
Abstract
Coordination engineering of high-valent Fe(IV)-oxo (FeIV=O) is expected to break the activity-selectivity trade-off of traditional reactive oxygen species, while attempts to regulate the oxidation behaviors of heterogeneous FeIV=O remain unexplored. Here, by coordination engineering of Fe-Nx single-atom catalysts (Fe-Nx SACs), we propose a feasible approach to regulate the oxidation behaviors of heterogeneous FeIV=O. The developed Fe-N2 SACs/peroxymonosulfate (PMS) system delivers boosted performance for FeIV=O generation, and thereby can selectively remove a range of pollutants within tens of seconds. In-situ spectra and theoretical simulations suggest that low-coordination Fe-Nx SACs favor the generation of FeIV=O via PMS activation as providing more electrons to facilitate the desorption of the key *SO4H intermediate. Due to their disparate attacking sites to sulfamethoxazole (SMX) molecules, Fe-N2 SACs mediated FeIV=O (FeIVN2=O) oxidize SMX to small molecules with less toxicity, while FeIVN4=O produces series of more toxic azo compounds through N-N coupling with more complex oxidation pathways.
Similar content being viewed by others

Introduction
The intensification of organic micropollutants has caused severe threats to clean water supply, one of the Sustainable Development Goals (SDGs)1,2,3. Fenton-like reactions can produce highly reactive oxygen species (ROSs), providing a promising solution for water pollution control4,5. Efficient water treatment needs to satisfy selectivity and safety requirements simultaneously6,7. The former means the selective degradation of target pollutants from complex water matrix, while the latter requires lower toxicity of degradation products. Radicals including hydroxyl radicals (•OH) and sulfate radicals (SO4•−) feature strong oxidizing ability and can detoxify pollutants via mineralization, while the practical application of these radicals is severely hindered by poor selectivity8,9,10. Singlet oxygen (1O2), an important non-radical ROSs with mild oxidation ability can selectively degrade target pollutants, but the products or intermediates are sometimes highly toxic11,12,13. As such, the trade-off between the activity and selectivity of traditional ROSs is the current bottleneck in the field of water treatment.
High-valent Fe(IV)-oxo (FeIVO), a key ROSs ubiquitously existing in bio-enzyme14,15, has been extensively probed in Fenton-like reactions in the last few years16,17. Sharply different from these ROSs prevailing in bulk water18, FeIV=O is an interfacial ROSs and the properties of FeIV=O greatly depend on its coordination microenvironment19. As such, coordination regulation of FeIV=O is expected to improve oxidation ability and manipulate oxidation pathways without sacrificing selectivity. Besides, as compared with the mentioned ROSs above, FeIV=O features lower oxidation potential (E0(Fe4+/Fe3+) = 2.0 V), longer lifetime (∼7 s, at pH = 3), and higher steady-state concentration (>9.76 × 10−9 M)20. These features endow FeIV=O with great promise for efficiently and selectively degrading target pollutants with minimized interference from the water matrix. To date, homogeneous FeIV=O with multivariate coordination structures has been harvested by engineering the ligands of Fe complexes, which demonstrates ligands-depend oxidation behaviors21. However, the wide application of homogeneous FeIV=O endures the dilemmas of poor stability and recyclability. Consequently, the controllable generation of heterogeneous FeIV=O with the desired structure is of great significance for efficient water treatment, yet remains an unexplored field.
Single-atom catalysts (SACs), characterized by the utmost atomic utilization efficiency and approximately uniform atomic environment, are one of the most promising candidates for Fenton-like reactions22,23,24. In the classical SACs, the metal sites (M) are usually coordinated by N atoms in a formula of M-Nx. Hence, the number of N atoms plays a decisive role in regulating the catalytic performance25,26. However, for the most popular SACs coordinated with four N atoms, the symmetrical planar structure of M-N4 leads to a symmetrical electron distribution, which is not beneficial to the adsorption and activation of reactants, thus deteriorating the catalytic performance27,28,29. For instance, Zhang et al. demonstrated that only Fe-N4 sites with the rigorous Fe1-Fe1 distance (4–5 Å) can transform the Fe-N4-PMS* to FeIVO, and the dosing enough PMS is another prerequisite30. Reducing coordination number is capable of creating unsaturated metal structures (M-Nx, x = 2∼3) and breaking the symmetry, thus facilitating the formation of key reaction intermediates (e.g. *COOH) and the desorption of target products via lowing the free energy barriers31,32,33, which is expected to manipulate the catalytic properties of Fe sites. Besides, Fe-Nx SACs mediated FeIV=O with various coordination numbers (denoted as FeIVNx=O) provide ideal models for unraveling the relationship between the coordination and oxidation behaviors of heterogeneous FeIV=O. However, attempts to systematically regulate the generation of ROSs in Fenton-like reactions by microenvironment engineering of Fe SACs remain scarce.
Here, by altering the coordination number of Fe-Nx SACs, we propose a feasible approach to regulate the generation and oxidation behaviors of heterogeneous FeIV=O. Three Fe-Nx SACs (x = 4, 3, 2) have been synthesized via an H2-assisted pyrolysis strategy at different temperatures. Fe-N2 SACs deliver remarkable performance for FeIV=O generation via PMS activation, outperforming its counterparts with higher coordination numbers and most systems reported so far. Hence, the Fe-N2 SACs/PMS system can selectively remove a range of pollutants from water within tens of seconds, featuring satisfactory resistance to environmental interference and desirable compatibility with water matrices. Theoretically, Fe-Nx SACs with lower coordination numbers are calculated to feature stronger PMS adsorption affinity and lower energy barrier for FeIV=O generation. Impressively, the oxidation behaviors of heterogeneous FeIV=O can be regulated by altering their chemical environments. Originating from their disparate interaction sites with sulfamethoxazole (SMX) molecules, FeIVN2=O tends to oxidize SMX to small intermediates with less toxicity, while FeIVN4=O produces a series of more toxic azo compounds through N-N coupling and the oxidation pathways are more complex. This work highlights how the microenvironment of Fe-Nx SACs can be used to significantly tune the generation and oxidation behaviors of heterogeneous FeIV=O, paving new ways to design powerful heterogeneous FeIV=O for various oxidation processes.
Results
Synthesis and characterization of Fe-Nx SACs
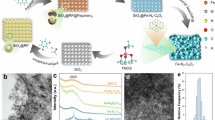
Target Fe-Nx SACs have been constructed by an H2-assisted pyrolysis strategy. As illustrated in Fig. 1a, Fe@ZIF-8@C precursor was first synthesized by the in-situ assembly reaction among zeolitic imidazolate framework (ZIF-8), ferric acetylacetonate and carbon black, which acts as a template, Fe source, and substrate, respectively. Then, Fe-Nx SACs with diverse N coordination numbers were synthesized by the pyrolysis of Fe@ZIF-8@C at disparate temperatures (800 ℃, 900 ℃, and 1000 ℃), followed by the acid treatment to remove any possible Fe nanoparticles. Of note, it is difficult to disperse evenly in water for the catalysts prepared without carbon black, implying that carbon black is essential to obtain highly hydrophilic samples. For comparison, N-doped carbon (NC) has also been prepared under the similar condition of Fe-N2 SACs, except for the addition of ferric acetylacetonate.

