
Abstract
The impact of microplastics on antibiotic resistance has attracted widespread attention. However, previous studies primarily focused on the effects of individual microplastics. In reality, diverse microplastic types accumulate in soil, and it remains less well studied whether microplastic diversity (i.e., variations in color, shape or polymer type) can be an important driver of increased antibiotic resistance gene (ARG) abundance. Here, we employed microcosm studies to investigate the effects of microplastic diversity on soil ARG dynamics through metagenomic analysis. Additionally, we evaluated the associated potential health risks by profiling virulence factor genes (VFGs) and mobile genetic elements (MGEs). Our findings reveal that as microplastic diversity increases, there is a corresponding rise in the abundance of soil ARGs, VFGs and MGEs. We further identified microbial adaptive strategies involving genes (changed genetic diversity), community (increased specific microbes), and functions (enriched metabolic pathways) that correlate with increased ARG abundance and may thus contribute to ARG dissemination. Additional global change factors, including fungicide application and plant diversity reduction, also contributed to elevated ARG abundance. Our findings suggest that, in addition to considering contamination levels, it is crucial to monitor microplastic diversity in ecosystems due to their potential role in driving the dissemination of antibiotic resistance through multiple pathways.
Similar content being viewed by others

Introduction
As a potential hallmark of the Anthropocene, contamination of microplastics (MPs, <5 mm in size) has become a critical challenge worldwide1. MP pollution has been detected in various environmental media including water, sediment, soil, air, and food2,3,4,5. Among various environments, soil ecosystems are severely impacted by MP pollution, and some studies have shown that soil has an estimated 4–23 times as many MPs as the ocean6. Agricultural land has become a major pollution sink for MPs in terrestrial environments due to the massive introduction of production activities (e.g., plastic film, organic fertilizers, and reclaimed water) and transport through environmental media (e.g., atmospheric deposition), up to 500,000 particles kg−1 soil7,8,9. Given the universal persistence and the increasing production of plastics, there is an increasing need to understand the ecological and health risks of MP contamination in soils.
Antibiotic resistance genes (ARGs) in the soil environment pose serious threats to human health and food security as they could transfer from soil to plants, animals and humans10,11. In recent years, ARGs have been recognized as emerging biological contaminants and prioritized in the latest One Health operational framework12,13. Accordingly, the effects of MPs on soil antibiotic resistance dissemination have been studied14,15,16. Different types of MPs, both conventional and biodegradable, significantly increase the abundance of ARGs17. Different particle sizes, degrees of weathering, and duration time in soils can also affect the enrichment of ARGs to different magnitudes18. Moreover, rather than being present as one single type, MPs in soils accumulate in diverse colors, shapes, and polymer types19. A single selective pressure often has limited effects on the bacterial community, while multiple pressures yield far greater impact in terms of negative and synergistic effects, highlighting the role of the diversity of pressure types20. Unlike conventional organic pollutants, MPs almost always occur as a collection of particles of many different types. However, most studies are currently focusing on the effects of specific types of MPs, and thus the occurrence, fate, and dynamics of ARGs in soils contaminated with a range of MPs remain largely unexplored.
Risk assessment of ARGs is complex, and in addition to considerations of types and abundance, factors such as “motility” based horizontal gene transfer and “human pathogenicity”, depending on whether they are present in pathogenic hosts, are critical21. The evaluation of microbial risk is increasingly focusing on virulence factor genes (VFGs), as these genes could enable bacterial pathogens to invade and cause infectious diseases22. Pathogens that contain ARGs are known as pathogenic antibiotic resistant bacteria; these organisms carry both ARGs and VFGs, and thus can cause illness and withstand antibiotic treatment23. Zhu et al. suggested that plastispheres in soil are habitats in which an increased potential pathogen abundance is spatially co-located with an increased abundance of ARGs under global change14. Therefore, revealing the coexistence status and patterns of ARGs and VFGs in the organisms and soil environment is a key task in evaluating the effects of diverse MPs on ARGs. Additionally, the motility of ARGs based on horizontal gene transfer, mediated by mobile gene elements (MGEs), should not be neglected. For instance, when ARGs, VFGs, and MGEs coexist in the genome (such as on plasmids and transposons), the risk to humans and animals would increase24. Nevertheless, a comprehensive evaluation of the joint occurrence VFGs and MGEs is missing from the current health risk assessment of environmental ARGs under MP pressure.
Multiple global change factors are increasingly threatening ecosystems, a circumstance that is attracting increasing attention worldwide25. In addition to MP pollution, soils are exposed to a range of different anthropogenic pressures26,27. In particular, there has been widespread concern about the consequences of land use change. Increasing harvest frequency requires increasing fungicide use, leading to more residues in soil28. For instance, a large amount of mefenoxam, which is used to prevent plant diseases, enters the soil environment, leading to residual contamination29. Effects of mefenoxam on microbial activity have been observed29. In addition, intensive land use can render habitats more susceptible to plant invasions, thereby reducing plant diversity30. This reduction in diversity is critical as it plays a significant role in maintaining belowground microbial functioning31. Therefore, more work is needed to decipher the effects and health risks of MP diversity and global change factors on the soil microbial resistome.
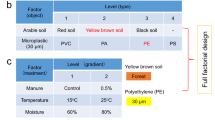
Based upon our current understanding of the soil microbial resistome and its responses to MPs, we hypothesize that a higher diversity of MPs would enrich the diversity and abundance of ARGs in soil, and thus exhibit high health risks. The microbial responses to multiple stressors induced by MP diversity might have evolutionary consequences, which makes it possible to increase the abundance of antibiotic resistance. We established a gradient of MP diversity, following an experimental design incorporating various global change factors20,32. While the identity and composition of MPs are known to influence response variables, including the soil microbiome and resistome14,15,16,17,18, we minimized this focus by randomly sampling from an MP pool. This approach allowed us to primarily address the simplified question regarding the impact of MP diversity. Here, we selected 12 MP polymers (powders sieved through a 180 μm mesh) commonly found in the environment and constructed an exposure system with three levels of MP diversity of 1, 3, and 6 different types, in combination with the fungicide application. Furthermore, considering the impact of land use intensity on the reduction of plant diversity30, we also set up mono- and multispecies plant community treatments to (1) characterize the profiles of ARGs, VFGs, and MGEs in the soil, evaluate environmental health risk of increased diversity of MPs, (2) reveal the host of ARGs and VFGs, and the elements co-exist patterns, (3) decipher the potential mechanisms of MP diversity on the soil antibiotic resistome. Our findings illuminate the effects of MP diversity on the soil antibiotic resistome and health risk, and offer theoretical foundations for understanding the ecological consequences of MPs in soil environments.
Results
Effects of MP diversity on soil ARG composition and abundance
A profile of ARGs across all soil samples comprised 29 types and 780 subtypes, of which the most diverse ARG type was identified as beta-lactam resistance genes containing 261 subtypes, followed by aminoglycoside, multidrug, macrolide-lincosamide-streptogramin (MLS), tetracycline, vancomycin and trimethoprim resistance genes (Fig. 1a). Notably, significant enrichment in the total abundance of ARGs occurred alongside the increased diversity of introduced MPs in soils (D3 vs. D1: P = 0.003; D6 vs. D1: P = 0.00012, Kruskal-Wallis test, Fig. 1b). Conversely, the fungicide application (P = 0.567, t test) and plant community (P = 0.336, t test) had no significant effects on the total abundance of ARGs (Fig. 1b). The linear mixed-effects model revealed significant impacts of MP diversity on the total relative abundance of ARGs (standard regression coefficient = 0.32, χ² = 5.5007, P = 0.019, Type II Wald chi-square test, Table 1). However, no significant interaction effects were observed among MP diversity, fungicide application, and plant community (all P > 0.05, Table 1). Furthermore, the introduction of six diverse MPs (P = 0.001, LSD-test) and mono-species community cultivation (P = 0.018, t test) strikingly increased the richness of soil ARGs (Fig. 1c). Among 29 types, the increasing diversity of MPs significantly increased the abundance of ARGs encoding resistance to bacitracin, multidrug, polymyxin, aminoglycoside, sulfonamide, and trimethoprim (all P < 0.05, Kruskal-Wallis test, Supplementary Fig. 1). With the application of fungicide, rifamycin, vancomycin, novobiocin, quinolone, and mupirocin resistance genes were substantially enriched in soils compared to soils not treated with fungicide (all P < 0.05, Kruskal-Wallis test, Supplementary Fig. 1). In soils cultivated with mono-species community, the relative abundance of mupirocin resistance was higher compared to soils with multi-species cultivation (P = 0.016, Kruskal-Wallis test, Supplementary Fig. 1). For the subtypes of ARGs, 58 Rank-1 high risk ARGs were identified (Supplementary Fig. 2). Compared to the low diversity of MPs, the increasing diversity of MPs tended to enhance the abundance of risk ARGs in soils. Similarly, fungicide application showed a contribution to risk ARG abundance. The increasing diversity of MPs significantly increased three risk aminoglycoside resistance genes, including APH(6)-Id, AAC(3)-IIg and ANT(2”)-Ia, an MLS resistance gene (lun(B)), a florfenicol resistance gene (floR), and a tetracycline resistance gene (tet(M)) (all P < 0.05, Kruskal-Wallis test, Supplementary Fig. 3). Aminoglycoside resistance APH(6)-Ic and AAC(6’)-Isa were greatly enriched in soils through the application of fungicide (all P < 0.05, Kruskal-Wallis test, Supplementary Fig. 3). Two aminoglycoside resistance genes (AAC(6’)-Ib, AAC(6’)-Ib10) were significantly more abundant in soils cultivated with mono-species community compared to the soils planted with multi-species (all P < 0.05, Kruskal-Wallis test, Supplementary Fig. 3).

