
Abstract
APOBEC3G, part of the AID/APOBEC cytidine deaminase family, is crucial for antiviral immunity. It has two zinc-coordinated cytidine-deaminase domains. The non-catalytic N-terminal domain strongly binds to nucleic acids, whereas the C-terminal domain catalyzes C-to-U editing in single-stranded DNA. The interplay between the two domains is not fully understood. Here, we show that DNA editing function of rhesus macaque APOBEC3G on linear and hairpin loop DNA is enhanced by AA or GA dinucleotide motifs present downstream in the 3’-direction of the target-C editing sites. The effective distance between AA/GA and the target-C sites is contingent on the local DNA secondary structure. We present two co-crystal structures of rhesus macaque APOBEC3G bound to ssDNA containing AA and GA, revealing the contribution of the non-catalytic domain in capturing AA/GA DNA. Our findings elucidate the molecular mechanism of APOBEC3G’s cooperative function, which is critical for its antiviral role and its contribution to mutations in cancer genomes.
Similar content being viewed by others


Introduction
Human APOBEC3G (hA3G), a member of AID/APOBEC family of zinc-containing cytidine deaminases, catalyzes the conversion of cytidine (C) to uridine (U) on DNA. This process generates DNA mutations from unrepaired uridines. hA3G is a well-known host restriction factor that plays a crucial role in restricting human immunodeficiency virus type 1 (HIV-1)1. In the absence of HIV viral infectivity factor (Vif), hA3G can catalyze excessive C to U editing on the HIV-1 negative cDNA strand, leading to hypermutation in HIV-1 genome2,3,4,5,6,7,8,9,10. hA3G can also impair HIV-1 replication through deaminase-independent mechanisms11,12,13,14,15,16,17.
Deaminases can also induce mutations in the host genome in the context of pathological misregulation in tumorigenesis18,19,20,21,22,23,24. Analysis of human cancers revealed hA3G’s contribution to mutational signatures in multiple cancer types25,26. In a murine bladder cancer model, transgenic expression of hA3G promotes mutagenesis and genomic instability25. Additionally, hA3G performs C to U editing on certain types of human and viral RNA27,28,29,30.
A3G is composed of two zinc-containing cytidine deaminase (CD) domains in tandem: the N-terminal CD1 domain or NTD (referred to as CD1 hereafter) and the C-terminal CD2 domain or CTD (referred to as CD2 hereafter). Multiple CD1-CD2 domain orientations in full-length A3G have been observed in protein crystal structures and cryo-electron microscopy (cryo-EM) structures31,32,33,34,35,36. Despite their similar tertiary structures, individual CD1 and CD2 domains have evolved to carry out distinct functions37,38. The CD1 domain is non-catalytic but binds strongly to nucleic acids39,40. Recent studies show that RNA purine dinucleotide sequence motifs rArA and rGrA are preferred RNA binders for the primate rhesus macaque A3G33. Cryo-EM studies have revealed that the rArA- or rGrA-RNA bound by A3G is a critical part recognized by HIV Vif-E3 ligase for A3G ubiquitination and proteasome degradation34,35,36. These studies provide evidence that CD1, with the assistance of CD2, engages in direct RNA binding. On the other hand, the CD2 domain carries out DNA target-C editing, although it has weak affinity to DNA4,5,41,42,43,44,45. CD2 favors 3′ target-C to U editing in the motifs CC or CCC (the target-C is underlined) on single-stranded DNA (ssDNA). X-ray protein crystallography studies of the catalytic CD2 domain bound to the DNA substrate or DNA oligonucleotide inhibitor have revealed the molecular details of the editing motif CCC selection and deamination44,46.
Efficient A3G editing requires cooperativity between its two domains. The catalytic CD2 domain on its own displays nearly three orders of magnitude lower editing efficiency than the full-length protein47,48. Furthermore, full-length A3G processively edits target-C in two CCC motifs located on a ssDNA substrate during one binding event and preferentially edits target-C in the CCC motif near the 5′ end of ssDNA substrates48,49,50,51. These two editing properties are impaired in the absence of the non-catalytic CD1 domain48. Data from experiments with optical tweezers show that A3G binds in multiple steps and conformations to search and deaminate single-stranded DNA52. Despite these advances, the precise molecular mechanism used by the two domains to coordinate DNA binding and editing has remained elusive.
In this study, we find that purine dinucleotide AA or GA motif downstream of the target-C editing sites in the 3′-direction facilitates rhesus macaque A3G (rA3G) DNA editing function in linear and hairpin loop DNA. The effective distance between AA/GA motifs and the target-C sites depends on the local DNA secondary structure. We present two co-crystal structures of rA3G in complex with ssDNA containing AA or GA motif, providing a mechanistic understanding of AA/GA motif recognition predominantly through the non-catalytic CD1 domain, and its impact on the target-C selection and editing efficiency. These structures also explain how RNA inhibits DNA editing. Our findings reveal molecular insights into the cooperativity between the two domains of A3G in facilitating efficient DNA editing, which is critical for its antiviral function against foreign pathogens and its mutagenic effects on genomic DNA.
Results
Purine dinucleotide motifs facilitate DNA editing of rA3G
Previously, we have shown that rA3G has a strong binding affinity to rArA dinucleotide containing RNA with KD between 10 to 17 nM, followed by rGrA dinucleotide with KD of ~47 nM, and other combinations of dinucleotide containing RNA with KD of ~124 nM or much worse33. It turns out that rA3G also binds AA-containing DNA (5′-FAM TTTTAATTTT) with KD of ~318 nM, and GA-containing DNA (5′-FAM TTTTGATTTT) with KD of ~473 nM (Supplementary Fig. 1). Based on this information, we hypothesized that the presence of AA or GA motifs in DNA may enhance substrate capture by A3G and facilitate the presentation of nearby target-Cs to the active site of the catalytic A3G-CD2 domain.
To study whether and how the AA motifs on DNA can facilitate A3G editing function, we compared editing efficiency between control DNA substrates that carry one editing motif CCC (the target-C is underlined) and AA-DNA substrates that carry both CCC and AA motifs. For simplicity, the control DNA contains only mixed pyrimidine bases, or a combination of mixed pyrimidine and guanine bases, but no adenine base. When designing single-stranded linear DNA substrates (Fig. 1a inset), two variables are considered: (1) substrate length and (2) distance of the editing motif CCC from 3′-end48,49,50. It has been reported that when distance of the editing motif CCC from 3′-end is less than 30 nt, the editing motif CCC falls into a weakly deaminated ‘dead’ zone at the 3′-end linear DNA with a specific activity of human A3G less than 1 pmol μg−1 min−1 48,50. We wished to study whether AA motifs could facilitate A3G editing efficiency under such situation.

The target-C (in pink) is underlined. Reaction conditions are indicated in the relevant data panels. Each plot is presented as mean values ± SD from three independent trials. a Design of a control DNA containing mixed pyrimidine bases with one editing motif CCC placed close to the 5′-end. It is designated as L1-CCC0-N22 with the target-C at position ‘0’. The subscript ’22’ specifies the 3’-end nucleotide position relative to the target-C. It also represents the distance (22 nt) between the target-C and the 3′-end, and the length of the editing product. ‘L1’ represents the linear DNA substrate 1 set. Three 28-nt DNA with a single adenine dinucleotide (AA) motif placed in a distance from the target-C are designated as L1-CCC0-AxAx+1-N22, where the subscript ‘x’ follows the nucleotide numbering pattern depicted in the inset. b Gel image and c plot of product formation by the four DNA substrates in a time course assay. d Calculated specific enzyme activities using the linear-range data from the first 8 min of each reaction. e Design of 25 3′ 6-FAM labeled 28-nt DNA substrates. RR denotes AA, GG, GA, or AG. f Gel image and plot of product formation of each DNA substrate in the L1 set. g Linear DNA substrate 2 (L2) set contains four groups of unlabeled DNA substrates with 32 nt, 38 nt, 44 nt or 50 nt in length. The corresponding distances between the target-C and the 3′-end are 26 nt, 32 nt, 38 nt, and 44 nt. Each group contains four DNA substrates: a control DNA substrate with no AA motifs and three DNA substrates containing individual A5A6, A15A16, or a combination of the two AA motifs (A5A6 and A15A16). h Gel images of the product formation of each DNA substrate in the L2 set. n = 3 independent trials. The quantification data of product formation and specific enzyme activity are presented in Supplementary Fig. 3. Source data are provided as a Source Data file.
A linear, 28-nt control DNA in the linear DNA substrate 1 (L1) set was designed with mixed pyrimidine bases (5′-TTTCCCTTTCTTCTTCTTCTTCTTCTTC-FAM 3′, Fig. 1a). A single A3G editing motif, CCC, was placed near the 5′-end with a 22-nt distance from 3′-end, reflecting the polarity preference of the A3G deaminase and falling within the ‘dead’ zone49,50. Multiple TC motifs were also scattered throughout the sequence. These TC motifs are known to be disfavored by A3G5,53 and are unlikely to interfere with editing assays. The nucleotides in the DNA substrate are numbered with the target-C at position ‘0’. Therefore, we designated this control DNA as L1-CCC0-N22, where the subscript ‘22’ specifies the 3′-end nucleotide position from the target-C (Fig. 1a inset). Importantly, this value also represents the distance of the editing motif CCC from the 3′-end, as well as the length of the hydrolyzed deamination product (simplified as “product” in this study) from the in vitro UDG-dependent deaminase assay (Supplementary Fig. 2). A 6-carboxyfluorescein dye (FAM) attached at the 3′ end facilitates assay quantification.
