
Abstract
Live vaccines are ideal for inducing immunity but suffer from the need to attenuate their pathogenicity or replication to preclude the possibility of escape. Unnatural amino acids (UAAs) provide a strategy to engineer stringent auxotrophies, yielding conditionally replication incompetent live bacteria with excellent safety profiles. Here, we engineer Pseudomonas aeruginosa to maintain auxotrophy for the UAA p-benzoyl-l-phenylalanine (BzF) through its incorporation into the essential protein DnaN. In vivo evolution using an Escherichia coli-based two-hybrid selection system enabled engineering of a mutant DnaN homodimeric interface completely dependent on a BzF-specific interaction. This engineered strain, Pa Vaccine, exhibits undetectable escape frequency (<10−11) and shows excellent safety in naïve mice. Animals vaccinated via intranasal or intraperitoneal routes are protected from lethal challenge with pathogenic P. aeruginosa PA14. These results establish UAA-auxotrophic bacteria as promising candidates for bacterial vaccine therapy and outline a platform for expanding this technology to diverse bacterial pathogens.
Similar content being viewed by others


Introduction
Heat-killed, fixed, and live-attenuated bacterial vaccines are clinically used for prophylaxis against endemic bacterial infections. The advantages of live bacteria as prophylactic vaccines compared to fixed or lysed bacteria has been well documented in recent years1,2,3,4, and at present, live-attenuated bacterial vaccines against cholera (Vaxchora), tuberculosis (BCG), and typhoid (Vivotif) are approved and administered globally5. While live-attenuated vaccines are excellent immunogens, they pose a clinical safety risk due to their potential to revert or ‘escape’ to an unsafe form, particularly for immunocompromised individuals who are more susceptible to infection. These bacteria are typically genetically modified to reduce cytotoxicity by mutating or removing genes associated with pathogenesis and virulence6,7. The underlying challenges of engineering attenuated bacteria include the robust nature of bacteria to remain viable in environmental circumstances that threaten survival (i.e., host immune response), and their ability to rapidly evolve mechanisms to overcome attenuated pathogenicity. One can potentially overcome these challenges by engineering auxotrophic bacteria in which replication is dependent on a natural or unnatural metabolite not found in the host, or ideally in Nature entirely. This approach has the advantage that the vaccination strain may differ from the wild-type pathogen by a single amino acid substitution. However, the requirement remains that auxotrophic bacterial vaccines must be highly resistant to escape mechanisms.
Unnatural amino acids (UAAs) have been used as a tool to engineer bacterial auxotrophy, whereby growth is impaired in the absence of the UAA8,9. Introducing a modified orthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pair into a bacterial strain enables efficient and selective incorporation of an UAA into an essential protein in response to nonsense codons10, resulting in an UAA auxotroph. However, mutations can arise during DNA replication that revert the nonsense codon to a sense codon, allowing the bacteria to survive in the absence of the UAA. To prevent bacterial escape, early efforts to incorporate up to three nonsense codons into a single essential gene or one into three separate essential genes yielded bacteria and viruses with significantly lower escape frequency8,9,11. However, the level of functional protein expression and the bacterial growth rate was also significantly affected due to the lower efficiency of incorporating an UAA at three sites.
In an effort to generate an auxotrophic bacteria incapable of escape from incorporation of a UAA at only one site, we previously engineered UAA-dependent enzyme active sites, metalloenzyme metal coordination sites, and orthogonal dimer interfaces within essential proteins12,13,14. The culmination of these studies shed light on the importance of selecting an essential protein required for a non-metabolic core life process. In a recent report, we evolved the dimer interface of the β-sliding clamp DnaN, an essential DNA replication protein, to exclusively oligomerize in the presence of the UAA p-benzoyl-l-phenylalanine (BzF) (Fig. 1a)15. The engineered Escherichia coli strain EV2.BzF.h5 grows only in the presence of exogenously added BzF and exhibited exceptionally low escape frequencies (<2 × 10−10) in its absence
Herein, we have extended this approach to develop a live UAA conditional vaccine of the human pathogen Pseudomonas aeruginosa. Infections by P. aeruginosa are the leading cause of death in individuals with cystic fibrosis and are responsible for 10–20% of nosocomial infections with heighted mortality rates in debilitated or immunocompromised patients16,17. The first sign of infection is often overwhelming Gram-negative bacterial sepsis, where surgery or heavy antibiotic dosing is the only reasonable recourse. Since the clinical emergence of multidrug-resistant strains of P. aeruginosa, even last resort treatment methods have proven ineffective, designating P. aeruginosa as a prime candidate for bacterial vaccine development18,19. Moreover, the methods reported here are readily generalizable to other bacterial pathogens given that DnaN is highly conserved across bacterial lineages.
Results
BzF incorporation in P. aeruginosa
To engineer a live Pseudomonas vaccine conditionally dependent on an UAA for replication first required us to incorporate the bulky UAA BzF into a target protein in response to the amber nonsense codon in P. aeruginosa PAO120. At the time of this work, a pyrrolysyl-based UAA incorporation system was reported for P. aeruginosa using the Methanosarcina barkeri pyrrolysyl-tRNA synthetase (MbPylRS)/tRNA pair21, which had been previously evolved to incorporate BzF in E. coli22,23. To test the feasibility of using this system, we first prepared an E. coli and P. aeruginosa shuttle vector that expressed the BzF MbPylRS/tRNA pair with and without codon optimization. We evaluated production of full-length prolyl-tRNA synthetase (ProS, a model protein for amber suppression analysis) containing a C-terminal hexa-histidine tag and known UAA permissive site Y174X in the presence of BzF and Ala-BzF, a dipeptide which can improve cellular uptake. No BzF incorporation was observed in either condition (Supplementary Fig. 1). We then turned to a BzF-incorporating variant of the Methanocaldococcus jannaschii tyrosyl-tRNA synthetase (MjTyrRS)/tRNA pair24 and did observe successful suppression of the amber nonsense codon in ProS (Supplementary Fig. 1), in agreement with a recent study in Pseudomonas putida KT244025.
