
Abstract
EWS fusion oncoproteins underlie several human malignancies including Desmoplastic Small Round Cell Tumor (DSRCT), an aggressive cancer driven by EWS-WT1 fusion proteins. Here we combine chromatin occupancy and 3D profiles to identify EWS-WT1-dependent gene regulation networks and target genes. We show that EWS-WT1 is a powerful chromatin activator controlling an oncogenic gene expression program that characterizes primary tumors. Similar to wild type WT1, EWS-WT1 has two isoforms that differ in their DNA binding domain and we find that they have distinct DNA binding profiles and are both required to generate viable tumors that resemble primary DSRCT. Finally, we identify candidate EWS-WT1 target genes with potential therapeutic implications, including CCND1, whose inhibition by the clinically-approved drug Palbociclib leads to marked tumor burden decrease in DSRCT PDXs in vivo. Taken together, our studies identify gene regulation programs and therapeutic vulnerabilities in DSRCT and provide a mechanistic understanding of the complex oncogenic activity of EWS-WT1.
Similar content being viewed by others


Introduction
Fusion oncoproteins underlie the molecular pathogenesis and clinical behavior of many human cancers, including sarcomas, which constitute more than 20% of all pediatric malignancies1,2. Among them, Desmoplastic Small Round-Cell Tumor (DSRCT) is an extremely aggressive malignancy of unknown origin that predominantly arises in the abdominal cavity of young male adults3,4. The five-year survival rate for DSRCT patients remains below 15% due to poor responses to multimodal therapies and lack of targeted treatments, rendering DSRCT one of the deadliest forms of human cancer5,6,7.
DSRCT is genetically characterized by translocations between chromosomes 11 and 22 t(11;22)(p13;q12) that produce the EWSR1-WT1 fusion oncogene8,9. EWS-WT1 is a member of a large family of fusion oncoproteins composed of the prion-like disordered domain of EWS, a ubiquitously expressed RNA binding protein, and one of several possible transcription factors (TFs)2. Given that EWS fusions operate as aberrant transcriptional regulators, the DNA binding domain of the partner TF plays a critical role in establishing an oncogenic gene expression program that is specific to each tumor type. In addition, as demonstrated for EWS-FLI1 fusions in Ewing sarcoma, the presence of EWS allows the fusion protein to function as a pioneer factor and bind previously inaccessible genomic sites10,11. Remarkably, these effects are dependent on the phase transition properties of the EWS prion-like domain and yield a related but distinct binding and activation profile12.
Because EWS-WT1 shares key functional domains with EWS-FLI1, analogous mechanisms may underlie the tumor promoting activity of both fusion proteins. Nevertheless, our current understanding of the role of EWS-WT1 in DSRCT remains limited. This fusion protein is composed of the disordered, prion-like, domain of EWS fused to the DNA binding domain of WT1, a zinc finger TF that plays prominent roles in kidney and gonad development. However, in sharp contrast with the oncogenic role of EWS-WT1 in DSRCT13, WT1 operates as a tumor suppressor in other cancers, including Wilms tumors and acute myeloid leukemia14,15,16,17,18, suggesting that the inclusion of EWS results in substantial reconfiguration of WT1 activity19. WT1 function is regulated by alternative splicing mechanisms, resulting in the expression of two isoforms that differ by the inclusion or exclusion of three amino acids between zinc fingers 3 and 4 (lysine, threonine, and serine: KTS). These events are highly conserved throughout evolution and provide WT1 isoforms with distinct biochemical and biological properties, including a preference for RNA binding by the +KTS isoform14,20,21,22,23. Although these splicing mechanisms also alter the DNA binding ability of EWS-WT1, their impact on the underlying tumor regulatory networks in DSRCT remains poorly understood8,24,25,26.
To gain insight into the mechanisms that underlie EWS-WT1-mediated transformation, we set out to annotate the epigenetic regulatory networks, chromatin remodeling mechanisms, and direct transcriptional targets controlled by EWS-WT1 in DSRCT, as well as to define the relative contribution of each fusion protein isoform to the final oncogenic program. To do so, we combine EWS-WT1 chromatin profiles and 3D connectivity maps with loss of function studies and derive a regulatory model for the oncogenic activity of the fusion protein. Moreover, given the complexity of EWSR1-WT1 splicing, we also model the function of the +KTS/-KTS isoforms in human mesothelial cells, considered as candidate tumor precursor cells given the frequent occurrence of these tumors on serosal surfaces4. We show that EWS-WT1 operates as a potent transcriptional activator that controls a tumor-specific gene regulation program through a large 3D looping network. Strikingly, these functions are not only highly dependent on the EWS prion-like domain but also on the coordinated activity of both EWS-WT1 isoforms, which are required to generate tumor xenografts that resemble primary DSRCT. In addition to revealing the extent of the regulatory networks centered on each EWS-WT1 isoform, our results also point to critical downstream target genes such a CCND1, whose pharmacological inhibition through CDK4/6 inhibitors leads to marked decrease in the growth of DSRCT cells in vitro and in vivo, and illustrates how these mechanistic insights can translate into rationale for therapeutic intervention.
Results
EWS-WT1 is associated with highly active distal regulatory sites in DSRCT tumors
The mechanisms by which EWS-WT1 regulates its target gene repertoire in DSRCT cells are not fully understood, and their elucidation requires a detailed understanding of the chromatin organization of tumor cells. To this end, we first used ChIP-seq to annotate the fusion protein binding sites in the well-established DSRCT cell line JN-DSRCT127. Given that wild-type WT1 is not expressed in these cells28 (Supplementary Fig. S1A), anti-WT1 antibodies were used to generate the fusion protein DNA occupancy profiles. This approach yielded a total of 6823 genomic locations that were consistently enriched for WT1 signals using two different antibodies and provided the core set of EWS-WT1 binding sites for further analyses. Among these, 6238 peaks (91%) were present at distal regulatory elements, whereas 585 (9%) were found at TSS regions in the JN-DSRCT1 cell line (Fig. 1a).

a Left: pie chart shows genomic locations of EWS-WT1 binding sites in the DSRCT cell line JN-DSRCT1. Two antibodies targeting the WT1 C-terminal region present in the fusion protein were used for the EWS-WT1 ChIP-seq profiling (WT1 ab1 and WT1 ab2). Consensus peaks were defined as those shared by both WT1 antibodies tested. Right: whole genome distribution of promoter, intragenic and intergenic regions in the human genome. b Left: Heatmaps of WT1, H3K4me1, H3K4me3, H3K27ac and H3K27me3 ChIP-seq signal densities in the DSRCT cell line JN-DSRCT1. Right: Heatmaps of H3K4me1 and H3K27ac in DSRCT primary tumors. 10-kb windows in each panel are centered on EWS-WT1 binding sites identified in the JN-DSRCT1 cell line as in Fig. 1a. c DNA Motif enrichment analysis at distal EWS-WT1 binding sites from the JN-DSRCT1 cell line as in Fig. 1a. The consensus WT1 motif CTCCC(A/C)C and the TCC repeat motif score first and third respectively. Associated transcription factors for the motifs were EGR1 and ZNF263 respectively. See also Supplementary Fig. S1D. p values were calculated based on the binomial test by the Homer software. d Boxplots show ChIP-seq signal intensity for H3K27ac and EWS-WT1 in JN-DSRCT1 cells at subsets of EWS-WT1 binding sites identified. Each category contains 5871 and 952 EWS-WT1 binding sites (n), respectively. H3K27ac (2 kb window) and EWS-WT1 (500 bp window). Median value is shown as a line within the boxplot, which spans from the 25th to 75th percentiles. p values were calculated using two-sided t tests. e Representative example of EWS-WT1 binding sites containing CTCCC clusters near the PDGFA locus, enriched in H3K27ac ChIP-seq signals in the JN-DSRCT1 cell line and DSRCT primary tumors. Regions of interest are highlighted in light gray. Each CTCCC motif detected at EWS-WT1 binding site is depicted as a box below.
To determine the chromatin states associated with EWS-WT1 binding sites we next profiled the major histone modifications H3K4me1, H3K27ac, H3K4me3, H3K9ac and H3K27me3 in JN-DSRCT1 cells as well as in four primary DSRCT tumors. The majority of EWS-WT1 distal binding sites were found to be associated with active enhancer elements, marked by the presence of H3K4me1 and H3K27ac signals (Fig. 1b and Supplementary Figs. S1B, C). Similarly, EWS-WT1 binding sites at TSSs were associated with marked H3K4me3, H3K9ac and H3K27ac signals (Fig. 1b and Supplementary Figs. S1B, C). Consistent with the notion that the fusion protein behaves as a transcriptional activator, no overlap between EWS-WT1 binding sites and the Polycomb repressive mark H3K27me3 was observed. Importantly, all four primary DSRCTs analyzed displayed similar chromatin state profiles at our consensus EWS-WT1 binding sites (Fig. 1b and Supplementary Figs. S1B, C).
The top enriched sequence at distal EWS-WT1 bound sites was the consensus WT1 binding motif CTCCC(A/C)C (Fig. 1c and Supplementary Fig. S1D)29. Ranked second, was a forkhead motif, but further analysis revealed that most of these sites also contained the main CTCCC motif (Supplementary Figs. S1D-E) suggesting that a forkhead transcription factor may co-bind a fraction of EWS-WT1 binding sites. Interestingly, the third most enriched motif corresponded to a repetitive TCC sequence (Fig. 1c and Supplementary Fig. S1D) that has been previously observed at individual EWS-WT1 binding sites30. This observation is analogous to that in Ewing sarcoma, where EWS-FLI1 binding to canonical single ETS motifs or GGAA repeat sequences11 results in distinct epigenetic activities and transcriptional outputs, prompting us to investigate whether similar mechanisms may be at play in DSRCT. Although the fusion protein was found to be associated with the canonical WT1 motif in the majority of cases (62%, Supplementary Fig. S1E), we identified three additional DNA features at EWS-WT1 binding sites: CTCCC clustered motifs (609 sites containing three or more motifs, 8.9%), TCC repeats (181 sites, 2.6%) and co-occurrence of both CTCCC clusters and TCC repeats (162 sites, 2.4%) (Supplementary Figs. S1E and S2A). The analysis of the chromatin states associated with these different EWS-WT1 binding patterns revealed their association with higher EWS-WT1 and H3K27ac signals compared to those of EWS-WT1 binding sites devoid of CTCCC clusters and TCC repeats, with the category of co-occurring CTCCC clusters and TCC repeats sites displaying the highest levels of both (Fig. 1d and Supplementary Figs. S2B-C).
