
Abstract
Direct carbonylation of CH4 to CH3COOH provides a promising pathway for upgrading of natural gas to transportable liquid chemicals, in which high-efficiency CH4 activation and controllable C–C coupling are both critical but challenging. Herein, we report that highly efficient photo-driven carbonylation of CH4 with CO and O2 to CH3COOH is achieved over MoS2-confined Rh-Zn atomic-pair in conjunction with TiO2. It delivers a high CH3COOH productivity of 152.0 μmol gcat.−1 h−1 and turnover frequency of 62.0 h−1 with a superior selectivity of 96.5%, outperforming previous photocatalytic CH4 carbonylation processes. Mechanistic investigations disclose the key effect of Rh-Zn synergy in combination with photo-excited electrons from TiO2 for CH3COOH formation. The active OH species produced from O2 photoreduction on the Zn site through proton-coupled electron transfer promotes CH4 dissociation to CH3 species, which then facilely couples with adsorbed CO on the adjacent Rh site forming the key CH3CO intermediate for CH3COOH formation.
Similar content being viewed by others
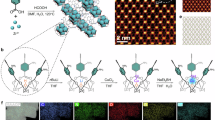


Introduction
Conversion of methane (CH4) to high value-added multi-carbon (C2+) oxygenates, such as acetic acid (CH3COOH), is a significant pathway for the utilization of natural gas1,2,3, which typically goes through the indirect and energy-intensive syngas route with severe carbon emission and poor product selectivity4,5,6,7. Direct conversion of CH4 to CH3COOH under mild conditions is highly attractive and environmental-friendly. However, it suffers from not only the difficulty in the C–H activation of the inert CH4, but also the trade-off in the formation of key CH3 and CO surface intermediates with opposite carbon valance states. Introducing carbon monoxide (CO) as a co-reactant can bypass the generation of CO* intermediate from the complex and sluggish dehydrogenation and oxygenation of CH4. Hence, combined with utilization of O2 as an inexpensive oxidant, oxidative carbonylation of CH4 with CO offers an economical and sustainable route for producing CH3COOH.
The oxidative carbonylation of CH4 with CO and O2 to CH3COOH involves three critical steps, including (i) activation and dissociation of O2 to generate active oxygen species, (ii) activation and dissociation of CH4 to CH3 species, and then (iii) C–C coupling between adsorbed CO and CH3. A variety of catalysts with supported transition metal (Rh, Pd, Ir, Pt, Au, Fe) single atoms or nanoparticles were developed for the oxidative carbonylation of CH4 with CO under mild conditions8,9,10,11,12,13,14,15,16,17,18,19,20,21. However, these processes are still limited by the insufficient productivity or selectivity of CH3COOH, which are mostly due to the difficulty in controlling the complex reaction network with a single type of active phase. Achieving both high activity and selectivity for the conversion of CH4, CO and O2 to CH3COOH requires precise regulation of above three key steps while suppressing the competing CO oxidation and product overoxidation to CO2, which remains a great challenge.
Herein, we report a high-efficiency photo-driven carbonylation of CH4 with CO and O2 to CH3COOH (CCOC2) process over a heterostructure catalyst with Rh-Zn atomic pair dual sites confined in MoS2 lattice integrated with TiO2 (RhZn-MoS2/TiO2). It exhibits a high CH4 turnover frequency (TOF) of 62.0 h−1 and CH3COOH productivity of 152.0 μmol gcat.−1 h−1 with a superior selectivity of 96.5%, significantly surpassing those of previously-reported photocatalytic CH4 conversion to CH3COOH processes without additional energy input. Systematic experimental and computational studies propose a synergy at the MoS2-confined Rh-Zn dual sites in combination with photoexcitation of the RhZn-MoS2/TiO2 for the selective production of CH3COOH, which enables much higher productivity and selectivity of CH3COOH than those over the catalysts with only Rh or Zn confined in the MoS2 parts. The photoexcited electrons from TiO2 prominently promotes the O2 reduction at the confined Zn-Mo bridge site via a proton-coupled electron transfer (PCET) mechanism to generate Mo=O and Zn–OH species, in which the highly reactive Zn–OH species can activate CH4 and enable dissociation of CH4 to CH3 species and then form the Zn–OH2 species. The preferentially adsorbed CO on the adjacent Rh site could then facilely couple with the generated CH3 forming CH3CO species. Then, after the desorption of the generated H2O from the Zn site and the PCET-based hydrogenation of the neighboring Mo=O to Mo–OH, the CH3CO on the Rh site could be facilely transferred to the adjacent Zn site coupled with the adsorption of CO on the vacated Rh site, followed by the formation of CH3COOH via the combination of the CH3CO and OH species (Fig. 1a). In this regard, this photo-driven CH4 carbonylation process is analogous to the CH4 conversion process with O2 by methane monooxygenase (MMO) in the aid of reduced nicotinamide adenine dinucleotide (NADH) which supplies protons and electrons22 (Fig. 1b). The construction of Rh-Zn atomic-pair dual sites separates the catalytic sites for the C–H activation and C–C coupling while establishes a synergy between them, thereby breaking the trade-off between the activity and selectivity in the CCOC2 process.

The catalytic cycle for the photo-driven CH4 carbonylation with CO and O2 to CH3COOH over the RhZn-MoS2/TiO2 (a), in comparison with the biocatalytic CH4 conversion with O2 by the MMO (Protein Data Bank 1FYZ) in the aid of NADH (b).