a ARG composition colored by ARG type (n = 144). The outer and inner circles represent ARG types and subtypes, respectively. Genes encoding resistance to macrolide-lincosamide-streptogramin are labeled as MLS. b, c Total normalized abundance and richness of ARGs across different treatments with three experimental factors. D1 (n = 48), diversity level one of microplastic (MP); D3 (n = 48), diversity level three of MPs; D6 (n = 48), diversity levels six of MPs; Non-fungicide (n = 72), non-fungicide application; Fungicide (n = 72), fungicide application; Mono-species (n = 72), one species planted alone; Multispecies (n = 72), five species planted in one pot. The tops of the boxes represent the 75th percentile, the bottoms indicate the 25th percentile, and the center lines denote the median. The whiskers extend to the maximum and minimum non-outlier values. d Total normalized abundance of VFGs in three MP diversity treatments (n = 48 per group). e Responders of the VFG types to different MP diversity treatments (n = 48 per group). Yellow nodes represent types with no significant difference. Biomarkers were identified using the LDA effect size at LDA > 2, P < 0.05. P values for the biomarkers were calculated using the two-sided Wilcoxon rank sum test. f Total normalized abundance of MGEs in three MP diversity treatments (n = 48 per group). g Normalized abundance of MGE types in three MP diversity treatments (n = 48 per group). h, i Correlations between copies of MGEs per cell and copies of ARGs/VFGs per cell across all samples (n = 144). The lines indicate the line of best fit, while the shaded areas represent the 95% confidence interval. A linear regression model with a two-sided test was used for statistical analysis. P values were calculated using Kruskal-Wallis test, one-way ANOVA with LSD-test and two-tailed unpaired Student’s t test. Significance among treatments is denoted as * P < 0.05, ** P < 0.01, *** P < 0.001. The unit of genes is copies per cell. Source data are provided as a Source Data file.
The composition of ARG subtypes in different treatments was visualized using principal coordinates analysis (PCoA), and three distinct groups dominated by MP diversity were clustered for ARGs (Supplementary Fig. 4a), as confirmed by the PERMANOVA test (P = 0.001, Supplementary Table 1). Furthermore, PCoA combined with the PERMANOVA test demonstrated significant influences of MP diversity (P = 0.001) and fungicide application (P = 0.021) on the composition of high-risk ARGs (P < 0.05, Supplementary Fig. 4b).
Effects of the MP diversity on the composition and abundance of soil VFGs and MGEs
We identified 14 VFG types and 2635 subtypes that render the pathogen invasive. The total abundance of VFGs was notably enriched in soils with increasing diversity of MPs (D3 vs. D1: P = 0.009; D6 vs. D1: P = 0.021, LSD-test, Fig. 1d), soil without fungicide application (P = 0.002, t test, Supplementary Fig. 5a) and in soils cropped with mono-species community (P = 0.013, t test, Supplementary Fig. 5b). Consistent results were observed in the linear mixed-effects model, though the effect of MP diversity on the total relative abundance of VFGs was minor and insignificant (standard regression coefficient = 0.17, χ² = 3.1398, P = 0.076, Type II Wald chi-square test, Table 1). Additionally, no significant interactions were found between MP diversity, fungicide application, and plant community (all P > 0.05, Table 1). For the types of VFGs, the diversity level six of MPs significantly increased the antimicrobial activity/competitive advantage, biofilm, immune modulation and motility related VFGs (LEfSe test, P < 0.05, LDA > 2, Fig. 1e). The application of fungicide greatly enriched the regulation and nutritional/metabolic factor associated VFGs (LEfSe test, P < 0.05, LDA > 2, Supplementary Fig. 5c). For the VFG composition, MP diversity, fungicide application and plant community both exerted substantial effects (PERMANOVA test, P < 0.05, Supplementary Fig. 6). Additionally, the abundance of ARGs was correlated with antimicrobial activity/competitive advantage (R2 = 0.624, P < 0.0001), biofilm (R2 = 0.125, P < 0.0001), motility (R2 = 0.481, P < 0.0001), and immune modulation VFG (R2 = 0.761, P < 0.0001) (Supplementary Fig. 7).
MGEs that would facilitate the prevalence and persistence of soil ARGs and VFGs through horizontal gene transfer were identified in 106 subtypes and 37 types. The significantly highest total abundance of MGEs was detected in soils exposed to the diversity level six of MPs (P = 0.002, Kruskal-Wallis test, Fig. 1f). Significant effects of MP diversity on the total relative abundance of MGEs were also detected from the linear mixed-effects model (standard regression coefficient = 0.29, χ² = 3.8474, P = 0.049, Type II Wald chi-square test, Table 1). Similar to the patterns observed for ARGs and VFGs, significant interaction effects between factors on total MGE relative abundance were not detected (all P > 0.05, Table 1). For all types of MGEs, increasing diversity of MPs substantially increased their abundance (P < 0.05, Kruskal-Wallis test, Fig. 1g). Additionally, MGE abundance was significantly positively correlated with ARG (R2 = 0.532, P < 0.0001, Fig. 1h) and VFG abundance (R2 = 0.118, P < 0.0001, Fig. 1i). Specifically, the compositional structure of ARGs was significantly correlated with all MGE types, particularly with transposase, IS91, and integrase (Supplementary Fig. 8). Transposase and IS91 were also found to be significantly correlated with VFG community (Supplementary Fig. 9).
We also characterized the coexistence of ARGs, VFGs, and MGEs of the assembled contigs based on metagenomic analysis (Supplementary Fig. 10). The multidrug related ARGs co-located with MGEs were observed in Azonexus, Pseudomonas, and Azospirillum. Besides, Rank IV high-risk sul1 frequently coexisted with MGEs. In Pseudomonas, ARG-VFG-MGE was detected to be co-located on contigs.
Potential mechanisms of ARG enrichment in the increasing diversity of MP-exposed soils
SEM analysis was conducted to assess the direct and indirect effects of MP diversity, plant biomass, soil properties, bacterial community and MGE abundance on ARG profiles in all soil samples (Fig. 2a). The SEM explained 64% of the variance of soil ARGs abundance. MGE abundance (β = 0.272, P = 0.002) and bacterial community (β = 0.616, P < 0.001) showed significant direct effects on ARG abundance. MP diversity demonstrated enhancement of ARG abundance through significant direct effects on bacterial community (β = 0.089, P = 0.050). Besides, plant biomass (β = −0.797, P < 0.001) and soil properties (β = −0.199, P < 0.001) constituted a strong direct effect on bacterial community. MGE abundance was also significantly and directly affected by the bacterial community (β = 0.895, P < 0.001). Furthermore, the bacterial community (standardized total effects = 0.859, Fig. 2b) had the highest total effect on the relative abundance of ARGs in soils, followed by the plant biomass (−0.657), MP diversity (0.336) and MGE abundance (0.272). Procrustes tests also revealed a significant correlation between ARG profiles and bacterial community (P < 0.001, Fig. 2c).

a Structural equation model (SEM) evaluating the direct and indirect effects of MP diversity, plant biomass, soil properties, bacterial community, and MGE abundance on ARG abundance in all soil samples (n = 144). Blue and orange arrows indicate positive and negative effects, respectively. Solid and dotted lines show significant and insignificant effects, respectively. Path coefficients are shown next to the arrows, and R2 denotes the proportion of variance explained for each variable. Significance levels of regression weight are calculated using two-sided t test and denoted as * P < 0.05, ** P < 0.01, *** P < 0.001. b Standardized total effect size of ARGs abundance based on SEM. c Procrustes test demonstrating the significant correlation between ARG profiles and bacterial community (n = 144). The length of the line connecting two points quantifies the dissimilarity between the bacterial community composition (green) and the ARG profile (red) within the same sample. P value was determined using Procrustean randomization tests. The M2 value indicates the sum of squared distances between matched sample pairs. Source data are provided as a Source Data file.
