
Abstract
Peptides are valuable for therapeutic development, with multicyclic peptides showing promise in mimicking antigen-binding potency of antibodies. However, our capability to engineer multicyclic peptide scaffolds, particularly for the construction of large combinatorial libraries, is still limited. Here, we study the interplay of disulfide pairing between three biscysteine motifs, and designed a range of triscysteine motifs with unique disulfide-directing capability for regulating the oxidative folding of multicyclic peptides. We demonstrate that incorporating these motifs into random sequences allows the design of disulfide-directed multicyclic peptide (DDMP) libraries with up to four disulfide bonds, which have been applied for the successful discovery of peptide binders with nanomolar affinity to several challenging targets. This study encourages the use of more diverse disulfide-directing motifs for creating multicyclic peptide libraries and opens an avenue for discovering functional peptides in sequence and structural space beyond existing peptide scaffolds, potentially advancing the field of peptide drug discovery.
Similar content being viewed by others


Introduction
Multicyclic peptides constrained through covalent crosslinkers can maintain stable 3D structures without recourse to integrating non-covalently interactive cores, thus possessing a greater surface-to-volume ratio compared to proteins1,2,3,4,5,6. This makes multicyclic peptides an attractive molecular modality for the development of pharmacological tools and therapeutic agents7,8,9,10,11,12,13,14,15. An especially intriguing example is the naturally occurring disulfide-rich peptides (DRPs) with conserved cysteine patterns, in which the interior is largely composed of disulfide bonds16,17,18,19,20,21. Most of their non-cysteine residues can be mutated without damaging structural integrity, enabling functional diversification of these DRPs through molecular evolution to bind various biological targets22,23,24,25. However, the conservativeness of their disulfide frameworks limits the structural diversity of naturally occurring DRPs to several most ubiquitous folds, including the inhibitor cystine knot, the cystine-stabilized α/β fold, and the three-finger toxin fold26,27,28. Recently, computational methods have been developed to de novo design DRPs with stable structures and useful functions29,30,31,32. Although powerful, most of these designed DRPs rely extensively on non-covalent interactions to configure globular cores, thus exhibiting a lesser surface-to-volume ratio compared to naturally occurring ones, which in turn leads to lower effective utilization of amino acid residues while designing functional binders to targets.
In addition to naturally occurring and computationally designed DRPs, in recent years, we have developed a class of DRPs by taking advantage of disulfide-directing motifs, including CXC, CPPC, and CPXXC (C: cysteine; P: proline; X: any amino acid) to direct the oxidative folding of random sequences33,34,35,36,37,38. A large diversity of DRPs have been designed by simply altering the biscysteine motif/cysteine patterns and the number of residues separating them35,36,37,38. Owing to their high tolerability to extensive sequence manipulations, these DRPs are suitable for serving as templates for developing peptide libraries, based on which selective and high-affinity multicyclic peptide ligands to cell-surface receptors have been developed36,37,38. Thus, these studies have opened an avenue for designing and discovering multicyclic peptides in sequence and structure space inaccessible previously by naturally occurring scaffolds and computational designs. Despite this, our capability of designing multicyclic peptide scaffolds that are not only diverse in structure but also tolerant to extensive sequence manipulation is still limited. Notably, only three disulfide-directed motifs were successfully identified over the past decade. The potential for identifying additional motifs with robust capability of directing the oxidative folding of peptides remains uncertain. This limitation poses a significant obstacle to the development of more multicyclic peptide binders and therapeutics. We believe that the design or discovery of more disulfide-directing motifs holds the key to expanding the sequence and structural diversity of multicyclic peptide libraries essential for peptide ligand and drug discovery.
In this work, we studied the interplay of disulfide pairing between each disulfide-directing motif (i.e., CXC, CPPC, and CPXXC) and developed a range of tandem motifs with unique disulfide-directing effects for designing diverse DRPs (or termed as disulfide-directed multicyclic peptides: DDMPs; Fig. 1). These DDMPs contain up to four disulfide bonds, which however are highly efficient in oxidative folding with precise disulfide pairing. By taking these DDMPs as templates, peptide libraries were developed using a phage display system, with which peptide binders with nanomolar affinities to several challenging targets, including tumor antigens and immune costimulatory receptors, have been discovered. This study provides an approach to design and discover multicyclic peptides with stable 3D structures and useful functions by rationally combining different triscysteine disulfide-directing motifs, thus inspiring the discovery of other disulfide-directing motifs and more diverse combinations between them.

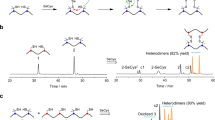
a Chromatograms showing the mixed oxidation of 1 (SCKCGW), 2 (SCPPCGW), and 3 (GCPIECKWG). Experiments were repeated three times independently (Supplementary Figs. 1–6). b Percentages of the heterodimers (orange), homodimers (blue), and monomer (green) forming from mixed oxidation of peptides 1–3. c Mixed oxidation to homodimers (up) and heterodimers (down). *denotes that parallel and antiparallel dimers are abbreviated as dimers here. d The tandem combination of disulfide-directing motifs and the design of nine tandem motifs. e Percentages of the parallel dimers (blue), antiparallel dimers (orange), and other products (green) forming from oxidation of peptides 4–12 containing a tandem motif. The cases of parallel and antiparallel dimers are shown after the column graph. Experiments were repeated three times independently (Supplementary Figs. 7–16). f The overall conception of this work from the design of triscysteine motifs to the discovery of functional DDMPs.
Results and discussion
Three peptides (1–3) bearing the CXC, CPPC, and CPXXC motif, respectively, were synthesized, and their oxidation in 30% (v/v) aqueous dimethyl sulfoxide (DMSO) were examined using high-performance liquid chromatography (HPLC) and mass spectrometry (MS). Upon mixing any two of these peptides for oxidation, both homodimers and heterodimers were observed (Fig. 1a and Supplementary Figs. 1–6). Notably, the yields of heterodimers were lower than those of homodimers, indicating that the heterodimeric pairing of disulfide bonds is less favored but permissible (Fig. 1b and c). Based on this finding, peptides bearing any two of the three disulfide-directing motifs arranged in tandem (Fig. 1d, e.g., CPPCXC and CXCPXXC; referred to as tandem motifs) were then designed and synthesized to investigate oxidative disulfide pairing (4−12; Fig. 1e). We refrained from exploring tandem motifs with two central cysteine residues (e.g., CPPCCXC) due to the presence of four cysteine residues, which would complicate disulfide pairing considerably. Consequently, we opted to design motifs in concatenated tandem linkages, strategically sharing a central cysteine residue between the two tandem motifs. As these tandem motifs contain three cysteine residues, in theory, there will be up to eleven dimeric isomers with different disulfide connectivities that can form after complete oxidation. However, in all cases, we observed the formation of both parallel and antiparallel dimers as major products (disulfide connectivity: [I–I’, II–II’, III–III’] and [I–III’, II–II’, III–I’]; Fig. 1e and Supplementary Figs. 7–15), highlighting the high propensity of disulfide pairing between each biscysteine motif. Moreover, the formation of parallel dimers is significantly more favorable than that of antiparallel dimers, facilitated by the homogenous pairing between each biscysteine motif (i.e., CXC-to-CXC, CPPC-to-CPPC, and CPXXC-to-CPXXC). The CPPC motif, known for its strong propensity to pair parallelly35,37, contributed to high yields of parallel dimers for CPPC-bearing tandem motifs, including CPPCXC (4; 100%), CXCPPC (5; 83%), CPPCPXXC (6; 94%), and CPXXCPPC (7; 100%). In contrast, the CXCPXXC (8) and CPXXCXC (9) motifs are particular for their capability of forming other disulfide pairing products and the relatively high yield of the antiparallel CPXXCXC dimer (9; 23%). Considering that the CPXXC motif has a relatively high propensity to form antiparallel disulfide pairing36, the oxidation of a mixture of 8 and 9 was further examined (Fig. 1e and Supplementary Fig. 16), showing the generation of heterodimers with a yield of 32%, which is notably higher compared to the formation of homodimers. In addition, the predominant product among these heterodimers is the antiparallel dimer, and the formation of other disulfide pairing products is significantly suppressed. We also examined tandem motifs featuring two identical biscysteine motifs (peptides 10−12). All of these peptides were oxidized to produce parallel dimers in relatively high yields (Fig. 1e), though the formation of antiparallel dimers was also observed for 10 and 11. These results clearly show that rational combinations of previously identified biscysteine motifs in tandem can generate triscysteine motifs with distinct and unique capabilities for directing disulfide pairing. The findings from these studies serve as the foundation for the subsequent development of multicyclic peptide scaffolds and libraries (Fig. 1f).
