
Abstract
Annotating active sites in enzymes is crucial for advancing multiple fields including drug discovery, disease research, enzyme engineering, and synthetic biology. Despite the development of numerous automated annotation algorithms, a significant trade-off between speed and accuracy limits their large-scale practical applications. We introduce EasIFA, an enzyme active site annotation algorithm that fuses latent enzyme representations from the Protein Language Model and 3D structural encoder, and then aligns protein-level information with the knowledge of enzymatic reactions using a multi-modal cross-attention framework. EasIFA outperforms BLASTp with a 10-fold speed increase and improved recall, precision, f1 score, and MCC by 7.57%, 13.08%, 9.68%, and 0.1012, respectively. It also surpasses empirical-rule-based algorithm and other state-of-the-art deep learning annotation method based on PSSM features, achieving a speed increase ranging from 650 to 1400 times while enhancing annotation quality. This makes EasIFA a suitable replacement for conventional tools in both industrial and academic settings. EasIFA can also effectively transfer knowledge gained from coarsely annotated enzyme databases to smaller, high-precision datasets, highlighting its ability to model sparse and high-quality databases. Additionally, EasIFA shows potential as a catalytic site monitoring tool for designing enzymes with desired functions beyond their natural distribution.
Similar content being viewed by others
Introduction
As catalysts for biochemical reactions, enzymes play a crucial role in accelerating chemical reactions both inside and outside living systems. They are essential for facilitating life-sustaining processes such as growth, metabolism, and disease prevention. The enzymatic activity is primarily determined by the three-dimensional structures of the active sites, which enable enzymes to specifically bind to certain substrates and catalyze chemical transformations. Despite the advancement in DNA sequencing technology that allows researchers to obtain a vast number of enzyme sequences from different species and sources on a daily basis, accurately annotating active sites remains a significant challenge. The UniProt database reveals that despite the identification of over forty million enzyme sequences, less than 0.7% of these sequences have high-quality annotations on their active sites1. Given the enormous annual growth rate of sequenced enzymes, it is unrealistic to annotate all of them through experimental techniques. Although reliable methods have been developed for annotating enzyme functions (e.g., predicting enzyme commission numbers)2, and considerable research has been devoted to developing algorithms for predicting protein active sites3, there remains a lack of a dependable, rapid, and robust tool for annotating enzyme active sites. This is primarily due to the inherent complexity of predicting enzyme active sites, because the tools need to precisely understand the relationship between enzymes and their specific substrates, as well as the types of reactions, and differentiate between various types of active sites, such as binding sites and catalytic sites directly involved in reactions. Furthermore, high-quality enzyme active site annotation data are scarce. These above factors pose significant challenges to conventional protein active site prediction tools. Therefore, accurate methods for predicting enzyme active sites are crucial for various scientific investigations in biology, pharmacology, and bioengineering. A proper understanding of enzymatic reactions significantly contributes to advancing drug design and discovery, clarifying disease mechanisms, and facilitating progress in enzyme engineering.
The difficulty of building a reliable annotation method may finally see substantial progress due to recent AI-led profound transformations in computational biology4,5,6,7,8, as exemplified by the introduction of protein language model (PLM) and AlphaFold2. PLMs treat amino acid sequences as an analog to natural language and employ masked language modeling methods under the framework of self-supervised training on extensive protein sequence data to derive learnable features that reflect the properties of proteins. The current state-of-the-art PLMs, exemplified by the Transformer-based ESM model6, have improved the performance of methodologies related to enzyme function prediction. For instance, Yu et al. utilized ESM in conjunction with contrastive learning strategies to develop an enzyme function classification model, CLEAN, surpassing BLASTp in achieving optimal accuracy for enzyme function classification tasks2. Furthermore, in predicting enzyme specificity for substrates, Kroll et al. developed the ESP model based on ESM, achieving the highest reported accuracy9. To fully leverage the rich information encoded in protein structural features, Zhang et al. proposed ESM-GearNet (PLMs-Structure), a feature fusion network10,11. This network utilizes ESM to extract amino acid residue node features in protein graphs, combined with the message-passing mechanisms of graph neural networks for enhanced protein feature extraction, thus leading to improved prediction accuracy in enzyme function classification tasks (prediction of EC numbers) compared to using PLMs representation models alone. The recent successes of PLMs in predicting enzyme functions and substrate specificities have sparked further exploration into the more challenging task of annotating enzyme active sites.
Existing algorithms for predicting enzyme active sites can be broadly categorized into three groups: homology and template-based methods12 (i.e., those based on empirical rules13,14), and machine learning-based approaches15,16,17,18. For a long time, homology and template-based methods have been the standard choice due to their reliable performance in identifying active residues of enzymes. A representative and robust algorithm in this category is BLASTp12, which identifies enzyme sequences annotated in databases that closely resemble the query sequence through sequence alignment, thus providing critical reference information for the identification of active sites. However, these methods require a large database that covers most sequences similar to the queried enzyme sequence for better prediction results. If the target enzyme differs significantly from those in the knowledge base, accurate prediction becomes challenging. Among methods based on empirical rules, a well-established commercial algorithm is Schrödinger’s SiteMap13,14, which predicts important catalytic sites and binding sites based on the physicochemical characteristics of protein residues and surfaces. While SiteMap offers valuable reference information for predicting the active sites of enzymes to some extent, it is not specifically tailored for the properties of enzymes. Furthermore, the empirical rules it employs significantly differ from the task of annotating enzyme catalytic sites. Therefore, its applicability for enzyme active site annotation is rather limited, and cannot be adjusted to different annotation databases.
In recent years, deep learning (DL)-based approaches have shown initial success in annotating enzyme active sites. For example, Gligorijević et al.18 proposed a graph convolutional neural network for protein function prediction based on structures. Although it was not explicitly trained on active site datasets, the model trained on enzyme function prediction datasets can use the gradient weighted Class Activation Maps (grad-CAMs)19 method to analyze the residue weights related to enzyme function. This allows for the inference of enzyme active sites. Since existing databases contain a substantial amount of data on enzyme active sites, direct modeling of enzyme active site issues could further enhance predictive performance. Subsequently, Shen et al. proposed AEGAN, a structure-based protein graph network DL method, which achieved the best prediction performance on multiple enzyme active site benchmark datasets15. However, its method of extracting enzyme features heavily relies on the computation of Positional Specific Scoring Matrix19 (PSSM), which requires substantial amount of time and computational power, greatly limiting its application in large-scale annotation tasks. Beyond these methods, Teukam et al. have explored an approach using attention mechanism-based self-supervised learning language models to directly explore the intrinsic relationship between enzyme sequences and their catalytic chemical reactions20. By analyzing the distribution of attention weights across enzyme sequences and chemical reactions, the active sites of enzymes can be unambiguously inferred. Although this scheme is creative, the pure sequence-based self-supervised learning approach struggles to capture some key structural information and overlooks the knowledge accumulated over the years, leaving room for further improvements. However, the approach taken by Teukam et al., which integrates knowledge about enzymes and the specific reactions they catalyze, is indeed worth further exploration. Given the high specificity of enzymes for their substrates, certain enzymes are often responsible for catalyzing only one or a few specific chemical transformations. Therefore, information on enzyme-catalyzed reactions can serve as additional features of enzymes in the prediction of active sites. This data can be utilized to enrich the feature set available to deep learning (DL) models.
In addressing the challenges faced by existing algorithms for annotating active sites of enzymes, this study introduces a DL-based algorithm named EasIFA for annotating enzyme active sites. The innovations of EasIFA are: (1) integrating PLMs with structure-based representation information to generate a more comprehensive description of enzyme structural information; (2) developing an atom-wise distance-aware attention mechanism-based, lightweight graph neural network, self-supervisedly pretrained on a broader dataset of organic chemical reactions, to represent the relatively limited enzyme reaction information; and (3) designing an attention-based, interpretable information interaction network that combines the representations of enzymes and their catalyzed biochemical reactions for the task of active site annotation. Through multiple computational validations, our proposed EasIFA algorithm not only outperformed all benchmark algorithms in prediction accuracy for both (1) locating active sites and (2) annotating their types, but it also demonstrated exceptional prediction speed. Compared to the mainstream BLASTp algorithm, EasIFA achieves a 10-fold increase in inference speed and an additional 7.85% improvement in recall. Furthermore, compared to graph network algorithms based on PSSM features that exhibit similar performance in catalytic site prediction tasks, the EasIFA algorithm boasts an approximate 1400-fold increase in inference speed. Thanks to the high quality and exceptional fast speed of EasIFA in annotating enzyme active sites, this study has also developed a user-friendly web server computational tool based on this algorithm, which is freely available at http://easifa.iddd.group. Furthermore, to overcome the significant differences in annotation trends and standards for the same enzyme active sites across various databases, we have also employed a transfer learning approach to attempt knowledge transfer among enzyme active site repositories with distinct annotation characteristics and tendencies. EasIFA was able to transition from training on large-scale, relatively coarse annotation data to high-quality, manually annotated datasets of enzyme catalytic site mechanisms, maintaining high levels of prediction accuracy. The transfer-trained EasIFA model is expected to work in conjunction with automatic enzyme catalytic mechanism prediction methods like EzMechanism21, enhancing the coverage of enzyme reactions catalytic mechanisms databases. This method surpasses the limitations of sequence-based BLASTp, illustrating remarkable adaptability and transfer capabilities. We also explored the potential of the EasIFA algorithm as a catalytic site monitoring tool in enzyme design, and developed data augmentation strategies to extend the knowledge of enzyme catalytic sites to a broader protein space.