Results
Photocatalytic performance of CH4 carbonylation
The photo-driven CCOC2 process was carried out in a windowed autoclave with water (H2O) as solvent under UV-visible light irradiation. To investigate the distribution and origin of reaction products, a series of controlled experiments were conducted by using CH4, CO and O2 (CH4 + CO + O2), isotopically labeled 13CH4, CO and O2 (13CH4 + CO + O2), isotopically labeled 13CO, CH4 and O2 (CH4 + 13CO + O2) and isotopically labeled 18O2, CH4 and CO (CH4 + CO + 18O2) as the feed gas, respectively, over the RhZn-MoS2/TiO2 catalyst. 1H and 13C nuclear magnetic resonance (NMR) spectra and gas chromatography-mass spectrometry (GC-MS) analysis of the products show that the CH4 was mainly converted to CH3COOH with a small amount of CH3OH and HOCH2OH (Fig. 2a and Supplementary Fig. 1a). Only 13C-labeled products (13CH3COOH, 13CH3OH and HO13CH2OH) were produced in the case of 13CH4 + CO + O2, and no liquid C1-2 oxygenated products was detected in the control experiments using pure Ar, CO + O2, or CH4 + CO as the feed gas (Supplementary Fig. 1c). Moreover, to track the source of specific carbon atoms in the product, isotopically labeled experiment was conducted by using CH4 + 13CO + O2 as the reactant and an obvious signal of CH313COOH can be observed in 1H NMR spectra (Fig. 2a). These results suggest that the carbon atom in the CH3 part and CO part of CH3COOH comes from the CH4 and CO, respectively, rather than from possible C-containing contaminants in the environment or catalyst itself. Furthermore, isotopically labeled experiment using CH4 + CO + 18O2 as the reactant showed that obvious peaks at m/z = 62 and m/z = 47 assigned to CH3CO18OH and CO18OH fragment were detected by GC-MS analysis, indicating that the oxygen atom in the OH part of CH3COOH comes from O2 (Supplementary Fig. 1b). In addition, no obvious signal of 13CO2 was observed in the GC-MS of gaseous products and 13C NMR spectra of liquid products when using 13CH4 + CO + O2 as the feed gas (Supplementary Fig. 2), suggesting that the overoxidation of CH4 is efficiently suppressed.

a 1H NMR spectra of products when using 13CH4 + CO + O2, CH4 + 13CO + O2 and CH4 + CO + O2 as the feed gas over the RhZn-MoS2/TiO2 catalyst, respectively. b Comparison of catalytic activity for different catalysts. c The effect of power density on the photocatalytic activity over RhZn-MoS2/TiO2 catalysts. d The catalytic activity over RhZn-MoS2/TiO2 under visible light irradiation (wavelength > 420 nm), UV-visible light irradiation (wavelength = 200–800 nm) and thermal catalysis at 65 °C with a similar reaction temperature to that of induced by the UV-visible light irradiation, respectively. Data in b and d are all presented as mean ± s.d. Each reaction was repeated for three times to obtain the error bars (n = 3). e Two-dimensional contour snapshot of CH3COOH selectivity under different partial pressure of CH4, O2 and CO. f The productivity and selectivity of CH3COOH in comparison with the previously reported photocatalytic systems for CH4 conversion to CH3COOH (see Supplementary Table 2 for more details). Reaction tests in (a-e) were conducted by using 5 bar CH4 (or 13CH4), 1 bar O2 (or 18O2), 11 bar CO (or 13CO), 5 mL H2O and 25 mg catalyst with a stirring rate of 1000 revolutions per minute (rpm) for 5 h under a 300 W Xe lamp irradiation (wavelength = 200–800 nm) with a light intensity of 1500 mW cm−2.
The CH3COOH productivities over different catalysts under UV-visible light irradiation are summarized in Fig. 2b. Compared with the undoped MoS2/TiO2 and pure TiO2 catalysts, Rh-doped or Zn-doped MoS2 in conjunction with TiO2 (Rh-MoS2/TiO2 and Zn-MoS2/TiO2) notably enhances the production of CH3COOH, which indicates the important effect of Zn-doping and Rh-doping in promoting the CCOC2 process. More importantly, co-doping Rh and Zn atoms into the MoS2 part (RhZn-MoS2/TiO2) with optimized mass ratio of 2.7 and 2.8 w.t.%, respectively, further, significantly increase the CH3COOH productivity and selectivity to 152.0 µmol gcat.−1 h−1 and 96.5%, respectively, compared with those of 56.3 µmol gcat.−1 h−1 and 87.7% over the Rh-MoS2/TiO2 catalyst, and 33.6 µmol gcat.−1 h−1 and 59.5% over the Zn-MoS2/TiO2 catalyst (Fig. 2b and Supplementary Fig. 3 and 4). These results demonstrate the existence of a synergy effect between the MoS2-confined Rh and Zn atoms in promoting the CCOC2 process. In addition, with the increase of UV-visible light (λ = 200–800 nm) intensity from 300 to 1500 mW cm−2, a notable enchantment on the CH3COOH productivity and selectivity from 40.5 µmol gcat.−1 h−1 and 86.0% to 152.0 µmol gcat.−1 h−1 and 96.5% was observed over the RhZn-MoS2/TiO2 catalyst with an optimized TiO2 mass ratio of 50 w.t.% (Fig. 2c and Supplementary Fig. 5a). However, the thermal-catalytic and visible light driven CCOC2 process by RhZn-MoS2/TiO2 presents much lower CH3COOH productivity and selectivity (Fig. 2d), implying that the UV-visible light can significantly promote the CH4 carbonylation to CH3COOH. Moreover, replacing TiO2 with SiO2 (RhZn-MoS2/SiO2) leads to a notably low CH3COOH productivity of 28.4 µmol gcat.−1 h−1 with a lower selectivity of 73.3% (Supplementary Fig. 5b). These results suggests that the photoexcited charge carriers from TiO2 can prominently facilitate the production of CH3COOH over RhZn-MoS2/TiO2 heterostructure under UV-visible light irradiation. Besides, the partial pressure ratio of CH4, O2 and CO also has a great influence on the CH3COOH productivity and selectivity. Under a total pressure of 17 bar with CH4/O2/CO partial pressure ratio of 5/1/11, the CH3COOH productivity reaches up to 152.0 µmol gcat.−1 h−1 with a selectivity of 96.5% (Fig. 2e and Supplementary Fig. 6), which is significantly superior to the previously reported photo-driven CH4 carbonylation process without external energy input (Fig. 2f). Moreover, the RhZn-MoS2/TiO2 catalyst offers significantly better selectivity and comparable productivity than that of photo-driven CCOC2 process when operated at similar reaction temperatures above 110 °C (Supplementary Table 1). The rising of CH4 pressure to 30 bar further promotes the productivity of CH3COOH up to 426.0 µmol gcat.−1 h−1 with a TOF of around 192.0 h−1 (Supplementary Table 2). The RhZn-MoS2/TiO2 catalyst also displays good reusability without obvious deactivation in six consecutive reaction cycles (Supplementary Fig. 7). The morphology and valence states of the used RhZn-MoS2/TiO2 catalyst are well maintained in contrast with the fresh catalyst (Supplementary Fig. 8), indicating the high stability of this catalyst.