We further investigated the disulfide-directing effect of the tandem motifs when they were incorporated into peptides in pairs (Fig. 2). Initially, three peptides were synthesized, each containing two CPPCXC or CXCPPC motifs separated by a 9-mer flexible peptide segment (13−15). Their oxidations in buffers containing oxidized glutathione (GSSG) were then analyzed using HPLC and MS (Fig. 2a and Supplementary Figs. 17–19). All three peptides exhibited efficient oxidation, yielding both parallel and antiparallel inter-motif pairings as major products (i.e., disulfide connectivity: [I–IV, II–V, III–VI] and [I–VI, II–V, III–IV]), which were characterized through trypsin digestion analysis and standard product comparisons as described previously (Supplementary Figs. 17–19)35,36. In brief, the peptides were digested by trypsin under mildly acidic conditions (pH 6.0) to avoid disulfide shuffling, and subsequently, the digested fragments were monitored by HPLC and MS. To characterize the digested fragments containing two disulfide bonds, we need to synthesize standard products of these fragments using an orthogonal protecting group strategy. Interestingly, while peptide 14, bearing two CXCPPC motifs (or 15, bearing a CPPCXC and a CXCPPC motif), produced both folds in comparable yields (30−40%), peptide 13, bearing two CPPCXC motifs predominantly formed the parallelly-paired fold (yield: 83%). We further found that the yield can be increased to 95% when the central peptide segment was extended to 15 residues (16; Fig. 2b and Supplementary Fig. 20). These results align with previous findings that the backbone loop-tension resulting from central peptide segments can facilitate the antiparallel inter-motif pairing35, providing an additional route for regulating oxidative folding of peptides. Similar results were observed from peptides with other tandem motifs that have a strong propensity of pairing parallelly, such as the CPPCPPC and CXCXC motif (peptides 17 and 18; Supplementary Figs. 21, 22). Furthermore, we synthesized and examined peptides containing two CPXXCXC or CXCPXXC motifs (19−23). While the oxidation of 19, featuring two CPXXCXC motifs, led to the formation of at least six different folds with variable yields (Supplementary Fig. 23), other peptides with either one or two CXCPXXC motifs could be efficiently oxidized to the two expectedly paired folds (i.e., the parallel and antiparallel inter-motif pairings; Fig. 2a, b and Supplementary Figs. 24–27). Notably, peptide 20, bearing two CXCPXXC motifs, and 21, bearing a CXCPXXC and a CPXXCXC motif, could both be oxidized to a third inter-motif paired fold (i.e., disulfide connectivity: [I–V, II–VI, III–IV]) with yields comparable to the parallelly paired fold. However, peptides 22 and 23, bearing a CPXXCXC and a CXCPXXC motif, only produced the two expected folds without other major products (Supplementary Figs. 26, 27), mirroring the results obtained from the oxidation of a mixture of peptides 8 and 9 containing a CXCPXXC and a CPXXCXC motif, respectively. To examine the disulfide-directing capability of these triscysteine motifs on peptides featuring random amino acid sequences, peptides 24−26 were designed and synthesized. These peptides were strategically designed to incorporate a random sequence (–DISLFDE–) using peptides 13, 15, and 22 as templates. All three peptides demonstrated remarkable efficiency in folding into the expected disulfide connectivities (Fig. 2b and Supplementary Figs. 28–30). Although not all possible combinations of tandem motifs were explored herein, these results collectively demonstrate the unique disulfide-directing effect of these triscysteine motifs in regulating the oxidative folding of peptides.

a Topological drawing shows major products obtained from the oxidation of peptides directed by different pairs of tandem motifs and chromatograms showing the oxidation of these peptides (13, 15, and 20). b Yields of different disulfide pairing products obtained from the oxidation of peptides 13−26 directed by different pairs of tandem motifs. nAA* denotes the number (n) of amino acid (AA) residues in central sequences. n.d. denotes not detected in HPLC. c The sequence (left, up) and major oxidative product drawing (left, down) of peptides 27–29 directed by two CPPCXC motifs. Chromatograms (right) showing the oxidation of peptides 27–29.
As described above, peptides with a pair of tandem motifs can spontaneously fold into multicyclic structures constrained through three inter-motif disulfide bonds. We further examined if the directed peptide folding by tandem motifs can still be retained when an additional pair of cysteine residues is introduced into peptides. This addition allows for the design of multicyclic peptides with more constrained and complex structures, which could serve as valuable templates for the de novo creation of multicyclic peptide libraries. However, it has long been a formidable challenge to direct the oxidative folding of peptides with up to four disulfide bonds due to the potential formation of a large number of isomers with varying disulfide connectivities. To illustrate this, we take a peptide template with two CPPCXC motifs separated by a 15-mer segment as an example. In this template, we introduced two cysteine residues at several different positions, resulting in the design and synthesis of peptides 27−29 (Fig. 2c). Remarkably, all three peptides can be oxidized to a single major product with negligible formation of other isomers (yields: 92%, 83% and 93%, respectively; Fig. 2c). In this major product, the two CPPCXC motifs are parallelly paired, and the two additional isolated cysteines formed an additional disulfide bond (Supplementary Figs. 31–33). Therefore, the oxidative folding of peptides directed by tandem motifs proved to be highly efficient, which makes these DDMPs with up to four disulfide bonds useful scaffolds for functional peptide designs and the development of peptide libraries. Given that the arrangement of CPPCXC motifs and other cysteine residues in peptides can be rationally manipulated, and considering the availability of other tandem motifs, numerous DDMPs with diverse motif/cysteine frameworks can be conveniently designed and synthesized in the future, which would expand the structural diversity of DDMPs.
To expedite the discovery of functional peptides with the ability to bind proteins, we extended our efforts to develop peptide libraries using phage display technology36,39,40,41. Peptides 27−29 serve as templates, wherein all residues except prolines and cysteines were randomly encoded using NNK codons (ran-CPPCXC, library size: ~1.0 × 109; Fig. 3a and Supplementary Table 2). To demonstrate the library is qualified for the discovery of peptide binders, we employed Kelch-like ECH-associated protein 1 (Keap1) as a model protein target for screening37,39. Following three rounds of biopanning and enrichment of phages (Supplementary Table 3), we identified eight sequences containing an ETGE motif, known to mediate the interaction of nuclear factor erythroid 2-related factor 2 (Nrf2) with Keap142, from a pool of 28 phage clones. It is noteworthy that the two most abundantly enriched sequences do not contain the ETGE motif (Fig. 3a and Supplementary Table 4), suggesting that the binding site of Keap1 for these peptides is different from that for Nrt2. To further validate this observation, we synthesized a sequence found most abundantly (K1) and a sequence containing the ETGE motif (K2), and subjected them to oxidation in redox buffers. The oxidation of K1 and K2 resulted in the formation of a major DDMP each, denoted as K1-Ox (93%) and K2-Ox (87%), respectively (Fig. 3b and Supplementary Figs. 34, 35). K2-Ox can bind to Keap1 with a nanomolar affinity (Fig. 3c, Ki = 62 nM), as determined by a reported fluorescence polarization (FP) competition assay39. However, the affinity of K1-Ox cannot be determined under the same condition, where an ETGE-containing fluorescent peptide served as the reporter. Then, we synthesized fluorescein-labeled versions of K1-Ox and K2-Ox as reporters (i.e., F-K1-Ox and F-K2-Ox, respectively). The oxidative folding yields of F-K1-Ox and F-K2-Ox are still high (Supplementary Figs. 36, 37), indicating the fluorescence labeling had minimal impact on the oxidation of these DDMPs. The dissociation constant (KD) of F-K1-Ox was determined to be 58 nM by fluorescence polarization (Fig. 3d), which exhibited a relatively higher affinity compared to the KD of F-K2-Ox (176 nM). Furthermore, we assessed the binding of K1-Ox and K2-Ox to Keap1 in competition assays with F-K1-Ox and F-K2-Ox as reporters, respectively. K1-Ox and K2-Ox exhibited submicromolar Ki values when competing against their respective fluorescent counterparts, but mutual competitions between the two pairs were not observed (Fig. 3e and f). These findings strongly suggest that the binding sites of the two DDMPs with Keap1 are distinct. Thus, our approach, which combines peptide design and multicyclic peptide library construction guided by CPPCXC motifs, not only validates known protein-binding sites but also enables the rapid discovery of peptide binders with the ability to bind both familiar and previously unknown binding sites of proteins. This significantly broadens the scope for exploring protein–peptide interactions and the development of functional peptides. We will further demonstrate the robust applicability of our multicyclic peptide libraries by selecting peptide binders to some other extracellular protein receptors of broad therapeutic interest.

