
Abstract
Electroreduction of CO2 to valuable multicarbon (C2+) products is a highly attractive way to utilize and divert emitted CO2. However, a major fraction of C2+ selectivity is confined to less than 90% by the difficulty of coupling C-C bonds efficiently. Herein, we identify the stable Cu0/Cu2+ interfaces derived from copper phosphate-based (CuPO) electrocatalysts, which can facilitate C2+ production with a low-energy pathway of OC-CHO coupling verified by in situ spectra studies and theoretical calculations. The CuPO precatalyst shows a high Faradaic efficiency (FE) of 69.7% towards C2H4 in an H-cell, and exhibits a significant FEC2+ of 90.9% under industrially relevant current density (j = −350 mA cm−2) in a flow cell configuration. The stable Cu0/Cu2+ interface breaks new ground for the structural design of electrocatalysts and the construction of synergistic active sites to improve the activity and selectivity of valuable C2+ products.
Similar content being viewed by others


Introduction
The electrosynthesis of multicarbon (C2+) products such as ethanol (C2H5OH) and ethylene (C2H4) from CO2 is highly attractive because of the versatility of these products in the chemical and energy industries1,2,3,4. However, the selective production of C2+ products from CO2 reduction reaction (CO2RR) is challenging, with competition from the hydrogen evolution reaction (HER) and C1 products (e.g., CO, HCOOH, and CH4) production5,6,7. Among all the nanostructured electrocatalysts reported thus far, copper-based materials are known to be the most selective for CO2-to-C2+ production8,9. Up to now, a variety of strategies have been proposed to improve CO2-to-C2+ selectivity, including controlling oxidation states10,11, constructing nanostructures12,13, alloying14,15, doping16, and molecular decorating1. Unfortunately, the selectivity of C2+ products is still low, especially at high current densities (j)17.
Recently, manipulating oxidation states for constructing a synergistic Cu0/Cu1+ interface has been demonstrated as an effective way to promote C2+ product conversion via stabilizing Cu1+ species11,12,17,18,19,20,21,22. For example, it’s reported that a high concentration of Cu1+ (8%) in the iodine-modified Cu catalyst was observed by the linear combination fitting (LCF) analysis of operando X-ray absorption spectra (XAS), which contributed to the high FEC2+ of about 80% at the j lower than 40 mA cm−2 (ref. 12). Our group also constructed the stable Cu0/Cu1+ interface via isolating Cu-S motif with C2+ selectivity over 80%22. By combining Cu0 and Cu1+ synergistic species, the adsorption of *CO can take place at different active sites, resulting in the improvement of CO dimerization, i.e., the Cu0 site can activate CO2 and facilitate the following electron transfers, while the Cu1+ site strengthens the *CO adsorption and boosts C-C coupling, further endowing a lower Gibbs free energy for *OCCOH formation11,21,23,24,25. Similarly, it was revealed that over the two adjacent Cu0 and positively charged Cuδ+ atoms, the Cu0 site can adsorb CO2 and the neighboring Cuδ+ site is conducive to H2O adsorption, thus promoting the activation of CO226. Moreover, theoretical studies have shown that the subsurface O stabilized surface Cuδ+ species indeed enhances the C2 products selectivity through increasing the *CO coverage27.
In addition to the widely reported Cu1+ species in the field of CO2RR, Cu2+ species with higher oxidation states than Cu0 or Cu1+ sites feature the structure characteristics to easily bind CO or H2O, which have been demonstrated in other heterogenous catalysis fields28,29,30. Recently, Qiao et al. have proved the feasibility of Cu2+ sites for promoting *CO hydrogenation in the Cu-Ce-Ox solid solutions, which facilitate the generation of *CHO intermediates instead of *CO dimerization on the single Cu2+ sites31. As a consequence, Cu-Ce-Ox delivered a high selectivity for CH4 with significant suppression of the C2 products. The above findings suggest the possibility of retaining Cu2+ sites through material structure design and promoting *CHO intermediate formation on Cu2+ catalytic active sites. Inspired by these works, we anticipate that the construction of Cu0/Cu2+ interface may provide the possibility of direct OC–CHO coupling process compared with single Cu2+ sites; however, it still remains a grand challenge to build stable Cu0/Cu2+ interface under operando CO2RR conditions.
Herein, we rationally screened the Cu-based precatalysts for constructing stable Cu0/Cu2+ interfaces, and elucidated their synergic roles in CO2-to-C2+ conversion. Using Materials Project Database (MPD) and density functional theoretical (DFT) calculations therewith, we screened different Cu2+-containing compounds and found Cu2+ phosphorus oxysalts of Cu2P2O7 and Cu3(PO4)2 (collectively named CuPO) to be the most promising candidate for exhibiting the highest stability of Cu0/Cu2+ interface under the electroreduction condition; in consequence, the peculiar Cu0/Cu2+ interfaces facilitate a low-energy pathway of *CHO coupling with *CO to form *OCCHO intermediate. Experimentally, we synthesized CuPO catalysts through a facile and scalable method, which achieved 69.7% FE for C2H4 in the neutral electrolyte, and 90.9% FEC2+ with a C2+ partial current density (jC2+) of over 300 mA cm−2 in a flow cell using the alkaline electrolyte. Our operando experimental characterizations unambiguously demonstrate the robust existence of Cu0/Cu2+ interfaces derived from CuPO during CO2RR, and in situ surface-enhanced infrared absorption spectroscopy (SEIRAS) in conjunction with DFT calculation testify that the Cu2+ sites are conducive to the formation of *CHO, which then facilely coupled with *CO on Cu0 surface to form *OCCHO intermediate, leading to high-efficiency CO2-to-C2+ conversion performance.
Results
Theoretical calculation
The formations of *CO, *COH, and *CHO intermediates on Cu0, Cu1+ and Cu2+ sites induce different coupling manners and probabilities for C2+ species formation on Cu0/Cu1+ and Cu0/Cu2+, which provide guidance on designing the Cu-based catalysts for CO2RR to C2+ products11,31. Firstly, we explored the formation energy of *CHO or *COH (*CO + H+/e → *CHO or *COH) on classical Cu0 (metallic Cu), Cu1+ (Cu2O), and Cu2+ (CuO) sites, respectively, which play key roles in the C2+ products. From Fig. 1a, one can see that on the Cu0 and Cu1+ sites, the *CHO or *COH formation is relatively endothermic at the potential U = 0 V (all potentials were calibrated to the reversible hydrogen electrode (RHE) if not mentioned), indicating that *CO could be the primary intermediates. By comparison, the Cu2+ site has an excellent ability to hydrogenate *CO to form *CHO (ΔG = 0.10 eV) rather than *COH (ΔG = 1.33 eV). Accordingly, it can be expected that the Cu0 and Cu1+ sites could be covered by CO intermediates, while the Cu2+ site contributes to *CO hydrogenation to form *CHO intermediates that can facilitate the direct OC–CHO coupling process to increase the selectivity of C2+ product (Fig. 1b).