Results
Problem formulation for enzyme active site prediction
In previous studies of enzyme active site identification, the prediction task was typically defined as either (1) amino acid residue token classification in a given enzyme’s amino acid sequence \({{{\mathcal{A}}}}^{E}=\{{a}_{1},\,\ldots,\, {a}_{n}\}\), or (2) graph node \({{{\mathcal{V}}}}_{i}\) classification in a given enzyme’s graph \({{{\mathcal{G}}}}^{E}=\left({{\mathcal{V}}}{{\mathcal{,\, }}}{{\mathcal{E}}}\right)\). These studies mainly emphasized the sequence or structural information of the enzyme, while the consideration of reaction information was insufficient. In this work, we aim to further improve the task of enzyme active site identification by taking into account of the inclusion of the corresponding enzymatic reactions. The input for this task not only includes the structural information of the enzyme (stored in PDB format) but also the chemical reaction sequence information (stored in reaction SMILES22 format). The structural information of the enzyme is converted into a graph representation through data processing, denoted as \({{{\mathcal{G}}}}^{E}=({\widetilde{{{\mathcal{V}}}}}^{E},\, {{{\mathcal{E}}}}^{E},\,{{{\mathcal{R}}}}^{E})\), where the \({{{\mathcal{V}}}}_{i}^{E}\) node represents the input feature of the i-th amino acid residue, incorporating its sequence information \({a}_{i}\). This can extract the amino acid sequence \({{{\mathcal{A}}}}^{E}=\{{a}_{i}\}\) from the graph structure of the enzyme, and \({{{\mathcal{E}}}}^{E}\) and \({{{\mathcal{R}}}}^{E}\) denote the sets of edges and edge relational types, respectively. The enzyme reaction SMILES is processed and converted into a graph representation, denoted as \({{{\mathcal{G}}}}^{R}=\{{{{\mathcal{G}}}}^{S},\,{{{\mathcal{G}}}}^{P}\}\) where \({{{\mathcal{G}}}}^{S}\) is the substrates graph of the enzyme reaction, denoted as \({{{\mathcal{G}}}}^{S}=({{{\mathcal{V}}}}^{S},\, {{{\mathcal{E}}}}^{S})\), and \({{{\mathcal{G}}}}^{P}\) is the products graph of the enzyme reaction, denoted as \({{{\mathcal{G}}}}^{P}=({{{\mathcal{V}}}}^{P},\, {{{\mathcal{E}}}}^{P})\).
In the context of the given enzyme structure graph \({{{\mathcal{G}}}}^{E}\) and reaction graph \({{{\mathcal{G}}}}^{R}\), the objective of the enzyme active site prediction task is to train a model \({{\mathcal{M}}}\). This model is capable of mapping the joint feature representation space \({{{\mathcal{G}}}}^{E}\times ({{{\mathcal{G}}}}^{S}\cup {{{\mathcal{G}}}}^{P})\) to a binary probability vector \({{\boldsymbol{P}}}=({p}_{1},\, {p}_{2},\,\ldots,\, {p}_{n})\), where \({p}_{i}\,\in \,[0,\,1]\) and \(n\) represents the length of the enzyme sequence. Alternatively, the model can map to a multi-class probability matrix:
where \(m\) indicates the potential functional roles (including scaffold, binding site, catalytic site, or other specific sites) for each amino acid node, \(i\) is the index of the amino acid, and \(j\) is the index for the type of functional role.
EasIFA framework
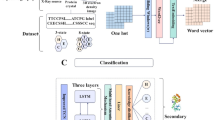
The EasIFA framework proposed is shown in Fig. 1. Given an enzyme-reaction pair as the input, we use two branches to represent the features of the enzyme and the reaction, respectively. The feature representation of the enzyme is divided into three stages, as illustrated in Supplementary Fig. S2a. In the initial stage, ESM-223 is used to convert the sequence of amino acid residues into a protein language representation. In the second stage, this protein language representation of each amino acid residue serves as the node feature in the enzyme graph \({{{\mathcal{G}}}}^{E}\), which is then input into GearNet24. Here, a message passing mechanism updates the node features of the enzyme. Subsequently, the node features of the enzyme graph are created by concatenating the protein language representation of the amino acid residues with the updated features of the enzyme graph. In the third stage, BridgeNet25 is used to map these features to the same feature size as that of the reaction information.

The black dashed circled modules represent parts of this model that by default inherit the pretrained model states. The reaction representation branch is pretrained on an organic chemical reaction dataset, with ESM-2 inheriting model state from PLM pretraining. The GearNet modules were always trained from scratch, without using pretrained model states.
The feature representation of the reaction is divided into two branches as displayed in Fig. 1, which independently represent the features of substrate molecules and product molecules. After utilizing an MPNN26 to update the features of the substrate and product molecules, a substrate-product interaction network based on the attention mechanism is employed to merge the features of the product molecules with those of the substrate molecules, resulting in a substrate molecule graph that carries product information. Following the embedding of features from both the enzyme and the reaction, the information on the substrate molecule graph is merged onto the enzyme graph via an enzyme-reaction interaction network based on the attention mechanism. It is worth noting that the enzyme-reaction interaction network based on the attention mechanism differs from the substrate-product interaction network, which uses atom-wise distance-aware global attention25,27 in its self-attention mechanism.
Once the information integration is complete, the amino acid residue activity annotation network known as Multi-layer Perceptron Residue Activity Predictor, as shown in Fig. 1, is utilized to predict the activity type of each amino acid residue. We evaluated two variations of Multi-layer Perceptron Residue Activity Predictor for accomplishing two tasks: (1) the identification of active sites and (2) the assignment of active site types.
Performance evaluation strategies and metrics
To conduct the main benchmark test, we constructed the SwissProt-ECReact enzyme-reaction active site annotation dataset (SwissProt E-RXN ASA dataset) based on the UniprotKB/Swiss-Prot28 and ECReact dataset29. Additionally, we retrieved all enzyme and reaction data from the MCSA30 database to establish the MCSA enzyme catalytic site annotation dataset (MCSA E-RXN CSA dataset) for knowledge base transfer experiments. Using enzyme sequences from Swiss-Prot, we selected experimentally validated structures (with PDB records) as the validation and test sets for enzyme structures. To ensure the independence of the training, validation, and test sets, we excluded any enzyme structures with more than 80% amino acid sequence similarity to the enzymes in the training set. After dividing the ECReact dataset into the training, validation, and test sets in an 8:1:1 ratio, we utilized the EC Number as a bridge to match enzyme catalytic reactions with enzyme structures. This matching process was conducted with a maximum match multiplier of 100, generating enzyme-reaction pairs. The final dataset numbers were training set: validation set: test set = 102944: 4711: 892. We then collected and standardized the active site annotations from UniProt, which includes three categories of enzyme active sites and the index of the active sites as the prediction labels, The three types of active sites are: binding sites, catalytic sites, and other sites. It is worth noting that in UniProt, amino acid residues that directly participate in chemical reactions are referred to as the ‘active site’, which is synonymous with the ‘catalytic site’ mentioned in this paper. In the test set, we evaluated and reported the model’s performance across different sequence identity intervals to the training set sequences (0~40%, 40~50%, 50~60%, 60~70%, and 70~80%), and compared it with benchmark methods. Additionally, in the Supplementary Section 5, we provided the evaluation results of the model’s performance based on the maximum TM-Score31 intervals with the training set (0~0.2, 0.2~0.5, and 0.5~1), and conducted comparisons with benchmark methods as well.
The MCSA E-RXN CSA dataset was also divided into the training, validation, and test sets with a maximum amino acid sequence similarity threshold of 80%, and the validation and test sets did not include any data with more than 80% similarity to the training set of the SwissProt E-RXN ASA dataset. In this dataset, all categories of active sites were classified as “catalytic site”, The MCSA E-RXN CSA dataset was then divided into the training set: validation set: test set = 781:88:82, serving as the evaluation dataset for the knowledge base transfer experiment. For more detailed dataset processing steps, please refer to the Dataset Curation subsection in the Methods section.
In the performance evaluation of algorithm, we conducted separate assessments for the model on two tasks: the active site location annotation task (a binary classification task to distinguish whether an amino acid residue is an active site) and the active site type annotation task (a multi-class classification task to predict the type of activity of amino acid residues). For the active site location annotation task, we utilized five metrics: precision, recall, false positive rate (FPR), F1 score, and Matthews correlation coefficient (MCC). For the active site type annotation task, we reported the recall for each activity category and the average MCC across multiple activity categories. Each metric was calculated individually for each validation/test sample, with the final report presenting the average value across all samples. The specific formulas used are as follows:
Active site location annotation task:
Here, \({TP}\) refers to the number of correctly predicted active sites in an enzyme sample, \({FP}\) represents the number of incorrectly predicted active sites, \({TN}\) stands for the number of correctly predicted inactive sites, and \({FN}\) is the number of incorrectly predicted inactive sites. The final reported scores are the average values across all test set samples.
Active site type annotation task:
Here, for the calculation of MCC in multi-classification, it is necessary to first define a confusion matrix C for K categories, where \({t}_{k}={\sum }_{i}^{K}{C}_{{ik}}\) represents the number of times class k truly occurred, \({p}_{k}={\sum }_{i}^{K}{C}_{{ki}}\) represents the number of times class k was predicted, \(c={\sum }_{k}^{K}{C}_{{kk}}\) represents the total number of samples correctly predicted, and \(s={\sum }_{i}^{K}{\sum }_{j}^{K}{C}_{{ij}}\) represents the total number of samples. \(T{P}_{k}\) represents the number of samples belonging to category k that are correctly predicted as category k. \(F{N}_{k}\), which represents the number of samples from category k that are misclassified into other categories. \(F{P}_{k}\) represents the number of samples that do not belong to k but are incorrectly predicted as category k. \(T{N}_{k}\), represent the number of samples that are neither actually nor predicted as category k.
Improved annotation accuracy and speed
In this study, we compared the EasIFA-ESM and EasIFA-SaProt algorithm with three other representative algorithms (as the baselines) on the SwissProt E-RXN ASA dataset, namely, BLASTp12, AEGAN15, and Schrodinger-SiteMap13,14. The primary difference between EasIFA-ESM and EasIFA-SaProt lies in the enzymatic sequence representation modules. Specifically, EasIFA-ESM utilizes ESM-2-650M as the model for representing enzyme sequences, whereas EasIFA-SaProt employs the SaProt-650M-AF model. The SaProt32 model, as an enhancement of ESM-2, integrates 3D structural encodings calculated by Foldseek33. Our evaluation encompassed not only the algorithms’ capability in labeling the active site positions of enzymes (a binary classification task for amino acid residues) but also their ability to predict the categories of enzyme active sites (a multiclass classification task for amino acid residues). The comparison results are concisely presented in Table 1. It is worth noting that: (1) The EasIFA-ESM/EasIFA-SaProt algorithm comes in two versions: EasIFA-ESM-bin/EasIFA-SaProt-bin, which solely focuses on annotating the positions of catalytic sites in amino acid residues, and EasIFA-ESM-multi/EasIFA-SaProt-multi, which not only annotates active site positions but also identifies the categories of active sites, including binding site, catalytic site and other sites; (2) Given the challenge associated with retraining AEGAN, we utilized the model state published in the original paper for testing purposes. Since a portion of our test set overlaps with their training set (approximately 25.22% of the test set), we report not only the average performance across the entire test set but also the average performance on the non-overlapping test data. To compare the computational capabilities of the algorithms on large-scale annotation tasks, we also compared the time consumption for inference by each algorithm under the same conditions, with results presented in Table 2.