Contribution of the genetics and function of the soil microbial community to the enrichment of soil ARGs
Nucleotide diversity and GC content were used to characterize the genetic evolution of the soil microbiome exposed to MPs. Compared with the single type of MPs, MP diversity levels three (P = 0.004, Kruskal-Wallis test) and six (P < 0.0001, Kruskal-Wallis test) significantly decreased the microbial nucleotide diversity (Fig. 3a), while a significant reduction in GC content was only shown in the three diversity MPs treatment (P = 0.043, LSD-test, Fig. 3b). In addition, a pronounced negative correlation between GC content and ARGs or MGEs abundance was noted, and this relationship was more obvious in MGEs (Fig. 3c, d, P < 0.05).

a Nucleotide diversity of the soil microbiome across three MP diversity treatments (n = 48 per group). b GC content of soil microbial genome in three MP diversity treatments (n = 48 per group). c, d Correlations between GC content and copies of MGEs/ARGs per cell across all samples (n = 144). e, f Shannon index of KOs and CAZyme genes in three MP diversity treatments (n = 48 per group). g, h Correlations between the Shannon index of KOs/CAZyme genes and copies of ARGs per cell across all samples (n = 144). i Relative abundance of microbial features essential for antibiotic resistance derived from KEGG C level (in transcripts per million reads, TPM). Statistical analysis was performed using linear regression with two-sided tests. P values are calculated using Kruskal-Wallis test and one-way ANOVA with LSD-test. Significance among treatments is denoted as * P < 0.05, ** P < 0.01, *** P < 0.001. For boxplots, the tops of the boxes represent the 75th percentile, the bottoms indicate the 25th percentile, and the center lines denote the median. The whiskers extend to the maximum and minimum non-outlier values. For scatterplots, the lines indicate the line of best fit, while the shaded areas represent the 95% confidence interval. Source data are provided as a Source Data file.
Based on the Shannon index of the KEGG pathway (D6 vs. D1: P = 0.002, LSD-test) as well as CAZyme genes (D3 vs. D1: P = 0.004; D6 vs. D1: P < 0.0001, Kruskal-Wallis test), we found a significant increase in the functional diversity of the soil microbiome in response to the increasing diversity of MPs (P < 0.05, Fig. 3e, f). Functional diversity was significantly positively correlated with the abundance of ARGs across all samples (P < 0.01, Fig. 3g, h). Certain microbial features associated with the proliferation of antibiotic resistance were significantly influenced by MP diversity, suggesting potential mechanisms by which increasing diversity of MPs promotes the enrichment of ARGs in soil (Fig. 3i). For the formation of biofilm, the biochemical pathways including biofilm formation of Escherichia coli (D3 vs. D1: P = 0.003; D6 vs. D1: P < 0.0001, Kruskal-Wallis test), Pseudomonas aeruginosa (D3 vs. D1: P = 0.015; D6 vs. D1: P < 0.0001, Kruskal-Wallis test), and Vibrio cholerae (D3 vs. D1: P = 0.027; D6 vs. D1: P < 0.0001, Kruskal-Wallis test) were more abundant in soils with the three and six diversity levels of MPs compared to the single MP type exposure. Features that encode for membrane structure and transport, involving lipopolysaccharide biosynthesis (D3 vs. D1: P = 0.018; D6 vs. D1: P < 0.0001, Kruskal-Wallis test), flagellar assembly (D3 vs. D1: P = 0.026; D6 vs. D1: P = 0.012, Kruskal-Wallis test), bacterial secretion system, and ABC transporters were also found to have a high abundance in the treatments with an increasing diversity of MP. Furthermore, SOS response pathways such as folate biosynthesis were significantly enriched under the three and six diversity levels of MP exposure (D3 vs. D1: P = 0.006; D6 vs. D1: P < 0.0001, Kruskal-Wallis test). Procrustes tests revealed a significant correlation between ARG profiles and microbial features predicted by KEGG (P = 0.001, Supplementary Fig. 11). The abundance of all features was significantly associated with the copies of ARGs per cell (P < 0.0001).
Identification of key driving bacterial taxa
A total of 1440 metagenome-assembled genomes (MAGs) were identified in the soil, 255 of which carried ARGs (Fig. 4a). The ARG-carrying MAGs were assigned to eight phyla among which Proteobacteria, Actinobacteriota, and Firmicutes were dominant. The 255 MAGs mainly carried 11 ARG types, among which multidrug, bacitracin, trimethoprim, MLS, and vancomycin were most frequently detected. Moreover, 232 ARG-carrying MAGs could be pathogenic ARG hosts due to the presence of diverse VFGs (214 subtypes). Of these ARG-VFG-containing bacteria, 99.57% (231 MAGs) contained a diverse array of MGEs (358 subtypes), and 95.26% (221 MAGs) carried CAZyme genes. Simultaneously, a significant positive correlation was observed between the number of CAZyme genes and ARGs in these bacteria (P = 0.005, Supplementary Fig. 12). The increasing diversity of MPs substantially shifts the community of ARG-VFG-containing bacteria (P = 0.001, Supplementary Fig. 13), mainly related to the changed soil physicochemical properties including TC, OM and DOC (P < 0.001, Supplementary Fig. 13). The linear mixed-effects model revealed significant impacts of MP diversity on the ARG-VFG-containing bacterial community (standard regression coefficient = 0.43, χ² = 8.4339, P = 0.004, Type II Wald chi-square test, Supplementary Table 2). Moreover, the diversity of ARG-VFG-containing bacteria indicated by the Shannon (D3 vs. D1: P = 0.001; D6 vs. D1: P < 0.0001, Kruskal-Wallis test, Fig. 4b) and Simpson (D3 vs. D1: P = 0.004; D6 vs. D1: P < 0.0001, Kruskal-Wallis test, Fig. 4b) index was significantly enhanced by increasing diversity of MPs. The correlation among ARG-VFG-containing bacteria was also altered by MP diversity, manifesting as a decrease in negatively correlated linkages and a decline in the stability of the co-occurrence network (Fig. 4c, d, e).

a Phylogenetic tree of the overall MAGs in the soil. The number of MAGs carrying CAZyme genes, ARGs, VFGs, and MGEs are shown on the outer edge of the tree, from the inner circle to the outer circle. The outermost green circles represent MAGs that carry ARGs, VFGs, MGEs, and CAZyme genes simultaneously. b Alpha diversity of ARG-VFG containing bacterial community in three MP diversity treatments, as denoted by Shannon and Simpson index (n = 48 per group). The tops of the boxes represent the 75th percentile, the bottoms indicate the 25th percentile, and the center lines denote the median. The whiskers extend to the maximum and minimum non-outlier values. c Co-occurrence network of ARG-VFG-containing bacteria (n = 144). Dot size indicates degree, and red and blue lines indicate positive and negative correlations, respectively. d Ratio (%) of negative links in the co-occurrence network of MP diversity treatments. e Robustness of the ARG-VFG containing bacterial network for different diversity levels of MP exposure. P values were calculated using Kruskal-Wallis test. Significance among treatments is denoted as * P < 0.05, ** P < 0.01, *** P < 0.001. Source data are provided as a Source Data file.
Among all MGE-carrying bacteria that containing ARG-VFG, Proteobacteria and Firmicutes phyla were significantly enriched in the highest MP diversity treatment (LEfSe test, P < 0.05, LDA > 2, Supplementary Fig. 14). In terms of genera, the abundance of Phaeospirillum, Aquabacterium, Giesbergeria, Rhizobacter, SYFN01, Azonexus, Azospira, Methyloversatilis, unclassified_ Burkholderiaceae, and unclassified_Peptostreptococcales was strikingly increased by MP diversity level six (LEfSe test, P < 0.05, LDA > 2). MP diversity level three significantly increased the abundance of Xanthobacter, Paucimonas, CAJAWW01, UBA2250, and Pseudoxanthomonas (LEfSe test, P < 0.05, LDA > 2). Through random forest analysis, 16 indicator genera associated with increased diversity of MPs were identified, 14 of which belonged to the phylum Proteobacteria (Fig. 5a). Among these, the relative abundance of Aquabacterium and Xanthobacter was strongly positively associated with soil ARG abundance (P < 0.001, Fig. 5b). Besides, these were significantly enriched by the increasing diversity of MPs (P < 0.05, Fig. 5c). We then gathered the distribution and genomic data on these two bacteria from NCBI, revealing that strains from global isolates generally carry a high abundance of ARGs (Supplementary Fig. 15). Resistance gene abundance was significantly higher in Aquabacterium (P = 0.025, LSD-test) and Xanthobacter (P < 0.0001, LSD-test) compared to Actinophytocola (D1 vs. D3/D6: P < 0.05, Fig. 5c) that enriched in the single MP exposure treatment (Fig. 5d). For the ARG types, the abundance of genes encoding resistance to bacitracin, MLS, and multidrug was significantly higher in Aquabacterium and Xanthobacter (all P < 0.05, Fig. 5e).

