
Abstract
In recent years, the application of deep learning models to protein-ligand docking and affinity prediction, both vital for structure-based drug design, has garnered increasing interest. However, many of these models overlook the intricate modeling of interactions between ligand and protein atoms in the complex, consequently limiting their capacity for generalization and interpretability. In this work, we propose Interformer, a unified model built upon the Graph-Transformer architecture. The proposed model is designed to capture non-covalent interactions utilizing an interaction-aware mixture density network. Additionally, we introduce a negative sampling strategy, facilitating an effective correction of interaction distribution for affinity prediction. Experimental results on widely used and our in-house datasets demonstrate the effectiveness and universality of the proposed approach. Extensive analyses confirm our claim that our approach improves performance by accurately modeling specific protein-ligand interactions. Encouragingly, our approach advances docking tasks state-of-the-art (SOTA) performance.
Similar content being viewed by others
Introduction
In the intricate journey of drug development, protein-ligand docking and affinity prediction tasks have been important components of the drug discovery process for years1,2. As a crucial task in optimization of drug molecular structures, protein-ligand docking is to predict the position and orientation of a ligand (a small molecule) when it binds to a protein receptor or enzyme. Leveraging accurate binding poses (protein-ligand binding complex conformations), the affinity prediction task offers a computational estimation of the binding strength between a ligand and its target protein, facilitating the screening of ligand with potential affinity.
Recent years have witnessed a surge of interest in using deep learning (DL) approaches for molecular modeling3,4. Treating docking as a generative modeling problem, ref. 5 introduced DiffDock, a graph neural network (GNN) based model that has established a benchmark in binding pose generation. However, the existing DL models often overlook the modeling of non-covalent interactions between protein and ligand atoms, which is essential for interpretability and generalization. As illustrated in Fig. 1 Left, the docking conformation produced by DiffDock remains closely resembles crystal structures but fails to capture the non-covalent interactions. Moreover, although traditional affinity prediction methods excel with crystal structures, their performance dramatically drops when dealing with the less precise binding pose, posing challenges for real-world applications6,7,8.

Left: The incorrect case generated by DiffDock. Pink and orange arrows indicate improperly formed hydrogen bond and hydrophobic interactions (RMSD: 1.13, PDB ID: 6QMT). Right: The correct binding pose generated by Interformer, where the predicted interaction energy function recovers almost all hydrogen bonds and hydrophobic interactions (RMSD: 0.67).
In this work, we present Interformer, a computational AI model designed to alleviate the interaction-aware problems on the protein-ligand docking and employs constructive learning for affinity prediction in real-world applications. Firstly, we propose an interaction-aware mixture density network (MDN) to model non-covalent interactions, explicitly focusing on the hydrogen bonds and hydrophobic interactions present in the protein-ligand crystal structure. As illustrated in Fig. 1 Right, Interformer can accurately produce specific interactions in the binding pose. Secondly, we introduce a pseudo-Huber loss function, leveraging the capabilities of contrastive learning to instruct the model in discriminating between favorable and unfavorable binding poses. Thirdly, the proposed model is based on the Graph-Transformer framework9,10, which has demonstrated its superior performance compared to GNN-based models in various graph representation learning tasks11. An additional advantage of Interformer is to interpret the internal mechanisms of protein-ligand interactions by examining the fusion coefficients of the MDN12. When evaluated on protein-ligand docking using two widely-used benchmarks, Interformer achieves a top-1 prediction performance with 84.09% accuracy on the Posebusters benchmark and 63.9% on the PDBbind time-split benchmark with the Root Mean Square Deviation (RMSD) less than 2 Å. The improvement is attributed to the model’s enhanced ability to capture non-covalent interactions between ligands and proteins, which is crucial for generating less ambiguous conformations and essential for successful performance in downstream tasks. Furthermore, the interformer can predict plausible affinity values even when the binding poses are less accurate. Evaluation of our in-house real-world benchmark demonstrates comparable performance to the other models, confirming its pose-sensitive and robust generalization capabilities. While applying to a real-world internal pharmaceutical pipeline, we successfully identify two small molecules, each with affinity IC50 values of 0.7 nM and 16 nM in their respective projects, thus demonstrating its practical value in advancing therapeutic development.
Results
The Interformer model
Interformer is a deep learning model trained on crystal structure data for protein-ligand docking task, and it redocks docking poses along with corresponding affinity values for the affinity prediction task. Its architecture is inspired by the Graph-Transformer, initially proposed for graph representation learning tasks.
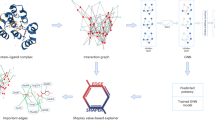
Firstly, the model takes a single initial ligand 3D conformation and protein binding site from the crystal structure as input. Graphs are widely used in various methods to illustrate ligands and proteins, as shown in Fig. 2a, in which nodes represent atoms and edges indicate the proximity between two atoms. We employ pharmacophore atom types13 as node features and use the Euclidean distance between two atoms as edge features. These pharmacophore atom types provide essential chemical information, thus enabling the model to better comprehend specific interactions such as hydrogen bonding or hydrophobic interactions. A more detailed description of these features is provided in Supplementary Table S3.

a Graph representation, atoms are represented as nodes, and the proximity between two atoms is represented as edges. b Docking pipeline, utilizes node and edge features as inputs, which are processed through Intra and Inter Blocks to update features. These features are then fed into an edge output layer to produce an inter representation, for predicting an interaction-aware MDN. Subsequently, a Monte Carlo sampling procedure utilizes the energy score function to sample multiple docking poses. c Pose Score and Affinity prediction pipeline, leverages the docking pose generated in (b) to update new edge features, which are then fed into Intra and Inter Blocks to update node features. Finally, a virtual node aggregates all node features to predict a pose score and affinity value for the corresponding docking pose.
