
Abstract
Enzyme catalysis is used to generate approximately 50,000 tons of value-added chemical products per year. Nearly a quarter of this production requires a stoichiometric cofactor such as NAD+/NADH. Given that NADH is expensive, it would be beneficial to regenerate it in a way that does not interfere with the enzymatic reaction. Water electrolysis could provide the proton and electron equivalent necessary to electrocatalytically convert NAD+ to NADH. However, this form of electrocatalytic NADH regeneration is challenged by the formation of inactive NAD2 dimers, the use of high overpotentials or mediators, and the long-term electrochemical instability of the enzyme during electrolysis. Here, we show a means of overcoming these challenges by using a bioelectrocatalytic palladium membrane reactor for electrochemical NADH regeneration from NAD+. This achievement is possible because the membrane reactor regenerates NADH through reaction of hydride with NAD+ in a compartment separated from the electrolysis compartment by a hydrogen-permselective Pd membrane. This separation of the enzymatic and electrolytic processes bypasses radical-induced NAD+ degradation and enables the operator to optimize conditions for the enzymatic reaction independent of the water electrolysis. This architecture, which mechanistic studies reveal utilizes hydride sourced from water, provides an opportunity for enzyme catalysis to be driven by clean electricity where the major waste product is oxygen gas.
Similar content being viewed by others
Introduction
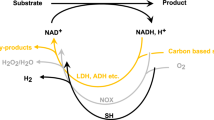
Enzyme catalysis enables challenging chemical reactions to be performed with a high activity and selectivity at ambient conditions1,2,3. The industrial use of enzymes for chemical production currently represents a 100 billion USD industry4. A major application of enzyme catalysis is enantioselective synthesis in the pharmaceutical sector, with two thirds of commercial chiral products involving enzyme catalysis5. Up to 25% of known classes of enzymes require costly stoichiometric cofactors such as nicotinamide adenine dinucleotide (NAD+) and its reduced form (NADH)6,7,8. These stoichiometric cofactors provide hydride as a reagent (Fig. 1) and must be continuously regenerated in situ for viable catalysis9,10. Cofactor regeneration in the industry is currently on performed with secondary enzymes (e.g. formate and glucose dehydrogenases) that catalyze NADH formation from NAD+ using stoichiometric co-substrates such as formate or glucose (Fig. 1b)11,12. These systems require the separation of water-soluble co-substrates and their byproducts, which results in high regeneration costs and limits their application to the synthesis of high-value products13,14. The development of alternative cofactor regeneration methods that do not require the separation of co-substrates and byproducts is a challenge for the design of low-cost enzymatic catalytic systems.

The BioPMR functions as follows: (i) proton reduction occurs on the Pd membrane to form H atoms; (ii) H atoms diffuse through the Pd membrane; and (iii) surface-bound H atoms react with NAD+ to form NADH which is subsequently used for enzyme catalysis. Orange arrows indicate the NADH regeneration step(s) in each method.
An alternative method for regenerating NADH is electrocatalytic regeneration. Protons and electrons sourced from water electrolysis can produce reduced cofactors (e.g., NAD+ + 2e– +H+ → NADH) directly at a cathode surface, without the use of secondary enzymes or co-substrates (Fig. 1b)15,16,17,18,19,20,21. This process occurs in two steps: (i) A one-electron reduction of NAD+ to NAD• beginning at –0.6 V vs. standard hydrogen electrode (SHE); and (ii) a proton-coupled reduction of NAD• to NADH at –1.4 V vs. SHE (Fig. 1b)22. This two-step process ideally produces NADH at the cathode and O2 at the anode as the sole byproduct. However, NAD• formed during step (i) rapidly dimerizes to form inactive NAD222. Dimerization can be mitigated by applying a high cathode voltage (–1.4 to –1.8 V vs. SHE) to accelerate step (ii)23. However, this high voltage introduces new complications in the form of heat and a strong electric field at the electrode surface, both of which can inactivate and/or denature enzymes24,25,26,27. A mediator can be added to prevent NAD+ dimerization and bypass the need for a high cathode voltage28,29, but this mediator reintroduces separation challenges similar to those found in enzymatic regeneration10, unless the mediator is immobilized on the electrode surface30. The development of electrocatalytic cofactor regeneration methods that completely circumvent NAD• formation would enable NADH regeneration to be performed without high cathode voltages or the use of a mediator, while producing O2 as the sole byproduct. A system that meets these criteria could potentially supplant the use of secondary enzymes for cofactor regeneration.
Here, we demonstrate that a bioelectrocatalytic palladium membrane reactor (BioPMR) represents such a system (Fig. 1c). The BioPMR consists of an electrochemical compartment and an enzymatic compartment, with a palladium foil separating both compartments. The palladium foil functions as: (i) the cathode of a 3-electrode electrochemical cell; (ii) a hydrogen-selective membrane; and (iii) a catalyst for cofactor regeneration. Electrolysis performed in the electrochemical compartment generates hydrogen at the palladium foil, which then permeates through the foil and reacts with NAD+ to regenerate NADH for consumption by the enzyme in the enzymatic compartment (Fig. 1c). We find that the NADH regeneration process in the BioPMR occurs through direct hydride (H−) transfer. This mechanism is distinctive from the 2-step electron-transfer processes characteristic of previously reported electrochemical cells where regeneration is driven by the electrochemical bias at the electrode. A benefit of NADH formation through hydride transfer is that NAD2 formation is bypassed even at low reductive potentials (–0.3 V vs. SHE). Moreover, the physical separation of the compartments enables conditions for the electrolytic (i.e., highly acidic or basic) and enzymatic (i.e., physiological pH) processes to be independently optimized. This separation also increases the durability of the system by isolating the enzyme from the electrochemical processes. We demonstrate these advantages herein by performing the following enzymatic processes at their optimal pH in the BioPMR: (i) aldehyde hydrogenation; (ii) ketone hydrogenation, and; (iii) reductive amination. The outcomes of this study show that this platform represents an approach for achieving low-waste, enzyme-catalyzed reactions.