a Schematic diagram displaying the preparation of the Fe-N4 SACs, Fe-N3 SACs, and Fe-N2 SACs (The golden sphere represents Fe, and the blue, brown, and pink spheres represent N, C, and H, respectively). b The AC HAADF-STEM image of Fe-N2 SACs (Fe atoms were marked with bright yellow circles). c The Fe K-edge XANES spectra of Fe-Nx SACs. d WT-EXAFS plots of Fe-Nx SACs. e FT k3-weighted Fe K-edge EXAFS spectra of Fe-Nx SACs.
The X-ray diffraction (XRD) patterns of the as-synthesized samples (Supplementary Fig. 1a) depict only two diffraction peaks corresponding to the (002) and (101) planes of carbon, and no diffraction peaks attributed to metal nanoparticles can be observed. The Fourier transform infrared spectrometer (FT-IR) spectra of NC and Fe-Nx SACs manifest characteristic peaks of N-H stretching vibration at 3430 cm−1 and 588 cm−1 (Supplementary Fig. 1b), and other peaks at 2924 cm−1, 1618 cm−1 and 1112 cm−1 correspond to C-H, C=O, and C-N, respectively34. As compared with NC, Fe-Nx SACs exert an additional peak at 1260 cm−1, which is attributed to the Fe-N bond35, indicating that Fe atoms are coordinated with N atoms. The N 1 s spectra (Supplementary Fig. 1c) of NC and Fe-Nx SACs show four characteristic peaks at ca. 398.8, 400.8, 401.5, and 403.4 eV, which are ascribed to the pyridinic N, pyrrolic N, graphitic N, and oxide N, respectively36. As compared with NC, a characteristic peak at 399.62 eV appears in all Fe-Nx SACs, which is attributed to the Fe-N bond37. As the pyrolysis temperature increases, the intensity of the Fe-N bond gradually decreases, indicating a continuous decrease in the number of N in Fe-Nx SACs. No characteristic peak for Fe-O (ca. 530 eV) can be detected in the O 1 s spectra of Fe-Nx SACs (Supplementary Fig. 1d), ruling out the presence of FeOx38. There are two peaks at ca. 710.9 and 714.9 eV that can be labeled in Fe 2p spectra (Supplementary Fig. 1e), corresponding to Fe2+ 2p3/2 and Fe3+ 2p3/2, respectively, suggesting that the valance of Fe lies in +2 to +3. As the coordination number decreases, the valence state of Fe gradually tends to +2, suggesting that the Fe atoms in Fe-N2 SACs own more electrons than those in their counterparts. The Fe contents were detected to be 0.37 wt% (Fe-N4 SACs), 0.33 wt% (Fe-N3 SACs), 0.35 wt% (Fe-N2 SACs) by inductively coupled plasma mass spectrometry (ICP-MS), and Zn species are barely detectable in the above samples (Supplementary Table 1). Besides, the three Fe-Nx SACs share similar BET surface areas and pore size distribution (Supplementary Table 2). The similar Fe content and physics structure are beneficial to study the role of Fe-Nx sites.
As revealed in transmission electron microscopy (TEM) images (Supplementary Fig. 2), NC and Fe-Nx SACs share a similar morphology with pristine carbon black, while typical dodecahedral nanoparticle morphology can be observed for Fe@ZIF-8@C. Only wrinkled carbon substrates without prominent bright spots for metal nanoparticles could be observed in the TEM and high-resolution TEM images (Supplementary Figs. 3–5) of the three kinds of Fe-Nx SACs. The energy-dispersive X-ray spectroscopy (EDS) mapping indicates that Fe, C, N, and O are uniformly distributed over the carbon substrate. As revealed in the aberration-corrected high-angle annular dark-field scanning TEM (AC HAADF-STEM) images (Fig. 1b and Supplementary Figs. 3–5), abundant bright metal sites are atomically dispersed on the substrate, demonstrating the atomic-level dispersion of Fe sites in the resultant Fe-Nx SACs.
To confirm the electronic structures and coordination environment of Fe-Nx SACs, the Fe K-edge X-ray absorption near-edge structure (XANES) and Fe K-edge extended X-ray absorption fine structure (EXAFS) were investigated. As shown in Fig. 1c, the XANES curves of Fe-Nx SACs manifest that the Fe near-edge absorption energies are located between Fe foil and Fe2O3, suggesting that the valance of Fe atoms is between 0 and +3. Two small pre-edge peaks at 7112.6 and 7114.8 eV are assigned to Fe2+ and Fe3+, respectively, further substantiating that the oxidized state of Fe atoms is located between +2 and +339. As shown in Fig. 1d, the main peaks at 5 Å−1 are observed from the wavelet transform (WT)-EXAFS contour plots of Fe-Nx SACs, corresponding to the Fe-N bond, which is different from the 7.8 Å−1 of Fe-Fe bond in Fe foil40, implying the coordination of Fe and N in Fe-Nx SACs. As compared with the Fourier-transformed EXAFS curve of Fe standard samples (e.g., Fe foil, FeO, and FePc, Supplementary Fig. 6), significant peaks at 1.52 Å are observable in Fe-Nx SACs (Fig. 1e), which can be ascribed to the scattering path of the Fe-N bond41, and the peaks for Fe-Fe bond (2.2 Å) is absence. These results further verify the single-atom-level dispersion characteristics of the Fe sites, and the configuration is Fe-Nx. Noticeably, the intensity for the Fe-N peak weakens with pyrolysis temperature, which is in line with the gradually declining N content and Fe-N bond intensity in Fe-Nx SACs by XPS (Supplementary Table 3), further indicating the decreasing coordination numbers of N in Fe-Nx SACs42. To distinctly unravel the coordination configurations, the quantitative EXAFS fitting analysis in k and R spaces has been conducted. As shown in Supplementary Table 4, the average N coordination numbers of Fe atoms in Fe-Nx are 3.9, 3.1, and 2.3, respectively. The bond length of Fe-N is ca. 2.01 Å, 1.97 Å and 1.90 Å for Fe-N4 SACs, Fe-N3 SACs, and Fe-N2 SACs, respectively. Based on this, the configurations of Fe-Nx SACs with the coordination number of 4, 3, and 2 are simulated (Supplementary Fig. 7). Moreover, XANES simulations of the Fe-Nx moiety have been carried out. As shown in Supplementary Fig. 8, all five features (labeled as 1–5) of the detected XANES spectra for Fe-Nx are accurately replicated in the simulation, confirming the rationality of the built models. Therefore, by regulating the pyrolysis temperature in the H2-assisted strategy, Fe-Nx SACs with different coordination numbers (4, 3, and 2) were harvested.