Interestingly, EWS-WT1-bound enhancers containing large clusters of CTCCC motifs were found proximal to known regulators of DSRCT biology, such as the Platelet Derived Growth Factor Subunit A gene (PDGFA), previously shown to play critical functions in shaping the stromal architecture of DSRCT (Fig. 1e)30. These sites displayed consistent H3K27ac levels in JN-DSRCT1 cells, as well as four primary DSRCT tumors, suggesting that EWS-WT1 functions as a potent chromatin regulator whose activity may be regulated by different types of recognition sequences.
DSRCT 3D tumor connectivity is dominated by EWS-WT1-centered chromatin interactions
Given that the vast majority of EWS-WT1 binding sites are located at distal genomic regions well outside of promoters, we reasoned that the identification of the direct targets of the fusion protein would require a better understanding of its 3D chromatin connectivity. To this end we used H3K27ac-mediated Hi-ChIP to profile all active chromatin interactions in JN-DSRCT1 cells and integrated Hi-ChIP with WT1 occupancy profiles to generate an EWS-WT1-centric network. This approach yielded a total of 37439 chromatin loops in JN-DSRCT1 cells (Fig. 2a), of which 59% were associated with EWS-WT1 (Fig. 2a, b). Strikingly, EWS-WT1 binding sites were involved in significantly more looping interactions than H3K27ac peaks not bound by the fusion protein. EWS-WT1-associated loops displayed higher read counts (Fig. 2c) and involved regions separated by longer distances (Fig. 2d, e and Supplementary Fig. S3A). A noticeable difference in loops crossing TAD boundaries was also observed in association with EWS-WT1 (15% vs 6%; Supplementary Fig. S3B). Interestingly, the strongest looping was observed at the subset of EWS-WT1 binding sites associated with TCC repeats and/or CTCCC clusters, pointing to the implication of these genomic regions in shaping DSRCT topology (Fig. 2c, d). These observations suggest that the fusion protein plays a prominent role in shaping chromatin conformation and activity in DSRCT cells, particularly since EWS-WT1 binding sites constitute a minority of H3K27ac peaks in tumor cells but a large fraction of the chromatin loops detected by Hi-ChIP (Fig. 2a, b). In support of this notion we identified a robust set of EWS-WT1-centered chromatin loops connecting the fusion protein binding sites to the promoters of genes involved in DSRCT biology, including Vascular Endothelial Growth Factor A (VEGFA) and PDGFA (Fig. 2f and Supplementary Fig. S3C)4,30.

a Circos plot shows genome-wide loops identified in JN-DSRCT1 cells by H3K27ac Hi-ChIP. Loops associated (red) or not (blue) with EWS-WT1 binding sites were determined based on EWS-WT1 ChIP-seq occupancy profiles. Inset: Barplot shows an enrichment of EWS-WT1 bound sites in Hi-ChIP loops genome-wide. b Distribution of loop interactions based on the presence or absence of EWS-WT1 binding in the JN-DSRCT1 cell line. c Boxplots show the number of loops (top) and interactions (bottom) between anchor sites in H3K27ac Hi-ChIP in absence or presence of EWS-WT1 binding in JN-DSRCT1 cells. Both the number of loops and interactions are increased at CTCCC clusters and TCC repeat sites bound by EWS-WT1 when compared to all EWS-WT1 binding sites, which includes other EWS-WT1 binding sites, and sites not bound by the fusion protein. Median value is shown as a line within the boxplot, which spans from the 25th to 75th percentiles. Whiskers indicate a 1.5× inter-quartile range. p values were calculated using two-sided t tests. n represents the number of chromatin loops in each category. d Plots showing the distribution of H3K27ac Hi-ChIP loops distances in presence or absence of EWS-WT1 binding at each anchor. The categories are combinations of: anchors not bound by the fusion protein (n = 15205), CTCCC clusters and/or TCC repeat sites bound by EWS-WT1 (both sides n = 645; one side only n = 4985), other EWS-WT1 binding sites (one side only n = 11940; both sides n = 2587), CTCCC clusters and/or TCC repeat sites bound by EWS-WT1 on one side and other EWS-WT1 binding site on the other side (n = 2077). The number of sites (n) for each category is indicated. Median distances for each category are shown as red dotted vertical lines. e Example shows representative image of the increases in H3K27ac Hi-ChIP looping distance for sites associated with EWS-WT1 binding (red, top) when compared to other H3K27ac sites (blue, bottom) on chromosome 8. See also Supplementary Fig. S3A. f Example shows H3K27ac Hi-ChIP looping maps and EWS-WT1/H3K27ac ChIP-seq signals at the VEGFA locus in JN-DSRCT1 cells. H3K27ac Hi-ChIP in JN-DSRCT1 cell line was performed once.
EWS-WT1 depletion leads to strong changes in chromatin activity and 3D connectivity
We next sought to determine the direct functional effect of EWS-WT1 on chromatin activity and 3D conformation. To this end, we used a lentiviral-based shRNA strategy to deplete the fusion transcript in JN-DSRCT1 cells. A virtually complete knock-down (KD) of the fusion protein was observed 96 h post-transduction (Fig. 3a right), and the chromatin profiles of corresponding EWS-WT1-depleted cells showed an almost complete loss of signals associated with the fusion protein across all binding sites (Fig. 3a left). Loss of EWSR1-WT1 expression led to marked reduction in H3K27ac and H3K4me1 signals at a large majority of its binding sites (Fig. 3b, c and Supplementary Fig. S4A), confirming its powerful chromatin activation properties. A similarly robust remodeling was observed for chromatin interactions by Hi-ChIP (Supplementary Fig. S4B), with the strongest decreases in connectivity being associated with loops bound by EWS-WT1 at both anchors (Fig. 3d, e). Conversely, increases in chromatin interactions were mostly observed for loops independent of the fusion protein binding sites (Fig. 3d and Supplementary Fig. S4C).

a Left: Composite plot shows EWS-WT1 ChIP-seq signals in JN-DSRCT1 cells lentivirally induced with a shRNA targeting WT1. 10 kb window centered on EWS-WT1 binding sites (n = 6823). Right: Immunoblotting shows EWS-WT1 protein levels in the same conditions. The experiment was independently repeated twice with similar results. Source data are provided as a Source Data file. b Histogram depicts changes in H3K27ac ChIP-seq signals at EWS-WT1 binding sites upon the fusion protein knockdown in JN-DSRCT1 cells as in Fig. 3a. 4711 EWS-WT1 binding sites show H3K27ac signal decrease of at least twofold in both experiments. Twofold changes are highlighted in red (decreases) and blue (increases). c Left: Representative ChIP-seq tracks showing chromatin changes at the FGFR4 locus upon EWS-WT1 knockdown in the JN-DSRCT1 cell line. Right: Corresponding changes in FGFR4 expression levels as detected by RNA-seq. d Histograms show changes in H3K27ac Hi-ChIP looping signals in JN-DSRCT1 cells upon EWS-WT1 knockdown at sites initially bound by EWS-WT1 on one side (n = 19606), both sides (n = 4929) or none (n = 27426). H3K27ac Hi-ChIP in shRNA transduced JN-DSRCT1 was performed once. e Left: Example shows marked changes in H3K27ac Hi-ChIP signal at the VEGFA locus upon EWS-WT1 depletion in JN-DSRCT1 cells. Right: Corresponding changes in VEGFA expression levels as detected by RNA-seq. f Left: Volcano plot depicting gene expression changes in JN-DSRCT1 cells upon EWS-WT1 knock-down. y axis is capped at a maximum adjusted p value of −100. Genes connected to EWS-WT1 with twofold changes and adjusted p value of 0.05 are shown in red. Right: Boxplots show gene expression levels in control (red) and EWS-WT1-depleted cells (blue) for genes initially connected to CTCCC and/or TCC repeats bound by EWS-WT1 (n = 2176), other EWS-WT1 sites (n = 2584) or sites not associated with EWS-WT1 (n = 7150). n represents the number of genes in each category. p value in CTCCC and/or TCC repeats = 2.16e–73, in other EWS-WT1 = 0.08, in not connected to EWS-WT1 = 0.24. Median value is shown as a line within the boxplot, which spans from the 25th to 75th percentiles. Whiskers indicate a 1.5× inter-quartile range. p values were calculated using two-sided t tests. H3K27ac Hi-ChIP in shRNA transduced JN-DSRCT1 was performed once. Expression profiling was performed four times in shRNA transduced JN-DSRCT1.
The alterations in chromatin activity and conformation that followed EWS-WT1 depletion were associated with decreased expression of a large set of direct target genes, as analyzed by RNA-seq (Fig. 3f, left), including a panel of genes previously reported to be directly regulated by EWS-WT1 (Supplementary Fig. S4D)30,31,32,33,34. Consistent with our previous observations, genes connected to EWS-WT1-bound CTCCC clusters and/or TCC repeats were most strongly down-regulated upon depletion of the fusion protein (Fig. 3f, right). This suggests that these sites have stronger regulatory effects and is in keeping with the observation that they have higher levels of H3K27ac and higher loop counts. In addition, we also observed that targets of CTCCC clusters and/or TCC repeats show a decrease in total loop connectivity arising from dramatic decreases in EWS-WT1 loops and modest reductions in non-EWS-WT1 loops. In contrast, promoters targeted by other EWS-WT1 binding sites show decreased EWS-WT1 looping but increased non-EWS-WT1 connectivity from other locations, resulting in an overall higher total connectivity (Supplementary Fig. S4E). Finally, functional annotation of the down-regulated target genes revealed their enrichment for terms related to DNA organization and cell-cycle (Gene Ontology Biological Process, Supplementary Fig. S4F), suggesting a direct role for EWS-WT1 in regulating the genomic organization and proliferation of DSRCT cells. Interestingly, the list of EWS-WT1 target genes identified in our study partially differs from previous datasets31,35,36, pointing to a refined repertoire of direct targets related to our integrated analysis of chromatin and topological profiles associated with EWS-WT1 binding sites.