a Biomarkers associated with ARGs abundance identified through Random Forest analysis. The area map illustrates the relative abundance of biomarkers in samples (n = 144). b Correlations between the relative abundance of Aquabacterium/Xanthobacter and copies of ARGs per cell across all samples (n = 144). The lines indicate the line of best fit, while the shaded areas represent the 95% confidence interval. A linear regression model with a two-sided test was used for statistical analysis. c Relative abundance of Actinophytocola, Aquabacterium, and Xanthobacter in three MP diversity treatments (n = 48 per group). P values were calculated using Kruskal-Wallis test. Significance among treatments is denoted as * P < 0.05, ** P < 0.01, *** P < 0.001. d Total normalized abundance of ARGs in Actinophytocola (n = 12), Aquabacterium (n = 68), and Xanthobacter (n = 36). Different letters above the bars represent significant differences in copies of ARGs per cell between bacteria (P < 0.05). P values were calculated using Kruskal-Wallis test. The tops of the boxes represent the 75th percentile, the bottoms indicate the 25th percentile, and the center lines denote the median. The whiskers extend to the maximum and minimum non-outlier values. e Heatmap showing the abundance of ARGs types in Actinophytocola, Aquabacterium, and Xanthobacter. Solid red circles indicate significant differences in ARGs abundance across bacteria. Source data are provided as a Source Data file.
Results of Microcosm Experiment 2
To minimize bias from MP composition, we selected a clustered group of MPs, as identified in the PCoA plot (Supplementary Fig. 13, where the most red dots aggregate), for the validation Microcosm Experiment 2 (Fig. 6a). In alignment with the results from Microcosm Experiment 1, treatments with higher MP diversity led to a significant increase in the total abundance of ARGs (D5 vs. CK: P = 0.001; D5 vs. D1: P = 0.011, Kruskal-Wallis test, Fig.6b) and heightened risk levels associated with VFG abundance (D5 vs. CK: P = 0.014; D5 vs. D1: P = 0.025, Kruskal-Wallis test, Supplementary Fig. 16) in the soil, compared to both the control and single MP type treatments. We also observed a trend where ARG richness (Fig.6b) and MGE abundance increased in correlation with higher levels of MP diversity (Supplementary Fig. 16). The composition of ARG subtypes was strikingly changed by increasing MP diversity (P = 0.043, PERMANOVA test, Supplementary Fig. 16). For bacterial community, the richness (D5 vs. CK: P = 0.009, Kruskal-Wallis test) and patterns (P = 0.019, PERMANOVA test) were influenced by the increasing diversity of MPs (Supplementary Fig. 17). The strong association between bacterial community and ARG profiles persists as well (P = 0.002, Supplementary Fig. 17).

a Schematic illustration of the design for Microcosm Experiment 2, depicting MP combinations across three levels of MP diversity: 1, 3, and 5 different types (D1, D3, D5). A control group without MP pollution (CK), with six replicates, was also included. b Total normalized abundance and alpha diversity of ARGs (n = 6 per group). c GC content of soil microbial genome (n = 6 per group). d Shannon index of KOs and CAZyme genes (n = 6 per group). e Correlations between GC content, Shannon index of KOs/CAZyme genes and copies of ARGs per cell across all samples (n = 24). f Heatmap showing the relative abundance of microbial features essential for antibiotic resistance derived from KEGG C level (in transcripts per million reads, TPM). g Correlations between Aquabacterium, and Xanthobacter communities and MGE types (n = 24). Edge width represents to Mantel’s r value, and edge color indicates statistical significance. Pairwise correlations of these variables were shown with a color gradient corresponding to spearman’s correlation coefficient. h Correlation between the relative abundance of Aquabacterium and copies of ARGs per cell across all samples (n = 24). A linear regression model with a two-sided test was used for statistical analysis. P values were calculated using Kruskal-Wallis test. Significance among treatments is denoted as * P < 0.05, ** P < 0.01, *** P < 0.001. The unit of genes is copies per cell. For boxplots, the tops of the boxes represent the 75th percentile, the bottoms indicate the 25th percentile, and the center lines denote the median. The whiskers extend to the maximum and minimum non-outlier values. For scatterplots, the lines indicate the line of best fit, while the shaded areas represent the 95% confidence interval. Source data are provided as a Source Data file.
The results of genetic (Fig. 6c) and functional traits (Fig. 6d) showed significant negative (P = 0.023) and positive (P < 0.001 for Shannon index of KOs; P = 0.018 for Shannon index of CAZyme genes) correlations with the copies of ARGs per cell (Fig. 6e), consistent with the findings from Microcosm Experiment 1. The heatmap showed that KEGG pathways related to biofilm formation, membrane structure, and the SOS response, which are key factors in the proliferation of antibiotic resistance, were expanded with increasing MP diversity (Fig. 6f). Furthermore, the key bacterial strains identified in Microcosm Experiment 1 remained significantly positively correlated with the copies of ARGs per cell in the Microcosm Experiment 2 (P = 0.044, Fig. 6h). The Aquabacterium likely played a crucial role in driving the enrichment of bacitracin and multidrug resistance genes (all P < 0.05, Fig. 6g).
Discussion
Our results showed that with an increasing diversity of MPs, the richness and abundance of ARGs in the soil environment were significantly increased. Notably, high-risk ARGs encoding resistance to aminoglycoside, tetracycline, and florfenicol were substantially elevated. The abundance of MGEs reflecting ARG motility and VFGs representing human pathogenicity were significantly increased along with increasing levels of MP diversity, posing a health risk. These changes are driven by gene changes, community compositional shifts and functional trait alterations in the soil microbiome (Fig. 7). This study uncovers the critical role of MP diversity in the dissemination of ARGs, expanding our knowledge of soil environmental MP contamination. In addition to examining different MP types, further research is crucial to explore other dimensions of their diversity, including aging, size, shape, and the impacts of their coexistence1. For example, the natural weathering of MPs might increase surface roughness, providing more niches for microbial attachment and potentially enhancing the establishment of biofilms. Comprehensive investigations into these dimensions will be crucial for developing effective strategies to mitigate the ecological impacts associated with MP contamination.

The model depicts genetic responses, shifts in microbial community and altertions in functional traits. For the shifts in microbial community, the increasing diversity of ARG-VFG-containing bacteria (pathogenic antibiotic resistant bacteria), and the enrichment of MP degrading bacteria were shown.
In this study, we attempt to decipher the increased ARG abundance from the perspective of microbial genome evolution. Nucleotide diversity and genomic GC content served as gene change indicators to assess microbial adaptation to the increasing MP diversity. Consistent with our study, Escherichia coli and Enterococcus faecium respond with a sharp decrease in nucleotide diversity under exogenous stress33. At the genome level, environmental adaptation may promote homologous recombination in a large number of soil bacteria34, leading to reduced nucleotide diversity. This decrease may reflect the potential selection of specific microbial genotypes under the pressure of increasing MP diversity33. The soil microbiome may experience unique evolutionary pressures exerted by different diversities of MPs (including the large number of additives contained in these plastic particles), driving microbial adaptation through mutation emergence and selection. Additionally, increasing diversity of MPs significantly reduced GC content. Compared to the core genomes, genomic islands typically have lower GC content35,36. This implied that ARGs may undergo extensive horizontal gene transfer among microbiome in soil contaminated with high MP diversity, as evidenced by the significant association linking GC content to the relative abundance of MGEs and ARGs.
With increasing MP diversity, we found a significant tendency for the metabolic diversity of the microbial community to increase. Changes in microbial metabolism, especially degradation, were observed with MP exposure in soil and animal intestines37,38. Carbohydrate digestion and absorption functions were enriched as MP diversity increased. Besides, carbohydrate metabolism related enzymes, such as polysaccharide lyases and glycoside hydrolases, were enhanced by high diversity of MPs (Supplementary Fig. 18). As the number of MP species rises, the emergence of various types, including degradable plastics, becomes more prevalent. Unlike non-degradable plastics, biodegradable plastics can serve as a potential carbon source for soil bacteria, inducing a stronger priming effect in scenarios of increasing MP diversity17. The exposure to MPs would cause intense metabolic activity, which would impact the propagation of ARGs39,40. In this study, we found that bacteria with a higher number of CAZyme genes also tend to carry more ARGs. Regarding microbial community interactions, the observed increase in positive interactions indicates an overall enhancement in metabolic efficiency. We further investigated KEGG categories contributing to antibiotic resistance41, including the biofilm formation, membrane structure and transport, and SOS response. These pathways are the crucial in the enrichment of ARGs caused by MPs1,14,42. The increasing diversity of MPs promoted the biofilm formation of Escherichia coli, Pseudomonas aeruginosa, and Vibrio cholerae. Biofilms, surface-attached microbial communities, can develop antibiotic resistance up to 1000-fold greater than planktonic cells43. Different MPs possess varying specific surface areas1,44. As MP diversity increases, it may alter the total surface area, influencing microbial biofilm formation. Flagellar assembly was also observed to be strengthened by the high diversity of MPs. The flagellum motility of bacteria has been reported to promote the effect of three disinfectants on transformation, probably benefiting from an accelerated uptake of external plasmids45. Furthermore, the increasing diversity of MPs enhanced folate biosynthesis, which is known to regulate oxidative stress41. SOS response reveals reactive oxygen species (ROS) overproduction, which could facilitate horizontal gene transfer through the increases of the cell membrane permeability42. Overgeneration of ROS within the microbiome likely drivers the increase in ARGs caused by MPs42. Varying MP types induce different levels of oxidative stress, and higher MP diversity may amplify this stress, playing a critical role in the increased abundance of ARGs in the soil.