P. aeruginosa strain design and engineering
As an essential protein in DNA replication, DnaN is conserved across diverse bacteria26. Comparing the sequence and structure of E. coli and P. aeruginosa DnaN reveals a 56% sequence identity and a highly similar tertiary fold, including at the homodimeric interface (Fig. 1a)27,28. In our previous work, we replaced E. coli DnaN L273 with BzF, one of the residues responsible for packing between the two subunits. Subsequent directed evolution of the adjacent interface by mutation of seven amino acids led to the generation of three BzF-dependent orthogonal dimers. Due to the homology between the E. coli and P. aeruginosa proteins, we first overlaid the mutations from three of the E. coli variants (M3, M5, and M8) onto the P. aeruginosa protein. Except for a single amino acid shift of BzF273 and the adjacent amino acid at 272, each amino acid overlaid precisely onto the P. aeruginosa protein at the same corresponding position. To determine if any P. aeruginosa variants (M3, M5, M8) produced functional DnaN in vivo, we next engineered a temperature-sensitive (TS) strain of P. aeruginosa PAO1 by introducing wild-type dnaN into a P. aeruginosa shuttle vector with a TS P. aeruginosa origin of replication. With wild-type DnaN now expressed from the TS complement plasmid at 30 °C, we were able to delete the genomic dnaN gene (Fig. 1b). The resulting strain exhibited a temperature-dependent growth phenotype, and dnaN deletion from the genome was confirmed by PCR (Supplementary Fig. 2). Next, we transformed the homologous P. aeruginosa variants carrying the E. coli M3, M5, or M8 mutations into our TS P. aeruginosa ΔdnaN strain. However, none of these variants supported growth at the non-permissive temperature 42 °C, indicating that they are non-functional..

a X-ray structure overlay of the homodimeric interface of P. aeruginosa (gold; PDB: 6AMS) and E. coli (blue; PDB: 1MMI) DnaN, with side chains involved in dimer interaction shown as sticks. The dotted line indicates the boundary of the homodimeric interface, with residues labeled according to the P. aeruginosa structure. Residues for wild-type (WT) and mutant (M3, M5, M8) amino acid sequences of E. coli DnaN evolved to form a BzF-dependent dimer are provided in the table, as well as the structure-based consensus sites in P. aeruginosa wild-type DnaN. b Outline of P. aeruginosa engineering campaign. An origin-less knock-in/knock-out plasmid (KI/KO) integrates up or downstream of genomic dnaN. A wild-type copy of dnaN is supplied on a plasmid containing a temperature sensitive origin (TS ori), allowing the KI/KO cassette to be excised and genomic dnaN deleted to afford a TS strain. This strain is transformed with a vector encoding a BzF aminoacyl-tRNA synthetase (BzFRS/tRNA) and the evolved BzF-dependent DnaN, and the TS plasmid encoding wild-type DnaN is cured at elevated temperature resulting in a BzF-dependent Vaccine strain. c Design of the directed evolution platform for P. aeruginosa dnaN in E. coli using a cAMP generating bacterial two-hybrid system. Four independent sequence clusters were obtained and are shown in the table. A western blot of DnaN variant containing Pa M2 and Ec M3 mutations formed a photocrosslink-trapped dimer.
Directed evolution of P. aeruginosa DnaN for BzF dependence
Due to the inherently poor transformation efficiency of the TS P. aeruginosa strain (~10−3 efficiency compared to the wild-type PAO1 strain), we utilized an E. coli-based two-hybrid evolution system to improve the function of BzF-dependent P. aeruginosa DnaN29. Each member of the DnaN homodimer is translationally fused to a separate domain (T25 or T18) of a split adenylate cyclase. Dimerization of DnaN directs assembly of adenylate cyclase, which generates cAMP to induce downstream expression of maltose catabolism genes and growth on maltose-containing minimal media in the adenylate cyclase deficient BTH101 E. coli strain (Fig. 1c, Supplementary Fig. 3).
Two libraries were constructed: a single-site saturation mutagenesis (NNK) library at position I273 on the Pa DnaN-T18 fusion to allow for flexibility at the end of the alpha helix containing BzF274, and a 218 bp random mutagenesis library comprising the entire BzF-accepting pocket of the homologous P. aeruginosa M3 variant on the T25-Pa DnaN fusion. These two libraries were introduced into BTH101 cells harboring the E. coli pUltra plasmid encoding the BzF MjTyrRS/tRNA pair30. Following selection on maltose minimal media, 26 BzF-dependent clones were identified whose DnaN sequences converged into four distinct clusters: (i) P65S, Q95H, variable I273 mutations; (ii) I273P; (iii) L111I, E120V, variable I273 mutations; and (iv) K80BzF, N114S, I273C (Supplementary Table 1). To confirm dimerization, one mutant from each group was cloned with a C-terminal hexa-histidine tag into a P. aeruginosa expression plasmid31,32. Western blot revealed that only the I273P variant exhibited significant crosslinking following UV irradiation (BzF also functions as a photocrosslinker when irradiated with UV light; Supplementary Fig. 4); however, this clone did not support growth of the TS ΔdnaN strain at an elevated temperature, which we hypothesized was due to its relatively low expression level. To increase expression, several combinations of strong promoters and plasmid replication origins were screened by Western blot (Supplementary Fig. 5)31,32,33,34,35,36. Maximum DnaN expression and dimerization (Fig. 1c) was observed using a vector (dnaN.L274X.M2) encoding a tac promoter and a RSF1010 origin (Fig. 2a). The TS ΔdnaN P. aeruginosa strain was transformed with this plasmid, which supported growth at non-permissive temperature. This BzF auxotrophic P. aeruginosa strain encodes the P. aeruginosa M2 DnaN variant with the following mutations: V70A, I78F, L97I, F106V, L108G, I273P, L274BzF. This strain is hereafter referred to as Pa Vaccine.

a Plasmid map encoding DnaN M2 and the Mj BzFRS/tRNA pair used in Pa Vaccine. b Bacterial growth curves of Pa Vaccine in the presence of LB supplemented with varying concentrations of BzF compared to wild-type P. aeruginosa at 37 °C. Data is plotted as mean ± standard deviation. c Bacterial growth curves of Pa Vaccine following removal of BzF in media at 37 °C. Data is plotted as mean ± standard deviation. All source data are provided in the Source Data file.