In contrast, we also observed that a substantial number of genes was induced following EWS-WT1 knock-down (Fig. 3f, left). This led us to investigate whether these changes are due to a repressive role of the fusion protein, as observed for EWS-FLI1 in Ewing sarcoma, or are related to changes in cellular phenotype that follow the loss of the fusion protein. We first observed that only 36% of up-regulated genes (n = 672) were initially connected to EWS-WT1 (Supplementary Fig. S4G), suggesting that indirect effects account for most gene upregulation. Deeper investigation of the 672 connected genes showed that most were targeted by a set of EWS-WT1-independent enhancers either arising de novo (57%) or showing increases in activity (11%) upon knock-down of the fusion protein (Supplementary Fig. S4H). Furthermore, de novo enhancers showed enrichment for the DNA motif recognized by the transcription factor AP-1 (Supplementary Fig. S4I). These results suggest that the majority of increases in gene expression that follow EWS-WT1 depletion reflect cellular phenotypic changes. Nevertheless, a yet unidentified mechanism of transcriptional repression directly controlled by EWS-WT1 may still apply to a minority of target genes (Supplementary Fig. S4H).
EWS-WT1 + KTS/-KTS isoforms control both shared and specific chromatin binding sites and transcriptional programs
Similar to wild-type WT1, EWS-WT1 also displays two splice isoforms that differ by the inclusion/exclusion of three amino acids (KTS) between the last two zinc fingers of its WT1 portion8. Although the function of both +KTS and -KTS isoforms has been previously investigated by exogenous expression in different cellular models, the specific oncogenic contribution of each isoform to DSRCT initiation and development remains elusive24,25,26. Our RNA-seq analysis revealed that 50 to 80% of EWSR1-WT1 transcripts correspond to the +KTS isoform in eleven primary human DSRCT (Supplementary Fig. S5A), consistent with previous studies that suggested the +KTS to be the more abundant of the two isoforms in DSRCT8. However, given that commercially available WT1 antibodies cannot distinguish between the two isoforms, the precise expression pattern of +KTS and -KTS in DSRCT cells still remains to be defined. To address this issue, we turned to RNA in situ hybridization (Basescope) to detect signals from each alternative transcript separately37. We designed probes targeting the EWSR1-WT1 RNA region that either include or exclude the 9 nucleotides coding for the KTS tripeptide, allowing selective detection of each splice variant. Using this approach we detected co-expression of both EWSR1-WT1 isoforms in JN-DSRCT1 cells (Supplementary Fig. S5B), suggesting that they may act in concert to initiate and maintain the global tumor oncogenic program.
To address the specific role of each isoform, we turned to a primary immortalized human mesothelial cell-line (MeT-5A), in which we expressed the two EWSR1-WT1 isoforms, either independently or together. Mesothelial cells have been proposed as possible DSRCT cells of origin4 and we reasoned that our experimental strategy may help define the relative contribution of each isoform to the early stages of DSRCT development. To distinguish between the two protein isoforms, MeT-5A cells were transduced with either V5- (+KTS) or HA-tagged (-KTS) EWS-WT1 constructs and used to generate chromatin occupancy profiles for each isoform by ChIP-seq, using either anti-V5 or -HA antibodies. We identified 43686 and 19185 peaks for the -KTS isoform and +KTS isoform, respectively (Fig. 4a), suggesting that the -KTS isoform displays a higher DNA binding capacity, although this difference may also be partly due to distinct ChIP antibody efficiencies. However, the genomic distribution of the two isoforms was similar, involving mainly distal regulatory regions, as initially observed for endogenous EWS-WT1 in JN-DSRCT1 cells (Fig. 1a).

a Pie charts show genomic distribution of EWS-WT1 binding sites in MeT-5A cells transduced with either the EWS-WT1 -KTS or +KTS isoforms. b Top enriched DNA motifs identified in MeT-5A cells expressing either EWS-WT1 + KTS or -KTS as in Fig. 4a. Associated transcription factors for the motifs were EGR2 (top) and ZNF263 (bottom). p values were calculated based on the binomial test by the Homer software. c Pie chart shows the percentage of consensus EWS-WT1 binding sites initially identified in JN-DSRCT1 cells (as in Fig. 1a), and that are bound by either EWS-WT1 + KTS, EWS-WT1 -KTS or both upon their expression in MeT-5A cells. d Heatmaps of V5 (EWS-WT1 + KTS), HA (EWS-WT1 -KTS) and H3K27ac ChIP-seq signal densities in MeT-5A cells expressing either EWS-WT1 -KTS or +KTS, illustrating that the two isoforms bind to a majority of sites identified in JN-DSRCT1 cells (as in Fig. 1a). 10-kb windows in each panel are centered on EWS-WT1 binding sites as defined in JN-DSRCT1 cells. The categories of sites are: bound by either EWS-WT1 + KTS (n = 240), EWS-WT1 -KTS (n = 3139) or both (n = 1083). Chromatin profiling was performed twice in EWS-WT1 transduced MeT-5A cell line. e Composite plots of V5 (EWS-WT1 + KTS) and HA (EWS-WT1 -KTS) ChIP-seq signals in MeT-5A cells at each category of EWS-WT1 binding sites shown in panel (d). 10-kb windows in each panel are centered on EWS-WT1 binding sites. f Venn diagram depicting the number of genes which are shared between the ones induced by EWS-WT1 + KTS/-KTS in MeT-5A cells, and the ones connected to EWS-WT1 sites and downregulated upon EWS-WT1 knockdown in JN-DSRCT1 cells. g Pie chart showing that the vast majority (86%) of genes induced upon expression of EWS-WT1 + KTS or -KTS in MeT-5A cells are also downregulated upon EWS-WT1 knockdown in JN-DSRCT1 cells. h Heatmap shows expression levels for genes induced by either EWS-WT1 + KTS (n = 70), EWS-WT1 -KTS (n = 174) or both EWS-WT1 isoforms (n = 148) upon their expression in MeT-5A cells. Expression profiling was performed thrice in EWS-WT1 transduced MeT-5A cell line.
Motif enrichment analyses of +KTS/-KTS binding sites, when expressed in isolation, indicated that both isoforms recognized similar albeit slightly different DNA sequences centered on the CTCCC core motif (Fig. 4b and Supplementary Fig. S5C). Both isoforms combined bound a majority of EWS-WT1 binding regions identified in the DSRCT tumor cell line JN-DSRCT1 (65%, Fig. 4c). Interestingly, each isoform expressed separately in MeT-5A cells showed the ability to bind both shared (1083 sites, 15.9%) and specific sites (+KTS only: 240 sites, 3.5%; -KTS only: 3139 sites, 46%)(Fig. 4c–e), suggesting that EWS-WT1 occupancy profiles in DSRCT cells may reflect a combination of +KTS/-KTS binding patterns.
Further integration of EWS-WT1 profiles with chromatin states revealed activation at the vast majority of specific and shared +KTS/-KTS sites, as demonstrated by strong H3K27ac signals (Fig. 4d). Closer examination, however, revealed that whereas both +KTS and -KTS isoforms successfully bound to and induced H3K27 acetylation at TCC repeat containing regions identified in DSRCT cells, only the -KTS isoform succeeded in inducing robust and consistent H3K27 acetylation at a majority of CTCCC cluster regions (Supplementary Fig. S6A).
Finally, expression of EWS-WT1 + KTS/-KTS isoforms in MeT-5A cells also resulted in the induction of a large fraction of the direct target genes identified in JN-DSRCT1 cells (32%; Fig. 4f). Conversely most genes induced in MeT-5A were also regulated by EWS-WT1 in JN-DSRCT1 cells (86%; Fig. 4g and Supplementary Fig. S6B) supporting the biological relevance of our model. In line with our observations based on chromatin activity, both isoform-specific and shared transcriptional programs were also observed (Fig. 4h).
EWS-WT1 DNA occupancy profiles are distinct from wild type WT1, and depend on EWS prion-like domains
We previously investigated the functional contribution of wild-type EWS and FLI1 proteins to the function of EWS-FLI1 in Ewing’s sarcoma, and identified an important role of the EWS prion-like domains (PrLD) for the pioneering properties of EWS-FLI112. We therefore reasoned that similar mechanisms may be shared by EWS-WT1 and interrogated the contribution of EWS PrLD to the DNA binding patterns of the fusion protein. To this end, we generated EWS-WT1-V5 mutants for each KTS isoform, by converting all 37 tyrosines present in the EWS PrLD into serines (EWS(YS37)-WT1-V5, Fig. 5a), and expressed the two mutants in MeT-5A cells (Supplementary Fig. S6C). In parallel, we expressed both wild-type WT1 + KTS and -KTS isoforms in MeT-5A cells (Supplementary Fig. S6C) and profiled all models by ChIP-seq using an anti-V5 antibody.

a Schematics of the constructs encoding for EWS, WT1, EWS-WT1 and the EWS(YS37)-WT1 mutant proteins. Tyrosines (Y) mutated into serines (S) are depicted as black bars within the EWS N-terminal prion-like domain of the EWS(YS37)-WT1 mutant. b Barplot depicts the number of genome-wide ChIP-seq binding sites observed for each protein as in Fig. 5a (+and -KTS isoforms for WT1, EWS-WT1 and EWS(YS37)-WT1) after their individual expression in MeT-5A cells. c Pie charts show the genomic distribution for the binding sites of the + and -KTS isoforms of WT1 and EWS(YS37)-WT1 upon their individual expression in MeT-5A cells. d Heatmaps depict V5 (WT1 + KTS or -KTS) and H3K27ac ChIP-seq signal densities in MeT-5A cells expressing either WT1 isoform, at the subset of consensus EWS-WT1 binding sites bound by either EWS-WT1 -KTS or +KTS as in Fig. 4d. 10-kb windows in each panel are centered on EWS-WT1 binding sites as defined in JN-DSRCT1 cells. Chromatin profiling was performed twice in WT1 transduced MeT-5A cell line. e Heatmaps showing V5 (EWS(YS37)-WT1 + KTS or -KTS) and H3K27ac ChIP-seq signal densities in MeT-5A cells expressing either EWS(YS37)-WT1 -KTS or +KTS mutant isoforms, at the subset of consensus EWS-WT1 binding sites bound by either EWS-WT1 -KTS or +KTS as in Fig. 4d. 10-kb windows in each panel are centered on EWS-WT1 binding sites as defined in JN-DSRCT1 cells. Chromatin profiling was performed twice in EWS(YS37)-WT1 transduced MeT-5A cell line. f EWS-WT1 + KTS and -KTS isoforms precipitate in a dose-dependent manner upon b-isox treatment in lysates from HEK293T-transfected cells. The experiments were independently repeated twice with similar results. Source data are provided as a Source Data file. g, h Wild-type WT1 and mutant EWS(YS37)-WT1 isoforms show weak precipitation properties when b-isox is added to lysates from HEK293T-transfected cells. * non-specific binding of WT1 proteins is detected in absence of b-isox. The experiment was independently repeated twice with similar results. Source data are provided as a Source Data file.