Microbial community compositional shifts occurred in response to the increasing diversity of MPs. The community of ARG-VFG-containing bacteria was strikingly altered by the high diversity of MP exposure, correlating with soil TC, DOC, and OM. We previously found that MP pollution significantly increased the concentration of DOC46,47. This indicated that the high diversity of MP addition facilitated the release of soil nutrients into the soil solution and the accumulation of DOC46. The microbial metabolism response to MP addition may enhance soil carbon cycling and bioavailability. As hypothesized, the diversity of ARG-VFG-containing bacteria dramatically increased with a high diversity of MPs, exhibiting high ecological risk. Numerous studies in terrestrial and aquatic ecosystems have revealed that contamination selectively enriches antibiotic-resistant bacterial assemblages in the plastisphere1,14,48,49. The pathogen load in the plastisphere may result in an enhanced pathogen-carrying capacity of ecosystems50. Random forest analysis indicated that the overwhelming majority of microbes contributing to soil resistance gene abundance changes due to increasing diversity of MPs belong to the phylum Proteobacteria. Significant enrichment of Proteobacteria was also observed in soils with the addition of six diversity levels of MPs. It appears that Proteobacteria are often enriched in the soil plastisphere14,51,52, playing an essential role in the presence of a wide range of contaminants53,54. Among Proteobacteria, changes in Aquabacterium and Xanthobacter abundance were significantly and positively correlated with increased ARG abundance. Data collected globally from different habitats show that these genera inherently carry higher ARG abundances than Actinophytocola, which is enriched in soils with low MP diversity. Interestingly, genes encoding resistance to bacitracin, multidrug, and sulfonamide, which were notably abundant in these two genera, were also detected in high abundance in soil with a high diversity of MPs. This strongly suggests that the enrichment of specific bacteria with adaptive advantage under high MP diversity stress accelerated the development and spread of soil ARGs. In a previous study, polyvinyl chloride MPs enriched pathogenic bacteria including Aquabacterium from surrounding sewage55. This genus is a common strain of pathogenic bacteria that carry ARGs (i.e., efflux pump-coding genes and quinolone resistance genes) and is widely found in soil and wastewater effluent56,57. Furthermore, Aquabacterium is shown to be a widespread host for class 1 integrons58, adding more insight into the ecological risks of high diversity of MP contamination. Xanthobacter is identified as a bacterial genus harboring various ARGs, primarily those associated with conferring aminoglycoside and multidrug resistance59. Notably, strains of these genera have been found to have the ability to degrade MPs60,61. These two genera belong to Proteobacteria, which have been reported to be the main carriers of MP degradation genes62. This implies that the enrichment of ARG-carrying pathogens may be associated with microbial metabolic activity in response to multiple MP contaminants. Further isolation and cultures are needed to robustly verify the ARG-promoting effects of bacteria under MP diversification stress.
The increasing diversity of MPs not only elevated ARG abundance but also increased health risks, as indicated by the enhanced abundance of VFGs. A study of Rhodococcus equi revealed that contact with plastics significantly increased VFG expression, with various plastic types regulating the same genes63. Taken together with our results, these findings demonstrate that environmental MP contamination could elevate health risks. Additionally, significantly positive relationships between ARGs and increased VFG categories (immune modulation, biofilm, motility and antimicrobial activity) were observed. Studies have shown that ARGs and VFGs were found to have a strong correlation and an intimate genetic linkage in human/animal habitats64. These elements are co-selected under selective pressure to adapt to harsh environments with the increasing diversity of MPs. Simultaneously, ARGs and VFGs could co-spread through MGEs-mediated horizontal transfer, as confirmed by discernible co-occurrence patterns among these elements.
Moreover, global change factors frequently co-occurring with MPs in terrestrial ecosystems have a significant impact on ARG changes. Fungicide applications tend to increase the abundance of high risk ARGs, including resistance to aminoglycoside. Studies have shown that fungicide mancozeb exposure could exert oxidative and osmotic stress on microbes, further facilitating plasmid-mediated ARG transfer65. Mono-species community significantly enhanced the richness of ARGs and expanded ARG risks as a consequence of the increased VFG abundance compared the soils cultivated with multi-species. While studies directly investigating the effects of plant diversity on resistance genes are limited, research has observed that monocultures may increase the abundance of plant pathogens in soil66. These findings indicated that soils are amid an anthropogenic multifactorial crisis20,32, with implications for the propagation and hazard of the antibiotic resistome.
In summary, our study highlights the importance of incorporating the diversity of MPs into risk assessment. We find that the increasing diversity of MPs significantly enriched the ARG richness and abundance, elevating health risks as a consequence of increased VFGs and MGEs compared to the low diversity level of MPs. Gene changes, shifts in community composition and alterations in functional traits likely contribute to the microbial response when exposed to multiple types of MPs, thus inducing higher abundance and diversity of ARGs. In addition, global change factors (fungicide application and plant diversity reduction) in the presence of MPs could also increase ARG abundance and health risks according to VFGs. Our study highlights the importance of diversity in the ecological risk assessment of MPs, especially under conditions of global change, providing novel perspectives to understand the risk of ubiquitous ARGs in terrestrial ecosystems. Considering that multiple MPs are increasingly threatening ecosystems worldwide and that the concentration of microplastics is commonly relatively low in soil environments, there is an urgent need to evaluate the diversity of MP types rather than just their total abundance.
Methods
Soils, plants, and microplastics
Soils were collected from the Ecological Research Station for Grassland Farming (ERSGF), Changling, Chinese Academy of Sciences (44°33′N, 123°31′E, altitude 145 m). The collected soils were immediately sieved through 2 mm mesh and then transported to the laboratory. The soil was Meadow solonchaks from grassland, with a pH of 7.78, an EC of 84.56 μs cm−1, an organic matter of 1.15% and total nitrogen, phosphorus, potassium content of 0.10%, 0.02%, 1.40% respectively (Supplementary Table 3).
In this study, five herbaceous plant species from three families were chosen as members for the plant community: Bidens pilosa L. and Artemisia stechmanniana Bess. (Compositae), Gypsophila licentiana Hand.-Mazz. (Caryophyllaceae), and Poa annua L. and Leymus chinensis (Trin.) Tzvel. (Poaceae) (Supplementary Table 4). All of the species are frequently co-occurring together in the grasslands of China. Seeds of each species were collected from a minimum of four distinct natural grassland populations (separated by at least 1 km) and combined into a single bulk sample for each species.
Microplastics were selected from 12 types commonly found in soil environments, and purchased from Suzhou Xin Su Yu Co. (Suzhou, China). The 12 MP pool are polycaprolactone (PCL), polyhydroxybutyrate (PHB), polybutylene succinate (PBS), polyhydroxyalkanoate (PHA), polylactic acid (PLA), polybutylene adipatecoterephthalate (PBAT), ethylene-vinyl acetate (EVA), polyvinyl chloride (PVC), polyethylene terephthalate (PET), polyoxymethylene (POM), polyamide 66 (PA66), and polypropylene (PP). Manufacturer and biodegradability of each MP type are shown in Supplementary Table 5. The MPs were milled and sieved to obtain powders, sieving through 180 μm mesh. Prior to the incubation, each MP was microwaved for 20 min at 500 W to eliminate microbial contaminants67, ensuring the temperature remained below the melting point. This process also facilitated the aging of the MPs, as microwave treatment induces hydrolysis, thermal degradation, and UV irradiation effects simultaneously68.