a Diagram showing phage surface-displayed peptides directed by tandem motifs (a pair of CPPCXC) and selected sequences obtained from biopanning of the library (ran-CPPCXC, Supplementary Table 2) against Keap1. b Chromatograms showing the oxidation of K1 and K2 obtained from CPPCXC-directed library (ran-CPXXC, Supplementary Table 2). c Binding of K1-Ox and K2-Ox to Keap1 was determined by competition with F-ETGE (FITC-[β-A]-DEETGEF-OH) in a FP assay. Data are presented as mean ± s.d. from three independent experiments. d Binding of F-K1/2-Ox to Keap1 recorded using a fluorescence polarization assay. Data are presented as mean ± s.d. from three independent experiments. e and f Binding of K1-Ox and K2-Ox to Keap1 determined by competition with F-K1/2-Ox in an FP assay. Data are presented as mean ± s.d. from three independent experiments. g Chromatograms showing the oxidation of EphA2-binding E1 obtained from CPPCXC-directed library (ran-CPXXC, Supplementary Table 2). h Design of a secondary library (sec-EphA2, Supplementary Table 2) against EphA2 to generate DDMPs with improved affinity. i Chromatograms showing the oxidation of E5 obtained from sec-EphA2. j Yields and dissociation constants of EphA2-binding DDMPs. SPR sensorgrams and other information on E1-Ox to E5-Ox are shown in Supplementary Figs. 38–42.
Overexpression of ephrin type-A receptor 2 (EphA2) has been associated with metastasis in solid tumors, highlighting the potential of targeting this receptor for cancer therapy43. This has spurred the development of drugs, including small molecule compounds, antibodies, and peptides44. Recently, we have successfully created EphA2-binding DDMPs with low micromolar affinities and few isomers through our CPXXC-directed phage library36. Herein we identified an additional DDMP, designated as E1-Ox, from the CPPCXC-directed phage library (Supplementary Tables 5, 6). Through HPLC analysis, we confirmed that E1-Ox, containing four disulfide bonds, can be obtained with an impressive nearly 100% yield by the direct oxidation of E1 in redox buffers (Fig. 3g and Supplementary Fig. 38). However, E1-Ox only exhibited a low micromolar affinity to EphA2 (KD = 4.9 μM; Fig. 3j and Supplementary Fig. 38) as determined by surface plasmon resonance (SPR) sensorgram. To improve the binding affinity, we divided the E1 sequence into three segments and constructed a secondary library (sec-EphA2), which was subsequently screened against EphA2 (Fig. 3h and Supplementary Table 2). The results revealed that sequences randomized at the N-terminus were significantly more enriched than the other two types of sequence randomizations (Supplementary Table 7). Following this discovery, we synthesized four peptides and evaluated their oxidation and binding to EphA2. Notably, three of these peptides (E2, E4, and E5) underwent oxidation to produce a singer product, as confirmed by HPLC analysis (Fig. 3i, Supplementary Figs. 39, 41 and 42). In contrast, 76% of E3 was successfully oxidized to E3-Ox without yielding other major products (Supplementary Fig. 40). Then, these oxidized products (E2/3/4/5-Ox) were purified and assessed for their affinity to EphA2 through SPR (Supplementary Figs. 39–42). While the dissociation constants of E2/3/4-Ox closely resembled that of E1-Ox, E5-Ox exhibited a notable improvement, binding to EphA2 with a KD of 492 nM, indicating an approximately 10-fold increase compared to E1-Ox (Fig. 3j). Our study, moving forward, will focus on conducting multiple rounds of affinity maturation experiments aimed at further enhancing the affinity of our CPPCXC-directed DDMPs.
HER3, a member of the epidermal growth factor receptor family, holds promise as a therapeutic target in various cancer types45. Notably, HER3 tends to form heterodimers with HER2, rendering HER2-targeted therapy ineffective when HER3 is overexpressed46. As a result, there is a growing interest in the development of drugs that target HER3, potentially serving as an effective strategy to overcome resistance encountered in HER2-targeted treatments47. By applying the CPPCXC-directed phage library to HER3, we achieved significant enrichment of phages following three rounds of biopanning. From the next-generation sequencing (NGS) data for the enriched phages (Supplementary Table 3), we selected the top two sequences for the synthesis of DDMPs, which were named H1-Ox and H2-Ox (Fig. 4a and Supplementary Tables 8, 9). The reducing peptide H1 successfully underwent oxidative folding into a single product (H1-Ox) with an impressive 91% yield (Fig. 4b), which can bind to HER3 with a KD value of 1.1 μM (Supplementary Fig. 43). However, it is worth noting that the yield of the reducing H1 during solid-phase peptide synthesis (SPPS) was very low due to the difficulty in coupling V10 after R11. This issue, which was unique to this sequence, could potentially affect the further application of H1-Ox. Despite a relatively lower yield for the oxidative folding of H2-Ox (61%), this peptide can bind to HER3 with a KD value of 1.8 μM that is comparable to that of H1-Ox (Fig. 4b and Supplementary Fig. 44). Subsequently, we conducted two rounds of affinity maturation experiments using H2 as a template (Fig. 4a).

a The flowchart of libraries designing and biopanning to generate HER3-binding DDMPs with improved affinity. b Chromatograms showing the oxidation of H1 and H2 obtained from CPPCXC-directed library (ran-CPXXC, Supplementary Table 2). c Solution structures of the lowest-energy NMR structures of H2’-Ox (PDB ID: 8WW1) and 4-Ox (PDB ID: 8WVS). d Chromatograms (top) showing the oxidation of H3 obtained from a secondary library (sec-HER3, Supplementary Table 2) and SPR sensorgrams (down) showing the interaction of H3-Ox with HER3. RU response unit. e Selected sequences obtained from biopanning of soft-randomization library (trd-HER3, Supplementary Table 2). Highlighting of soft-randomization difference with template H3. f Chromatograms (top) showing the oxidation of H4 obtained from trd-HER3 against HER3 and SPR sensorgrams (down) showing the interaction of H4-Ox with HER3.