In the second stage, the docking pipeline, as illustrated in Fig. 2b, processes the node features and edge features from the protein and ligand through Intra-Blocks. Intra-Blocks are designed to update node features for each atom by capturing the intra-interactions within the same molecule. These updated node features are then fed into Inter-Blocks, which are designed to capture inter-interactions between protein and ligand atom pairs, leading to further updated node and edge features. The edge output layer subsequently combines these two sets of features to generate an Inter-representation for each protein-ligand atom pair. Subsequently, the Inter-representation is processed by an Interaction-aware MDN. This network predicts the parameters of four Gaussian functions for each protein-ligand atom pair, which are constrained separately by different possible specific interactions. The first two Gaussian functions encapsulate all types of pair interactions, while the third one signifies the hydrophobic interactions and the fourth one denotes the hydrogen bond interactions. By integrating these four Gaussian functions, we derive a mixture density function (MDF), representing the conditional probability density function of distance for any given protein-ligand atom pair. This MDF can serve as an energy function to estimate the most probable distance between the protein atom and its corresponding ligand atom. Hydrogen bonds and hydrophobic interactions play a significant role in the binding free energy14. The distribution of these specific interactions differs markedly from that of other typical interactions. We designed individual modeling of each specific interaction distribution, such as the third term for hydrophobic and the fourth term for hydrogen bonding. Consequently, the docking poses generated by our MDF inherently display these specific interactions, much like most natural crystal structures. In contrast, other methods like refs. 15, 16 utilize ten MDFs to model all types of protein-ligand pairs, thereby mixing all types of pairs and overlooking the importance of specific interactions. Alternatively, the methods like DiffDock solely minimize the RMSD as the loss function, resulting in the docking process merely approximating the crystal structure rather than capturing critical specific interactions. Finally, the MDF of all protein-ligand pairs is aggregated into a sum of energy functions, which is then introduced into a Monte Carlo (MC) sampling method17 for generating top-k candidate ligand conformations relative to its protein target. The MC sampling initially positions the ligand in various locations within the protein binding sites and assigns random torsion angles, then seeks to minimize the given energy function with respect to the ligand conformation. By aggregating all candidates sorted by energy values, we can obtain top-k candidate docking poses. A more detailed description of the sampling method is provided in Supplementary Information Section 2.
In the third stage, pose score and affinity prediction pipeline is illustrated in Fig. 2c. The distances and specific interactions between protein and ligand atoms from the generated docking pose update new edge features. The node and edge features are then processed through Intra and Inter-Blocks to create implicit interactions. A virtual node collects all the information about the binding pose through the self-attention mechanism. Finally, the binding embedding of a virtual node is fed into the affinity and pose layer to predict the binding affinity value and the confidence pose score for the corresponding docking pose. By incorporating poor poses, a contrastive pseudo-Huber loss function is utilized to guide the model in discerning whether a pose is good or poor. The training objective ensures that the model predicts a lower value for the poor pose and a higher value for the good pose. The primary distinction between the good and poor pose lies in their interactions. This strategy assists the model in learning crucial interactions rather than artificial features. This characteristic, we refer to as pose-sensitive, has demonstrated superior performance in real-world drug development projects.
Performance for protein-ligand docking accuracy
We use the PDBBind time-split test set to determine the success rate of docking poses with an RMSD of less than 2 Å, initially introduced in the DiffDock paper5. We examine two primary scenarios for docking: the first involves providing the entire protein structure as input, termed “blind docking”, and the second is based on a known reference ligand, from which we extract nearby residues using a distance cut-off, termed “pocket residues specified”. Table 1 illustrates the results that our proposed method significantly surpass all previous methods, achieving a top-1 success rate of 63.9%, significantly higher than the SOTA methods, DiffDock and GNINA18 on both scenarios. Including a pose score model, the top-1 success rate decreases to 62.1%. Despite the seemingly decreased RMSD, the selected poses tend to exhibit more accurate specific interactions. A more detailed discussion of this observation is provided in Section “Results”.
Moreover, we consider a recently published PoseBusters benchmark19, which emphasizes the importance of physical plausibility in docking simulations. We evaluate our proposed method against this benchmark, and the results are shown in Fig. 3a. In the conventional docking sampling procedure adopted from the preceding studies, such as those by Vina and other methods5,16,17,20, the initial ligand conformation is based on the reference structure (the ligand structure from crystal structure), but with random position and torsion angles for benchmark evaluations. To ensure fairness, we report two sets of results, each using either the reference or starting conformation (provided by PoseBusters benchmark) as the initial ligand conformation during docking pose sampling. Noteworthy, we apply the starting conformation as the input for energy function prediction in both sets of results to make sure no data leakage. Our method significantly outperforms various SOTA AI and traditional models, achieving a success rate of 84.09%. Nonetheless, 7.8% of the generated poses do not pass the posebusters-validity check, primarily due to steric clashes between the protein and ligand atoms. Despite this, our method notably mitigates this issue of physical plausibility compared to all other AI models. A significant performance decrease is observed when using the starting conformation. We attribute this primarily to incorrect stereochemistry, specifically wrong chiral centers and inaccuracies in the force field. Once these issues are rectified, the performance should align with the result of using reference ligand.