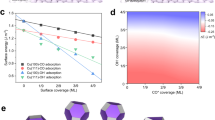
a Reaction energy of *CO hydrogenation to *COH or *CHO on Cu, Cu1+ and Cu2+ sites, respectively. b Schematic diagram of different intermediates (CO or CHO) on the Cu0/Cu1+ and Cu0/Cu2+ interfaces. c Thermodynamic stabilities of materials as a function of Ef and Ehull. d Electrochemical stability of candidates, where Udiss is the dissolution potential of materials, larger and greener circles represent more stable materials (i.e., larger Udiss), and the structures of the most stable Cu2P2O7 and Cu3(PO4)2. e–g Energy profiles of different C–C coupling processes (* and # represent Cu0 and Cu2(1)+ sites, respectively), e CO-CO, f COH-CO, and g CHO-CO at the Cu0/Cu2(1)+ interfaces and Cu(100), and the related transition state structures (TS1 ~ TS9) of different C-C coupling processes.
Facing various Cu-based catalysts with Cu2+ sites, it is difficult and time-consuming to examine each one and locate the optimal one with superior electrochemical stability and catalytic performance using complex experimental methods. High-throughput screening as an efficient forecast method for a large group of candidate materials has been widely used to predict promising materials for experimental synthesis. Here, we extracted 83 Cu2+-containing compounds from Materials Project, and various criteria were utilized to filtrate candidates (details in the Supplementary Note 1). Firstly, the thermodynamic stability that is the intrinsic property of materials was considered, in which two criteria were used: (i) the formation energy (Ef) should be less than 0 eV, and the more negative Ef means the higher thermodynamic stability; (ii) the energy above the convex hull (Ehull) should be less than 70 mV/atom32, which can further quantify the structural stability of materials, and the smaller Ehull indicates that the corresponding material is more stable. Accordingly, 26 candidates were screened out (Fig. 1c). Then, their electrochemical dissolution potential Udiss was further assessed (see details in the Supplementary Note 2), and a more positive Udiss means higher electrochemical stability. As shown in Fig. 1d, it can be found that 9 Cu2+-containing compounds exhibit good electrochemical stability, in which Cu2P2O7 (Udiss = 0.94 V) and Cu3(PO4)2 (Udiss = 0.77 V) process the optimal electrochemical stability. Meanwhile, the high formation energy of the Ovac on Cu3(PO4)2 (0.82 eV) indicated that the phosphate group could play an important role in stabilizing the O atoms, thus limiting the reduction of Cu2+ (Supplementary Fig. 3). Therefore, Cu2P2O7 and Cu3(PO4)2 could be two promising candidates to construct Cu0/Cu2+ interface for CO2RR to C2+ products under the suitable reaction conditions.
In order to clarify the performance of the Cu0/Cu2+ interface in catalyzing CO2RR to produce C2+ products, we selected Cu2P2O7 and Cu3(PO4)2 as examples to construct the Cu0/Cu2+ interface (Supplementary Fig. 1), and explored its ability to promote the C–C bond coupling in comparison with the common Cu0/Cu1+ interface and the classical Cu(100) surface (Fig. 1e, f). Here, we considered all three general collaborative pathways: (i) *CO + #CO → OC–CO, (ii) *CO + #COH → OC–COH and (iii) *CO + #CHO → OC–CHO (* and # represent Cu0 and Cu2(1)+ sites, respectively)8, and calculated their reaction energy energies, aiming to understand the C–C bond coupling mechanism in depth. As shown in Fig. 1e–g, CO2RR on the Cu0/Cu2+ interface always has a lower energy profile than that on the Cu0/Cu1+ one, regardless of the C-C bond coupling pathways, implying the better acceleration of the Cu0/Cu2+ interface for the C-C coupling. In addition, although the OC–COH coupling process has a relatively low barrier of 0.71 eV at the Cu0/Cu2+ interfaces compared with the OC-CO coupling process (Ea = 0.95 eV), COH formation is difficult and requires amount of energy, leading the high effective energy barrier (Fig. 1f). The formation of CHO is much easier compared with that of COH on Cu2+ sites; importantly, the #CHO intermediate only needs to overcome a low-energy barrier of only 0.46 eV to couple with *CO at the Cu0/Cu2+ interface (Fig. 1g), which is also lower than the energy barrier (0.54 eV) for OCCO dimerization on classical Cu(100). These indicate that the OC–CHO coupling process at the Cu0/Cu2+ interface constructed by Cu2P2O7 (or Cu3(PO4)2, Supplementary Fig. 2) has an evident superiority over the OC–CO or OC–COH coupling processes, and at the same time, the Cu2+ site can well facilitate the *CO hydrogenation to the CHO intermediate for high C2+ yield instead of C1 (Supplementary Fig. 4).
Catalysts synthesis and structure investigation
Based on the theoretical studies, CuPO catalysts were synthesized using a facile and scalable method, as illustrated in the “Methods”, Supplementary Fig. 5 and Note 3. The XRD patterns of the catalyst powders investigated in this study are shown in Fig. 2a, b, with diffraction peaks well indexed to the triclinic phase of Cu3(PO4)2 (PDF#97-006-8811) and the monoclinic Cu2P2O7 (PDF#97-015-7107), respectively, without any impurities. After 1 h (h) of CO2RR at a potential of −1.40 V, both Cu3(PO4)2 and Cu2P2O7 are partially reduced to metallic Cu, indicating the coexistence of CuPO and metallic Cu components during CO2RR. Furthermore, X-ray absorption fine structure (XAFS) analysis in conjunction with the XRD results proved the coexistence of Cu2+ and Cu0 species as the CO2RR goes on for at least 10 h (Supplementary Figs. 6 and 7). CuO as a control sample was synthesized using the same method, and XRD characterizations were also carried out to identify the material components (Supplementary Fig. 8). The shape and microstructure of Cu3(PO4)2, Cu2P2O7, and CuO were examined via transmission electron microscopy (TEM), and all the three nanoparticles possess irregular shapes (Supplementary Figs. 9–11a). Supplementary Figs. 9–11b give the related high-resolution TEM (HRTEM) images of Cu3(PO4)2, Cu2P2O7, and CuO, and the clear lattice fringes with the spacing of 0.408, 0.316, and 0.252 nm are observed, corresponding to the (110) plane of triclinic phase Cu3(PO4)2, the (\(\bar{2}\)02) plane of monoclinic phase Cu2P2O7 and the (111) plane of monoclinic phase CuO, respectively. Further high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, Supplementary Figs. 9c and 10c) and corresponding energy-dispersive spectroscopy (EDS) maps (Supplementary Fig. 9d–f and Supplementary Fig. 10d–f) verify that Cu, O, and P elements are homogeneously distributed along the Cu3(PO4)2 and Cu2P2O7 nanoparticles. Notably, after CO2RR, the HAADF-STEM image of Cu3(PO4)2 shows the agglomeration of small-size nanoparticles (Supplementary Fig. 12), and the HRTEM image exhibits that metallic Cu nanocrystal with a d-spacing of (111) plane is closely contacted to Cu3(PO4)2 with a d-spacing of (210) and (012) plane (Fig. 2c). False color further highlights the close contact between metallic Cu and Cu3(PO4)2 (Supplementary Fig. 13a). As shown in the enlarged HAADF- STEM image (Fig. 2d) and the element mappings (Fig. 2e–g), the segregated Cu nanoparticles are observed, which is in agreement with the XRD result. As for Cu2P2O7, the TEM images show that metallic Cu nanoparticles with sizes ranging from 3 ~ 25 nm that are featured by the d-spacing of (111) planes are distributed on Cu2P2O7 that are featured by the d-spacing of (022) planes after the CO2RR process (Fig. 2h and Supplementary Fig. 12b). The distribution of metallic Cu and Cu2P2O7 was further confirmed by the false-color HRTEM (Supplementary Fig. 13b) and HAADF-STEM images (Fig. 2i) overlapped with the corresponding EDS element mappings (Fig. 2j–l). By contrast, the TEM images of the CuO control sample after CO2RR show that the pristine CuO phase has been reduced to Cu0 during the test (Supplementary Fig. 14).