Three linear AA-containing 28-nt ssDNA in the L1 set were designed with a single AA motif placed at three different locations downstream (in the 3’ direction) of the editing motif CCC (Fig. 1a, inset). We designated these AA-containing substrates as L1-CCC0-A8A9-N22, L1-CCC0-A11A12-N22, and L1-CCC0-A14A15-N22, where the numbers following the adenine bases specify the adenine base positions from the target-C.
We utilized a soluble variant of rA3G protein, which was purified from Escherichia coli, for the deaminase assay. As documented in our previous studies, this purified variant is monomeric and largely free from RNA contamination31,33. It carries a replacement of N-terminal domain loop 8 (139-CQKRDGPH-146 to 139-AEAG-142, designated as rA3GR8) to enhance solubility and has been shown to be catalytically active31.
A time course assay was conducted with the control DNA and three AA-containing DNA (Fig. 1b, c). We observed a significant difference in the editing level among the four DNA substrates. The control DNA L1-CCC0-N22 has only ~4% edits. L1-CCC0-A8A9-N22 has even lower editing, with ~0.8% edits. However, L1-CCC0-A14A15-N22 has ~91% edits, followed by L1-CCC0-A11A12-N22 with ~30% edits. Corresponding specific enzyme activities were calculated in the linear product range (Fig. 1d), and they varied dramatically from 0.01 pmol μg−1 min−1 (L1-CCC0-A8A9-N22) to 11.81 pmol μg−1 min−1 (L1-CCC0-A14A15-N22). The best and the worst rA3GR8 specific activity are comparable to those reported for human A3G, about 12 to 15 pmol μg−1 min−1 with a 69-nt single stranded DNA, and about 0.07 pmol μg−1 min−1 when the editing motif CCC falling within the 30-nt ‘dead’ zone48,50.
Next, we extended substrates in the L1 set to include each of the four purine dinucleotide motifs RR (R denotes A or G) and additional RR positions on DNA, while keeping the DNA sequence surrounding the editing motif CCC (5′-TTTCCCTTT) the same in all substrates. Collectively, a panel of 24 ssDNA substrates were derived with six RR positions: R5R6, R8R9, R11R12, R14R15, R17R18, and R20R21 (Fig. 1e). Evaluation in the linear product range shows that the top edited substrates were with AA motif, followed by GA motif. The substrates with AG motif also showed low but above-background editing. All GG substrates are poorly edited (Fig. 1f). In addition, a pattern of editing efficiency per function of RR position was observed among AA/GA substrates. R14A15 and R17A18 are in the best productive positions to promote target-C editing, whereas R5A6 and R8A9 are in the non-productive positions.
Following that, we investigated whether increasing distance of the editing motif CCC from 3′-end on longer ssDNA substrates could further facilitate rA3G editing efficiency. We took a low-cost approach and used a panel of unlabeled DNA substrates in combination with a fluorescent SYBR Gold Nucleic Acid Gel Stain detection. Due to relatively weak SYBR Gold signal with pyrimidine only DNA, individual guanine bases (G) were inserted in DNA to boost the staining signal (Fig. 1g). Four groups of unlabeled DNA substrates in the linear DNA substrate 2 (L2) set were designed with the distance of CCC from 3’-end increased to 26, 32, 38, or 44 nt. Their substrate lengths were 32, 38, 44, or 50 nt, respectively. Each group contains four substrates including a control DNA without AA motifs and three AA-containing DNA carrying A5A6 (in the non-productive position), A15A16 (in the productive position), or both AA motifs (Fig. 1g, h). The results confirmed that substrates with A5A6 are poorly edited, whereas substrates with A15A16 are efficiently edited. The enzyme specific activities are improved to ~15.14 pmol μg−1 min−1 with L2-CCC0-A15A16-N32, and then it stays close to this value as the distance of the editing motif CCC from 3′-end increases (such as ~14.31 pmol μg−1 min−1 with L2-CCC0-A15A16-N44, Supplementary Fig. 3). Additionally, a combination of A5A6 and A15A16 generates a reduced editing output. The inhibitory effect of A5A6 diminishes as the DNA length increases (Fig. 1h, Supplementary Fig. 3a).
We also observed that, as the distance of the editing motif CCC from 3′-end increases, a substantial number of edits are generated even in the control DNA that contains no AA motifs (such as L2-CCC0-N44 with the specific enzyme activity of 6.59 pmol μg−1 min−1, Supplementary Fig. 3b). Consequently, AA-facilitated editing is less pronounced, with the specific enzyme activity being about two-fold higher (14.31 pmol μg−1 min−1, Supplementary Fig. 3b). These results suggest that with increasing distance of CCC from 3′-end, AA-independent interactions between rA3G and substrate DNA also increase, leading to efficient DNA capture and target-C deamination in the absence of AA dinucleotide motifs.
Control experiments with the purified catalytically inactive protein rA3GR8/E259A, presented in later sections (also see Supplementary Fig. 4a, b), confirmed that the observed editing is solely attributable to rA3G activity and not to other co-purified factors. Additional experiments were conducted to examine whether the substitution in the N-terminal domain loop 8, made to improve solubility, affected AA-facilitated rA3G’s editing function. These results are also presented in later sections.
In summary, we find that a single AA or GA motif can facilitate rA3GR8 editing efficiency on its target-C. AA/GA-facilitated editing is dictated by their position from the target-C with R14A15 to R17A18 in the best productive positions (specific enzyme activities ~11.8 to ~15.14 pmol μg−1 min−1), and with R5A6 to R8A9 in the non-productive (or inhibitory) positions (specific enzyme activities ~0.01 to 0.41 pmol μg−1 min−1). The magnitude of AA-facilitated editing is also influenced by the distance of the editing motif CCC to the 3′-end. As this distance increases, AA-facilitated editing is attenuated, while AA-independent editing is boosted. Lastly, two adjacent AA motifs can generate a combined effect on a single CCC motif.
Overall structures of rA3G bound with ssDNA containing AA or GA motif
Prior to crystallization trials, we determined the minimal productive AA position of deamination on a target-C. We compared a panel of 17 substrates in the linear DNA substrate 3 (L3) set, each carrying a single AA motif placed from 5 nt to 21 nt downstream of the editing motif CCC (Fig. 2a). The results show that 10 nt (A10A11) is the minimal distance to elicit AA-facilitated editing function (Fig. 2b). Control experiments with catalytically inactive rA3GR8/E259A showed no detectable activity on the DNA substrate containing A14A15 (Supplementary Fig. 4a, c). With this information, crystallization trials were carried out using the catalytically inactive rA3GR8/E259A and ssDNA with AA or GA positioned at R10A11, R11A12, or R14A15. The lengths of the DNA substrate were shortened by removing the last four nucleotides at the 3’-end (Fig. 2c, d). Additionally, GA-containing DNA sequences were further modified to replace guanine bases outside of the GA motif with thymine bases (Fig. 2d). The best diffracting crystals were obtained with A10A11- and G11A12-containing DNA (Fig. 2c, d), and their structures were determined (Supplementary Table 1, Supplementary Fig. 5).

a Linear DNA substrate 3 (L3) set contains 17 unlabeled 28-nt DNA substrates with a single AA motif placed stepwise downstream from the target-C. A control DNA with no AA motif (L3-CCC0-N22) is also shown. b Gel images of product formation. n = 3 independent trials. c, d Surface and stick representation of the structure of rA3GR8/E259A in complex with a short DNA sequence 5’-CAATC (in marine sticks) or 5′-TGAT (in light orange sticks). Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. Nucleotides in black are resolved. e Superimposition of the two models. f, g 2Fo-Fc electron density map of the resolved short DNA sequence 5′-CAATC or 5′-TGAT contoured at 1.5σ level. h Comparative modeling of rA3G bound to AA-DNA (this study) and the editing motif CCC-DNA (modeled from PDB 6BUX44). The straight-line distance between A1 in the CCC-DNA (in pink sticks, modeled from PDB 6BUX44) and C9 in the AA-DNA (in marine sticks) is indicated by a black dotted line and shown in the inset. Length per nucleotide ranging from 6.3−6.76 Å54,55 is used to convert distance to number of nucleotides. i Linear DNA substrate 4 (L4) set contains 10 unlabeled 47-nt DNA substrates with a single AA motif placed systematically upstream and downstream from the target-C. A control DNA with no AA motif is also shown. In the three TTT controls, the editing motif CCC was replaced by TTT. j Gel images of product formation. n = 3 independent trials. Source data are provided as a Source Data file.
Both structures of the rA3GR8/E259A-DNA complexes are monomers, each with one rA3GR8/E259A molecule bound to one DNA molecule. The resolutions were determined to be 1.93 Å and 1.89 Å, respectively (Fig. 2c, d). Five nucleotides spanning over the AA motif (5′-C9A10A11T12C13) and four nucleotides over the GA motif (5′-T10G11A12T13) were built into the electron density unambiguously (Fig. 2f, g). However, the editing motif CCC and the remainder of the 5’-end DNA were unresolved in both structures, likely due to their flexibility, as the CCC motif does not strongly bind to the CD2 domain. Superimposition of the two complex structures yielded a root-mean-square deviation (rmsd) of 0.388 Å (2839 to 2839 atoms, Fig. 2e), indicating they are essentially the same structure. A subtle but noticeable difference is seen between A10 of the AA motif (A10A11) and G11 of the GA motif (G11A12) (Fig. 2e, Supplementary Fig. 6), which is expected for the guanine base G11 of the GA motif to fit into the groove that tightly binds the adenine base A10 of the AA motif. The structure of rA3GR8/E259A bound to the AA-containing DNA (resolution 1. 93 Å) is used to describe the protein-DNA interactions in the following sections.