a The bar plot of the successful docking rate on PoseBusters version 2 benchmark, * denotes using the provided start conformation as initial ligand sampling conformation. b The docking performance bar plot on three levels of homology sequence similarity subsets. c The histogram depicts the number of specific interactions recovered by various methods within the five recovery rate ranges. d The predicted five MDN distributions, the x-axis is the VdW radius distance of the atoms pair d, and the y-axis is the probability of the MDN. Yellow is a hydrogen bond pair, orange is hydrophobic, and brown are other non-interaction pairs. e The upper diagram illustrates the predicted fusion coefficients α for the hydrogen bond interaction between the 20th ligand atom (a hydrogen bond acceptor) and all other protein atoms that are hydrogen bond donors. The model predicts the top-2 highest α value for the true hydrogen bonds on 20-143 and 20-280. A similar analysis applies to the lower diagram but for another hydrophobic pair set. f Two docking poses were generated by Interformer. The white represents the crystal structures, the green indicates the docking poses, and the yellow dashed lines signify the hydrogen bond interactions. As can be observed, the poses can directly form hydrogen bond interactions.
For a more comprehensive understanding of the generalizability of our method, we assess the maximum protein sequence similarity to the training set for each protein within the PDBBind time-split test set. Subsequently, we divide the test set into three subsets, each indicative of low, medium, and high levels of homology. We evaluate the docking accuracy within these subsets, and the results are shown in Fig. 3b. Interformer with pose score achieved an accuracy rate of 63.4% on the low homology subset. This result demonstrates that the model does not merely recall the location of other homologous proteins in the training set but also has the capability of identifying the actual binding position. For the medium and high homology subsets, Interformer with pose score achieves accuracy rates of 56.7% and 63.5%, respectively.
Interformer captures specific interactions of molecule docking
The primary objective of most deep learning methods is to minimize the RMSD between the docked ligand and the crystal ligand. However, the generated docking poses tend to closely resemble the crystal ligand rather than adhering to physical principles such as non-covalent interaction. The sample we have previously provided demonstrates the ability of our proposed method to predict a reasonable energy function. For a quantitative analysis, we identify the number of the same hydrogen and hydrophobic pairs presented both in the crystal structure and the docking poses. We use the Protein-Ligand Interaction Profiler (PLIP)21 for evaluation, an open-source tool that determines the formation of specific interactions based on physical rules. Upon the evaluation of the PDBBind time-split test set, DiffDock and DeepDock were only capable of recovering an average of 29.42%, 23.55% of hydrogen bonds and 19.36%, 16.26% of hydrophobic interactions. In contrast, Interformer with pose score could recover an average of 57.25% of hydrogen bonds and 43.7% of hydrophobic interactions. However, the average recovery rate slightly decreases to 52.7% and 41.6% for hydrogen bonds and hydrophobic interactions, respectively, without the pose score. This finding suggests that the pose score model can further enhance the accuracy of specific interactions. Therefore, we propose using the docking pose selected by the pose score model instead of the energy model. For a more comprehensive view of this statistic, Fig. 3c displays a histogram that outlines the count of different hit rate ranges. The results further confirm that Interformer relies on non-covalent interactions rather than a simplistic data-driven strategy.
Visualization of energy score function
In drug development processes such as small molecule optimization or virtual screening, it is essential to consider specific protein-ligand interactions as shown in Fig. 3e, the predicted fusion coefficients, denoted as α, are shown for all potential hydrogen bond and hydrophobic pairs. The α values control the weight of the hydrogen bond and hydrophobic terms. The Interformer accurately predicts various interactions between the ligand and protein atoms. Specifically, it identifies the existence of two hydrogen bonds that occur between ligand atom 20 and protein atoms 143 and 280. In addition, it also predicts the presence of hydrophobic interactions between ligand atom 25 and protein atoms 66, 186, and 232. Figure 3d presents the five predicted MDNs. The x-axis represents the probability in this figure, while the y-axis corresponds to the Van der Waals radius distance between two atoms, denoted as d. The MDNs associated with the actual hydrogen bond and hydrophobic pairs are noticeably positioned on the left side of the figure, suggesting the specific interactions and close distance between the two atoms. On the contrary, the remaining three MDNs, which represent potential hydrophobic pairs, hydrogen bonds, and other pairs, are positioned on the right side of the figure. These three MDNs imply that these atom pairs are likely to be relatively distant from each other and are not expected to exhibit any specific interactions. The result indicates that the Interformer can effectively predict reasonable MDNs. Figure 3f showcases two examples of binding poses generated by the Interformer, which exhibit direct hydrogen bonds and hydrophobic interactions. These binding poses can be forwarded seamlessly to Computer-Aided Drug Design software, such as MMGBSA and FEP22,23, without requiring any post-optimization procedures like force-field minimization with OPLS4 or AMBER24,25.
Negative samples enhance pose-sensitive on affinity prediction
Most methods perform well in predicting affinity based on crystal structure. However, it is hard to obtain such realistic conformation in practical drug development scenarios. The predictions based on poor binding poses often lead to substantial overfitting. To address this issue, we incorporate bad binding poses (negative samples) into the training set and employ a contrastive loss mechanism, as detailed in Method Affinity Module. This strategy brings an ability termed as “pose-sensitivity” that can help to distinguish the different interactions between good and bad poses rather than memorizing the shape of the ligand or binding sites. We evaluate two strategies for our model: one that includes negative samples and another that solely utilizes the crystal structures without incorporating them. On the PDBBind time-split test set, the affinity model using only crystal structures in the training presents a Pearson correlation coefficient R = –0.174 between the predicted affinity value and the RMSD. However, when negative samples are used, the affinity model achieves a correlation of R = –0.562, and the pose score model achieves a higher correlation of R = –0.659. Additionally, Fig. 4a demonstrates that the affinity model without negative samples maintains consistent predictions for both good and poor binding poses because the model does not leverage any non-covalent interaction features. In contrast, when negative samples are incorporated, both affinity and pose score models predict lower values for binding poses with larger RMSD values. The result showcases the capacity of the Interformer to distinguish between good and poor poses.