a, b XRD patterns of a Cu3(PO4)2 and b Cu2P2O7 before and after CO2RR at the potential of −1.40 V, indicating the coexistence of CuPO and metallic Cu components in both Cu3(PO4)2 and Cu2P2O7 samples during CO2RR. c HRTEM images of the Cu3(PO4)2 sample after CO2RR at the potential of −1.40 V. d Enlarged HAADF-STEM image of Cu3(PO4)2 after reaction and e–g its corresponding EDS elemental mapping images of e Cu, f P and g mixed elements, respectively. h HRTEM images of the Cu2P2O7 sample after CO2RR at the potential of −1.40 V. i Enlarged HAADF-STEM image of Cu2P2O7 after reaction and i–l its corresponding EDS elemental maps of j Cu, k P and l mixed elements, respectively, displaying the nanometric Cu/CuPO architecture in both Cu3(PO4)2 and Cu2P2O7.
In order to identify the variation of surface valence states and chemical compositions of CuPO and CuO samples after CO2RR, X-ray photoelectron spectroscopy (XPS) was conducted (Supplementary Figs. 15–18). As observed in Supplementary Fig. 15, the peaks detected at about 933.2 and 935.5 eV of initial Cu2P2O7 and CuO can be ascribed to the Cu2+ component33. After 1 h’s CO2RR at −1.40 V, the characteristic Cu0/Cu1+ (932.5 eV) peak appears and occupies a dominant position in CuO, indicating the surface Cu2+ components of the CuO sample are mainly reduced to Cu0/Cu1+ (ref. 33). While the Cu2P2O7 sample continues to exhibit the main component of Cu2+ species after CO2RR, which benefits from the stability at a negative potential of Cu2P2O7. Due to the fact that the binding energies of the Cu0/Cu1+ states are difficult to distinguish in the Cu 2p region, we further performed a Cu LMM auger peak analysis (Supplementary Fig. 16). The peaks at around 567.7 and 570.0 eV demonstrate that both Cu0 and Cu1+ are formed in Cu2P2O7 and CuO after CO2RR34. Furthermore, the feature around 570.4 eV confirms the dominant existence of Cu(OH)2 (ref. 34), indicating the component Cu2+ species are preserved in the Cu2P2O7 catalyst after CO2RR. For the spectrum in the P 2p region (Supplementary Fig. 17a), the characteristic P 2p1/2 and 2p3/2 peaks for P2O74− are detected at the BE of 134.8 and 133.8 eV, indicating the phosphate group persists during the test35. Further evidence is derived from the O 1s spectrum, the peak located at 531.3 eV can be assigned to the non-bridging O in the phosphate group (P=O), and the peak at the BE of 533.1 eV can be attributed to the symmetric bridging O in P–O–P group (Supplementary Fig. 17b)35. Fourier-transformed infrared (FTIR) and Raman spectroscopy are two powerful methods to analyze low-frequency modes in the phosphates system. As shown in the FTIR spectrum of Cu2P2O7, characteristic peaks that appear in 1100–900 cm−1 region are attributed to symmetric and asymmetric P–O bonds, as well as the asymmetric P–O–P bridge vibration, confirming the phosphate group in the sample before and after CO2RR (Supplementary Fig. 19)36. The Raman spectrum of Cu2P2O7 is displayed in Supplementary Fig. 20a, peaks located at the frequency areas of 1250–950 cm−1 are known to be assigned to the P–O stretching modes of [P2O7]4−. The symmetric and asymmetric stretch of P–O–P bridge appear in 930–970 and 680–760 cm−1 regions respectively, and characteristic peaks observed in the area of 600–500 cm−1 (PO22- radical) and 500–370 cm−1 (P–O–P bridge) correspond to the P–O–P bending vibration37. In addition, the P–O–P deformations, the PO3 rocking and deformation modes, and the external and torsional modes are detected in the region of 430–180 cm−1 (ref. 38). These characteristic peaks are confirmed as pyrophosphate compounds, and they further demonstrate the durability of the pyrophosphate group during CO2RR. For comparison, the Raman spectra of CuO are given in Supplementary Fig. 20b, and the bands found at 293, 345, and 633 cm−1 are attributed to the standard Ag, Bg (1), and Bg (2) mode, respectively39. After being applied at −1.40 V for 1 h, no obvious peak could be detected in the Raman spectra, which proves that the CuO was totally reduced to Cu(0) during CO2RR. We then implemented in situ Raman spectroscopy on the Cu3(PO4)2 catalysts, which were measured under a constant potential of −1.40 V in the CO2-saturated 0.1 M KHCO3 electrolyte. Figure 3a shows the time-dependent in situ Raman spectra of Cu3(PO4)2. The stretching of the PO43− unit is observed at around 1000–1100 cm−1, and the bands around 450–650 cm−1 are attributed to the bending vibration of the PO43- unit38. Considering that a moderate decrease in Raman spectra is observed in 60 minutes (min), we concluded that metallic Cu0 formed as the reduction of Cu3(PO4)2 started, whereas residual Cu3(PO4)2 remained present in the bulk. Noticeably, the atop-adsorbed CO (COatop) peak, acting as an important intermediate for the C–C coupling process, was detected at about 2060 cm−1 (ref. 40). As for the CuO control sample in Fig. 3b, the Raman peaks of CuO disappeared after 30 min, which indicated its entire reduction; moreover, no obvious *CO peak was detected. The in situ Raman measurement result suggests that sufficient coverage of CO* could be built on the Cu/Cu3(PO4)2 catalyst, promoting further dimerization during CO2RR.

In situ Raman spectra of a Cu3(PO4)2 and b CuO catalysts, respectively, during CO2RR at −1.40 V in CO2-saturated 0.1 M KHCO3, suggesting the existence of Cu3(PO4)2 and the disappear of CuO during CO2RR. XAFS characterization of c the normalized Cu K-edge operando XANES spectra, d Fourier-transformed Cu K-edge EXAFS spectra, and e, f Calculated ratio of Cu oxidation states in e CuO and f Cu3(PO4)2 catalysts from linear combination fitting with respect to time during 30 min of reaction at −1.45 V. The in situ Raman spectra along with operando XAFS analysis demonstrate the coexistence of Cu2+ and Cu0 components in Cu3(PO4)2 and the reductive process of oxide Cu species to the metallic Cu0 states in the CuO control sample. g Morlet WT of the k3-weighted operando EXAFS data for the Cu3(PO4)2 and CuO samples with standard Cu foil and CuO powder as controls, suggesting the coexistence of Cu–O and Cu–Cu bond in Cu3(PO4)2 during CO2RR.