Results
The BioPMR cell (Fig. 2) consists of an electrochemical compartment and an enzyme compartment. The electrochemical compartment contains a 3-electrode cell with a Ag/AgCl reference electrode, a Pt mesh counter electrode (where water oxidation occurs), a 25-µm thick Pd foil working electrode (where proton reduction and hydrogen absorption occur), and electrolyte (8 mL of 1 M H2SO4)31. The Pd foil also functions as a hydrogen-selective membrane separating both compartments. The enzyme compartment (where the hydrogen that diffuses through the membrane reacts with NAD+) contains buffer solution (10 mL of 0.1 M phosphate or tris buffer), 1.5 mM NAD+, enzyme, and 1.5 mM substrate. The side of the palladium membrane facing the enzyme compartment is coated with electrodeposited Pd black to increase the active area31. The Pd black layer is further coated with sputtered Pt (10-nm thick). Details regarding the cell construction can be found in the SI. Details regarding the choice of enzyme and substrate are found below.

The 2-compartment cell contains an electrochemical compartment filled with 1 M H2SO4 and an enzyme compartment filled with buffer solution. The electrochemical compartment contains a platinum anode, a Ag/AgCl reference electrode, and the palladium cathode/membrane. Catalyst (Pd black or Pt-coated) is coated on the side of the membrane exposed to the enzyme compartment.
We first demonstrated that the BioPMR enables the generation of NADH from NAD+ using water as a proton and electron source. A solution of 1.5 mM NAD+ and 0.1 M tris buffer (pHmeasured = 9.0) was added to the enzyme compartment and 1 M H2SO4 was added to the electrochemical compartment (Fig. 3a). The cell was configured as described above (Fig. 2) and heated to physiological temperature (37 °C) in an oven. Galvanostatic electrolysis was performed (Iapplied = –50 mA cm–2) under Ar gas. We initially confirmed the formation of the biologically active NADH isomer (i.e. 1,4-NADH) in the enzyme compartment with a Pd black catalyst by UV–Vis absorption spectroscopy (λmax = 340 nm, Supplementary Fig. 1) and a lactate dehydrogenase (LDH) assay (Supplementary Fig. 2). The percentage of 1,4-NADH formed was then quantified by 1H NMR spectroscopy as a function of time (Fig. 3b, Supplementary Fig. 3). This experiment was performed for both a Pd black catalyst and a Pt-coated catalyst (Fig. 3a). The yield of 1,4-NADH after 3 h was ~26% with the Pd black catalyst, and ~67% with the Pt-coated catalyst (Fig. 3b). 1,6-NADH was the primary byproduct observed by 1H NMR (Supplementary Fig. 3b). 1,6-NADH is an inactive form of cofactor commonly observed as a byproduct within electrochemical and chemical regeneration. The faradaic efficiency for NADH formation was ~1%, indicating that the majority of hydrogen is lost through H2 generation at the surface of the membrane. This low faradaic efficiency is likely a result of the low quantity of NAD+ (0.015 mmol) relative to the high quantity of hydrogen permeating through the membrane (~4.7 mmol h–1). The faradaic efficiency could be increased to ~16% with a lower applied current density and increased NAD+ concentration (see Methods for experimental details). No NAD2 dimer was observed by 1H NMR (Supplementary Fig. 3) and ESI-MS (Supplementary Fig. 4)32,33. The higher rate and greater yield observed with the Pt-coated catalyst is consistent with previous studies identifying Pt as a highly active 1,4-NADH regeneration catalyst34. The higher selectivity of the Pt-coated catalyst to 1,4-NADH is likely due to the preferential binding of carbonyl to Pt35, which renders the C4’ position of NAD+ more electrophilic and accessible to surface-bound hydrogen. The mechanism of hydrogen permeation through Pd with subsequent migration onto Pt is discussed in depth in our previous work35. The turnover frequency of the Pt-coated catalyst was 1.5 µmol1,4-NADH h–1 cm–2. Five cycles of NADH formation experiments showed high stability of the Pt-coated catalyst with <5% decrease in yield after 3 h (Supplementary Fig. 5).

a Cell setup and idealized process for NADH generation. b Reaction profile for 1,4-NADH generation with Pd black catalyst and Pt-coated catalyst (Pt/Pd black) in tris buffer at pH 9. c 1,4-NADH yield in 3 h with Pt-coated catalyst as a function of buffer pH (phosphate buffer for pH 6.5, tris for pH 7–9). Error bars represent the standard deviation of triplicate measurements. The center point of each error bar represents the mean of the triplicate measurements.
We also examined 1,4-NADH formation with the Pt-coated catalyst as a function of pH (Fig. 3c). Basic conditions gave a greater yield of 1,4-NADH over the range of typical enzyme-relevant pH values (pH 6.5–9). This observation is consistent with previous studies that demonstrated that basic conditions mitigate acid-catalyzed decay pathways for 1,4-NADH36,37. We also observed that 20‒30% of 1,4-NADH decays in a buffer at both pH 6.5 and 7 at 37 °C over 3 h in the absence of catalyst and applied current (Supplementary Fig. 6). This instability suggests the disparate yields of 1,4-NADH at different pH values is a result of 1,4-NADH degradation over the course of the 3 h experiment at lower pH values. Such degradation will not be a significant issue if the NADH generated is quickly consumed. It should also be noted that if a polar organic solvent such as dimethylsulfoxide (DMSO) was used in lieu of water, no NADH was formed (Supplementary Fig. 7). Our results demonstrate that the BioPMR can regenerate 1,4-NADH from aqueous NAD+ with high yield (67%) and minimal byproduct formation (Supplementary Fig. 3) using water electrolysis.