a We utilize RMSD intervals of 1 Å to calculate the average prediction values of all conformations for various methods. PoseRank is determined by sorting the energy from 1 to 10. To facilitate display, we multiplied the predicted values of PoseScore by 10. b The box plot presents the affinity prediction performance across four real-world test sets, categorized by the maximum similarity of ligands found in the training set. The ChEMBL-Kinase dataset (n = 2539), and the Mpro covalent test (n = 142), are derived from ChEMBL and patent databases. The LSD1 project (n = 55) and the Mpro project (n = 22) originate from our internal real-world drug design projects. c (left) The most potent compound, Cpd27 in the LSD1 project achieves an affinity of 0.7 nM. c (middle, right) The correlation plot between the Interformer predicted affinity value and experimental pIC50 in LSD1 project and prediction of Interformer without negative samples for training. d (left) The most potent compound, TAD 6-ref, in the Mpro project achieves an affinity of 16 nM. d (middle, right) The correlation plot in the Mpro project and prediction of Interformer without negative samples for training. e (left) The docking pose of Cpd27 aligns with the PDB ID: 6W4K, it holds crucial hydrogen bond interaction with LYS-661 and creating two additional hydrogen bond interactions with GLN-358 and ALA-539. e (right) The docking pose of TAD 6-ref aligns with the PDB ID: 7RFS and is designated to form a macrocyclic to stabilize the ligand. For the box plots in (b), the lower limit represents lower quartile, the center line represents the median and the upper limit represents the upper quartile. The whiskers do not include outliers. For the regression plot in 4c and 4d, the blue line indicates linear regression fit. The light-blue region indicates the corresponding 95% confidence interval computed via bootstrapping mean.
Evaluation of affinity prediction on real-world test sets
In drug development, the accurate prediction of binding poses holds significant importance. However, it is equally crucial to predict the affinity based on these poses, as it directly influences the performance of virtual screening and small molecule optimization processes. As the CASF2016 benchmark is often used for comparison, there is a question of the persuasiveness of the benchmark because many models tend to overfit this test set. Therefore, we consider using our internal test set for affinity performance comparison as follows:
-
ChEMBL-Kinase test: We curated data solely from the kinase family available on ChEMBL26. The data points for each target should be no less than 30, resulting in 27 distinct protein targets and 2539 data points. The final assessment was based on the average affinity correlation across each target.
-
LSD1 project: Our in-house pharmaceutical pipeline is guided by the Interformer model. It focuses on optimizing small molecules for the LSD1 target27. The project produced 54 small molecules, with the most potent one achieving an affinity level of 0.7 nM.
-
Mpro covalent test: We sourced data on small molecules involving four types of covalent bonds from patents for the SARS-CoV-2 main protease (Mpro) target28, yielding 142 data points. The final assessment was based on the average affinity correlation for each covalent bond type.
-
Mpro project: Our in-house pharmaceutical pipeline, guided by the Interformer model, concentrates on optimizing covalent-type small molecules for the SARS-CoV-2 Mpro target29,30. This project has developed nine small molecules, with the most potent one achieving an affinity level of 16 nM. We have also incorporated 12 small molecule data points from the Shionogi31 publication for a comprehensive evaluation.
Table 2 presents the performance of various models on these four real-world internal test sets. All conformations in these test sets, except for the large kinase test set, were generated by various docking programs and verified by humans. On average, of all test sets, the affinity module that does not utilize negative samples training achieves a correlation of 0.124, while the affinity module incorporating negative samples reaches a correlation of 0.454. Within the ChEMBL-Kinase dataset, the Interformer model achieves a correlation coefficient of 0.229, presenting a comparative performance with the SOTA GNINA model. In the LSD1 internal test set, our method achieves a correlation of 0.523, outperforming GNINA. In the Mpro covalent test, we did not test GNINA as it can not handle docking with covalent bonds, and our method achieves a correlation of 0.460, outperforms CovDock32,33. In the Mpro project test set, our method significantly outperforms CovDock by a correlation of 0.604. Due to pose sensitivity, the model substantially outperforms models trained only on crystal structures in terms of generalization.
In order to demonstrate effectively the practical applicability of our test set, we utilize MMSeq2 to assess the maximum protein sequence similarity within our internal test sets. The average protein sequence similarity are found to be 82.2% for Kinase, 100% for LSD1, and 96% for Mpro. While Mpro shares homology with the SARS-CoV 3C-like protease, it presents notable differences within the binding pocket site. Despite similar homologous proteins in the PDBBind training set, we further examine the ligand similarity using Tanimoto Similarity of Morgan Fingerprint. As depicted in Fig. 4b, the Kinase and Mpro covalent test sets, which are sourced online, exhibit a similarity median of 38% and 50%, respectively. In contrast, the molecules derived from our internal drug development demonstrate a lower homology to the training set, with a similarity median of 33% and 28%.
For reference, on the Public Benchmark CASF2016, as shown in Supplementary Information Table S5, the affinity model also demonstrates strong predictive capabilities, achieving a performance of Pearson correlation coefficient R=0.809 and R=0.810 when predicting crystal structures and docking poses.
Application to real world drug design
In order to validate the effectiveness of Interformer in real-world scenarios and to demonstrate our team’s drug development capabilities, we have independently developed two drug development pipelines. Both projects involve small molecule optimization, where a list of candidate small molecules is designed by medicinal chemistry experts based on the crystal structures and binding modes of reference small molecules. These candidates are then ranked by Interformer for affinity scoring and subjected to various ADMET molecule property prediction models to ensure that the final designed small molecules perform well in terms of properties and affinity.