The results in Table 1 demonstrate that the EasIFA-ESM/EasIFA-SaProt models outperform other baseline methods in annotation quality. In the active site location annotation task, EasIFA displays remarkable precision, recall, false positive rate (FPR), F1 score, and Matthews Correlation Coefficient (MCC). In the active site type annotation task, EasIFA-ESM/EasIFA-SaProt achieves recall comparable to the specialized AEGAN model for ‘catalytic sites’, but with a significantly lower FPR, indicating a reduced number of false positives. In the enzyme binding site identification task, the performance of EasIFA-ESM/EasIFA-SaProt is markedly superior to that of Schrodinger-SiteMap. The performance of EasIFA relative to Schrodinger-SiteMap is attributed to differences in algorithm design, Schrodinger-SiteMap’s reliance on empirical rules, and variations in the knowledge bases associated with the evaluation datasets. Moreover, when utilizing sequence alignment, BLASTp, which leverages larger databases and knowledge bases, significantly reduces FPR and notably enhances precision, f1 score, and MCC. However, it still lags significantly behind the EasIFA-ESM/EasIFA-SaProt models, with a 10.15% gap in f1 score and a 0.1012 gap in MCC. Comparing the two variants of the EasIFA model, EasIFA-ESM and EasIFA-SaProt, their performance in active site location annotation is quite similar. EasIFA-ESM-bin shows slightly higher precision, FPR, F1 score, and MCC, albeit with a marginally lower recall. In comparisons between the multi-class versions of the models, EasIFA-SaProt-multi demonstrates a slight advantage over EasIFA-ESM-multi in FPR, but EasIFA-ESM-multi slightly outperforms in other metrics. For the active site type annotation task, EasIFA-ESM-multi is slightly superior to EasIFA-SaProt-multi in all metrics except FPR.
To more clearly demonstrate the predictive capabilities of the EasIFA model and baseline methods across various sequence identity levels between the test and training samples, we used CD-HIT (version 4.8.1) to divide the test set into five subsets, each with a different level of sequence identity compared to the enzyme sequences in the training set: 0~40%, 40~50%, 50~60%, 60~70%, and 70~80%. We subsequently calculated the F1 scores, MCC, Recall, and FPR for each test subset, and the results are displayed in Fig. 2a–d. It is evident from the figures that the predictive performances of EasIFA-ESM-bin, EasIFA-SaProt-bin, and BLASTp are significantly better than those of AEGAN and Schrodinger-SiteMap across all sequence identity intervals. Moreover, the performance of these three algorithms declines as sequence identity decreases. Notably, the performance drop is more pronounced for the two variants of BLASTp, each utilizing a different sequence alignment database (see notes ① and ④ in Table 1), compared to the other algorithms. In the 0~40% sequence identity subset, the gap in F1 score and MCC between EasIFA-SaProt-bin and BLASTp widens to 15.23% and 0.1629, respectively. Except for the highest sequence identity subset of 70~80%, where the performance of the two EasIFA variants is comparable to that of BLASTp, EasIFA demonstrates a clear advantage in other sequence identity subsets. Additionally, EasIFA maintains a significantly lower false positive rate across all sequence identity intervals, consistently outperforming BLASTp and other baseline methods. The performance data based on the protein topological similarity metric TM-Score for the test set reveals consistent trends with the sequence identity-based analysis, as seen in the Supplementary Information Fig. S3.

Comparison of a F1 score, b MCC, c Recall, and d FPR between EasIFA and the baseline methods in test set subsets with different levels of sequence identity compared to enzyme sequences in the training set, and comparison of (e) average ROC and (f) average PRC of different EasIFA variants on the test set. The numbers in the square brackets in the figures a–d represent the number of test samples in that subset.
Table 2 demonstrates that the EasIFA algorithm has exceptional inference speed, averaging only 0.144 s (EasIFA-ESM-bin) to annotate active sites for one enzyme. The BLASTp algorithm also exhibits relatively fast annotation speed, which can be significantly boosted by increasing the number of CPU threads used for sequence alignment. Nevertheless, even when utilizing the same structural database of enzymes, it is still marginally slower compared to the EasIFA algorithm. Although enhancing the annotation quality with a large-scale comparison database improves its quality, it still falls significantly short of the EasIFA algorithm, and the time required is approximately 10 times that of the EasIFA algorithm. The annotation quality of AEGAN is similar to that of EasIFA for catalytic site annotation tasks, but it relies heavily on the computation of PSSM, leading to lower inference efficiency. It takes about 1300 times longer than the EasIFA algorithm. Similarly, Schrodinger-SiteMap has significantly lower computational efficiency compared to the EasIFA algorithm. These results highlight the active sites annotation quality of the EasIFA algorithm, coupled with its high inference efficiency. Moreover, although most sequences in UniProt can now be linked to 3D structures inferred by AlphaFold, we still provide the inference speed of the EasIFA pure sequence version (EasIFA-NG-bin) in Table 2. This variant requires only 0.127 s on average to complete the inference of a single sample, offering a rapid solution for annotating active sites without 3D structures. Although its performance is not as high as that of EasIFA-ESM-bin and EasIFA-SaProt-bin, it still significantly outperforms other benchmark methods, achieving an F1 score 5.42% higher and an MCC 0.0588 higher than BLASTp. In scenarios lacking enzyme 3D structures, one can either utilize the swift EasIFA-NG-bin or rapidly infer 3D structures using ESMfold2 and then apply the complete EasIFA-ESM-bin version. In such cases, the average total inference time per sample in the test set (including prediction of 3D structures and annotation of active sites) is 19.4 s. In most real-world scenarios, the 3D structures of enzymes stored in the AlphaFold Protein Structure Database are readily accessible for inference, eliminating the need for individual 3D structure prediction. Although the EasIFA-ESM-bin model was trained on AlphaFold2-inferred structures, using ESMfold2-inferred structures for active site prediction only leads to a minor decline in annotation quality (a 0.38% drop in F1 score and 0.0005 decrease in MCC compared to EasIFA-ESM-bin). Specific performance details are provided in the Ablation Study section. We also provided a comparison of the number of trainable parameters for various variants of EasIFA and AEGAN in Supplementary Table S3.
Ablation study
In this section, we conducted ablation experiments to evaluate the influence of various factors on the annotation of enzyme active sites: the inclusion or exclusion of chemical reaction information, the pretraining of chemical reaction branches, different pretrained reaction representation schemes, the utilization of 3D graph structures to represent enzymes, and different sequence representations of enzymes. The variations in model configurations for each ablation experiment are illustrated in Supplementary Fig. S6. The results are summarized in Table 3, and the ROC and PRC curves for different EasIFA variants are visualized in Fig. 2e, f. To evaluate the predictive capabilities of models without chemical reaction information, we introduced EasIFA-E-bin, a variant that excludes the reaction representation branch and the enzyme-reaction interaction network from the EasIFA algorithm. We also examined EasIFA-RS-bin, a variant trained solely on the SwissProt E-RXN ASA dataset for reaction representation. Furthermore, to explore the impact of different pretrained reaction representations, we replaced the reaction embedding branch with RXNFP34, resulting in EasIFA-RXNFP-bin. Additionally, we explored the importance of the enzyme’s 3D structure representation module, GearNet, by excluding it to create EasIFA-NG-bin. Finally, to investigate the impact of 3D structured sequence representations calculated by Foldseek, we replaced the enzyme sequence representation method with SaProt, yielding the variant EasIFA-SaProt-bin.
Our study reveals that incorporating reaction branch information significantly enhanced the predictive performance of the EasIFA model, with a 3.79% increase in F1 score, 0.0388 improvement in MCC, and 0.0375 gain in AUPRC. This indicates the value of reactions related to enzyme specificity in annotating enzyme active sites, and the effective integration of the enzyme-reaction interaction network also improves the quality of model annotations. However, the ablation experiments on the reaction branch trained from scratch (EasIFA-RS-bin) suggest that inadequate representation of reaction information could confuse the predictions of the EasIFA model, leading to a significant decline in active site annotation quality. Specifically, this configuration achieves the lowest performance among all variants, with F1 score, MCC, and AUPRC lower by 4.27%, 0.0602, and 0.0666 respectively compared to EasIFA-ESM-bin. This suggests that accurately representing reactions based solely on a limited set of enzyme reactions may be challenging. Therefore, pretraining on a broader dataset of organic reactions is crucial for enhancing the representational capabilities of the reaction branch in annotating enzyme active sites. For the EasIFA-RXNFP-bin variant, where the reaction representation was switched to RXNFP, its performance is very close to that of EasIFA-E-bin, which does not include a reaction representation branch. This suggests that the RXNFP representation neither augmented nor hindered the model’s capabilities, indicating that a pretrained graph network representation based on atom-wise distance awareness is more suitable for this prediction task compared to the purely sequence-based RXNFP representation. The EasIFA-NG-bin model, which lacks the GearNet, exhibited a decline in performance compared to EasIFA-ESM-bin, with drops of 3.32% in F1 score, 0.0333 in MCC, and 0.0326 in AUPRC. However, this reduction was compensated by a reduced computational load and faster inference speed, with an average inference time per test sample reduced by 0.017 s. For the EasIFA-SaProt-bin variant, which changed the sequence representation to SaProt, the recall improved by 1.54% compared to EasIFA-ESM-bin. While other scores were slightly lower, the differences were negligible, indicating that the 3D structured representation information from Foldseek had a minor impact on EasIFA, which already included GearNet.
Beyond examining the aforementioned EasIFA variants, we also investigated the impact of using different PDB structure sources that represent the same structure. We replaced the structures of enzymes inferred by AlphaFold2 in the test set with the structures inferred by ESMFold2. The test results, shown in Table 3, indicate a slight decrease in model performance. However, the decline is negligible, demonstrating the strong robustness of EasIFA against variations in structural data distributions.