Scanning electron microscopy (SEM) images of the Pt-coated catalyst surface before (Supplementary Fig. 8a) and after five NADH generation experiments (Supplementary Fig. 8b) showed that the high-surface morphologies of the Pt-coated Pd catalyst were retained during five cycles of NADH regeneration. Top-view SEM images of the Pt-coated catalyst with an energy-dispersive X-ray spectroscopy (EDX) analysis showed a minimal change in atomic % Pt (ΔPt% ≈ 0.3%) and Pd (ΔPt% ≈ 1.5%), which suggested negligible degradation of the Pt-coated catalyst after five cycles of NADH regeneration (Supplementary Fig. 9).
We proceeded to study enzyme catalysis in the BioPMR with concurrent 1,4-NADH regeneration. Three different relevant enzymatic processes were examined: (i) aldehyde reduction (Fig. 4a); (ii) ketone reduction (Fig. 4b), and; (iii) reductive amination (Fig. 4c). These enzymatic reactions were chosen because each has a different optimum operating pH38,39,40. Each reaction mixture consisted of 5 units of enzyme and 1 mg/mL bovine serum albumin (BSA) with 0.1 M of the appropriate buffer in the enzyme compartment. The electrochemical compartment was filled with 1 M H2SO4. All enzymatic studies were performed at 37 °C for 3 h with an applied current density of ‒50 mA cm–2 (Vavg = ‒0.3 ± 0.05 V vs. SHE, Supplementary Fig. 10a–c). Product conversion was tracked as a function of time by 1H NMR (Fig. 4, Supplementary Figs. 11–13). All enzymes show high yields (65–100%) and conversion rates for their respective substrates. Control experiments with no added NAD+ and no enzyme confirmed that the NADH-dependent enzymes were responsible for product formation (Fig. 4). We also performed the enzymatic aldehyde reduction with 1:5 and 1:25 equivalents of substrate relative to NAD+, and observed ~100% conversion in 4 h and >95% conversion 24 h, demonstrating a turnover number of ~2.2 h–1 (Supplementary Fig. 14).

a Reduction of propionic aldehyde with alcohol dehydrogenase (ADH) in phosphate buffer at pH 6.5. b Reduction of pyruvic acid with lactate dehydrogenase (LDH) in tris buffer at pH 7. c Reductive amination of pyruvic acid with alanine dehydrogenase (AlaDH) in tris buffer at pH 9. Each enzymatic reaction mixture contains 1 mg/mL BSA.
We hypothesized that the isolation of the enzyme from the electrochemical process in the BioPMR would increase the enzyme stability. We tested this hypothesis by performing two experiments with LDH under similar conditions, one in the BioPMR (Fig. 5a) and one under conventional electrocatalytic regeneration conditions (Fig. 5b). Both experiments were performed under electrolysis with LDH in 0.1 M tris (pH 7). The appropriate potential (–0.8 V vs. SHE) for NAD+ reduction in the electrocatalytic experiment was identified by cyclic voltammetry (CV, Supplementary Fig. 15). The activity of LDH was lost after 30 min for electrocatalytic regeneration conditions while BioPMR conditions retained activity comparable to the control in which no electrolysis was performed (Fig. 5c).

Cell setup for a BioPMR regeneration conditions and b conventional electrocatalytic regeneration conditions. c LDH activity as a function of time for both setups. Each enzyme solution contains 1 mg/mL BSA. Error bars represent the standard deviation of triplicate measurements. The center point of each error bar represents the mean of the triplicate measurements.
We conducted pH-dependent hydrogen permeation studies to probe the mechanism of NADH regeneration in the BioPMR (Fig. 6). We hypothesized that the mechanism of formation of the reduced product (NADH) involved disproportionation of H atoms (i.e., H· + H· → H+ + H−) on the catalyst surface. These charged H species (i.e., H+ + H-) will desorb as H2 (i.e., H+ + H– → H2(g)) if H− does not react with NAD+. Our previous study demonstrated that the amount of H2(g) formed on the Pt-coated Pd catalyst in the enzyme compartment is governed by the desorption process34. This desorption process would result in a pH-dependence of the hydrogen generation in the enzyme compartment while H2 desorption that does not involve disproportionation of H atoms (i.e., H· + H· → H2(g)) would not be pH-dependent.

a Cell setup. b H2 gas evolution at steady-state with a Pt-coated catalyst as a function of pH for tris (teal) and phosphate (orange) buffer. Error bars represent the standard deviation of triplicate measurements. The center point of each error bar represents the mean of the triplicate measurements.
A BioPMR cell was connected to an atmospheric mass spectrometer (atm–MS), with gas sampling of the enzyme compartment (Fig. 6a). The cell was filled with electrolyte (1 M H2SO4) in the electrochemical compartment and buffer only (1 M tris or phosphate) in the enzyme compartment. Electrolysis was performed (Iapplied = –50 mA cm–2) and the amount of H2 gas generated at the catalyst surface was allowed to reach steady state (~1 h). We confirmed that the H2 generated in the chemical compartment was stable (0.76 mmol cm–2 h–1) after reaching steady-state (Supplementary Fig. 16c). This stability was retained even if the foil was repeatedly used to regenerate NAD+ (Supplementary Fig. 16b). A series of these measurements were performed at sequential buffer pH values (pHtris = 3.4, 7, 8, 9; pHphosphate = 3.4, 6, 7, 8). We observed that lower pH values resulted in higher H2 gas generation for both tris and phosphate buffer (Fig. 6b). A detailed discussion of the results is provided below. It should be noted that the pH 3.4 data points extend outside of the buffering pH range.
Discussion
This work demonstrates that the BioPMR can be used to: (i) electrochemically regenerate NADH from NAD+, and; (ii) perform NADH regeneration in parallel with enzyme catalysis. The BioPMR has several advantages over conventional enzymatic and electrochemical cofactor regeneration methods. First, the BioPMR does not require any secondary enzymes, co-substrates, or immobilized mediators. Second, the only major waste product in the BioPMR is oxygen gas generated at the anode. Both advantages are in contrast to formate and glucose dehydrogenase systems that require separation of water-soluble co-substrates and byproducts13,14, or electrochemical cofactor regeneration systems that often require immobilized mediators to suppress inactive NAD2 formation22. Both advantages minimize complexity and the purification steps required for isolation of the desired product.