Analysis of the genomic distribution of wild-type WT1 binding sites revealed a limited DNA binding capacity of the +KTS isoform (Fig. 5b, c), suggesting that similar to its effect on EWS-FLI1 in Ewing’s sarcoma, the EWS PrLD provides neomorphic DNA binding properties to +KTS EWS-WT1 (Fig. 4a). Next, we coupled the occupancy profiles for wild-type WT1 and mutant EWS(YS37)-WT1 + KTS/-KTS isoforms with chromatin states across EWS-WT1 binding sites identified in DSRCT cells and observed that despite binding a significant fraction of sites, the -KTS WT1 isoform did not induce chromatin activation, as indicated by the absence of H3K27ac deposition at these genomic regions (Fig. 5d and Supplementary Fig. S6D). The same held true for both mutant EWS(YS37)-WT1 isoforms, which displayed neither chromatin binding nor activation ability at all tested sites (Fig. 5e and Supplementary Fig. S6E).
As previously shown in the context of EWS-FLI1 in Ewing sarcoma, chromatin binding and activation properties correlated with the ability of EWS-WT1, wild-type WT1 and the EWS(YS37)-WT1 mutants to precipitate or not in presence of biotinylated isoxazole (b-isox) as a proxy for their phase-transition properties12,38. Our results revealed that in JN-DSRCT1 cells, EWS-WT1 exhibits robust and b-isox concentration-dependent precipitation, which was even stronger than that of wild-type EWS (Supplementary Figs. S7A, B). The same held true for both EWS-WT1 + KTS and -KTS isoforms expressed in HEK293T cells (Fig. 5f), but not for wild-type WT1, which showed limited ability to precipitate even at the highest b-isox concentration (Fig. 5g). Finally, neither of the EWS(YS37)-WT1 mutant isoforms precipitated, irrespective of b-isox concentrations (Fig. 5h). Altogether these observations support the notion that the DNA binding ability displayed by EWS-WT1 + KTS is largely a neomorphic property acquired by the fusion protein, and that the EWS PrLD is essential for the phase transition ability of both EWS-WT1 isoforms.
Both EWS-WT1 +KTS and -KTS isoforms are required to induce DSRCT in vivo
Based on these results, we next sought to test the relative contribution of each EWS-WT1 isoform to in vivo DSRCT tumorigenesis by assessing the tumorigenic capacity of MeT-5A cells transduced with either an empty vector (control), V5-tagged EWS-WT1 + KTS, HA-tagged EWS-WT1 -KTS, or both EWS-WT1 + KTS/-KTS isoforms simultaneously. Isoform-specific expression of the EWS-WT1 protein alone or in combination was confirmed by Western blot using anti-V5 and -HA antibodies (Fig. 6a). 2 × 106 MeT-5A cells from each condition were injected subcutaneously into the flank of 5–7 week-old NOD/SCID c Gamma (NSG) mice. Remarkably, tumor formation was observed only when mice were injected with MeT-5A cells expressing both EWS-WT1 + KTS and -KTS isoforms (Fig. 6b). MeT-5A cells expressing EWS-WT1 -KTS alone developed small lesions that failed to progress into full-blown tumors (Fig. 6b) suggesting that tumor formation may require the combined expression of the transcriptional programs controlled by each isoform (Fig. 4h).

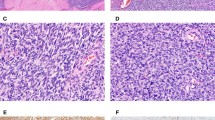
a Immunoblots of whole cell extracts from MeT-5A cells lentivirally transduced with either control vector, V5-EWS-WT1 + KTS, HA-EWS-WT1 -KTS or both EWS-WT1 isoforms concomitantly before subcutaneous injection into immunocompromised mice. GAPDH confirms equal protein loading. Source data are provided as a Source Data file. b MeT-5A cells co-expressing EWS-WT1 -KTS and +KTS isoforms generate tumors in immunocompromised mice. Boxplots show tumor weight 9 months after subcutaneous injection of 6 week-old female mice. 7 tumors were assayed per condition. (**) two-sided p value Wilcox rank-sum test = 0.001058. Median value is shown as a line within the boxplot, which spans from the 25th to 75th percentiles. Whiskers indicate minimum to maximum value range. The displayed results were obtained from a single in vivo experiment. Source data are provided as a Source Data file. c Micrographs show H&E staining of a primary DSRCT tumor (Top) and a tumor derived from MeT-5A cells co-expressing EWS-WT1 -KTS and +KTS isoforms implanted in mice as in (b) (Bottom). Scale bar: 300 μm. d UMAP plot based on RNA-seq showing clustering of MeT-5A-derived tumor xenografts with the JN-DSRCT1 cell line and primary DSRCT tumor samples. Comparison with dedifferentiated Liposarcoma (DDLPS), Myxofibrosarcoma (MFS), Malignant Peripheral Nerve Sheath Tumor (MPNST), SARC (other Sarcoma), Synovial Sarcoma (SS), Leiomyosarcoma arising in gynecologic location (uterine ULMS) and soft tissue (STLMS) sites, and Undifferentiated Pleomorphic Sarcoma (UPS) described in ref. 31.
Importantly, the histology of the tumors generated by MeT-5A cells expressing both EWS-WT1 isoforms recapitulated the defining morphological features of primary human DSRCT, including a small blue round cell phenotype and the presence of an abundant stromal reaction surrounding the tumor cells (Fig. 6c and Supplementary Fig. S8)4. To assess this phenotypic similarity at the molecular level, we profiled MeT-5A-derived tumors by RNA-seq, and compared their gene expression profiles to those derived from a cohort of 9 different primary sarcoma subtypes31, including DSRCT, as well as to JN-DSRCT1 cells. The tumors that developed from EWS-WT1-transformed MeT-5A cells clustered with both primary DSRCT and JN-DSRCT1 cells, underscoring their morphological and molecular resemblance to patient-derived tumors (Fig. 6d).
Combined analysis of chromatin occupancy and looping profiles identify CDK4/6 as a new potential therapeutic target for DSRCT tumors
Our analyses so far highlighted the prominent role of EWS-WT1 in regulating the epigenetic and transcriptional landscape of DSRCT and showed that the fusion protein controls its target gene repertoire through a complex network of chromatin interactions. We therefore reasoned that direct transcriptional targets required for tumor cell survival may be regulated by a high number of EWS-WT1-centered loops to maintain robust and constant expression.
To address this possibility, we ranked the top 1000 EWS-WT1-connected target promoters by the number of EWS-WT1-associated loops involved and selected those displaying 5 or more chromatin interactions (Fig. 7a, Supplementary Data 1). This analysis identified a total of 323 promoters corresponding to genes that are highly expressed in primary DSRCT compared to other human sarcoma subtypes (Fig. 7b). A large fraction of these genes was connected to EWS-WT1 bound CTCCC clusters and/or TCC repeats (94%). We next ranked this gene set based on their expression levels in primary DSRCT and their transcriptional changes upon EWS-WT1 depletion in JN-DSRCT1 cells (Supplementary Fig. S9A). Finally, to increase the potential translational reach of the analysis, target genes were filtered according to their susceptibility to inhibition by clinically-approved drugs based on the Drug Gene Interaction database (Fig. 7c and Supplementary Fig. S9A)39. This strategy identified 9 actionable targets (Fig. 7c) from which we selected the CCND1 gene, based on its role in regulating tumor cell proliferation and the possibility to target its function by potent and selective inhibitors, which are currently used in the clinic.

a Hockey plot shows the number of EWS-WT1 associated loops connected to genes transcriptionally down-regulated upon EWS-WT1 knockdown in the JN-DSRCT1 cell line (n = 1000). Out of most connected 1000 promoters, 323 target genes displaying more than 5 EWS-WT1-associated loops were selected for further analysis. H3K27ac Hi-ChIP in JN-DSRCT1 cell line was performed once. Expression profiling was performed four times in shRNA transduced JN-DSRCT1. b Heatmap depicts gene expression levels in primary DSRCT tumors for the 323 genes connected to EWS-WT1 binding sites and whose expression decreases upon the fusion protein knockdown identified in Fig. 3f. Comparison with Dedifferentiated Liposarcoma (DDLPS), Myxofibrosarcoma (MFS), Malignant Peripheral Nerve Sheath Tumor (MPNST), SARC (Sarcoma), Synovial Sarcoma (SS), Leiomyosarcoma arising in gynecologic location (uterine ULMS) and soft tissue (STLMS) sites, and Undifferentiated Pleomorphic Sarcoma (UPS) described in ref. 31. c Top 9 target genes that can be targeted by at least 1 clinically-approved compound inhibiting their activity amongst the 323 that are connected to EWS-WT1 sites and show high expression in primary DSRCT tumors (as in Fig. 7a, b). d CCND1 expression levels across a panel of primary sarcoma samples described in ref. 31. Number of samples: DDLPS n = 51, MFS n = 17, MPNST n = 5, DSRCT 10, SARC n = 53, SS n = 10, STLMS n = 54, ULMS n = 29, UPS n = 45. Median value is shown as a line within the boxplot, which spans from the 25th to 75th percentiles. Whiskers indicate a 1.5× inter-quartile range. e Left: Example shows marked changes in H3K27ac Hi-ChIP connectivity at the CCND1 locus upon EWS-WT1 knockdown in the JN-DSRCT1 cell line. Right: changes in CCND1 expression levels in JN-DSRCT1 cells upon EWS-WT1 knockdown, as assessed by RNA-seq. f Absolute Palbociclib IC50 value for JN-DSRCT1 cells, as compared to a panel of 492 tumor cell lines described in ref. 41. g Boxplots show weight of JN-DSRCT1-derived tumors in Palbociclib- or solvent-treated-immunocompromised mice at the end of the treatment. 6 week-old male mice were injected at the beginning of the study. The total number of treated tumors were 15 and 14 respectively. Median value is shown as a line within the boxplot, which spans from the 25th to 75th percentiles. Whiskers indicate minimum to maximum value range. p values were calculated using two-sided t tests. (**) p value = 0.0033. Source data are provided as a Source Data file. h Tumor growth progression over time of patient-derived DSRCT xenografts (SJDSRCT046151_X1 = SJ-PDX1), in solvent and Palbociclib-treated immunocompromised mice. 6 week-old male mice were injected at the beginning of the study. The total number of treated tumors were 16 and 12 respectively. p values were calculated using two-sided t tests. (***) p value at endpoint = 5.7 e-6. Source data are provided as a Source Data file. i Boxplots show weight of SJ-PDX1-derived tumors in solvent- or Palbociclib-treated-immunocompromised mice at the end of the treatment. The total number of treated tumors were 16 and 12, respectively. p values were calculated using two-sided t tests. (***) p value = 3.3 e–6. Median value is shown as a line within the boxplot, which spans from the 25th to 75th percentiles. Whiskers indicate minimum to maximum value range. Source data are provided as a Source Data file.