Growth and escape frequencies of synthetic P. aeruginosa Vaccine
To determine the escape frequency of Pa Vaccine, 1011 cells were plated on LB agar lacking BzF. No growth was observed after 7 days, demonstrating that the escape frequency is below the experimental detection limit of 10−11. Additionally, mutants of the M2 DnaN plasmid (Fig. 2a) were constructed in which BzF274 was replaced with each of the 20 canonical amino acids. Each variant failed to support growth in P. aeruginosa ΔdnaN, verifying that a BzF274 mutational revertant will not allow escape (Supplementary Fig. 6). Next, growth characteristics of Pa Vaccine were analyzed. When grown at 37 °C in LB supplemented with 1 mM BzF, Pa Vaccine exhibits a doubling time of 100 min, compared to 35 min for the parent PAO1 strain (Fig. 2b). Minimal growth is observed at 0.125 mM BzF, while no growth is observed in the absence of BzF. Furthermore, Pa Vaccine undergoes 2.6 additional rounds of replication after BzF is removed (Fig. 2c). Presumably, depletion of intracellular reserves of BzF during cell division leads to insufficient concentrations for growth after 2–3 cycles of replication.
Vaccine efficacy of synthetic P. aeruginosa
Next, we determined the safety and efficacy of Pa Vaccine as a live vaccine in a pathogenic P. aeruginosa mouse infection model. To determine its safety profile, Pa Vaccine was grown in the presence of 1 mM BzF, washed to remove any toxins, resuspended in PBS, and injected intraperitoneal (IP) into BALB/c mice. Formalin inactivated PAO1 (FI PAO1) cells were used as a control. No deaths were observed at doses up to 6 × 108 CFU for Pa Vaccine, or the highest dose of FI PAO1 at 2 × 109 CFU (Supplementary Fig. 7). Both Pa Vaccine and FI PAO1 dosed mice exhibited similar mild symptoms of discomfort at higher doses. We surmise that the death caused by the highest Pa Vaccine dose of 2 × 109 CFU is due to a strong and lethal immune reaction to lipopolysaccharide (LPS) and other immune-dominant bacterial matter rather than an established infection, consistent with previous reports37.
After establishing the safety profiles of Pa Vaccine and FI PAO1, we evaluated their ability to induce production of anti-P. aeruginosa IgG and IgA serum antibodies and provide protection against the pathogenic P. aeruginosa strain PA14 in both IP and intranasal (IN) infection models (Fig. 3a). Mice were immunized via IN or IP with 6 × 107 CFU of Pa Vaccine (prepared as described above) or the equivalent amount of FI PAO1 on days 0, 14, 21, and 28. A slight and transient reduction in body weight was observed in animals immunized with either vaccine in both IP and IN immunization models (Fig. 3b, Supplementary Fig. 8). In both IN and IP vaccination models, both Pa Vaccine and FI PAO1 immunization generated a strong anti-PAO1 immune response in a whole-cell ELISA assay as measured by serum IgG titers (Fig. 3c). Notably, for IN vaccinated mice, Pa Vaccine induced a stronger IgG response against pathogenic PA14 compared to FI PAO1 (Fig. 3d). This difference was evident 1 week after the first booster dose (Fig. 3c). In mice immunized via IP, Pa Vaccine and FI PAO1 generated a similar IgG antibody response against PA14. A moderate and transient increase in anti-PA14 serum IgA antibody titers was observed following the second immunization in both IN Pa Vaccine and FI PAO1 vaccinated animals, while IP immunized mice exhibited no increase in IgA titers (Supplementary Fig. 9).

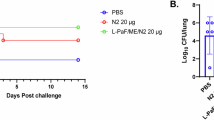
a Experimental timeline. Mice were immunized via intranasal (IN) or intraperitoneal (IP) routes and blood collected on days 0, 14, 21, 28. Additional blood was collected on days 35 and 42 for ELISA analysis of IgG titers. Animals were challenged with a lethal dose of PA14 (8 × 106 CFU via IN or 3 × 107 CFU via IP) on day 43 and observed for seven additional days. b Percent change in animal body weights following the first immunization via IP. N = 4 for all groups. p > 0.05 for Pa Vaccine vs. PBS groups at all time points. Data is plotted as mean ± standard deviation. c Time course IgG titers for animals in each experimental group. Enzyme-linked immunosorbent assays (ELISA) were used to determine IgG binding to whole cells of each indicated coating strain. * p < 0.05 for Pa Vaccine vs. FI PAO1 immunized groups at day 42. d Whole-cell ELISAs with serum from day 42. N = 8 for all groups. * p < 0.05 for Pa Vaccine vs. FI PAO1 groups. Data is plotted as mean ± standard deviation. e Survival following PA14 challenge. IN vaccinated mice were challenged via IN, and IP vaccinated animals challenged via IP. N = 8 for all groups. f Whole-cell ELISAs against a panel of pathogenic P. aeruginosa strains. O-antigen serogroup and isolation source for each strain are provided in Table S2. * p < 0.05 for Pa Vaccine vs. FI PAO1 groups. Data is plotted as mean ± standard error of the mean. Welch’s two-tailed t-tests were used for all pairwise statistical comparisons. All source data, including p values, are provided in the Source Data file. a Created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license (https://creativecommons.org/licenses/by-nc-nd/4.0/deed.en).
To determine whether Pa Vaccine could provide protection against P. aeruginosa infection, Pa Vaccine and FI PAO1 immunized mice were challenged on day 43 with lethal doses of 8 × 106 CFU PA14 via IN, or 3 × 107 CFU PA14 via IP (for IN or IP vaccinated groups respectively. Lethal doses determined experimentally in Supplementary Fig. 10). All mice from both FI PAO1 and Pa Vaccine immunized groups survived this infection challenge, demonstrating that the Pa Vaccine strain and FI PAO1 effectively induce a comparably protective immune response in this model (Fig. 3e).
