 (2020).
Massasso, G. et al. Molecular iodine adsorption within Hofmann-type structures M(L)[M′(CN)4] (M = Ni, Co; M′ = Ni, Pd, Pt): impact of their composition. Dalton Trans. 44, 19357–19369 (2015).
Xie, Y. et al. Electrostatically driven selective adsorption of carbon dioxide over acetylene in an ultramicroporous material. Angew. Chem. Int. Ed. 17, 9604–9609 (2021).
Peng, Y.-L. et al. Robust ultramicroporous metal-organic frameworks with benchmark affinity for acetylene. Angew. Chem. Int. Ed. 34, 10971–10975 (2018).
Zhang, L. et al. Ultramicroporous building units as a path to bi-microporous metal-organic frameworks with high acetylene storage and separation performance. Angew. Chem. Int. Ed. 29, 15995–16002 (2021).
Ye, Y. et al. Pore space partition within a metal-organic framework for highly efficient C2H2/CO2 separation. J. Am. Chem. Soc. 9, 4130–4136 (2019).
Xiang, S. et al. Open metal sites within isostructural metal-organic frameworks for differential recognition of acetylene and extraordinarily high acetylene storage capacity at room temperature. Angew. Chem. Int. Ed. 27, 4615–4618 (2010).
Zeng, H. et al. Induced Fit of C2H2 in a flexible MOF through cooperative action of open metal sites. Angew. Chem. Int. Ed. 25, 8515–8519 (2019).
Mukherjee, S. & Zaworotko, M. J. Crystal engineering of hybrid coordination networks: from form to function. Trends Chem. 6, 506–518 (2020).
Niu, Z. et al. A MOF-based ultra-strong acetylene nano-trap for highly efficient C2H2/CO2 separation. Angew. Chem. Int. Ed. 10, 5283–5288 (2021).
Yang, L. et al. Adsorption site selective occupation strategy within a metal-organic framework for highly efficient sieving acetylene from carbon dioxide. Angew. Chem. Int. Ed. 9, 4570–4574 (2021).
Wang, L. et al. Interpenetration symmetry control within ultramicroporous robust boron cluster hybrid MOFs for benchmark purification of acetylene from carbon dioxide. Angew. Chem. Int. Ed. 42, 22865–22870 (2021).
Li, P. et al. A rod-packing microporous hydrogen-bonded organic framework for highly selective separation of C2H2/CO2 at room temperature. Angew. Chem. Int. Ed. 2, 574–577 (2015).
Li, Y.-P. et al. Ultramicroporous building units as a path to bi-microporous metal-organic frameworks with high acetylene storage and separation performance. Angew. Chem. Int. Ed. 38, 13590–13595 (2019).
Li, B. et al. An ideal molecular sieve for acetylene removal from ethylene with record selectivity and productivity. Adv. Mater. 47, 1704210 (2017).
Wang, Y. et al. One-step ethylene purification from an acetylene/ethylene/ethane ternary mixture by cyclopentadiene cobalt-functionalized metal-organic frameworks. Angew. Chem. Int. Ed. 20, 11350–11358 (2021).
Ye, Y. et al. Metal-organic framework based hydrogen-bonding nanotrap for efficient acetylene storage and separation. J. Am. Chem. Soc. 4, 1681–1689 (2022).
Hao, H.-G. et al. Simultaneous trapping of C2H2 and C2H6 from a ternary mixture of C2H2/C2H4/C2H6 in a robust metal-organic framework for the purification of C2H4. Angew. Chem. Int. Ed. 49, 16067–16071 (2018).
Li, H. et al. An unprecedented pillar-cage fluorinated hybrid porous framework with highly efficient acetylene storage and separation. Angew. Chem. Int. Ed. 14, 7547–7552 (2021).
Krap, C. P. et al. Enhancement of CO2 adsorption and catalytic properties by Fe-doping of [Ga2(OH)2(L)] (H4L = biphenyl-3,3′,5,5′-tetracarboxylic acid), MFM-300(Ga2). Inorg. Chem. 3, 1076–1088 (2016).
Smith, B. C. Fundamentals of Fourier transform infrared spectroscopy. pp, 55–84 (CRC press, 2011).
Dutta, A. Fourier transform infrared spectroscopy. Spectroscopic methods for nanomaterials characterization. pp, 73–93 (2017).
Acknowledgements
The research work was supported by the National Natural Science Foundation of China (no. 21908090 and 22168023 (J.W.), 22008101 (S.C.), and 22108243 (J.C.)).
Author information
Authors and Affiliations
Contributions
Y.L. and J.W. conceived the project and designed the research, and co-wrote the manuscript. Y.L. designed the MOF materials. Y.L. and J.L. carried out the materials synthesis and adsorption experiments. Y.L. conducted the column breakthrough measurements. Y.L. and H.X. performed the IAST calculations and DFT simulations. Y.L., J.L., and H.X. collected and analyzed the X-ray diffraction data. J.C. and S.C. validated and visualized the results. J.W., Z.Z., and S.D. contributed to the discussion of the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shane Telfer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Liu, J., Xiong, H. et al. Negative electrostatic potentials in a Hofmann-type metal-organic framework for efficient acetylene separation. Nat Commun 13, 5515 (2022). https://doi.org/10.1038/s41467-022-33271-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-022-33271-3