Consistent with our in-silico analyses, CCND1 showed high expression levels in a cohort of primary DSRCT compared to 8 other human sarcoma subtypes (Fig. 7d)31 and was strongly dependent on EWS-WT1 expression in JN-DSRCT1 cells (Supplementary Figs. S9B and 7e right inset). EWS-WT1 depletion also resulted in a robust decrease in 3D connectivity at the CCND1 genomic locus, supporting the strong dependency of the expression of this gene on the fusion protein (Fig. 7e). Moreover, CCND1 depletion by shRNAs in JN-DSRCT1 cells reduced their proliferation to an extent similar to EWS-WT1 depletion itself, supporting the relevance of targeting CCND1 as a therapeutic strategy for DSRCT (Supplementary Fig. S9C).
Recently, several pharmacological inhibitors that affect CCND1 activity have been generated. These compounds target the enzymatic activity of CDK4/6, and therefore reduce the ability of the CCND1-CDK4/6 complex to promote cell cycle progression through the G1 phase. In particular, three inhibitors have been approved by the Food and Drug Administration (FDA) for the treatment of metastatic breast cancer40: Palbociclib, Abemaciclib, and Ribociclib, prompting us to assess their therapeutic potential in DSRCT cells and mouse xenografts. To assess the sensitivity of DSRCT cells to CDK4/6 inhibition, we took advantage of a recent study that screened over 400 different tumor cell lines for their sensitivity to Abemaciclib and Palbociclib41. Since this cohort lacked DSRCT cell lines, we followed the same protocol to treat JN-DSRCT1 cells and assessed their sensitivity to both CDK4/6 inhibitors. Our results confirmed that JN-DSRCT1 cells are highly sensitive to both Abemaciclib and Palbociclib in vitro, with absolute IC50 values of 0.18 µM and 1.09 µM, respectively (Supplementary Figs. S9D, E), which ranked them in the top 5% most sensitive cell lines (Fig. 7f and Supplementary Fig. S9F).
To expand our in vitro results to more clinically relevant models, we next investigated the effect of CDK4/6 inhibitors on DSRCT growth in vivo. To this end, we generated JN-DSRCT1-derived mouse xenografts, and treated them daily with 100 mg/kg Palbociclib in cycles of 5 out of 7 days for a total of 26 days. This drug concentration is close to that administered in human clinical settings, which may vary between 125 mg to 200 mg/kg per day, during 14 to 28 days40, and was well tolerated by all mice with no obvious sign of distress or weight loss (Supplementary Fig. S10A). Mice were monitored for tumor growth three times per week and at the end of the experiment (Supplementary Fig. S10B). Our results confirmed the marked anti-tumor effect of Palbociclib in vivo, with all treated mice showing a significant decrease in tumor burden (Fig. 7g, Supplementary Figs. S10B, S10C). To further corroborate these results, we generated a second in vivo model (SJ-PDX1), using a patient-derived xenograft obtained from the Childhood Solid Tumor Network42. These additional xenografts were treated and monitored following the same protocol during 19 days. Similar to the results obtained with JN-DSRCT1-derived tumors, patient-derived xenografts treated with 100 mg/kg of Palbociclib also showed marked growth reduction (Fig. 7h, i, Supplementary Figs. S10D, S10E), highlighting the clinical relevance of our findings.
Discussion
EWS fusion proteins constitute a large set of related oncogenes that are responsible for the generation of diverse tumor types whose spectrum continues to widen with the application of molecular diagnostic technologies capable of detecting novel chromosomal translocations43. However, our understanding of the corresponding fusion protein function and precise role in cell transformation has not paralleled the progress in their detection and discovery, limiting the development of effective therapies. To further elucidate the oncogenic function of EWS fusion proteins, here we investigated the mechanistic implications of EWS-WT1 in the formation of DSRCT and found that it operates as a powerful chromatin and transcriptional activator, responsible for shaping the transcriptional and chromatin interaction landscapes of DSRCT cells. Previous studies, most notably in Ewing sarcoma, have shown that the N-terminal prion-like domains (PrLDs) of EWS can mediate nuclear condensate formation and enable the TF to which they are fused to bind otherwise inaccessible genomic sites, endowing the corresponding fusion protein with pioneer factor properties. Our present results show that these principles are operative in diverse cellular contexts, and suggest that they may represent a common feature required for cellular transformation across EWSR1-translocations. They also point to the importance of the partner TF in establishing distinctive oncogenic programs.
The generation of two distinct, functionally complementary, oncogenic fusion protein isoforms from a single genetic alteration is a salient feature of DSRCT that is linked to the developmental splicing patterns of WT1. Our study shows that both oncogenes are co-expressed within single DSRCT cells and further define the dramatic increase in EWS-WT1 + KTS genomic occupancy compared to its wildtype counterpart, allowing both isoforms to contribute to the oncogenic transcriptional program. Given that wild type WT1 primarily operates as a tumor suppressor and that its +KTS isoform has a strong preference for RNA-binding14, it is remarkable that addition of the N-terminal domains of EWS endows WT1 with new oncogenic properties. Moreover, our observations indicate that the distinct target gene repertoires of the two fusion protein isoforms cooperate to shape the transcriptional and oncogenic properties of DSRCT. We find that +KTS and -KTS EWS-WT1 display both shared and private chromatin binding profiles and transcriptional targets, and that the combined expression of their complementary transcriptional outputs in precursor cells is required for tumor initiation and progression, each isoform providing a key portion of the required pro-tumorigenic signals.
Finally, we have combined chromatin profiling with 3D nuclear mapping to identify direct EWS-WT1 target genes of potential therapeutic value. Among them we selected CCND1, based on its well-known oncogenic functions, as well as its pharmacological targetability using CDK4/6 inhibitors. Inactivation of the CCND1-CDK4/6 axis has been previously assessed in other sarcoma subtypes, including Ewing44 and synovial sarcoma45, with encouraging preliminary results. These agents are also currently approved to treat estrogen receptor-positive metastatic breast cancers in combination with endocrine therapies, with surprisingly low toxicity profiles. Intriguingly, the high male prevalence of DSRCT tumors alongside their robust expression of the androgen receptor raise the possibility that combining hormonal therapy with CDK4/6 inhibitors may also be applicable in DSRCT46.
In summary our study shows how a single genetic alteration can result in the expression of two complementary oncogenes through post-transcriptional and gain of function events, elucidates the mechanisms by which these aberrant TFs induce strong chromatin and topological changes, and identifies CDK4/6 as a potentially attractive therapeutic target for the clinical management of these tumors. Given the importance of fusion proteins as drivers in cancer, we expect that similar mechanisms may play a role in variety of cancer types and that their detailed characterization may point to new therapeutic opportunities.
Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to the Lead Contacts Nicolò Riggi (riggin@gene.com) and Miguel N Rivera (mnrivera@mgh.harvard.edu).
Experimental model and subject details
DSRCT tumor samples
Primary tumor specimens were collected with ethical approval from the Institutional review Boards (IRB) of Center Hospitalier Universitaire Vaudois (CHUV, University of Lausanne, Switzerland), and Massachusetts General Hospital (MGH, Boston, USA). Samples were anonymized prior to analysis. For samples collected after 2015, informed written consent was obtained from patient and/or their legal representatives under the Commission Cantonale d’éthique de la recherche sur l’être humain (CER-VD, protocol number 260/15). Tumor samples collected before 2015 were obtained from the tumor bank as discarded material with IRB approval, without informed consent.
Cell lines
The JN-DSRCT1 cell line was a generous gift from Daniel Haber. Other cell lines were obtained from the ATCC and media from Life Technologies. JN-DSRCT127 (male gender), MeT-5A (male gender, ATCC CRL-9444) and LentiX HEK293T (female gender, Takara #632180) cells were respectively grown in Dulbecco’s Modified Eagle Medium (DMEM/F12), Medium 199 and DMEM. All media were supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin/Streptomycin. All cells were cultured at 37 °C with 5% CO2. Cells were maintained and split every 2–4 days (at approximately 80% confluence). When needed, FBS was replaced with Tet-Free FBS from Pan Biotech (P30-3602). Cells were cryopreserved in cryogenic medium [90% FBS with 10% dimethyl sulfoxide (DMSO)] into liquid nitrogen for long-term storage. Cells were authenticated by the provider. All cell cultures were periodically tested negative for mycoplasma contamination by PCR-based methods.
Lentiviral generation
Lentivirus was produced in LentiX HEK293T either by LT1 (Mirus Bio) or by FuGene6 (Promega) transfection with gene delivery vector and packaging vectors GAG/POL and VSV plasmids. Growth medium was changed after 6–8 h from the initial transfection. Viral supernatants were collected 72 h after transfection and concentrated using LentiX concentrator (Clontech-Takara), following the manufacturer’s instructions. Virus containing pellets were resuspended in PBS and added dropwise on cells in presence of media supplemented with 6 μg/ml polybrene. JN-DSRCT1 and Met-5A lentivirally-infected cells were selected using 1 μg/ml puromycin or 0.25 μg/ml G418. Induction of gene expression in lentivirally-infected cells was achieved adding 250 μg/ml doxycycline to the culture medium. Overexpression or knockdown efficiency was determined by Western blot analysis and RT-qPCR.