The P. aeruginosa strains in our mouse model came from two different serogroups, distinguished by LPS composition; PAO1 is O-antigen serogroup 2/5 (O2/5), while PA14 is serogroup O1938. Based on our observation that IN Pa Vaccine elicits a higher level of cross-reactive IgG antibodies against PA14 compared to FI PAO1 (Fig. 3d), we hypothesized that Pa Vaccine could provide superior antibody cross-reactivity against strains spanning a wider spectrum of O-antigen serogroups. Using serum collected at day 42 from Pa Vaccine and FI PAO1 immunized mice, we analyzed IgG binding against a panel of pathogenic P. aeruginosa strains spanning several serogroups and isolation sites (Fig. 3f, Supplementary Table 2). Indeed, serum from Pa Vaccine immunized mice dosed IP and IN contained similar or higher IgG titers against all pathogenic strains in the panel. Interestingly, IN immunized mice receiving Pa Vaccine exhibited substantially increased IgG antibody levels compared to those in the IP cohort.
Discussion
To engineer a UAA-based auxotrophic strain of P. aeruginosa, we adapted a two-hybrid selection system in E. coli to evolve the DnaN β-sliding clamp of P. aeruginosa to be functionally dependent on incorporation of BzF at position 274. The high structural conservation of DnaN from these bacteria allowed us to overlay a previously engineered functional E. coli dnaN mutant onto the sequence of P. aeruginosa dnaN to identify productive sites for mutagenesis. The final strain, Pa Vaccine, exhibits complete dependence on the UAA BzF, a chemical structure not found in nature. We confirmed this essential characteristic by an undetectable auxotrophic escape frequency (<10−11) and the inability of all P. aeruginosa DnaN M2 variants containing a canonical amino acid at position 274 to support growth. Owing to the high sequence identity of DnaN across Gram-negative and -positive bacteria, this two-hybrid platform will facilitate the development of other strictly UAA-dependent bacteria for use as live conditional vaccines and other biotechnology applications. For clinical deployment of a strain using this system, genomic integration and expression optimization of dnaN M2, BzF-MjYRS, and MjYtRNA-CTA genes would be required to remove antibiotic resistance markers associated with a plasmid-based system. Furthermore, despite the excellent in vitro auxotrophic characteristics and in vivo safety profile demonstrated here, the potential of this strain to revert to wild-type would need to be carefully monitored in a clinical setting. For example, horizontal gene transfer of dnaN that can complement P. aeruginosa would provide an avenue of escape for this strain and would need to be considered in future efforts, particularly for immunocompromised patients who are more susceptible to chronic infections and negative side effects of attenuated vaccines.
As a live conditional vaccine, Pa Vaccine generated similar IgG titers against parent strain PAO1 over the course of the 42-day experiment compared with FI bacteria in both IN and IP vaccination models. In comparison, Pa Vaccine induces higher IgG titers against pathogenic PA14 in the IN model compared to FI bacteria even after a single boost, which were sustained to the end of the experiment (Fig. 3c). It has been shown that various immunization routes elicit distinct immune responses following vaccination against Pseudomonas38. Mucosal immunity plays a crucial role in protection from this pathogen, involving mucosal antibodies, T helper 17 cells that can activate local inflammation, and tissue-resident memory T cells39,40. These published studies support the superiority of mucosal immunization through IN pathways over intramuscular or IP routes against acute Pseudomonas infection. We attribute our IgG titer results in part to the ability of Pa Vaccine to survive and replicate through multiple cell divisions before intracellular concentrations of depleted BzF can no longer support cell growth.
This vaccination paradigm, in which a live immunization strain differs from the wild-type bacterium by as little as a single mutation, more closely resembles an active bacterial infection compared to fixed or heat-lysed bacterial vaccines. Indeed, previous studies suggest that live-attenuated P. aeruginosa vaccines, such as those based on cell wall constituents or amino acid auxotrophies39,41, may elicit better protection in mouse models than protein subunit-based vaccines, particularly for cross protection. This may be due to the ability of live cells to stimulate a stronger immune response in the mucosa. We contend that the excellent safety profile and minimal risk of pathogenic reversion provided by our UAA-auxotrophy platform will allow us to more effectively leverage the benefits of live-attenuated vaccines.
Pa Vaccine elicits comparable or higher cross-reactive IgG titers than FI PAO1 against all pathogenic strains tested. The strains in this panel belong to several O-antigen serogroups, including O2/5 (Boston 41501), O6 (ATCC 10145), O11 (PA15678), O12 (PA8915), and O19 (PA14) (Fig. 3f, Table S2). These groups encompass ~50% of all pathogenic Pseudomonas isolates tested in a survey of clinical cases of invasive P. aeruginosa infection42, and constitute isolates sourced from wound, respiratory, blood, and urinary-tract infections. We hypothesize that this is in part due to Pa Vaccine retaining intact and immunogenic LPS, which is likely highly crosslinked on the surface of FI PAO1. The O-specific antigen, which distinguishes O-antigen serogroups, is the immunodominant epitope on the surface of P. aeruginosa43. Interestingly, Pa Vaccine (O2/5) elicits nearly identical antibody titers against Boston 41501, PA14, ATCC 10145, and PA8915, despite only Boston 41501 sharing the same O-serogroup as Pa Vaccine. These data suggest the cross-reactive IgGs generated by immunization with Pa Vaccine recognize shared O-antigen epitopes, or other immunogenic cell surface factors such as Lipid A or Common polysaccharide antigen, that are distorted or rendered non-immunogenic in the case of chemically modified FI PAO1. We envision that further development and optimization of a broadly protective and safe Pa Vaccine would have a substantial clinical impact given the increasing prevalence of multi-drug resistant P. aeruginosa, and the increased risk of Pseudomonas infection to cystic fibrosis patients. In immunocompromised individuals, attenuated vaccines have historically been avoided due to the potential risk of life-threatening infection. This population may particularly benefit from a vaccine that is first and foremost highly safe, as well as effective and broadly protective.
