
Abstract
G-protein coupled receptors help regulate cellular function and communication, and are targets of small molecule drug discovery efforts. Conventional techniques to probe these interactions require labels and large amounts of receptor to achieve satisfactory sensitivity. Here, we use frequency-locked optical microtoroids for label-free characterization of membrane interactions in vitro at zeptomolar concentrations for the kappa opioid receptor and its native agonist dynorphin A 1-13, as well as big dynorphin (dynorphin A and dynorphin B) using a supported biomimetic membrane. The measured affinity of the agonist dynorphin A 1-13 to the κ-opioid receptor was also measured and found to be 3.1 nM. Radioligand assays revealed a dissociation constant in agreement with this value (1.1 nM). The limit of detection for the κOR/DynA 1-13 was calculated as 180 zM. The binding of Cholera Toxin B-monosialotetrahexosyl ganglioside was also monitored in real-time and an equilibrium dissociation constant of 1.53 nM was found. Our biosensing platform provides a method for highly sensitive real-time characterization of membrane embedded protein binding kinetics that is rapid and label-free, for drug discovery and toxin screening among other applications.
Similar content being viewed by others


Introduction
The cell membrane is comprised of lipids and transmembrane proteins and is fundamental for controlling the signal transduction events essential for cellular communication and regulatory responses. As much as 30% of the cell interface consist of integral membrane proteins1, which serve a wide variety of functions. This includes G protein coupled receptors (GPCRs, currently the most common therapeutic target), various channels and pumps, immunoglobins, and virulence receptors. In addition, lipids within the membrane act as receptors as well, such that the cell membrane serves as a key interface for recognizing and responding to extracellular phenomena. The lipid composition of the membrane itself can also affect the cellular consequences of activation or blockade, due to its influence on transmembrane protein conformation2. While a significant portion of pharmaceutical compounds are designed to target membrane components such as receptors, they are often difficult to label. Radioisotopes can break bonds and induce conformational changes, while fluorescent labels are large in comparison to small molecules and can alter their chemical behaviors3. Typical screening methods are also limited by the ability to control the membrane environment of the protein. The ability to observe membrane receptor–drug interactions in real-time without the use of labels could enhance the drug discovery process as well as increase our understanding of physiological events.
Conventional methods for studying membrane-protein binding events include the use of in vivo animal models4 (typically functional readouts with implied affinities) or selected cells in vitro5. Enzyme linked-immunosorbent assays (ELISA)6, liposome microarrays7,8, radioisotope labeling assays9, and fluorescent imaging10 are also methods used with cells or tissue homogenates to ascertain these interactions, each having limitations such as those described above. Of particular interest are label-free sensing schemes, which can directly and rapidly detect the binding of molecular ligands to receptors. Several examples that can quantify binding kinetics are surface plasmon resonance11 (SPR) with a limit of detection (LOD) down to 1 pg/mm2, electrochemical biosensors12,13, and electrochemical impedance spectroscopy (EIS) (LOD < 10 pM)14. Complex and potentially interfering signal enhancement tags are often required to boost the sensitivity of these systems15,16,17.
Whispering gallery mode (WGM) optical biosensors are ultra-sensitive, label-free, and capable of observing single molecule binding events. They confine photons in a path circumscribing the cavity; the photons can circulate many times thus allowing small changes in the optical path, such as those caused by biomolecular binding18,19,20,21,22,23. Here, we measure lipid membrane formation and membrane binding events by utilizing an ultra-sensitive, label-free biosensing system known as FLOWER (frequency locked optical whispering evanescent resonator)18,19,24,25,26,27. FLOWER is able to detect a single macromolecule; the high quality (Q)-factor and the evanescent field of WGM microtoroid resonators are exploited so that any local refractive index changes caused by analyte binding events to the resonator’s surface can be measured in real-time as a shift in the cavity’s resonance frequency (Fig. 1)24,25,28,29. Previous applications of WGM microresonators include detecting performance enhancing drugs28, nucleic acids30,31,32, proteins25,32,33,34, volatile organic compound sensing26, protein interaction screening35,36, and frequency comb generation37,38. Although previous studies have demonstrated the use of optical microcavities for studying the formation of lipid membranes39 and protein–lipid interactions35, to the best of our knowledge, optical microcavities have not been used to observe membrane receptor–ligand binding kinetics and associated affinity measurements. The ability to detect these interactions in a real-time, label-free, and ultra-sensitive manner will significantly advance biomarker screening as well as drug discovery, among other applications.

a A tunable laser is coupled to the microtoroid cavity through a tapered optical fiber. b Schematic diagram of the constructed biosensing chamber (top cover glass not shown). The tapered fiber is glued to the support wall with a polymer adhesive. Inset shows the structure of a membrane protein (pink) incorporated in a lipid bilayer (blue) on the toroid’s glass surface. c Sketches of how the resonant wavelength red-shifts when analytes adsorb onto the functionalized toroid surface (inset). d SEM image of microtoroid array alongside a support wall. The scale bar is 500 µm. e SEM side view image of a microtoroid structure. The scale bar is 40 µm. The inset picture shows a COMSOL simulation of an optical mode of the lipid coated cavity in a buffered aqueous solution.
In our approach, we establish a methodology for directly investigating membrane binding events on a WGM microtoroid resonator (Fig. 1) using two different ligand-receptor schemes: Cholera Toxin B (CTB) binding to monosialotetrahexosyl ganglioside (GM1) membrane receptors and Dynorphin A 1-13 (DynA 1-13) binding to the κ-opioid GPCR (κOR). Respectively, these represent a pathologic signaling cascade leading to the symptoms of cholera and a neuronal target for treating mood and other stress reponses40,41 To mimic a native lipid environment for studying membrane binding events, a synthetic lipid bilayer was first self-assembled by rupturing lipid vesicles onto the microtoroid’s dielectric silica surface. The GM1 glycolipid receptors were prepared together with the lipid membrane8, while κORs were incorporated by means of micelle dilution42. The binding kinetics of CTB to GM1 and Dynorphin A 1-13 to κOR, respectively, were measured without labels, in real-time (i.e., minutes) with minimal (30 μl) sample consumption and low LOD (180 zM for the Dynorphin A 1-13 assay) offering a significant advantage over other molecular assays. To confirm the specificity of the binding assay, fluorescent imaging, and radioligand competitive binding assays were performed. Our results demonstrate a platform for rapid, label-free, ultra-sensitive measurements of lipid biomembrane formation, small molecule binding, and protein binding kinetics, among other applications.