There are additional advantages of the BioPMR that stem from the separation of the electrochemical and enzymatic processes. Typical electrochemical cofactor regeneration systems with both processes occurring in the same reaction mixture necessitate compromise with regard to the ideal operating conditions (e.g., pH, temperature, ionic strength, and catalyst choice) for each process. Enzyme inactivation or denaturation can also occur from the electric field or resistive heating at the electrodes in these systems22. In the BioPMR, the Pd foil separates the two compartments and enables the optimal conditions for each process to be used while also isolating the enzyme from the electric field of the cell. Moreover, the separation of the BioPMR enables a lower overpotential for NADH formation at biologically relevant pH values (pH 6.5–9.0) relative to a typical electrochemical cofactor regeneration system (–0.3 V vs SHE (Supplementary Fig. 10) and –0.6 to –1.7 V vs. SHE41,42, respectively). The operating voltage also showed no change for >10 h of electrolysis (Supplementary Fig. 17). In summary, the BioPMR can perform a wide range of NADH-dependent enzymatic transformations on different reagents under ideal operating conditions with water and electricity as the primary inputs and benign gasses as the primary outputs.
NADH formation from NAD+ proceeds through one of the following mechanisms: (a) electron transfer and proton transfer (2e– + H+); (b) electron transfer and hydrogen transfer (e– and H), or; (c) hydride transfer (H–). Electrocatalytic NADH generation follows (a) or (b)22. NADH formation in the BioPMR is unlikely to undergo mechanisms (a) and (b) because the surface of the catalyst is isolated from the electrochemical potential of the cell (Fig. 1c), thus any pathway involving direct e– transfer is unlikely. This assertion is also supported by the absence of NAD2 formation in the BioPMR, which occurs following direct electron transfer.
The presence of surface-bound hydride is supported by the pH-dependence of H2 gas generation experiments. These experiments demonstrate that higher H+ concentrations result in faster H2 gas generation. We conjecture that the presence of charged hydrogen species (i.e., H+ and H−) on the catalyst surface is responsible for the accelerated H2 gas generation at low pH. This acceleration would not be expected if the desorption-limited hydrogen evolution35 was solely occurring through charge neutral processes (e.g., H· + H· → H2). The formation of charged hydrogen species on precious metal surfaces has been previously demonstrated43. This process occurs through water binding with surface hydrogen followed by heterolytic cleavage of the Pt–H bond, which ultimately forms H3O+ and transfers an e- to the Pt bulk. Such a mechanism can be conceptualized as a reverse Volmer process (H+ + e– → H). We confirmed that water is necessary for NADH regeneration to occur by performing comparable experiments in DMSO (Supplementary Fig. 7). We propose a mechanism here (Fig. 7) where the formation of hydride occurs through water-assisted disproportionation of H atoms (e.g., H· + H· → H+ + H–) on the Pt-coated catalyst.

(1) Protons are reduced at the Pd foil to form atomic hydrogen; (2) atomic hydrogen permeates through the Pd foil to form surface-boundhydrogen; (3) surface-bound hydrogen undergoes water-assisted charge separation, producing hydronium and a reduced metal surface; (4) electron transfer from the reduced metal to surface-bound hydrogen forms hydride; (5) hydride transfers to NAD+, forming 1,4-NADH.
In conclusion, we demonstrate a strategy for performing NADH-driven enzymatic reactions. This platform enables 1,4-NADH to be electrochemically regenerated without the formation of inactive NAD2. The system uses a Pd foil as both a cathode and selective membrane that enables both enzymatic and electrolytic processes to be physically separated by the membrane. This architecture increases the stability of the enzyme relative to conventional electrochemical NADH regeneration methods and requires a lower overpotential NAD+/NADH conversion at physiological pH (6.5–9.0). A variety of enzymatic reactions are performed using the BioPMR at high yield under their optimum enzyme pH (Fig. 4) conditions using in situ regenerated 1,4-NADH. We also show that this system regenerates 1,4-NADH through direct hydride transfer to NAD+. This hydride transfer likely involves water-assisted charge separation of H atoms at the catalyst surface. The BioPMR is a method for 1,4-NADH regeneration that with further study (e.g., Pd membranes with immobilized enzyme and cofactor for faster reaction rates44) could result in an expansion of the use of NADH-dependent enzymes for industrial chemical production.
Methods
Materials
Pd (99.95%) was obtained from Silver Gold Bull. β-Nicotinamide adenine dinucleotide hydrate (≥99%), β-Nicotinamide adenine dinucleotide, reduced disodium salt hydrate (≥97%, HPLC), propionic aldehyde (reagent grade, 97%), ammonium chloride, bovine serum albumin, L-lactic dehydrogenase from rabbit muscle, alcohol dehydrogenase from Saccharomyces cerevisiae, L-alanine dehydrogenase from Bacillus subtilis, sulfuric acid (95.0–98.0%), H2O2 solution (30 wt.% in H2O) and dimethylsulfone (quantitative NMR standard, TraceCERT) were purchased from Sigma Aldrich and used as received. Sodium pyruvate (≥99%) was purchased from Fisher Scientific. 68–70% Nitric acid was obtained from VWR. Pt gauze (52 mesh, 99.9%) and Pt wire (0.5 mm, 99.95%) were obtained from Alfa Aesar. Ag/AgCl reference electrodes (RE5B) were purchased from BASi.