Methods
Ethics statement
All animal studies were performed in compliance with the protocol approved by the Institutional Animal Care and Use Committee at Scripps Research (#22-0010). Mice were housed in AAALAC-accredited animal facilities at Scripps Research at ambient temperature (67–75 °F) and humidity (40–60%) with 12 hour light-dark cycles. Animals were fed a standard rodent diet and given free access to water, and monitored daily for weight loss, lethargy, or signs of distress. Those exhibiting significant weight loss (>20%) or signs of severe infection were euthanized according to American Veterinary Medical Association Guidelines for Euthanasia.
Culture conditions and reagents
All E. coli (Ec) and P. aeruginosa (Pa) cultures were grown in Lysogeny Broth (LB) media unless otherwise indicated. Cloning was carried out in E. coli DH10B or DH5-alpha. Antibiotics were used at the following concentrations unless specified otherwise: kanamycin (50 µg/mL Ec), spectinomycin (100 µg/mL Ec), carbenicillin (100 µg/mL Ec, 150 µg/mL Pa), streptomycin (500 µg/mL Pa), gentamycin (200 µg/mL Pa). All oligonucleotides and gene blocks were purchased from Integrated DNA Technologies, USA. para-benzoyl phenylalanine (BzF) was purchased from Combi-blocks, USA and solubilized in 1 M NaOH. Alanine-BzF was synthesized following established methodology.
Western blots for assessment of UAA suppression in P. aeruginosa
To assess the suppression ability of the BzF-variants of both the Methanosarcina barkeri pyrrolysyl-tRNA synthetase (MbPyrRS)/tRNA and Methanocaldococcus jannaschii tyrosyl-tRNA synthetase (MjTyrRS)/tRNA, we designed a Pa shuttle vector using the backbone of pACRISPR (Addgene ID: 113348) as a template encoding an ampicillin resistance marker that functions in Ec and Pa, a ColE-1 Ec origin of replication (ori), and a pRO1600 oriV/msF Pa ori24. The pRO1600 oriV/msF Pa origin was selected because of known point mutations in msF that impart temperature sensitivity to the origin, which was utilized later in the study. We prepared three plasmids, one encoding a Pa optimized codon sequence of the BzF-variant of MjTyrRS and tRNA, a second encoding the BzF-variant of MbPyrRS and tRNA, and the third encoding a Pa optimized codon sequence of the MbPyrRS and tRNA. Each aaRS was cloned downstream of the Pa PAO1 wild-type tyrosyl-tRNA synthetase promoter. The MbPyr tRNA was cloned downstream of a modified Pa PAO1 tyrosyl-tRNA promoter, while the MjTyrR tRNA was cloned downstream of the wild-type Pa PAO1 methionyl-tRNA promoter. To test amber suppression, we used the Prolyl-tRNA synthetase (ProS) as a model protein with known permissive site Y174X (X represents the TAG amber codon) and a C-terminal hexa-histidine tag, under the control of a strong constitutive promoter, psbA32. Samples were analyzed by Coomassie staining and western blot (Supplementary Fig. 1) with anti-hexa-histidine antibody and horseradish peroxidase-conjugated secondary antibody (BioXCell #RT0266, 1:10,000; Sigma-Aldrich #A9044, 1:3000).
Temperature sensitive compliment plasmid and genomic dnaN knockout
A pEX18Gm plasmid was used to delete Pa dnaN from the genome44. pDD091 containing 500 bp up and downstream of the Pa dnaN gene in the pEX18Gm plasmid template was prepared. In parallel, pDD108, a temperature-sensitive dnaN complement plasmid containing WT dnaN controlled by the constitutive glnS promoter was prepared. To impart temperature sensitivity into the mSF ori, point mutations G100C and S204R were added to yield mSFts145. Additionally, the plasmid contained a Pa streptomycin resistance marker. To delete the genomic dnaN gene, a modified version of the literature protocol was used wherein each instance of a temperature >30 °C was performed at 30 °C, and incubation times in each case were scaled 1.5x. The dnaN knockout was validated by PCR, gene sequencing, and phenotypic temperature-sensitive growth (Supplementary Fig. 2).
Demonstration of amber suppression in Pa DnaN and western blot assessment of dimerization
The dnaN mutants with C-terminal hexa-histidine tags were cloned into an expression plasmid encoding the BzFRS/tRNA pair under constitutive tyrS and metT promoters by Gibson Assembly. Cultures of PAO1 cells containing constitutive dnaN expression vectors were inoculated at OD600 = 0.1 into fresh LB (with 1 mM BzF and carbenicillin) and grown overnight at 20 °C. Cells were pelleted, washed, resuspended in Dulbecco’s phosphate buffered saline (pH 7.4; PBS), and lysed by sonication (Branson, SFX150). Debris was removed by centrifugation (15,000 g for 15 min) and the protein concentration of each lysate sample was normalized to 1.33 mg/mL. For UV crosslinking, 50 µL of cell lysate in 200 µL PCR tubes were irradiated in a 368 nm UV crosslinker (Boekel Scientific, model 234100) for 15 min at 4 °C (0.12 Joules).
Bacterial two-hybrid library design and construction
A two-plasmid system was used for the bacterial two-hybrid library. pBK-SC-T18 contained a ColE1 origin and kanamycin resistance cassette. A gene encoding the DnaN-T18 fusion protein was constructed by overlap extension PCR between a gBlock fragment encoding an E. coli optimized version of the P. aeruginosa dnaN and the T18 gene from pUT1846. This fusion gene was inserted into the pBK-SC-T18 backbone under control of the constitutive glnS promoter and ribosome binding site by Gibson Assembly. The second plasmid, pBK-T25-SC, encoded a p15A origin and ampicillin resistance marker. Similarly, a T25-dnaN fusion gene was created by overlap extension PCR between a gBlock fragment encoding a P. aeruginosa optimized version of dnaN and the T25 gene from pKT2546, and cloned into pBK-T25-SC under the glnS promoter by Gibson Assembly. The L274X mutation (X denotes TAG amber codon) was introduced into dnaN on each plasmid by site-directed mutagenesis.