In early 2022, we initiated our first project targeting LSD127,34, a potential therapeutic target for cancer. We developed two series of small molecules, resulting in 54 compounds. The most potent demonstrates an affinity of 0.7 nM, as shown in Fig. 4c (left). This compound (Cpd 27 pg 64) was subsequently evaluated for metabolic stability in oral and intravenous mouse trials, exhibiting half-lives (t1/2) of 5.86 and 8.33 h and AUC(0-t) of 6,528 and 11,502 h.ng/mL, respectively. Amid the COVID-19 pandemic that swept across China in late 2022, our focus shifted towards the widely recognized SARS-CoV-2 viral, main protease (Mpro)29,30. We adopted a strategy of macrocyclic modifications35 to small molecules and designed nine small molecules. The most potent among these exhibit an affinity of 16 nM, as shown in Fig. 4d (left). The correlation between actual pIC50 and predicted pIC50 value of Interformer in the LSD1 and Mpro projects was 0.523 and 0.604, respectively, as shown in Fig. 4c (middle) and 4d (middle). Figure 4c (right) and 4d (right) display the correlation of affinity prediction models trained exclusively on crystal structures, which are a mere 0.330 and 0.097. These real-world pharmaceutical scenarios underscore that models trained solely on crystal structures deliver subpar performance when the actual docking pose may not be precise.
In our LSD1 project, we undertake a structure-activity relationship (SAR) of the docking pose, and analyze the crystal structure of a reference small molecule PDB ID: 6W4K36. It is crucial to consider the presence of another small molecule, the co-factor FAD, within the pocket, as it could interact with the inhibitor. As shown in Fig. 4e (left), the Cpd27 maintains the critical hydrogen bond interaction with LYS-661 of the reference molecule while modifying other functional groups to form two additional hydrogen bond interactions with GLN-358 and ALA-539, both metabolic stability property and affinity are better than the reference molecule. Within our Mpro project, we analyze the crystal structure of a reference small molecule PDB ID: 7RFS37. This inhibitor is a covalent inhibitor. Hence, our series of small molecules also have to consider the positioning of the warhead nitrile group, which must form a covalent bond with CYS-145. Our strategy involve opening the five-membered ring of the reference small molecule to form a macrocyclic modification, thereby circumventing chemical patent protection. As shown in Fig. 4e (right), the TAD 6-ref, a macrocyclic molecule with a six-carbon length, is spatially reasonable through SAR analysis. Nearly all other interactions remain consistent with the reference molecule. Finally, this approach enable us to break through patent protection and find a small molecule that achieves a comparative affinity result with 16 nM.
Discussion
Determining the structure of protein-ligand complexes represents a significant challenge in the field of drug development.
In response, we have demonstrated Interformer, a deep-learning generative model specifically designed for protein-ligand docking and affinity prediction. This model integrates a powerful interaction-aware MDF that successfully recovers specific interactions at a high rate. Moreover, the mechanism underlying Interformer can be easily interpreted by users, effectively addressing the common shortfall in state-of-the-art deep learning models, which tend to overlook the critical non-covalent interactions.
Beyond the importance of elucidating binding modes in drug design, the capacity to rank or screen ligands based on their affinity for a particular target is equally critical. Recognizing that many state-of-the-art deep learning models are prone to over-fitting on crystal structures, Interformer employs a training strategy that leverages contrastive learning with negative sampling to enhance pose sensitivity. This approach enables Interformer to distinguish between less accurate and more favorable docking poses by focusing on the specific interactions between protein and ligand atom pairs. Such robust capability allows the model to enhance generalizability for predictions in real-world scenarios.
Interformer has demonstrated consistent improvements in protein-ligand docking across two widely recognized benchmarks, generating physically plausible and reasonable docking poses that enhance the potential for downstream applications. In the realm of affinity prediction, Interformer has shown consistent advancements on four in-house, real-world affinity benchmarks. Further application of Interformer within two internal drug development pipelines has led to the successful identification of two high-potency molecules at the nanomolar level.
Our study underscores the considerable potential of Interformer to impact computational biology and accelerate the drug design process. In the future, we aim to expand the application of Interformer to a broader spectrum of real-world biological challenges and to enhance its performance for diverse molecular interaction types, including protein-protein and protein-nucleic acid interactions. For additional considerations regarding future directions, please refer to Supplementary Information Section 3.
Methods
Preliminary
Graph representation in protein-ligand complex
Graph representation is well-suited for depicting the structure of a protein-ligand complex, as shown in Fig. 2a. A complex can be represented as a graph \({{{\mathcal{G}}}}=({{{\mathcal{V}}}},{{{\mathcal{E}}}})\), where \({{{\mathcal{V}}}}={{{{\mathcal{V}}}}}_{{{{\mathcal{L}}}}}+{{{{\mathcal{V}}}}}_{{{{\mathcal{P}}}}}\). Here \({{{{\mathcal{V}}}}}_{{{{\mathcal{L}}}}}\) is all atoms of the ligand, and \({{{{\mathcal{V}}}}}_{{{{\mathcal{P}}}}}\) = {\({v}_{j}| {v}_{i}\in {{{{\mathcal{V}}}}}_{{{{\mathcal{L}}}}},{v}_{j}\in {{{{\mathcal{V}}}}}_{{{{\mathcal{W}}}}}\,;{{{\mathcal{D}}}}({v}_{i},{v}_{j})\, < \, 7\) Å} denotes protein pocket atoms. \({{{{\mathcal{V}}}}}_{{{{\mathcal{P}}}}}\) is determined by the Euclidean distance \({{{\mathcal{D}}}}(\cdot )\) between reference ligand atoms and \({{{{\mathcal{V}}}}}_{{{{\mathcal{W}}}}}\) protein atoms. Furthermore, \({{{\mathcal{E}}}}=\{{e}_{ij}| {v}_{i},{v}_{j}\}\) denote the set of all edges between each pair of nodes. \({{{\bf{X}}}}\in {{\mathbb{R}}}^{n\times {d}_{x}}\) represent the node features, where Xi corresponding to the node features of the ith atom in the complex. The edge features are denoted as \({{{\bf{E}}}}\in {{\mathbb{R}}}^{n\times n\times {d}_{e}}\), where n denotes the number of nodes in \({{{\mathcal{G}}}}\). The edge feature \({e}_{LP}=\{{e}_{ij}| {v}_{i}\in {{{{\mathcal{V}}}}}_{{{{\mathcal{L}}}}},{v}_{j}\in {{{{\mathcal{V}}}}}_{{{{\mathcal{P}}}}}\}\) between ligand and protein pocket atoms are set to zero in the docking pipeline and to the Euclidean distance when a docking pose is available in the pose score and affinity prediction pipeline.