FLOWER biosensing system
Figures 1a and 1b illustrate the approach, including the microtoroid functionalized with a proteolipid membrane. The tuning range of the laser used in these studies was 765 to 781 nm, conditions where light absorption in water is minimal. Light from the laser is evanescently coupled into the toroid using a tapered optical fiber, although free-space coupling of light into the toroid can be done as well43,44. The tunable laser is frequency-locked to the microtoroid resonator so that the laser wavelength matches the microtoroid resonance. To calculate the Q-factor of a resonance, the spectral dip was fitted to a Lorentzian curve and the resonant wavelength was divided by the full-width-half-max (FWHM)39. The dip in the transmitted intensity occurs when the optical path length of the resonator is equal to an integer multiple of the circulating light’s wavelength:
where m is an integer representing the number of wavelengths per round trip, \(\lambda\) is the free-space wavelength of the laser, \(r\) is microtoroid major radius, and \({n}_{{eff}}\) is the effective refractive index (RI) of the guided mode. As protein or lipid materials land on the microtoroid surface within the evanescent light field, the effective refractive index of the optical mode increases, causing a shift in resonance frequency proportional to the number and size of analytes bound (Fig. 1c)25,30,45. A two-dimensional axisymmetric COMSOL simulation was performed to aid in visualizing the effective sensing region at the equator of the lipid coated microtoroid (Fig. 1d, e) (see “Methods” for details). By choosing RIs in our simulations comparable to experimental values for the microtoroid cavity, lipid materials, and surrounding aqueous solution, we can see that a portion of the optical mode extends past the surface and into the lipid layer and aqueous environment. FLOWER has an advantage over plasmonic sensors46 in that the toroid has a larger capture area thus enabling faster detection times29.
Results
GM1-DOPC lipid functionalization of microtoroid optical resonators
A synthetic phospholipid membrane composed of DOPC lipids doped with 2% mol GM1 receptors was used to functionalize the silica microtoroids for quantifying CTB binding affinity (Fig. 2a). DOPC is a zwitterionic lipid membrane that can be used as an antifouling membrane to resist non-specific binding of proteins to silica (Fig. S1). Unilamellar GM1-DOPC lipid vesicles were produced by extruding the GM1-DOPC lipid suspension through a 100 nm pore filter (see “Methods”) and their size was confirmed using dynamic light scattering (Fig. S3a). A GM1-DOPC lipid bilayer can be quickly formed on the silica toroid surface as the hydrophilic head of the lipid vesicles readily rupture and adsorb onto the toroid (Fig. 2b). Membrane fluidity on the toroid surface was measured by fluorescent recovery after photobleaching (FRAP) (Fig. S3b), which shows the lipid bilayer retaining 80% membrane fluidity with a diffusion coefficient, D, of 3.38 ± 0.26 μm2 s−1 which is in the range typically observed for supported lipid bilayers formed on glass47,48,49.

a Schematic model of pentameric CTB bound to a GM1-DOPC lipid membrane silica microtoroid. b Wavelength shift from GM1-DOPC lipid vesicles adsorbing onto the silica toroid. c Fluorescent images of 50 nM CTB-AF647 binding to varying % mol fraction GM1 in DOPC coated-microtoroids. d Fluorescent intensity determined from the fluorescent assay in (c). Bars and whiskers show the mean ± SD from biological repeats (for 1% (n = 21), 2% (n = 13), 4% (n = 15), and 10% (n = 5)). Statistical significance was determined using a one-way ANOVA, *p < 0.0001. e Wavelength shift as CTB binds to a GM1-DOPC functionalized toroid. f Dose–response binding curve generated based on the maximum wavelength shift from (e). Data are mean ± SEM from 3 independent experiments. g Control experiment attempting to detect an irrelevant protein (human chorionic gonadotropin, hCG) binding to the GM1-DOPC coated microtoroid.
For bare microtoroids, we generally obtained Q-factors in the range of 106−107 in aqueous buffer solution. The Q-factors of optical microresonator cavities in aqueous solutions are typically lower than in air due to the smaller difference in refractive index between the resonator material and surrounding environment (Fig. S3c). After lipid functionalization, microtoroids still maintained a high Q-factor in the range of ~106 which is suitable for high-sensitivity biosensing (Fig. S3d).
FLOWER detects CTB binding
Cholera toxin B is a subunit of the cholera toxin secreted by the bacterium Vibrio cholera and binds to GM1 glycolipid receptors presented on the surface of cells. The GM1 receptor is responsible for the binding and internalization of this virulent protein, resulting in the subsequent rapid loss of fluids from the intestine and severe diarrhea presented in cholera12,50.
CTB binding to a GM1-DOPC functionalized toroid was demonstrated in vitro by sequentially injecting increasing concentrations of unlabeled CTB prepared in buffer into a fluidic chamber containing the microtoroid and tracking the wavelength shift with our FLOWER system (Fig. 1a)24,25,28. The shape and kinetics of the WGM response was consistent, exhibiting a rapid and positive wavelength shift and peak at t ~ 120 s after the start of the sample injection, followed by a gradual decline back to the baseline for CTB concentrations <10 nM (Fig. 2e). The WGM response exhibited a net positive wavelength shift at steady state for higher CTB concentrations (>10 nM). It should be noted that CTB is being continuously injected (~1.2 μL/s) when the WGM response is being recorded in Fig. 2e (t = 0 s to t = 400 s). Between each CTB injection, buffer is injected for 10 min to rinse the fluidic chamber and bring the sensor back to a steady state (data not shown). As expected, the peak of the WGM response increased along with the CTB concentration (Fig. 2e). The shape of our WGM response is similar to the dynamic mass redistribution (DMR) response from resonant waveguide grating biosensors, which also rely on the evanescent field for label-free detection51,52,53,54.
The binding curve was generated using the peak of the WGM response for each CTB concentration and fitted using the Hill-Waud binding model (See ‘Constructing Binding Curves’ in the Supplementary Information) (Fig. 2f)55,56. The dissociation constant, Kd, of the GM1-CTB binding interaction was 1.1 nM ± 0.17 nM. For our particular system, which uses a 2% mol doping density for GM1, our Kd value of 1.1 nM is in reasonable agreement with the Kd value of 0.73 nM that was obtained via surface plasmon resonance for the same doping density57. Surprisingly, the Hill coefficient of the fit was n = 0.52, suggesting some level of negative binding cooperativity between the CTB and GM1 receptor, while previous literature reported positive cooperativity (i.e., n > 1)55,58. However, the previously reported experiments were performed using supported lipid bilayers on flat surfaces, while the curvature of the toroid geometry at the sensing region may decrease the cooperative effect between the CTB and GM1 receptor.