A site-saturated library at dnaN I273 on pBK-SC-T18 was introduced by Q5 site-directed mutagenesis Kit (KLD kit, New England Biolabs, Ipswich, MA) using NNK degenerate primers (N = A, T, G, or C; K = G or T). Separately, a fragment comprising the 218 base pairs of the N-terminal DnaN dimer interface was randomly mutagenized according to manufacturer’s instructions (GeneMorphII, Agilent, USA), and inserted into pBK-T25-SC by Gibson Assembly. The average mutations per gene from the error prone PCR was optimized to 2.5 bp and the library size was 2×109 transformants. Both libraries were sequentially transformed into BTH101 cells46 containing IPTG-inducible pUltra-BzFRS15 to achieve a total library size of 1.1 × 1010.
Two-hybrid BzF-DnaN library selection
Bacterial two-hybrid selection was performed as described previously29. BTH101 cells containing the two-hybrid library were cultured for 2 h at 30 °C in LB containing 1 mM BzF, 0.5 mM IPTG (for BzFRS induction), and appropriate antibiotics. Cells were then washed with PBS, spread onto selective media (M63 minimal media, 0.25 mg/mL MgSO4, 0.2% maltose, 1 mM BzF, 0.5 mM IPTG, 75 µg/mL carbenicillin, 37 µg/mL kanamycin, 75 µg/mL spectinomycin, pH 7.0), and grown at 30 °C. Surviving clones were rescreened on selective media with and without BzF, and BzF-dependent clones were sequenced. A complete list of mutation sequences is provided in Supplementary Table 1.
Assembly of vaccine plasmid and WT complement plasmid curing
Putative vaccine plasmids were prepared by a combination of Gibson assembly and KLD with a dnaN gene sequence containing mutations V70A, I78F, L97I, F106V, L108G, I273P, L274X, and additional mutations Pa M1, M2, M3 and M4 (Fig. 1c) downstream of either a psbA, tac, 14 g, or T7 promoter32,33,34,35. Each plasmid encoded ampicillin resistance and either an mSF or RSF1010 ori31,36. Western blot analysis of each plasmid with and without UV exposure indicated that the RSF1010 origin and tac promoter produced optimal gene expression and dimer formation (Supplementary Fig. 5). The corresponding plasmid, named dnaN.L274BzF.M2, was electroporated into the engineered temperature sensitive ΔdnaN strain. Transformed colonies were streaked on LB agar (with 1 mM BzF and carbenicillin) and grown at 42 °C for three consecutive passages. Surviving colonies were plated on LB agar with and without streptomycin at 30 °C to confirm TS complement plasmid curing, and no growth was observed.
Growth and auxotrophic characterization of Pa Vaccine
Cultures were inoculated to a starting OD600 of 0.0025 in fresh media with appropriate supplements. For BzF wash out experiments, cells were grown to OD600 = 0.1, washed in PBS, and resuspended in the appropriate media. Growth curves were collected using a BioTek Logphase 600 plate reader (Agilent, CA, USA) in 96 well plates in triplicate.
To calculate auxotrophic escape frequency, Pa Vaccine cells were cultured in LB supplemented with 0.5 mM BzF at 37 °C to OD600 = 1.0. Cells were washed and resuspended in PBS, and 1011 cells spread onto LB agar without BzF. Ten-fold serial dilutions were plated on LB agar containing 1 mM BzF to quantify the colony forming units and confirm that 1011 cells were plated.
Using the Pa vaccine plasmid as a template, a codon for all 20 canonical amino acids was separately cloned into the 274 position by KLD. The temperature-dependent P. aeruginosa strain was individually transformed with plasmid encoding each dnaN mutant, and cultured in LB at 20 °C. Saturated cultures were diluted and spotted onto replica LB plates and grown at 20 °C or 42 °C.
Mouse immunization and challenge experiments
Female BALB/c mice (5–6 weeks old; Jackson Laboratories) were used in all experiments unless otherwise indicated. Mice were vaccinated four times according to the schedule shown in Fig. 3a. Unless otherwise noted, for all vaccination and challenge inoculations, P. aeruginosa strains were cultured in LB medium overnight at 37 °C, diluted into fresh media and grown to OD600 = 0.6–0.7. Cells were centrifuged, washed twice in PBS, then resuspended in PBS to the appropriate cell density. Cell density, expressed as colony forming units per mL (CFU/mL), was determined for each strain by serial dilution plating of an OD600 = 1.0 solution. For intraperitoneal injections, 100 µL of cell solution were injected using a 26-gauge needle. For intranasal administrations, animals were anesthetized with isoflurane and 20 µL of cell solution pipetted into one nostril while in an upright position. Mice were challenged on day 43 with 3 × 107 CFU of PA14 via IP (for IP vaccinated groups), or 8 × 106 CFU of PA14 via IN (for IN vaccinated groups). PA14 challenge doses were determined experimentally with naïve age-matched mice (Supplementary Fig. 10).
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were collected on days 0, 14, 21, 28, 35, and 42 by retroorbital sinus puncture using heparin-coated tubes. Whole blood was centrifuged at 4 °C for 15 min at 1500 g, and the resulting plasma transferred to a fresh tube and stored at –80 °C. P. aeruginosa strains were cultured and prepared as described above. Cells were diluted to a concentration of 2 × 107 CFU/mL (for longitudinal and day 42 ELISA studies) or OD600 = 0.01 (for P. aeruginosa panel ELISA) and coated onto 384-well ELISA plates at 4 °C. Bovine serum albumin (BSA)-coated wells were prepared as a negative control by incubation with 1% BSA in PBS. After blocking all wells with 1% BSA in PBS for 1 hour, plasma from immunized mice was serially diluted 1:102, 1:103, 1:104, and 1:105 in 1% BSA in PBS and incubated for an additional hour. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (H + L) antibody (1:5,000, #115-035-146, Jackson Immuno Research Laboratories, West Grove, PA, USA) or HRP-conjugated goat anti-mouse IgA antibody (1:5,000, #1040-05, Southern Biotech, Birmingham, AL) was added in 1% BSA in PBS and incubated at room temperature for 1 hour. One-Step Ultra TMB-ELISA (#34028, Thermo Fisher Scientific, Inc., Waltham, MA, USA) substrate was used according to manufactures instruction for color development, and the reaction was quenched by adding 2 N H2SO4. Absorbance was measured at 450 nm using a plate reader (BioTek Instruments, Winooski, VT, USA). Plates underwent three washes before each incubation step. Titers are expressed as Absorbance summation47.