To place the editing motif CCC into the context of the full-length A3G structure, we carried out comparative modeling based on our rA3G bound to AA-DNA structure, alongside a previously determined structure of human A3G-CD2 bound to the editing motif CCC (referred to as ‘CCC-DNA’, modelled from PDB 6BUX44). The comparative model predicted that the AA-DNA fragment (predominately bound by rA3G-CD1) positions its 5′-end (C9, Fig. 2h) in the general direction of the 3′-end of the editing motif CCC, as modeled with rA3G-CD2 (Fig. 2h). This prediction is consistent with the inherent directionality of CCC and AA motifs in our DNA substrates (Fig. 1e, g). To further validate the polar arrangement, we utilized a panel of 11 unlabeled 47-nt DNA substrates in the linear DNA substrate 4 (L4) set with a single AA motif systematically positioned upstream or downstream of the editing motif CCC (Fig. 2i). Our observations show that AA-facilitated editing occurs only in positions A13A14, A17A18, and A21A22 downstream of the editing motif CCC (Fig. 2j), aligning with the spatial organization predicted in the comparative model. Control experiments with catalytically inactive rA3GR8/E259A showed no detectable activity on the DNA substrate containing A13A14 (Supplementary Fig. 4a, d).
We further estimated the distance between the editing motif CCC and the AA motif by measuring the straight-line distance between A1 in CCC-DNA and C9 in AA-DNA (Fig. 2h and its inset), and determined it to be ~36 Å. Using a nucleotide length of 6.3 or 6.76 Å for ssDNA54,55, this distance corresponds to roughly 6 nt between A1 and C9. In a real situation, the distance is expected to be longer than 6 nt as it should not follow a straight line connecting A1 and C9 due to the protein surface features. Therefore, this estimation aligns well with our experimentally determined minimal distance of 7 nt between A1 and C9 for the productive configuration, which is equivalent to A10A11 (Fig. 2b).
Detailed interactions between rA3G and DNA
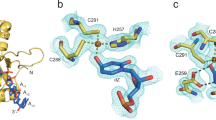
In the co-crystal structure of rA3GR8/E259A bound to AA-containing DNA, the short 5-nt DNA centered around the AA dinucleotide (5′-C9A10A11T12C13) out of the 21-nt ssDNA are clearly visible (Fig. 2f). The AA dinucleotide bases (A10 and A11) are inserted deep inside the protein, the nucleotides before and after A10A11 (i.e. C9, T12 and C13) are bound on the protein surface. The rA3G binding interface for the 5-nt DNA is composed of 15 amino acid residues, 13 residues of which are located on the CD1 loops near CD1 Zn-center (loops 1, 3, 5, and 7), with the remaining 2 residues coming from CD2 (Fig. 3a-e). A hydrophobic groove conformed between CD1 and CD2 binds to the 5’-A (A10), and a hydrophobic cave-like pocket on CD1 binds to the 3′-A (A11) of the AA dinucleotide. The groove donates five residues (I26, F126, W127 on CD1, and F268 and K270 on CD2) to interact with A10 through mostly hydrophobic interactions and only one hydrogen bond (Fig. 3b). The cave-like pocket of CD1 interacts with A11 via hydrophobic packing and four strong hydrogen bonds through eleven CD1 residues, including 25-PILS-28 on loop 1, Y59 on loop 3, W94 on loop 5, and 123-LYYFW-127 on loop 7 (Fig. 3c, e). Additionally, five CD1 residues, 24-RPILS-28 (loop 1), form a small surface area that interacts with C9 (Fig. 3d). Two CD1 residues, 59-YP-60 (CD1 loop 3), have weak interactions with T12C13 (Fig. 3e).

a Surface and ribbon representation of the CD1 (in light blue surface) and the CD2 (in gray ribbon) bound to AA-DNA 5′-C9A10A11T12C13 (in sticks). CCC-DNA (in gray sticks) bound to CD2 is modeled from PDB 6BUX44. Three surface patches surrounding the A10A11 binding pocket/groove are formed by residues (26ILS28, 124YY125, and 126FW127). An inactive mutation E259A (in pink surface) on CD2 is drawn. CD2 residues F268 and K270 are drawn in magenta sticks. Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. b–e Detailed interactions between rA3G and DNA. The color scheme is the same as in (a). Hydrogen bonds are indicated by dashed lines. f Sequences of the L1 DNA substrates containing a single AA motif and a control DNA containing no AA motif. g Gel images and plot of product formation. The plot shows the mean values ± SD from three independent trials. h Two groups of L2 DNA substrates with 32-nt and 50-nt in length, respectively. Each group contains a control DNA substrate with no AA motifs and three DNA substrates containing individual A5A6, A15A16, or a combination of the two AA motifs (A5A6 and A15A16). i Gel images of product formation with 32-nt and 50-nt DNA. n = 3 independent trials. Source data are provided as a Source Data file.
To verify the importance of AA binding residues in AA-facilitated editing efficiency, we generated alanine mutations on seven CD1 amino acid residues that engage with the AA dinucleotide of the bound DNA, including I26A/L27A/S28A, Y124A/Y125A, and F126A/W127A. A wild type (rA3GR8) and a catalytically inactive mutant (rA3GR8/E259A) were used as positive and negative controls (Supplementary Fig. 7a). Using a panel of 3’ FAM-labeled AA-containing substrates (in the L1 set), the results show that while the catalytically inactive rA3GR8/E259A exhibited no detectable editing, two of the three CD1 mutants, Y124A/Y125A and F126A/W127A, displayed only basal-level editing function (Fig. 3f, g). These results indicate the critical importance of AA binding residues 124-YYFW-127 in CD1 for facilitating efficient editing by CD2. However, AA-facilitated editing is only partially lost in the mutant I26A/L27A/S28A, suggesting that these residues are less critical, and the mutant may still retain partial binding to AA.
Further validation was carried out using two groups of long DNA substrates (in the L2 set) with increased distances between the editing motif CCC and the 3′-end (26 nt and 44 nt, Fig. 3h). Similar results were obtained that theses mutants show defective in AA-facilitated editing (Fig. 3i). However, significant number of edits are generated by these mutants in the long DNA substrates that have 44 nt between the editing motif CCC and the 3′-end, indicating that AA-independent editing is largely unaffected in these mutants.
We also generated alanine mutations on CD2 residues F268 and K270 that participate in binding to the adenine base A10 (Fig. 4a, Supplementary Fig. 7b) and tested it with a panel of 3′ FAM labeled RR-containing substrates (Fig. 4b). Comparing to the wild type, the mutant F268A/K270A has an overall reduced editing efficiency when RR is in the productive positions (R11R12, R14R15, R17R18, and R20R21, Fig. 4c, d), indicating that the impairment in AA-binding leads to an impaired AA-facilitated editing. Interestingly, it has an overall slightly increased editing efficiency when RR is in the non-productive positions (R5R6 and R8R9, Fig. 4c, d). Further examination of the minimal AA register using eight DNA substrates in the L3 substrate series (Fig. 4e, f) shows that the minimal AA register for productive DNA editing is shortened to ~6 nt (A6A7). These observations suggest that the physical barrier between the CCC motif and AA motif has changed, possibly due to weakened interface rigidity between the CD1-CD2 domains. This allows CD2 greater freedom to rotate relative to CD1, enabling it to interact with DNA more flexibly and reach the CCC motif at a shorter distance.

Each plot is presented as mean values ± SD from three independent trials. a Surface and ribbon representation of the CD1 domain (in light blue surface) and the CD2 domain (in gray ribbon) bound to AA-DNA 5′-CAATC (in marine sticks). CCC-DNA (in light gray sticks) is modeled from PDB 6BUX44. Two CD2 residues F268 and K270 are shown (in magenta surface). Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. b Sequences of the L1 DNA substrates, where RR denotes AA, GG, GA, or AG. c Gel image and plot of product formation by the wild-type rA3GR8. d Gel image and plot of product formation by the mutant rA3GR8/F268A/K270A. e Sequences of the first 8 DNA substrates in the L3 DNA substrate set. f Gel image of the product formation. n = 3 independent trials. Source data are provided as a Source Data file.
Editing property of a hyperactive rA3G variant
We investigated whether rA3G carrying a hyperactive catalytic domain could override or escape from the AA-facilitated editing. We constructed a rA3GR8 variant carrying two mutations on its CD2 domain, P247K and Q317K (Fig. 5a, b). Their corresponding mutations from human A3G, P247K and Q318K, have shown to contribute to the hyperactivity of the human A3G CD2 catalytic domain44. The rA3GR8/P247K/Q317K variant displays much enhanced editing efficiency on an AA-containing DNA substrate L1-CCC0-A14A15-N22 (Fig. 5c, d, g). Its enzyme specific activity reached 101.82 pmol μg−1 min−1, about 8.6-fold higher than that of the wild-type rA3GR8. Dramatic increase in editing efficiency was also observed in other AA-containing DNA and in the control DNA (Fig. 5g). Despite the overall enhanced efficiency, the substrate rank order remained similar to that of the wild type: L1-CCC0-A14A15-N22 is still the best substrate, followed by L1-CCC0-A11A12-N22, control DNA, and L1-CCC0-A8A9-N22. Using the complete panel of 25 FAM labeled DNA (Fig. 5e, f), we show that the hyperactive rA3G variant displays a pattern comparable to that of the wild type, albeit under a much lower enzyme concentration. GG-containing substrates remained to be poor substrates. Of note, the editing efficiency on AG substrates were disproportionally enhanced.