Cell design
The three cell cores (Fig. 2) are printed by a stereolithography 3D printer. The cores are composed of high-temperature resistant resin (Formlabs proprietary resin). The membrane is sealed between the cores by Viton ring gaskets, and the assembly is clamped together by two waterjet cut ¼ in thick aluminum end plates connected by four M4 stainless steel bolts. Quick-turn polycarbonate couplings (¼−28 in) were used to feed gas inlet/outlet ports on the enzyme cell (threads were printed in the core).
Electrochemistry
A Metrohm Autolab PGSTAT302N/PGSTAT204M potentiostat was used for electrochemical experiments. Electrochemical data was analyzed both with Metrohm Autolab NOVA (Version 2.1) and Origin (Version 9.3) palladium foil was fitted between the enzyme and cathode electrochemical compartment to assemble the electrocatalytic palladium membrane reactor. Viton ring gaskets were used to seal the palladium foil in place and prevent leaking. The enzyme compartment was filled with 10 mL of tris or phosphate buffer. The electrochemical compartment was filled with 8 mL of 1 M H2SO4. A Ag/AgCl reference electrode (3.0 M NaCl) and a Pt mesh counter electrode were housed in the electrochemical compartment. All potentials are reported versus SCE unless stated otherwise. Electrolysis was conducted galvanostatically at –50 mA cm–2 (Vavg = –0.3 V vs. SCE) for the BioPMR experiments. The uncompensated resistance was 1–2 Ω for all experiments, and no IR correction was used. The thickness of the foil was 25 μm and the geometric surface area of the foil was 2.5 cm2. Cyclic voltammetry (CV) measurements were performed under an Ar atmosphere with a Pt-coated Pd working electrode, a Ag/AgCl reference electrode, a Pt mesh counter electrode, and a scan rate of 250 mV/s. The faradaic efficiency (FE) was calculated assuming a reaction stoichiometry of 2 H· per H− equivalent:
Pd foil and Pd black catalyst preparation
Pd foils were rolled from a palladium wafer bar to a thickness of 25 μm. The thickness of the foil was measured by a Mitutoyo digital micrometer to an accuracy of ±1 μm. The freshly rolled foil was annealed in Ar at 850 °C for 1.5 h and stored in air. Prior to use, the annealed foils were cleaned by soaking the foil in a 0.5:0.5:1 (by volume) conc. HNO3:H2O:30% H2O2 for ~45 min, and subsequently cleaned with Milli-Q ultrapure water. The Pd black catalyst was then electroplated onto the surface. The cell was assembled as described above, except the cathode compartment was filled with an electrodeposition solution (15.9 mM PdCl2 in 1 M HCl) in lieu of the 1 M H2SO4 electrolyte. The solution was stirred at a constant rate (400 rpm) and a voltage of –0.1 V vs. SCE was applied to the Pd foil working electrode to reduce the Pd ions in solution. The electrodeposition was stopped when 18.5 C of charge (7.38 C/cm2) had been passed. The mass loading for all electrodepositions was ~10 mg. The sputter-deposited Pt-coated samples were within error of this mass loading. The foils were then thoroughly rinsed with Milli-Q ultrapure water, covered in a 4” diameter petri dish to maintain cleanliness, and stored in ambient conditions.
Pt-coated membrane preparation
The catalyst-coated membranes were prepared by sputter deposition. The electrodeposited palladium foils were placed in a Leica EM MED020 coating system at a working distance of 6 cm, the chamber was sealed, and a vacuum applied to achieve a base pressure of 1 × 10–4 mbar. After the base pressure was reached, argon was continuously flowed into the chamber to maintain a pressure of 1 \(\times\)10–2 mbar, the plasma was ignited, and voltage was adjusted to maintain a constant sputter current of 30 mA. Following a 30 s presputter, the target shutter was opened and 10-nm platinum was sputtered onto the electrodeposited palladium catalyst. The sputtering rate was 0.2 nm/s, as determined by in situ quartz crystal microbalance. The foils were used for NADH regeneration and enzymatic reduction/reductive amination experiments without any further processing. The catalyst-coated membranes were used for up to 20 reactions. The entire catalyst layer of Pt-coated Pd black was removed with concentrated nitric acid, then the foil was cleaned (described above), replated, and reused. Each palladium foil was used for ~5 cleaning cycles before being discarded.
Buffer preparation
Phosphate buffers were prepared by dissolving the appropriate amounts of disodium and monosodium phosphate salts in deionized water. Tris (tris(hydroxymethyl)aminomethane) buffer was prepared by dissolving tris salts in deionized water. The pH for these buffers was then adjusted with the addition of the appropriate amounts of 1 M aqueous solutions of NaOH and/or HCl.
NADH generation
NADH regeneration was performed in a 0.1 M phosphate or tris buffer at pH 6.5, 7, and 9. All reactions were carried out in an oven at 37 °C under Ar. Enzyme compartment of the 3D-printed cell with a magnetic stir bar was filled with 1.5 mM NAD+ and 10 mg bovine serum albumin (BSA) in buffer (10 mL) or dry DMSO. 1 M H2SO4 electrolyte solution (8 mL) was added to an electrochemical compartment and a constant reductive current of –50 mA was applied for 3 h. NADH reaction mixture was stirred at a constant rate (400 rpm) throughout the experiment. Reaction aliquots were sampled every 0.5 h to monitor the reaction progression of NADH generation by UV/Vis and 1H NMR spectroscopy. UV/Vis absorption spectra of reaction mixture were collected by using a Jasco J-815 spectrometer without dilution. All measurements were performed at room temperature with the sample perpendicular to the light path. A tris buffer blank was used as the baseline measurement. Solution temperature and pH were checked before and after the reaction. Electrospray mass spectrometry was recorded with a Waters ZQ mass spectrometer equipped with ESI ion source without sample dilution. The faradaic efficiency could be increased from ~1% to ~16% by decreasing the current density (–50 mA/cm2 to –5 mA/cm2) and increasing the NAD+ concentration (1.5 mM to 15 mM).