PMS catalytic activation on Fe-Nx SACs
Sulfamethoxazole (SMX), a typical sulfonamide antibiotic, was selected as the target pollution to study the performance of the synthesized Fe-Nx SACs in PMS-based Fenton-like reactions. As shown in Supplementary Fig. 9, in the absence of PMS, the catalysts can only absorb 40% of SMX after the adsorption equilibrium, and there is almost no difference in their adsorption properties. The efficiency of SMX oxidation by PMS alone is poor (Fig. 2a), while adding NC can improve the oxidation performance to a certain extent. Notably, SMX can be completely removed within 60–160 s in the Fe-N2 SACs/PMS and Fe-N3 SACs/PMS systems, and efficiency increases significantly as coordination numbers decrease. The first-order kinetic rate constant (kobs) for SMX degradation in the Fe-N2 SACs/PMS system was calculated to be 0.088 s−1 (Fig. 2b), which is 5.5, 9.8, and 44 folds higher than that of the Fe-N3 SACs/PMS, Fe-N4 SACs/PMS and NC/PMS system, respectively, indicating the crucial role of coordination numbers of Fe-Nx SACs in PMS activation. The tremendously different oxidation properties between NC and Fe-N2 SACs also demonstrate that it is the Fe-Nx sites rather than the N sites or C sites that serve as the active centers in the Fe-NxSACs/PMS systems. In different reaction conditions (e.g., the concentration of PMS and the dosage of catalysts), the Fe-N2 SACs/PMS system delivers higher performance for SMX removal than its counterparts under corresponding conditions (Supplementary Figs. 10 and 11), implying the advantage of lower-coordination Fe-N2 SACs in PMS-based Fenton-like reactions. Then, with the mentioned reaction conditions as parameters, the catalytic performance of the developed systems is comparatively analyzed. As shown in Fig. 2c and Supplementary Table 5, the Fe-N2 SACs/PMS system delivers the highest kvalue (the modification value of reaction rate constant) with relatively less PMS and catalysts for SMX degradation among the existing advanced systems, further corroborating the outstanding catalytic property of Fe-N2 SACs in PMS-based Fenton-like reactions.

a, b The SMX removal efficiencies and the corresponding reaction rate constant in Fe-Nx SACs/PMS (Inset: the fitting plot). c The kvalue of different catalysts in various PMS-based Fenton-like systems. d The oxidation activity of Fe-N4 and Fe-N2 SACs in the existence of various pollutants. e The removal rate of SMX within 1 and 4 min in Fe-N2 SACs/PMS systems with various anions and NOM. f SMX removal efficiencies in the presence of different actual wastewater in Fe-N2 SACs/PMS. [organic]0 = 10 ppm, [catalysts]0 = 0.1 g L−1, [PMS]0 = 1.0 mM, [Cl−]0 = [SO42−]0 = 20 mM, [HCO3−]0 = 10 mM, [CO32−]0 = 5 mM, [HA]0 = 5 ppm, pH0 = 6.3, pHover = 5.8. The error bars in the figures represent the standard deviations from triplicate tests.
To further evaluate the potential of the Fe-Nx SACs/PMS systems in practical application, Fe-N4 SACs and Fe-N2 SACs were selected as target catalysts owing to their tremendously different performance. As displayed in Fig. 2d, both Fe-N4 SACs and Fe-N2 SACs exert excellent elimination performance for other pollutants, including phenol, bisphenol A (BPA), p-chlorophenol (4-CP), and tetracycline (TC), indicating that they can degrade a broad spectrum of micro-pollutants. However, for both systems, the degradation rate of atrazine (ATZ) was much lower than that of the other pollutants, possibly because ATZ is an electron-deficient pollutant. For all selected pollutants, the Fe-N2 SACs/PMS system exhibits a higher performance than its counterparts, demonstrating that the enhancement of Fe-N2 SACs in pollutant elimination is universal. For the Fe-N2 SACs/PMS system, almost all selected pollutants can be rapidly and completely degraded within one minute, while there are significant differences in removing disparate pollutants in the Fe-N4 SACs/PMS system, indicating that the ROSs generated in the Fe-N2 SACs/PMS system are more powerful. The activity of the Fe-N2 SACs/PMS system in degrading SMX is hardly affected by the pH value (2–10) of water (Supplementary Fig. 12). As shown in Fig. 2e, f, Fe-N2 SACs exhibit excellent resistance to coexisting anions (Cl−, SO42−, CO32−, and HCO3−) and natural organic matter (NOM, e.g., humic acid), demonstrating high compatibility with water matrices, including tap water, lake water (Guangzhou, China), and Pearl River water (Guangzhou, China). Besides, there is only slight attenuation in SMX degradation performance and Fe content for Fe-N2 SACs after four cycles (Supplementary Fig. 13 and Supplementary Table 1), while the chemical composition remained nearly unaltered (Supplementary Fig. 14) and the atomically scattered Fe sites were preserved (Supplementary Fig. 15), jointly implying good stability of Fe-N2 SACs in this system. The above results distinctly verify the great promise of the Fe-N2 SACs/PMS system in practical water treatment.
Identification of ROSs
To unravel the origin of the catalytic performance, the PMS activation pathways on Fe-Nx SACs have been explored by quenching experiments. First, the addition of potassium thiocyanate (KSCN), a typical probe to shield Fe atoms43, leads to the deactivation of the Fe-Nx SACs/PMS systems (Fig. 3a and Supplementary Fig. 16), further demonstrating that the Fe atoms rather than C or N sites act as the active centers. Then, methyl alcohol (MeOH), furfuryl alcohol (FFA), L-histidine (L-His), and p-benzoquinone (p-BQ) are employed to identify the contribution of each ROSs44. As shown in Fig. 3a and Supplementary Fig. 16, almost no discount of SMX removal performance can be detected after adding MeOH and p-BQ, implying the negligible contribution of •OH, SO4•−, and O2•−. After the addition of FFA or L-His, the removal of SMX slightly falls in the Fe-N4 SACs/PMS and Fe-N3 SACs/PMS systems, while that of Fe-N2 SACs/PMS system hardly changed. However, FFA or L-His causes a certain amount of consumption of PMS (Supplementary Fig. 17), suggesting that the inhibition by FFA or L-His may be derived from the depletion of PMS rather than the quenching of 1O2. To further explore the contribution of 1O2, deuterium oxide (D2O) was employed as the solvent due to the longer lifetime of 1O2 in D2O (22–70 μs) than in H2O (2.9–4.6 μs)45. However, the existence of D2O inhibits the SMX degradation rather than promoting it (Supplementary Fig. 18), further validating the negligible contribution of 1O2 in the Fe-Nx SACs/PMS systems. The inhibited performance in D2O may be because D2O slows the overall kinetics of aqueous processes46. Furthermore, the tiny difference in PMS consumption with and without SMX (Supplementary Fig. 19) indicates the absence of direct electron transfer pathways in these systems47.