To explore the oxidation state evolution of the electrocatalysts during CO2RR, we carried out operando XAFS measurements under the electrochemical condition at −1.45 V (details in the Methods, Supplementary Fig. 21). The operando X-ray absorption near edge structure (XANES) spectra clearly show that the CuO sample appears metallic Cu0 during CO2RR and kept its metallic Cu0 state after test, while Cu3(PO4)2 stays in higher valence states than metallic Cu(0) in the whole course of CO2RR (Fig. 3c). Figure 3d exhibits the operando extended region of the XAS (EXAFS) spectra, and peaks at about 1.5 Å and 2.1 Å are attributed to the coordination of Cu–O and Cu–Cu, respectively41,42. A distinct peak of Cu−O coordination can be detected in CuPO at about 1.4 Å, indicating the existence of oxygen-bearing Cu species, and the peak shifts to a smaller radial distance gradually when CO2RR takes place, implying the partial reduction of Cu2+. The enhancement of peak (Cu−Cu) in the samples (solid lines) suggests the emergence of Cu0, and Cu3(PO4)2 catalyst shows a higher Cu–O coordination number and a lower Cu–Cu coordination number compared with CuO indicating the stability of Cu3(PO4)2 at a negative potential to a certain extent. Meanwhile, we fitted the EXAFS data of Cu3(PO4)2 during CO2RR, and the fitting results further show that Cu–O coordination and Cu–Cu coordination exist simultaneously in the Cu3(PO4)2 catalyst under reaction conditions (Supplementary Fig. 22 and Table 1). To quantitatively analyze the evolution of oxidation states during CO2RR, we processed in situ spectra using an LCF of the CuO and Cu3(PO4)2 samples (Fig. 3e, f). The LCF spectra and data prove that the fitting results are reliable (Supplementary Fig. 23 and Table 2). From the LCF results, we calculated the ratios of Cu oxidation species presented at each 6 min under −1.45 V. After 6 min, the majority of CuO (96%) and Cu3(PO4)2 (95%) were both in the Cu2+ oxidation state, while they had respectively decreased to 25% and 72% after a further 12 min, which revealed that the transition between Cu2+ and Cu0 for CuO was more rapid, while the reduction for Cu3(PO4)2 was slower. After the catalysts had been electrochemically reduced via CO2RR for 30 min, the majority of the CuO sample had been almost entirely reduced to Cu0 (73%), while the Cu3(PO4)2 sample was still 49% composed of the Cu2+. These results show that Cu2+ of Cu3(PO4)2 may be stabilized so that Cu2+ and Cu0 could be coexistent under −1.45 V for over 30 min. Solid support for the coexistent metallic Cu0 and Cu2+ during the CO2RR process in Cu3(PO4)2 was also provided through the additional analysis of the Morlet wavelet transform (WT). As shown in Fig. 3g, a WT maximum at 6–8 Å−1 assigned to Cu–Cu bond is visible in the CuO sample during CO2RR at −1.45 V, which indicates the metallic Cu0 species occupy the main composition in the bulk during the test. By contrast, the Cu3(PO4)2 catalyst exhibits a distinct feature at 5–10 Å−1 besides the WT maximum at 6–8 Å−1, which represents the coexistence of Cu–O and Cu–Cu bond, demonstrating the component of Cu oxidation species still remained under the same reaction condition41. The operando XAFS analysis in conjunction with in situ Raman results clearly proved the specific coexistence of Cu2+ and Cu0 species in CuPO samples under CO2RR condition which could affect the selectivity.
Electrochemical CO2 conversion
The activity of CuPO catalysts for electrocatalytic CO2RR was evaluated in an H-cell with CO2-saturated 0.1 M KHCO3 solution as an electrolyte, and the electrodes were prepared by dropping casting ink solution onto the glassy carbon electrodes (GCE, see more details in the “Methods”). The linear sweep voltammetry (LSV) curves of the Cu3(PO4)2/GC, Cu2P2O7/GC, and CuO/GC in the CO2-saturated (full line) or Ar-saturated (dotted line) environments are shown in Fig. 4a. The Cu3(PO4)2 and Cu2P2O7 catalysts exhibited larger geometric reduction j in the CO2 atmosphere, which demonstrated the reaction priority to CO2RR. Thereafter, Cu3(PO4)2/GC, Cu2P2O7/GC, and CuO/GC were measured over a range of potentials for catalytic activity (Supplementary Fig. 24). Cu3(PO4)2/GC shows a marked selectivity for C2H4 (FEC2H4 > 60%) under potentials from −1.25 to −1.50 V, with a topmost FEC2H4 reaching 69.7% at −1.45 V (Fig. 4b), corresponding to a jC2H4 of −23.0 mA cm−2, similar to what is tested on Cu2P2O7/GC (FEC2H4 of 64.0% and jC2H4 of −17.6 mA cm−2 at −1.40 V (Fig. 4c). As a comparison, the CuO/GC is less selective and its CO2RR catalysis yields CO (<8% FE) and CH4 (<18% FE), as well as C2H4 (<8% FE) in −1.20 ~ −1.50 V (Fig. 4c and Supplementary Fig. 24). In particular, the ratios of C2H4 and C1 products for Cu3(PO4)2/GC and Cu2P2O7/GC reach about 12.4 and 7.8 at −1.5 V, which is overwhelmingly superior to CuO/GC (Fig. 4d). Specific values for all products are given in Supplementary Tables 3 and 4.

a LSV curves of CuPO/GC and CuO/GC catalysts with respect to RHE at a scan rate of 1 mV/s in the CO2- or Ar-saturated 0.1 M KHCO3 electrolyte, indicating the occurrence of CO2RR. b FEs of C2H4 of catalysts at different applied potentials in CO2-saturated 0.1 M KHCO3, suggesting the superior C2H4 selectivity for CuPO. Error bars above were all based on the standard deviation of three measurements at each potential. c Potential dependence of jC2H4 on CuPO/GC and CuO/GC, exhibiting higher C2H4 activity for CuPO. d FEC2H4/FEC1 ratios of catalysts at different applied potentials, suggesting the superior C2H4 selectivity for CuPO/GC. e FEs for products over Cu3(PO4)2/Cu/PTFE at various applied current densities in a flow cell reactor with 2.0 M KOH as the electrolyte. f FEs of various products produced by Cu3(PO4)2/Cu/PTFE at the applied j of −300 mA cm−2 for 18 h in the 2.0 M KOH electrolyte (refresh electrolyte for every 2 h), and the liquid products were collected after the reaction run for more than 30 min. g Maximum FEC2+ of Cu3(PO4)2/Cu/PTFE and recently reported Cu-based CO2RR catalysts (details in Supplementary Table 5). Blue rhombus: alkaline medium; yellow circle: neutral medium; violet triangle: acidic medium.