Morphology and electronic structure of RhZn-MoS2/TiO2
To reveal the active sites of RhZn-MoS2/TiO2 catalyst for the CCOC2 process, comprehensive characterizations were performed to study the structural and electronic properties of the heterostructure. X-ray diffraction (XRD) analysis shows the obvious crystallographic structure and lattice parameters of 2H-MoS2 and anatase and rutile mixed phase TiO2 in the RhZn-MoS2/TiO2 catalyst (Supplementary Fig. 9)23. In addition, the Raman spectra of RhZn-MoS2/TiO2 displays two characteristic peaks at 378 cm−1 and 402 cm−1 assigned to in-plane (E12g) and out-of-plane (A1g) vibration peaks of the 2H phase MoS2, respectively, which further demonstrates the existence of 2H-MoS2 in RhZn-MoS2/TiO2 (Supplementary Fig. 10). The HRTEM presents that the TiO2 nanoparticles with a diameter of 20-30 nm are coated by 2-4 layers of MoS2 nanosheets (Fig. 3a and Supplementary Fig. 11). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image presents a distinct interface between the MoS2 and TiO2 (Fig. 3b, c and Supplementary Fig. 12), which suggests the existence of Ti–S bonds at the interface of MoS2 and TiO2 based on the intensity profile analysis (Fig. 3d)24. This interface could work as the transport channels to promote the separation and transfer of the photogenerated electrons and holes, which have an important impact on the photocatalytic activity.

High-resolution TEM images (a) and high-resolution HAADF-STEM images (b) of RhZn-MoS2/TiO2. c-f The wide-field HAADF-STEM, the geometrical configuration and atomic intensity profile analysis of RhZn-MoS2/TiO2. g Simulated image of the monolayer RhZn-MoS2. h The measured and simulated atomic intensity profile along the corresponding dotted boxes in f and g, respectively. i, j Normalized XAFS spectra of the Zn K-edge and Rh K-edge for obtained RhZn-MoS2/TiO2 and referenced Zn foil, ZnS, Rh foil and Rh2O3. k The EXAFS signal in R-space for the Zn foil, ZnS, Rh foil, Rh2O3 and RhZn-MoS2/TiO2 catalysts. l Wavelet transform of Zn K-edge and Rh K-edge of the RhZn-MoS2/TiO2 catalysts, respectively.
Beyond that, the high magnification HAADF-STEM image combined with the extended X-ray absorption fine structure (EXAFS) and wavelet transform reveal that the Zn and Rh were atomically confined in the MoS2 nanosheets via replacing the Mo atoms and the confined Zn and Rh were coordinated with S atoms without the formation of Rh–Rh and Zn–Zn bonds (Fig. 3e, k, l and Supplementary Fig. 13)25. The intensities of spots are counted through a cross-line scan (Fig. 3e, f), and the relative statistical intensities suggest the existence of adjacent Zn-Rh pairs confined in MoS2 lattice, which is further confirmed by the simulated image of the atomic model and the line intensity profiles (Fig. 3g, h and Supplementary Fig. 14). The detailed analysis of the X-ray adsorption near-edge structure (XANES) spectra show that the Zn atoms are electron-deficient while the Rh atoms are electron-rich in the RhZn-MoS2/TiO2 compared with those in the Zn-MoS2/TiO2 and Rh-MoS2/TiO2, respectively (Fig. 3i, j), which could be due to the interaction between the adjacent Zn and Rh atoms in the RhZn-MoS2/TiO2 catalysts, leading to charge transfer from the less electronegative Zn to the more electronegative Rh. Based on the above analysis, we deduce that the enhanced photocatalytic CH4 conversion could be originated from the Zn-Rh pair confined in MoS2 in combination with the Ti–S bonds at the MoS2-TiO2 interface.
Furthermore, the light absorption properties, charge separation and transport efficiency were investigated to understand the photo-effect on the CCOC2 process over the RhZn-MoS2/TiO2 catalyst. Compared with the TiO2 and RhZn-MoS2, the RhZn-MoS2/TiO2 catalyst presented a stronger light absorption on the UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS) measurement, indicating a better ability of broad-spectrum light trapping from UV to infrared region (Fig. 4a). The increased optical absorption can significantly enhance the photothermal catalytic effect, which render an enhanced CH4 conversion. Besides, the photoluminescence (PL) intensity and average lifetime (ave. τ) derived from the time-resolved PL (TRPL) spectroscopy decreased in the following sequence: RhZn-MoS2/TiO2 < RhZn-MoS2 < TiO2, which indicates that RhZn-MoS2/TiO2 possesses a better ability to inhibit the recombination of photogenerated electrons and holes (Fig. 4b and Supplementary Fig. 15a)26. In addition, the RhZn-MoS2/TiO2 presents a higher photocurrent density than the TiO2 and RhZn-MoS2, which suggests that the RhZn-MoS2/TiO2 has a stronger photo response ability and greater transfer efficiency of the photogenerated carriers (Fig. 4c)26. Based on the above results, we propose that the enhancement in the light absorption, separation and transfer of photogenerated excitons over the RhZn-MoS2/TiO2 effectively promote photocatalytic CCOC2 process26,27.