Each plot is presented as mean values ± SD from three independent trials. a Surface and ribbon representation of the CD1 domain (in light blue surface) and the CD2 domain (in gray ribbon) bound to AA-DNA 5′-CAATC (in marine sticks). CCC-DNA (in light gray sticks) is modeled from PDB 6BUX44. Two previously characterized residues P247 and Q317 (in purple surface) are shown on the CD2 domain (in gray ribbon). Corresponding mutations P247K and Q318K cause a hyperactive phenotype in human A3G CD2 domain44. Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. b SDS-PAGE gel image showing the purified rA3GR8/ P247K/Q317K and the wild-type rA3GR8. n = 1 trial. c Gel image and d plot of product formation in a dose response assay. The 3′ FAM labeled DNA substrate sequence, L1-CCC0-A14A15-N22, is also shown. e Sequences of the L1 DNA substrates, where RR denotes AA, GG, GA, or AG. f Gel image and plot of product formation. g Calculated specific enzyme activity of four 3′ FAM labeled DNA substrates by rA3GR8/P248K/Q317K. Source data are provided as a Source Data file.
AA-facilitated DNA editing on editing sites TC and CC
A3G favors the target-C in the context of CCC over CC or TC5,53. We investigated AA-facilitated editing on editing sites TC, CC, and CCC using the hyperactive variant rA3GR8/P247K/Q317K. A panel of three linear AA-DNA substrates containing a single editing site (TC, CC, or CCC) and a single 3’ downstream AA motif was designed: L3-TTC0-A15A16-N22, L3-TCC0-A15A16-N22, and L3-CCC0-A15A16-N22 (Supplementary Fig. 8a). Their corresponding control DNA substrates were also included, wherein A15A16 was replaced with a non-AA motif G15T16. A time course assay was conducted, and we observed AA-facilitated editing at all three editing sites (Supplementary Fig. 8b–g). The substrates were ranked from best to worst as CCC-A15A16, CC-A15A16, TC-A15A16/CCC-G15T16, CC-G15T16, and TC-G15T16. A negative control using the catalytically inactive rA3GR8/P247K/E259A/Q317K showed no detectable editing activity. These results demonstrate that under the experimental conditions, editing sites TC or CC with 3′ downstream AA motif could be as efficient as, or even more efficient than, editing sites CCC without 3′ downstream AA motif.
RNA inhibition of AA-facilitated DNA editing
When comparing the rA3G structures in complex with the AA-DNA (5′-C9A10A11T12C13) vs rArA-RNA (5′-rU4rA5rA6rU7rU8)33, the first four DNA nucleotides (C9A10A11T12) align well with the four RNA nucleotides (rU4rA5rA6rU7) and show nearly identical interactions with rA3G, with the fifth DNA nucleotide C13 adopting different interactions with rA3G from the corresponding RNA nucleotide rU8 (Fig. 6a, Supplementary Fig. 9a). These results indicate that rA3G binds to the AA dinucleotide and the immediate 5′ and 3′-side nucleotides similarly for both DNA and RNA. The noticeable differences are that RNA forms hydrogen bonds with the S28 sidechain, the main-chain N, and the G29 main-chain N through the 2′-OH of the sugar moiety of rU4 and rA6 (yellow sticks in Supplementary Fig. 9b), which are absent in DNA due to the lack of 2′-OH. For the fifth DNA nucleotide C13, it turns in a different direction from its equivalent RNA nucleotide rU8 in such a way that C13 packs with T12. Such packing interaction between C13 and T12 should not allowed in RNA due to the presence of 2′-OH of rU7 that would clash with rU8. Another difference includes the 2′-OH of the sugar moiety of the fifth RNA nucleotide rU8 forming an additional hydrogen bond with the R99 sidechain (Supplementary Fig. 9b).

DNA and RNA were mixed at the indicated molar ratio in the reaction mixture, and the assays were performed using the hyperactive variant rA3GR8/P247K/Q317K and its S28A mutant (rA3GR8/S28A/P247K/Q317K). a Overlapping binding surfaces for the AA motif on DNA and the rArA motif on RNA with rmsd 0.193 (2660 atom pairs). See Supplementary Fig. 9 for further details. Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. b Sequences of six L1-AA DNA substrates and one 10-nt rArA-RNA competitor used in the inhibition assay. c Gel image and plot of product formation. d Inhibition assay with six 10-nt RNA competitors. Each plot is presented as mean values ± SD from three independent trials. The P values shown for the indicated sets in (d) were calculated using a two-tailed Student’s t-test. Source data are provided as a Source Data file.
These observations together with the difference in rA3G affinity between AA-DNA (Supplementary Fig. 1) and AA-RNA33 suggest that the hydrophobic pocket favors AA-RNA over AA-DNA. It provides a plausible structural explanation to prior data on RNA inhibition of A3G DNA editing function56,57,58,59,60. To test this, we conducted rArA-RNA (5′-rUrUrUrUrArArUrUrUrU) inhibition study on a panel of AA-DNA substrates (A5A6, A8A9, A11A12, A14A15, A17A18, and A20A21). We used the hyperactive variant rA3GR8/P247K/Q317K and its S28A mutant rA3GR8/S28/P247K/Q317K (Supplementary Fig. 7c) to perform in the competition assay (Fig. 6b, c). The results show that (1) rArA-RNA inhibits AA-facilitated DNA editing with the hyperactive variant rA3GR8/P247K/Q317K; (2) The S28A mutation alleviated the rArA-RNA inhibition but not to the full extent. This is likely caused by the loss of hydrogen bonding to 2’-OH of the sugar moiety of rU4 in RNA and the remaining RNA-specific interactions in the S28 mutant. In addition, its DNA editing efficiency is slightly reduced. This is likely caused by the loss of hydrogen bonding to O3′ of the sugar moiety of C9 in DNA (depicted in Fig. 3d). Human A3G carrying the S28A mutation displays reduced packaging efficiency61,62, supporting the involvement of S28 in RNA binding.
We further tested RNA inhibition of AA-facilitated DNA editing using one of the DNA substrates, L1-CCC0-A17A18-N22, and a set of six 10-nt RNA competitors (5′-rUrUrUrUrNrNrUrUrUrU, where rNrN denotes rArA, rUrA, rUrU, rGrG, rGrA, or rArG. The results show that rArA or rGrA-containing RNA competitors cause substantial reduction in the AA-facilitated DNA editing (Fig. 6d), which supports that rArA/rGrA RNA compete with AA-DNA for the same binding site on rA3G. Because many cellular RNAs contain unpaired rArA motifs, the cellular rArA-containing RNA bound to A3G-CD1 is expected to inhibit the editing activity of A3G if they are not displaced.
AA-facilitated DNA editing in hairpin forming sequences
A3A and A3B have been shown to edit the target-C presented in both linear and short hairpin loop DNA, displaying a preference for the hairpin loop substrates. However, A3G-DNA structures with linear DNA show no base-paring, as seen in A3A/DNA structures44,63,64. On the other hand, it has been reported that A3G can edit the target-C in the hairpin loop of RNA hairpin substrates27,28,29,30.
From comparative modeling of our rA3G bound to AA-DNA structure and a previously determined structure of human APOBEC3A bound to a tetraloop DNA hairpin (PDB 8FIK)64, we hypothesized that a DNA hairpin substrate with a long stem-length and a 3’overhang (for presenting AA motifs to the non-catalytic CD1 domain) could potentially be edited by rA3G (Fig. 7a). We used the hyperactive variant rA3GR8/P247K/Q317K to test this hypothesis.

a Modeling of the editing motif CCC-DNA (in pink, modeled from PDB 6BUX44) and a DNA hairpin structure with a tetraloop and a 12-bp stem (in green, based on PDB 8FIK64) mapped on to the AA-bound rA3G structure (this study). A potential connection path (~46 Å) between the hairpin DNA (with a 10-bp stem) and the AA motif is indicated by a black dotted line and labeled as ‘portion of a 3′overhang’. Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. b Location of the AA motif on 3′-side affects editing efficiency. Deaminase activity was monitored on hairpin DNA 1 substrates carrying a 10-bp hairpin stem, a tetraloop CCCC, and a 3′overhang. A single AA motif was placed at various locations on the 3′-side. A negative control with 5′-TTTT in the hairpin loop and a single A23A24 motif in the 3′-side was included. n = 3 independent trials. c Effect of stem length on editing efficiency. Deaminase activity was monitored on hairpin DNA 2 substrates with varying stem lengths from 4 bp to 12 bp. The position of the AA motif was kept the same in all substrates as the position of A23A24 in the 10-bp hairpin DNA substrate. Three linear DNA substrates were included as controls. n = 3 independent trials. Source data are provided as a Source Data file.
We designed a control DNA substrate (hairpin DNA substrate 1 or HP1) carrying an editing motif CCC in the loop region of a hairpin structure with a 10-bp hairpin stem, 5′-GCAGCAAGCG(CCCC) CGCTTGCTGC. The hairpin DNA also carries a 21-nt 3′overhang to boost its interaction with CD1 (Fig. 7b). Seven AA-containing DNA were designed with the AA motifs placed at various locations in the 3′overhang. All annealed hairpin DNA were essentially monomeric (Supplementary Fig. 10a) with the estimated Tm between 75.4 to 76.9 °C (Fig. 7b). The results show that the hairpin DNA are not efficiently edited without AA motif or with AA motif located from 11–12 nt to 17–18 nt downstream from the target C (substrates with A11A12, A14A15, or A17A18). Instead, efficient editing was only observed when the AA motif is positioned at a distance longer than 20-21 nt between A20A21 and A29A30. Control experiments using catalytically inactive rA3GR8/E259A showed no detectable activity on the hairpin DNA substrate containing A20A21 (Supplementary Fig. 4a, e).