a SMX degradation performance in the presence of radical scavengers. b The EPR signals of 5,5-dimethyl-1-pyrrolidone-N-oxyl (DMPOX) in the Fe-N2 SACs/PMS systems under different reaction conditions with H2O as the solution (blank, or with MeOH, FFA, and PMSO, respectively). c The oxidation of trans-stilbene in the reactions of Fe-N2 SACs/PMS. d In-situ Raman spectra in Fe-N2 SACs/PMS. e The PMSO consumption and PMSO2 production as well as calculated η(PMSO2) values. f Mass spectra of PMSO and PMSO2 generated in 18O-labeled or unlabeled Fe-N2 SACs/PMS systems. [SMX]0 = 10 ppm, [catalysts]0 = 0.1 g L−1, [PMS]0 = 1.0 mM, [KSCN]0 = 10 mM, [MeOH]0 = 1000 mM, [L-His]0 = [FFA]0 = [p-BQ]0 = 4 mM, [TEMP]0 = [DMPO]0 = 200 mM, [trans-stilbene]0 = 0.5 mM, [PMSO]0 = 2 mM, pH0 = 6.3, pHover = 5.8. The error bars in the figures represent the standard deviations from triplicate tests.
Then, electron paramagnetic resonance spectroscopy (EPR) was carried out to monitor the ROSs generated in the Fe-Nx SACs/PMS systems. As displayed in Supplementary Fig. 20, either in water or methanol solvents, no signal of the typical DMPO-•OH or DMPO- SO4•− adduct can be detected in the EPR spectra, indicating the absence of radicals (•OH, SO4•−, and O2•−) in this work, which is in line with the results of quenching experiments. The characteristic signal with an intensity of about 1:2:1:2:1:2:1 can be attributed to 5,5-dimethyl-1-pyrrolidone-2-oxyl (DMPOX), which may be attributable to DMPO oxidation by high-valent metals or excessive amount of •OH or 1O217. However, adding MeOH or FFA has little effect on the characteristic peaks of DMPOX in the Fe-N2 SACs/PMS system (Fig. 3b), further indicating that •OH and 1O2 generated in this system were negligible43. Besides, for 1O2, all Fe-Nx SACs/PMS systems emerge equally strong characteristic triplet peaks of 2,2,6,6-tetramethylproline-1O2 (1:1:1, TEMP-1O2), which is similar to PMS alone. As such, 1O2 may originate from PMS self-cleavage rather than catalytic activation, which contributes little to the SMX degradation in these systems. Besides, the degradation of 1,3-diphenylisobenzofuran (DPBF, a chemical probe of 1O2) has been conducted to further verify the role of 1O248. As shown in Supplementary Fig. 21, DPBF can be hardly removed in all Fe-Nx SACs/PMS systems. Therefore, the above results jointly corroborate that PMS activation over Fe-Nx SACs may not produce radicals (e.g., •OH, SO4•−, and O2•−) or 1O2.
Adding PMSO can completely quench the DMPOX signal (Fig. 3b) and Fe atoms serve as the active sites for PMS activation, it is therefore reasonable to speculate that FeIV=O may be the dominant ROSs. The transfer reaction of trans-stilbene towards trans-stilbene oxide, a traditional oxygen atom transfer (OAT) reaction, was used to identify FeIV=O49. As shown in Fig. 3c and Supplementary Fig. 22, as the reaction progresses, the intensities of trans-stilbene in the Fe-Nx SACs/PMS systems become weaker while the signal for trans-stilbene oxide gradually increases. However, PMS alone cannot cause signal changes, verifying the generation of FeIV=O in the Fe-Nx SACs/PMS systems. The maximum values for the consumption of trans-stilbene and the generation of its oxide are observed in the Fe-N2 SACs/PMS systems, indicative of its merits for the production of FeIV=O. Then, the generation of FeIV=O in Fe-Nx SACs/PMS systems has been further monitored by in-situ Raman spectra. As shown in Fig. 3d, the PMS solution alone shows three characteristic peaks at ca. 1060, 980 and 878 cm−1, corresponding to SO3− (HSO5−), SO42 and O-O (HSO5−) respectively. After adding Fe-Nx SACs, two new characteristic peaks appeared at ca. 847 cm−1 and ca. 830 cm−1, which are assigned to FeIV=O and Fe-Nx-PMS*, respectively50. When H216O was replaced by H218O, a redshift of the characteristic peaks for FeIV=O could be observed (Supplementary Fig. 23), further indicating the genaration of heterogeneous FeIV=O51. Noticeably, as the coordination number decreases, the Raman shift of the characteristic peaks for FeIV=O and Fe-Nx-PMS* gradually increases, indicating that heterogeneous FeIV=O with different coordination environments are obtained.
Furthermore, since methyl phenyl sulfoxide (PMSO) can be selectively converted into methyl phenyl sulfone (PMSO2) via FeIV=O according to OAT reaction52, the concentration of FeIV=O was quantified by the consumption (Δ) of PMSO and the yield (η) of PMSO2, in which η(PMSO2) indicates the yield of FeIV=O while the ratio of η(PMSO2)/Δ(PMSO) relates to the selectivity of FeIV=O in PMS activation53. As displayed in Fig. 3e and Supplementary Fig. 24, with the decrease in coordination number, the production and selectivity of FeIV=O increased significantly. Noticeably, the Fe-N2 SACs/PMS system consumes 0.673 mM PMSO and generates 0.669 mM PMSO2, corresponding to the selectivity of FeIV=O up to 99.4%, demonstrating the admirable yield and selectivity of heterogeneous FeIV=O in PMS-based Fenton-like reactions (Supplementary Fig. 25 and Supplementary Table 6). Introducing SMX into the Fe-Nx SACs/PMS system significantly inhibits the yield of PMSO2 (Supplementary Fig. 26), indicating the crucial role of FeIV=O in SMX degradation. 18O isotope-labeling experiments were conducted to provide additional convincing evidence for FeIV=O according to the special oxygen atom exchange reaction between FeIV=O and H2O (Supplementary Fig. 27a)37. As showned in Fig.3f, the Mass spectra (MS) of Fe-N2 SACs/PMS/H218O system exhibits two characteristic peaks at m/z 157.03 and 159.04, which correspond to the PMS16O16O and PMS16O18O, respectively54, verifying the successful integration of 18O to PMSO2. However, only a peak at m/z 15.03 can be detected in the Fe-N2 SACs/PMS/H2O and PMS/H218O systems (Fig. 3f and Supplementary Fig. 27b–d). Thus, the formation of 18O-labeled PMSO2 in the Fe-N2 SACs/PMS/H218O system provides conclusive evidence for FeIV=O.
Then, the steady-state concentration of •OH, SO4•−, O2•−, 1O2 and FeIV=O in the Fe-Nx SACs/PMS systems were further quantified benzoic acid (BA), p-hydroxybenzoic acid (p-HBA) nitrotetrazolium blue chloride (NBT), FFA and the kinetics of PMSO-to-PMSO2 oxidation, respectively55. As shown in Supplementary Figs. 28 and 29 and Supplementary Table 7, the concentration of FeIV=O in the Fe-N2 SACs/PMS system is calculated to be 2.81 × 10−5 mM, which is 2 and 4 times higher than that of the Fe-N3 SACs and the Fe-N4 SACs systems, respectively. Of note, other ROSs are almost undetectable and the contribution of FeIV=O in each Fe-Nx SACs/PMS system is up to 100%, providing reliable platforms to investigate the coordination-generation/oxidation relationship of heterogeneous FeIV=O.