Transient transfections
HEK293T cells were grown to 80% confluency prior to transfection using LT1 (Mirus Bio) according to the manufacturer recommendations and were collected after 48 h.
Real-time quantitative RT-PCR
For gene expression assays, total RNA was isolated from cells using either NucleoSpin RNA Plus (Clontech) or RNeasy mini kit (Qiagen, cat#74104), DNAse treatment was performed following the manufacturer’s instructions. cDNA was obtained using a high-capacity RNA-to-cDNA kit (Applied Biosystems). 500 ng to 1 μg of template total RNA and random hexamers were used for each reaction. Real-time qPCR amplification was performed using fast SYBR Green Master Mix (Life Technologies), or PowerUp SYBR® Green PCR Master Mix (Applied Biosystems) and specific PCR primers in a Lightcycler 480 instrument (Roche) or QuantStudio 5 Real-Time PCR System (ThermoFisher Scientific). Oligonucleotides used are provided in Supplementary Table 1. Relative quantification of each target, normalized to an endogenous control (GAPDH), was performed using the comparative Ct method (Applied Biosystems). Error bars indicate SD of three technical replicates and represent two independent biological experiments. Statistical analyses were performed by Student’s t-test.
Western blot analysis
Cell lysis, SDS-PAGE and immunoblotting were performed using standard procedures. Primary antibodies used for Western blotting are listed in Supplementary Table 2. Primary antibodies HA (clone 3F10, cat#11867423001, Roche), V5 (SV5-Pk1, cat#ab27671, Abcam), WT1 (cat#12609-1-AP, Proteintech) were used at a final 1:1000 dilution; β-actin (AC-15, cat#ab6276, Abcam), GAPDH (HRP-conjugated: clone GAPDH-71.1, cat#G9295, Sigma), GAPDH (clone 6C5, cat# MAB374, Millipore) were used at a final 1:10 000 dilution; WT1 (C19, cat#sc-192, Santa Cruz Biotechnology) was used at a final 1:200 dilution and EWSR1 (G5, cat# sc-28327 Santa Cruz Biotechnology) was used at a final 1:500 dilution. Secondary antibodies were goat anti-mouse, goat anti-rabbit and goat anti-rat horseradish peroxidase-conjugated immunoglobulin G. Membranes were developed using Western Lightning Plus-ECL enhanced chemiluminescence substrate (PerkinElmer) and visualized using photographic film. Alternatively, protein signals were revealed by SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and captured with the Fusion FX device (Vilber Lourmat) and analyzed with the FusionCapt Advance FX7 software.
Cell proliferation assays
Cell Proliferation Assays were performed as previously described41. Briefly, the day before the experiment, JN-DSRCT1 cells were plated in complete media in white-walled flat-bottom plates (ThermoFisher Scientific) at the predetermined optimal density (2000 cells per well to keep cells in a logarithmic growth phase during the experiment timeline). 16 h after plating, different concentrations of Abemaciclib (Selleckchem, LY2835219) or Palbociclib (Selleckchem, PD0332991), ranging from 0.001 μM to 20 μM were added to the cells. CellTiter-Glo® (Cat# G7571, Promega Corporation) was prepared according to the manufacturer’s protocols. 76 h after compound addition (corresponding to two JN-DSRCT1 cell doubling times27), CellTiter-Glo® reagent was added to each well. Plates were incubated at room temperature for 10 min and luminescence measured using a plate reader (Synergy Mx, BioTek Instruments, Cat# 7191000). Half maximal inhibitory concentration (IC50) values were calculated using the GraphPad Prism software. Absolute IC50 for each drug was determined as the concentration reducing cells viability by 50%, as compared to control cells, using GraphPad Prism Software file (https://www.graphpad.com/guides/prism/7/curvefitting/index.htm?reg_fitting_the_absolute_ic50.htm).
Immunoprecipitations
Immunoprecipitations were performed as previously described12. Briefly, cells were resuspended in IPH Buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 5 mM EDTA, 0.5 % NP-40 and 10% glycerol supplemented with protease and phosphatase inhibitors (Pierce) and 1 mM PMSF) before sonication in a QSONICA 800 R instrument. Protein supernatant was then collected after centrifugation for 15 min at 21,000 × g and 4 °C. Proteins were quantified using a Bradford assay (Pierce) and 500 μg to 1 mg of lysate were diluted in IPH buffer to a final volume of 1 mL and incubated overnight at +4 °C with 2 μg of EWS antibody (Bethyl A300-418A), in the presence of magnetic G-Dynabeads (Life Technologies) and 100 μg/mL of Ethidium Bromide (SIGMA-ALDRICH). Beads were washed 5 times with IPH buffer and eluted by boiling in loading Laemmli buffer.
ChIP-seq
ChIP assays were carried out on approximately 2–5 million cells per sample and per epitope, following the protocol described previously47. Briefly, chromatin from formaldehyde-fixed cells was fragmented to a size range of 200–700 bases using a Branson 250 sonicator. Solubilized chromatin was immunoprecipitated with the indicated antibodies (listed in Supplementary Table 3) overnight at 4 °C. 3 μg of HA (clone 3F10, cat#11867423001, Roche) or V5 (D3H8Q, cat#13202, Cell signaling) antibodies per 3–5 million cells were used. 5 μg per 3–5 million cells were used for WT1 (cat#12609-1-AP, Proteintech), WT1 (C19, cat#sc-192, Santa Cruz Biotechnology), H3K4me3 (cat#07-473, Millipore), H3K27ac (cat#39133, Active motif), H3K4me1 (cat#ab8895, Abcam), H3K27me3 (C36B11, cat#9733, Cell signaling or cat#07-449, EMD Millipore), H3K9ac (cat#ab4441, Abcam) and H3K36me3 antibodies (cat#ab9050, Abcam). Antibody-chromatin complexes were pulled down with protein G-Dynabeads (Invitrogen), washed, and then eluted. After crosslink reversal, RNase A, and proteinase K treatment, immunoprecipitated DNA was extracted with AMP Pure beads (Beckman Coulter). ChIP DNA was quantified with Qubit (Life technologies). 1 to 5 ng of ChIP DNA was used to prepare sequencing libraries, and ChIP DNA and input controls were sequenced with the Nextseq 500 Illumina genome analyzer.
ChIP-seq bioinformatic processing
Reads of ChIP-seq sequencing were aligned against GRCH37 (hg19) genome using bwa v.0.7.1248 with default settings. Aligned reads were then filtered to exclude PCR duplicates using picard-tools v.1.95 and were extended to 200 bp to approximate fragment sizes. Density maps were generated by counting the number of fragments overlapping each position using igvtools, and normalized to 10 million reads using BEDtools v.2.17.0. Average ChIP-seq signals across intervals were calculated using bwtool49.
Peakcall
ChIP-seq peaks were identified by using MACS2 v.2.2.7.150 using matching input controls with a q-value threshold of 0.01 and narrow peak calling setting for transcription factors. Peaks were filtered to exclude blacklisted regions as defined by the ENCODE consortium51 and peaks within 200 bp of each other were merged.
Peak intersections
Peak intersections were identified using bedtools52. Consensus peaks were defined as those common to both WT1 antibodies in JN-DSRCT1 cells (Fig. 1a) or both biological replicates for either HA or V5 in Met-5A cells (Figs. 4a and 5c) using the Bioconductor genomic ranges package53. de novo H3K27ac peaks were defined as peaks with four-fold signal increases upon EWS-WT1 knock-down and a signal of less than 5 relative units in control conditions (Supplementary Fig. S4H).
Peak annotation
Peaks within 1 kb of RefSeq transcription start sites or with strong H3K4me3 ChIP-seq signals (average normalized signal above 8) were considered as promoters, peaks overlapping a RefSeq gene body were considered as distal intragenic and the remaining peaks were considered distal extragenic.
Heatmaps and composite plots
Signals shown in heatmaps (100 bp windows) and composite plots (10 bp window) were calculated using bwtool49. Heatmap signals were plotted by heatmap.2 of gplot2 (2.16.0) package, centered on the indicated peaks and capped at the 99th percentile.
DNA Motif enrichment analysis
For motif discovery, we used the Homer suite of tools v.4.7 (Heinz et al., 2010). “findMotifsGenome.pl” with parameters: -size given -len 4,5,6,7,8,9,10,12,16 was used to identify de novo motifs within indicated peaks.
The fimo v.4.11.2 from MEME54 package was used for motif annotation of EWS-WT1 peaks. EWS-WT1 peaks containing ≥ 3 CTCCC motifs were defined as CTCCC cluster EWS-WT1 peaks, and ≥1 (TCC)4 were defined as TCC repeat EWS-WT1 peaks.
RNA-Seq
Total RNA was isolated from cells using NucleoSpin RNA Plus (Clontech). 0.5–1 ug of total RNA was treated with Ribogold zero to remove ribosomal RNA. Illumina sequencing libraries were constructed using random primers according to the manufacturer’s instructions using the Truseq Stranded RNA LT Kit.
RNA-seq processing and gene expression analysis
RNA-seq reads were aligned against GRCH37 (hg19) using STAR v.2.4.0 h55. Gene expression was quantified with featureCounts56. Sample scaling and differential gene analysis were performed with the R package DESeq257. Aligned fragments were normalized by RPM for hg19 RefSeq genes. Function enrichment (GO) analysis was performed using GSEA.
The primary tumor RNA-seq public data were downloaded from EGAD0000100398031. To compare gene expression to TCGA datasets (https://xenabrowser.net/)58, we used kallisto (v0.43.0)59 for the gene expression quantification. Then, Seurat (v.3.2.2)60 was used for expression normalization. The scaled expression (z-scores for each gene) matrix was used for dimensional reduction. The top 25 principal components were used for visualization with UMAP.