Comparing with linear DNA substrates, the minimal AA register for the productive DNA editing has changed from A10A11 to be longer than A17A18. This is likely caused by the difference between the rigid form of the hairpin duplex and the flexible linear DNA, as well as their spatial arrangement with the rA3G protein (Fig. 7a). In the comparative model, the straight-line distance between the 3’-end of the hairpin stem and the 5′-end of the AA-DNA is about ~46 Å, which is equivalent to 7 or 8 nt. This model shows that A17A18 does not have sufficient linear space between A17A18 bound at CD1 and the target CCC in the stem-loop bound at CD2 active site. It requires at least A20A21 to cover the distance and support the AA-facilitated editing of the target-C in this hairpin DNA.
We further tested the effect of stem-length on editing efficiency. Nine unlabeled hairpin DNA substrates were designed to carry varies stem-lengths from 4 bp to 12 bp (hairpin DNA 2 or HP2 set, Fig. 7c). Additionally, they all have the same 3’overhang sequence with one AA motif placed at the 13–14 nt position from the 3′-end of the hairpin stem. All annealed hairpin DNA of different hairpin stem lengths were monomeric with the estimated Tm between 61.8 to 77.4 °C (Fig. 7c Supplementary Fig. 10b). Three unlabeled linear DNA from the L3 set, L3-CCC0-N22, L3-CCC0-A5A6-N22, and L3-CCC0-A14A15-N22 (Figs. 2a and 7c) were also included as the linear DNA controls. Our results show that the linear DNA displayed the expected editing pattern with near complete editing in L3-CCC0-A14A15-N22 and very little editing in L3-CCC0-A5A6-N22. Nine hairpin DNA2 substrates also displayed a dramatic difference in editing efficiency. Hairpin DNA with short stem lengths is poorly edited (4 bp and 5 bp), whereas hairpin DNA with long stem lengths is efficiently edited (11 bp, 10 bp, and 9 bp). DNA with the longest hairpin stem tested (12 bp) was less efficiently edited.
Additional tests were carried out with hairpin DNA substrates containing a fixed stem length of 4 bp (HP3 set) or 11 bp (HP4 set), with varying AA positions in the 3′overhang (Supplementary Figs. 11 and 12). The results indicate that hairpin DNA with a 4-bp stem length are generally poor substrates, whereas hairpin DNA with an 11-bp stem length yield results similar to those with a 10-bp stem length (Fig. 7b). These findings align with our model predictions (Supplementary Fig. 13), which suggest that helix rotation of the hairpin stem, in conjunction with the AA motif on the 3′overhang DNA, is likely responsible for the proper orientation (or positioning) of the target C to the CD2’s catalytic cavity. It appears that the target C on longer stems, together with the AA motif on the 3′overhang DNA, can effectively reach the catalytic cavity, whereas the target C on shorter stems cannot. Consequently, the principle governing the spatial requirement between the AA motif recognized largely by CD1 and the target cytosine edited by CD2’s active site is consistent across both linear DNA and hairpin DNA, despite differences in the number of nucleotides between the AA motif and the target cytosine.
Editing characteristics of the wild-type rhesus macaque A3G and human A3G
Due to the challenging nature of purifying the wild-type rhesus macaque A3G (rA3GWT, with native loop 8), we used whole-cell extracts from HEK293T cells expressing rA3GWT to investigate its AA-facilitated editing function and to evaluate whether the substitution in the CD1 domain loop 8, made to improve solubility, affected this function. Structural analysis reveals that the substituted loop 8 is located remotely from the DNA binding sites, suggesting minimal perturbation on protein-DNA interaction upon substitution of loop 8 (Fig. 8a).

a Surface and ribbon representation of rA3G. The rA3G CD1 domain (in light blue surface) is bound to AA-DNA 5′-CAATC (in marine sticks). The rA3G CD2 domain (in gray ribbon) is bound to CCC-DNA (in light gray sticks, modeled from PDB 6BUX44). The four substituted residues in the CD1 domain loop 8, 139AEAG142, are shown as a surface patch (in teal). Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. b Western blot of HEK293T whole-cell extracts expressing rA3GR8, rA3GWT or rA3GE259A (catalytically inactive). HEK293T whole-cell extract with empty vector pcDNA served as a negative control. α-Tubulin served as a loading control. rA3GR8 migrated slightly faster than rA3GWT because the substituted loop 8 in rA3GR8 is four residues shorter. M indicates protein molecular weight standards. n = 3 independent trials. c DNA sequences of 3′ 6-FAM labeled 28-nt DNA substrates (L1 set). RR denotes AA, GG, GA, or AG. d Deaminase activity of HEK293T whole-cell extract expressing rA3GWT, rA3GR8, or rA3GE259A on linear DNA substrates (L1 set). n = 3 independent trials. e Deaminase activity of HEK293T whole-cell extracts expressing rA3GWT or rA3GE259A on hairpin DNA 1 substrates (HP1 set) carrying a 10-bp hairpin stem, a tetraloop CCCC, and a 3′overhang. A single AA motif was placed at various locations on the 3′ overhang. A negative control with 5′-TTTT in the hairpin loop and a single A23A24 motif in the 3′overhang was included. Three linear DNA substrates were included as linear DNA controls. Deaminase activity was monitored using 1.6 μg of HEK293T whole-cell extract on 500 nM DNA substrates in a 20 μl reaction volume. n = 3 independent trials. Source data are provided as a Source Data file.
Experiments with the 3′ FAM-labeled linear DNA confirmed that rA3GWT and rA3GR8 in the HEK293T whole-cell extracts (Fig. 8b–d) displayed similar editing characteristics to each other and to the purified rA3GR8 protein (Fig. 1f, Fig. 4c). Experiments with the unlabeled hairpin DNA substrates also showed that rA3GWT in the HEK293T whole-cell extracts (Fig. 8e, Supplementary Fig. 14) displayed similar editing patterns as those from the purified rA3GR8 protein (Fig. 7b, c). A negative control using the HEK293T whole-cell extract expressing the catalytically inactive rA3GE259A (with native loop 8) showed no detectable deaminase activity (Fig. 8b, d, e, Supplementary Fig. 14).
Human A3G (hA3G) protein is closely related to rA3G, comprising approximately 77% identical and 85% similar residues (Supplementary Fig. 15a). Comparative modeling of hA3G (PDB 8CX0)64 bound to AA-DNA (PDB 8TVC, this study) predicted that the putative binding surface for the AA-DNA motif remains largely the same in hA3G, along with multiple amino acid substitutions at the periphery of the AA-DNA binding area (Fig. 9a). This area also overlaps with the experimentally defined rArA- or rGrA-RNA binding area35,36. HEK293T whole-cell extracts expressing the wild-type hA3G (hA3GWT, Fig. 9b) was used to investigate AA-facilitated editing function due to difficulty of purifying the hA3GWT protein. HEK293T whole-cell extract carrying the empty vector pcDNA was used as a negative control (Fig. 9b).

a Comparative modeling of hA3GWT (PDB 8CX035) bound to DNA. The hA3G CD1 domain (in mosaic-color surface, color key is shown) is bound to 5′-CAATC (in light gray sticks, modeled from PDB 8TVC, this study). The hA3G CD2 domain (in gray ribbon) is bound to CCC-DNA (in light gray sticks, modeled from PDB 6BUX44). Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. b Western blot of HEK293T whole-cell extracts expressing hA3GWT or containing an empty vector pcDNA. α-Tubulin served as a loading control. M indicates protein molecular weight standards. n = 4 independent trials. c DNA sequences of 3′ 6-FAM labeled 28-nt DNA substrates (L1 set). RR denotes AA, GG, GA, or AG. Deaminase activity of hA3GWT on linear DNA substrates (L1 set). The plot is presented as mean values ± SD from three independent trials. d Deaminase activity of hA3GWT on hairpin DNA 1 substrates (HP1 set) carrying a 10-bp hairpin stem, a tetraloop CCCC, and a 3′overhang. A single AA motif was placed at various locations on the 3′ overhang. A negative control with 5′-TTTT in the hairpin loop and a single A23A24 motif in the 3′overhang was included. Three linear DNA substrates were included as linear DNA controls. n = 3 independent trials. e Deaminase activity of hA3GWT on hairpin DNA 2 substrates (HP2 set) with varying stem lengths from 4 bp to 12 bp. The position of the AA motif was kept the same in all substrates as the position of A23A24 in the 10-bp stem loop DNA substrate. Three linear DNA substrates were included as linear DNA controls. Deaminase activity was monitored using 0.18 μg of HEK293T whole-cell extract on 24 nM DNA substrates in a 20 μl reaction volume. n = 3 independent trials. See Supplementary Fig. 15 for control experiments with a catalytically inactive mutant hA3GE259A on four key unlabeled-DNA substrates. Source data are provided as a Source Data file.
Experiments with the 3′ FAM-labeled linear DNA and unlabeled hairpin DNA substrates revealed that hA3GWT in the HEK293T whole-cell extract can effectively edit the target-C on both linear and hairpin loop DNA. Its editing efficiency exhibited a similar dependence on the AA position on both linear and hairpin DNA substrates (Fig. 9c, d). Additionally, it shows a similar dependence on the stem-length of the hairpin DNA substrates (Fig. 9e). Despite the observed similarity between the two A3G proteins, hA3GWT in the HEK293T whole-cell extract also exhibited its distinct editing features. One notable feature is the elevated AA-independent editing activity towards DNA substrate HP1-N-13-CCC0-N31 (Fig. 9d, lanes 7 and 8). Finally, HEK293T whole-cell extracts carrying an empty vector pcDNA (Fig. 9b-e) or expressing a catalytically inactive hA3GE259A (Supplementary Fig. 15b–g) showed no detectable deaminase activity.