Determination of optimum concentration of GM1 in the lipid membrane
A high concentration of GM1 in the lipid membrane results in clustering of the receptor or other effects, and decreased CTB binding efficiency58,59. Thus, we investigated the optimum concentration of GM1 doping on the lipid coated microtoroid. A fluorescent assay was performed using multiple microtoroid substrates doped with varying GM1 doping ratios (0 to 10% mol). For each substrate, 50 nM of CTB labeled with Alexa Fluor 647 (CTB-AF647) was incubated and the binding was measured using fluorescent imaging (Fig. 2c, d). The amount of doped GM1 glycolipid in the membrane has an effect on the association of CTB molecules. We observed that at 10% mol GM1 the fluorescent signal on the toroidal microcavity was significantly decreased, whereas a maximum in the fluorescent signal was observed with 2 and 4% mol of doped GM1. This phenomenon is in agreement with existing literature58,59. Alternatively, the weakened binding of CTB to GM1 can also be explained by the electrostatic repulsion between negatively charged CTB and increasing negative charge of lipid membrane by increased GM1 ligand density (each GM1 headgroup has (−1) charge)58. The specificity of the detection was confirmed by introducing an irrelevant protein (HCG, human chorionic gonadotropin) onto a GM1-functionalized microtoroid (Fig. 2g). hCG has very low affinity for the GM1 receptor60. The injection of 100 nM hCG produced a red-shift near the baseline noise (not statistically different than buffer) which was several thousand fold lower than the CTB response.
FLOWER detects DynA 1-13–κOR interactions
κ-opioid receptors (κOR), together with μ- and δ-opioid receptors are an important mediator system for emotional and behavioral responses to stress and pain. The binding of the endogenous ligand dynorphin during stress activates κOR to produce analgesia and dysphoria61. The dysregulation of central nervous system responses to stress can elevate anxiety, which can lead to depression and drug seeking behavior61,62,63. Here, we explore the potential use of our lipid membrane coated optical microcavity for drug screening purposes, demonstrated by a κOR proteolipid functionalized microtoroid sensor for the detection of unlabeled DynA 1-13. The proteolipid membrane formation used in this study is shown in Fig. 3. The microtoroids were incubated in DOPC lipid (0.5 mg/mL) to form an artificial lipid bilayer on the silica sensing surface of the sensor. The formation of a lipid bilayer on the microtoroid was confirmed by measuring the resonant wavelength shift as DOPC lipid was injected into the fluidic chamber; at t = 1750 s buffer is injected to wash away excess lipid vesicles (Fig. 4a). HEK-293T cells were transfected with the cDNA encoding the human κOR, with expression verified by immunoblots of lysed cells and intact cell confocal imaging (Fig. S9). κOR-expressing cells were solubilized using the detergent CHAPS in a lysis buffer to form κOR micelles prior to sensor deposition (see “Methods”). The κOR micelles were incorporated into the lipid bilayer on the microtoroid by means of micelle dilution64. Successful incorporation of κOR was confirmed by measuring the resonant wavelength shift (Fig. 4b) and by fluorescence imaging (Fig. S4). Note that a relatively low concentration of κOR was used in Fig. 4b, yet a large spectral shift was produced immediately following receptor addition, suggesting a strong incorporation efficiency.

a, b Just before lipid adsorption, c, d after lipid coating process, e, f after κOR reconstitution by means of micelle dilution.

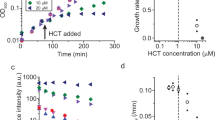
a WGM shift as lipid vesicles adsorb onto a bare silica toroid. (b) WGM shift as κOR is incorporated into the lipid bilayer on the toroid. c WGM shifts as a function of increasing concentrations of unlabeled DynA 1-13 binding to the κOR-lipid toroid sensor. d Binding curve of DynA 1-13 (solid line) and [Leu5]-Enkephalin (dashed line) to the κOR-lipid toroid. Results are mean ± SEM from 3 experiments. e DynA 1-13 competes for binding of the κOR radioligand 3H-naloxone in the membranes from κOR-transfected cells. Results are mean ± SEM from 3 experiments.
DynA 1-13 is a tridecapeptide, a major metabolite from the endogenous Dynorphin A65. The study of DynA 1-13 binding kinetics has been significantly challenging due to its inaccessibility to labeling techniques. Here, the assay was performed by injecting increasing concentrations of unlabeled DynA 1-13 into the experimental fluidic chamber containing the κOR-lipid functionalized toroid (Fig. S8). The WGM response consistently exhibited a rapid red-shift and peak (t ~ 100–200 s) after the initial injection of the κOR sample followed by a decline back to the baseline (Fig. 4c). Note that DynA was continuously injected for the entire duration of recording the WGM response (t = 0 to 400 s). Buffer was injected for 10 min between each DynA injection to rinse the chamber and bring the sensor back to a steady state. The peaks of the WGM shifts were fitted to the Hill-Waud specific binding model yielding a Kd of 3.1 ± 0.3 nM (Fig. 4d). Depending on the experimental methods, the Kd has variously been reported to range from 0.25 to 6.1 nM for DynA 1-13 at the κOR63,66,67. A fluorescent imaging assay was conducted to ascertain non-specific interactions. We conducted the fluorescent assay experiment using biotinylated DynA 1-17 and streptavidin labeled AF647. Fig. S4 shows the fluorescent imaging results of DynA 1-17 binding to the κOR reconstituted lipid coated microtoroid. In the absence of κOR in the lipid membrane, changes in the fluorescent intensity were insignificant. This data suggests that the κOR binding function to DynA 1-13 molecule is retained using the micelle dilution method, and the observed binding kinetics of κOR receptors characterized by our microtoroid sensing system is specific to the DynA 1-13 molecule. To demonstrate rank-order specificity of the κOR toroid sensor, the experiment was repeated with the low-affinity κOR agonist [Leu5]-enkephalin. [Leu5]-enkephalin is another endogenous opioid peptide which is known to have agonist action at both the μ- and δ-opioid receptors, but weakly binds to the κOR subtype (compared to DynA 1-13)63,68. The Kd of [Leu5]-enkephalin at the κOR was determined experimentally to be 947 ± 92 nM (Fig. 4d).
To confirm our DynA 1-13 affinity determination in a parallel manner, we performed radioligand binding assays using the same recombinantly expressed κOR in HEK-293T cells as was used in the microtoroid studies. Here, 3H-naloxone was used as a label for the receptor. Cell membranes were incubated with 5 nM of this radiolabel and varying concentrations of DynA 1-13, and the bound 3H-naloxone captured by filtration and washing over glass fiber filters (see “Methods”). The Kd for DynA 1-13 derived from these competition studies was 1.1 ± 0.84 nM (Fig. 4e), in good agreement with the WGM data.