To further improve the CO2RR current density, we constructed gas diffusion electrodes (GDE) with the Cu3(PO4)2 catalyst deposited on the Cu-coated polytetrafluoroethylene gas diffusion layers (Cu3(PO4)2/Cu/PTFE) and evaluated CO2RR performance utilizing a flow cell (details in the “Methods”). The 2.0 M KOH alkaline electrolyte was used to enhance the conductivity and improve the CO2RR kinetics by suppressing H2 evolution. As shown in Fig. 4e, the Cu3(PO4)2/Cu/PTFE catalyst was screened at different current densities and reached a maximum FEC2+ of 90.9% (52.8% ethylene, 30.7% ethanol, 5.9% acetic acid, and 1.5% n-propyl alcohol) with jc2+ of −318.2 mA cm−2 corresponding to experiments performed at jtot = −350 mA cm−2. Supplementary Fig. 25 exhibits the chronopotential curves obtained at different j. Meanwhile, the bare Cu/PTFE showed FEC2+ of 58.9 ~ 77.2% with the jtot ranging from −100 to −400 mA cm−2 (Supplementary Fig. 26), proving the Cu3(PO4)2 catalyst has excellent CO2-to-C2+ conversion performance intrinsically. Electrochemical stability is also a key parameter for CO2RR performance. Remarkably, Cu3(PO4)2/Cu/PTFE unveils a desirable stability in the flow cell during CO2RR for 18 h (Fig. 4f). The total current density keeps at −300 mA cm−2, and FEC2+ remains above 80% after 18 h of reaction, representing that the catalyst is stable during CO2RR. Furthermore, we compared the maximum FEC2+ of 90.9% for Cu3(PO4)2/Cu/PTFE with that for other Cu-based electrocatalysts (Fig. 4g and Supplementary Table 5), which was found to possess a remarkable C2+ production selectivity compared to the reported Cu-based catalysts10,12,17,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57.
Insights into CO2-to-C2+ electroreduction
The CO2RR intermediates chemisorbed on CuPO and CuO were assessed via SEIRAS to determine the mechanism for boosted C2+ selectivity (details in the Methods, Supplementary Fig. 27). As shown in Fig. 5a, the in situ SEIRAS differential spectra of Cu3(PO4)2 exhibit peaks at about 2065 cm−1 and 1025 cm−1, which are associated with the absorbed *CO and the nonplanar vibration (O=C–H) of *CHO on the catalyst surface, respectively58,59. Additionally, the peaks detected at about 1255 and 1385–1410 cm−1 could be attributed to the C–O stretch and symmetric vibration (vibration of O–C=O) of *COOH, respectively59. Prominently, based on reported experimental and theoretical studies, the distinctive 1180 and 1520 cm−1 peaks can demonstrate the existence of *OCCHO intermediate; it increased on scanning to more negative potentials, which is consistent with the trend of C2+ products’ formation rates59,60,61. To support the band assignments of the IR to *OCCHO theoretically, we simulated the IR of *OCCHO intermediates that adsorbed at the Cu0/Cu2+ (Cu3(PO4)2) interface. As shown in Supplementary Fig. 28, a band peak with strong oscillator strength is located at 1189 cm−1, which is close to our experiment result (1180 cm−1) for *OCCHO. Moreover, the peak intensity of *CO peaks on Cu3(PO4)2 enhances first as *CO increases, and then gradually decreases as the applied potential continues to turn negative, suggesting the *CO is consumed as dimerization accelerates. In comparison, the absorbed *CO continues to accumulate and the C–C coupling bond is not detected on the CuO sample, which would be entirely reduced to Cu0 during CO2 reduction (Fig. 5b). As illustrated in the schematic diagram (Fig. 5c), the in situ SEIRAS study in conjunction with theoretical research reveals that the stable and abundant Cu0/Cu2+ interfaces derived from Cu phosphate-based electrocatalysts can facilitate the pathway of *CHO coupling with *CO to form *OCCHO, thus improving the selectivities of C2+ products during CO2 reduction.

In situ SEIRAS differential spectra of the a Cu3(PO4)2 and b CuO sample in CO2 purged 0.1 M KHCO3 electrolyte in real-time condition. c Schematic illustration on proposed role of Cu(II)/Cu(0) interfaces in CO2-to-C2+ conversion. Red: oxygen; gray: carbon; white: hydrogen; origin: Cu0; golden: Cu2+.
Discussion
In summary, we have theoretically predicted and experimentally constructed the in situ formation of nanometric Cu/CuPO with rich and stable Cu0+/Cu2+ interfaces to be the efficient electrocatalyst for highly selective CO2-to-C2+ products conversion. The resultant nanometric Cu/CuPO could obtain a high FEC2H4 of 69.7% in a neutral medium and perform a maximum FEC2+ above 90% with industrially relevant current densities in a flow cell configuration. The Cu2+ sites of Cu0+/Cu2+ interfaces were revealed to be available for the formation of *CHO, which then facilely coupled with *CO on the adjacent Cu0 surface to form *OCCHO, leading to high-efficiency CO2-to-C2+ conversion performance. Our work highlights the role of the peculiar Cu0/Cu2+ interfaces in promoting selectivity toward C2+ products, and we believe that this finding will contribute to the design of improved Cu-based electrocatalysts for future CO2RR, with high selectivity and underlying mechanisms for its conversion to valuable products.
Methods
DFT calculation
All the calculations were performed using the Vienna Ab-initio Simulation Package (VASP) package62,63,64. The exchange-correlation functional was described by the Perdew–Burke–Ernzerhof (PBE) functional65 within the generalized gradient approximation (GGA)66. The project-augmented wave (PAW) method67 was employed to treat core electrons, and the cut-off energy of the plane-wave basis was set to 450 eV. A vacuum layer of 15 Å was applied to separate each periodic unit cell. In the structural optimizations, the Brillouin zone was sampled by 2 × 2 × 1 Monkhorst–Pack mesh k-points, and the bottom two layers of the slab were fixed, and the top three layers and adsorbates were fully relaxed. The empirical correction in Grimme’s scheme was used to describe the van der Waals interactions68. Considering the solvation environment, we constructed an explicit solvation model with one layer H2O molecules in calculation models.
Catalyst preparation
For Cu3(PO4)2, 12 mmol Cu(NO3)2·3H2O and 8 mmol NH4H2PO4 were dissolved in H2O and stirred well to form a suspension in a beaker. Then, added 6 mmol C6H10O8 to beaker and stirred until the suspension was clear. The beaker was completely covered with tin foil, with some small holes punched in the top, then placed in the oven and dried at 120 °C. Finally, the dried material was transferred to a crucible and calcined at 700 °C for 1 h in a muffle furnace at a heating rate of 5 °C/min. The preparation method of Cu2P2O7 was the same as the above procedure except for changing the mass of Cu(NO3)2·3H2O to 8.5 mmol. The preparation method of CuO was the same as that of Cu3(PO4)2 except that NH4H2PO4 was not added. All reagents were commercially available as analytical grade (Supplementary Note 3).
Working electrodes preparation
For the Cu3(PO4)2/CP, Cu2P2O7/CP, and CuO/CP used for XRD, XPS, Raman, FTIR, and XAFS measurements. The sample ink was prepared by dispersing 10 mg of catalyst (Cu3(PO4)2, Cu2P2O7, or CuO) and 100 μL of Nafion solution into 0.5 mL of H2O and 1.5 mL of iso-propanol, followed by sonication for more than 1 h. The sample ink was sprayed on carbon paper using an air-brush, and the total catalyst loading was about 2.0 mg cm−2. For the GCE working electrode used in H-Cell electrochemical measurements, the sample ink was prepared by dispersing 5 mg catalyst (Cu3(PO4)2, Cu2P2O7 or CuO) and 50 μL Nafion solution into 0.125 mL H2O and 0.375 mL iso-propanol followed by sonication for more than 30 min. 2.5 μL ink was dropped on GCE (with a diameter of 3 mm) and dried in the air for 2 times, with the total loading amount of catalyst about 0.7 mg cm−2.