Graph-Transformer
Leveraging a powerful self-attention mechanism9, Graph-Transformer10,11 excels at learning node relationships and has achieved superior performance in various graph tasks. Graph-Transformer adds a bias term to the Multi-Head Self-Attention of Transformer9 to incorporate graph structure information. It takes the node features X and the edge features E as inputs, and the modified self-attention mechanism can be described as follows:
where \({{{\bf{Q}}}}\in {{\mathbb{R}}}^{n\times d}\), \({{{\bf{K}}}}\in {{\mathbb{R}}}^{n\times d}\), and \({{{\bf{V}}}}\in {{\mathbb{R}}}^{n\times d}\) are linear transformations of X, referred as queries, keys, and values. \({{{\bf{Z}}}}({{{\bf{E}}}})\in {{\mathbb{R}}}^{n\times n}\) is the bias term where Z is a linear layer that transforms the edge features E from the dimension de into dimension 1, when two nodes connects; otherwise we set it to − inf. \({{{\bf{A}}}}\in {{\mathbb{R}}}^{n\times n}\) is a matrix that captures the similarity between the queries Q and the keys K, A updates the query representation through a weighted sum of the values V. For simplicity of illustration, we consider the single-head self-attention mechanism in this literature.
Interformer architecture overview
Masked self-attention (MSA)
The classical self-attention mechanism9 permits each node to attend to all other nodes through attention weights, which cannot distinguish between the internal information of ligands and proteins and the information between them. Thus, we use Masked self-attention that restricts certain nodes to attend only to specific designated nodes by a mask M, which is shown in Fig. 5a and can be described as follows:
where M is applied to A through element-wise multiplication denotes by ⊙.

a Masked self-attention: is a modified self-attention mechanism that uses an attention mask M to restrict nodes from attending to only certain other nodes. Besides, it can incorporate edge features through element-wise addition. ⨀ denotes as Hadamard product; ⨁ denotes element-wise summation; ⊠ denotes dot product. b Edge Output Layer, a module integrates node and edge features into a comprehensive protein-ligand inter-representation. This representation is achieved by summing the node features from protein and ligand with the mean of a learned edge feature, thus forming a two-dimensional feature. Subsequently, the two-dimensional feature is input into a FFN to learn the inter-presentation, which is essential for predicting the MDN.
Intra-blocks
We propose Intra-Blocks to capture the intra-interactions of ligands and proteins better. The Intra-Blocks outputs updated node features \({{{\bf{H}}}}\in {{\mathbb{R}}}^{n\times d}\) through applying MSA to the node features X and the edge features E by an Intra-mask \({{{{\bf{M}}}}}^{{\prime} }\):
Inter-Blocks
To capture the inter-interactions and predict relationships between the atoms of ligand and protein, we employ an Inter-mask M″ to promote the information exchange between the atoms of ligand and protein:
The Inter-Blocks outputs updated edge features \({{{{\bf{E}}}}}^{{\prime} }\) by a residual of A from the previous layer, and both layer normalization (LN) and feed-forward network (FFN) are applied to A in each block. Finally, we obtain the updated node feature \({{{{\bf{H}}}}}^{{\prime} }\) by applying \({{{{\bf{E}}}}}^{{\prime} }\) with MSA.
Edge output layer for protein-ligand docking
To predict the energy between protein-ligand atom pairs, the fusion of node and edge features can provide a more comprehensive inter-feature. As shown in Fig. 5b, this can be described as follows:
where \({{{{\bf{E}}}}}^{o}={{{\rm{LN}}}}({{{{\bf{E}}}}}^{{\prime} }{{{\bf{W}}}})\), W is a learnable weight matrix. Initially, we compute the mean of protein-ligand pairs edge feature \({{{{\bf{e}}}}}_{ij}^{o}\) and \({{{{\bf{e}}}}}_{ji}^{o}\) from the semi-positive matrix Eo. Subsequently, this is enhanced through the addition of pair-wise node features, which are obtained by multiplying each ligand node feature \({{{{\bf{h}}}}}_{i}^{{\prime} }\) with every other protein node feature \({{{{\bf{h}}}}}_{j}^{{\prime} }\) from \({{{{\bf{H}}}}}^{{\prime} }\). At the end, we obtain the protein-ligand inter-features \({{{{\bf{H}}}}}^{a}\in {{\mathbb{R}}}^{| {{{{\mathcal{V}}}}}_{{{{\mathcal{L}}}}}| \times | {{{{\mathcal{V}}}}}_{{{{\mathcal{P}}}}}| }\) by applying a FFN.
Training
The training procedure consists of two stages. Initially, we train the energy model using the crystal structure, which is subsequently employed to generate negative sample poses. Following this, both positive and negative sample poses are used to train the pose score and affinity prediction model. The training objectives of the three modules will be elaborated in greater detail below (additional details related to training data, training protocol, ablation study and hyper-parameters can be found in Supplementary Information Section 1).