a UV-vis diffuse reflectance spectra. b Time-resolved photoluminescence spectra. c Transient photocurrent responses. d–f Quasi in-situ high-resolution Zn 2p, Rh 3d and Mo 3d XPS spectra with and without UV-visible light irradiation. g, h Normalized XAFS spectra of the Zn K-edge and Mo K-edge in RhZn-MoS2/TiO2 catalysts with and without UV-visible light irradiation. i Schematic energy band diagrams and charge transfer route of the heterojunction between TiO2 and RhZn-MoS2 (Evac = Vacuum energy, CB = Conduction band, VB = Valence band, EF = Fermi level).
Quasi-in-situ X-ray photoelectron spectroscopy (XPS) and in-situ X-ray absorption fine structure (XAFS) were further investigated to analyze the photo-generated electron transfer direction by measuring the variation of the valence state of metal sites before and after light irradiation of the RhZn-MoS2/TiO2 catalyst. Quasi-in-situ XPS results display the obvious shifts towards lower binding energy for Zn 2p, Rh 3d and Mo 3d but towards higher binding energy for Ti 2p after UV-visible light, which indicates that the Zn, Rh and Mo atoms receive electrons from TiO2 (Fig. 4d–f and Supplementary Fig. 12e). This is in accordance with the results from in-situ XAFS spectroscopy, where the Zn and Mo K-edge shifts to a lower energy level but the Ti K-edge shifts to a higher energy level under light irradiation (Fig. 4g, h and Supplementary Fig. 12f)28. Furthermore, we have conducted UV-vis diffuse reflectance spectrum (UV-vis DRS), ultraviolet photoelectron spectroscopy (UPS) and femtosecond transient absorption to clarify the band structure, contact type and the separation of photoinduced carriers between of TiO2 and RhZn-MoS2. The UV-vis DRS and UPS analysis indicate that RhZn-MoS2 has a smaller work function but a higher Fermi level than that of TiO2 (Fig. 4i and Supplementary Fig. 16). This demonstrates that the electrons could transfer from RhZn-MoS2 to TiO2 to attain the Fermi level equilibrium, thus leading to the generation of an electric field pointing from RhZn-MoS2 to TiO2 at the interface. The built-in electric field induces the transfer of photogenerated electrons from the conduction band of TiO2 to the valence band of RhZn-MoS2, thus resulting in the photogenerated electrons and holes left on the RhZn-MoS2 and TiO2, respectively. Such a direct Z-scheme heterojunction between RhZn-MoS2 and TiO2 is not only beneficial for the electron mobility by shortening the charge transfer time and distance (Supplementary Fig. 15b), but also favorable for enhancing O2 activation by using the photogenerated electrons, thus promoting the photocatalytic CH4 conversion to CH3COOH.
Mechanism analysis for the photocatalytic CH4 conversion
To understand the reaction mechanism for the photocatalytic CCOC2 process over the RhZn-MoS2/TiO2 catalyst, we constructed models of MoS2 with Rh and Zn atoms co-confined into the MoS2 lattice substituting the Mo atoms and performed density functional theory (DFT) studies. Calculation results show that introducing Rh and Zn atoms into the MoS2 lattice notably decreases the average formation energy of sulfur vacancies (SVs) by 1.17 and 1.57 eV for the edge and in-plane SVs, respectively (Supplementary Fig. 17), which is favorable for the generation of SVs during the H2 reduction treatment of the RhZn-MoS2 catalyst at 300 °C. Among the models with different Rh-Zn co-doping configurations, the SVs confining an adjacent Rh-Zn pair is relatively more stable by over 1.42 eV than those confining separated Rh and Zn atoms (Supplementary Fig. 18), which suggests that the formation of SVs at the Rh-Zn pair-doped sites is energetically more favorable. Then, the adsorption-free energies (∆Gads) of CO and O2 on different sites of edge SVs confining a Rh-Zn pair were investigated, which shows that CO tends to be adsorbed on the Rh top sites with ∆Gads of –1.10 eV while O2 can be facilely co-adsorbed on the Zn-Mo bridge site with ∆Gads of –1.01 eV (Supplementary Fig. 19). However, as shown in Fig. 5a and Supplementary Fig. 20, direct dissociation of the adsorbed O2 (O2*) to form Zn=O* and Mo=O* species for CH4 activation (the symbol ‘=’ denotes top site adsorption configuration for O*) is endergonic by 0.92 eV and thus is thermodynamically unfavorable. In contrast, combined with protons in the water solvent and photoexcited electrons from the TiO2, O2* can be facilely hydrogenated to OOH* with a free energy change of –0.66 eV through a proton-coupled electron transfer (PCET) mechanism, followed by readily dissociation of OOH* to Zn–OH* and Mo=O* species with a free energy gain of –0.99 eV (Fig. 5a). In addition, a CO-assisted O2* dissociation path without involving the PCET process was also considered, in which the O2* reacts with another CO to generate Zn–O*–Mo (the symbol ‘–’ denotes bridge site adsorption configuration for O*) and CO2 with an energy barrier of 0.57 eV and free energy change of –2.74 eV (Fig. 5a). Electronic structure analysis shows that the Zn–OH* species possesses notably higher electronic density of O 2p states near the Fermi level compared with those of the Zn–O*–Mo and Mo=O* species, suggesting that the Zn–OH* species has a higher activity for the following CH4 activation (Fig. 5b).

a Comparison of the formation energy for different O species. b Projected density of states (PDOS) of the O sites in the Mo=O*, Zn–O*–Mo and Zn–OH* structure. c Formation pathways for CH3OH and CH3COOH at Zn–O*–Mo and Zn–OH* site, respectively. Insets show the atomic structures of reaction intermediates.