For Cu/PTFE, bare PTFE with an average pore diameter of 0.22 μm was used. Approximately 600 nm nominal thick Cu films were constructed by vacuum evaporation method on the PTFE substrate using Cu target material (99.999%) at an evaporation rate of around 0.5 Å s−1 in an OMV FS300-S6 evaporating tool at a base pressure of <6*10−4 Torr. For Cu3(PO4)2/Cu/PTFE, the sample ink was prepared as above mentioned on carbon paper and sprayed on the Cu/PTFE films electrodes using air-brush, with the total loading amount of catalyst about 1.0 mg cm−2.
Materials characterizations
The crystal structure was determined using X-ray diffraction (Bruker D8 Advanced Diffractometer with Cu Kα radiation). The morphology and structure were characterized by scanning electron microscope (Hitachi S4800) and transmission electron microscopy (TEM, JEOL JEM 2010, operated at 200 kV). Scanning transmission electron microscopy (STEM) characterization was performed using a ThermoFisher Talos F200X. High-angle annular dark-field (HAADF)-STEM images were recorded using a convergence semi angle of 11 mrad, and inner- and outer collection angles of 59 and 200 mrad, respectively. Energy-dispersive X-ray spectroscopy (EDS) was carried out using 4 in-column Super-X detectors. The chemical state was analyzed by X-ray photoelectron spectroscopy (XPS, Thermo Escalab 250), and the binding energy of C 1s peak at 284.8 eV was taken as an internal standard. Raman analysis was carried out using a Leica DMLM microscope (Renishaw) with the 514 nm laser. FTIR spectroscopy was characterized on a Nicolet 6700 spectrometer with a spectral range of 4000–400 cm−1. XAFS spectra at the Cu K-edge were performed on the 1W1B beamline station of the Beijing Synchrotron Radiation Facility (BSRF), China. Cu foil, Cu2O and CuO were used as references. LCF were processed using the ATHENA module implemented in the IFEFFIT software packages.
The in situ XAS measurement was performed in a homemade plastic electrolytic cell. The graphite rod (spectral purity, 3 mm in diameter) and the Ag/AgCl electrode acted as the counter electrode and the reference electrode in the three-electrode system, respectively. The carbon paper coated with specific catalysts was used for the working electrode, and a gas inlet for purging CO2 into the electrolyte (0.1 M KHCO3) was also contained in the cell. A window (1.1 cm × 1.8 cm) was designed on the cell for the working electrode that enabled XAS measurement under a sensitive fluorescence model while the working potentials were applied. A spectrum can be acquired on average every 1.5 min, and the in situ curves of the samples in Fig. 3c, d were taken at around 36 min of reaction.
The in situ Raman measurement was performed on the Raman spectrometer (LabRAM HR) utilizing an excitation laser with a wavelength of 514 nm and a 50× microscope objective with a numerical aperture of 0.5, 10.6 mm. Before the experiments, calibration was carried out based on the peak at 520 cm−1 of a silicon wafer standard. To acquire information about the electroreduction process of Cu3(PO)4 and CuO, we utilize a spectroelectrochemical cell and detect the in situ Raman of the cathode GDE through a quartz window. The GDEs of Cu3(PO)4 and CuO were taken as the working electrodes, which were prepared by the catalyst sprayed on carbon paper. Platinum wire and Ag/AgCl were used as the counter electrode and reference electrode, respectively. During the in situ experiment, a peristaltic pump was used to control the flow rate of CO2-saturated 0.1 M KHCO3 electrolyte at 10 mL/min, while the flow rate of CO2 was kept at 10 sccm with a mass flow controller.
In situ surface-enhanced infrared absorption spectroscopy (SEIRAS) was recorded in a homemade reflection accessory with internal reflection configuration using an FTIR spectrometer equipped with a PerkinElmer spectrum 100 detector. FTIR spectra were obtained by averaging 16 scans with a resolution of 8 cm−1 at the selected potential. Every spectrum was obtained by applying a single potential step compared to the reference potential (0 V). For the spectroelectrochemical measurements, a thin Au film with a thickness of ∼10 nm is prepared on a Si prism using electroless deposition69. The sample ink was prepared as mentioned above on GCE. 25 μL ink was dropped on Au underlayer and dried in the air for 2 times. The catalyst-coated Si crystal was placed in a three-electrode spectroelectrochemical cell as the working electrode, and the counter and reference electrodes were platinum wire and Ag/AgCl electrode (3.5 M KCl), respectively. A gas inlet for purging CO2 was also contained in the cell. Electrolyte (0.1 M KHCO3) was injected into the cell and gassed with high-purity CO2 (99.9999%) prior to electrochemical measurements. A CHI1242C potentiostat was used to record electrochemical responses. Spectra were expressed as absorbance, with positive and negative peaks showing an increase and decrease in signal, respectively.
Electrochemical measurements
All electrochemical studies were performed using an electrochemical station (CHI 660E). The H-type gas-tight reactor consisted of two compartments separated by a Nafion 117 membrane. In the three-electrode system, a custom-made GCE was used as working electrode with a surface area of 0.07 cm2 and a catalyst mass loading of 0.7 mg cm−2. Ag/AgCl electrode (3.5 M KCl) and platinum mesh were used as reference electrode and counter electrode, respectively. CO2-saturated 0.1 M KHCO3 (pH ≈ 6.75) was used as electrolyte. All measured potentials were calibrated to the RHE reference scale using ERHE = EAg/AgCl + 0.059 × pH + 0.205 (all potentials were not iR-corrected if not mentioned). Linear sweep voltammetry (LSV) at a scan rate of 1 mV s−1 were performed in Ar and CO2-saturated 0.1 M aqueous KHCO3. The current density was calculated by normalizing the current to the corresponding geometric surface area.
The electrocatalytic performance of Cu3(PO4)2 was determined in a flow cell configuration. The device consists of Cu3(PO4)2/Cu/PTFE as the working electrode, nickel foam as the anode and an anion exchange membrane. They were assembled using PTFE gaskets with 2.0 M KOH as the liquid electrolyte flowing (10 mL per min) in the chambers between membrane and working electrode, and membrane and anode. CO2 gas was flowed behind the PTFE layer at a rate of 20 sccm, which was decreased to 19 sccm at the outlet due to the consumption of the alkaline electrolyte. Chronopotentiometry experiments were performed at currents of −100 mA cm−2, −150 mA cm−2, −200 mA cm−2, −250 mA cm−2, −300 mA cm−2, −350 mA cm−2, and −400 mA cm−2, showing consistent gas product distributions. The liquid products were collected after the reaction run for more than 30 min.
Products analysis
The gas products of CO2 electroreduction were analyzed by gas chromatography (GC online test, RAMIN, GC2060), equipped with a flame ionization detector (FID to detect CO, CH4 and C2H4) and a thermal conductivity detector (TCD to detect H2). Ar was used as a carrier. CO2 was continuously sparged through the electrolyte (30 mL of catholyte and anolyte in each compartment) at a rate of 20 sccm and was routed into the gas chromatograph. The Faradic efficiencies were tested online and averaged for multiple data.