Microcosm Experiment 1: impacts of MP diversity on the soil microbiome and resistome
The experiment was conducted in 1-L circular plastic pots (13.5 cm diameter × 12 cm height), each containing 0.8 L of substrate, which was a 3:1 (v/v) mixture of soil and gravel. The sieved MP powders (180 μm) were mixed at a concentration of 3% (v/v, 24 mL, corresponding to 0.5–1.0% w/w), and three levels of MP diversity were established 1, 3, and 6 different types. There were 12 replicates of each level of the treatment (Supplementary Fig. 19). For the pollution level with one type of MP, we utilized each of the 12 MPs (i.e., PCL, PHB, PBS, PHA, PLA, PBAT, EVA, PVC, PET, POM, PA66, and PP). For the pollution level involving three MP types, we created 12 distinct combinations of MPs, ensuring that each MP type appeared in three different combinations. Similarly, for the pollution level with six MP types, we also generated 12 distinct combinations, with each MP type included in six different combinations. The total volume of MPs was kept constant at 24 mL across all diversity levels, with 24 mL (24/1) for one type, 8 mL (24/3) for three types, and 4 mL (24/6) for six types. The MPs were thoroughly mixed with the soil substrate in pots, and 48 pots were made for each level of diversity. The mixing was in the high-density polyethylene boxes (30 cm × 30 cm × 20 cm) with smooth inner surfaces, one for each MP treatment. Each mixture was stirred five times. Once the substrate and MPs were thoroughly mixed and showed no clumping, the pots were filled. Here, we assigned the 48 pots of each MP diversity to four treatments of two levels of a fungicide application treatment (fungicide vs. non-fungicide) fully crossed with two levels of a plant community treatment (multi-species vs. mono-species). For each pot of fungicide application treatment, environmentally relevant concentrations of 1.65 g mefenoxam L−1 soil (Syngenta Crop Protection, Inc.) were sprayed on the surface of potted soil29. For the mono-species community, we used Bidens pilosa L. for planting, while the multi-species community comprised all five species (Supplementary Table 4). For each plant species, only one individual was planted per pot, positioned into the center of 1-L circular plastic pots. To ensure uniform seedling size for the experiments, we sowed seeds of each species in separate trays (20 cm × 20 cm × 4.5 cm, filled with commercial potting soil) on different dates for nursery cultivation.
On 12 July 2022, individual similar-sized seedlings (~3 cm) of every plant species were transplanted into the 1-L pots. The pots were randomly placed on four benches in a greenhouse with a temperature range of 22–28 °C and a 14:10 light cycle. After 7 days, we started the soil fungicide application treatment. In the fungicide-applied treatments, we sprayed a mefenoxam solution into each pot to achieve a final concentration of 1.65 g per liter of soil. For the non-fungicide treatment, we added an equivalent amount of water to the pots. To ensure no water limitation for plant growth during the experiment, the pots were watered three times a week with 100 mL of water at a time during the first five weeks of growth. As the plants grew up, the pots were watered four times a week with 100 mL of water at a time during the second 4 weeks of growth. After 5 weeks, the pots were redistributed randomly. The incubation lasted 60 days (starting from 12 July) and bulk soil samples were collected after plant harvest. The harvested plants were directly dried at 65 °C for 72 h and weighed to obtain biomass. The collected soil was randomly divided into two parts, one was placed at room temperature to air dry and another at −80 °C for subsequent DNA extraction.
Microcosm Experiment 2: impacts of MP diversity on the soil microbiome and resistome under identical composition
The study emphasized the impact of MP diversity on soil microbiome and resistome, acknowledging that the identity and composition of MPs can exert significant influences. Therefore, we selected types of conventional non-degradable MPs commonly found in soil for the validation experiment of the diversity gradient. The MP pool consisted of six types of conventional non-degradable MPs (EVA, PET, POM, PA66, PP, and PVC), establishing three levels of MP diversity with one, three, and five different types. There were six replicates of each level of the treatment. For the pollution level with one type of MP, we utilized each of the six MPs (i.e., EVA, PET, POM, PA66, PP, and PVC). For the pollution level involving three MP types, we created six distinct combinations of MPs, ensuring that each MP type appeared in three different combinations (Fig. 6). Similarly, for the pollution level with five MP types, we also generated six distinct combinations, but with each MP type being included in five different combinations. The specific experimental details, including substrate composition and MP content, was consistent with Microcosm Experiment 1. Meanwhile, we established a control group without MP pollution for six replicates. For the plant community, all five species were cultivated by planting one similarly-sized seedling of each species in the center of a 1-L circular plastic pot. No fungicide applications were made in this experiment.
Soil physicochemical properties analyses
Soil physicochemical properties were determined according to a previous study69. Specifically, soil pH, total carbon (TC), total nitrogen (TN), total phosphorus (TP), available phosphorus (AP), organic matter (OM), and dissolved organic carbon (DOC) were determined in each soil sample.
DNA extraction, metagenome sequencing, and quality filtration
Soil DNA was extracted by applying FastDNA Spin Kit (MP Bio, USA) following the manufacturer’s instructions. Around 0.5 g of soil was weighed, lysed, purified, and eluted to obtain total genomic DNA. The purity of the extracted DNA was assessed using 1.0% agarose gel electrophoresis and Nanodrop spectrophotometry (Nanodrop ND-1000, Thermo Scientific, USA). Subsequently, the DNA was kept frozen at −20 °C until analysis. The extracted DNA was transported to Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) for metagenomic analysis. Approximately 5 μg of each DNA sample was utilized for library construction with the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, USA). Metagenomic sequencing was conducted on the Illumina HiSeq4000 platform, employing the PE150 strategy. The average raw reads obtained for each DNA sample were ~10 Gb. Finally, adapter sequences, trimming, and low-quality reads (including reads with >10% ambiguous nucleotides (“N”), and >50% nucleotides with a quality value lower than 10) were filtered.
Metagenomic assembly and bioinformatics analysis
After quality filtering, high-quality metagenomic reads were assembled to contigs using MEGAHIT (v.1.2.9) with default parameters70. Assembled contigs with length > 800 bp were subsequently used for predicting open reading frames (ORFs) through Prodigal (v.2.6.3) with the -p meta option71, and non-redundant gene catalog was acquired using CD-HIT (v.4.8.12) with a 95% identity and 90% coverage threshold72. Quality-controlled reads after were mapped to the non-redundant gene catalog at 95% identity using Bowtie 2 (v.2.4.2)73, and gene abundance in each sample was evaluated by SAMtools74. The representative sequences from the gene catalog were annotated with BLASTp against the NCBI NR database, using DIAMOND (v.2.0.14) at an e-value cutoff of 1e−5. KEGG annotations were conducted with DIAMOND (v.2.0.14) against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database75 with an e-value cutoff of 1e−5. Carbohydrate-active enzyme (CAZyme) annotation followed the same approach using the CAZy database76. For each sequence, the highest-scoring hit (HSP > 60 bits) was chosen for further analysis.
To characterize ARGs, predicted genes were identified as ARG-like genes by searching nucleotide sequences against the Structured ARG (SARG) database77 using BLASTX with an e-value ≤1e−10 and ≥ 80% identity. The abundance of ARG-like genes in each sample was calculated as copies per cell by ARGs-OAP (v.3.2)78. We furthermore identified high-risk ARGs based on the list of high-risk ARGs constructed by Zhang et al.79. Similarly, VFGs were determined using the Virulence Factor Database (VFDB)22, and the bacteria harboring VFGs were regarded as potential pathogens. To further classify MGEs, a database of bacterial MGE hallmark genes, mobileOG-db80, was used to assign element class labels of transposable elements (defined as sequences derived from ISfinder), integrative elements (such as integrases, transposases, which are not in ISfinder, and which do not encode conjugation machinery), or conjugative element (reads with hits to conjugation machinery). Reads were also annotated at 80% identity and e-value <1e−10. The co-occurrence of ARGs, VFGs, and MGEs was further explored to identify if these genes were simultaneously located on the same contig.
MAGs were recovered using the Vamb (v.4.1.1)81. The qualities of derived MAGs were examined using CheckM (v.1.0.12)82, and only those with ≥50% completeness and ≤10% contamination were retained for further analysis. GTDB-Tk (v.2.1.1) was used to predict MAG taxonomy and build the phylogenetic tree83. ORFs from MAGs were annotated against SARG, VFDB, MGE, and CAZy databases with a cutoff of ≥80% identity and e-value ≤10−10. GC content was calculated based on all assembled contigs, and nucleotide diversity analyses were carried out with InStrain84. We used GC content and nucleotide diversity to reflect the gene adaptation of microbes to changes in stress85,86.
Statistical analysis
The alpha diversity (measured as richness, Shannon, and Simpson index) was calculated to characterize the species diversity of ARGs, ARG-VFG-containing microbes, and microbial function in all samples employing the “vegan” package (v.2.6-4) in R (v.4.2.1). Based on Bray-Curtis distances, Principal Coordinates Analysis (PCoA) in the “vegan” R package (v.2.6-4) was used to visualize the dissimilarity of ARG, high-risk ARG and VFG composition, and ARG-VFG-containing microbial community. PERMANOVA was conducted to analyze the explanatory power of different treatments on the sample variance using the “vegan” package (v.2.6-4). With IBM SPSS (v.22.0), the nonparametric Kruskal-Wallis test, one-way analysis of variance (one-way ANOVA) with LSD-test, and two-tailed unpaired Student’s t test were used to test the differences between treatments at a significance level of 0.05. The changes in the VFGs and ARG-VFG-containing microbes generated among treatments were examined with the linear discriminant analysis effect size (LEfSe, LDA score (Log10) = 2 as the cutoff value) approach. Partial Least Squares Discriminant Analysis (PLS-DA) in the “ropls” package (v.1.20.0) was conducted to reveal differences in functions between high MP diversity and single MP exposure treatments. The linear fit between data was used to construct correlations by the “ggplot2” package (v.3.5.0). Mantel tests were used to examine the correlation between MGE abundance and ARG/VFG distance matrices with 999 permutations in the “vegan” package (v.2.6-4). The correlations between the community of ARG and bacterial community/functional features were determined in the “vegan” package (v.2.6-4) by Procrustes analysis.