After the PCET-assisted formation of Zn–OH* or the CO-assisted formation of Zn–O*–Mo species at the edge SVs-confined Rh-Zn pair sites, the CH4 activation and C–C coupling steps were further studied and compared to understand the formation of CH3COOH. At the Zn–OH* site, the breakage of the first C–H bond of CH4 forming Zn–OH2* and CH3 followed by the direct coupling of the generated CH3 with the pre-adsorbed CO on the adjacent Rh site (Rh–CO*) to form the key CH3CO* intermediate, requires an overall activation energy of 0.70 eV, which is notably lower than the CH4 activation energies of 0.90 eV and 1.30 eV at the Zn–O*–Mo and Mo=O* sites, respectively (Fig. 5c). Then, after the desorption of the generated H2O* from the Zn site and the PCET-based hydrogenation of the neighboring Mo=O* to Mo–OH*, the CH3CO* on the Rh site could be facilely transferred to the adjacent Zn site coupled with the adsorption of CO on the vacated Rh site, followed by the formation of CH3COOH via the combination of the CH3CO* and OH* with a low activation energy of 0.13 eV. Such a PCET-involved reaction mechanism with one oxygen of O2 entering H2O and the other entering CH3COOH (Supplementary Fig. 21), was further verified by isotope-labeling experiments using CH4 + 18O2 + H2O as the reactants, in which H218O was observed in the products under the photocatalytic condition while no H218O was observed without light irradiation (Fig. 6a).

a GC-MS spectra of H2O generated from CH4 conversion with and without UV-visible light irradiation. b Impact of electrons and holes scavengers on photocatalytic CH4 conversion. Reaction tests in a were conducted by using 5 bar CH4, 1 bar O2 (or 18O2), 5 mL H2O, and 25 mg catalyst with a stirring rate of 1000 rpm for 5 h.
Furthermore, we have conducted the in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) experiments to identify the role of Rh and Zn sites during the reaction process (Supplementary Fig. 22). In-situ DRIFTS using CO as a probe molecule was first performed on various catalysts under ambient conditions (Supplementary Fig. 22a). On the MoS2/TiO2 and Zn-MoS2/TiO2, similar peaks were observed at 2051 cm−1 corresponding to adsorbed CO on the Mo site (Mo-CO*) rather than on the Zn site. In contrast, on the Rh-MoS2/TiO2 and RhZn-MoS2/TiO2 catalysts, larger peaks appear at 2083 cm−1 beside the attenuated peak of Mo-CO*, which can be attributed to the adsorbed CO on the Rh site (Rh-CO*)15. These results suggest that CO prefers to be adsorbed at the Rh sites, which is in accordance with the DFT calculations that Rh sites possess the lowest CO adsorption free energy in contrast to the Zn and Mo sites (Supplementary Fig. 19).
Additionally, in-situ DRIFTS was performed to capture the reaction intermediates by controlling the reaction gas for CH4 conversion over the RhZn-MoS2/TiO2 catalyst (Supplementary Fig. 22b and c). Firstly, the RhZn-MoS2/TiO2 catalyst is treated by 11 bar CO for 30 min, which leads to an absorption peak at 2083 cm−1 corresponding to the vibrations of Rh-CO* being detected. As the feed gas is switched to 1 bar O2 + H2O for 1 min (Supplementary Fig. 22b), notable increase in the Mo=O* and Mo–O*–Mo signals owing to dissociation of O2 is observed under light irradiation compared with the reference case without introducing O2 (Supplementary Fig. 22c). It is worth noting that the signal of Zn-O species is hard to be observed because the infrared signals of Zn-O species are generally below 600 cm−1. When RhZn-MoS2/TiO2 catalyst is further treated with 5 bar CH4 for 10 min, the peak intensity of Mo=O* and Mo-O*-Mo gradually decreases but the intensity of the CH3* peak at 1436 cm−1 increases, indicating the reaction of CH4 with the surface oxygen species. Meanwhile, the C=O vibration of CH3CO*(1743 cm−1) and the stretching vibration of the C-H bond in CH3CO* (2974 and 2872 cm−1) can be clearly observed, suggesting that the generated CH3 species can facilely couple with the adsorbed CO on the adjacent Rh site to form the key intermediate CH3CO* for CH3COOH formation.
To interpret the product selectivity, the formation path of CH3OH was also explored and compared with that of the CH3COOH. At the Mo=O* site adjacent to Zn–OH*, the activation of CH4 for CH3OH formation is kinetically unfavorable due to its notably high activation energy of 1.30 eV as mentioned above (Fig. 5c). In contrast, at the Zn–O*–Mo site, dissociation of CH4 to Zn–CH3* and Mo–OH* requires a lower activation energy of 0.90 eV, followed by the formation of CH3OH via the combination of CH3* and OH* with an energy barrier of 1.02 eV or the formation of CH3CO* intermediate via the combination of CH3* with a pre-adsorbed CO* on the adjacent Rh site with an energy barrier of 1.13 eV (Fig. 5c). This indicates that the formation of CH3OH at the Zn–O*–Mo site is kinetically more favorable with a relatively lower energy barrier than the formation of CH3COOH.
The effect of TiO2 on the reaction mechanism was also considered by constructing a heterojunction model with RhZn-MoS2 integrated with TiO2 (RhZn-MoS2/TiO2) (Supplementary Fig. 23). The energetics of several key steps including the adsorption of CO and O2 and the activation of CH4 were calculated using DFT (Supplementary Fig. 24 and 25), which shows that the adsorption energies of CO and O2 and the CH4 activation energy on the sites of RhZn-MoS2/TiO2 have no significant difference from those on the similar sites of the RhZn-MoS2 (Supplementary Table 3 and 4). Moreover, the Zn, Rh, and Mo sites in the RhZn-MoS2/TiO2 possess the same activity trend compared with those in the RhZn-MoS2 for the key adsorption and activation steps. On both structures, CO tends to be adsorbed on the Rh top sites and O2 can be facilely co-adsorbed on the Zn-Mo bridge site, and the Zn–OH* sites exhibit higher activity than the Zn-O*-Mo sites for the C-H bond activation of CH4. These calculation results indicate that TiO2 in the heterojunction has little effect on the intrinsic activity of the lattice-confined catalytic sites in the MoS2.