The liquid products were quantified by 1H nuclear magnetic resonance (NMR) (Varian 700 MHz spectrometer, 16.4 T). In a typical analysis, the mixture of 500 mg of the electrolyte and 100 mg of 149 ppm DMSO (used as internal standard) in D2O solution was used as measured sample after 2 h of CO2RR with j applied at −300 mA cm−2. The 1H spectra were obtained by suppressing the water peak using the pre-saturation method.
Assuming that 12 electrons are required to generate one C2H4 molecule, the FE and the partial current densities of C2H4 formation were calculated as below:
where v = volume concentration of C2H4 obtained from gas chromatography (GC) data, p0 = 1.013 bar and T = 298.15 K, G = 20 sccm is the CO2 flow rate (19 sccm for 2 M KOH in a flow cell), itotal (mA) = steady-state cell current, F = 96485 C mol−1, R = 8.314 J mol−1 K−1. Then:
Data availability
Source data are provided with this paper.
References
Li, F. et al. Molecular tuning of CO2-to-ethylene conversion. Nature 577, 509–513 (2019).
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
Zhou, Y. et al. Long-chain hydrocarbons by CO2 electroreduction using polarized nickel catalysts. Nat. Catal. 5, 545–554 (2022).
Xu, Y. et al. A microchanneled solid electrolyte for carbon-efficient CO2 electrolysis. Joule 6, 1333–1343 (2022).
Li, Y. et al. Atomically dispersed single Ni site catalysts for high-efficiency CO2 electroreduction at industrial-level current densities. Energy Environ. Sci. 15, 2108–2119 (2022).
Zhang, X. Y. et al. Selective methane electrosynthesis enabled by a hydrophobic carbon coated copper core–shell architecture. Energy Environ. Sci. 15, 234–243 (2022).
Dey, S., Masero, F., Brack, E., Fontecave, M. & Mougel, V. Electrocatalytic metal hydride generation using CPET mediators. Nature 607, 499–506 (2022).
Wang, Y., Liu, J. & Zheng, G. Designing copper-based catalysts for efficient carbon dioxide electroreduction. Adv. Mater. 33, 2005798 (2021).
Kim, C. et al. Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings. Nat. Energy 6, 1026–1034 (2021).
Zhou, Y. et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 10, 974980 (2018).
Yuan, X. et al. Controllable Cu0-Cu+ sites for electrocatalytic reduction of carbon dioxide. Angew. Chem. Int. Ed. 60, 15344–15347 (2021).
Gao, D. et al. Selective CO2 electroreduction to ethylene and multicarbon alcohols via electrolyte-driven nanostructuring. Angew. Chem. Int. Ed. 58, 17047–17053 (2019).
Scholten, F., Sinev, I., Bernal, M. & Roldan Cuenya, B. Plasma-modified dendritic Cu catalyst for CO2 electroreduction. ACS Catal. 9, 5496–5502 (2019).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Sultan, S. et al. Interface rich CuO/Al2CuO4 surface for selective ethylene production from electrochemical CO2 conversion. Energy Environ. Sci. 15, 2397–2409 (2022).
Wang, X. et al. Efficient upgrading of CO to C3 fuel using asymmetric C-C coupling active sites. Nat. Commun. 10, 5186 (2019).
Yang, P.-P. et al. Protecting copper oxidation state via intermediate confinement for selective CO2 electroreduction to C2+ fuels. J. Am. Chem. Soc. 142, 6400–6408 (2020).
Chou, T.-C. et al. Controlling the oxidation state of the Cu electrode and reaction intermediates for electrochemical CO2 reduction to ethylene. J. Am. Chem. Soc. 142, 2857–2867 (2020).
Kim, D. et al. Insights into an autonomously formed oxygen-evacuated Cu2O electrode for the selective production of C2H4 from CO2. Phys. Chem. Chem. Phys. 17, 824–830 (2015).
Lee, S., Kim, D. & Lee, J. Electrocatalytic production of C3-C4 compounds by conversion of CO2 on a chloride-induced Bi-phasic Cu2O-Cu catalyst. Angew. Chem. Int. Ed. 54, 14701–14705 (2015).
Wang, J., Tan, H.-Y., Zhu, Y., Chu, H. & Chen, H. M. Linking the dynamic chemical state of catalysts with the product profile of electrocatalytic CO2 reduction. Angew. Chem. Int. Ed. 60, 17254–17267 (2021).
Wen, C. F. et al. Highly ethylene-selective electrocatalytic CO2 reduction enabled by isolated Cu−S motifs in metal–organic framework based precatalysts. Angew. Chem. Int. Ed. 61, e202111700 (2022).
De Luna, P. et al. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat. Catal. 1, 103–110 (2018).
Xiao, H., Goddard, W. A., Cheng, T. & Liu, Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl Acad. Sci. USA 114, 6685–6688 (2017).
Chang, C.-C., Li, E. Y. & Tsai, M.-K. A computational exploration of CO2 reduction via CO dimerization on mixed-valence copper oxide surface. Phys. Chem. Chem. Phys. 20, 16906–16909 (2018).
Zheng, Y. et al. Understanding the roadmap for electrochemical reduction of CO2 to multi-carbon oxygenates and hydrocarbons on copper-based catalysts. J. Am. Chem. Soc. 141, 7646–7659 (2019).
Favaro, M. et al. Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl Acad. Sci. USA 114, 6706–6711 (2017).
Woertink, J. S. et al. A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc. Natl Acad. Sci. USA 106, 18908–18913 (2009).
Xu, S. et al. Sustaining metal–organic frameworks for water–gas shift catalysis by non-thermal plasma. Nat. Catal. 2, 142–148 (2019).
Wang, W. et al. Photocatalytic C–C coupling from carbon dioxide reduction on copper oxide with mixed-valence copper(I)/copper(II). J. Am. Chem. Soc. 143, 2984–2993 (2021).
Zhou, X. et al. Stabilizing Cu2+ ions by solid solutions to promote CO2 electroreduction to methane. J. Am. Chem. Soc. 144, 2079–2084 (2022).
Liu, H. et al. Screening stable and metastable ABO3 perovskites using machine learning and the materials project. Comp. Mater. Sci. 177, 109614 (2020).
Platzman, I., Brener, R., Haick, H. & Tannenbaum, R. Oxidation of polycrystalline copper thin films at ambient conditions. J. Phys. Chem. C 112, 1101–1108 (2008).
Lee, S. Y. et al. Mixed copper states in anodized Cu electrocatalyst for stable and selective ethylene production from CO2 reduction. J. Am. Chem. Soc. 140, 8681–8689 (2018).
Chang, Y. et al. Coralloid Co2P2O7 nanocrystals encapsulated by thin carbon shells for enhanced electrochemical water oxidation. ACS Appl. Mater. Interfaces 8, 22534–22544 (2016).
Gupta, S. K., Mohapatra, M., Godbole, S. V. & Natarajan, V. On the unusual photoluminescence of Eu3+ in α-Zn2P2O7: a time resolved emission spectrometric and Judd–Ofelt study. RSC Adv. 3, 20046–20053 (2013).