Discussion
In this study, we demonstrated that the editing of target-C by rA3G in both linear and hairpin loop sequences is significantly influenced by the presence of AA and GA dinucleotides at a certain distance downstream (but not upstream) of the target-C. We also provided the mechanistic understanding for these biochemical observations through determination of two co-crystal structures of the full-length rA3G in complex with AA- or GA-containing ssDNA sequences. These structures reveal how rA3G predominately uses its non-catalytic CD1 domain to capture the substrate through recognition of the AA/GA motifs in a specific orientation, thereby presenting the target-C to the distally located active site on the catalytic CD2 domain for deamination. Although most of the work presented here is based on purified rA3GR8 (and its derivatives) carrying a substituted loop 8 on the N-terminal domain, we show that rA3GWT in HEK293T whole-cell extracts displays similar editing characteristics as the purified protein rA3GR8. Additionally, hA3GWT in HEK293T whole-cell extracts displays a similar editing property as rA3GWT in terms of AA motif influence on target-C deamination. However, hA3GWT also displays distinct editing features that warrant further investigation.
The AA/GA-facilitated DNA editing is directional and dynamic, supporting previous findings regarding the directionality and processivity features of A3G catalysis on ssDNA. Multiple AA, GA motifs present in the DNA substrates in previous reports would allow A3G to bind at multiple locations on these DNA substrates, thus rationalizes A3G “promiscuous” DNA binding property. The biochemical and structural data described here, together with the prior information from other studies44,48,50,52, support a mechanism (Fig. 10) in which CD1, with the assistance of CD2, scans, recognizes, and captures exposed AA/GA motifs in ssDNA region, which allow CD2 sufficient time to hit on those CCC motifs located 5′-upstream within a 9–20 nt linear distance window (optimal 12–16 nt), for deamination. If CCC motifs are located either at the 5′-side of AA/GA within a 7 nt distance or at the 3′-side of AA/GA, they cannot efficiently reach the catalytic cavity on CD2 for deamination. Additionally, DNA secondary structure can affect the geometric distance between AA/GA and CCC, which in turn, affects the register of the productive distance between AA/GA and the target-C (Fig. 10, Supplementary Fig. 13). Specifically, the editing efficiency of the hairpin loop CCC is influenced by both the stem length and its spatial reach to the CD2 active site from the AA/GA on the 3′ overhang, which is primarily captured by CD1.

a Cartoon depicts the domain organization of rA3G. Location of the zinc-catalytic residue E259 is marked by a pink star in the schematic diagram. b Cartoon depicts how rA3G predominantly uses its CD1 domain to capture the AA/GA dinucleotide on linear ssDNA and presents the editing motif CCC, located at the 5′-side (or upstream) of AA/GA, to its CD2 active site within a 9–20 nt window, with an optimal linear distance of 12−16 nt, for deamination. The editing motif CCC located at the 5′-side of AA/GA within 1–7 nt distance or at the 3′-side of the AA/GA cannot reach to the CD2 active site efficiently. c Diagram depicts that editing efficiency of CCC on hairpin loop DNA is affected by both stem length (h) and 3′ overhang distance (m) to the AA/GA on the 3′-side (downstream). The fate of the editing sites, CCC, whether located on a hairpin loop or on linear ssDNA, will be as follows: the CCC located right next to the 5′-side of AA/GA within 1–7 nt linear distance remains unedited, whereas both the middle CCC on the hairpin loop and the CCC on the 5′-side of the hairpin loop are within the editing distance from the AA/GA for efficient deamination.
rA3G also carries out AA/GA independent DNA editing at a reduced efficiency, which requires further study. The editing on ssDNA devoid of AA/GA may be carried out by the action of CD2 without much contribution from CD1, or there could be other DNA binding sites present on CD1 in either sequence specific or non-specific manner. Indeed, the editing level of ssDNA devoid of AA/GA is similar to those by individual CD2 protein alone65. However, the editing efficiency of AA/GA-independent editing significantly increases as the DNA length 3′-downstream of the CCC motif increases (Fig. 1h and Fig. 3i), suggesting that the increase of the length of the ssDNA can enhance DNA binding by CD1 to facilitate the substrate capturing and target-C deamination. Additionally, rA3G mutants disrupting the AA/GA specific binding are shown to retain AA-independent DNA editing (Fig. 3g, i).
Previous studies have shown that, with the exception of CpG and UpA, other RNA dinucleotides, including UpU and UpC (which translate to ApA and GpA on -cDNA), have normal representation levels in the HIV-1 RNA genome66,67,68. Therefore, we didn’t see evidence to suggest that viruses undergo depletion of the AA/GA motif in the viral -cDNA genome during evolution to evade A3G activity. We speculate that a balanced level of dinucleotides is likely a requirement for maintaining both the secondary and tertiary RNA structural features as well as efficient transcription and translation66. Another concern is that the catalytic enhancement by the AA/GA sequence decreased when longer ssDNA substrates were tested. Our preliminary analysis of the frequency of AA/GA occurrence on the 3′ side of most (hot) and least (cold) mutated sites in HIV and SIV cDNA did not reveal any difference between the hot and cold spots. These data suggest that, compared to our controlled experiments with oligos containing a single target site, mutations with the HIV and SIV cDNA during revers transcription are influenced by several factors, including not only distances from the AA and GA sites but also complex structural features in the RNA-cDNA hybrid, variations in the number of packaged APOBEC3 molecules69, presence of other APOBEC3 enzymes such as APOBEC3D, APOBEC3F, and APOBEC3H70,71, interaction between APOBEC3G and RT17, among other unknown factors. Accurately identifying all these factors and their contributions to the level of C > T mutations warrant further investigation.
We consider the following two factors to be important for AA/GA-facilitated DNA editing: (1) differential binding affinity for purine and pyrimidine motifs on DNA, and (2) a physical barrier between the catalytic cavity and the motif binding pocket. Variations in both binding affinity and flexibility degree between the two domains could influence the enzyme function among A3G homologs. Additionally, multiple factors could potentially shape the contribution of AA/GA-facilitated editing under physiological and pathological situations. Due to the largely shared binding interactions between AA/GA-containing ssDNA and ssRNA, mutants defective in binding to AA/GA ssDNA are also defective in binding to AA/GA ssRNA33. Cellular and viral ssDNA binding proteins could compete with A3G in binding to ssDNA72,73,74,75. Furthermore, temporary formed DNA secondary structures may cause deviation from expected productive configuration for AA-facilitated editing7,76,77. Finally, selection pressure in living cells may promote certain mutations over others and shape the eventual mutational outcome78. Further investigation is needed to determine the mechanisms of CD1/CD2 cooperativity and the outcome of target-C editing/mutation carried out by A3G in vivo. Structural studies of double-domain APOBECs in complex with nucleic acids can also provide valuable molecular insights into the cooperative mechanisms that have evolved during conflicts between host restriction factors and retroviruses.
Methods
Protein expression and purification
A soluble variant of rhesus macaque APOBEC3G (rA3G, accession code: AGE34493) with a replacement in the N-terminal domain loop 8 (139CQKRDGPH146 to 139AEAG142, designated as rA3GR8) was constructed in the pET-sumo vector (K30001, Thermo Fisher Scientific). This construct generated a SUMO fusion with an N-terminal 6xHis tag and a PreScission protease cleavage site. Protein expression and purification followed previously published protocols33. In brief, E. coli Rosetta™(DE3) pLysS cells (70956, Millipore SIGMA) expressing rA3G were cultured at 37 °C in LB medium with 50 μg/ml kanamycin. Temperature was lowered to 16 °C when OD600 reached ~0.3. Protein expression was induced by 0.1 mM IPTG when OD600 reached 0.7 to 0.9. After overnight growth at 16 °C, cells were harvested by centrifugation. The resulting cell pellet was lysed in buffer A (25 mM HEPES at pH 7.5, 500 mM NaCl, 20 mM MgCl2, 0.5 mM TCEP, and 60 μg/ml RNase A) using sonication. The protein purification process included Ni-NTA agarose chromatography, RNase A/T1 treatment and PreScission protease cleavage, size-exclusion chromatography, heparin chromatography, and a second-round of size-exclusion chromatography. The final protein samples were quantified, verified for purity, and stored in buffer B (50 mM HEPES at pH 7.5, 250 mM NaCl, and 0.5 mM TCEP) at −80 °C until use. Sequences of all mutant constructs were verified by Sanger sequencing (Azenta Life Sciences). Mutant proteins were purified using the same protocol.
Electrophoretic mobility shift assay (EMSA)
DNA labeled with 5′ 6-FAM (Integrated DNA Technologies) at 10 nM was titrated by rA3G in 20 μl reaction volume containing 50 mM HEPES pH 7.5, 250 mM NaCl, 1 mM DTT, 0.1 mg/ml recombinant albumin (B9200S, New England Biolabs), 0.1 mg/ml RNase A (19101, QIAGEN), and 10% glycerol. Reaction mixtures were incubated on ice for 10 min and analyzed by 8% native PAGE in 4 °C. A solution with acrylamide:bis-acrylamide ratio of 72.5:1 was used in preparing 8% native gels. TyphoonTM Biomolecular Imager (Cytiva) was used to visualize gel images. ImageQuant TL (v8.1, Cytiva) was used for image quantification. Dissociation constant KD was calculated using GraphPad Prism version 8.0.0 for Windows. Three independent trials were carried out for each DNA molecule.