Theoretical simulation of PMS activation on Fe-Nx SACs
To unravel the origination of different performances for the generation of heterogeneous FeIV=O in the Fe-Nx SACs/PMS systems, density functional theory (DFT) calculations were conducted to understand the PMS activation over the resultant Fe-Nx SACs. Based on the models obtained by EXAFS (Supplementary Fig. 7), the electron density distributions of these Fe-Nx SACs (Fig. 4a) demonstrate that the Bader charge of Fe atoms in Fe-N2 SACs (+0.95 e) is smaller than that of Fe-N4 SACs (+1.09 e) and Fe-N3 SACs (+1.05 e), implying that the Fe atoms coordinated with fewer N atoms carry more electrons56. The Partial density of states (PDOS) of Fe centers and N atoms in Fe- Nx SACs were calculated, in which the up and down refers to two channels (spin-up and spin-down) of electrons in the d orbit of the Fe center and p orbit of N atoms, respectively57. After coordination, the bonding orbital and antibonding orbital of Fe 3 d and N 2p would be redistributed. As shown in Fig. 4b, the d-bond center of Fe atoms in Fe-N2 SACs (−0.28 eV) is closer to the fermi-level than that of Fe-N3 SACs (−0.75 eV) and Fe-N4 SACs (−0.80 eV), suggesting that the overlap of Fe 3 d and N 2p orbit decreases with the number of N atoms58. The more abundant vacant orbitals (lower than the Fermi level) in Fe-N2 SACs are capable of providing extra opportunities for the subsequent adsorption of PMS.

a The structure models and two-dimensional electron density distribution images of Fe-Nx SACs. b PDOS of Fe atoms and N atoms in Fe-Nx SACs. c Free energy of the possible reaction paths in different adsorption sites. d The paths on the reaction of PMS and Fe-N2 SACs. e Charge density difference of Fe-Nx sites after PMS adsorption. The blue and yellow isosurfaces refer to electron depletion and accumulation, respectively. The golden sphere represents Fe, and the blue, gray, red, and pink spheres represent N, C, O, and H, respectively. We set LVHAR = TRUE for calculation to obtain the LOCPOT file and ESP.
Then, the possible interaction process between PMS and Fe-Nx SACs has been simulated. When PMS is close to the single-atom Fe sites, there are two potential adsorption configurations, in which its Oα and Oβ serve as the adsorption sites, respectively (Fig. 4c and Supplementary Fig. 30). When Oγ directly acts as the adsorption site, the charge transfer between Fe-O will greatly weaken the O-O bond, resulting in the direct fracture of the O-O bond, thus unable to obtain a stable adsorption configuration. It is found that two adsorption configurations on Fe-Nx SACs feature approximate adsorption energy, while the adsorption energy of the Oα one was slightly higher (Fig. 4c). Noteworthily, Fe-N2 SACs deliver the largest adsorption energy than Fe-N4 SACs and Fe-N3 SACs in both configurations. Based on these initial adsorption models, the possible PMS activation pathways have been further simulated (Supplementary Fig. 31). For reaction route 1 with Oα as adsorption sites, the adsorbed PMS needs to undergo two successive thermodynamically unfavorable processes with insurmountable energy barriers, especially the dissociation of *SO4 (Fig. 4c). Thus, path 2 is the favorable reaction pathway for PMS activation on the Fe-Nx sites.
In path 2, the Oβ of PMS molecular is first adsorbed on the Fe sites. Then, owing to the strong hydrogen-bond interaction between H atoms and the adjacent O atoms, the adsorption site of PMS is transferred from Oβ to Oγ, followed by the formation of FeIV=O-*SO4H intermediates via an energetically downhill process (Fig. 4d and Supplementary Fig. 32). Finally, FeIV=O can be generated by the detachment of the *SO4H fragment (resulting in HSO4−)59, which acts as the rate-determining step. In this pathway, the adsorption affinity of Fe-Nx sites to PMS increases with the decrease of the coordination number, while the energy barrier for the desorption of *SO4H shows the opposite trend (Fig. 4c), implying that Fe-Nx SACs with lower coordination numbers are more conducive to the activation of PMS to generate FeIV=O. Charge density difference analysis has been conducted to explore the underlying mechanism of the above rules. As shown in Fig. 4e, after the adsorption of PMS, the charge density of Fe-Nx sites declined while that of PMS increased, indicating the charge transfer from Fe-Nx sites to PMS60. Therefore, Fe-Nx SACs with lower coordination numbers transfer more electrons to PMS, facilitating the desorption of *SO4H and the subsequent generation of FeIV=O. Of note is that FeIV=O may exhibit multiconfigurational character61,62, which is of importance to their catalytic performance.
Controllable oxidation reaction pathways by heterogeneous FeIVNx=O
Inspired by the fact that the oxidation behaviors of homogeneous FeIV=O complexes are significantly affected by their ligands63, and the huge difference between the Fe-N4 SACs/PMS and Fe-N2 SACs/PMS systems in eliminating disparate organic pollutants, SMX was selected as typical organics to analyze the oxidation behaviors of generated FeIVN2=O and FeIVN4=O. According to the results of Ultra-performance liquid chromatography coupled to hybrid Quadrupole-Exactive Orbitrap mass spectrometry (UPLC-Q-Orbitrap HRMS), SMX can be oxidated to various organic intermediates (Supplementary Table 8), and some of them can be detected in both FeIVN2=O and FeIVN4=O systems (Supplementary Fig. 33). In addition, as shown in Fig. 5a, SMX tends to be oxidized to small molecules by FeIVN2=O, while in the FeIVN4=O system, it tends to form complex molecules via N-N coupling and the oxidation pathways are more diverse. Noticeably, reducing the amount of Fe-N2 SACs leads to a declined degradation rate for SMX, but did not affect the distribution of the intermediate products. Therefore, the disparate intermediate products in these two systems were mainly due to their diverse FeIVNx=O rather than their different degradation rate. Then, the toxicity of products in these two systems is further studied by the ECOSAR system with QSAR analysis64. Noticeably, as shown in Fig. 5b and Supplementary Table 9, from the point of the acute and chronic toxicity for fish, daphnids, and green algae, the intermediates of SMX degradation in the FeIVN2=O system are less toxic than those in the FeIVN4=O one. Therefore, it can reasonably be inferred that the oxidation behaviors of heterogenous FeIVNx=O can be manipulated by facilely regulating the coordination number FeIVNx=O, providing great potential to satisfy the needs of different oxidation reaction pathways.

a The possible degradation pathway of SMX in Fe-N4 SACs/PMS or Fe-N2 SACs/PMS systems alone. b The toxic analysis of various products generated in Fe-N4 SACs/PMS or Fe-N2 SACs/PMS systems. c The Charge density difference of FeIVN4=O and FeIVN2=O. The light blue and light yellow isosurfaces refer to electron depletion and accumulation, respectively. d The adsorption configuration and corresponding adsorption energy of FeIVN4=O on the amido gen sites (-NH2) of SMX and FeIVN2=O on imino (-NH) sites of SMX. e Schematic illustration for different attacking pathways of SMX in FeIVN4=O and FeIVN2=O sites.