Statistics and reproducibility
Doubling times for Pa Vaccine and PAO1 were calculated by fitting log-phase growth curves to the exponential growth equation Y = Y0*e(kt), where Y is the final OD600 value, Y0 is the initial OD600 value at time t = 0, and k is the growth rate constant. The doubling time is calculated as the ln(2)/k. Welch’s two-tailed t-tests were used for all pairwise statistical comparisons. All calculations were performed using Microsoft Excel or GraphPad Prism. Experiments were generally performed once unless otherwise indicated.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all relevant data supporting the findings of this study are available within the paper and its Supplementary Information files, or available upon request. Data generated in this study are provided in the Supplementary Information/Source Data files. Source data are provided with this paper.
References
Cabral, M. P. et al. Design of live attenuated bacterial vaccines based on D-glutamate auxotrophy. Nat. Commun. 8, 15480 (2017).
Belshe, R. B. et al. Live attenuated versus inactivated influenza vaccine in infants and young children. N. Engl. J. Med. 356, 685–696 (2007).
Killeen, K., Spriggs, D. & Mekalanos, J. Bacterial mucosal vaccines: Vibrio cholerae as a live attenuated vaccine/vector paradigm. Curr. Top. Microbiol. Immunol. 236, 237–254 (1999).
Tennant, S. M. & Levine, M. M. Live attenuated vaccines for invasive Salmonella infections. Vaccine 33(Suppl 3), C36–C41 (2015).
Hussein, I. H. C. et al. A. vaccines through centuries: major conerstones of global health. Front. Public Health 3, 269 (2015).
Detmer, A. & Glenting, J. Live bacterial vaccines—a review and identification of potential hazards. Micro. Cell Fact. 5, 23 (2006).
Mazzolini, R. et al. Engineered live bacteria suppress Pseudomonas aeruginosa infection in mouse lung and dissolve endotracheal-tube biofilms. Nat. Biotechnol. 41, 1089–1098 (2023).
Mandell, D. J. et al. Biocontainment of genetically modified organisms by synthetic protein design. Nature 518, 55–60 (2015).
Rovner, A. J. et al. Recoded organisms engineered to depend on synthetic amino acids. Nature 518, 89–93 (2015).
Diercks, C. S., Dik, D. A. & Schultz, P. G. Adding new chemistries to the central dogma of molecular biology. Chem 7, 2883–2895 (2021).
Si, L. et al. Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science 354, 1170–1173 (2016).
Xuan, W. & Schultz, P. G. A strategy for creating organisms dependent on noncanonical amino acids. Angew. Chem. Int. Ed. Engl. 56, 9170–9173 (2017).
Gan, F., Liu, R., Wang, F. & Schultz, P. G. Functional replacement of histidine in proteins to generate noncanonical amino acid dependent organisms. J. Am. Chem. Soc. 140, 3829–3832 (2018).
Koh, M., Nasertorabi, F., Han, G. W., Stevens, R. C. & Schultz, P. G. Generation of an orthogonal protein-protein interface with a noncanonical amino acid. J. Am. Chem. Soc. 139, 5728–5731 (2017).
Koh, M., Yao, A., Gleason, P. R., Mills, J. H. & Schultz, P. G. A general strategy for engineering noncanonical amino acid. Depend. Bact. Growth J. Am. Chem. Soc. 141, 16213–16216 (2019).
Malhotra, S., Hayes, D. Jr. & Wozniak, D. J. Cystic fibrosis and Pseudomonas aeruginosa: the host-microbe interface. Clin. Microbiol. Rev. 32, e00138–18 (2019).
Reynolds, D. & Kollef, M. The epidemiology and pathogenesis and treatment of Pseudomonas aeruginosa infections: an update. Drugs 81, 2117–2131 (2021).
Dik, D. A., Fisher, J. F. & Mobashery, S. Cell-wall recycling of the gram-negative bacteria and the nexus to antibiotic resistance. Chem. Rev. 118, 5952–5984 (2018).
Kunz Coyne, A. J., El Ghali, A., Holger, D., Rebold, N. & Rybak, M. J. Therapeutic strategies for emerging multidrug-resistant Pseudomonas aeruginosa. Infect. Dis. Ther. 11, 661–682 (2022).
Held, K., Ramage, E., Jacobs, M., Gallagher, L. & Manoil, C. Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J. Bacteriol. 194, 6387–6389 (2012).
Zheng, H., Lin, S. & Chen, P. R. Genetically encoded protein labeling and crosslinking in living Pseudomonas aeruginosa. Bioorg. Med. Chem. 28, 115545 (2020).
Lacey, V. K., Louie, G. V., Noel, J. P. & Wang, L. Expanding the library and substrate diversity of the pyrrolysyl-tRNA synthetase to incorporate unnatural amino acids containing conjugated rings. Chembiochem 14, 2100–2105 (2013).
Avila-Cobian, L. F. et al. Amber-codon suppression for spatial localization and in vivo photoaffinity capture of the interactome of the Pseudomonas aeruginosa rare lipoprotein A lytic transglycosylase. Protein Sci. 32, e4781 (2023).
Chin, J. W., Martin, A. B., King, D. S., Wang, L. & Schultz, P. G. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl Acad. Sci. USA 99, 11020–11024 (2002).
He, X. et al. Genetic code expansion in Pseudomonas putida KT2440. ACS Synth. Biol. 11, 3724–3732 (2022).
Noirot-Gros, M. F. et al. An expanded view of bacterial DNA replication. Proc. Natl Acad. Sci. USA 99, 8342–8347 (2002).
McGrath, A. E. et al. Crystal structures and biochemical characterization of DNA sliding clamps from three Gram-negative bacterial pathogens. J. Struct. Biol. 204, 396–405 (2018).
Oakley, A. J. et al. Flexibility revealed by the 1.85 A crystal structure of the beta sliding-clamp subunit of Escherichia coli DNA polymerase III. Acta Crystallogr. D. Biol. Crystallogr. 59, 1192–1199 (2003).