At first, A DDMP similar to H2-Ox, named H2’-Ox, was synthesized and purified for NMR structural analysis. Solution structures were then determined using 1H, 1H distance constraints derived from 2D NOESY experiments (Supplementary Fig. 59 and Supplementary Table 15). H2’-Ox has disulfide connectivity of [I–VI, II–VII, III–VIII, IV–V], as identified by abundant cross-peaks between Cys2-Hβ and Cys25-Hβ, Cys7-Hβ, and Cys30-Hα, as well as Cys11-Hβ and Cys17-Hβ, indicating the parallel orientation pairing of the two CPPCXC motifs as anticipated (Fig. 4c). The disulfide pairing in H2’-Ox was further confirmed using chymotrypsin digestion and HPLC analysis (Supplementary Fig. 59). This parallel arrangement resulted in the formation of a knot structure, which exhibited certain similarities but remained distinct from the structure of the homodimer of peptide 4 determined by solution NMR (Supplementary Fig. 58). This observation implies that the dimeric CPPCXC motifs offer both rigidity and adjustability in shaping the overall DDMP structures. Notably, the peptide ring between the two parallel motifs exhibited some degree of twisting in space, creating a roller-coaster-shaped cysteine pattern. Moreover, we observed the secondary α-helical structure (from P15 to R23) and a hydrophobic core (L12, L18, and V21) in H2’-Ox, providing valuable insights for designing secondary libraries for further affinity maturation.
Then, we constructed a phage library that incorporated randomizations, either within or flanking the α-helical segment, while preserving the cysteine pattern and the hydrophobic core (sec-HER3, Fig. 4a and Supplementary Table 2). After four rounds of biopanning (Supplementary Table 3), we selected and sequenced 20 phage clones, of which 17 shared the same amino acid sequence (Supplementary Table 10). This peptide was then synthesized and oxidated into H3-Ox in a redox buffer (Fig. 4d and Supplementary Fig. 45). SPR sensorgrams revealed that H3-Ox exhibited a nanomolar affinity to HER3 (KD = 344 nM; Fig. 4d), representing a ~ 5-fold improvement compared to H2-Ox. Following this success, we further constructed a soft-randomization phage library (trd-HER3) and applied it to HER3 biopanning (Supplementary Tables 2, 3). 20 enriched-phage clones were sequenced, yielding 14 unique amino acid sequences, each with 3–7 mutant amino acids (Fig. 4e and Supplementary Table 11). Four of the most enriched sequences were synthesized, folded, and purified, and their affinity to HER3 was assessed using SPR. Among these, the most enriched peptides H4 can be folded into H4-Ox with a yield of 82%. H4-Ox can bind to HER3 with a KD value of 56 nM (Fig. 4f and Supplementary Fig. 46), representing a remarkable ~30-fold of increase compared to H2-Ox. Other peptides (H5/6/7-Ox) selected from the soft-randomization library also possessed higher affinity than their precursors, with KD values ranging from 170 to 990 nM (Supplementary Fig. 47-49). Currently, several HER3-targeted antibodies have been explored, including those that bind to HER3-EDC (seribantumab and patritumab), trap inactive HER3 (elgemtumab), and improve ADCC (lumretuzumab and TrasGex)48. Additionally, bispecific antibody zenocutuzumab is under investigation for its ability to inhibit HER2 and HER3 dimerization. However, most of these antibodies are no longer in clinical development for various reasons. This represents an opportunity for the development of HER3-targeted peptide-based therapeutics. H4-Ox, with the highest affinity among the CPPCXC-bearing DDMPs and peptides we have reported previously36, holds great potential as a hit compound for further developments and applications.
The efficient oxidative folding of DDMPs binding to various proteins can be attributed to the powerful parallel pairing ability of the CPPCXC motifs. We then sought to select functional multicyclic peptides from DDMP libraries directed by other tandem motifs. A 7-Cys DDMP library containing the CPXXCXC and CXCPXXC motifs and an isolated cysteine (i.e., ran-CPXXCXC, library size: ~5.5 × 108) was constructed (Fig. 5a and Supplementary Table 2). When an additional cysteine residue appears in the randomized sequences, these peptides can oxidize and form four pairs of disulfide bonds. This implies that this phage library contains 22 different eight-cysteine patterns, which are evenly distributed according to NGS results. Then, an inducible T cell costimulator (ICOS) was chosen as the target49, and we conducted four rounds of biopanning. The peptide I1 was identified from Sanger sequencing of 30 enriched-phage clones and was subsequently synthesized (Supplementary Tables 12, 13). Interestingly, I1 can be oxidized into three major products (Supplementary Fig. 50). The highest-yield isomers, named I1-Ox1, was purified and its binding ability was assessed by SPR, yielding a KD value of 1.8 μM (Fig. 5b). It seems that identifying peptide binders for ICOS is quite challenging, as selections using the ran-CPPCXC library and mixed libraries previously reported in our lab35,36,37,38 did not yield obvious phage enrichment (Supplementary Table 3), suggesting the lack of efficient peptide binders in these libraries. This finding highlights the need to expand the sequence and structure diversity in libraries by incorporating different disulfide-directing motifs for library construction.

a Diagram showing phage-surface-displayed peptides directed by tandem motifs (CPXXCXC and CXCPXXC). b Chromatograms (top) showing the oxidation of I1 obtained from a 7-Cys DDMP library (ran-CPXXCXC, Supplementary Table 2) and SPR sensorgrams (down) showing the interaction of I1-Ox with ICOS. c The flowchart of library design and biopanning to generate ICOS-binding DDMPs with improved affinity. d Chromatograms showing the oxidation of I2 obtained from a secondary library (sec-ICOS, Supplementary Table 2) and SPR sensorgrams (down) showing the interaction of I2-Ox with ICOS. e Solution structures of the lowest-energy NMR structures of I2’-Ox (PDB ID: 8WW0). f Confocal fluorescence images of F-I2-Ox and F-H4-Ox bound on the cell surfaces. Green fluorescence was from FITC-labeled peptides, and magenta fluorescence was from the mCherry-fusion protein. Experiments were repeated three times independently and showed similar results. Scale bars are 10 μm.
Using the ICOS binder I1-Ox1 as a template, we further designed a convergent secondary library (sec-ICOS, Fig. 5c), similar to the design of the secondary library for EphA2 affinity maturation. The experimental group exhibited substantial phage enrichment after two rounds of biopanning, with the majority of sequences originating the N-terminus randomizations, as indicated by NGS results. Then, we synthesized two of the most abundantly enriched sequences (I2 and I3; Supplementary Table 14), both of which can efficiently fold into major products. While a small number of isomers can still form during the folding process, the yield of the desired product is notably high (Supplementary Figs. 51 and 52). This is particularly noteworthy when considering that theoretically, there could be 105 isomers with varying disulfide connectivities generated for peptides containing up to four disulfides. This result underscores the crucial role of the disulfide-directing triscysteine motifs in directing the oxidative folding of peptides toward the intended products. The main oxidation product of I3, I3-Ox2, exhibited binding to ICOS with a KD value of 127 nM, as recorded by SPR, while the binding of another isomer was negligible (Supplementary Fig. 52). What is even more interesting is the remarkable ability of I2 to fold accurately, directed very likely by the cooperation of motifs and primary amino acid sequences. The single major product, I2-Ox, exhibited a 22-fold higher affinity (KD = 58 nM; Fig. 5d and Supplementary Fig. 51) than its precursor, I1-Ox. We further characterized the structures of I2’-Ox using NMR. The 3D NMR structures revealed that the CPXXCXC and CXCPXXC motifs are paired together to form antiparallel dimeric loops (Fig. 5e and Supplementary Fig. 60), which aligns with the major product formed during the oxidation of peptides 8 and 9. The structures also revealed a unique β-sheet fold stabilized by the antiparallel pairing of CPXXC motifs and the precise integration of two additional disulfide bonds within the β-sheet framework. To evaluate the impact of the three inter-motif disulfide bonds on the binding affinity of the peptide to ICOS and its oxidative folding efficiency, we further designed and synthesized peptide I2-CXC and I2-CPXXC, in which one of the cysteine residues in the tandem motifs was substituted with serine, thus transforming the tandem motif into CXC and CPXXC motifs, respectively. We found that the oxidative folding of I2-CXC led to the formation of two major products, indicating a somewhat reduced folding directionality when compared to its precursor peptide I2, though the ICOS-binding affinity of the oxidized peptides was only marginally affected (Supplementary Fig. 53). In addition, though the folding of I2-CPXXC is as efficient as that of the precursor peptide, the folding product lost its binding ability to ICOS (Supplementary Fig. 54). These results underscore the crucial role of the tandem motif in both the structure and function of DDMPs. Given the pivotal role of ICOS in the T- and B-cell co-signaling pathway, it has emerged as a promising target for immunotherapy49. Notably, the remarkable high affinity of I2-Ox to ICOS, in sharp contrast to the natural ICOS ligand (ICOSL; KD: 722 nM)50, opens up intriguing opportunities for harnessing I2-Ox in the development of tailored ICOS binding agents or modulators aimed at precisely targeting and regulating the ICOS-related signaling pathway.