HiChIP data processing
The paired-end H3K27ac HiChIP reads were aligned against the GRCH37 (hg19) genome using the HiC-Pro pipeline (v 2.7.6)61 with default settings. The valid interactions were identified, and the interaction matrices were generated. Next, the hichipper tool (v 0.7.7)62 was used for loop calling with H3K27ac ChIP-seq peak (q-value < 0.00001) as the anchors, with parameter: --max-distance 100000000. Loops with counts number greater than 5 were kept. When comparing shGFP and shEWS-WT1 JN-DSRCT1 HiChIP samples, we generated the union H3K27ac peaks from both shGFP and shEWS-WT1 ChIP-seq peaks. The loops were called using the union H3K27ac peaks by hichipper as described above. Loops were further normalized by the method described in diffloop63. We aligned the paired-end HiC reads against the GRCH37 (hg19) genome using the HiC-Pro pipeline (v 2.7.6) (Servant et al., 2015) with default settings. The valid interactions were identified, and the interaction matrices were generated. Then, O/E matrix was generated by dividing each interaction of the matrix by the expected interaction frequency (means) for a given diagonal distance. TADs were defined with 40-kb bin matrix using the analysis method described by ENCODE (https://github.com/dekkerlab/cworld-dekker; version v0.0.1) for each chromosome. In brief, the boundary strength for each chromosome from the O/E matrix by using matrix2insulation.pl with default settings. Further, insulation2tads.pl was applied to define TAD boundaries by using boundary strength.
Mapping EWS-WT1 network with differentially expressed genes
Genes connected to EWS-WT1 binding sites by loops in their promoter and gene body regions were defined as EWS-WT1 connected genes. EWS-WT1 connected genes which were down-regulated after EWS-WT1 depletion with at least 2-fold decreases and q-value < 0.01 in JN-DSRCT1 cells were selected as EWS-WT1 target genes.
According to the differences between +KTS and -KTS EWS-WT1 ChIP-seq binding peaks in MeT-5A over-expression samples, we separated the JN-DSRCT1 EWS-WT1 connected and down-regulated genes (1.5-fold down-regulated and p value < 0.05) after EWS-WT1 depletion into +KTS/-KTS EWS-WT1 associated genes. Those genes which were up-regulated (1.5-fold up-regulated and p value < 0.05) in MeT-5A over-expression samples were defined as +KTS/-KTS EWS-WT1 targeted genes.
Cell viability assays
JN-DSRCT1 cells were seeded in triplicates and grown under log phase growth conditions in 96 well cell culture plates. After the indicated incubation times, cell viability was measured using the CellTiter-Glo® luminescent cell viability assay (Cat# G7571, Promega Corporation), as described by the manufacturer. Endpoint luminescence was measured on a SpectraMax M5 plate reader (Molecular Devices). The data displayed are representative of two biological experiments. Statistical analyses were performed by Student’s t-test.
Biotinylated isoxazole-mediated precipitation
These assays were performed as previously described38 with slight modifications. Biotinylated isoxazole (b-isox, Sigma-Aldrich) was reconstituted in DMSO. Briefly, 5-10 million cells were resuspended in 1 mL lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM MgCl2, 0.5% NP-40 and 10% glycerol supplemented with 1X Protease/Phosphatase inhibitors (Pierce), 0.1 mM PMSF and 20 mM beta-mercaptoethanol) and incubated for 30 min with rotation at +4 °C. Protein supernatant was then collected after centrifugation for 15 min at 21,000 g + 4 °C. A 5 % whole cell extract (WCE) control was collected and the remaining proteins were divided into four aliquots before addition of biotinylated isoxazole at 0, 10, 30 100 uM final concentrations. The Reaction solutions were incubated at +4 °C for 1 h under rotation and centrifuged for 15 min at 21,000 g + 4 °C. Supernatant was saved for further analysis and pellets were washed twice in supplemented Lysis buffer before resuspension in Laemmli buffer. WCE, pellets and supernatants were analyzed by 4%–12% Tris-Glycine gradient gels (Life Technologies) and Western blotting was performed using standard protocols.
In vivo studies
All mice experiments were approved by the Cantonal Veterinary Office of the Canton de Vaud, authorization number VD3021 and VD3437. 5–7 weeks old NOD/SCID c Gamma (NSG) mice were purchased from Jackson Laboratory, USA (stock number 005557). Light cycle (7AM-7PM) and humidity was controlled at all times. Temperatures varied between 20 °C and 23 °C. Food and water was accessible at all times, autoclaved or irradiated prior use. When needed, mice were fed with doxycycline-containing food (SAFE, U8200P Version 0115, A03 at 0.625 g/kg Doxycycline, irradiated). All mice were monitored three times a week for tumor development, and euthanized using CO2 inhalation and cervical dislocation, when the tumor reached a maximum of 1500 mm3 volume or at the end of the experiment. Every time, an accurate necropsy of the mice was performed to assess local tumor growth and metastatic spread.
MeT-5A xenografts
4 × 106 MeT-5A cells were resuspended in 100 μL of 1X PBS, and injected subcutaneously into 6 week-old female NOD/SCID c Gamma (NSG) mice (Jackson Laboratory, USA). Mice were then monitored, and euthanized as described above. At the end of the experiment, a fragment of each collected tumors was mechanically dissociated using the Human Tumor Dissociation Kit (Miltenyi Biotec) in combination with the Gentle-MACS device, following the manufacturer’s instructions. The remaining fragments were stored at −80 °C or used for histopathological and molecular analyses. When needed, mice were fed with doxycycline-containing food (SAFE, U8200P Version 0115, A03 at 0.625 g/kg Doxycycline, irradiated).
Generation of DSRCT human PDXs
Primary patient-derived DSRCT xenografts (SJDSRCT046151_X1) were obtained from the Childhood Solid Tumor Network42. Samples were defrosted and washed in 1X PBS before immediate implantation into the flank of 6 week-old male NOD/SCID c Gamma (NSG) mice (Jackson Laboratory, USA). Mice were then monitored, and euthanized as described above. Upon collection, each tissue was maintained less than one hour at RT in DMEM with GlutaMax before implantation. Collected tumors were mechanically dissociated and used to generate FFPE blocks for histological analysis, or re-implanted into new NSG mice.
In vivo palbociclib treatment
For the SJ-PDX1 model (SJDSRCT046151_X1) tumors were harvested and minced with scissors into small fragments of 80-100 mg before immediate implantation into the flank of 6 week-old male NOD/SCID c Gamma (NSG) mice (Jackson Laboratory, USA). For the JN-DSRCT1 line, 2 × 106 cells were resuspended in 100uL of 1X PBS prior to subcutaneous injection into 6 week-old male NOD/SCID c Gamma (NSG) mice (Jackson Laboratory, USA). Each experiment included cohorts of eight mice, which were allocated between the control-treated (n = 8) or the drug-treated (n = 8) group to normalize their size before starting the treatment. Palbociclib (Selleckchem PD0332991) was prepared daily and administered by oral gavage as a solution of sodium lactate buffer (Aldrich, 71718, 50 mM Sodium lactate, pH 4.0) based on the mice body weight. When tumors reached an average volume of 50 mm3, mice were treated with either 100 mg/kg of Palbociclib or solvent during 4 weeks (5 days per week, with a 2 days drug holiday during the week end). All mice were monitored daily for signs of distress and were weighed three times a week. Tumor size was measured three times a week with a caliper, and tumor volume calculated according to the following equation: (length x width2)/2 = tumor volume [mm3]. Control-treated mice received the solvent used for oral administration. Mice were euthanized, as described above, the day after the last dose of Palbociclib.
Hematoxylin and eosin (H&E) staining
H&E staining was performed by the mouse pathology facility of the UNIL-CHUV, using standard protocols. Briefly, paraffin blocks were cut at 5 µm, mounted and dried on glass slides. All tissues were deparaffined in xylol, followed by progressive rehydration in EtOH (100%, 95%, 80%) and water. The tissues were stained with Harris hematoxylin (J.T. Baker, cat# 3873) for 4 min and destained with 1% acid ethanol (1% HCl 37% in 70% EtOH) for 3 sec. Tissues were then stained with Eosin (Merck, cat# 15936, 0,2% in dd water and 1/1000 formaldehyde) for 30 sec, dehydrated and mounted on coverslips.
RNA in situ hybridization (BaseScope)
The BaseScope in situ hybridization37 was used to assess specific expression of the +KTS and -KTS EWSR1-WT1 isoforms. To this goal, BaseScope paired oligo-probes were designed to bind either the +KTS or its equivalent region in the -KTS EWSR1-WT1 transcript, and the assay was performed following the manufacturer’s guidelines. Briefly, 5 μm thick tissue sections were mounted on slides, then heated for 2 h at 60 °C, deparaffinized and dehydrated. The tissues were pretreated with Hydrogen Peroxide for 10 min at room temperature and with the Target Retrieval Reagent for 30 min at 98 °C. Protease IV was then applied for 30 min at 40 °C. BaseScope ZZ probes (ACDbio) were hybridized for 2 h at 40 °C, followed by signal amplification. Tissues were counterstained with Gill’s hematoxylin, and mounted on slides using VectaMount mounting media (Vector Laboratories). Images were taken with a Hamamatsu NanoZoomer S60 Digital slide scanner, at 40X magnification.
Quantification and statistical analysis
All statistical details of experiments are included in the Figure legends or corresponding Methods section. Chromatin and expression profiling were performed once in untreated JN-DSRCT1 cell line and primary DSRCT tumor samples (Fig. 1 and Supplementary Figs. S1, S2). Chromatin profiling were performed twice in shRNA transduced JN-DSRCT1 and EWS-WT1 transduced MeT-5A cell lines; expression profiling were performed four times in shRNA transduced JN-DSRCT1 and thrice in EWS-WT1 transduced MeT-5A cell lines (Figs. 3, 4, 5, Supplementary Figs. S5, S6). B-isox precipitation assays and EWSR1 immunoprecipitation assays were performed twice and representative examples are shown (Fig. 5f-g-h and Supplementary Fig. S7). H3K27ac Hi-ChIP in untreated JN-DSRCT1 cell line (Fig. 2 and S3) and in shRNA transduced JN-DSRCT1 (Figs. 3, 7 and Supplementary Fig. S4) was performed once.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data accompanying this study have been deposited into National Center of Biotechnology Information Gene Expression Omnibus (GEO) under accession number “GSE212979 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE212979]”. It contains the datasets “GSE212976 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE212976]” (RNA-seq), “GSE212977 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE212977]” (ChIP-seq), “GSE212978 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE212978]” (Hi-ChIP) and “GSE242863 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE242863]” (HiC). Raw data for ChIP-seq profiles for two primary samples collected prior to 2015 were deposited under restricted access in dbGAP (database of Genotypes and Phenotypes) in order to protect the privacy of research participants (accession number phs003682, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003682.v1.p1). Access can be requested through dbGAP by investigators who are permanent employees of their institutions at a level equivalent to tenure-track professor or senior scientist. Requests are managed by NCI’s Data Access Committee, take approximately 2 weeks for approval and provide access for 12 months. To implement our analysis we used the following publicly available dataset “EGAD00001003980 [https://ega-archive.org/datasets/EGAD00001003980]”31. Source data are provided with this paper. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Mitelman, F., Johansson, B. & Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 7, 233–245 (2007).