Interaction aware mixture density network
To model specific interactions and output an energy function for the sampling algorithm to generate binding poses, we predict the interaction-aware MDN of each pair of ligand \(i\in {{{{\mathcal{V}}}}}_{{{{\mathcal{L}}}}}\) and protein \(j\in {{{{\mathcal{V}}}}}_{{{{\mathcal{P}}}}}\) atoms using the inter-feature Ha. This MDN is formed by the weighted sum of four mixed Gaussian functions. The first two can be considered regular interaction forces, whereas the third represents hydrophobic interactions modeled exclusively between hydrophobic atom pairs. The fourth represents hydrogen bonding, which is modeled specifically between hydrogen bond donors and acceptors. Further details are provided as follows:
where the fusion coefficient α, mean μ and variance σ are learnable parameters of each Gaussian distribution \({{{\mathcal{N}}}}\) in the MDN for each pair of atoms, which obtained by applying a linear weight W to the inter-feature Ha. Given the VdW radius distance d, we can compute the probability p of each term in MDN. In the third term p3, α is non-zero only on hydrophobic \({{{\mathcal{H}}}}\) pair, and in the fourth term p4, α is non-zero only on hydrogen bond donor \({{{\mathcal{D}}}}\) and acceptor \({{{\mathcal{A}}}}\) pair. The final probability of each atom pair P is derived by summing four \({{{\mathcal{N}}}}\) multiplied by their respective α. Lastly, the model can be optimized through the negative log-likelihood loss function \({{{{\mathcal{L}}}}}_{MDN}\).
Pose score module
MC sampling generates multiple candidate docking poses based on energy function E (see Supplementary Information Section 2). However, the best pose may not necessarily be at the top of the list, necessitating an additional model to further re-rank the docking poses to enhance docking success rate. In this work, we employ a FFN on the virtual node feature v to predict p, which indicates whether the input binding pose is correct. This is determined by calculating if RMSD is less than 2 Å between the docking pose and the crystal pose. The training objective is a binary loss function.
Affinity module
Docking algorithms typically employ energy functions to determine the affinity value of a ligand13,17,38. These energy functions are generally composed of linear combinations, with the weights of these combinations optimized to minimize the energy of crystal structures rather than directly modeling the experimental affinity values. This practice could result in generating accurate docking poses, but it often falls short in effectively ranking the affinity of the ligands. To address this challenge, we employ a FFN on the virtual node feature v to predict the experimental affinity value y. Affinity units used are IC50, Kd, and Ki, which are normalized by taking the negative logarithm, with higher values indicating stronger affinities. The training objective is a contrastive pseudo-Huber loss function, which ensures the model predicts a lower value for a poor pose and a higher value for a good pose. The primary difference between these two poses lies in their interactions. This strategy aids the model in learning essential interactions rather than artificial features, which we refer to as pose-sensitivity as follows:
where \(\widehat{y}\) is the predicted affinity value, we select Huber loss hyperparameter σ as 4.
Join training further enhance pose-sensitive by virtual node
We introduce an extra virtual node v to represent the entire information of the binding pose. v is learnable and is connected to all other nodes within the graph \({{{\mathcal{G}}}}\) via a mask Mv in Inter-Blocks as follows:
Pose Score and our contrastive learning affinity module share the same purpose of determining the correctness of a binding pose. Given the strong complementarity between these two tasks, we choose a single model, based on the same virtual node v, to train and predict both the pose score and affinity tasks simultaneously as follows:
Since the affinity value is approximately ten times greater than the binary value, the loss for training the affinity is reduced by a factor of ten.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Datasets used for all analysis were deposited via Zenodo at https://doi.org/10.5281/zenodo.10828798. Structures data for training are available from the PDBBind website at https://www.pdbbind-plus.org.cn/download. Accession codes for discussed structures from the PDB: 6QMT, 6W4K and 7RFS. The Interformer predicted data generated in this work are provided in Supplementary Data 1. Source data are provided with this paper.
Code availability
Installable source code, associated guidelines, various custom scripts, and interactive data analysis notebooks are available at GitHub https://github.com/tencent-ailab/Interformerand Zenodo https://doi.org/10.5281/zenodo.10828798.
Change history
12 February 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-56973-w
References
Öztürk, H., Özgür, A. & Ozkirimli, E. Deepdta: deep drug–target binding affinity prediction. Bioinformatics 34, i821–i829 (2018).
Stanzione, F., Giangreco, I. & Cole, J. C. Use of molecular docking computational tools in drug discovery. Prog. Med. Chem. 60, 273–343 (2021).
Stärk, H., Ganea, O., Pattanaik, L., Barzilay, R. & Jaakkola, T. Equibind: Geometric deep learning for drug binding structure prediction. In Proc. International Conference on Machine Learning, 20503–20521 (PMLR, 2022).
Zhang, Y., Cai, H., Shi, C., Zhong, B. & Tang, J. E3bind: an end-to-end equivariant network for protein-ligand docking. In Proc. International Conference on Learning Representations (ICLR, 2023).
Corso, G., Stärk, H., Jing, B., Barzilay, R. & Jaakkola, T. Diffdock: diffusion steps, twists, and turns for molecular docking. In Proc. International Conference on Learning Representations (ICLR, 2023).
Ballester, P. J. & Mitchell, J. B. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 26, 1169–1175 (2010).
Wang, C. & Zhang, Y. Improving scoring-docking-screening powers of protein–ligand scoring functions using random forest. J. Comput. Chem. 38, 169–177 (2017).
Karimi, M., Wu, D., Wang, Z. & Shen, Y. DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 35, 3329–3338 (2019).
Vaswani, A. et al. Attention is all you need. https://arxiv.org/abs/1706.03762 (2017).
Yun, S., Jeong, M., Kim, R., Kang, J. & Kim, H. J. Graph transformer networks. In Proc. Advances Neural Information Processing System 32 (Curran Associates, Inc., 2019).