Scanning electron microscopy (SEM) and X-ray energy-dispersive spectroscopy (EDX)
Scanning Electron Microscope (SEM) and energy-dispersive X-ray spectroscopy (EDX) experiments were performed using a Helios NanoLab 650 Focused Ion Beam SEM. EDX data was acquired from 600 μm2 regions over 50 s at 10 kV accelerating voltage and 120 nA.
Enzyme catalysis
All enzyme-catalyzed reactions were carried out in the oven at 37 °C under Ar. The enzyme compartment contained a magnetic stir bar and was filled with 1.5 mM of NAD+, 10 mg BSA, 1.5 mM of substrates (propionic aldehyde for aldehyde reduction, sodium pyruvate for ketone reduction, pyruvate and 25 mM NH4Cl for reductive amination), and 5 units of the corresponding enzyme (alcohol dehydrogenase for aldehyde reduction, lactate dehydrogenase for ketone reduction, and L-alanine dehydrogenase for reductive amination) in 10 mL of the buffer. Alcohol reduction was performed in 0.1 M phosphate buffer at pH 6.5; ketone reduction was performed in 0.1 M tris buffer at pH 7; reductive amination was performed in 0.1 M tris buffer at pH 9. 1 M H2SO4 electrolyte solution (8 mL) was added to the electrochemical compartment and a constant reductive current of –50 mA was applied for 3 h. The reaction mixture was stirred at a constant rate (400 rpm) throughout the experiment. Reaction aliquots were sampled every 0.5 h to monitor the reaction progression of product formation by 1H NMR spectroscopy. Aldehyde reduction was also performed with 1:5 and 1:25 mol equivalent of NAD+ (1.5 mM) and propionic aldehyde (7.5 mM or 37.5 mM) with 10 or 30 units of ADH in 0.1 mM phosphate buffer at pH 6.5, respectively.
Product quantification
Proton nuclear magnetic resonance (1H NMR) was used for product quantification. 460 μL of the reaction mixture (1.5 mM) and 40 μL 0.2 M dimethylsulfone internal standard in D2O were added to an NMR tube. 1H NMR spectra were acquired on a Bruker Avance 400dir, or 400inv spectrometer at 298 K. Relative concentrations were determined by comparing the integrated signal of the methyl group of dimethylsulfone.
Enzyme activity assay
All enzyme stability experiments were carried out in an oven at 37 °C under Ar. 10 uL (~245 units) LDH and 30 mg BSA in 30 mL Tris buffer at pH 7 were prepared and divided into 3×10 mL solutions. For the BioPMR platform, the enzyme compartment was filled with an LDH solution and a stir bar. 1 M H2SO4 electrolyte solution (8 mL) was added to the electrochemical compartment and a constant reductive current of –50 mA (Vave = –0.3 V vs. SCE) was applied for 3 h. For the electrocatalytic platform, the enzyme compartment was empty, and the Pt-coated side of the membrane was facing the electrochemical compartment. LDH solution was added to the electrochemical compartment and a constant potential of –0.8 V vs. SCE was applied for 3 h. The LDH solution was stirred at a constant rate (400 rpm) throughout the experiment. Reaction aliquots were sampled every 0.5 h to monitor the LDH stability in each platform. These samples were then diluted 100 times in the assay buffer (0.1 M Tris buffer pH 7, 2 mM NADH) and incubated for 2 min. The reactions were then initiated by the addition of sodium pyruvate (final concentration of 3.5 mM) and the rate of each reaction was determined based on the rate of the consumption of NADH, by monitoring the absorbance at 340 nm. The concentration of the remaining active enzyme after each time point was thus quantified, since the rate of these reactions are directly proportional to the enzyme concentration under these conditions. All the assays were performed in triplicate and in 96-well half-area plate [Corning] and monitored with a Synergy H1 plate reader [BioTek].
H2 gas permeation experiment
An ESS CatalySys atmospheric mass spectrometer was used for pH-dependent hydrogen permeation experiments. Hydrogen permeation experiments were conducted with 1 M H2SO4 in the electrochemical compartment and phosphate or tris buffer at their buffering pH in the enzyme compartment. H2 generation outside of their buffering pH (pH 3.4) was also measured to examine if the pH-dependency extends outside of their buffering pH. The palladium foil was placed between the electrochemical and enzyme compartments with the Pt-coated surface facing into the enzyme compartment and a constant reductive current of –50 mA was applied. The production of gaseous H2 (2 m/z) in the enzyme compartment with constant stirring was monitored by atmospheric mass spectrometry (atm–MS) with a flow rate of 10 mL/min into the instrument. The ion current value was taken once the signal had equilibrated (~1 h). The average value for at least three experiments is reported, with error bars representing one standard deviation of the mean. The hydrogen diffusion at steady-state was calculated as follows:
Where Hydrogenchem is the mols of hydrogen evolved in the chem compartment, Hydrogenechem is the mols of hydrogen evolved in the echem compartment, Iapplied is the total current in A, F is Faraday’s constant, and SAgeometric is the geometric surface area of the membrane in cm–2. For steady-state H2 flux measurements, electrolysis was performed at –50 mA/cm2 for 1 h to reach steady-state conditions. At 1 h, atm–MS measurements were performed and the H2 ion current was recorded for 10 h.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings in this study are available either within the paper or its Supplementary Information, or from the corresponding author on request.
References
Wu, S., Snajdrova, R., Moore, J. C., Baldenius, K. & Bornscheuer, U. T. Biocatalysis: enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. Engl. 60, 88–119 (2021).
Bornscheuer, U. T. et al. Engineering the third wave of biocatalysis. Nature 485, 185–194 (2012).
Chen, K. & Arnold, F. H. Engineering new catalytic activities in enzymes. Nat. Catal. 3, 203–213 (2020).
Hilterhaus, L., Liese, A., Kettling, U. & Antranikian, G. Applied Biocatalysis: From Fundamental Science to Industrial Applications (John Wiley & Sons, 2016).