In a separate experiment for determining the limit of detection (LOD), low levels of DynA 1-13 were injected while the wavelength shift was measured (Fig. 5a). For each DynA 1-13 concentration, the peak wavelength shift was taken and plotted on a calibration curve. The mean, ±1 SD, and ±3 SD values from repeated buffer (i.e., blank) injections are shown as a horizontal line and shaded areas in the calibration curve (Fig. 5b). The wavelength shift at + 3 SD (2180 fm) was used to define the LOD (Eq. S5). A wavelength shift higher than the LOD can be considered to be due to the existence of analytes at a level that can be distinguished from the blank. The DynA 1-13 concentration corresponding to the LOD wavelength shift was extracted by fitting the maximum wavelength shift vs. the concentration with a linear fit, indicated as the solid red line in Fig. 5b. The dashed red lines indicate the 95% confidence interval (CI) of the fitting. The DynA 1-13 concentration where the linear fit and confidence bands intersect the +3 SD line correspond to the LOD and 95% CI, giving the value of 180 zM [47 zM, 490 zM]. In practice, the maximum wavelength shift from one specific concentration will have deviations as indicated by the error bars in Fig. 5b. Thus, it does not necessarily mean that concentrations higher than 180 zM will always provide a response higher than the LOD. A sensor response at zeptomolar levels was also seen from big dynorphin (Fig. S10). The data near the LOD in Fig. 5b is also shown on a linear-linear scale with slope given in Fig. S11.

a Separate experiment showing zeptomolar DynA 1-13 binding to the κOR-lipid toroid. b Calibration response curve for zeptomolar DynA 1-13 binding experiment. Results are mean ± 1 SD from 3 experiments. Blue shaded areas show ±1 SD and ±3 SD from the mean of repeated buffer injections.
The limit of quantification (LOQ) is interpreted as the lowest concentration of analyte that can not only be detected, but reliably quantified. The value of the LOQ is higher than the value of the LOD and was calculated where the linear fit intersects the + 10 SD line (Eq. S6). The LOQ and 95% CI for the KOR/DynA pair was 5.5 aM [2 aM, 27 aM]. In comparison, the LOQ for opioid receptor antagonists and agonists using mass spectrometry lie in the range of pg/mL and ng/mL range, respectively69,70.
Two-drug competitive binding experiments with κOR functionalized-microtoroid
Another way to assess drug targeting to receptors is to bind a drug to the receptor and then ascertain the output effects of increasing concentrations of a second ligand (similar to the radioligand competition experiment in Fig. 4e). Using FLOWER, we considered that the wavelength shift from bound DynA 1-13 would decrease with increasing concentrations of the opiate antagonist naloxone, indicating its applicability in this type of pharmacologic assay. To test this, we varied naloxone concentrations from 100 pM to 1 μM while keeping the DynA 1-13 concentration constant (10 nM). The increasing concentrations of naloxone indeed competed with the binding of DynA 1-13 to κOR, resulting in the decreases in the peak resonant wavelength shifts as the naloxone concentration increased (Fig. 6a). A high and low IC50 was obtained by fitting the competitive response curve with a two-site competitive binding model (See ‘Constructing Binding Curves’ in the Supplementary Information) (Fig. 6b). The Ki was calculated from the IC50 using the Cheng-Prusoff equation (Eqn. S4). The low Ki was calculated to be 92 nM and the high Ki was calculated to be 0.26 nM. The basis of this 2-site fit is not altogether clear, but this has also been observed using radioligand binding with similar affinity ratios71. Although the response in Fig. 6a at 1 nM naloxone was higher than the 0.1 nM, repeated experiments resulted in higher responses from 0.1 nM than 1 nM (Fig. S12). We attribute this experimental deviation to the complexity of the competitive binding process between the small naloxone molecule and the larger DynA 1-13 molecule to the κOR, and variations in the final κOR concentration after surface functionalization.

a WGM shift showing the competitive binding of naloxone with 10 nM DynA 1-13 to κOR binding sites from a representative experiment. b Competitive binding curve of naloxone vs. 10 nM DynA 1-13 on a κOR-lipid toroid. Data shows points from repeated experiments.
Discussion
FLOWER offers ultra-sensitive, real-time, rapid characterization of biomimetic membrane formation, protein incorporation, and measurement of lipid membrane receptor–ligand binding kinetics and associated affinity measurements without the use of labels. An in vitro biomimetic membrane model can be formed on the dielectric microtoroid surface to serve as a sensitive detection interface for direct tracking of lipid fusion and membrane protein incorporation, as well as subsequent ligand binding events. Fluorescent imaging confirmed that the activity of reconstituted membrane proteins was preserved together with the respective ligand recognition ability. Without the complexity of a labeling tag, unlabeled ligands were directly detected resulting in a rapid sample-to-result diagnosis within several minutes. High affinity binding of CTB molecules to GM1 receptors, as well as the study of endogenous opioid peptide neurotransmitters such as Dynorphin A 1-13, [Leu5]-enkephalin and the opioid antagonist naloxone to the κ-opioid receptor were successfully characterized in a label-free, real-time manner. The limit of detection (zeptomolar) obtained in this study is several orders of magnitude lower than reported with other label-free technologies such as SPR, electrochemical impedance spectroscopy (EIS), and microring resonators. These technologies have a detection range usually in the nM to pM level. The present study not only demonstrates the potential of using FLOWER for drug and toxin screening research, but also provides a platform for advancing knowledge in membrane interactions in vitro, providing an approach for preliminary therapy and disease prevention.