Control experiments were also conducted by using AgNO3 as electron scavenger to interrupt the PCET process or using Na2SO3 as hole scavenger to prevent exciton recombination and prolong the lifetime of excited electrons, in which CH3COOH productivity was significantly decreased and increased, respectively (Fig. 6b). These results indicate the crucial role of photoexcited electrons for the CH3COOH production via the CH3CO* intermediate, which are supportive to the above mechanism analysis. In contrast to the edge SVs-confined Rh-Zn pair site, the in-plane SVs-confined Rh-Zn pair site tends to be occupied by oxygen due to the much stronger adsorption of O2 than CO and the generated O* species are less active for CH4 activation owing to the higher energy barriers of over 1.13 eV compared with that of 0.70 eV at the edge (Supplementary Fig. 26). Therefore, we propose that the photocatalytic promotion effect on the CH3COOH production is due to the lower energy barriers for the formation of active OH species from photoreduction of O2 on the Zn site and the C–C coupling on the adjacent Rh site for the formation of CH3CO* intermediate.
Discussion
In summary, an efficient RhZn-MoS2/TiO2 heterostructure catalyst is synthesized for the photo-driven CCOC2 process, which delivers a record-high CH3COOH selectivity of 96.5%, productivity up to 152.0 µmol gcat.−1 h−1 and a TOF of 62.0 h−1, outperforming previous photocatalytic CH4 conversion to CH3COOH processes. The Rh-Zn atomic-pair dual sites separates the catalytic sites for the C–H activation and C–C coupling while establishing a synergy between them, thereby breaking the trade-off between the activity and selectivity in the CCOC2 process and leading to an exceptional performance for CH3COOH production. Detailed in-situ characterization combined with theoretical investigations reveal that the photoexcited electrons from TiO2 significantly promote the O2 dissociation at the confined Zn site to generate highly reactive Zn–OH* species via a PCET mechanism, which can efficiently activate the C–H bond of CH4 to CH3 species, followed by facile C–C coupling between the CH3 and adsorbed CO on the adjacent Rh site forming CH3CO* species for CH3COOH production. This work provides a reference to design an efficient catalyst for photocatalytic CH4 carbonylation to CH3COOH.
Methods
Catalyst preparation
The RhZn-MoS2 catalysts were prepared via a one-pot solvothermal synthesis method. Typically, 900 mg of (NH4)6Mo7O24·4H2O, 58.6 mg of (NH4)3RhCl6, and 118.0 mg Zn(NO3)2·6H2O were dissolved into 20 mL deionized water to form a homogeneous solution. Then, the homogeneous solution and 10 mL of CS2 were added together in a 40 mL stainless steel autoclave in a glovebox under argon. The autoclave was then transferred to a muffle furnace and heated at 400 °C for 4 h. Subsequently, the formed RhZn-MoS2 was collected and treated by 100 mL 6 M KOH solution at a constant temperature of 60 °C with stirring for 3 h, then washed several times with water and ethanol, and finally dried at 60 °C overnight. Finally, the RhZn-MoS2 catalyst was transferred into a quartz tube reactor and reduced with H2 (>99.9%) at 300 °C for 2 h to create sulfur vacancies. The mass contents of atomic Zn and Rh in RhZn-MoS2 samples were measured by inductively coupled plasma optical emission spectrometry (ICP-OES).
The MoS2 nanosheets were prepared via the direct chemical reaction of 900 mg (NH4)6Mo7O24 and 10 mL CS2 under H2O-assisted (20 mL) environment at 400 °C. In detail, CS2 and H2O can react to produce H2S and CO2 (CS2 + H2O → CO2 + H2S), and then the MoO42- generated from the dissolution of (NH4)6Mo7O24 can be reduced by H2S to form MoS2 nanosheets (MoO42- + H2S → MoS2 + H2O).
The RhZn-MoS2/TiO2 catalysts were prepared through a solid phase synthesis method. Typically, the H2 pretreated RhZn-MoS2 (50 mg) and TiO2 nanoparticles (50 mg) were fully mixed and then the mixed sample was transferred into a quartz tube reactor and treated with argon at 200 °C for 2 h.
Evaluation of photocatalytic methane conversion performance
The photocatalytic CH4 carbonylation reaction was performed in a 50 mL autoclave equipped with a top quartz window to allow light irradiation. First, the autoclave was charged with 25 mg catalyst and 5 mL distilled water and then sealed. Before the reaction, the autoclave was flushed with argon for several times and then pressurized to 5 bar CH4, 1 bar O2 and 11 bar CO. The reaction mixture was stirred at an optimized speed of 1000 rpm to promote mass transfer under a 300 W Xe lamp irradiation (wavelength = 200–800 nm) with a light intensity of 1500 mW cm−2 for a fixed reaction time (typically 5 h). During the reaction, a thermocouple was inserted into the solution to detect the temperature of the liquid solution. After the reaction, the autoclave was cooled in an ice bath to room temperature. The gaseous products were analyzed by gas chromatograph (GC, Shimadzu) and the liquid products were collected by filtration and then analyzed by 600 M 1H and 13C NMR.
The reusability test of the catalyst
The reusability test of the catalyst was carried out as follows. After the first reaction cycle, the catalyst (25 mg) was separated from the reaction solution by a high-speed rotary dryer under vacuum conditions at room temperature. The dried sample was subsequently evaluated under the same reaction conditions to obtain the performance of the second cycle of the catalyst. The remaining cycles were carried out in the same way and six reaction cycles were repeated.
Products analysis
Liquid-State Nuclear Magnetic Resonance (NMR) experiments were performed on Bruker Avance III 600 MHz equipped with pulsed field gradient and ultra-low temperature probes, which is a highly accurate approach with high detection sensitivity and high reproducibility. The assignment of oxygenated products (CH3COOH, CH3OH, and HCOOH) was identified by 1H NMR and 13C NMR spectroscopy. The quantitative analysis was based on the linear relationship between CH3COOH, CH3OH or HCOOH concentration and relative area versus 2 mM dimethyl sulfoxide (DMSO) as internal standard (Supplementary Fig. 27). Typically, 850 μL liquid product was firstly mixed with 50 μL of D2O and 100 μL 2 mM DMSO, and then 600 μL of the mixture was sealed into the NMR tube for testing. The productivity (µmol gcat.−1 h−1), CH3COOH selectivity in oxygenated products, and the turnover frequency (TOF) were calculated as the following Eqs. (1), (2) and (3). The calculation of CH3COOH selectivity in this system is based on the generated products from CH4 conversion.