Boonchom, B. & Baitahe, R. Synthesis and characterization of nanocrystalline manganese pyrophosphate Mn2P2O7. Mater. Lett. 63, 2218–2220 (2009).
Baitahe, R. & Vittayakorn, N. Phase formation and evolution of Cu:Zn partials in binary metal pyrophosphates Cu(2−x)Zn(x)P2O7; x≈1. Thermochim. Acta 596, 21–28 (2014).
Mukherjee, N. et al. CuO nano-whiskers: electrodeposition, Raman analysis, photoluminescence study and photocatalytic activity. Mater. Lett. 65, 3248–3250 (2011).
Deng, Y. et al. On the role of sulfur for the selective electrochemical reduction of CO2 to formate on CuSx catalysts. ACS Appl. Mater. Interfaces 10, 28572–28581 (2018).
Zhang, W. et al. Atypical oxygen-bearing copper boosts ethylene selectivity toward electrocatalytic CO2 reduction. J. Am. Chem. Soc. 142, 11417–11427 (2020).
Weng, Z. et al. Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction. Nat. Commun. 9, 415 (2018).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Zhong, D. et al. Coupling of Cu(100) and (110) facets promotes carbon dioxide conversion to hydrocarbons and alcohols. Angew. Chem. Int. Ed. 60, 4879–4885 (2021).
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
Kibria, M. G. et al. A surface reconstruction route to high productivity and selectivity in CO2 electroreduction toward C2+ hydrocarbons. Adv. Mater. 30, 1804867 (2018).
Zhang, X. et al. Selective and high current CO2 electro-reduction to multicarbon products in near-neutral KCl electrolytes. J. Am. Chem. Soc. 143, 3245–3255 (2021).
Wang, P. et al. Boosting electrocatalytic CO2–to–ethanol production via asymmetric C–C coupling. Nat. Commun. 13, 3754 (2022).
Wang, P. et al. Synergized Cu/Pb core/shell electrocatalyst for high-efficiency CO2 reduction to C2+ liquids. ACS Nano 15, 1039–1047 (2020).
Song, Y. et al. B-Cu-Zn gas diffusion electrodes for CO2 electroreduction to C2+ products at high current densities. Angew. Chem. Int. Ed. 60, 9135–9141 (2021).
Pham, T. H. M. et al. Enhanced electrocatalytic CO2 reduction to C2+ products by adjusting the local reaction environment with polymer binders. Adv. Energy Mater. 12, 2103663 (2022).
Miao, R. K. et al. Electroosmotic flow steers neutral products and enables concentrated ethanol electroproduction from CO2. Joule 5, 2742–2753 (2021).
Wu, Z.-Z. et al. Identification of Cu(100)/Cu(111) interfaces as superior active sites for CO dimerization during CO2 electroreduction. J. Am. Chem. Soc. 144, 259–269 (2022).
Zheng, M. et al. Electrocatalytic CO2-to-C2+ with ampere-level current on heteroatom-engineered copper via tuning *CO intermediate coverage. J. Am. Chem. Soc. 144, 14936–14944 (2022).
Sang, J. et al. A reconstructed Cu2P2O7 catalyst for selective CO2 electroreduction to multicarbon products. Angew. Chem. Int. Ed. 61, e202114238 (2022).
Li, H. et al. High-rate CO2 electroreduction to C2+ products over a copper-copper iodide catalyst. Angew. Chem. Int. Ed. 60, 14329–14333 (2021).
Niu, Z.-Z. et al. Hierarchical copper with inherent hydrophobicity mitigates electrode flooding for high-rate CO2 electroreduction to multicarbon products. J. Am. Chem. Soc. 143, 8011–8021 (2021).
Gunathunge, C. M., Ovalle, V. J., Li, Y., Janik, M. J. & Waegele, M. M. Existence of an electrochemically inert CO population on Cu electrodes in alkaline pH. ACS Catal. 8, 7507–7516 (2018).
Qiu, X.-F., Zhu, H.-L., Huang, J.-R., Liao, P.-Q. & Chen, X.-M. Highly selective CO2 electroreduction to C2H4 using a metal-organic framework with dual active sites. J. Am. Chem. Soc. 143, 7242–7246 (2021).
Chen, X.-M. et al. A porous π–π stacking framework with dicopper(I) sites a nd adjacent proton relays for electroreduction of CO2 to C2+ products. J. Am. Chem. Soc. 144, 13319–13326 (2022).
Wei, Z. & Sautet, P. Improving the accuracy of modelling CO2 electroreduction on copper using many-body perturbation theory. Angew. Chem. Int. Ed. 61, e202210060 (2022).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmiiller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, JohnP., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Heyes, J., Dunwell, M. & Xu, B. CO2 reduction on Cu at low overpotentials with surface-enhanced in situ spectroscopy. J. Phys. Chem. C 120, 17334–17341 (2016).
Acknowledgements
This work was financially supported by the Key Program of National Natural Science Foundation of China (22239001), the International (Regional) Cooperation and Exchange Projects of the National Natural Science Foundation of China (51920105003), the National Natural Science Foundation of China (22379043, 22309053), the Science and Technology Commission of Shanghai Municipality (21DZ1207101, 22ZR1416400, 23YF1408500, 23ZR1416800), the Innovation Program of Shanghai Municipal Education Commission (E00014), the China Postdoctoral Science Foundation Funded Project (2022M721137), the Shanghai Engineering Research Center of Hierarchical Nanomaterials (18DZ2252400), and the Fundamental Research Funds for the Central Universities. The authors also thank the Frontiers Science Center for Materiobiology and Dynamic Chemistry. The authors also thank the crew of the BL14W1 beamline at the Shanghai Synchrotron Radiation Facility (SSRF) and the 1W1B beamline of Beijing Synchrotron Radiation Facility (BSRF) for their constructive assistance with the XAFS measurements and data analyses.
Author information
Authors and Affiliations
Contributions
H.G.Y. and P.F.L. directed the research. X.Y.Z. performed the experiments and data analyses. Z.X.L. performed the DFT calculations. M.Z. and J.C. assisted with the in situ SEIRAS tests and data analyses. S.D. and X.W. provided assistance with the HAADF-STEM characterization. Y.L. and J.Y.Z. performed the XAFS test and analyses. C.S., H.F.W. and H.Y.Y. guided the DFT calculations. All the authors participated in writing and editing the manuscript, and contributed their efforts to the discussion.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X.Y., Lou, Z.X., Chen, J. et al. Direct OC-CHO coupling towards highly C2+ products selective electroreduction over stable Cu0/Cu2+ interface. Nat Commun 14, 7681 (2023). https://doi.org/10.1038/s41467-023-43182-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-023-43182-6
This article is cited by
-
Dual-confinement of reconstructed covalent organic framework for enhanced CO2 electrolysis in acid
Science China Materials (2026)
-
In situ stabilization of Cu+ for CO2 Electroreduction via Environmental-molecules-induced ZnO1-x shield
Nature Communications (2025)
-
Strain engineering of single-site Cu on SWCNTs for highly efficient diene cyclopropanation
Nature Communications (2025)
-
A chelate to break diffusion limits on Helmholtz plane for CO2 electroreduction to ethanol
Nature Communications (2025)
-
Advanced X-ray absorption spectroscopy for probing dynamics in CO2 reduction reaction electrocatalysts
Communications Materials (2025)