Moreover, we compared the differences of the ROC and PRC curves between EasIFA-ESM-bin and EasIFA-E-bin for the test samples with varying levels of sequence identity to the enzymes in the training set. As shown in Fig. 3, the AUPRC gap between EasIFA-ESM-bin and EasIFA-E-bin widens as enzyme sequence identity increases. This suggests that the enzyme-reaction interaction network can improve model performance more effectively when it can assess more similar information from the reaction branch. This is due to the fact that as enzyme sequence identity increases, the reaction patterns catalyzed by the enzymes also become more similar. The attention mechanism within the enzyme-reaction interaction network can then extract this information more easily, thereby enhancing model performance. Therefore, reaction information not only aids in identifying distant relatives but also enhances performance for all samples. In fact, the higher the enzyme similarity, the more valuable the information extracted by the interaction network, thereby further improving active site annotation performance.

Comparison of (a) average ROC and (b) average PRC between EasIFA-ESM-bin and EasIFA-E-bin in test set subsets with different levels of sequence identity compared to enzyme sequences in the training set, and (c) the difference in AUPRC between the two models in test set subsets with different levels of sequence identity compared to enzyme sequences in the training set. “Enzyme Sequence Identity” is abbreviated as “E.S.I.”.
Case study
We visualized the active site annotations generated by the EasIFA algorithm on the Swiss-Prot E-RXN ASA test set, presented in Fig. 4a. The left panel of Fig. 4a depicts the model’s annotation of protein tyrosine phosphatases (UniProt ID: Q4G0W2, EC Number: 3.1.3.48), enzymes that catalyze tyrosine dephosphorylation. The EasIFA model (default as EasIFA-ESM-bin) accurately predicted the active site cysteine residue at position 103, which is recorded in the UniProt database and functions as a nucleophile that reacts with the phosphate group. The right panel illustrates the annotation results for carnosine N-methyltransferase (UniProt ID: P53934, EC Number: 2.1.1.22), a transferase enzyme that catalyzes the conversion of S-adenosyl-L-methionine and carnosine to S-adenosyl-L-homocysteine. For this enzyme, UniProt only provides information on its binding sites, which are widely distributed across the amino acid residue sequence, but our model was able to accurately identify all the substrate binding sites within the active pocket. Notably, even though the ASN274 residue is not annotated in the UniProt database, it also positioned in the active pocket and holds some relevance.

a Visualization of the enzyme active site annotation results of the EasIFA model in the test set. The left panel shows EasIFA’s annotation results for the active site of protein tyrosine phosphatases (UniProt ID: Q4G0W2, EC Number: 3.1.3.48), with the table below detailing the predicted active site amino acids information. In the ‘Correct’ column, ‘Y’ indicates that the prediction completely coincides with the recorded results in the dataset, while ‘R’ represents redundant results not recorded in the dataset (same below). The right panel displays EasIFA’s annotation results for the active site of carnosine N-methyltransferase (UniProt ID: P53934, EC Number: 2.1.1.22), with the table above showing the predicted active site amino acids information. b Visualization of the enzyme active site annotation results of the EasIFA model in the test set, which contain TIM Barrel structures but have completely different functions. The left panel shows the annotation results of the active site for Malate synthase A (UniProt ID: P08997, EC Number: 2.3.3.9), and the table below provides detailed information of the predicted active site amino acids. The right panel shows the annotation results of the active site for Ketose 3-epimerase (UniProt ID: A0A1L7NQ96, EC Number: 5.1.3.-), and the table above shows the information of the predicted active site amino acids. c Confusion matrix obtained using EasIFA on a test set of pseudoenzyme-enzyme pairs. d Visualization of a differentiated pair of kinase and pseudoenzyme using EasIFA. For the kinase (upper), serine/threonine-protein kinase BIK1 (UniProt ID: O48814, EC Number: 2.7.11.1), EasIFA accurately predicted all the active sites recorded in UniProt. For the pseudoenzyme (lower), inactive serine/threonine-protein kinase BKN1 (UniProt ID: Q9LFL7), EasIFA did not predict any catalytic active sites. The E-value between the two proteins is \(1.64\times {10}^{-58}\), calculated by BLASTp.
EasIFA can accurately identify the catalytic active residues in TIM Barrel structures with different catalytic functions. The TIM Barrel, composed of eight strands and eight helices, is one of the most abundant folds in nature and participates in numerous biological processes. We visualized all EasIFA predictions for the SwissProt E-RXN ASA test set containing TIM Barrel structures. Figure 4b shows two enzymes with significantly different functions but shared TIM Barrel structures. The active sites predicted by EasIFA are marked on the enzymes’ 3D structures, and detailed information on the active residues is displayed in the table in the middle of Fig. 4b. The left panel shows the EasIFA’s annotation for Malate synthase A (UniProt ID: P08997, EC Number: 2.3.3.9), which catalyzes malate synthesis. This enzyme has two crucial catalytic sites: ARG166 and ASP447 residues, respectively acting as the proton acceptor and proton donor in the reaction, respectively. EasIFA accurately predicted these two key catalytic residues. The right panel displays the EasIFA’s active site annotation for Ketose 3-epimerase (UniProt ID: A0A1L7NQ96, EC Number: 5.1.3.-), which catalyzes the reversible C-3 epimerization of several ketoses (the enzymatic reaction visualized here is the conversion of L-ribulose to L-xylulose). Similarly, EasIFA accurately predicted all binding sites and catalytic residues. We also provide visualized cases of the EasIFA prediction results for four other enzymes containing TIM Barrel structures in Supplementary Fig. S7. Overall, EasIFA exhibits remarkable precision in annotating the active sites of enzymes with different functions but similar 3D structures.
Furthermore, we constructed an enzyme and pseudokinase pairing dataset to assess the discriminatory ability of EasIFA against unknown pseudoenzymes. In this experiment, we used “Pseudokinase” as a keyword to search for pseudoenzyme sequences with a length of less than 600 in the SwissProt subset of the UniProt database. Next, we used BLASTp, with an E-value threshold of 0.001, to locate the closest matching enzyme for each pseudoenzyme in the SwissProt E-RXN ASA validation and test sets. Pseudoenzymes without a similar enzyme match were excluded. Through these steps, we assembled a paired dataset consisting of pseudokinases and their corresponding true enzymes. For each pair, we hypothesized that the enzyme’s catalytic reaction could be attributed to the pseudoenzyme, and used EasIFA-ESM-multi to predict active sites. The differentiation criteria were as follows: 1. Successful differentiation was achieved when no active sites were predicted for a pseudoenzyme. 2. If a binding site was predicted for a pseudoenzyme and its paired enzyme had a documented catalytic site, differentiation was also considered successful. However, if the paired enzyme did not have a recorded catalytic site, this data was discarded due to uncertainty regarding whether the predicted protein lacked a recorded catalytic site or if it was simply a pseudoenzyme. 3. If a catalytic site was predicted for a pseudoenzyme, it was considered a failure. This prediction task was treated as a binary classification problem, where pseudoenzymes were labeled as negative data and their paired enzymes as positive data. The final dataset comprised 27 valid pairs, including 27 pseudokinases and 27 enzymes. The confusion matrix for classifier on this test set is shown in Fig. 4c, revealing that EasIFA correctly identified all enzyme samples and 21 pseudoenzyme samples. Further visualization of the EasIFA-ESM-multi’s predictions for serine/threonine-protein kinase BIK1 (UniProt ID: O48814, EC Number: 2.7.11.1) and inactive serine/threonine-protein kinase BKN1 (UniProt ID: Q9LFL7) is provided in Fig. 4d. These results demonstrate that EasIFA precisely predicted all active sites of serine/threonine-protein kinase BIK1 while making no such predictions for inactive BKN1. These findings indicate that EasIFA possesses a certain ability to discriminate between unknown pseudoenzymes.
Knowledge base transfer experiment
The primary source of enzyme active site annotation data currently comes from the UniProt database. While UniProt offers comprehensiveness and extensive collection of manually annotated protein data, its annotations in the specific field of enzymes are often lacking in detail and significantly differ from specialized enzyme mechanism databases. Professionally hand-annotated enzyme datasets like MCSA30 boast high annotation quality and detailed catalytic mechanisms, but they are costly to annotate and cannot be rapidly expanded to a wider range of enzyme entities on a large scale. Furthermore, the MCSA dataset primarily comprises exemplar and iconic instances, resulting in a limited number of entries under the same EC number. This leads to low similarity among enzymes within the dataset. Consequently, modeling on such small-scale, yet high-quality datasets like MCSA poses significant challenges due to their representative but limited data coverage.
In this study, we used the EasIFA algorithm, which was initially pretrained on the SwissProt E-RXN ASA dataset, and employed transfer learning to model the MCSA E-RXN CSA dataset. Prior to transfer learning, the MCSA E-RXN CSA dataset underwent rigorous sequence identity processing with CD-HIT to guarantee that no test set samples had a sequence identity above 80% with those in the training set or the SwissProt E-RXN ASA dataset. The detailed steps used to process the MCSA E-RXN CSA dataset are described in the Experimental Setting subsection within the Methods section. In our experiments, transfer learning studies were conducted using both EasIFA-ESM-bin and EasIFA-SaProt-bin, where the model states trained on the SwissProt E-RXN ASA dataset were directly utilized on the MCSA E-RXN CSA test set. Furthermore, we compared the performance of EasIFA with the sequence alignment-based BLASTp method, and the detailed results are presented in Table 4.
The results on the MCSA E-RXN CSA dataset indicated that the EasIFA-SaProt-bin model achieved the best performance with a precision of 66.59%, recall of 65.32%, a false positive rate of 0.54%, F1 score of 61.33%, MCC of 0.6295, AUROC of 0.9511, and AUPRC of 0.6798. The EasIFA-ESM-bin model exhibited a precision of 61.09%, recall of 63.68%, a slightly higher false positive rate of 0.71%, F1 score of 58.56%, MCC of 0.5977, AUROC of 0.9584, and AUPRC of 0.6479. When the models trained on the SwissProt E-RXN ASA dataset were directly applied to the MCSA E-RXN CSA test set, EasIFA-SaProt-bin recorded a precision of 56.60%, recall of 57.63%, a false positive rate of 1.36%, F1 score of 50.55%, MCC of 0.5271, AUROC of 0.9343, and AUPRC of 0.5501. Meanwhile, EasIFA-ESM-bin recorded a precision of 53.96%, recall of 51.64%, a false positive rate of 1.36%, F1 score of 46.04%, MCC of 0.4829, AUROC of 0.9449, and AUPRC of 0.5292. We have also provided the ROC and PRC curves for EasIFA-ESM-bin and EasIFA-SaProt-bin, obtained via transfer learning and directly using the model states trained on the SwissProt E-RXN ASA dataset (Supplementary Fig. S13).