Gaseous products were quantified by gas chromatography (GC-2014, Shimadzu), which was equipped with flame ionization detector (FID) coupled with a methanizer (UMTR unit). The generated CO2 was quantified by FID detector with a methane reformer (Supplementary Fig. 28).
Isotope labeling experiment
For isotopic reaction, 13C enriched CH4 (13CH4, Sigma-Aldrich, ≥99%), 13C enriched CO (13CO, Sigma-Aldrich, ≥99%) and 18O enriched O2 (18O2, Sigma-Aldrich, ≥97%) were used to trace the source of C and O in the products, respectively. The products were analyzed by 600 M 1H NMR, 13C NMR spectra and GC-MS. The detailed reaction conditions for isotope labeling experiment were shown in the notes of Fig. 2.
Catalyst characterizations
Transmission electron microscopy (TEM) measurements were performed on a Phillips Analytical FEI Tecnai 20 electron microscope operated at an acceleration voltage of 200 kV. The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images were collected on a JEOL JEM-2300F field-emission transmission electron microscope operating at 200 kV accelerating voltage.
Quasi in-situ XPS measurements were carried out on an Omicron XPS System using Al Kα X-rays as the excitation source at a voltage of 15 kV. Before the collection of spectrums, the catalyst was transferred into the pretreatment chamber and illuminated for 10 min by a 300 W Xe lamp (wavelength = 200–800 nm, light intensity of 1500 mW cm−2).
Zn K-edge XAFS spectra, Rh K-edge XAFS spectra, and Mo K-edge XAFS spectra for RhZn-MoS2/TiO2 were measured at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF). For the in-situ XAFS test, the spectrum was collected when the RhZn-MoS2/TiO2 catalyst was exposed to a 300 W Xe lamp (wavelength = 200–800 nm, light intensity of 1500 mW cm−2).
Quantitative identification of active sites
To quantitatively identify the coordination unsaturated Rh-Zn pair at the Mo edge of MoS2 that serve as the active sites for CH4 conversion, in-situ O2 pulse adsorption method29,30,31 in combination with in-situ DRIFTS characterizations of the Mo=O and Mo–O–Mo species were applied to determine the density of sulfur vacancies (SVs) at the edge of the prepared MoS2-based catalysts. The total density of SVs was calculated on the basis of specific adsorption capacity of O2, where one O2 molecule corresponds to one SV. The relative ratio of the integrated areas from DRIFTS was used to distinguish the proportion of edge and in-plane SVs, so the density of SVs at the edge of catalysts can be approximately obtained. Three edge SVs correspond to one active site for CH4 carbonylation to CH3COOH production based on the results of DFT calculations (Supplementary Fig. 29 and 30).
Computational details
DFT calculations were performed using the Vienna Ab-initio Simulation Package (VASP) with the projector augmented-wave (PAW) pseudopotential for describing the ionic cores32,33,34. All calculations were based on the generalized gradient approximation (GGA) method with the Perdew-Burke-Ernzerhof (PBE) functional for the exchange-correlation term with a plane-wave cutoff energy of 400 eV35,36,37. DFT-D3 method of Grimme was used to calculate the van der Waals correction38,39. The spin-polarize calculation was carried out in all cases. A nanoribbon model of RhZn-MoS2 with six repeating units along the ribbon direction was built to simulate the edges, with the Mo edge saturated with S monomers40,41. A heterojunction model was built with the nanoribbon RhZn-MoS2 integrated with one oxygen vacancy anatase TiO2 (101), corresponding to a 20.66 × 22.69 × 25.79 Å3 large supercell. The vacuum thicknesses were set larger than 15 Å. A Monkhorst-Pack k-point sampling of 1 × 1 × 1 was selected. In structural optimizations, the residual forces between atoms were converged to below 0.05 eV Å−1. Formation energies of SVs at the edge are calculated by using H2 and H2S as reference states. The transition states were searched using the fixed bond-length method as implemented in the Atomic Simulation Environment (ASE)42 and VASP. All transition states were confirmed to have only one imaginary frequency. The free energies (G) of the adsorption intermediates on the surface were calculated as Etotal + ZPE + Hvib − TSvib, where Etotal is the DFT-calculated total energy, ZPE is the zero-point energy, T is the temperature and Hvib and Svib are the enthalpy and entropy parts.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data that support the findings of this study are available within the paper and its Supplementary Information files. Source data are provided with this paper.
References
Schwach, P., Pan, X. & Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: challenges and prospects. Chem. Rev. 117, 8497–8520 (2017).
Meng, X. et al. Direct methane conversion under mild condition by thermo-, electro-, or photocatalysis. Chem 5, 2296–2325 (2019).
Periana, R. A. et al. Catalytic, oxidative condensation of CH4 to CH3COOH in one step via CH activation. Science 301, 814–818 (2003).
Besenbacher, F. et al. Design of a surface alloy catalyst for steam reforming. Science 279, 1913–1915 (1998).
Pakhare, D. & Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 43, 7813–7837 (2014).
Laudenschleger, D., Ruland, H. & Muhler, M. Identifying the nature of the active sites in methanol synthesis over Cu/ZnO/Al2O3 catalysts. Nat. Conmun. 11, 3898 (2020).
Qi, J. et al. Selective methanol carbonylation to acetic acid on heterogeneous atomically dispersed ReO4/SiO2 catalysts. J. Am. Chem. Soc. 142, 14178–14189 (2020).
Lin, M. & Sen, A. Direct catalytic conversion of methane to acetic acid in an aqueous medium. Nature 368, 613–615 (1994).
Dong, C. et al. Direct photocatalytic synthesis of acetic acid from methane and CO at ambient temperature using water as oxidant. J. Am. Chem. Soc. 145, 1185–1193 (2023).