Finally, we examined the binding of DDMPs with target proteins expressed on the cell surface using confocal fluorescence imaging. HEK293T cells transfected with a plasmid expressing the extracellular domain of ICOS (or HER3) fused with a mCherry tag were constructed for the experiments. Fluorescein-labeled DDMPs were synthesized and purified, with labeling occurring at the N-terminus (F-I2-Ox and F-H4-Ox; Supplementary Figs. 55 and 57). These labeled peptides were subsequently incubated with the transfected cells for a duration of 30 min. Following this incubation, unbound fluorescein-labeled DDMPs were removed through PBS washing, and confocal fluorescence images were recorded. As shown in Fig. 5f, fluorescein-labeled I2-Ox demonstrated a remarkable ability to selectively bind to the surface of the transfected cell expressing ICOS. Additionally, fluorescein-labeled H4-Ox can bind with HER3 expressed on the cell surface, as evidenced by the colocalization of the fluorescent mCherry tag and fluorescein (Fig. 5f). Thus, these results demonstrated that the DDMPs exhibited a remarkable specificity in binding to their targets on the live cell surface without interacting with other cell-surface proteins, making the DDMPs highly promising lead compounds for further development and diverse applications.
Inspired by the divalent interaction between antibodies and antigens, we tried to further improve the binding affinity of DDMPs to ICOS by chemically linking two F-I2-Ox through an organic crosslinker (Fig. 6a). Sensorgrams from SPR experiments revealed that the F-I2-Ox dimer, designated as DI-F-I2-Ox, exhibited a subnanomolar affinity for the chip surface-immobilized ICOS (0.20 nM, Fig. 6b and Supplementary Fig. S56), representing a ~200-fold improvement compared to the monomer F-I2-Ox. This improvement mainly resulted from the significantly reduced dissociation rate of DI-F-I2-Ox from the binding ICOS. Moreover, utilizing DI-F-I2-Ox enabled convenient fluorescent imaging of cell-surface ICOS at concentrations as low as 1 nM, contrasting with the unsuccessful imaging attempts using 100 nM F-I2-Ox (Supplementary Fig. 62). The binding of F-I2-Ox and DI-F-I2-Ox with extracellular ICOS was further confirmed on a cancer cell line (HuT-78) through flow cytometry (Fig. 6c). Initially, we validated ICOS expression on HuT-78 and Jurkat cell lines using APC-labeled anti-ICOS antibody (Fig. 6c and Supplementary Fig. 64)51. ICOS expression was obvious on HuT-78 cells (median fluorescence intensity (MFI) ratio = 24.6) and nearly absent on Jurkat cells (MFI ratio = 3.2). Thus, these two cell lines can serve to assess the binding specificity of F-I2-Ox/DI-F-I2-Ox to natively expressed ICOS on the cell surface. Following ~30 min incubation of HuT-78 cells with F-I2-Ox and DI-F-I2-Ox, we observed concentration-dependent increases in MFI values (Fig. 6c, Supplementary Figs. 65, 66). In contrast, no obvious fluorescence increase was observed on Jurkat cells for both F-I2-Ox and DI-F-I2-Ox (Fig. 6c and Supplementary Fig. 67), indicating the specific binding of the FITC-labeled DDMPs to ICOS on the cell surface. The KD values for F-I2-Ox and DI-F-I2-Ox were determined to be 773 and 180 nM, respectively (Fig. 6e). The relatively high apparent affinity for the dimeric peptide suggests the presence of bivalent cooperation on the cancer cell surface. We also noticed that the fluorescent intensity of DI-F-I2-Ox on cells was relatively weak in both confocal cell imaging and flow cytometry analysis. This resulted from the self-quenching effect of the two fluorescein dyes in DI-F-I2-Ox, which led to a ~2.3-time decrease in fluorescent intensity compared to F-I2-Ox (Fig. 6d). Thus, the corrected MFI ratio of DI-F-I2-Ox significantly surpassed that of F-I2-Ox across all tested concentrations (Fig. 6e), indicating the superior binding capability of DI-F-I2-Ox. These results illustrated the efficacy of our fluorophore-labeled DDMPs as molecular tools for quantitatively evaluating the expression level of endogenous ICOS on the cell surface.

a Synthesis of DI-F-I2-Ox. b SPR sensorgrams showing the interaction of DI-F-I2-Ox with ICOS. Experiments are repeated three times independently (Supplementary Fig. 56). c Flow cytometry showing the binding of APC-anti-ICOS antibody and DI-F-I2-Ox to HuT-78 and Jurkat cells. d Standard curves showing the fluorescence quenching of FITC in DI-F-I2-Ox (F = 172.6 c-453.8, r2 = 0.97) compared to that in F-I2-Ox (F = 286.4c−574.8, r2 = 0.99). Data are presented as mean ± s.d. from three independent experiments. FI fluorescence intensity. e Flow cytometry analysis to characterize the affinity of peptides F-I2-Ox and DI-F-I2-Ox to ICOS expressed on HuT-78 cells. The corrected values are included for comparison. Data are presented as mean ± s.d. from three biologically independent replicates (Supplementary Figs. 65 and 66). MFI: median fluorescence intensity.
In summary, our work explored the complex interplay of the three previously identified biscysteine motifs in the context of selective disulfide bond formation. We demonstrated that by combining these biscysteine motifs in tandem, we can create triscysteine motifs with distinct and unique capabilities for directing the disulfide pairing of peptides. A rich diversity of multicyclic peptides (DDMPs) with very few isomers can be conveniently obtained by the incorporation of a couple of the tandem triscysteine motifs and additional cysteine residues without being confined to rigid design rules. These DDMP templates provide us with abundant resources for the construction of DNA-encoded peptide libraries, such as phage-display libraries. Though our focus in this work was primarily on exploiting CPPCXC motifs and coupled CPXXCXC and CXCPXXC motifs for the development of phage-display peptide libraries, it is worth noting that these biscysteine motifs represent varying degrees of disulfide-directing capability, thus implying that many other triscysteine motifs investigated in this work can also be harnessed for future library design. We demonstrated the robust applicability of our DDMP libraries by successfully selecting functional peptides with nanomolar affinities to protein targets of great therapeutic interest, including EphA2, HER3, and ICOS. We recognize the significant challenge in further improving the binding affinity of these multicyclic peptides through phage display, potentially attributed to the limited library size (typically ~109), which may not adequately cover the extensive sequence space required for our peptides. However, in the future, affinity maturation strategies combining structural analyses and computational designs could be employed to further enhance the binding affinity. This study thus opens an avenue for discovering functional peptides with stable 3D structures, greatly expanding the scope of accessible sequence and structural space beyond the constraints of naturally occurring and computationally designed scaffolds.