For the BLASTp method relying on sequence alignment, two evaluation strategies were employed: (1) sequence alignment and active site annotation on the MCSA E-RXN CSA dataset using the MCSA’s enzyme catalytic site data for scoring; (2) sequence alignment with the larger SwissProt database followed by annotation using the SwissProt catalytic site data and scoring based on MCSA tags. However, the first strategy yielded poor predictive quality due to low homology between the test set and the knowledge base, achieving only a precision of 19.95%, recall of 18.54%, an F1 score of 18.12%, and an MCC of 0.1838. The second strategy did not significantly enhance annotation quality, hampered by notable differences in catalytic site labels between SwissProt and MCSA, with an F1 score of only 22.99% and an MCC of 0.2394. In contrast, the EasIFA algorithm, trained on a large yet coarse dataset of enzyme activity site annotations, successfully transferred its knowledge to a high-quality, small-scale dataset.
These results clearly demonstrate that introducing 3D structure data from Foldseek significantly enhances the EasIFA’s ability to transfer knowledge across data spaces. Moreover, the transfer learning strategy implemented on the MCSA E-RXN CSA dataset, which has substantial differences in data distribution, notably improves the model’s predictive performance on the test set. In this dataset with considerable sample variability, the EasIFA algorithm shows a significant advantage over the sequence alignment-based BLASTp method, regardless of whether a transfer learning strategy is implemented.
Exploration of potential as a catalytic sites monitor for artificially designed enzymes
In nature, enzymes with the same function typically exhibit structural patterns and catalytic mechanisms. However, with significant advancements in protein design and enzyme engineering, more artificial proteins and enzymes are being created. These artificially designed enzymes may possess structural patterns that are completely different from those of naturally occurring enzymes, posing challenges in predicting their properties of these artificial enzymes. To address these challenges, reliable in silico activity validation methods could greatly enhance the success rate of enzyme design, thereby reducing experimental costs.
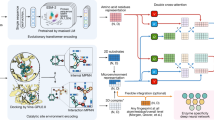
In this section, we focused on the task of scaffolding active sites in enzyme design, exploring the potential applications of the EasIFA algorithm (default as EasIFA-ESM-bin) as a catalytic site monitor. The object of scaffolding active sites task is to design and construct enzymes with minimalist yet functional active sites. However, predicting the activity sites of artificially designed enzymes poses significant challenges due to their distinct amino acid sequence distribution compared to natural enzymes. We refer to these artificially designed enzyme structures as scaffolding enzymes. We tested the performance of BLASTp and AEGAN in predicting the enzyme active sites of four categories designed by Watson et al. using RFdiffusion, categorized under EC2, EC3, EC4, and EC5. Due to the multifunctional nature of the EC1 class enzymes used in the original study, we excluded this category from our analysis. BLASTp requires alignment with similar enzyme structures to predict active sites, while AEGAN relies on computing PSSM to represent enzymes. Since the scaffolding enzymes designed by RFdiffusion differ significantly from natural enzymes with the same function, except at the active sites, both algorithms struggled to accurately predict these challenging active sites accurately. Neither method detected any of the active sites in the four designed enzyme classes. The detailed prediction results can be found in the Supplementary Table S4.
To address the aforementioned issue, we developed a workflow that enables the EasIFA algorithm to annotate catalytic sites of enzymes that fall outside the natural distribution of enzymes. We studied several enzymes, including 4-alpha-glucanotransferase (UniProt ID: O87172, EC Number: 2.4.1.25), ribonuclease alpha-sarcin (UniProt ID: P00655, EC Number M-CSA: 3.1.27.10, EC Number UniProt: 4.6.1.23), deoxyribose-phosphate aldolase (UniProt ID: P0A6L0, EC Number: 4.1.2.4), and galactose mutarotase (UniProt ID: Q96C23, EC Number: 5.1.3.3), along with their corresponding artificially constructed scaffolding enzyme structures designed by Watson et al.33 using RFdiffusion35. Figure 5a shows the structure of 4-alpha-glucanotransferase and its triad active site Asp293-Glu340-Asp395. Figure 5b displays the structural alignment of the same active site across 20 other natural enzymes with the same EC number, revealing high overlap, indicating similar structures among naturally occurring enzymes with the same function. Figure 5c presents the overlapped conformations of 7 artificially scaffolded enzymes designed for 4-alpha-glucanotransferase by RFdiffusion, aligned at the triad catalytic site, demonstrating low overlap and high diversity. Figure 5d displays the sequence logo plot of the 20 natural enzymes surrounding the triad catalytic site, revealing a relatively fixed pattern, while Fig. 5e’s sequence logo plot of the artificially scaffolded enzymes exhibits almost no fixed pattern. We also present similar analysis results for ribonuclease alpha-sarcin, deoxyribose-phosphate aldolase and galactose mutarotase in Supplementary Figs. S8–S10, all of which show huge differences between the artificially constructed enzymes and natural counterparts. Furthermore, we employed CD-HIT to cluster these artificially constructed enzymes with the MCSA E-RXN CSA dataset and SwissProt E-RXN ASA dataset using a 40% sequence identity threshold. All the artificial enzymes did not cluster with sequences from the MCSA E-RXN CSA dataset and SwissProt E-RXN ASA dataset, indicating a sequence identity of less than 40%. Additionally, sequence alignments conducted with BLASTp at an E-value threshold of 0.001 failed to find any similar sequences. This explains why BLASTp and AEGAN are unable to predict the active sites of these unique artificial enzymes. To enhance EasIFA’s prediction capabilities for artificial enzymes, we augmented the MCSA E-RXN CSA dataset with sequence and structure-based data augmentation. Please refer to the MCSA E-RXN CSA dataset augmentation subsection for detailed dataset curation methods. Following the implementation of this augmentation strategy, EasIFA proved effective in accurately identifying the catalytic sites of artificially designed scaffold enzymes, thus demonstrating its superiority over BLASTp and AEGAN in recognizing the activity of the aforementioned four types of artificial enzymes. The left panel of Fig. 6a illustrates the active sites annotated by the EasIFA algorithm on an artificially scaffolded enzyme structure. The algorithm accurately identified the key triad catalytic sites, and the additional predicted Asp93 residue, located near these sites, holds potential relevance. The right panel of Fig. 6a shows the active sites annotated by the EasIFA algorithm on the artificially constructed deoxyribose-phosphate aldolase structure, accurately identifying the ternary active site of ASP35-LYS61-LYS82. We also added the prediction results of the corresponding artificial enzymes of ribonuclease alpha-sarcin and galacto-mutarotase in Supplementary Fig. S11. Th detailed predictions of EasIFA, BLASTp and AEGAN on four types of artificial active scaffolding enzymes can be found in Supplementary Table S4. In general, EasIFA has successfully identified the active sites of these artificial enzymes through our tailored data enhancement process, which is currently challenging for comparable algorithms.

a Structure and ternary active site of the naturally occurring 4-alpha-glucanotransferase (UniProt ID: O87172, EC Number: 2.4.1.25). b Alignment results of 20 enzymes with the same catalytic site amino acid types, all having EC Number 2.4.1.25. c Alignment results of 6 enzymes designed by RFdiffusion. d Sequence logo of the catalytic site (index = 11) and surrounding 10 amino acids for 20 enzymes with the same catalytic site amino acid types and EC Number 2.4.1.25. e Sequence logo of the active site (index = 11) and surrounding 10 amino acids for seven enzymes designed by RFdiffusion.

a EasIFA’s annotation results for the active site of an in silico designed 4-alpha-glucanotransferase (left panel) and deoxyribose-phosphate aldolase (right panel) by Watson et al.53 b Visualization of attention weights in the enzyme-reaction information interaction network.
Attention weight visualization of interpretable information interaction network
The integration of an attention mechanism into the enzyme-reaction information interaction network has greatly improved the high interpretability of the EasIFA model. By analyzing a few cases in the validation set of MCSA E-RXN CSA, we identified attention layers and heads that specifically focus on critical enzyme-reaction interactions. We visualized the attention weights for enzyme active residue sites on substrate atoms (marked in blue) that exceeded a threshold (determined based on the validation set). This visualization offered strong interpretability for many test set samples. Figure 6b displays the annotation results for cysteine lyase (left) and the visualization of the active site weights on substrate molecules (right). We also visualized the entire cystine cleavage reaction, highlighting the reaction center (middle), using the RDChiral’s reaction template36). It is noteworthy that His144 exhibited a high attention weight towards the reaction center of the L-cystine zwitterion, particularly the amino group37. The alignment with the enzyme’s catalytic mechanism highlights the crucial role played by His144 in deprotonating the amino group of L-cystine zwitterion. Due to the symmetrical structure of L-cystine zwitterion, the EasIFA model focuses on both sides of the reaction center. However, it is worth noting that the model’s interaction network pays less attention to water molecules, a trend that was also observed in other samples.
EasIFA webserver
We have developed a webserver for EasIFA (accessible at http://easifa.iddd.group), which offers more than just the conventional input of enzyme structure and corresponding enzyme-catalyzed reaction equation to annotate catalytic active sites. It also features an automated workflow that retrieves enzyme structure and catalyzed chemical reaction equations from UniProt, followed by automatic annotation of the enzyme’s active sites using EasIFA. This web-based graphical interface provides users with a convenient way to obtain higher quality annotations of enzyme active sites. Supplementary Fig. S12a–d demonstrates the two modes of input and the results interface. Supplementary Fig. S12a shows the interface for inputting the reaction equation and uploading the enzyme structure. Users can either paste the reaction’s SMILES in the left panel or draw the corresponding enzyme-catalyzed reaction in the JSME molecular editor. The right panel serves as an interface for uploading and previewing the enzyme structure. After uploading, users can preview the enzyme structure. Once all inputs are complete, click the ‘Start Prediction’ button, and users will be presented with the results interface as shown in Supplementary Fig. S12b. The upper left of the interface displays the enzyme’s sequence structure, with different types of catalytic active site amino acid residues marked in different colors. The upper right features an interactive enzyme structure viewing interface, with different types of active sites marked in different colors. In the center is the catalyzed reaction equation, and the detailed information about the active amino acid residues is provided at the bottom. Supplementary Fig. S12c illustrates the prediction workflow starting from a UniProt ID. Users can enter the UniProt ID of an enzyme they are interested in, and after clicking the ‘Start Prediction’ button, EasIFA Web will automatically retrieve the enzyme’s structure data and corresponding catalytic reaction data from the database for prediction. The returned results interface, as shown in Supplementary Fig. S12d, typically presents multiple sets of results, each set organized in a drawer format, with content similar to that shown in Supplementary Fig. S12b.