Methods
Materials
The silicon substrate with 2 µm thermal oxide layer was purchased from University Wafer. Buffered oxide etchant 6:1 (Catalog #040889, KMG), ethylenediaminetetraacetic acid (EDTA, Catalog #E9884, Sigma-Aldrich), isopropanol (Catalog # 190764, Sigma-Aldrich), methanol (Catalog# 179337), unlabeled Cholera toxin B subunits (Catalog #C9903, Sigma-Aldrich), Dynorphin A 1-13 (Catalog #D7017, Sigma-Aldrich), and naloxone (Catalog #N7758, Sigma-Aldrich). 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, Catalog #850375C, Avanti Polar Lipids); Ganglioside GM1 from ovine brain (Catalog #860065P, Avanti Polar Lipids); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(Lissamine rhodamine B sulfonyl) (ammonium salt) (LissRhod:PE, Catalog #810150C, Avanti Polar Lipids); 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)2000-N’-carboxyfluorescein] (ammonium salt) (PEG2000-PE-CF, Catalog # 810120C, Avanti Polar Lipids); 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000-Amine, Catalog #880128C). Cholera Toxin B subunit labeled with Alexa Fluor 647 (Catalog # C34778, ThermoFisher), and Alexa Fluor™ 488 NHS Ester (Succinimidyl Ester) (Catalog # A20000, ThermoFisher). [Leu5]-enkephalin (Catalog # AS-24333, Anaspec), biotinylated Dynorphin A 1-17 (Catalog # AS-23978, Anaspec). All solutions were prepared with 18.2 MΩ∙cm water passed through a 0.2 μm polycarbonate Whatman filter and degassed to remove bubbles before use.
Microtoroid fabrication
Microtoroid structures were fabricated using photolithography, chemical etching, and thermal reflow19. In brief, fabrication of a microtoroid started with a 2 µm thick layer of SiO2 on a Si wafer (universitywafer.com). Disks with diameter 150 µm along with a 350 µm wide support wall were patterned onto the SiO2 surface with Microposit S1813 photoresist. Afterward, buffered oxide etchant (BOE) 6:1 was used to etch the exposed SiO2 down to the Si substrate, followed by resist stripping with acetone and isopropanol. After a dehydration bake at 130 °C, a XeF2 dry chemical etcher (Xactix e2, Orbotech, Yavne, Israel) was used to etch the exposed Si substrate. The microdisks and support wall were then reflowed using a CO2 laser (Synrad 48-1, WA, USA) to create SiO2 microtoroids with major diameter around 100 µm on a 50 µm Si pillar.
Lipid preparation
Lipid vesicles were prepared according to a well-established protocol42. For GM1-CTB studies, a controlled mol% of methanolic GM1 ganglioside was mixed with DOPC lipid and dried under argon gas. For κOR pharmacokinetic studies, DOPC lipid vesicles were prepared using a 100 nm membrane filter and mini extruder kit from Avanti Lipids. DOPC lipid vesicles were prepared and then stored at 1 mg/mL in PBS at 4 °C. Fluorescent doped lipid vesicles were prepared by adding 1% mol of either Liss-RhodPE or PE-PEG2000-CF into a GM1/DOPC lipid mixture (Fig. S2). The average size of the lipid vesicles was characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). Lipid vesicles were stained using 1 mg/mL uranyl acetate and air-dried overnight before TEM imaging.
Fluorescent recovery after photobleaching (FRAP) assay
To assess the mobility of the lipid membrane on the microtoroid, FRAP experiments were performed with a Leica SP5-II confocal microscope using GM1 doped DOPC labeled with AF488. A 488 nm Argon laser line at 50 mW was used for photobleaching. Images of fluorescent recovery were taken using a 20x/0.75NA objective and the data was analyzed using ImageJ.
Microtoroid biosensing setup
An optical fiber (Thorlabs SM600, Newton, NJ) was tapered using a stationary hydrogen flame and a custom fiber-pulling stage (Newport, CA, USA). Light from a tunable laser (New Focus TLB-6712, Newport, CA, USA) was coupled into the fiber and the transmission was measured using an auto-balanced photodetector (Nirvana 2007, Newport, CA, USA). After surface functionalization, the microtoroid chip was affixed into a 3D-printed fluidic chamber (internal volume ~120 μl) using double-sided tape (Fig. S8). A glass cover slip was cut to size and placed on top of the fluidic chamber to retain the fluid. Fluid samples were perfused into the chamber using a 16-channel syringe rack and electric rotary valve system (ASP-ERV-O1.2-16, Aurora Pro Scientific). The microtoroid was evanescently coupled with the tapered fiber using a 3-axis micrometer stage and a 3-axis piezo nanopositioning stage (P-611.3 NanoCube, PI, MA).
GM1-CTB bioassay
For lipid bilayer formation on the microtoroid, DOPC vesicles (0.5 mg/mL in Tris-HCl 25 mM, 100 nm in diameter) consisting of different mol % of GM1 were introduced onto the microtoroid chip. The vesicle rupture and fusion process was induced immediately by the edge of the hydrophilic silica microtoroid44,45. The lipid coating process was quantified in real-time by tracking the resonance frequency shifts, then washed thoroughly with the binding buffer (Tris-HCl 25 mM, NaCl 150 mM, EDTA 1 mM, pH 7.4). For the CTB binding assay, increasing concentrations of CTB prepared in binding buffer were sequentially introduced into the microtoroid sensing chamber. A control experiment was conducted using fluorescently labeled lipids to confirm lipid stability upon multiple cycles of buffer washing (Fig. S5).
κOR studies
For FLOWER, the lipid bilayer was formed on the microtoroid by incubating the toroid chip in a DOPC vesicle solution (0.5 mg/mL, 100 mM phosphate buffered saline) for 1 h at room temperature. Afterward, the toroid chip was washed with PBS and then transferred to Tris buffer (Tris-HCl 25 mM, NaCl 150 mM, EDTA 1 mM, BSA 0.05% w/v, pH 7.5). κOR were generated by transfecting HEK-293T cells (American Type Culture Collection, CRL-3216) with an expression vector consisting of the human κOR coding sequence with an amino-terminal peptide tag (HA) in the expression vector pcDNA3. Transfections were performed using Lipofectamine 2000, and after 2 days the cells were washed with PBS, pelleted, and frozen at −80 °C72,73,74. Non-transfected HEK-293T cells were used as a negative control. Expression in cell membranes was confirmed by immunoblotting using an HA-specific antibody (Sigma H6908, at a titer of 0.5 μg/mL) as described73. The same antibody was used for fluorescent confocal microscopy (titer 10 μg/mL) with fixed cells as described74. κOR were isolated from the HEK-293T crude membranes using a combination of shearing with a syringe and detergent solubilization64. Briefly, 100 μl of lysis buffer (CHAPS 10 mM, Tris-HCl 25 mM, NaCl 150 mM, EDTA 1 mM, AEBSF 1 mM, pH 7.5) was added to ~80 mg of cell membranes in a 1 mL centrifuge tube. κOR extraction from the membrane was aided by repeated passage of the suspension through a 23-gauge needle attached to a 1 mL syringe. The cell suspension was incubated on ice for 1 h and then centrifuged for 30 min at 15,000 × g at 4 °C. The resulting supernatant with the κOR-detergent micelles, was reconstituted into the lipid coated microtoroid by the detergent dilution method42,64. In brief, 100 μL of the KOR supernatant was added to a 1 mL centrifuge tube containing 100 μL of Tris buffer and the lipid coated toroid chip. Detergent dilution below the CHAPS CMC (8–10 mM) results in spontaneous insertion of the κOR into the lipid membrane. Successful incorporation of κOR onto the microtoroid sensor was confirmed by measuring the resonant wavelength shift and with fluorescence imaging (Figs. 3 and 4b, Fig. S4). κOR-DynA 1-13 dose–response and competitive binding experiments were conducted entirely in Tris-HCl 25 mM, NaCl 150 mM, EDTA 1 mM, BSA 0.05% (w/v), pH 7.5. For radioligand binding75, cell membranes were incubated in 12 × 75 mm glass tubes with 5 nM 3H-naloxone with varying concentrations of DynA 1-13 at 25 °C for 30 min. Receptor bound radiolabel was separated from free radiolabel by dilution and vacuum filtration over Whatman GF/C filters. Filters were counted in a liquid scintillation counter.