In vitro UDG-dependent deaminase activity assay
DNA and RNA oligonucleotides were synthesized by Integrated DNA Technologies. Hairpin DNA substrates are annealed overnight in the DNA annealing buffer (10 mM Tris at pH 8, 50 mM NaCl), and their size exclusion chromatography (SEC, Cytiva) elution profiles were checked to ensure no self-dimer formation.
DNA deamination activity assays were performed as described31 with minor modifications. Reactions (20 μl) containing the purified protein (rA3GR8 or indicated mutants, with specified concentrations) and individual DNA substrates (with indicated sequences and concentrations) were incubated at 37 °C for an indicated duration in DNA deamination buffer [25 mM HEPES at pH 7, 250 mM NaCl, 1 mM DTT, 0.1% Triton X-100, 0.1 mg/ml recombinant albumin (B9200S, New England Biolabs), and 0.1 mg/ml RNase A (19101, QIAGEN)]. Reactions were stopped by heating to 90 °C for 5 min. Uracil was removed by 0.025 U/μl uracil DNA glycosylase (M0280, New England Biolabs) at 37 °C for 15 minutes, followed by abasic site hydrolysis at 90 °C for 10 minutes in 0.13 M NaOH. Reactions were mixed with equal volume of 2× gel loading buffer (95% formamide, 25 mM EDTA) and heated to 95 °C for 5 minutes. DNA fragments was separated on 20% denaturing acrylamide gel (5% crosslinker, 7 M urea, 1× TBE buffer) using Criterion™ cell apparatus (1656001, Bio-Rad) at 300 V for 40 to 60 min. For unlabeled DNA, gels were stained with 1× SYBR™ Gold Nucleic Acid Gel Stain (S11494, Thermo Fisher) for 10 min. Gel images were visualized by TyphoonTM Biomolecular Imager (Cytiva) and quantified by ImageQuant TL image analysis software (v8.1, Cytiva). The percent product formation was calculated by dividing the intensity of the lower product band by the sum of the intensities of the product and substrate bands. RNA competition experiments were conducted with both RNA and DNA present in the reaction mixture, following the same protocol with the addition of an RNase inhibitor (M0314, New England Biolabs).
Crystal growth, data collection, structure determination, and analysis
rA3GR8 carrying the inactive mutation E259A (rA3GR8/E259A) was purified using the same protocol as described above. The rA3GR8/E259A-DNA complexes were prepared by mixing protein (4 mg/ml) with DNA at 1 to 1 molar ratio. After incubating on ice for 1 h, precipitation was removed by centrifugation (21,000 × g, 2 min, 4 °C). Initial screening was conducted using the sitting-drop vapor diffusion method with the ARI Crystal Gryphon Robot (ARI) and crystallization screening kits (QIAGEN) at 18 °C. Crystallization hits were further optimized using the hanging-drop vapor diffusion method at 18 °C. High-quality crystals of the rA3GR8/E259A-AA DNA complex were obtained with a reservoir solution consisting of 0.1 M Bis-Tris Propane at pH 7.3, 0.2 M Na/KPO4, and 18% PEG 3350. Similarly, high-quality crystals of the rA3GR8/E259A-GA DNA complex were obtained with a reservoir solution consisting of 0.1 M Bis-Tris Propane at pH 7.5, 0.2 M Na/KPO4, and 16% PEG 3350. These crystals were transferred to synthetic mother liquor supplemented with suitable amounts of glycerol for cryoprotection and then flash-cooled in liquid nitrogen. X-ray diffraction data were collected at the Advanced Photon Source (GM/CA@APS, Argonne National Laboratory) beamline 23ID-D and at the Advanced Light Source (ALS, Lawrence Berkeley Laboratory) beamline 5.0.3.
Data were processed by automated data processing pipelines at the beamlines. Initial phase information was obtained by molecular replacement method with PHENIX (v1.20.1) using the rA3G crystal structures (PDB 7UU4 or 8EDJ)33 without bound RNA. DNA was built manually in COOT. The structural models were refined using PHENIX (v1.20.1) and modified with COOT (v0.9.8.7). Data collection statistics and refinement parameters are summarized in Supplementary Table 1. Hydrogen bonding predictions were done by QtPISA (v2.1.0). Structure images were prepared with PyMOL (v2.5.3).
HEK293T whole-cell extract preparation and Western blot
HEK293T cells were seeded in 12-well plates 24 h before transfection. FLAG-tagged A3G constructs or empty vector pcDNA were transfected using Lipofectamine 3000 Transfection Reagent (L3000008, Thermo Fisher) following the manufacturer’s recommendations and incubated for 48 h before harvesting. HEK293T whole-cell extracts were prepared in 100 μl M-PER™ Mammalian Protein Extraction Reagent (78501, Thermo Fisher Scientific) supplemented with 1× Halt™ Protease Inhibitor Cocktail (78429, Thermo Fisher Scientific) and 150 mM NaCl. Total protein concentration in the extracts was estimated using Pierce™ BCA Protein Assay Kit (23225, Thermo Fisher Scientific) following the manufacturer’s recommendations, with a typical yield of around 3 mg/ml. Extracts were stored in small aliquots at −80 °C.
For western blot, proteins in HEK293T whole-cell extracts were separated by SDS-PAGE and transferred onto Immobilon® -FL PVDF membrane (IPFL00010, EMD Millipore). The membranes were blotted with primary antibodies: anti-FLAG M2 mAb (F3165, SIGMA, 1:3000) and anti-alpha tubulin mAb (GT114, GeneTex, 1:5000). ECL Plex Goat-α-Mouse IgG-Cy3 (PA43009, Cytiva, 1:3000) was used as the secondary antibody. Cy3 signals were detected using the TyphoonTM Biomolecular Imager (Cytiva).
Statistical analysis
We assessed the assumption of equal variances using an F-test. Data analysis was performed using a two-tailed Student’s t-test in Excel (v16.82), assuming homoscedasticity. A significance level of α = 0.05 was applied to determine statistical significance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information files, and available from the corresponding author upon request. Atomic coordinates and structure factors have been deposited in the PDB database under accession codes 8TVC (rA3GR8/E259A in complex with DNA 5′-CAATC) and 8TX4 (rA3GR8/E259A in complex with DNA 5′-TGAT). The atomic models used in this study are available in the PDB database under accession codes 6BUX, 7UU4, 8CX0, and 8FIK. Source data are provided with this paper.
References
Sheehy, A. M., Gaddis, N. C., Choi, J. D. & Malim, M. H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650 (2002).
Lecossier, D., Bouchonnet, F., Clavel, F. & Hance, A. J. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300, 1112 (2003).
Mangeat, B. et al. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424, 99–103 (2003).
Zhang, H. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424, 94–8 (2003).
Yu, Q. et al. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11, 435–442 (2004).
Suspene, R. et al. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 32, 2421–2429 (2004).
Holtz, C. M., Sadler, H. A. & Mansky, L. M. APOBEC3G cytosine deamination hotspots are defined by both sequence context and single-stranded DNA secondary structure. Nucleic Acids Res. 41, 6139–6148 (2013).
Cuevas, J. M., Geller, R., Garijo, R., Lopez-Aldeguer, J. & Sanjuan, R. Extremely high mutation rate of HIV-1 in vivo. PLoS Biol. 13, e1002251 (2015).
Browne, E. P., Allers, C. & Landau, N. R. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology 387, 313–321 (2009).
Conticello, S. G., Harris, R. S. & Neuberger, M. S. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 13, 2009–2013 (2003).
Newman, E. N. et al. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15, 166–170 (2005).
Guo, F., Cen, S., Niu, M., Saadatmand, J. & Kleiman, L. Inhibition of tRNA(3)(Lys)-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 80, 11710–11722 (2006).
Iwatani, Y. et al. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 35, 7096–7108 (2007).
Bishop, K. N., Verma, M., Kim, E. Y., Wolinsky, S. M. & Malim, M. H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4, e1000231 (2008).
Wang, X. et al. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J. Virol. 86, 3777–3786 (2012).
Gillick, K. et al. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J. Virol. 87, 1508–1517 (2013).
Pollpeter, D. et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat. Microbiol. 3, 220–233 (2018).
Olson, M. E., Harris, R. S. & Harki, D. A. APOBEC enzymes as targets for virus and cancer therapy. Cell Chem. Biol. 25, 36–49 (2018).
Green, A. M. & Weitzman, M. D. The spectrum of APOBEC3 activity: from anti-viral agents to anti-cancer opportunities. DNA Repair (Amst.) 83, 102700 (2019).
Ito, J., Gifford, R. J. & Sato, K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc. Natl Acad. Sci. USA 117, 610–618 (2020).
Uriu, K., Kosugi, Y., Ito, J. & Sato, K. The battle between retroviruses and APOBEC3 genes: its past and present. Viruses 13, 124 (2021).
Mertz, T. M., Collins, C. D., Dennis, M., Coxon, M. & Roberts, S. A. APOBEC-induced mutagenesis in cancer. Annu. Rev. Genet. 56, 229–252 (2022).
Pecori, R., Di Giorgio, S., Paulo Lorenzo, J. & Nina Papavasiliou, F. Functions and consequences of AID/APOBEC-mediated DNA and RNA deamination. Nat. Rev. Genet. https://doi.org/10.1038/s41576-022-00459-8 (2022).
Petljak, M. et al. Mechanisms of APOBEC3 mutagenesis in human cancer cells. Nature 607, 799–807 (2022).
Liu, W. et al. The cytidine deaminase APOBEC3G contributes to cancer mutagenesis and clonal evolution in bladder cancer. Cancer Res. 83, 506–520 (2023).