To shed insights into the intrinsic difference between FeIVN2=O and FeIVN4=O, as well as the origin of the different oxidation behaviors of FeIVN2=O and FeIVN4=O towards SMX, the interaction modes between FeIVNx=O and SMX have been further stimulated by DFT calculation. Charge density difference and Bader charge analysis were calculated to explore the internal electron structure distribution of FeIVN2=O and FeIVN4=O. Because of the large charge density difference between Fe atoms and O atoms, accompanying the formation of FeIVNx=O, electrons are transferred from Fe atoms to O atoms. Due to the large electron negativity density of Fe atoms in Fe-N2 SACs than in Fe-N4 SACs (Fig. 4a), the O atoms in FeIVN2=O can gain more electrons than that in FeIVN4=O, as shown in Fig. 5c. Then, the electrostatic potential was calculated to analyze the electronegativity of SMX. As shown in Supplementary Fig. 34, it is found that the negative charges (red color area) are mainly located at the imino sites (-NH) of SMX, whereas the positive one sits at the amido gen sites(-NH2) of SMX, and thereby FeIVN2=O and FeIVN4=O may attack different sits of SMX. According to the results of DFT calculations (Fig. 5d and Supplementary Fig. 35), FeIVN4=O owns the larger adsorption energy to the amido gen sites of SMX (Eads = −1.35 eV) than to its imino sites (Eads = −1.04 eV), while FeIVN2=O features an opposite trend. Based on the above results, the different reaction pathways between FeIVN2=O and FeIVN4=O are illustrated. As revealed in Fig. 5e, FeIVN2=O tends to attack the imino group of SMX, resulting in the breakage of its S-N bond to form some small molecular compounds with low toxicity, and the reaction path is relatively simple. On the other hand, FeIVN4=O tends to interact with the terminal amino group of SMX, forming a series of highly toxic complex compounds via N-N coupling, and pathways are more diverse. Therefore, according to the characteristics of pollutants, the efficient and safe Fenton-like systems can be customized on-demand by regulating the charge properties of heterogeneous FeIV=O, which is crucial for green and sustainable water treatment.
Discussion
This work demonstrates that the production and oxidation behaviors of heterogeneous FeIV=O are closely related to the coordination microenvironment of Fe sites, and outlines the fundamental principle of the design of powerful heterogeneous FeIV=O. As the coordination number decreases, the performance of the Fe-Nx SACs/PMS systems in degrading organic pollutants gradually increases. FeIV=O is identified as the sole ROSs and makes a nearly 100% contribution to pollutant degradation. The concentration of Fe = O increases with the decreased coordination number, and the Fe-N2 SACs/PMS system delivers admirable concentration and selectivity for heterogeneous FeIV=O generation in Fenton-like reactions. In-situ spectra and DFT calculations revealed that Fe-Nx SACs with lower coordination numbers feature stronger PMS adsorption affinity, and favor the generation of FeIV=O by providing more electrons to facilitate the desorption of the key *SO4H intermediate. Due to the different attacking sites to SMX, FeIVN2=O can oxidize SMX to small molecules with lower toxicity, while larger and highly toxic intermediates can be induced by FeIVN4=O. These findings unravel the coordination-generation/oxidation relationship of heterogeneous FeIV=O at the atomic level, opening new horizons for designing powerful heterogeneous FeIV=O towards various oxidation processes.
Methods
Synthesis of Fe-Nx SACs
Typically, carbon black (100 mg) was dispersed in 60 mL methanol with sonification for 20 min to form solution A. Zn(NO3)2•6H2O (1666 mg, 5.56 mmol) and Fe(acac)3 (99.60 mg, 0.28 mmol) were dissolved in 40 mL methanol to form solution B. Then, solution A and solution B were mixed. After sonification for 20 min, 2-methylimidazole (2-MI, 3700 mg, 45.07 mmol) was added to the hybrid solution and then stirred for 24 h at room temperature. The black sample named Fe@ZIF-8@C was obtained by washing it with methanol and drying it at 60 ℃ overnight, with a yield of 80 %. Then, Fe-N4 SACs, Fe-N3 SACs, and Fe-N2 SACs were obtained by pyrolysis of the Fe@ZIF-8@C precursor within a 10% H2/Ar atmosphere at 800, 900, and 1000 ℃ at a heating rate of 5 ℃ min−1, respectively. 0.5 M sulfuric acid was used to eliminate the possible Fe nanoparticles. The yield of Fe-N4 SACs, Fe-N3 SACs, and Fe-N2 SACs were calculated to be 80.2%, 74.7% and 65.3%, respectively.
Characterizations
The concentration of Fe was quantified by an inductively coupled plasma Mass spectrometry (ICP-MS) instrument (Thermo Fisher Scientific, USA). The BET surface area, Pore Volume, and Pore size were carried out by Brunauer-Emmett-Teller (BET) instrument (ASAP 2460, USA). The phase and surface properties of samples were characterized by X-ray diffraction (XRD, D8 Advance, Bruker Ltd., Germany) with Cu-Ka radiation, and the functional group of samples was performed by Fourier transform infrared spectrometer (FT-IR, Bruker TENSOR27, Germany). The molecular structure and valence states of samples were characterized by X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, USA), and all the peaks were revised by the C 1s spectrum at 284.8 eV. The transmission electron microscopy (TEM) and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) were carried out on FEI-Talos FX200X. The X-ray absorption fine structure (XAFS) spectra were collected at BL14W1 station in Shanghai Synchrotron Radiation Facility (SSRF). The FeIV=O was detected by Raman spectroscopy (HORIBA Jobin Yvon, France, equipped with a 532 nm laser) in both H216O and H218O. Note: Mossbauer spectra is powerful for identifying heterogenous FeIV=O, but it usually requires high-concentration Fe or isotopically labeled 57Fe as sources.
Experiment procedures
The activation of catalysis was performed by the removal of SMX. In a typical experiment, 5 mg catalysis was dispersed in a 50 mL SMX solution (10 ppm), and the solution was stirred for 20 min to acquire the adsorption-desorption equilibrium. To initiate the catalytic degradation reaction, a certain amount of PMS (The final concentration of PMS is 1 mM) was added. Then, at specific intervals, a 0.5 mL liquor sample was collected and followed by filtered through 0.22 µm polyether sulfone filters. The concentration of organics is tested by High-Performance Liquid Chromatography (HPLC, Waters, USA).
Concentration determination and toxicity evaluation
The intermediates produced from SMX were conducted by Ultra-performance liquid chromatography coupled to hybrid Quadrupole-Exactive Orbitrap mass spectrometry (UPLC-Q-Orbitrap HRMS, Thermo Fisher, USA), and the toxicity of the intermediate was evaluated by ECOSAR.
Data availability
All the data supporting the findings of this study are available within the paper and its supplementary information files. Source Data are provided with this paper. Additional data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Lotfi, S., Fischer, K., Schulze, A. & Schäfer, A. I. Photocatalytic degradation of steroid hormone micropollutants by TiO2-coated polyethersulfone membranes in a continuous flow-through process. Nat. Nanotechnol. 17, 417–423 (2022).