Methods
Peptide synthesis
All chemicals were bought from GL Biochem (Shanghai, China), Energy Chemical (Shanghai, China), Sigma-Aldrich (Beijing, China), Sinopharm Chemical Reagent (Beijing, China), and Thermo Fisher Scientific (Shanghai, China). Peptide synthesis was carried out by standard Fmoc-protected SPPS on a Liberty BlueTM automated microwave peptide synthesizer (CEM). Peptide sequences were provided in Supplementary Table 1. Initially, the resin was swollen in DMF for 15 min, followed by deprotection using a standard deprotection method. Then, the C-terminal protected amino acid (0.2 M in DMF), Oxyma (1.0 M in DMF), and DIC (0.5 M in DMF) were introduced into the reactor and reacted with the resin through a standard coupling method. The Fmoc group was then removed through standard deprotection procedures. These reaction steps were repeated until the target peptides were synthesized. Next, a cleavage cocktail (TFA:Thioanisole:Phenol:H2O:1,2-Ethanedithiol = 87.5:5:2.5:2.5:2.5) was applied to the resin to release the crude peptide products. After 4–6 h of peptide cleavage, the cocktail was filtered, and 30 mL of cold diethyl ether was added to precipitate the peptides. Finally, the crude peptides were purified using HPLC (SHIMADZU, equipped with DGU-20AD degassing device, LC-20AD pump device, SIL-20A sample injector, CTO-20A constant temperature column). The target peptides were collected, and the solvent was removed using a Freeze dryer (Labconco FreeZone 4.5L).
Oxidative folding of peptides
All peptides were dissolved in a 100 mM phosphate buffer (PB, pH 7.4), and their concentrations were measured using UV–Vis spectrophotometry (HITACHI, U-3900H) at a wavelength of 280 nm. The molar absorption coefficients were estimated via the bioSYNTHESIS peptide property calculator. The purification and analysis of the peptides and their products were carried out using HPLC (SHIMADZU, equipped with DGU-20AD degassing device, LC-20AD pump device, SIL-20A sample injector, CTO-20A constant temperature column). The chromatographic column employed was a Waters, X Bridge C18 (4.6 × 250 mm, 5 μm). The progress of the reactions was monitored using HPLC at a flow rate of 1.0 mL min−1 with a mobile phase consisting of H2O (+0.1% TFA) and ACN (+0.1% TFA). The elution method began with an isocratic flow of 5% ACN (+0.1% TFA) for the first 5.0 min, followed by a linear gradient increasing from 5% to 75% ACN (+0.1% TFA) over 30 min. The mass of the peptides was determined using a Bruker Autoflex max MALDI-TOF mass spectrometer. 2,5-Dihydroxybenzoic acid (DHB) and α-cyano-4-hydroxycinnamic acid (CHCA) were used as the matrix compounds.
A peptide solution comprising peptides (1−12), DMSO, and 100 mM PB (pH 7.4) was prepared in a low-protein-adsorption centrifuge tube (Eppendorf) to reach a final concentration of 100 μM for the peptides and 30% (v/v) for DMSO. Notably, when conducting oxidation experiments for mixed peptides, the final concentration of each peptide was adjusted to 50 μM. These tubes were then placed in a shaker at 37 °C, and the reaction was finally quenched using a 1:1 (v/v) solution of 10% HPO3. Other peptides (50 µM) were co-incubated with 500 µM GSSG (or an extra 50 µM GSH) in 100 mM phosphate buffer (pH 7.4). After the reaction proceeded at 37 °C, the reaction was quenched with HPO3, and the resulting samples were analyzed using HPLC and MS.
Synthesis of DI-F-I2-Ox
A reaction mixture containing 2.5 mM of F-I2-Ox and 0.6 mM of disuccinimidyl suberate (DSS) was co-incubated in DMF containing 3 mM of triethylamine (Et3N). After 24 h at 37 °C, the resulting sample was analyzed using HPLC and MS. The product, DI-F-I2-Ox, was then collected and lyophilized for use in further experiments.
Trypsin digestion of oxidized peptides
To analyze the disulfide pairings of peptides, a tryptic digestion approach was employed. Initially, the oxidized peptides were purified and lyophilized. Subsequently, ~10–30 μg of the peptide powder was dissolved in 190 μL of 100 mM PB (pH 6.0). Then, 10 μL of a trypsin solution (1 mg/mL in water) was added to the mixture. The digestion was carried out at 37 °C for 4 h, during which the resulting fragments were monitored using HPLC and MS.
Synthesis of digested fragments by orthogonal protection strategy
Disulfide pairings in certain digested peptide fragments were determined using an orthogonal protecting group strategy. In brief, specific cysteine residues within the peptides were substituted with Acm-protected cysteines. After this modification, the peptides underwent dimeric oxidation, and the resulting products were purified and recovered via lyophilization. The purified products were then dissolved in methanol containing 1% TFA and 10 equivalents I2 dissolved in methanol were introduced to enable the orthogonal formation of additional disulfide bonds (standard products). This method allowed for the confirmation of disulfide pairings in the digested fragments by comparing them against standard products synthesized through the orthogonal protecting group strategy.
Peptide library construction
This work involves the construction of two random libraries and four custom libraries (Supplementary Table 2). The construction of these peptide libraries followed the methodology outlined in Supplementary Fig. 61. Primes necessary for library construction were provided by Tsingke Biotech (Beijing, China), Sangon Biotech (Shanghai, China), and Generay Biotech (Shanghai, China). The synthesized nucleotide sequences encoding the peptide fragments and extension primers were co-incubated to form DNA fragments through an extension reaction with Klenow fragment (4 h at 37 °C; 15 min at 65 °C). The resulting DNA fragments and the phage vectors were digested by the restriction endonucleases SfiI and NotI, respectively, which were then ligated with T4 DNA ligase. The ligated products were recovered and transfected into competent cells through electroporation (200 Ω, 2.5 kV). Finally, the transfected competent cells were gently agitated in a shaker at 80 rpm and 37 °C to facilitate their resuscitation. For the soft randomization, a mixture containing 70% wild-type (wt) nucleotides and 10% of each of the other nucleotides was used during the oligosynthesis for each randomized nucleotide.
Phage biopanning and peptide sequencing
Exogenous peptide-encoded phages were recovered from the supernatant of 2-YT medium through PEG/NaCl precipitation followed by resuspension in PBS. Magnetic beads (either streptavidin or neutravidin-coated) were first washed twice with a binding buffer consisting of 10 mM Tris–HCl, 150 mM NaCl, 10 mM MgCl2, and 1 mM CaCl2 (pH 7.4). These beads were then incubated with biotinylated target proteins and subsequently treated with a blocking buffer (which included the binding buffer, 0.3% Tween-20, and 3% (w/v) BSA) for 120 min. The phages were similarly blocked in the same blocking buffer for 120 min. Afterward, the magnetic beads were incubated with the phage peptide library at room temperature for 30 min. After nine washes with a washing buffer (the binding buffer containing 0.1% Tween-20) and two additional washes with the binding buffer, the phages were eluted from the magnetic beads using a glycine–hydrochloric acid solution (50 mM glycine, pH 2.2). The phage amplification and titer measurement were performed in preparation for subsequent panning rounds. Phage titers were determined after each round of panning to monitor the progress of the screening. After three or four rounds of panning, the enriched phages underwent sequencing, either by Sanger sequencing or next-generation sequencing. Sequencing services were provided by Sangon Biotech and Novogene Biotech.
Affinity determination by fluorescence polarization
Fluorescence polarization measurements were performed using a Tecan Infinite® 200 PRO Microplate Reader. Both the fluorescence polarization binding assay and competition assay were conducted in PBS (10 mM, pH 7.4). FITC-labeled peptides, proteins, and peptides were incubated at room temperature for 10 min prior to measurement. Fluorescence anisotropy was then measured using a plate reader, with excitation at 485 nm and emission at 535 nm. All measurements were taken in triplicate, and the binding and competition polarization data were analyzed using Origin 2021 software.
For the preparation of the solution of F-ETGE (FITC-[β-A]-DEETGEF-OH), a 1.0 mM stock solution in DMSO was diluted with 1 × PBS (the final DMSO concentration is <1%). F-ETGE was then added to solutions of Keap1 protein, resulting in a final concentration of 20 nM and varying concentrations of Keap1 from 0 to 500 nM. The KD value for the interaction of F-ETGE with Keap1 was determined to be 72 nM, consistent with the previously published value39.