To explore the effects of three experimental factors (including MP diversity, fungicide application, and plant community) and their interactions on the soil resistome and ARG-VFG-containing microbial community, a full linear mixed-effects model as y ~ MP Diversity × Fungicide × Plant + (1|Mixture). In the model, the fixed effects of “MP Diversity (D)”, “Fungicide (F)” and “Plant (P)” represent the introduced types of MPs (as numeric values: 1, 3, and 6 for number of MP species introduced), whether the fungicide was application to soil (as numeric values: 1 for nonfungicide application and 2 for fungicide application), and mono- or multispecies plant community (as numeric values: 1 for monospecies and 2 for multispecies) respectively. A random factor “Mixture” represents the combination of different types of MPs (as a character, such as A and ABC for groups that contained PCL or both PCL, PHA, and POM, respectively). Conditional R square and marginal R square were calculated using “MuMIn” package (v.1.47.5), and statistical significance is based on Wald type II chi-square tests using “car” package (v.3.1-2).
To characterize the effect of soil properties on the ARG-VFG-containing microbial community, LM in the “stats” package (v.4.3.3) was conducted. A co-occurrence network of ARG-VFG-containing microbes was constructed using the “ggClusterNet” package (v.0.1.0) (Spearman’s r > 0.6, Padj < 0.05), and the network topological index was calculated. Random forest analysis in the “randomForest” package (v.4.7-1.1) (ntree = 1000) was performed to identify the biomarkers to indicate treatments with different MP diversity exposure levels. The world map of ARG abundance in Aquabacterium and Xanthobacter was generated using the “ggplot2” package (v.3.5.0). A structural equation model (SEM) was developed to evaluate the direct and indirect effects of MP diversity, plant biomass, soil properties, bacterial community, and MGE abundance on the soil ARG abundance using AMOS 21 (SPSS Inc., Chicago, USA). The value of principal coordinate axis 1 from the PCoA results was applied to represent soil properties and bacterial community. The overall model fit was assessed by the Chi-square test, goodness-of-fit index, and root means square error of approximation.
Reporting summary
Further information on research design is available in Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study have been deposited in the National Center for Biotechnology Information (NCBI) SRA database (https://www.ncbi.nlm.nih.gov/sra) under accession code PRJNA1066406. Source data are provided in this paper.
Code availability
The R script is available in a publicly accessible database (https://github.com/YifeiWangang/MP-diversity.git).
References
Rillig, M. C., Kim, S. W. & Zhu, Y. G. The soil plastisphere. Nat. Rev. Microbiol. 22, 64–74 (2024).
Koelmans, A. A. et al. Microplastics in freshwaters and drinking water: Critical review and assessment of data quality. Water Res. 155, 410–422 (2019).
Klein, S., Worch, E. & Knepper, T. P. Occurrence and spatial distribution of microplastics in river shore sediments of the Rhine-Main area in Germany. Environ. Sci. Technol. 49, 6070–6076 (2015).
Allen, S. et al. Atmospheric transport and deposition of microplastics in a remote mountain catchment. Nat. Geosci. 12, 339–344 (2019).
De-la-Torre, G. E. Microplastics: an emerging threat to food security and human health. J. Food Sci. Technol. 57, 1601–1608 (2020).
Horton, A. A., Walton, A., Spurgeon, D. J., Lahive, E. & Svendsen, C. Microplastics in freshwater and terrestrial environments: Evaluating the current understanding to identify the knowledge gaps and future research priorities. Sci. Total Environ. 586, 127–141 (2017).
Weithmann, N. et al. Organic fertilizer as a vehicle for the entry of microplastic into the environment. Sci. Adv. 4, eaap8060 (2018).
Evangeliou, N. et al. Atmospheric transport is a major pathway of microplastics to remote regions. Nat. Commun. 11, 3381 (2020).
Chen, L. et al. Spatial distributions, compositional profiles, potential sources, and intfluencing factors of microplastics in soils from different agricultural farmlands in China: a national perspective. Environ. Sci. Technol. 56, 16964–16974 (2022).
Zhu, Y. G. et al. Soil biota, antimicrobial resistance and planetary health. Environ. Int. 131, 105059 (2019).
Rohr, J. R. et al. Emerging human infectious diseases and the links to global food production. Nat. Sustain. 2, 445–456 (2019).
Pruden, A., Pei, R., Storteboom, H. & Carlson, K. H. Antibiotic resistance genes as emerging contaminants: studies in northern Colorado. Environ. Sci. Technol. 40, 7445–7450 (2006).
White, A. & Hughes, J. M. Critical importance of a one health approach to antimicrobial resistance. EcoHealth 16, 404–409 (2019).
Zhu, D., Ma, J., Li, G., Rillig, M. C. & Zhu, Y. G. Soil plastispheres as hotspots of antibiotic resistance genes and potential pathogens. ISME J. 16, 521–532 (2022).
de Souza Machado, A. A., Kloas, W., Zarfl, C., Hempel, S. & Rillig, M. C. Microplastics as an emerging threat to terrestrial ecosystems. Glob. Chang. Biol. 24, 1405–1416 (2018).
Boots, B., Russell, C. W. & Green, D. S. Effects of microplastics in soil ecosystems: above and below ground. Environ. Sci. Technol. 53, 11496–11506 (2019).
Zhou, Q. et al. Persistent versus transient, and conventional plastic versus biodegradable plastic? -Two key questions about microplastic-water exchange of antibiotic resistance genes. Water Res. 222, 118899 (2022).
Lu, X. M., Lu, P. Z. & Liu, X. P. Fate and abundance of antibiotic resistance genes on microplastics in facility vegetable soil. Sci. Total Environ. 709, 136276 (2020).
Li, C. et al. “Microplastic communities” in different environments: differences, links, and role of diversity index in source analysis. Water Res. 188, 116574 (2021).
Yang, G. et al. Multiple anthropogenic pressures eliminate the effects of soil microbial diversity on ecosystem functions in experimental microcosms. Nat. Commun. 13, 4260 (2022).
Zhang, Z. et al. Assessment of global health risk of antibiotic resistance genes. Nat. Commun. 13, 1553 (2022).
Liu, B., Zheng, D., Zhou, S., Chen, L. & Yang, J. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D9172022 (2022).
Lee, Y. J. et al. The PathoChip, a functional gene array for assessing pathogenic properties of diverse microbial communities. ISME J. 7, 1974–1984 (2013).
Liang, J. et al. Identification and quantification of bacterial genomes carrying antibiotic resistance genes and virulence factor genes for aquatic microbiological risk assessment. Water Res. 168, 115160 (2020).
Rillig, M. C. et al. Increasing the number of stressors reduces soil ecosystem services worldwide. Nat. Clim. Chang. 13, 478–483 (2023).
Smith, P. et al. Global change pressures on soils from land use and management. Glob. Chang. Biol. 22, 1008–1028 (2016).
Rillig, M. C., Ryo, M. & Lehmann, A. Classifying human influences on terrestrial ecosystems. Glob. Chang. Biol. 27, 2273–2278 (2021).
Komárek, M., Čadková, E., Chrastný, V., Bordas, F. & Bollinger, J. C. Contamination of vineyard soils with fungicides: a review of environmental and toxicological aspects. Environ. Int. 36, 138–151 (2010).
Monkiedje, A., Ilori, M. O. & Spiteller, M. Soil quality changes resulting from the application of the fungicides mefenoxam and metalaxyl to a sandy loam soil. Soil Biol. Biochem. 34, 1939–1948 (2002).
Carboni, M., Livingstone, S. W., Isaac, M. E. & Cadotte, M. W. Invasion drives plant diversity loss through competition and ecosystem modification. J. Ecol. 109, 3587–3601 (2021).
Chen, C., Chen, H. Y. H., Chen, X. & Huang, Z. Meta-analysis shows positive effects of plant diversity on microbial biomass and respiration. Nat. Commun. 10, 1332 (2019).
Rillig, M. C. et al. The role of multiple global change factors in driving soil functions and microbial biodiversity. Science 366, 886–890 (2019).
Li, J. et al. Antibiotic treatment drives the diversification of the human gut resistome. Genom. Proteom. Bioinform. 17, 39–51 (2019).
Crits-Christoph, A. et al. Soil bacterial populations are shaped by recombination and gene-specific selection across a grassland meadow. ISME J. 14, 1834–1846 (2020).
Gao, F. & Zhang, C. T. GC-Profile: a web-based tool for visualizing and analyzing the variation of GC content in genomic sequences. Nucleic Acids Res. 34, W686–W691 (2006).