Li, H. et al. Selective formation of acetic acid and methanol by direct methane oxidation using rhodium single-atom catalysts. J. Am. Chem. Soc. 145, 11415–11419 (2023).
Wang, W. et al. Selective oxidation of methane to methanol over Au/H-MOR. J. Am. Chem. Soc. 145, 12928–12934 (2023).
Yuan, Q., Zhang, Q. & Wang, Y. Direct conversion of methane to methyl acetate with nitrous oxide and carbon monoxide over heterogeneous catalysts containing both rhodium and iron phosphate. J. Catal. 233, 221–233 (2005).
Wu, B. et al. Fe binuclear sites convert methane to acetic acid with ultrahigh selectivity. Chem 8, 1658–1672 (2022).
Tang, Y. et al. Single rhodium atoms anchored in micropores for efficient transformation of methane under mild conditions. Nat. Commun. 9, 1231 (2018).
Shan, J. et al. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 551, 605–608 (2017).
Li, B. et al. Direct conversion of methane to oxygenates on porous organic polymers supported Rh mononuclear complex catalyst under mild conditions. Appl. Catal. B, Environ. 293, 120208 (2021).
Li, M. et al. Single-step selective oxidation of methane to methanol in the aqueous phase on iridium-based catalysts. Appl. Catal. B, Environ. 292, 120124 (2021).
Qi, G. et al. Au-ZSM-5 catalyses the selective oxidation of CH4 to CH3OH and CH3COOH using O2. Nat. Catal. 5, 45–54 (2022).
Li, H. et al. Selective methane oxidation by heterogenized iridium catalysts. J. Am. Chem. Soc. 145, 769–773 (2023).
Zhang, W. et al. Light-driven flow synthesis of acetic acid from methane with chemical looping. Nat. Commun. 14, 3047 (2023).
Wang, J. et al. Multi-radicals mediated one-step conversion of methane to acetic acid via photocatalysis. Appl. Catal. B, Environ. 337, 122983 (2023).
Banerjee, R. et al. Structure of the key species in the enzymatic oxidation of methane to methanol. Nature 518, 431–434 (2015).
Shi, Z. et al. Phase-dependent growth of Pt on MoS2 for highly efficient H2 evolution. Nature 621, 300–305 (2023).
Wu, Y. et al. Photocatalytically recovering hydrogen energy from wastewater treatment using MoS2@TiO2 with sulfur/oxygen dual-defect. Appl. Catal. B Environ. 303, 120878 (2022).
Meng, X. et al. Distance synergy of MoS2-confined rhodium atoms for highly efficient hydrogen evolution. Angew. Chem. Int. Ed. 59, 10502–10507 (2020).
Xie, S. et al. Visible light-driven C–H activation and C–C coupling of methanol into ethylene glycol. Nat. Commun. 9, 1181 (2018).
Zhang, X. et al. Heterostructural CsPbX3-PbS (X = Cl, Br, I) quantum dots with tunable Vis–NIR dual emission. J. Am. Chem. Soc. 142, 4464–4471 (2020).
Kim, K. H. et al. Triphasic metal oxide photocatalyst for reaction site‐specific production of hydrogen peroxide from oxygen reduction and water oxidation. Adv. Energy Mater. 12, 2104052 (2022).
Hu, J. et al. Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol. Nat. Catal. 4, 242–250 (2021).
Mao, J. et al. Direct conversion of methane with O2 at room temperature over edge-rich MoS2. Nat. Catal. 6, 1051–1062 (2023).
Hu, J. et al. Edge-rich molybdenum disulfide tailors carbon-chain growth for selective hydrogenation of carbon monoxide to higher alcohols. Nat. Commun. 14, 6808 (2023).
Hafner, G. K. A. J. Ab. initio molecular dynamics for liquid metals. Phys. Rev. B. 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 54, 11169–11186 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B. 50, 17953–17968 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Tsai, C., Abild-Pedersen, F. & Nørskov, J. K. Tuning the MoS2 edge-site activity for hydrogen evolution via support interactions. Nano Lett 14, 1381–1387 (2014).
Wilson, J. A. & Yoffe, A. D. Transition metal dichalcogenides discussion and interpretation of observed optical, electrical and structural properties. Adv. Phys. 18, 193–335 (1969).
Hjorth Larsen, A. et al. The atomic simulation environment-a python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017).
Acknowledgements
This work received financial support from the National Key R&D Program of China (2022YFA1504500 to D.D. and 2022YFA1503100 to X.C.), the National Natural Science Foundation of China (21988101, 22225204 to D.D., 22272174 to X.C., and 22272170 to L.Y.), the Strategic Priority Research Program of the Chinese Academy of Science (XDB36030200 to D.D.), the Fundamental Research Funds for the Central Universities (20720220008 to D.D.), and the CAS Project for Young Scientists in Basic Research (YSBR-028 to X.C.). We thank the staff at XAFS beamline (BL14W1) of the Shanghai Synchrotron Radiation Facilities for assistance with the EXAFS and XANES measurements.
Author information
Authors and Affiliations
Contributions
D.D. and X.C. conceived and designed the experiments. Y.L. performed the materials synthesis, characterization, and performance test. H.L. and L.Y. contributed to the DFT calculations. M.G., Y.Z., Q.Z., W.Zhou and W.L. conducted HAADF-STEM test and simulation. M.L. and K.W. assisted TA test. W.Zhang and J.M. assisted TEM test., Y.S. and J.H. assisted the NMR test. J.M. and R.H. assisted in-situ DRIFTS test and data analysis. Y.L., H.L., J.M., L.Y., X.C. and D.D. co-wrote the paper. All the authors discussed and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Vitaly Ordomsky, Jing Gu and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Liu, H., Mao, J. et al. MoS2-confined Rh-Zn atomic pair boosts photo-driven methane carbonylation to acetic acid. Nat Commun 16, 487 (2025). https://doi.org/10.1038/s41467-024-54061-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54061-z
This article is cited by
-
Study on the preparation of cobalt catalyst and its catalytic reaction of diisobutene hydroesterification
Research on Chemical Intermediates (2026)