COMSOL multiphysics simulation
A two-dimensional axisymmetric COMSOL simulation was used to compute the interaction between the microtoroid dielectric surface (refractive index n = 1.45) and the functionalized GM1-DOPC lipid bilayer (n = 1.42, total thickness [lipid + hydration layer] = ~9 nm) in buffer solution (n = 1.3385). The circular cross-section of toroid has a major radius of 45 µm and a minor radius of 2 µm. The value of the azimuthal mode number m of Eq. (1) was set to 535 corresponding to a wavelength of 778.8 nm. The field distribution and Q-factor of the WGM were analyzed using an eigenfrequency solver at a minimal mesh size of 0.4 nm. An imaginary component (10−7) of the cavity refractive index was introduced to represent the scattering, absorption and other losses to lower the Q-factor to the actual experimental value. It is clearly seen that a portion of the optical field extends into the buffer sensing region.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon request.
References
Dupuy, A. D. & Engelman, D. M. Protein area occupancy at the center of the red blood cell membrane. PNAS 105, 2848–2852 (2008).
Oates, J. & Watts, A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol. 21, 802–807 (2011).
Fechner, P. et al. Size does matter! label-free detection of small molecule–protein interaction. Anal. Bioanal. Chem. 406, 4033–4051 (2014).
Sawasvirojwong, S., Srimanote, P., Chatsudthipong, V. & Muanprasat, C. An adult mouse model of Vibrio cholerae-induced diarrhea for studying pathogenesis and potential therapy of cholera. PLoS Negl. Trop. Dis. 7, e2293 (2013).
Spangler, B. D. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol. Mol. Biol. Rev. 56, 622–647 (1992).
Meza-Lucas, A. et al. Comparison of DOT-ELISA and standard-ELISA for detection of the Vibrio cholerae toxin in culture supernatants of bacteria isolated from human and environmental samples. Indian J. Microbiol. 56, 379–382 (2016).
Evers, D. L., He, J. & Mason, J. T. O’Leary, T. J. The liposome PCR assay is more sensitive than the Vibrio cholerae enterotoxin and Escherichia coli heat-labile enterotoxin reversed passive latex agglutination test at detecting cholera toxin in feces and water. J. Clin. Microbiol. 48, 4620–4622 (2010).
Aspinwall, C. A., Nguyen, P.-D., Wang, J. & Wang, X. Biofunctionalized Nanoshell Immobilized Microarrays and Applications Thereof. US20180327833A1, November 15, 2018. https://patents.google.com/patent/US20180327833A1/en (2018).
Felgueiras, H. P., Antunes, J. C., Martins, M. C. L. & Barbosa, M. A. In Peptides and Proteins as Biomaterials for Tissue Regeneration and Repair (eds Barbosa, M. A. & Martins, M. C. L.) 1–27 (Woodhead Publishing, 2018).
Fang, Y., Peng, J., Ferrie, A. M. & Burkhalter, R. S. Air-stable G protein-coupled receptor microarrays and ligand binding characteristics. Anal. Chem. 78, 149–155 (2006).
Puiu, M. & Bala, C. SPR and SPR imaging: recent trends in developing nanodevices for detection and real-time monitoring of biomolecular events. Sensors https://doi.org/10.3390/s16060870 (2016).
Karapetis, S. et al. Development of an electrochemical biosensor for the rapid detection of cholera toxin based on air stable lipid films with incorporated ganglioside GM1 using graphene electrodes. Electroanalysis 28, 1584–1590 (2016).
Mazaafrianto, D. N., Ishida, A., Maeki, M., Tani, H. & Tokeshi, M. Label-free electrochemical sensor for ochratoxin A using a microfabricated electrode with immobilized aptamer. ACS Omega 3, 16823–16830 (2018).
Chiriacò, M. S. et al. EIS microfluidic chips for flow immunoassay and ultrasensitive cholera toxin detection. Lab Chip 11, 658–663 (2011).
Miao, P., Tang, Y. & Yin, J. MicroRNA detection based on analyte triggered nanoparticle localization on a tetrahedral DNA Modified electrode followed by hybridization chain reaction dual amplification. Chem. Commun. 51, 15629–15632 (2015).
Lin, M. et al. Target-responsive, DNA nanostructure-based E-DNA sensor for microRNA analysis. Anal. Chem. 86, 2285–2288 (2014).
Zeng, D. et al. DNA tetrahedral nanostructure-based electrochemical miRNA biosensor for simultaneous detection of multiple miRNAs in pancreatic carcinoma. ACS Appl. Mater. Interfaces 9, 24118–24125 (2017).
Su, J. Label-free biological and chemical sensing using whispering gallery mode optical resonators: past, present, and future. Sensors 17, 540 (2017).
Su, J. Label-free single molecule detection using microtoroid optical resonators. JoVE 106, e53180 (2015).
Swaim, J. D., Knittel, J. & Bowen, W. P. Detection of nanoparticles with a frequency locked whispering gallery mode microresonator. Appl. Phys. Lett. 102, 183106 (2013).
Lu, T., Su, T.-T. J., Vahala, K. J. & Fraser, S. E. Split Frequency Sensing Methods and Systems. US8593638B2, November 26, 2013. https://patents.google.com/patent/US8593638B2/en (2013).
Li, C., Chen, L., McLeod, E. & Su, J. Dark mode plasmonic optical microcavity biochemical sensor. Photon. Res., PRJ 7, 939–947 (2019).
Chen, L., Li, C., Liu, Y., Su, J. & McLeod, E. Three-dimensional simulation of particle-induced mode splitting in large toroidal microresonators. Sensors 20, 5420 (2020).