Richards, T. et al. A residue-free approach to water disinfection using catalytic in situ generation of reactive oxygen species. Nat. Catal. 4, 575–585 (2021).
Deng, F. et al. Critical review on the mechanisms of Fe2+ regeneration in the electro-fenton process: Fundamentals and boosting strategies. Chem. Rev. 123, 4635–4662 (2023).
Zong, Y. et al. Unraveling the overlooked involvement of high-valent cobalt-oxo species generated from the cobalt(II)-activated peroxymonosulfate process. Environ. Sci. Technol. 54, 16231–16239 (2020).
Ren, W. et al. Hydroxyl radical dominated elimination of plasticizers by peroxymonosulfate on metal-free boron: Kinetics and mechanisms. Water Res. 186, 116361 (2020).
Chen, Z. et al. Single-atom Mo-Co catalyst with low biotoxicity for sustainable degradation of high-ionization-potential organic pollutants. Proc. Natl Acad. Sci. USA 120, e2305933120 (2023).
Chen, F. et al. Efficient decontamination of organic pollutants under high salinity conditions by a nonradical peroxymonosulfate activation system. Water Res. 191, 116799 (2020).
Chu, C. et al. Cobalt single atoms on tetrapyridomacrocyclic support for efficient peroxymonosulfate activation. Environ. Sci. Technol. 55, 1242–1250 (2020).
Liu, T. et al. Water decontamination via nonradical process by nanoconfined Fenton-like catalysts. Nat. Commun. 14, 2881 (2023).
Xie, L. et al. Pauling-type adsorption of O2 induced electrocatalytic singlet oxygen production on N-CuO for organic pollutants degradation. Nat. Commun. 13, 5560 (2022).
Zhang, C. et al. Modulating the site density of Mo single atoms to catch adventitious O atoms for efficient H2O2 oxidation with light. Adv. Mater. 35, 2208704 (2022).
Wang, Z. et al. Contact-electro-catalysis for the degradation of organic pollutants using pristine dielectric powders. Nat. Commun. 13, 130 (2022).
Weng, Z. et al. Site engineering of covalent organic frameworks for regulating peroxymonosulfate activation to generate singlet oxygen with 100% selectivity. Angew. Chem. Int. Ed. 62, e202310934 (2023).
Cho, J. et al. Structure and reactivity of a mononuclear non-haem iron(III)-peroxo complex. Nature 478, 502–505 (2011).
Huang, Y., Ren, J. & Qu, X. Nanozymes: classification, catalytic mechanisms, activity regulation, and applications. Chem. Rev. 119, 4357–4412 (2019).
Qian, K. et al. Single-atom fe catalyst outperforms its homogeneous counterpart for activating peroxymonosulfate to achieve effective degradation of organic contaminants. Environ. Sci. Technol. 55, 7034–7043 (2021).
Liu, H.-Z. et al. Tailoring d-band center of high-valent metal-oxo species for pollutant removal via complete polymerization. Nat. Commun. 15, 2327 (2024).
Zhang, Y.-J. et al. Distinguishing homogeneous advanced oxidation processes in bulk water from heterogeneous surface reactions in organic oxidation. Proc. Natl Acad. Sci. USA 120, e2302407120 (2023).
Cavazza, C. et al. Crystallographic snapshots of the reaction of aromatic C-H with O2 catalysed by a protein-bound iron complex. Nat. Chem. 2, 1069–1076 (2010).
Cui, J. et al. Regulating the metal-support interaction: double jump to reach the efficiency apex of the Fe-N4-catalyzed Fenton-like reaction. ACS Catal. 12, 14954–14963 (2022).
Wei, X.-Z. et al. Tracking a FeV(O) intermediate for water oxidation in water. Angew. Chem. Int. Ed. 62, e202308192 (2023).
Shang, Y., Xu, X., Gao, B., Wang, S. & Duan, X. Single-atom catalysis in advanced oxidation processes for environmental remediation. Chem. Soc. Rev. 50, 5281–5322 (2021).
Yu, X. et al. A green edge-hosted zinc single-site heterogeneous catalyst for superior Fenton-like activity. Proc. Natl Acad. Sci. USA 120, e2221228120 (2023).
Dong, X., Chen, Z., Tang, A., Dionysiou, D. D. & Yang, H. Mineral modulated single atom catalyst for effective water treatment. Adv. Funct. Mater. 32, 2111565 (2022).
Liu, S. et al. Atomically dispersed iron sites with a nitrogen-carbon coating as highly active and durable oxygen reduction catalysts for fuel cells. Nat. Energy 7, 652–663 (2022).
Qian, M. et al. Modulation of charge trapping by island-like single-atom cobalt catalyst for enhanced photo-Fenton-like reaction. Adv. Funct. Mater. 33, 2208688 (2023).
Mi, X. et al. Almost 100% peroxymonosulfate conversion to singlet oxygen on single-atom CoN2+2 sites. Angew. Chem. Int. Ed. 133, 4638–4643 (2021).
Jiao, L. et al. Non-bonding interaction of neighboring Fe and Ni single-atom pairs on MOF-derived N-doped carbon for enhanced CO2 electroreduction. J. Am. Chem. Soc. 143, 19417–19424 (2021).
Xu, S. et al. Highly efficient peroxymonosulfate activation on electron-enriched ruthenium dual-atom sites catalysts for enhanced water purification. Adv. Funct. Mater. 33, 2308204 (2023).
Cheng, C. et al. Generation of FeIV=O and its contribution to Fenton-like reactions on a single-atom iron-N-C catalyst. Angew. Chem. Int. Ed. 62, e202218510 (2023).
Feng, J. et al. A Mn-N3 single-atom catalyst embedded in graphitic carbon nitride for efficient CO2 electroreduction. Nat. Commun. 11, 4341 (2020).
Wang, X. et al. Regulation of coordination number over single Co sites: triggering the efficient electroreduction of CO2. Angew. Chem. Int. Ed. 57, 1944–1948 (2018).
Zhou, Y. et al. Asymmetric dinitrogen-coordinated nickel single-atomic sites for efficient CO2 electroreduction. Nat. Commun. 14, 3776 (2023).
Chen, F. et al. Single-atom iron anchored tubular g-C3N4 catalysts for ultrafast Fenton-like reaction: roles of high-valency iron-oxo species and organic radicals. Adv. Mater. 34, 2202891 (2022).
Sheng, B. et al. In situ hydroxylation of a single-atom iron catalyst for preferential 1O2 production from H2O2. ACS Catal. 12, 14679–14688 (2022).
Xiong, Y. et al. Single-atom Fe catalysts for Fenton-like reactions: Roles of different N species. Adv. Mater. 34, 2110653 (2022).
Cui, X. et al. Simultaneously crafting single-atomic Fe sites and graphitic layer-wrapped Fe3C nanoparticles encapsulated within mesoporous carbon tubes for oxygen reduction. Adv. Funct. Mater. 31, 2009197 (2021).