EasIFA’s structure-based prediction is rapid, requiring only a few seconds to annotate an enzyme’s active site with GPU support. Predictions starting from UniProt depend on the network environment of the server where EasIFA is deployed, typically resulting in an annotated enzyme and all its catalyzed reaction combinations under a UniProt ID within one minute. With sufficient database resources and GPU computational power, EasIFA has the potential to provide reliable active site annotation information for the majority of enzyme structures.
Discussion
In this study, we developed the EasIFA algorithm, an end-to-end model for annotating enzyme active sites. Our approach incorporates several key components: (1) using the PLMs-Structure fusion method to represent enzymes; (2) introducing specific enzyme reactions as additional features through a reaction representation branch based on graph attention networks, pretrained on large-scale organic chemistry datasets; (3) integrating enzyme reaction information into enzyme representation using an explainable cross-modal interaction network based on attention mechanism. Experimental results on the SwissProt E-RXN ASA dataset demonstrate that EasIFA significantly outperforms current mainstream algorithms (i.e., BLASTp, AEGAN, and SiteMap) in annotating enzyme active sites. Furthermore, EasIFA offers rapid annotation speed, being 1300 times faster than the state-of-the-art model AEGAN and 10 times faster than BLASTp using the entire SwissProt as the knowledge base. We developed a knowledge base transfer scheme to address the disparities in enzyme catalytic site databases. This allows the model trained on large, roughly annotated databases to be transferred to smaller, meticulously annotated datasets. The EasIFA model, trained on high-quality databases like MCSA, is expected to synergize with automatic enzyme mechanism annotation methods like EzMechanism21, expanding the knowledge domain of enzyme reaction catalytic mechanism databases. Moreover, we explored EasIFA’s potential as a catalytic site monitor in challenging enzyme design tasks and developed a workflow to extend the active site knowledge learnt from natural enzymes to the broader field of artificial enzymes, which may come from a completely different distribution. The EasIFA algorithm’s enzyme-reaction information interaction network can extract mechanistic information between the enzyme and its specific reactions through an attention mechanism. The visualization highlights the most relevant reaction substrate atoms to the catalytic residues, offering high interpretability. Overall, according to our assessment, EasIFA can readily replace the standard annotation tools commonly used in the industry and academic setting. It can robustly handle large-scale enzyme active site annotation tasks under most circumstances, reducing the burden and costs for researchers, and advancing drug design, disease mechanism elucidation, and enzyme engineering.
Methods
EasIFA architecture
Sequence and structure fusion representation branch of enzyme
As illustrated in Supplementary Fig. S2a, the enzyme representation branch comprises three computational stages: the sequence embedding stage based on PLMs, the embedding stage using a GeomEtry-Aware Relational Graph Neural Network (GearNet)9,10, and the node representation linear transformation stage. In the first stage, the enzyme is treated as a one-dimensional amino acid sequence similar to natural language, and we compute the representations of amino acid residues using PLMs models. These models learn evolutionary information through self-supervised learning from billions of protein sequences. In this study, we utilize the Transformer-based ESM-2 model (ESM-2-650M). This model processes the sequence of amino acid residues through 33 self-attention layers and feed-forward neural networks to capture the dependencies between residues. The input enzyme structure is denoted as \({{{\mathcal{G}}}}^{E}=\left({{{\mathcal{V}}}}^{E},\,{{{\mathcal{E}}}}^{E},\,{{{\mathcal{R}}}}^{E}\right)\), \({{{\mathcal{V}}}}^{E}=\left\{{x}_{i}^{E}\right\}\), where \({x}_{i}^{E}\) represents the input features of the i-th amino acid residue, including its amino acid residue type \({a}_{i}\) and the amino acid sequence \({{{\mathcal{A}}}}^{E}=\{{a}_{i}\}\) that can be extracted from the graph structure of the enzyme. \({{{\mathcal{E}}}}^{E}\) and \({{{\mathcal{R}}}}^{E}\) represent edges and types of edges, respectively. We use \({x}_{i}^{\left(l\right)}\) to denote the representation of the i-th amino acid residue in the l-th layer of the ESM-2 model. Initially, the representation of this residue is \({x}_{i}^{\left(0\right)}\,={Embedding}\left({a}_{i}\right)\in \,{{\mathbb{R}}}^{d}\), where d represents the dimension of hidden features. In the l-th layer, the process involves calculating the attention coefficients \({A}_{{ij}}\) through the self-attention layer, which represent the relevance between the i-th and j-th residues. Subsequently, a feed-forward neural network is used to update the representations of the residues. Overall, the process of computing the PLMs representation \({x}_{i}\) of the i-th amino acid using ESM-2 can be described as:
After the first stage of PLMs representation, we obtain an updated enzyme representation graph \({{{\mathcal{G}}}}^{E}=({\hat{{{\mathcal{V}}}}}^{E},\, {{{\mathcal{E}}}}^{E},\,{{{\mathcal{R}}}}^{E})\), where \({\hat{{{\mathcal{V}}}}}^{E}=\{{\hat{x}}_{i}^{E}\}\).\(\,{W}_{q}^{\left(l\right)}{,\,W}_{k}^{\left(l\right)},\,{W}_{v}^{\left(l\right)}\) represent the weights of queries, keys and values, respectively.
In the second stage, we use GearNet to integrate the enzyme representations based on sequence and structure. To construct the structural graph of the enzyme, three types of directed edges are taken into account: sequential edges, radius edges, and K-NN edges, represented as:
Here, \({d}_{{seq}}=3\) defines the sequence distance, \({d}_{{radius}}=10\,\)Å defines the spatial distance, and \({knn}(i)\) represents the K-nearest neighboring residues of the i-th amino acid residue, with \(k=10\). \({Po}{s}_{i}\) is the coordinate of the i-th amino acid residue’s α carbon. It is important to note that the sequential distance is directional, and edges traveling in different directions are considered as different types of edges. Consequently, the message passing process in the l-th layer of GearNet can be represented as:
where \({{{\mathcal{N}}}}_{i}^{r}\) denotes the set of neighbor indices of node i under relation \({r}\in \,{{{\mathcal{R}}}}^{E}\). \({W}_{r}^{\left(l\right)}\) and \({W}_{0}^{\left(l\right)}\) represent the linear layers, respectively, and ReLU stands for the Rectified Linear Unit (ReLU) activation function. Following the initial stage of GearNet representation, we obtain an updated enzyme representation graph \({{{\mathcal{G}}}}^{E}=({\check{{{\mathcal{V}}}}}^{E},\, {{{\mathcal{E}}}}^{E},\,{{{\mathcal{R}}}}^{E})\), where \({\check{{{\mathcal{V}}}}}^{E}=\{{\check{x}}_{i}^{E}\}\).
The third stage involves a feed-forward network used for scaling the dimensions of amino acid attributes, which can be represented as:
where \({||}\) represents the concatenation operator. At the end of this branch, the enzyme can be represented as \({{{\mathcal{G}}}}^{E}=({\widetilde{{{\mathcal{V}}}}}^{E},\,{{{\mathcal{E}}}}^{E},\,{{{\mathcal{R}}}}^{E})\), where \({\widetilde{{{\mathcal{V}}}}}^{E}=\{{\widetilde{x}}_{i}^{E}\}\).
Atom-wise distance-aware global attention interaction representation branch of reaction
As shown in Fig. 1, the representation branch of reaction comprises two subbranches, utilizing MPNN to represent the molecular graphs of the substrates (reactants) and the products separately. The reaction can be represented as\(\,{{{\mathcal{G}}}}^{R}=\{{{{\mathcal{G}}}}^{S},\,{{{\mathcal{G}}}}^{P}\}\) where \({{{\mathcal{G}}}}^{S}=({{{\mathcal{V}}}}^{S},\, {{{\mathcal{E}}}}^{S},\, {{{\mathcal{D}}}}^{S})\) and \({{{\mathcal{G}}}}^{P}=({{{\mathcal{V}}}}^{P},\, {{{\mathcal{E}}}}^{P},\,{{{\mathcal{D}}}}^{P})\). Consequently, the computation process for the substrates or products node features after the t-th message passing can be expressed as:
where \({{{\mathcal{N}}}}_{i}\) denotes the set of neighbor indices for node \(i\). The features of all edges adjacent to node \(i\) are denoted as \(\{{x}_{{ij}}\}\). Subsequently, the node features of the substrates and the products are denoted as \(\{{x}_{i}^{S}\}\) and \(\{{x}_{i}^{P}\}\), respectively.
Afterwards, we employed a reaction information interaction network module utilizing the attention mechanism, which consists of two components: an atom-wise distance-aware self-attention network and a reaction component cross-attention network. The computation process for the substrates or products node features after \(l\) iterations of information exchange can be represented as:
where \({tgt}\) represents the molecular graph that receives the information, and \({src}\) denotes the molecular graph that sends the information. During the pretraining of reaction representation branch, substrates and products take turns acting as the recipients and senders of information. When applying the EasIFA algorithm for embedding, only the substrate molecular graph is treated as the recipient of information, with the product molecular graph acting as the sender. In the reaction component cross-attention network, we used the original node features (calculated from MPNN and not yet updated), denoted as \({x}_{j}^{{src}\left(0\right)}\).
The architecture of the atomic-wise distance-aware self-attention module is shown in Supplementary Fig. S2b. When the input atomic features are denoted as \({x}_{i}\,\), its computation process can be represented as:
where \({r}_{{ij}}\) is the relative positional embedding, which can be obtained from a look-up table according to the distance \({{{\mathcal{D}}}}_{{ij}}\) between two atoms i and j. \({{{\mathcal{D}}}}_{{ij}}\) is obtained through the following division method:
where \({{\rm{distance}}}(i,\, {j})\) refers to the shortest number of bond linking atoms i and j, which is the topological distance between these atoms within the molecular graph.