Butler, K. & Banday, A. R. APOBEC3-mediated mutagenesis in cancer: causes, clinical significance and therapeutic potential. J. Hematol. Oncol. 16, 31 (2023).
Sharma, S., Patnaik, S. K., Taggart, R. T. & Baysal, B. E. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci. Rep. 6, 39100 (2016).
Sharma, S. & Baysal, B. E. Stem-loop structure preference for site-specific RNA editing by APOBEC3A and APOBEC3G. PeerJ 5, e4136 (2017).
Sharma, S. et al. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol. 20, 37 (2019).
Kim, K. et al. The roles of APOBEC-mediated RNA editing in SARS-CoV-2 mutations, replication and fitness. Sci. Rep. 12, 14972 (2022).
Yang, H. et al. Understanding the structural basis of HIV-1 restriction by the full length double-domain APOBEC3G. Nat. Commun. 11, 632 (2020).
Maiti, A. et al. Crystal structure of a soluble APOBEC3G variant suggests ssDNA to bind in a channel that extends between the two domains. J. Mol. Biol. 432, 6042–6060 (2020).
Yang, H., Kim, K., Li, S., Pacheco, J. & Chen, X. S. Structural basis of sequence-specific RNA recognition by the antiviral factor APOBEC3G. Nat. Commun. 13, 7498 (2022).
Ito, F. et al. Structural basis for HIV-1 antagonism of host APOBEC3G via Cullin E3 ligase. Sci. Adv. 9, eade3168 (2023).
Li, Y. L. et al. The structural basis for HIV-1 Vif antagonism of human APOBEC3G. Nature 615, 728–733 (2023).
Kouno, T. et al. Structural insights into RNA bridging between HIV-1 Vif and antiviral factor APOBEC3G. Nat. Commun. 14, 4037 (2023).
Hache, G., Liddament, M. T. & Harris, R. S. The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J. Biol. Chem. 280, 10920–10924 (2005).
Navarro, F. et al. Complementary function of the two catalytic domains of APOBEC3G. Virology 333, 374–386 (2005).
Kouno, T. et al. Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat. Struct. Mol. Biol. https://doi.org/10.1038/nsmb.3033 (2015).
Xiao, X., Li, S. X., Yang, H. & Chen, X. S. Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat. Commun. 7, 12193 (2016).
Nowarski, R., Britan-Rosich, E., Shiloach, T. & Kotler, M. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat. Struct. Mol. Biol. 15, 1059–1066 (2008).
Furukawa, A. et al. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 28, 440–451 (2009).
Rausch, J. W., Chelico, L., Goodman, M. F. & Le Grice, S. F. Dissecting APOBEC3G substrate specificity by nucleoside analog interference. J. Biol. Chem. 284, 7047–7058 (2009).
Maiti, A. et al. Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 9, 2460 (2018).
Iwatani, Y., Takeuchi, H., Strebel, K. & Levin, J. G. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 80, 5992–6002 (2006).
Maiti, A. et al. Structure of the catalytically active APOBEC3G bound to a DNA oligonucleotide inhibitor reveals tetrahedral geometry of the transition state. Nat. Commun. 13, 7117 (2022).
Holden, L. G. et al. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 456, 121–124 (2008).
Chelico, L., Prochnow, C., Erie, D. A., Chen, X. S. & Goodman, M. F. Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J. Biol. Chem. 285, 16195–16205 (2010).
Chelico, L., Pham, P., Calabrese, P. & Goodman, M. F. APOBEC3G DNA deaminase acts processively 3’ –> 5’ on single-stranded DNA. Nat. Struct. Mol. Biol. 13, 392–399 (2006).
Chelico, L., Sacho, E. J., Erie, D. A. & Goodman, M. F. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 283, 13780–13791 (2008).
Chelico, L., Pham, P. & Goodman, M. F. Stochastic properties of processive cytidine DNA deaminases AID and APOBEC3G. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 364, 583–593 (2009).
Morse, M. et al. HIV restriction factor APOBEC3G binds in multiple steps and conformations to search and deaminate single-stranded DNA. Elife https://doi.org/10.7554/eLife.52649 (2019).
Harjes, S. et al. Impact of H216 on the DNA binding and catalytic activities of the HIV restriction factor APOBEC3G. J. Virol. 87, 7008–7014 (2013).
Ambia-Garrido, J., Vainrub, A. & Pettitt, B. M. A model for structure and thermodynamics of ssDNA and dsDNA near a surface: a coarse grained approach. Comput. Phys. Commun. 181, 2001–2007 (2010).
Chi, Q., Wang, G. & Jiang, J. The persistence length and length per base of single-stranded DNA obtained from fluorescence correlation spectroscopy measurements using mean field theory. Physica A 392, 1072–1079 (2013).
Soros, V. B., Yonemoto, W. & Greene, W. C. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 3, e15 (2007).
McDougall, W. M. & Smith, H. C. Direct evidence that RNA inhibits APOBEC3G ssDNA cytidine deaminase activity. Biochem. Biophys. Res. Commun. 412, 612–617 (2011).
Polevoda, B. et al. RNA binding to APOBEC3G induces the disassembly of functional deaminase complexes by displacing single-stranded DNA substrates. Nucleic Acids Res. 43, 9434–9445 (2015).
Belanger, K. & Langlois, M. A. RNA-binding residues in the N-terminus of APOBEC3G influence its DNA sequence specificity and retrovirus restriction efficiency. Virology 483, 141–148 (2015).
Smith, H. C. RNA binding to APOBEC deaminases; Not simply a substrate for C to U editing. RNA Biol. 14, 1153–1165 (2017).
Bulliard, Y. et al. Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J. Virol. 85, 1765–1776 (2011).
Fukuda, H. et al. Structural determinants of the APOBEC3G N-terminal domain for HIV-1 RNA association. Front. Cell Infect. Microbiol. 9, 129 (2019).
Kouno, T. et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 8, 15024 (2017).
Harjes, S. et al. Structure-guided inhibition of the cancer DNA-mutating enzyme APOBEC3A. Nat. Commun. 14, 6382 (2023).
Prochnow, C., Bransteitter, R., Klein, M. G., Goodman, M. F. & Chen, X. S. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature 445, 447–451 (2007).
van der Kuyl, A. C. & Berkhout, B. The biased nucleotide composition of the HIV genome: a constant factor in a highly variable virus. Retrovirology 9, 92 (2012).
Ebrahimi, D., Anwar, F. & Davenport, M. P. APOBEC3 has not left an evolutionary footprint on the HIV-1 genome. J. Virol. 85, 9139–9146 (2011).
Alinejad-Rokny, H., Anwar, F., Waters, S. A., Davenport, M. P. & Ebrahimi, D. Source of CpG Depletion in the HIV-1 Genome. Mol. Biol. Evolut. 33, 3205–3212 (2016).
Desimmie, B. A. et al. APOBEC3 proteins can copackage and comutate HIV-1 genomes. Nucleic Acids Res. 44, 7848–7865 (2016).
McDonnell, M. M. et al. Highly-potent, synthetic APOBEC3s restrict HIV-1 through deamination-independent mechanisms. PLoS Pathog. 17, e1009523 (2021).
Yousefi, M., Annan Sudarsan, A. K., Gaba, A. & Chelico, L. Stability of APOBEC3F in the presence of the APOBEC3 antagonist HIV-1 Vif increases at the expense of co-expressed APOBEC3H haplotype I. Viruses 15, 463 (2023).
Adolph, M. B., Love, R. P., Feng, Y. & Chelico, L. Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res. 45, 11925–11940 (2017).
Wong, L., Vizeacoumar, F. S., Vizeacoumar, F. J. & Chelico, L. APOBEC1 cytosine deaminase activity on single-stranded DNA is suppressed by replication protein A. Nucleic Acids Res. 49, 322–339 (2021).
Brown, A. L. et al. Single-stranded DNA binding proteins influence APOBEC3A substrate preference. Sci. Rep. 11, 21008 (2021).
Wong, L., Sami, A. & Chelico, L. Competition for DNA binding between the genome protector replication protein A and the genome modifying APOBEC3 single-stranded DNA deaminases. Nucleic Acids Res. 50, 12039–12057 (2022).
Lada, A. G. et al. Replication protein A (RPA) hampers the processive action of APOBEC3G cytosine deaminase on single-stranded DNA. PloS ONE 6, e24848 (2011).
McDaniel, Y. Z. et al. Deamination hotspots among APOBEC3 family members are defined by both target site sequence context and ssDNA secondary structure. Nucleic Acids Res. 48, 1353–1371 (2020).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385.e318 (2018).
Acknowledgements
We thank Phuong Pham and Malgorzata Jaszczur for advice on functional biochemical analyses. Beamlines of GM/CA@APS have been funded by the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006, P30GM138396). The ALS-ENABLE beamlines are supported in part by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169. This work is supported by the NIH grant R01 AI150524 to X.S.C.
Author information
Authors and Affiliations
Contributions
H.Y. and X.S.C. designed the experiments. H.Y. performed crystallization, data collection, structural determination, and functional biochemical analyses. J.P. performed structural work and refinements. K.K. and F.I assisted the project. D.E. and A.B. analyzed the HIV-1 and SIV genomes. H.Y. and X.S.C. wrote the paper. All authors commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rémi Buisson, Yasumasa Iwatani, who co-reviewed with Hirotaka Ode and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, H., Pacheco, J., Kim, K. et al. Molecular mechanism for regulating APOBEC3G DNA editing function by the non-catalytic domain. Nat Commun 15, 8773 (2024). https://doi.org/10.1038/s41467-024-52671-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52671-1
This article is cited by
-
On the mechanism of NPM1 mutations in acute myeloid leukemia
Leukemia (2025)