Perry, J. A., Seong, B. K. A. & Stegmaier, K. Biology and therapy of dominant fusion oncoproteins involving transcription factor and chromatin regulators in sarcomas. Annu. Rev. Cancer Biol. 3, 299–321 (2019).
Gerald, W. L. et al. Intra-abdominal desmoplastic small round-cell tumor. Report of 19 cases of a distinctive type of high-grade polyphenotypic malignancy affecting young individuals. Am. J. Surg. Pathol. 15, 499–513 (1991).
Mora, J. et al. Desmoplastic small round cell tumor 20 years after its discovery. Fut. Oncol. 11, 1071–1081 (2015).
Hayes-Jordan, A., LaQuaglia, M. P. & Modak, S. Management of desmoplastic small round cell tumor. Semin Pediatr. Surg. 25, 299–304 (2016).
Lal, D. R. et al. Results of multimodal treatment for desmoplastic small round cell tumors. J. Pediatr. Surg. 40, 251–255 (2005).
Mello, C. A. et al. Desmoplastic small round cell tumor: A review of main molecular abnormalities and emerging therapy. Cancers (Basel) 13 https://doi.org/10.3390/cancers13030498 (2021).
Gerald, W. L., Rosai, J. & Ladanyi, M. Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc. Natl Acad. Sci. USA 92, 1028–1032 (1995).
Ladanyi, M. & Gerald, W. Fusion of the EWS and WT1 genes in the desmoplastic small round cell tumor. Cancer Res. 54, 2837–2840 (1994).
Gangwal, K. et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc. Natl. Acad. Sci. USA 105, 10149–10154 (2008).
Riggi, N. et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 26, 668–681 (2014).
Boulay, G. et al. Cancer-specific retargeting of BAF complexes by a prion-like domain. Cell 171, 163–178 e119 (2017).
Slotkin, E. K. et al. Comprehensive molecular profiling of desmoplastic small round cell tumor. Mol. Cancer Res. 19, 1146–1155 (2021).
Hastie, N. D. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development 144, 2862–2872 (2017).
Call, K. M. et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 60, 509–520 (1990).
Haber, D. A. et al. An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms’ tumor. Cell 61, 1257–1269 (1990).
King-Underwood, L. & Pritchard-Jones, K. Wilms’ tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood 91, 2961–2968 (1998).
King-Underwood, L., Renshaw, J. & Pritchard-Jones, K. Mutations in the Wilms’ tumor gene WT1 in leukemias. Blood 87, 2171–2179 (1996).
Kim, J., Lee, K. & Pelletier, J. The DNA binding domains of the WT1 tumor suppressor gene product and chimeric EWS/WT1 oncoprotein are functionally distinct. Oncogene 16, 1021–1030 (1998).
Haber, D. A. et al. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc. Natl. Acad. Sci. USA 88, 9618–9622 (1991).
Hammes, A. et al. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 106, 319–329 (2001).
Laity, J. H., Dyson, H. J. & Wright, P. E. Molecular basis for modulation of biological function by alternate splicing of the Wilms’ tumor suppressor protein. Proc. Natl Acad. Sci. USA 97, 11932–11935 (2000).
Larsson, S. H. et al. Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell 81, 391–401 (1995).
Kim, J., Lee, K. & Pelletier, J. The desmoplastic small round cell tumor t(11;22) translocation produces EWS/WT1 isoforms with differing oncogenic properties. Oncogene 16, 1973–1979 (1998).
Bandopadhayay, P. et al. The oncogenic properties of EWS/WT1 of desmoplastic small round cell tumors are unmasked by loss of p53 in murine embryonic fibroblasts. BMC Cancer 13, 585 (2013).
Reynolds, P. A. et al. Identification of a DNA-binding site and transcriptional target for the EWS-WT1(+KTS) oncoprotein. Genes Dev. 17, 2094–2107 (2003).
Nishio, J. et al. Establishment and characterization of a novel human desmoplastic small round cell tumor cell line, JN-DSRCT-1. Lab. Invest. 82, 1175–1182 (2002).
Ogura, K. et al. Therapeutic potential of NTRK3 inhibition in desmoplastic small round cell tumor. Clin. Cancer Res. 27, 1184–1194 (2021).
O’Brien, L. L. et al. Transcriptional regulatory control of mammalian nephron progenitors revealed by multi-factor cistromic analysis and genetic studies. PLoS Genet 14, e1007181 (2018).
Lee, S. B. et al. The EWS-WT1 translocation product induces PDGFA in desmoplastic small round-cell tumour. Nat. Genet 17, 309–313 (1997).
Hingorani, P. et al. Transcriptome analysis of desmoplastic small round cell tumors identifies actionable therapeutic targets: a report from the Children’s Oncology Group. Sci. Rep. 10, 12318 (2020).
Kang, H. J. et al. EWS-WT1 oncoprotein activates neuronal reprogramming factor ASCL1 and promotes neural differentiation. Cancer Res 74, 4526–4535 (2014).
Li, H. et al. Adenosine transporter ENT4 is a direct target of EWS/WT1 translocation product and is highly expressed in desmoplastic small round cell tumor. PLoS One 3, e2353 (2008).
Palmer, R. E. et al. Induction of BAIAP3 by the EWS-WT1 chimeric fusion implicates regulated exocytosis in tumorigenesis. Cancer Cell 2, 497–505 (2002).
Bleijs, M. et al. EWSR1-WT1 target genes and therapeutic options identified in a novel DSRCT in vitro model. Cancers (Basel) 13 https://doi.org/10.3390/cancers13236072 (2021).
Gedminas, J. M. et al. Desmoplastic small round cell tumor is dependent on the EWS-WT1 transcription factor. Oncogenesis 9, 41 (2020).
Wang, F. et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 14, 22–29 (2012).
Kato, M. et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 (2012).
Freshour, S. L. et al. Integration of the drug-gene interaction database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res 49, D1144–D1151 (2021).
Schettini, F. et al. CDK 4/6 inhibitors as single agent in advanced solid tumors. Front Oncol. 8, 608 (2018).
Gong, X. et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 32, 761–776 e766 (2017).
Stewart, E. et al. The childhood solid tumor network: A new resource for the developmental biology and oncology research communities. Dev. Biol. 411, 287–293 (2016).
Krystel-Whittemore, M. et al. Novel and established EWSR1 gene fusions and associations identified by next-generation sequencing and fluorescence in-situ hybridization. Hum. Pathol. 93, 65–73 (2019).
Kennedy, A. L. et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 6, 30178–30193 (2015).
Li, X. et al. Inhibition of cyclin-dependent kinase 4 as a potential therapeutic strategy for treatment of synovial sarcoma. Cell Death Dis. 9, 446 (2018).
Lamhamedi-Cherradi, S. E. et al. The androgen receptor is a therapeutic target in desmoplastic small round cell sarcoma. Nat. Commun. 13, 3057 (2022).
Mikkelsen, T. S. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Pohl, A. & Beato, M. bwtool: a tool for bigWig files. Bioinformatics 30, 1618–1619 (2014).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Amemiya, H. M., Kundaje, A. & Boyle, A. P. The ENCODE blacklist: Identification of problematic regions of the genome. Sci. Rep. 9, 9354 (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 9, e1003118 (2013).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–W208 (2009).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Vivian, J. et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 35, 314–316 (2017).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 e1821 (2019).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Lareau, C. A. & Aryee, M. J. hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat. Methods 15, 155–156 (2018).
Lareau, C. A. & Aryee, M. J. diffloop: a computational framework for identifying and analyzing differential DNA loops from sequencing data. Bioinformatics 34, 672–674 (2018).
Acknowledgements
We thank all members of the Rivera, Riggi, Stamenkovic, Bernstein and Suva labs for valuable guidance and advice. We would like to thank Francis Derouet and his staff at the animal facility of Epalinges (Thomas Derré, Antoine Job, Yamay Becerra Arango, Marylin Gaillard and Aurélie Chanthany) for their help and support with the in vivo experimental part of this study. This work was supported by the Swiss National Science Foundation (grants no. PP00P3_183724), the Swiss National Science Foundation Division Biology and Medicine (grant no. 310030_200569), the Fond’Action Contre le Cancer Foundation, the FORCE Foundation, the Liddy Shriver Early Career Research Award, the Emma Muschamp Award and The Novartis Foundation for Medical-Biological Research grant (N.R.) and the Bertarelli Rare Cancers Fund (M.N.R.). M.N.R is supported by the Thomas F. and Diana L. Ryan MGH Research Scholar Award.
Author information
Authors and Affiliations
Contributions
G.B., L.C.B., N.R. and M.N.R designed the study and wrote the manuscript. L.Z., R.R, I.C., G.P.N., I.L. and I.S. provided necessary reagents and conceptual advice. E.C. and G.M.C. contributed to study conception. G.B., L.C.B, S.R., Y-H.X, R.B., S.R., N.G., B.N., A.V., M.E.A., B.D., R.S. performed the experiments. R.D, S.I. and G.B. conducted bioinformatic analyses.
Corresponding authors
Ethics declarations
Competing interests
N.R is an employee of Genentech since 7 February 2022. M.N.R. receives support from ACD (Advanced Cell Diagnostics) and Merck-Serono for research unrelated to this study. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Berkley Gryder and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Boulay, G., Broye, L.C., Dong, R. et al. EWS-WT1 fusion isoforms establish oncogenic programs and therapeutic vulnerabilities in desmoplastic small round cell tumors. Nat Commun 15, 7460 (2024). https://doi.org/10.1038/s41467-024-51851-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51851-3
This article is cited by
-
Desmoplastic small round cell tumor: an update of current management practices
Journal of the Egyptian National Cancer Institute (2025)
-
Desmoplastic Small Round Cell Tumors and the Role of Androgen Receptors
Current Treatment Options in Oncology (2025)