When the atomic features of the receiving molecular graph are denoted as \({h}_{i}^{{tgt}}\) and the original features of the sending molecular graph are denoted as \({h}_{j}^{{src}}\), the computational process of the substrate-product cross-attention module, as shown in Supplementary Fig. S2c, can be represented as:
In the EasIFA algorithm, reaction information is integrated into the substrate molecular graph through the reaction representation branch. At this stage, the substrate molecular graph can be represented as \({{{\mathcal{G}}}}^{S}=({\widetilde{{{\mathcal{V}}}}}^{S},\, {{{\mathcal{E}}}}^{S},\,{{{\mathcal{D}}}}^{S})\), where \({\widetilde{{{\mathcal{V}}}}}^{S}=\{{\widetilde{x}}_{i}^{S}\}\).
Attention mechanism-based enzyme-reaction information interaction network
The architecture of the enzyme-reaction information interaction network is shown in Fig. 1. Similar to the method of information interaction with substrates and products, we use an attention mechanism-based information interaction network to integrate reaction information into the graphical representation of the enzyme. This interaction network comprises the enzyme self-attention module shown in Supplementary Fig. S2d and the enzyme-substrate cross-attention module shown in Supplementary Fig. S2e. After completing the computations of the enzyme branch and the reaction branch, we obtain the enzyme’s integrated representation graph \({{{\mathcal{G}}}}^{E}=({\widetilde{{{\mathcal{V}}}}}^{E},\, {{{\mathcal{E}}}}^{E})\), where \({\widetilde{{{\mathcal{V}}}}}^{E}=\{{\widetilde{x}}_{i}^{E}\}\), and the substrate molecular graph with enzyme reaction information \({{{\mathcal{G}}}}^{S}=({\widetilde{{{\mathcal{V}}}}}^{S},\, {{{\mathcal{E}}}}^{S})\), where \({\widetilde{{{\mathcal{V}}}}}^{S}=\{{\widetilde{x}}_{i}^{S}\}\). Then, the enzyme node attributes at the l-th layer can be calculated using the following formula:
The calculation process in the self-attention module of the enzyme can be expressed as:
The calculation process in the cross-attention module of enzyme and substrate can be expressed as:
We denote the enzyme structure that has been integrated with enzyme reaction information, obtained through the computation of the enzyme-reaction information interaction network computation, as \({{{\mathcal{G}}}}^{E}=\big({\widetilde{\bar{{{\mathcal{V}}}}}}^{E},\,{{{\mathcal{E}}}}^{E},\,{{{\mathcal{R}}}}^{E}\big)\), where \({\widetilde{\bar{{{\mathcal{V}}}}}}^{E}=\{{\widetilde{\bar{x}}}_{i}^{E}\}\).
Multi-layer perceptron residue activity annotation network
The multi-layer perceptron residue activity annotation network includes both binary and multi-class versions. In the binary version, the network predicts whether the residue is an active site, whil the multi-class version predicts the type of activity of the residue. Its computation process can be represented as:
Due to the significantly higher number of negative residues compared to positive residues, we employ weighted cross-entropy to optimize the parameters of the EasIFA model. The hyperparameters of the model are provided in the Supplementary Section 3.
Experimental setting
Dataset curation
USPTO reaction dataset curation
The USPTO-STEREO reaction dataset35,37,38 is used to pretrain the atom-wise distance-aware global attention interaction representation branch of reaction. We convert the original reaction SMILES to canonical SMILES and treat both reactants and reagents as reaction substrates. For example, ‘CS(=O)(=O)Cl.OCCCBr>CCN(CC)CC.CCOCC>CS(=O)(=O)OCCCBr’ is converted to ‘CCN(CC)CC.CCOCC.CS(=O)(=O)Cl.OCCCBr»CS(=O)(=O)OCCCBr’ for pretraining input. Ultimately, the dataset for pretraining the reaction representation branch contains 791,168 training samples, 43,932 validation samples, and 43,970 test samples.
SwissProt E-RXN ASA dataset curation
We downloaded the original SwissProt data from the UniProt database, selecting entries that include EC Numbers and also include information about “Binding Site”, “Active Site”, and “Site” (other site) to retrieve their amino acid sequences and structures predicted by AlphaFold2. To conserve computational resources, we selected enzymes with sequence lengths not exceeding 600 amino acids. We standardized the activity labels for the three types of sites, recording their active site residue indices and active site types. Ultimately, we obtained data on approximately 170,000 enzyme sequences/structures and their corresponding active activity labels. For the validation and test sets, we chose data with experimentally verified structures. Then, we used CD-HIT39 to cluster enzyme sequences with a threshold of 80% to prevent data leakage by discarding the training set data belonging to the same cluster as the validation and test sets. The enzyme catalysis reaction dataset ECReact was compiled by Probst et al.29, sourced from Rhea40, BRENDA41, PathBank42, and MetaNetX43. It comprises 62,222 enzyme catalysis reaction data. We randomly divided it into the training, validation, and test sets in an 8:1:1 ratio. Then, using the EC Number as a bridge, we matched this dataset with the divided SwissProt enzyme active site dataset. The maximum match for the training and validation sets was 100 entries. After matching, we obtained the SwissProt E-RXN ASA dataset, with the training set containing 102,944 enzyme-reaction pairs, the validation set containing 4,711 pairs, and the test set containing 892 pairs. Each enzyme-reaction pair includes the enzyme’s sequence, AlphaFold2 predicted PDB (44341 PDB files), the enzyme catalyzed reaction’s SMILES, as well as the index and active site type of the enzyme’s active site amino acid residues. Following the partitioning of the test set, CD-HIT was used again to divide it into five subsets, each representing a different level of consistency with the training set sequences: 0~40%, 40~50%, 50~60%, 60~70%, and 70~80%. We evaluated the predictive capabilities of EasIFA, its variants, and baseline methods across five subsets and the entire test set. Additionally, as a supplementary analysis, we used the TM-Score metric to assess the protein topology similarity between the test set and the training set. The test set was further divided into three subsets based on the maximum TM-Score values with the training set: 0~0.2, 0.2~0.5, and 0.5~1. The performance of each predictive method was then individually evaluated within these three subsets.
MCSA E-RXN CSA dataset curation
We further processed the Mechanism and Catalytic Site Atlas dataset30 to create the MCSA E-RXN CSA dataset. We downloaded the original data using its API and extracted the amino acid indices of the enzyme’s catalytic sites. Subsequently, we converted the recorded enzyme catalysis reactions into SMILES format using their corresponding ChEBI IDs. To divide the training, validation, and test sets, we clustered all enzyme sequences along with the SwissProt E-RXN ASA dataset using CD-HIT with a threshold of 80%. After division, the validation and test sets contain enzymes that are not in the same cluster as the training set of the SwissProt E-RXN ASA dataset or the MCSA training set. The final MCSA E-RXN CSA dataset consists of 781 enzyme-reaction pairs in the training set, 88 pairs in the validation set, and 82 pairs in the test set. Each enzyme-reaction pair includes the enzyme’s sequence, AlphaFold2 predicted PDB, the enzyme catalyzed reaction’s SMILES, and the index of the enzyme’s active amino acid residues.
MCSA E-RXN CSA dataset augmentation
To expand the knowledge base of the enzyme catalytic site prediction model and adapt it to the task of enzyme design for scaffolding enzyme active sites, we performed data augmentation on the training and validation sets of the MCSA E-RXN CSA dataset5. The process of data augmentation is as follows: 1. Firstly, all protein sequences from the MCSA E-RXN CSA training and validation sets are retrieved from UniProt. Active site residues are protected, ensuring no mutations are introduced at these positions. On the remaining residues, three types of mutations are sequentially executed to generate new amino acid sequences, with a maximum length of 150 residues. Each original sample can generate 20 modified sequences, and the mutations include: a. Replacing 20% of the residues: for the chosen residues, a random amino acid is selected from the 20 natural amino acids, with equal probability, to serve as the replacement; b. Randomly inserting amino acids at intervals of 10% of the residues, with each inserted amino acid also randomly selected from one of the 20 natural amino acids; c. Random deletion at 10% of the residues. 2. Following the sequence mutations, the newly generated sequences are predicted using ESMFold2 6 to determine their PDB structures. 3. Subsequently, these PDB structures undergo hydrogen completion and energy minimization using Amber20. 4. MDAnalysis is used to calculate the RMSD between the active sites of the original and mutated structures. If the motif RMSD (active site RMSD) is less than 1.5 Å, the active site is considered effective and this structure is classified as positive data with active sites; otherwise, it is regarded as negative data without active sites. 5. After data augmentation, the imbalance in positive and negative data samples is addressed by randomly undersampling structures without active sites. As a result, we obtained a training set of 4,786 samples (1759 positive: 3027 negative) and a validation set of 592 samples (211 positive: 381 negative).
Pretraining of atom-wise distance-aware global attention interaction representation branch of reaction
The pretraining strategy for the atom-wise distance-aware global attention interaction representation branch of reaction involves a masked graph network node modeling approach similar to Grover44. A certain proportion of nodes and their adjacent edges in the substrate and product molecular graphs of chemical reactions are randomly masked, enabling the model to learn how to predict the chemical environment of those nodes on the training set. The masking ratio of nodes is set at 0.15. The accuracy of atomic environment prediction on the validation and test sets of the USPTO-STEREO dataset is 98.59% and 98.57%, respectively. Detailed schematic diagrams and descriptions are provided in the Supplementary Information Section 1.
Implementation
The EasIFA algorithm is implemented in Python 3.8 and Pytorch 1.12.145, leveraging DGL46, DGL-LifeSci47, and TorchDrug48 for constructing small molecular graphs and protein graphs, and RDKit49 for molecular data processing tasks. In the model architecture, fair-esm was utilized, and the pretrained model weights from ESM-2-650M were inherited. The web-based graphical user interface was developed using Flask. During training, we used the Adam optimizer. Additional hyperparameters related to the model architecture and training are provided in the Supplementary Section 3. The model’s performance is not sensitive to hyperparameter selection. The model was trained in parallel on two NVIDIA Tesla A100-SXM4-80GB GPUs and 64 core AMD EPYC 7742 CPUs @ 2.25 GHz based on the recall on the validation set, achieving the performance reported in this study. To fairly compare the inference speed of various algorithms, we conducted all inference tasks on the test set using a Dell OptiPlex-7090 PC (8 core Intel(R) Core (TM) i7-11700 CPUs @ 2.50 GHz, 32GB RAM) equipped with a single NVIDIA RTX3060-12GB GPU. Additionally, we utilized a single Nvidia RTX8000-48GB GPU to complete the structural predictions using ESMFold2.