Ciriminna, R. & Pagliaro, M. Green chemistry in the fine chemicals and pharmaceutical industries. Org. Process Res. Dev. 17, 1479–1484 (2013).
McDonald, A. G., Boyce, S., Moss, G. P., Dixon, H. B. F. & Tipton, K. F. ExplorEnz: a MySQL database of the IUBMB enzyme nomenclature. BMC Biochem. 8, 14 (2007).
Wu, H. et al. Methods for the regeneration of nicotinamide coenzymes. Green. Chem. 15, 1773–1789 (2013).
Vidal, L. S., Kelly, C. L., Mordaka, P. M. & Heap, J. T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta- Proteins Proteom. 1866, 327–347 (2018).
De Wildeman, S. M. A., Sonke, T., Schoemaker, H. E. & May, O. Biocatalytic reductions: from lab curiosity to ‘first choice’. Acc. Chem. Res. 40, 1260–1266 (2007).
Wang, X. et al. Cofactor NAD(P)H regeneration inspired by heterogeneous. Pathw. Chem. 2, 621–654 (2017).
Schmid, A., Hollmann, F., Park, J. B. & Bühler, B. The use of enzymes in the chemical industry in Europe. Curr. Opin. Biotechnol. 13, 359–366 (2002).
Liese, A., Seelbach, K. & Wandrey, C. Industrial Biotransformations (John Wiley & Sons, 2006).
Lee, Y. S., Gerulskis, R. & Minteer, S. D. Advances in electrochemical cofactor regeneration: enzymatic and non-enzymatic approaches. Curr. Opin. Biotechnol. 73, 14–21 (2021).
Morthensen, S. T., Sigurdardóttir, S. B., Meyer, A. S., Jørgensen, H. & Pinelo, M. Separation of xylose and glucose using an integrated membrane system for enzymatic cofactor regeneration and downstream purification. J. Memb. Sci. 523, 327–335 (2017).
Immanuel, S., Sivasubramanian, R., Gul, R. & Dar, M. A. Recent progress and perspectives on electrochemical regeneration of reduced nicotinamide adenine dinucleotide (NADH). Chem. Asian J. 15, 4256–4270 (2020).
Yuan, M., Kummer, M. J., Milton, R. D., Quah, T. & Minteer, S. D. Efficient NADH regeneration by a redox polymer-immobilized enzymatic system. ACS Catal. 9, 5486–5495 (2019).
Zhang, L., Vilà, N., Kohring, G.-W., Walcarius, A. & Etienne, M. Covalent immobilization of (2,2′-bipyridyl) (pentamethylcyclopentadienyl)-rhodium complex on a porous carbon electrode for efficient electrocatalytic NADH regeneration. ACS Catal. 7, 4386–4394 (2017).
Megarity, C. F. et al. Electrocatalytic volleyball: rapid nanoconfined nicotinamide cycling for organic synthesis in electrode pores. Angew. Chem. Int. Ed. Engl. 58, 4948–4952 (2019).
Chen, H. et al. Upgraded bioelectrocatalytic N2 fixation: from N2 to chiral amine intermediates. J. Am. Chem. Soc. 141, 4963–4971 (2019).
Song, H.-K. et al. Electrochemical regeneration of NADH enhanced by platinum nanoparticles. Angew. Chem. 120, 1773–1776 (2008).
Chen, Y. et al. Stabilization of formate dehydrogenase in a metal-organic framework for bioelectrocatalytic reduction of CO2. Angew. Chem. Int. Ed. Engl. 58, 7682–7686 (2019).
Schmakel, C. O., Santhanam, K. S. & Elving, P. J. Nicotinamide adenine dinucleotide (NAD+) and related compounds. Electrochemical redox pattern and allied chemical behavior. J. Am. Chem. Soc. 97, 5083–5092 (1975).
Ali, I., Soomro, B. & Omanovic, S. Electrochemical regeneration of NADH on a glassy carbon electrode surface: the influence of electrolysis potential. Electrochem. Commun. 13, 562–565 (2011).
Pandelia, M.-E. et al. Membrane-bound hydrogenase I from the hyperthermophilic bacterium Aquifex aeolicus: enzyme activation, redox intermediates and oxygen tolerance. J. Am. Chem. Soc. 132, 6991–7004 (2010).
Ciaccafava, A. et al. An innovative powerful and mediatorless H2/O2 biofuel cell based on an outstanding bioanode. Electrochem. Commun. 23, 25–28 (2012).
Ho, S. Y., Mittal, G. S. & Cross, J. D. Effects of high field electric pulses on the activity of selected enzymes. J. Food Eng. 31, 69–84 (1997).
Armstrong, F. A. Probing metalloproteins by voltammetry. in Bioinorganic Chemistry 137–221 (Springer Berlin Heidelberg, 1990).
Kohlmann, C., Märkle, W. & Lütz, S. Electroenzymatic synthesis. J. Mol. Catal. B Enzym. 51, 57–72 (2008).
Chenault, H. K., Simon, E. S. & Whitesides, G. M. Cofactor regeneration for enzyme-catalysed synthesis. Biotechnol. Genet. Eng. Rev. 6, 221–270 (1988).
Zhang, C., Zhang, H., Pi, J., Zhang, L. & Kuhn, A. Bulk electrocatalytic NADH cofactor regeneration with bipolar electrochemistry. Angew. Chem. Int. Ed. Engl. 61, e202111804 (2022).
Sherbo, R. S., Delima, R. S., Chiykowski, V. A., MacLeod, B. P. & Berlinguette, C. P. Complete electron economy by pairing electrolysis with hydrogenation. Nat. Catal. 1, 501–507 (2018).
Kadowaki, J. T., Jones, T. H., Sengupta, A., Gopalan, V. & Subramaniam, V. V. Copper oxide-based cathode for direct NADPH regeneration. Sci. Rep. 11, 180 (2021).