Bohlin, J., Eldholm, V., Pettersson, J. H., Brynildsrud, O. & Snipen, L. The nucleotide composition of microbial genomes indicates differential patterns of selection on core and accessory genomes. BMC Genom. 18, 151 (2017).
Sheridan, E. A. et al. Plastic pollution fosters more microbial growth in lakes than natural organic matter. Nat. Commun. 13, 4175 (2022).
Zhao, Y. et al. Effects of polyethylene microplastics on the microbiome and metabolism in larval zebrafish. Environ. Pollut. 282, 117039 (2021).
Meng, L. et al. The effect of bacterial functional characteristics on the spread of antibiotic resistance genes in Expanded Granular Sludge Bed reactor treating the antibiotic wastewater. Ecotoxicol. Environ. Saf. 225, 112714 (2021).
Ren, Z. et al. P, N, and C-related functional genes in SBR system promoted antibiotics resistance gene transmission under polystyrene microplastics stress. Water Res. 235, 119884 (2023).
Algavi, Y. M. & Borenstein, E. A data-driven approach for predicting the impact of drugs on the human microbiome. Nat. Commun. 14, 3614 (2023).
Luo, G. et al. Determining the contribution of micro/nanoplastics to antimicrobial resistance: challenges and perspectives. Environ. Sci. Technol. 57, 12137–12152 (2023).
Mah, T. F. et al. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426, 306–310 (2003).
Li, R. et al. Impact of urbanization on antibiotic resistome in different microplastics: evidence from a large-scale whole river analysis. Environ. Sci. Technol. 55, 8760–8770 (2021).
Jia, Y., Wang, Z., Zhu, S., Wang, Z. & Liu, Y. Disinfectants facilitate the transformation of exogenous antibiotic resistance genes via multiple pathways. Ecotoxicol. Environ. Saf. 253, 114678 (2023).
Liu, H. et al. Response of soil dissolved organic matter to microplastic addition in Chinese loess soil. Chemosphere 185, 907–917 (2017).
Dong, Y., Gao, M., Qiu, W. & Song, Z. Effect of microplastics and arsenic on nutrients and microorganisms in rice rhizosphere soil. Ecotoxicol. Environ. Saf. 211, 111899 (2021).
Kaur, K. et al. Microplastic-associated pathogens and antimicrobial resistance in environment. Chemosphere 291, 133005 (2022).
Wang, J. et al. Evidence of selective enrichment of bacterial assemblages and antibiotic-resistant genes by microplastics in urban rivers. Water Res. 183, 116113 (2020).
Fackelmann, G. et al. Current levels of microplastic pollution impact wild seabird gut microbiomes. Nat. Ecol. Evol. 7, 698–706 (2023).
Bandopadhyay, S. et al. Soil microbial communities associated with biodegradable plastic mulch films. Front. Microbiol. 11, 587074 (2020).
Shi, Z., Xiong, L., Liu, T. & Wu, W. Alteration of bacterial communities and co-occurrence networks as a legacy effect upon exposure to polyethylene residues under field environment. J. Hazard. Mater. 426, 128126 (2022).
Zhang, Q. et al. Gammaproteobacteria, a core taxon in the guts of soil fauna, are potential responders to environmental concentrations of soil pollutants. Microbiome 9, 196 (2021).
Li, W. et al. Variations in soil bacterial composition and diversity in newly formed coastal wetlands. Front. Microbiol. 9, 3256 (2018).
Zhao, Y., Gao, J., Wang, Z., Dai, H. & Wang, Y. Responses of bacterial communities and resistance genes on microplastics to antibiotics and heavy metals in sewage environment. J. Hazard. Mater. 402, 123550 (2021).
Yan, L. et al. Bacterial plasmid-mediated quinolone resistance genes in aquatic environments in China. Sci. Rep. 7, 40610 (2017).
Wei, Z. et al. Organic fertilizer potentiates the transfer of typical antibiotic resistance gene among special bacterial species. J. Hazard. Mater. 435, 128985 (2022).
Gillings, M. et al. The Evolution of Class 1 Integrons and the rise of antibiotic resistance. J. Bacteriol. 190, 5095–5100 (2008).
Yan, W. et al. Antibiotic resistance genes are increased by combined exposure to sulfamethoxazole and naproxen but relieved by low-salinity. Environ. Int. 139, 105742 (2020).
Muenmee, S., Chiemchaisri, W. & Chiemchaisri, C. Enhancement of biodegradation of plastic wastes via methane oxidation in semi-aerobic landfill. Int. Biodeterior. Biodegrad. 113, 244–255 (2016).
Martínez-Campos, S. et al. Early and differential bacterial colonization on microplastics deployed into the effluents of wastewater treatment plants. Sci. Total Environ. 757, 143832 (2021).
Chen, B. et al. Global distribution of marine microplastics and potential for biodegradation. J. Hazard. Mater. 451, 131198 (2023).
Hansen, P., von Bargen, K., Jünger-Leif, A. & Haas, A. Laboratory plasticware induces expression of a bacterial virulence factor. mSphere 7, e00311–e00322 (2022).
Pan, Y. et al. Coexistence of antibiotic resistance genes and virulence factors deciphered by large-scale complete genome analysis. mSystems 5, e00821–19 (2020).
Song, J. et al. A novel bidirectional regulation mechanism of mancozeb on the dissemination of antibiotic resistance. J. Hazard. Mater. 455, 131559 (2023).
Latz, E. et al. Plant diversity improves protection against soil-borne pathogens by fostering antagonistic bacterial communities. J. Ecol. 100, 597–604 (2012).
de Souza Machado, A. A. Impacts of microplastics on the soil biophysical environment. Environ. Sci. Technol. 52, 9656–9665 (2018).
Liu, S. et al. Thermal aging of polystyrene microplastics within mussels (Mytilus coruscus) under boiling and drying processing. J. Hazard. Mater. 465, 133282 (2024).
Wang, Y. F. et al. The ecological clusters of soil organisms drive the ecosystem multifunctionality under long-term fertilization. Environ. Int. 161, 107133 (2022).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014).
Cantarel, B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–D238 (2009).
Yin, X. et al. ARGs-OAP v2.0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 34, 2263–2270 (2018).
Yin, X. et al. ARGs-OAP v3.0: Antibiotic-resistance gene database curation and analysis pipeline optimization. Engineering 27, 234–241 (2023).
Zhang, A. N. et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 12, 4765 (2021).
Brown, C. L. et al. mobileOG-db: a manually curated database of protein families mediating the life cycle of bacterial mobile genetic elements. Appl. Environ. Microbiol. 88, e0099122 (2022).
Nissen, J. N. et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 39, 555–560 (2021).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Matthew, R. O. et al. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat. Biotechnol. 39, 727–736 (2021).
Wang, C. et al. Bacterial genome size and gene functional diversity negatively correlate with taxonomic diversity along a pH gradient. Nat. Commun. 14, 7437 (2023).
Liao, J. et al. Microdiversity of the vaginal microbiome is associated with preterm birth. Nat. Commun. 14, 4997 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22193062 to D.Z. and 42307169 to Y.-F.W.), China Postdoctoral Science Foundation (BX20220289 and 2022M723081 to Y.-F.W.), Ningbo Yongjiang Talent Project (2022A-163-G to D.Z.), Ningbo S&T project (2021-DST-004 to D.Z.) and Youth Innovation Promotion Association, Chinese Academy of Sciences (2023321 to D.Z.).
Author information
Authors and Affiliations
Contributions
All authors contributed intellectual input and assistance to this study. D.Z. and M.Q. conceived and designed the experiments. Y.-F.W., Y.-J.L., and Y.-M.F. carried out the experiments. Y.-F.W., J.-Y.X. and T.-L.Z. analyzed the data. Y.-F.W. and H.-L.C. prepared the figures. Y.-F.W. wrote the first draft, and M.-C.R., Y.-G.Z., M.Q., and D.Z. reviewed and commented on the paper. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Olesia Dulia, Jennifer D. Rocca, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, YF., Liu, YJ., Fu, YM. et al. Microplastic diversity increases the abundance of antibiotic resistance genes in soil. Nat Commun 15, 9788 (2024). https://doi.org/10.1038/s41467-024-54237-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54237-7
This article is cited by
-
Effects of aging treatments on the biofilm formation on low density polyethylene microplastics in constructed wetland soils
Biology and Fertility of Soils (2026)
-
Soil microbiomes in urban green spaces: Foundation of ecosystem functions and human health
Soil Ecology Letters (2026)
-
Systematic review of microplastics and heavy metals co-contamination on soils: The triple-chain effects of soil properties, crop responses, and biological communities
Plant and Soil (2026)
-
Effects of predatory mite biocontrols on the dispersal of antibiotic resistomes and virulence factors in tea garden soils
Microbiome (2025)
-
Unraveling antibiotic resistance dynamics at the soil–plant interface under climate change for One Health
One Health Advances (2025)