F-K1-Ox and F-K2-Ox were prepared in PBS, and their concentration was determined using UV–vis spectrometry (εFITC, 495 nm = 72000 M−1). Then, the FITC-labeled peptides (20 nM) were added to Keap1 protein solutions with concentrations ranging 0–800 nM. In the FP competition assay, complexes of FITC-labeled peptides and Keap1 complexes (20/300 nM) were prepared and treated with varying concentrations of competing peptides from 1.0 nM to 10.0 μM.
Affinity determination using SPR
The biotin CAPture Kit (Cytiva) was employed to determine the binding affinity of peptides to various protein targets. A running buffer consisting of PBS with 50 μM EDTA and 0.05% P20 was selected for the assays. First, a chip pre-coated with single-stranded DNA molecules was treated with a capture reagent to introduce surface-immobilized streptavidin. Then, biotinylated target proteins were immobilized on the chip through biotin and streptavidin interaction. The reference channel was set up with the capture reagent and biotin–BSA for comparison purposes. Subsequently, a series of peptide samples at different concentrations were injected over the chip, and the Biacore T200 recorded the response curves. Finally, the collected data were analyzed using the Biacore T200 evaluation software.
NMR experiments
NMR experiments were conducted at a temperature of 298 K using Bruker AVANCE III 850 MHz, which is equipped with a cryogenic triple-resonance probe. Samples were prepared in a solvent mixture comprising 50% (v/v) H2O and 50% (v/v) perdeuterated acetonitrile, with the final concentration of all peptides (H2’-Ox, I2’-Ox, and 4-Ox) reaching ~1 mM. The experiments involved recording two-dimensional (2D) 1H, 15N/13C HSQC spectra to obtain the chemical shift information for heavy atoms in both the peptide backbone and side chains. To aid in the sequential assignment of the peptides, 2D 1H–1H TOCSY (with a mixing time of 80 ms) and 2D 1H–1H COSY spectra were collected. For structural calculation, 2D 1H–1H NOESY spectra were recorded with a mixing time of 300 ms. NMR data were processed using the NMRPipe/NMRDraw suite52 and analyzed with NMRFAM-SPARKY53. Cross-peaks from the NOE data were assigned manually. Dihedral angle restraints for the backbone were determined using the TALOS-N program, which relies on the chemical shifts of the backbone resonances54. Distance constraints were derived from the volume integration of NOE cross-peaks and processed using ARIA2.3.2 and CNS1.2155,56. From a total of 150 structures produced from the last iteration in each NMR run, the ensemble of the 15 lowest-energy structures was generated. The visualization of these structures was carried out using PyMol, and their quality was evaluated using PROCHECK57.
Cell culture
The HEK293T cell line and the Jurkat cell line were generous gifts from Dr. Jiahuai Han's group and Dr. Yuhsuan Tsai's group, respectively. HuT-78 cell line (Cs-011-008078) was purchased from Shanghai Tongwei (Shanghai, China). All three cell lines originated from ATCC cell lines. HEK293T cells were cultured in DMEM (VivaCell) supplemented with 10% FBS, while HuT-78 and Jurkat cells were maintained in RPMI 1640 medium (Gibco) with 10% FBS. All cell lines were incubated at 37 °C in a humidified environment with 5% CO2.
Confocal cell imaging
Plasmids that encode the extracellular domains of HER3 (Ser20-Thr643) and ICOS (Glu21-Phe141), both fused with a mCherry tag, were obtained from General Biol (Anhui, China). HEK293T cells were cultured at a density of 1.5 × 104 cells/dish in DMEM supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified environment containing 5% CO2. After 24 h of incubation, the cells were transiently transfected with either the HER3-mCherry or ICOS-mcherry plasmids. Another 24 h later, the cells were treated with 1 μM F-H4-Ox (or with 1 μM F-I2-Ox, or 10 nM DI-F-I2-Ox) in DMEM for a period of 30 min. The cells were then washed six times with 1 mL of DMEM to remove unbound peptides. As a control group, non-transfected cells underwent the same treatment for comparison. Following these treatments, confocal fluorescent images were acquired using a Leica TCS SP8 confocal microscope system. The FITC-labeled peptides were excited with a 488 nm laser, and emissions were recorded in the range of 506–571 nm. Meanwhile, the mCherry fluorescence was excited using a 512 nm laser, and emissions were collected between 605 and 670 nm.
Flow cytometry analysis
HuT-78 cells (approximately 1.0 × 106 cells/tube) were collected by centrifugation at 150 × g for 10 min and then resuspended in 200 μL of RPMI 1640 medium supplemented with 10% FBS. Different concentrations of F-I2-Ox or DI-F-I2-Ox or 5 μL APC anti-ICOS antibody (313509, BioLengend) were then added to the suspension. The mixture was incubated at room temperature for 30 min on a slowly rotating wheel. Following the incubation, the cells were again collected by centrifugation and washed three times with 200 μL of 1 × PBS. The final cell pellet was resuspended in 200 μL RPMI 1640 medium containing 2% FBS), preparing them for flow cytometry analysis. Flow cytometric analysis was performed using a CytoFLEX S (Beckman Coulter) instrument. FlowJo v10.10 was used to generate histograms of the flow cytometry results on a biexponential scale. The KD values were calculated using a one-site binding saturation equation (Y = BmaxX/(KD + X)) in Prism 8.0. Additionally, standard curves for F-I2-Ox and DI-F-I2-Ox were measured using an ELISA reader (PerkinElmer Enspire) and fitted in Prism 8.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data generated in this study are provided in the Source data file. The NMR structures of 4-Ox, H2’-Ox, and I2’-Ox are available in the Protein Data Bank (PDB IDs: 8WVS, 8WW1, and 8WW0). All additional data supporting the findings of this study are provided in the Supplementary Information. Source data are provided with this paper.
References
Wang, C. K. & Craik, D. J. Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat. Chem. Biol. 14, 417–427 (2018).
Gongora-Benitez, M., Tulla-Puche, J. & Albericio, F. Multifaceted roles of disulfide bonds. peptides as therapeutics. Chem. Rev. 114, 901–926 (2014).
Kale, S. S. et al. Cyclization of peptides with two chemical bridges affords large scaffold diversities. Nat. Chem. 10, 715–723 (2018).
Yin, Y. et al. Chemical and ribosomal synthesis of topologically controlled bicyclic and tricyclic peptide scaffolds primed by selenoether formation. Angew. Chem. Int. Ed. 58, 4880–4885 (2019).
Wong, J. Y. K. et al. Genetically-encoded discovery of proteolytically stable bicyclic inhibitors for morphogen NODAL. Chem. Sci. 12, 9694–9703 (2021).
Heinis, C., Rutherford, T., Freund, S. & Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 5, 502–507 (2009).
Muttenthaler, M., King, G. E., Adams, D. J. & Alewood, P. E. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 20, 309–325 (2021).
de Veer, S. J., Weidmann, J. & Craik, D. J. Cyclotides as tools in chemical biology. Acc. Chem. Res. 50, 1557–1565 (2017).
Liu, W. Y. et al. An ultrapotent and selective cyclic peptide inhibitor of human beta-factor XIIa in a cyclotide scaffold. J. Am. Chem. Soc. 143, 18481–18489 (2021).
Guo, Y. et al. Diaminodiacid bridges to improve folding and tune the bioactivity of disulfide-rich peptides. Angew. Chem. Int. Ed. 54, 14276–14281 (2015).
Cooper, B. M., Iegre, J., O’ Donovan, D. H., Ölwegård Halvarsson, M. & Spring, D. R. Peptides as a platform for targeted therapeutics for cancer: peptide–drug conjugates (PDCs). Chem. Soc. Rev. 50, 1480–1494 (2021).
Kintzing, J. R. & Cochran, J. R. Engineered knottin peptides as diagnostics, therapeutics, and drug delivery vehicles. Curr. Opin. Chem. Biol. 34, 143–150 (2016).