Su, J. Label-free single exosome detection using frequency-locked microtoroid optical resonators. ACS Photonics 2, 1241–1245 (2015).
Su, J., Goldberg, A. F. & Stoltz, B. M. Label-free detection of single nanoparticles and biological molecules using microtoroid optical resonators. Light Sci. Appl. 5, e16001 (2016).
Li, C. et al. Part-per-trillion trace selective gas detection using frequency locked whispering-gallery mode microtoroids. ACS Appl. Mater. Interfaces 14, 42430–42440 (2022).
Hao, S. & Su, J. Noise-induced limits of detection in frequency locked optical microcavities. J. Lightwave Technol. 38, 6393–6401 (2020).
Ozgur, E. et al. Ultrasensitive detection of human chorionic gonadotropin using frequency locked microtoroid optical resonators. Anal. Chem. 91, 11872–11878 (2019).
Suebka, S., Nguyen, P.-D., Gin, A. & Su, J. How fast it can stick: visualizing flow delivery to microtoroid biosensors. ACS Sens 6, 2700–2708 (2021).
Baaske, M. D., Foreman, M. R. & Vollmer, F. Single-molecule nucleic acid interactions monitored on a label-free microcavity biosensor platform. Nat. Nanotechnol. 9, 933–939 (2014).
Dell’Olio, F. et al. Photonic technologies for liquid biopsies: recent advances and open research challenges. Laser Photonics Rev. 15, 2000255 (2021).
Luu, G. T. et al. An integrated approach to protein discovery and detection from complex biofluids. Mol. Cell. Proteom. https://doi.org/10.1016/j.mcpro.2023.100590 (2023).
Luchansky, M. S., Washburn, A. L., McClellan, M. S. & Bailey, R. C. Sensitive on-chip detection of a protein biomarker in human serum and plasma over an extended dynamic range using silicon photonic microring resonators and sub-micron beads. Lab Chip 11, 2042–2044 (2011).
Muehl, E. M. et al. Multiplexed silicon photonic sensor arrays enable facile characterization of coagulation protein binding to nanodiscs with variable lipid content. J. Biol. Chem. 292, 16249–16256 (2017).
Sloan, Kuhnline, Marty, C. D., Sligar, M. T. & Bailey, S. G. R. C. Interfacing lipid bilayer nanodiscs and silicon photonic sensor arrays for multiplexed protein-lipid and protein-membrane protein interaction screening. Anal. Chem. 85, 2970–2976 (2013).
Byeon, J.-Y. & Bailey, R. C. Multiplexed evaluation of capture agent binding kinetics using arrays of silicon photonic microring resonators. Analyst 136, 3430–3433 (2011).
Choi, G. & Su, J. Impact of stimulated raman scattering on darksoliton generation in a silica microresonator. J. Phys. Photonics https://doi.org/10.1088/2515-7647/aca8e1 (2022).
Choi, G., Gin, A., & Su, J. Optical frequency combs in aqueous and air environments at visible to near-IR wavelengths. Opt. Express 30, 8690–8699 (2022).
Sun, V. & Armani, A. M. Real-time detection of lipid bilayer assembly and detergent-initiated solubilization using optical cavities. Appl. Phys. Lett. https://doi.org/10.1063/1.4908270 (2015).
Fricke, N. & Dimova, R. GM1 softens POPC membranes and induces the formation of micron-sized domains. Biophys. J. 111, 1935–1945 (2016).
Benady, A., Freidin, D., Pick, C. G. & Rubovitch, V. GM1 ganglioside prevents axonal regeneration inhibition and cognitive deficits in a mouse model of traumatic brain injury. Sci. Rep. 8, 1–10 (2018).
Alves, I. D. et al. Phosphatidylethanolamine enhances rhodopsin photoactivation and transducin binding in a solid supported lipid bilayer as determined using plasmon-waveguide resonance spectroscopy. Biophys. J. 88, 198–210 (2005).
Suebka, S., McLeod, E. & Su, J. Ultra-high-Q free-space coupling to microtoroid resonators. Light Sci. Appl. 13, 75 (2024).
Chen, L., Li, C., Liu, Y.-M., Su, J. & McLeod, E. Simulating robust far-field coupling to traveling waves in large three-dimensional nanostructured high-Q microresonators. Photon. Res. 7, 967–976 (2019).
Wilson, K. A. & Vollmer, F. In Encyclopedia of Nanotechnology (ed. Bhushan, B.) 2837–2849 (Springer Netherlands, 2012).
Nguyen, P.-D., Zhang, X. & Su, J. One-step controlled synthesis of size-tunable toroidal gold particles for biochemical sensing. ACS Appl. Nano Mater. 2, 7839–7847 (2019).
Kurniawan, J., Ventrici de Souza, J. F., Dang, A. T., Liu, G. & Kuhl, T. L. Preparation and characterization of solid-supported lipid bilayers formed by Langmuir–Blodgett deposition: a tutorial. Langmuir 34, 15622–15639 (2018).
Bao, P. et al. Controlling transmembrane protein concentration and orientation in supported lipid bilayers. Chem. Commun. 53, 4250–4253 (2017).
Faysal, K. M. R., Park, J. S., Nguyen, J., Garcia, L. & Subramaniam, A. B. Lipid bilayers are long-lived on solvent cleaned plasma-oxidized poly (dimethyl) siloxane (ox-PDMS). PLOS ONE 12, e0169487 (2017).
Baldauf, K. J., Royal, J. M., Hamorsky, K. T. & Matoba, N. Cholera toxin B: one subunit with many pharmaceutical applications. Toxins 7, 974–996 (2015).
Fang, Y., Ferrie, A. M., Fontaine, N. H., Mauro, J. & Balakrishnan, J. Resonant waveguide grating biosensor for living cell sensing. Biophys. J. 91, 1925–1940 (2006).
Forster, L. & Pockes, S. Investigating the ligand agonism and antagonism at the D2long receptor by dynamic mass redistribution. Sci. Rep. 12, 9637 (2022).
Servant, N. B. et al. A dynamic mass redistribution assay for the human sweet taste receptor uncovers G-protein dependent biased ligands. Front Pharm. 13, 832529 (2022).
Morse, M., Tran, E., Sun, H., Levenson, R. & Fang, Y. Ligand-directed functional selectivity at the Mu opioid receptor revealed by label-free integrative pharmacology on-target. PLoS ONE 6, e25643 (2011).