Chen, Z. et al. Fe1N4-O1 site with axial Fe-O coordination for highly selective CO2 reduction over a wide potential range. Energy Environ. Sci. 14, 3430–3437 (2021).
Pan, Y. et al. Regulating the coordination structure of single-atom Fe-NxCy catalytic sites for benzene oxidation. Nat. Commun. 10, 4290 (2019).
Yuan, K. et al. Boosting oxygen reduction of single iron active sites geometric and electronic engineering: Nitrogen and phosphorus dual coordination. J. Am. Chem. Soc. 142, 2404–2412 (2020).
Gu, H. et al. Adjacent single-atom irons boosting molecular oxygen activation on MnO2. Nat. Commun. 12, 5422 (2021).
Liang, X. et al. Coordination number dependent catalytic activity of single-atom cobalt catalysts for Fenton-like reaction. Adv. Funct. Mater. 32, 2203001 (2022).
Li, X. et al. Single cobalt atoms anchored on porous N-doped graphene with dual reaction sites for efficient Fenton-like catalysis. J. Am. Chem. Soc. 140, 12469–12475 (2018).
Zhou, X. et al. Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl Acad. Sci. USA 119, e2119492119 (2022).
Weng, Z. et al. Donor-acceptor engineered g-C3N4 enabling peroxymonosulfate photocatalytic conversion to 1O2 with nearly 100% selectivity. J. Hazard. Mater. 448, 130869 (2023).
Ren, W. et al. Origins of electron-transfer regime in persulfate-based nonradical oxidation processes. Environ. Sci. Technol. 56, 78–97 (2022).
Zhao, Z. et al. Enhanced interfacial electron transfer by asymmetric Cu-Ov-In sites on In2O3 for efficient peroxymonosulfate activation. Angew. Chem. Int. Ed. 135, e202216403 (2023).
Chen, Y.-Z. et al. Singlet oxygen-engaged selective photo-oxidation over Pt nanocrystals/porphyrinic MOF: The roles of photothermal effect and Pt electronic state. J. Am. Chem. Soc. 139, 2035–2044 (2017).
Bao, Y. et al. Generating high-valent iron-oxo FeIV=O complexes in neutral microenvironments through peroxymonosulfate activation by Zn. Angew. Chem. Int. Ed. 134, e202209542 (2022).
Wang, Z. et al. High-valent iron-oxo species mediated cyclic oxidation through single-atom Fe-N6 sites with high peroxymonosulfate utilization rate. Appl. Catal. B 305, 121049 (2022).
Li, X. et al. CoN1O2 single-atom catalyst for efficient peroxymonosulfate activation and selective cobalt(IV)=O generation. Angew. Chem. Int. Ed. 62, e202303267 (2023).
Zou, Y. et al. High-valent cobalt-oxo species triggers singlet oxygen for rapid contaminants degradation along with mild peroxymonosulfate decomposition in single Co atom-doped g-C3N4. Chem. Eng. J. 471, 144531 (2023).
Wang, Z. et al. Is sulfate radical really generated from peroxydisulfate activated by iron(II) for environmental decontamination? Environ. Sci. Technol. 52, 11276–11284 (2018).
Li, M. et al. Highly selective synthesis of surface FeIV=O with nanoscale zero-valent iron and chlorite for efficient oxygen transfer reactions. Proc. Natl Acad. Sci. USA 120, e2304562120 (2023).
Lee, J., von Gunten, U. & Kim, J.-H. Persulfate-based advanced oxidation: Critical assessment of opportunities and roadblocks. Environ. Sci. Technol. 54, 3064–3081 (2020).
Jin, H. et al. Unprecedentedly high activity and selectivity for hydrogenation of nitroarenes with single atomic Co1-N3P1 sites. Nat. Commun. 13, 723 (2022).
Guo, Z.-Y. et al. Mn-O covalency governs the intrinsic activity of Co-Mn spinel oxides for boosted peroxymonosulfate activation. Angew. Chem. Int. Ed. 60, 276–280 (2020).
Wu, Q.-Y., Yang, Z.-W., Wang, Z.-W. & Wang, W.-L. Oxygen doping of cobalt-single-atom coordination enhances peroxymonosulfate activation and high-valent cobalt-oxo species formation. Proc. Natl Acad. Sci. USA 120, e2219923120 (2023).
Song, J. et al. Asymmetrically coordinated CoB1N3 moieties for selective generation of high-valence Co-oxo species via coupled electron-proton transfer in Fenton-like reactions. Adv. Mater. 35, 2209552 (2023).
Zhang, H.-C. et al. Axial N ligand-modulated ultrahigh activity and selectivity hyperoxide activation over single-atoms nanozymes. Adv. Sci. 10, 2205681 (2022).
Sen, A., Ansari, A., Swain, A., Pandey, B. & Rajaraman, G. Probing the origins of puzzling reactivity in Fe/Mn-oxo/hydroxo species toward C-H bonds: A DFT and AB initio perspective. Inorg. Chem. 62, 14931–14941 (2023).
Sen, A. & Rajaraman, G. Can you break the oxo-wall? A multiconfigurational perspective. Faraday Discuss. 234, 175–194 (2022).
Kumar, R. et al. Role of oxidation state, ferryl-oxygen, and ligand architecture on the reactivity of popular high-valent FeIV=O species: A theoretical perspective. Coord. Chem. Rev. 419, 213397 (2020).
Huang, Y. et al. Abts as both activator and electron shuttle to activate persulfate for diclofenac degradation: Formation and contributions of ABTS•+, SO4•–, and •OH. Environ. Sci. Technol. 57, 18420–18432 (2023).
Acknowledgements
Financial support from the Basic Science Center Project of the National Natural Science Foundation of China (52388101), the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (No. 2019ZT08L213), and the National Natural Science Foundation of China (No. 42107074) are gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
B.H. and Z.Y. conceived and designed the project. Y.L. performed the experiments and collected the data. Y.W., Z.W., and Y.Z. participated in some experiments. X.O. conducted the DFT calculations and analyzed the data. B.H., Y.L., S.L. X.X., Y.C., and J.J. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fuqiang Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, Y., Wang, Y., Weng, Z. et al. Coordination engineering of heterogeneous high-valent Fe(IV)-oxo for safe removal of pollutants via powerful Fenton-like reactions. Nat Commun 15, 10032 (2024). https://doi.org/10.1038/s41467-024-54225-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54225-x
This article is cited by
-
Neutral microenvironment-driven catalytic polymerization for closed-loop wastewater treatment and resource recovery
Nature Water (2026)
-
From mineralization to low-carbon conversion: emerging paradigms in water decontamination
Science China Chemistry (2026)
-
High-valent iron-driven sulfamethoxazole removal during sludge dewatering process
ENGINEERING Environment (2026)
-
Dynamic identification of reactive iron-oxo species in heterogeneous fenton-like reaction via operando stopped-flow IR spectroscopy
Nature Communications (2025)
-
Curved interface-induced Jahn-Teller effect in single-atom catalysts for water purification
Nature Communications (2025)