Busby, R. W. et al. Pharmacologic properties, metabolism, and disposition of linaclotide, a novel therapeutic peptide approved for the treatment of irritable bowel syndrome with constipation and chronic idiopathic constipation. J. Pharmacol. Exp. Ther. 344, 196–206 (2013).
Cox, N., Kintzing, J. R., Smith, M., Grant, G. A. & Cochran, J. R. Integrin-targeting knottin peptide-drug conjugates are potent inhibitors of tumor cell proliferation. Angew. Chem. Int. Ed. 55, 9894–9897 (2016).
Yang, X. et al. A lanthipeptide library used to identify a protein–protein interaction inhibitor. Nat. Chem. Biol. 14, 375–380 (2018).
Hansen, S. et al. Directed evolution identifies high-affinity cystine-knot peptide agonists and antagonists of Wnt/beta-catenin signaling. Proc. Natl Acad. Sci. USA 119, e2207327119 (2022).
Lui, B. G. et al. Targeting the tumor vasculature with engineered cystine-knot miniproteins. Nat. Commun. 11, 295 (2020).
Clark, R. J. et al. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. 49, 6545–6548 (2010).
Akondi, K. B. et al. Discovery, synthesis, and structure activity relationships of conotoxins. Chem. Rev. 114, 5815–5847 (2014).
Getz, J. A., Rice, J. J. & Daugherty, P. S. Protease-resistant peptide ligands from a knottin scaffold library. ACS Chem. Biol. 6, 837–844 (2011).
Tamaoki, H. et al. Folding motifs induced and stabilized by distinct cystine frameworks. Protein Eng. 11, 649–659 (1998).
Undheim, E. A. B., Mobli, M. & King, G. F. Toxin structures as evolutionary tools: using conserved 3D folds to study the evolution of rapidly evolving peptides. Bioessays 38, 539–548 (2016).
Smith, J. J. et al. Unique scorpion toxin with a putative ancestral fold provides insight into evolution of the inhibitor cystine knot motif. Proc. Natl Acad. Sci. USA 108, 10478–10483 (2011).
Gunasekera, S., Daly, N. L., Clark, R. J. & Craik, D. J. Dissecting the oxidative folding of circular cystine knot miniproteins. Antioxid. Redox Signal. 11, 971–980 (2009).
Lahti, J. L., Silverman, A. P. & Cochran, J. R. Interrogating and predicting tolerated sequence diversity in protein folds: application to E. elaterium trypsin inhibitor-II cystine-knot miniprotein. PLoS Comput. Biol. 5, e1000499 (2009).
Pineda, S. S. et al. Structural venomics reveals evolution of a complex venom by duplication and diversification of an ancient peptide-encoding gene. Proc. Natl Acad. Sci. USA 117, 11399–11408 (2020).
Kessler, P., Marchot, P., Silva, M. & Servent, D. The three-finger toxin fold: a multifunctional structural scaffold able to modulate cholinergic functions. J. Neurochem. 142, 7–18 (2017).
Dash, T. S. et al. A centipede toxin family defines an ancient class of CS alpha/beta defensins. Structure 27, 315–326 (2019).
Bhardwaj, G. et al. Accurate de novo design of hyperstable constrained peptides. Nature 538, 329–335 (2016).
Yao, S. C. et al. De novo design and directed folding of disulfide-bridged peptide heterodimers. Nat. Commun. 13, 1539 (2022).
Chevalier, A. et al. Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79 (2017).
Crook, Z. R. et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 8, 2244 (2017).
Wu, C. L., Leroux, J. C. & Gauthier, M. A. Twin disulfides for orthogonal disulfide pairing and the directed folding of multicyclic peptides. Nat. Chem. 4, 1045–1050 (2012).
Zheng, Y. W., Zhai, L. X., Zhao, Y. B. & Wu, C. L. Orthogonal cysteine-penicillamine disulfide pairing for directing the oxidative folding. Pept. J. Am. Chem. Soc. 137, 15094–15097 (2015).
Lu, S. M. et al. Directed disulfide pairing and folding of peptides for the de novo development of multicyclic peptide libraries. J. Am. Chem. Soc. 142, 16285–16291 (2020).
Lu, S. M. et al. Disulfide-directed multicyclic peptide libraries for the discovery of peptide ligands and drugs. J. Am. Chem. Soc. 145, 1964–1972 (2023).
Wu, Y. et al. Structure-guided design of CPPC-paired disulfide-rich peptide libraries for ligand and drug discovery. Chem. Sci. 13, 7780–7789 (2022).
Li, J. J. et al. De novo discovery of cysteine frameworks for developing multicyclic peptide libraries for ligand discovery. J. Am. Chem. Soc. 145, 28264–28275 (2023).
Zha, M. R., Lin, P., Yao, H. W., Zhao, Y. B. & Wu, C. L. A phage display-based strategy for the de novo creation of disulfide-constrained and isomer-free bicyclic peptide affinity reagents. Chem. Commun. 54, 4029–4032 (2018).
Jafari, M. R. et al. Discovery of light-responsive ligands through screening of a light-responsive genetically encoded library. ACS Chem. Biol. 9, 443–450 (2014).
Chen, S. et al. Bicyclic peptide ligands pulled out of cysteine-rich peptide libraries. J. Am. Chem. Soc. 135, 6562–6569 (2013).
Hancock, R. et al. Peptide inhibitors of the Keap1-Nrf2 protein–protein interaction. Free Radic. Biol. Med. 52, 444–451 (2012).
Ireton, R. C. & Chen, J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr. Cancer Drug Targets 5, 149–157 (2005).
Mudd, G. E. et al. Identification and optimization of EphA2-selective bicycles for the delivery of cytotoxic payloads. J. Med. Chem. 63, 4107–4116 (2020).
Gandullo-Sánchez, L., Ocaña, A. & Pandiella, A. HER3 in cancer: from the bench to the bedside. J. Exp. Clin. Cancer Res. 41, 310 (2022).
Majumder, A. et al. The role of HER2 and HER3 in HER2-amplified cancers beyond breast cancers. Sci. Rep.-Uk 11, 9091 (2021).
Dolgin, E. HER3-addicted tumors: how biotechs are closing in. Nat. Biotechnol. 40, 1157–1159 (2022).
Haikala, H. M. & Jänne, P. A. Thirty years of HER3: from basic biology to therapeutic interventions. Clin. Cancer Res. 27, 3528–3539 (2021).
Solinas, C., Gu-Trantien, C. & Willard-Gallo, K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open 5, e000544 (2020).
Rujas, E., Cui, H., Sicard, T., Semesi, A. & Julien, J. P. Structural characterization of the ICOS/ICOS-L immune complex reveals high molecular mimicry by therapeutic antibodies. Nat. Commun. 11, 5066 (2020).
Amatore, F. et al. ICOS is widely expressed in cutaneous T-cell lymphoma, and its targeting promotes potent killing of malignant cells. Blood Adv. 4, 5203–5214 (2020).
Delaglio, F. et al. NMRPipe—a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Lee, W., Tonelli, M. & Markley, J. L. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015).
Shen, Y. & Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 (2013).
Rieping, W. et al. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23, 381–382 (2007).
Brunger, A. T. et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 (1998).
Laskowski, R. A., Rullmann, J. A. C., MacArthur, M. W., Kaptein, R. & Thornton, J. M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 (1996).
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (92356308 and 22174119), the Fundamental Research Funds for the Central Universities (20720210001 and 20720220005), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_17R66), and the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (21521004).
Author information
Authors and Affiliations
Contributions
Z.D. and C.W. designed the research; Z.D., C.K., and S.F. performed all experiments; Z.D. and C.W. wrote and revised the manuscript. C.W. supervised the research; All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Christian Heinis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Duan, Z., Kong, C., Fan, S. et al. Triscysteine disulfide-directing motifs enabling design and discovery of multicyclic peptide binders. Nat Commun 15, 7799 (2024). https://doi.org/10.1038/s41467-024-51723-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51723-w