Ying, C. et al. Do transformers really perform badly for graph representation? In Proc. Thirty-Fifth Conference on Neural Information Processing Systems https://openreview.net/forum?id=OeWooOxFwDa (2021).
Bishop, C. M. Mixture density networks (Aston University, 1994).
Wang, R., Lai, L. & Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 16, 11–26 (2002).
Cournia, Z., Allen, B. & Sherman, W. Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J. Chem. Inf. Model. 57, 2911–2937 (2017).
Zhang, X. et al. Efficient and accurate large library ligand docking with karmadock. Nat. Comput. Sci. 3, 789–804 (2023).
Méndez-Lucio, O., Ahmad, M., del Rio-Chanona, E. A. & Wegner, J. K. A geometric deep learning approach to predict binding conformations of bioactive molecules. Nat. Mach. Intell. 3, 1033–1039 (2021).
Trott, O. & Olson, A. J. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Ragoza, M., Hochuli, J., Idrobo, E., Sunseri, J. & Koes, D. R. Protein–ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 57, 942–957 (2017).
Buttenschoen, M., Morris, G. M. & Deane, C. M. Posebusters: Ai-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem. Sci. 15, 3130–3139 (2024).
Zhou, G. et al. Uni-mol: a universal 3d molecular representation learning framework. In Proc. International Conference on Learning Representations (ICLR, 2023).
Adasme, M. F. et al. PLIP 2021: expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 49, W530–W534 (2021).
Genheden, S. & Ryde, U. The mm/pbsa and mm/gbsa methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 (2015).
Fratev, F. & Sirimulla, S. An improved free energy perturbation fep+ sampling protocol for flexible ligand-binding domains. Sci. Rep. 9, 16829 (2019).
Lu, C. et al. Opls4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 17, 4291–4300 (2021).
Hornak, V. et al. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 65, 712–725 (2006).
Zdrazil, B. et al. The chembl database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 52, D1180–D1192 (2023).
Kim, D., Kim, K. I. & Baek, S. H. Roles of lysine-specific demethylase 1 (lsd1) in homeostasis and diseases. J. Biomed. Sci. 28, 41 (2021).
La Monica, G., Bono, A., Lauria, A. & Martorana, A. Targeting sars-cov-2 main protease for treatment of covid-19: covalent inhibitors structure-activity relationship insights and evolution perspectives. J. Med. Chem. 65, 12500–12534 (2022).
Jin, Z. et al. Structure of mpro from sars-cov-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Narayanan, A. et al. Identification of sars-cov-2 inhibitors targeting mpro and plpro using in-cell-protease assay. Commun. Biol. 5, 169 (2022).
Unoh, Y. et al. Discovery of s-217622, a noncovalent oral sars-cov-2 3cl protease inhibitor clinical candidate for treating covid-19. J. Med. Chem. 65, 6499–6512 (2022).
Zhu, K. et al. Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J. Chem. Inf. Model. 54, 1932–1940 (2014).
Toledo Warshaviak, D., Golan, G., Borrelli, K. W., Zhu, K. & Kalid, O. Structure-based virtual screening approach for discovery of covalently bound ligands. J. Chem. Inf. Model. 54, 1941–1950 (2014).
Maiques-Diaz, A. & Somervaille, T. C. Lsd1: Biologic roles and therapeutic targeting. Epigenomics 8, 1103–1116 (2016).
Yu, X. & Sun, D. Macrocyclic drugs and synthetic methodologies toward macrocycles. Molecules 18, 6230–6268 (2013).
Kanouni, T. et al. Discovery of cc-90011: a potent and selective reversible inhibitor of lysine specific demethylase 1 (lsd1). J. Med. Chem. 63, 14522–14529 (2020).
Owen, D. R. et al. An oral sars-cov-2 m pro inhibitor clinical candidate for the treatment of covid-19. Science 374, 1586–1593 (2021).
Halgren, T. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 (2004).
Lu, W. et al. Tankbind: Trigonometry-aware neural networks for drug-protein binding structure prediction. Adv. Neural Inf. Process. Syst. 35, 7236–7249 (2022).
Friesner, R. A. et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Knutson, C., Bontha, M., Bilbrey, J. A. & Kumar, N. Decoding the protein–ligand interactions using parallel graph neural networks. Sci. Rep. 12, 7624 (2022).
Acknowledgements
This work was supported by AI Lab, Tencent, Shenzhen, China, AI for science center. We acknowledge each colleague who helped in this work.
Author information
Authors and Affiliations
Contributions
H.L. conceived the project idea from scratch. H.L. designed and training the model. H.L. performed data analysis. H.L. and W.L. carried out all real world drug design applications. R.Q. and H.L. performed the design of Monte Carlo sampling algorithm. H.L. and L.W. wrote the paper. J.H. and P.Z. enhanced the accessibility of the paper. All authors, including X.Z., G.Y., Fandi W., and Fang W. read and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Neeraj Kumar, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lai, H., Wang, L., Qian, R. et al. Interformer: an interaction-aware model for protein-ligand docking and affinity prediction. Nat Commun 15, 10223 (2024). https://doi.org/10.1038/s41467-024-54440-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54440-6
This article is cited by
-
Advancing the modernization of traditional Chinese medicine through artificial intelligence and multimodal data integration
Chinese Medicine (2026)
-
Molecular docking in histological biomarker discovery and disease modeling: techniques, validation, and translational perspectives
Journal of Molecular Histology (2026)
-
MetaboGNN: predicting liver metabolic stability with graph neural networks and cross-species data
Journal of Cheminformatics (2025)
-
Support field neural representation learner framework for learning stability landscapes in molecular geometry
npj Biomedical Innovations (2025)
-
Development and Characterization of Psoralen-Loaded PVP/PCL Electrospun Nanofibers for Wound Healing Applications
BioNanoScience (2025)