Worstell, N. C., Krishnan, P., Weatherston, J. D. & Wu, H.-J. Binding cooperativity matters: a GM1-like ganglioside-cholera toxin B subunit binding study using a nanocube-based lipid bilayer array. PLoS ONE https://doi.org/10.1371/journal.pone.0153265 (2016).
Lin, H., Kitova, E. N. & Klassen, J. S. Measuring positive cooperativity using the direct ESI-MS assay. Cholera toxin B subunit homopentamer binding to GM1 pentasaccharide. J. Am. Soc. Mass Spectrom. 25, 104–110 (2014).
MacKenzie, C. R., Hirama, T., Lee, K. K., Altman, E. & Young, N. M. Quantitative analysis of bacterial toxin affinity and specificity for glycolipid receptors by surface plasmon resonance. J. Biol. Chem. 272, 5533–5538 (1997).
Shi, J. et al. GM1 clustering inhibits cholera toxin binding in supported phospholipid membranes. J. Am. Chem. Soc. 129, 5954–5961 (2007).
Patel, D. S. et al. Influence of ganglioside GM1 concentration on lipid clustering and membrane properties and curvature. Biophys. J. 111, 1987–1999 (2016).
Fishman, P. H., Bradley, R. M., Rebois, R. V. & Brady, R. O. The role of gangliosides in the interaction of human chorionic gonadotropin and cholera toxin with murine Leydig tumor cells. J. Biol. Chem. 259, 7983–7989 (1984).
Hang, A., Wang, Y., He, L. & Liu, J. The role of the dynorphin/κ opioid receptor system in anxiety. Acta Pharmacol. Sin. 36, 783–790 (2015).
Chavkin, C. & Koob, G. F. Dynorphin, dysphoria, and dependence: the stress of addiction. Neuropsychopharmacology 41, 373–374 (2016).
Ferré, G., Czaplicki, G., Demange, P. & Milon, A. In Vitamins and Hormones (ed. Litwack, G.) Vol. 111, 17–47 (Academic Press, 2019).
Calmet, P. et al. Real time monitoring of membrane GPCR reconstitution by plasmon waveguide resonance: on the role of lipids. Sci. Rep. 6, 36181 (2016).
Müller, S., Grundy, B. L. & Hochhaus, G. Metabolism of dynorphin A1–13 in human CSF. Neurochem Res. 21, 1213–1219 (1996).
Hansen, R. W. et al. Functional stability of the human kappa opioid receptor reconstituted in nanodiscs revealed by a time-resolved scintillation proximity assay. PLoS ONE 11, e0150658 (2016).
Bradford, C. S., Walthers, E. A., Searcy, B. T. & Moore, F. L. Cloning, heterologous expression and pharmacological characterization of a kappa opioid receptor from the brain of the rough-skinned newt, Taricha granulosa. J. Mol. Endocrinol. 34, 809–823 (2005).
Efremenko, I. & Fish, R. H. Quantum chemical and molecular docking studies of [(Η6-Cp*Rh-Tyr1)-Leu-enkephalin]2+ to G-protein-coupled μ-, ∂-, and κ-opioid receptors and comparisons to the neuropeptide [Tyr1]-Leu-enkephalin: conformations, noncovalent amino acid binding sites, binding energies, electronic factors, and receptor distortion forces. Organometallics 34, 4117–4126 (2015).
Liu, Y., Li, X., Xu, A., Nasser, A. F. & Heidbreder, C. Simultaneous determination of buprenorphine, norbuprenorphine and naloxone in human plasma by liquid chromatography/tandem mass spectrometry. J. Pharm. Biomed. Anal. 120, 142–152 (2016).
Fernández, MdelM. R. et al. Quantitative analysis of 26 opioids, cocaine, and their metabolites in human blood by ultra performance liquid chromatography–tandem mass spectrometry. Ther. Drug Monit. 35, 510 (2013).
Garzón, J., Sánchez-Blázquez, P., Gerhart, J., Loh, H. H. & Lee, N. M. Dynorphin1–13: interaction with other opiate ligand bindings in vitro. Brain Res. 302, 392–396 (1984).
Kim, D. et al. Identification and characterization of an atypical Gαs-biased β2AR agonist that fails to evoke airway smooth muscle cell tachyphylaxis. Proc. Natl Acad. Sci. USA 118, e2026668118 (2021).
Tokmakova, A. et al. Predicted structure and cell signaling of TAS2R14 reveal receptor hyper-flexibility for detecting diverse bitter tastes. iScience https://doi.org/10.1016/j.isci.2023.106422 (2023).
Woo, J.-A. A. et al. β-Arrestin2 oligomers impair the clearance of pathological tau and increase tau aggregates. Proc. Natl Acad. Sci. USA 117, 5006–5015 (2020).
Small, K. M., Forbes, S. L., Rahman, F. F., Bridges, K. M. & Liggett, S. B. A four amino acid deletion polymorphism in the third intracellular loop of the human α2C-adrenergic receptor confers impaired coupling to multiple effectors. J. Biol. Chem. 275, 23059–23064 (2000).
Acknowledgements
This work was supported in part by NIH R35GM137988 (J.S.), R21MH111109 (J.S.), R01HL155532 (S.L.), and R01HL114471 (S.L.).
Author information
Authors and Affiliations
Contributions
A.G. conducted experiments, analyzed data, and wrote the manuscript. P.-D.N. established experimental methods, analyzed data, and wrote the manuscript. J.M. wrote software for acquiring experimental data, C.L. performed the COMSOL simulation for the microtoroid field distribution, H.S. generated κOR transfected HEK-293T cells for the κOR studies, S.L. provided κOR transfected HEK-293T cells and provided feedback on results, J.S. conceived the presented work, supervised the findings of this work, and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests. J.S. owns a financial stake in Femtorays Technologies which develops label-free molecular sensors. The remaining authors declare no other competing interests.
Peer review
Peer review information
Nature Communications thanks Hans-Peter Loock, Hoang Nguyen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gin, A., Nguyen, PD., Melzer, J.E. et al. Label-free, real-time monitoring of membrane binding events at zeptomolar concentrations using frequency-locked optical microresonators. Nat Commun 15, 7445 (2024). https://doi.org/10.1038/s41467-024-51320-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51320-x
This article is cited by
-
Optical biosensors for diagnosing neurodegenerative diseases
npj Biosensing (2025)
-
Frequency locked whispering evanescent resonator (FLOWER) for biochemical sensing applications
Nature Protocols (2025)
-
Steviol rebaudiosides bind to four different sites of the human sweet taste receptor (T1R2/T1R3) complex explaining confusing experiments
Communications Chemistry (2024)