Baselines
In our comparative performance analysis for enzyme active site annotation, the EasIFA algorithm was benchmarked against three distinct methodologies. The first baseline method is BLASTp (version 2.9.0+), a mainstream homology and template-based approach. For its predictions, active sites were aggregated from the dataset’s top five aligned structures using Biopython50. The second baseline method is AEGAN, notable for its utilization of graph DL techniques and its advanced performance in multiple active site annotation tasks. The third baseline method is an empirical rule-based approach provided by Schrödinger (version 2023) computational suite, SiteMap (version 49012), for predicting enzyme binding sites. In this method, amino acids within an 8 Å radius of the predicted active pockets were identified as the predicted binding sites.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The EasIFA webservice is available for free access at http://easifa.iddd.group. The inference results of EasIFA and the benchmark model, as well as the source data for Figs. 2a–f, 3a–c, Supplementary Figs. S3a–d–S5a–d, S13a, b, and S14a–d, are available at https://doi.org/10.5281/zenodo.1281967451. Source data are provided with this paper.
Code availability
All code of EasIFA and its GUI are freely available on GitHub at https://github.com/wangxr0526/EasIFA and on Zenodo at https://doi.org/10.5281/zenodo.1281944052 with an MIT license. It takes just a few steps to make a prediction with EasIFA.
References
Bateman, A. et al. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Yu, T. et al. Enzyme function prediction using contrastive learning. Science 379, 1358–1363 (2023).
Chatterjee, A. et al. Improving the generalizability of protein-ligand binding predictions with AI-Bind. Nat. Commun. 14, 1989 (2023).
Elnaggar, A. et al. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2022).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. 118, e2016239118 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Elnaggar, A. et al. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2021).
Rao, R. et al. Transformer protein language models are unsupervised structure learners. In International Conference on Learning Representations (2021).
Kroll, A., Ranjan, S., Engqvist, M. K. M. & Lercher, M. J. A general model to predict small molecule substrates of enzymes based on machine and deep learning. Nat. Commun. 14, 2787 (2023).
Zhang, Z. et al. A systematic study of joint representation learning on protein sequences and structures. Preprint at http://arxiv.org/abs/2303.06275 (2023).
Zhang, Z. et al. Enhancing protein language model with structure-based encoder and pre-training. In ICLR Workshop on Machine Learning for Drug Discovery (2023).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Halgren, T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model 49, 377–389 (2009).
Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 69, 146–148 (2007).
Shen, X. et al. A highly sensitive model based on graph neural networks for enzyme key catalytic residue prediction. J. Chem. Inf. Model. https://doi.org/10.1021/acs.jcim.3c00273 (2023).
Zhang, T. et al. Accurate sequence-based prediction of catalytic residues. Bioinformatics 24, 2329–2338 (2008).
Gutteridge, A., Bartlett, G. J. & Thornton, J. M. Using a neural network and spatial clustering to predict the location of active sites in enzymes. J. Mol. Biol. 330, 719–734 (2003).
Petrova, N. V. & Wu, C. H. Prediction of catalytic residues using Support Vector Machine with selected protein sequence and structural properties. BMC Bioinforma. 7, 312 (2006).
Jones, D. T. Protein secondary structure prediction based on position-specific scoring matrices11Edited by G. Von Heijne. J. Mol. Biol. 292, 195–202 (1999).
Gaetan, Y. et al. Language models can identify enzymatic active sites in protein sequences. ChemRxiv https://doi.org/10.26434/CHEMRXIV-2021-M20GG-V3 (2023).
Ribeiro, A. J. M., Riziotis, I. G., Tyzack, J. D., Borkakoti, N. & Thornton, J. M. EzMechanism: an automated tool to propose catalytic mechanisms of enzyme reactions. Nat. Methods 20, 1516–1522 (2023).
Weininger, D. SMILES, a chemical language and information system: 1: Introduction to methodology and encoding rules. J. Chem. Inf. Comput Sci. 28, 31–36 (1988).
Verkuil, R. et al. Language models generalize beyond natural proteins. bioRxiv 2022.12.21.521521 https://doi.org/10.1101/2022.12.21.521521. (2022)
Zhang, Z. et al. Protein representation learning by geometric structure pretraining. In International Conference on Learning Representations (2023).
Chen, S. & Jung, Y. A generalized-template-based graph neural network for accurate organic reactivity prediction. Nat. Mach. Intell. 1–9 https://doi.org/10.1038/s42256-022-00526-z (2022).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In International conference on machine learning 1263–1272 (PMLR, 2017).
Tu, Z. & Coley, C. W. Permutation invariant graph-to-sequence model for template-free retrosynthesis and reaction prediction. J. Chem. Inf. Model. 62, 3503–3513 (2022).
Boutet, E., Lieberherr, D., Tognolli, M., Schneider, M. & Bairoch, A. UniProtKB/Swiss-Prot: the manually annotated section of the UniProt KnowledgeBase. In Plant bioinformatics: methods and protocols 89–112 (Springer, 2007).
Probst, D. et al. Biocatalysed synthesis planning using data-driven learning. Nat. Commun. 13, 1–11 (2022).
Ribeiro, A. J. M. et al. Mechanism and Catalytic Site Atlas (M-CSA): a database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 46, D618–D623 (2018).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Su, J. et al. SaProt: protein language modeling with structure-aware vocabulary. In International Conference on Learning Representations (2024).
Van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01773-0 (2023).
Schwaller, P. et al. Mapping the space of chemical reactions using attention-based neural networks. Nat. Mach. Intell. 3, 144–152 (2021).
Schwaller, P., Gaudin, T., Lányi, D., Bekas, C. & Laino, T. “Found in Translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 9, 6091–6098 (2018).
Coley, C. W., Green, W. H. & Jensen, K. F. RDChiral: An RDKit wrapper for handling stereochemistry in retrosynthetic template extraction and application. J. Chem. Inf. Model 59, 2529–2537 (2019).
Schwaller, P. et al. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 5, 1572–1583 (2019).
Lowe, D. Chemical reactions from US patents (1976-Sep2016). URL https://figshare.com/articles/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873 (2017).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Alcántara, R. et al. Rhea—a manually curated resource of biochemical reactions. Nucleic Acids Res. 40, D754–D760 (2012).
Schomburg, I., Chang, A. & Schomburg, D. BRENDA, enzyme data and metabolic information. Nucleic Acids Res. 30, 47–49 (2002).
Wishart, D. S. et al. PathBank: a comprehensive pathway database for model organisms. Nucleic Acids Res. 48, D470–D478 (2020).
Ganter, M., Bernard, T., Moretti, S., Stelling, J. & Pagni, M. MetaNetX.org: a website and repository for accessing, analysing and manipulating metabolic networks. Bioinformatics 29, 815–816 (2013).
Rong, Y. et al. Self-supervised graph transformer on large-scale molecular data. Adv. Neural Inf. Process Syst. 33, 12559–12571 (2020).
Paszke, A. et al. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process Syst. 32, 8026–8037 (2019).
Wang, M. et al. Deep graph library: A graph-centric, highly-performant package for graph neural networks. In ICLR Workshop on Representation Learning on Graphs and Manifolds (2019).
Li, M. et al. Dgl-lifesci: An open-source toolkit for deep learning on graphs in life science. ACS Omega 6, 27233–27238 (2021).
Zhu, Z. et al. Torchdrug: A powerful and flexible machine learning platform for drug discovery. Preprint at https://arxiv.org/abs/2202.08320 (2022).
Landrum, G. et al. RDKit: Open-source cheminformatics. http://www.rdkit.org (2006).
Chapman, B. & Chang, J. Biopython: Python tools for computational biology. ACM Sigbio Newsl. 20, 15–19 (2000).
Xiaorui, W. et al. Multi-modal deep learning enables efficient and accurate annotation of enzymatic active sites. EasIFA and baseline results. Zenodo, https://doi.org/10.5281/zenodo.12819674 (2024).
Xiaorui, W. et al. Multi-modal deep learning enables efficient and accurate annotation of enzymatic active sites. EasIFA. Zenodo, https://doi.org/10.5281/zenodo.12819440 (2024).
Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
Acknowledgements
This work was funded by the National Key R&D Program of China (2021YFE0206400), the Macao Science and Technology Development Fund (Project no: 0056/2020/AMJ, 0114/2020/A3), and the National Natural Science Foundation of China (22220102001, 92370130, 22373085).
Author information
Authors and Affiliations
Contributions
X.W. contributed to the main ideas, coding, and writing of the manuscript. X.Yin contributed to the collection of the dataset. D.J. and H.Z. helped implement the benchmark model, Z.W., O.Z., J.W., Y.L. and Y.D. participated in discussions on the method implementation. H.L. and P.L. assisted in revising the manuscript. Y.H. participated in the project discussions. T.H. contributed to the paper revisions and provided partial computational resources. X.Yao provided computational resources and the initial concept for the paper. C.H. contributed to the essential financial support and conception, and was responsible for the overall quality.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ziheng Cui, Diego del Alamo, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Yin, X., Jiang, D. et al. Multi-modal deep learning enables efficient and accurate annotation of enzymatic active sites. Nat Commun 15, 7348 (2024). https://doi.org/10.1038/s41467-024-51511-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51511-6
This article is cited by
-
Advancing cellulose degradation through synthetic biology: engineered pathways and microbial systems for sustainable biomass conversion
Journal of Animal Science and Biotechnology (2026)
-
A virtual platform for automated hybrid organic-enzymatic synthesis planning
Nature Communications (2025)
-
Accelerated enzyme engineering by machine-learning guided cell-free expression
Nature Communications (2025)
-
De novo multi-objective generation framework for energetic materials with trading off energy and stability
npj Computational Materials (2025)
-
Large language models transform biological research: from architecture to utilization
Science China Information Sciences (2025)