Molano-Arevalo, J. C. et al. Insights from ion mobility-mass spectrometry, infrared spectroscopy, and molecular dynamics simulations on nicotinamide adenine dinucleotide structural dynamics: NAD+ vs. NADH. Phys. Chem. Chem. Phys. 20, 7043–7052 (2018).
Wang, X. & Yiu, H. H. P. Heterogeneous catalysis mediated cofactor NADH regeneration for enzymatic reduction. ACS Catal. 6, 1880–1886 (2016).
Kurimoto, A. et al. Physical separation of H2 activation from hydrogenation chemistry reveals the specific role of secondary metal catalysts. Angew. Chem. Int. Ed. Engl. 60, 11937–11942 (2021).
Johnson, S. L. & Tuazon, P. T. Acid-catalyzed hydration of reduced nicotinamide adenine dinucleotide and its analogs. Biochemistry 16, 1175–1183 (1977).
Saba, T. et al. NADH regeneration: a case study of Pt-catalyzed NAD+ reduction with H2. ACS Catal. 11, 283–289 (2021).
Iitani, K. et al. Improved sensitivity of acetaldehyde biosensor by detecting ADH reverse reaction-mediated NADH fluoro-quenching for wine evaluation. ACS Sens. 2, 940–946 (2017).
Al-Onazi, M., Al-Dahain, S. & El-Ansary, A. Isolation and characterization of Thielaviopsis paradoxa L-alanine dehydrogenase. Asian J. Appl. Sci. 4, 702–711 (2011).
Fritz, P. J. Rabbit lactate dehydrogenase isozymes: effect of pH on activity. Science 156, 82–83 (1967).
Ali, I., Ullah, N., McArthur, M. A., Coulombe, S. & Omanovic, S. Direct electrochemical regeneration of enzymatic cofactor 1,4-NADH on a cathode composed of multi-walled carbon nanotubes decorated with nickel nanoparticles. Can. J. Chem. Eng. 96, 68–73 (2018).
Damian, A., Maloo, K. & Omanovic, S. Direct electrochemical regeneration of NADH on Au, Cu and Pt-Au electrodes. Chem. Biochem. Eng. Q. 21, 21–32 (2007).
Zhao, Z. et al. Solvent-mediated charge separation drives alternative hydrogenation path of furanics in liquid water. Nat. Catal. 2, 431–436 (2019).
Velasco-Lozano, S., Benítez-Mateos, A. I. & López-Gallego, F. Co‐immobilized phosphorylated cofactors and enzymes as self‐sufficient heterogeneous biocatalysts for chemical processes. Angew. Chem. Int. Ed. Engl. 56, 771–775 (2017).
Acknowledgements
Thank you to Maria Ezhova in the UBC nuclear magnetic resonance laboratory. Thank you to Dr. Yang Cao, Cameron Kellett, and Dr. Kyle Robinson for useful discussions and suggestions. The authors are grateful to the Natural Sciences and Engineering Research Council of Canada (RGPIN-2018-06748), Canadian Foundation for Innovation (229288), Canadian Institute for Advanced Research (BSE-BERL-162173), and Canada Research Chairs for financial support. These grants were obtained by C.P.B. S.A.N. was supported by a Vanier scholarship from NSERC. This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund, Quantum Materials and Future Technologies Program. Sputter coating was performed in the Centre for High-Throughput Phenogenomics at the University of British Columbia, a facility supported by the Canada Foundation for Innovation, British Columbia Knowledge Development Foundation, and the UBC Faculty of Dentistry. We thank Dr. Shao Ren and Dr. Arthur Fink for their expertise and support throughout the project.
Author information
Authors and Affiliations
Contributions
A.K. and C.P.B. devised the concept. A.K. performed NADH regeneration and enzyme catalysis experiments. S.A.N. performed LDH assay study and enzyme stability experiments. N.E.L. performed SEM and EDX analysis. M.R. and R.P.J. designed the 3D-printed cell and M.R. fabricated the cell. D.J.D. sputtered the palladium membrane foils. C.C.H. performed cyclic voltammetry experiments. A.K., C.C.H., S.G.W. and C.P.B. wrote the manuscript. S.G.W. and C.P.B. supervised the project.
Corresponding author
Ethics declarations
Competing interests
Patent applications based on the palladium membrane reactor technology described in this work have been filed and published: Berlinguette, C.P.; Sherbo, R.S. “Methods and Apparatus for Performing Chemical and Electrochemical Reactions”. US Patent Application No. 16964944 (PCT filed January 2019, national entry July 2020), Publication No. US20210040017A1 (published February 2021). Canadian Patent Application No. 3089508 (PCT filed January 2019, national entry July 2020), Publication No. CA3089508, (published August 2019). Priority data: US Provisional Patent Application No. 62/622,305, filed January 2018. A U.S. provisional patent application based on the enzyme catalysis technology described in this work has been filed: Berlinguette, C.P.; K.A. “Method for Performing Coupled Enzymatic Electrocatalytic Reactions.” US Provisional Patent Application No. 63/430,735. Filed December 2022. The remaining authors declare no other competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kurimoto, A., Nasseri, S.A., Hunt, C. et al. Bioelectrocatalysis with a palladium membrane reactor. Nat Commun 14, 1814 (2023). https://doi.org/10.1038/s41467-023-37257-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-023-37257-7
This article is cited by
-
Thermochemical heterolytic hydrogenation catalysis proceeds through polarization-driven hydride transfer
Nature Chemistry (2026)
-
Accelerating lithium-mediated nitrogen reduction through an integrated palladium membrane hydrogenation reactor
Nature Communications (2025)
-
Frontier Biological Electrodes for Cell Analysis
Chemical Research in Chinese Universities (2025)
-
Solar-driven sugar production directly from CO2 via a customizable electrocatalytic–biocatalytic flow system
Nature Communications (2024)
-
Electrochemical hydrogenation and oxidation of organic species involving water
Nature Reviews Chemistry (2024)