Karimova, G., Pidoux, J., Ullmann, A. & Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl Acad. Sci. USA 95, 5752–5756 (1998).
Chatterjee, A., Sun, S. B., Furman, J. L., Xiao, H. & Schultz, P. G. A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52, 1828–1837 (2013).
Chen, W. et al. CRISPR/Cas9-based genome editing in Pseudomonas aeruginosa and cytidine deaminase-mediated base editing in pseudomonas species. iScience 6, 222–231 (2018).
Wilton, R. et al. A new suite of plasmid vectors for fluorescence-based imaging of root colonizing pseudomonads. Front. Plant. Sci. 8, 2242 (2017).
Elmore, J. R., Furches, A., Wolff, G. N., Gorday, K. & Guss, A. M. Development of a high efficiency integration system and promoter library for rapid modification of Pseudomonas putida KT2440. Metab. Eng. Commun. 5, 1–8 (2017).
Zobel, S. et al. Tn7-based device for calibrated heterologous gene expression in Pseudomonas putida. ACS Synth. Biol. 4, 1341–1351 (2015).
Tang, X. et al. Chromosomal expression of CadR on Pseudomonas aeruginosa for the removal of Cd(II) from aqueous solutions. Sci. Total Environ. 636, 1355–1361 (2018).
Nagahari, K. & Sakaguchi, K. RSF1010 plasmid as a potentially useful vector in Pseudomonas species. J. Bacteriol. 133, 1527–1529 (1978).
Cryz, S. J. Jr., Friedman, R. L., Pavlovskis, O. R. & Iglewski, B. H. Effect of formalin toxoiding on Pseudomonas aeruginosa toxin A: biological, chemical, and immunochemical studies. Infect. Immun. 32, 759–768 (1981).
Hao, Y., Murphy, K., Lo, R. Y., Khursigara, C. M. & Lam, J. S. Single-nucleotide polymorphisms found in the migA and wbpX glycosyltransferase genes account for the intrinsic lipopolysaccharide defects exhibited by Pseudomonas aeruginosa PA14. J. Bacteriol. 197, 2780–2791 (2015).
Cabral, M. P. et al. A live auxotrophic vaccine confers mucosal immunity and protection against lethal pneumonia caused by Pseudomonas aeruginosa. PLoS Pathog. 16, e1008311 (2020).
Priebe, G. P. & Goldberg, J. B. Vaccines for Pseudomonas aeruginosa: a long and winding road. Expert Rev. Vaccines 13, 507–519 (2014).
Priebe, G. P., Meluleni, G. J., Coleman, F. T., Goldberg, J. B. & Pier, G. B. Protection against fatal Pseudomonas aeruginosa pneumonia in mice after nasal immunization with a live, attenuated aroA deletion mutant. Infect. Immun. 71, 1453–1461 (2003).
Nasrin, S. et al. Distribution of serotypes and antibiotic resistance of invasive Pseudomonas aeruginosa in a multi-country collection. BMC Microbiol. 22, 13 (2022).
Lam, J. S., Taylor, V. L., Islam, S. T., Hao, Y. & Kocincova, D. Genetic and functional diversity of Pseudomonas aeruginosa lipopolysaccharide. Front. Microbiol 2, 118 (2011).
Huang, W. & Wilks, A. A rapid seamless method for gene knockout in Pseudomonas aeruginosa. BMC Microbiol. 17, 199 (2017).
Silo-Suh, L. A., Elmore, B., Ohman, D. E. & Suh, S. J. Isolation, characterization, and utilization of a temperature-sensitive allele of a Pseudomonas replicon. J. Microbiol. Methods 78, 319–324 (2009).
Kjelstrup, S., Hansen, P. M., Thomsen, L. E., Hansen, P. R. & Lobner-Olesen, A. Cyclic peptide inhibitors of the beta-sliding clamp in Staphylococcus aureus. PLoS One 8, e72273 (2013).
Hartman, H., Wang, Y., Schroeder, H. W. Jr. & Cui, X. Absorbance summation: a novel approach for analyzing high-throughput ELISA data in the absence of a standard. PLoS ONE 13, e0198528 (2018).
Acknowledgements
We acknowledge Kristen Williams for her assistance in manuscript preparation. We thank the Department of Animal Resources at Scripps Research. We thank Prof. Matthew Parsek for providing plasmid pEX18gm, and Prof. Quanjiang Ji for providing pACRISPR (Addgene Plasmid #113348) and pCasPA (Addgene Plasmid #113347). We thank Prof. Anders Løbner-Olesen for providing BTH101, and Prof. Colin Manoil and Jeannie Bailey from the P. aeruginosa Transposon Mutant Library at the University of Washington for providing the P. aeruginosa PAO1 strain. We thank BEI Resources, NIAID, NIH for providing the following P. aeruginosa strains: Strain MRSN 8915, NR-51561; Strain MRSN 1617, NR-51528; Strain MRSN 1739, NR-51530; Strain MRSN 1925, NR-51534; Strain MRSN 15678, NR-51579; and Strain PA14, NR-50573. MRSN strains are part of the Pseudomonas aeruginosa Diversity Panel provided by the Multidrug-Resistant Organism Repository and Surveillance Network (MRSN) at the Walter Reed Army Institute of Research (WRAIR), Silver Spring, MD, USA. Boston 41501 (ATCC 27853) and ATCC 10145 were obtained through the American Type Culture Collection (Manassas, VA, USA). This work was supported by NIH Grant 5R35GM145323 (P.G.S.).
Author information
Authors and Affiliations
Contributions
M.P., Y.C.L., M.K., T.F.R., D.A.D. and P.G.S. designed research. M.P., Y.C.L., M.K., C.S.D. and D.A.D. performed research. M.P., Y.C.L., M.K., T.F.R., D.A.D. and P.G.S. analyzed data. M.P., D.A.D. and P.G.S. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Emilie Camberlein, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pigula, M., Lai, YC., Koh, M. et al. An unnatural amino acid dependent, conditional Pseudomonas vaccine prevents bacterial infection. Nat Commun 15, 6766 (2024). https://doi.org/10.1038/s41467-024-50843-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-50843-7
