
Abstract
Acylation stands as a fundamental process in both biological pathways and synthetic chemical reactions, with acylated saccharides and their derivatives holding diverse applications ranging from bioactive agents to synthetic building blocks. A longstanding objective in organic synthesis has been the site-selective acylation of saccharides without extensive pre-protection of alcohol units. In this study, we demonstrate that by simply altering the chirality of N-heterocyclic carbene (NHC) organic catalysts, the site-selectivity of saccharide acylation reactions can be effectively modulated. Our investigation reveals that this intriguing selectivity shift stems from a combination of factors, including chirality match/mismatch and inter- / intramolecular hydrogen bonding between the NHC catalyst and saccharide substrates. These findings provide valuable insights into catalyst design and reaction engineering, highlighting potential applications in glycoside analysis, such as fluorescent labelling, α/β identification, orthogonal reactions, and selective late-stage modifications.
Similar content being viewed by others
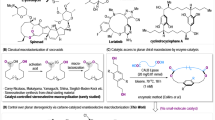
Introduction
Acylation and its related reactions1,2,3, involving the process of reacting a carboxylic acid and its derivatives with an alcohol to form the corresponding esters, represent fundamental processes in both biological systems and synthetic chemistry4,5,6,7. They constitute privileged methods for generating structurally diverse molecules8,9,10,11,12,13. Notably, acylsugars exemplify a significant class of these compounds, characterized by the esterification of various hydroxyl groups of saccharides with carboxylic acids. Acylsugars play crucial roles in biological processes as intrinsic components or secondary metabolites, serving as pharmaceuticals, food additives, and natural insecticides14,15,16,17 (Fig. 1a). For example, Taraxiroside B and its derivatives, derived from Taraxacum officinale, have demonstrated inhibitory activities against α-glucosidase18, while sucrose esters Peruvioses A to F exhibit α-amylase inhibitory properties, thereby exerting hypoglycemic effects19. Additionally, glucose ester chaenomeloidin exhibits promising therapeutic effects against stomach cancer20. On the other hand, synthetic chemists also employ acylation to modify sugars of interest and/or optimize their biopharmaceutical profiles (Fig. 1a). For instance, palmitoylation of clindamycin improves pediatric compliance due to the better taste and less gastrointestinal irritation. Sugars esterified with fatty acids have been established as safe and effective adjuvants for eliciting humoral responses21.Furthermore, Synthesis of natural products with multiple acyl groups is challenging such as Ellagitannin22,23,24. Despite their significance and the general ease of acylation, the facile assembly of acylsugars still presents challenges in selectively acylating specific hydroxyl groups within saccharides. Additionally, elucidating their biosynthesis also demands precise determination of the acylation site on sugar cores16.

a Significance of acylsugars. b Site selective modifications onto saccharides. c This work: carbene-controlled site-divergent acylation assisted by hydrogen bonding.
To address these challenges, the scientific community has devised various strategies for controlling the selective transformations of carbohydrates25,26,27,28,29,30,31,32. Among these approaches, one of the most successful methods involves covalent bond formation such as using organoboron/organotin (Fig. 1b)33,34. By selectively masking particular hydroxyl groups or enhancing their reactivity, site-selective acylation35,36, alkylation37,38,39, tosylation40, and glycosylation41 have been achieved. Notably, Miller introduced chiral ligands CuCl2(R)PhBOX into selective acylations onto 4, 6-O-benzylidene protected monosaccharides42. After that, Niu’s group developed the chiral copper-catalyzed divergent alkylation onto monosaccharides in the presence of borinic acid34 or hypervalent iodine43. Furthermore, structural modifications of small organocatalysts have yielded promising results (Fig. 1b). Histidine-based catalyst developed by Miller’s group pioneered the organocatalyzed regioselective functionalization of cabohydrates44,45,46. Hamachi’s tri-DMAP systems illustrated that achiral organocatalysts are also competent for site selective acylations47,48. Kirch’s group optimized the peptide-appended DMAP and achieved selectively acylation on polyhydroxylated compounds including partially protected monosaccharides49. Kawabata and colleagues have modified DMAP catalysts with chiral amino acids, enabling the selective acylation of unprotected glucoside at the 4-OH position with 6-OH serving as a directing group50. Likewise, Jacobsen’s group has designed a series of bis-thiourea catalysts with C–H/π interactions to glycosyl acceptors, resulting in impressive glycosylation onto minimally protected sugars in a selective manner51. Tang group investigated selective acylation on C2-OH/C3-OH of benzylidene protected glucosides in presence of chiral benzotetramisoles (BTM)52. Despite these notable advances, regioselective functionalization onto saccharides still suffer from poor generality and adaptability. addressing these limitations remains a key focus in the field to enable more versatile and efficient acylation methods. N-heterocyclic carbenes (NHCs) are important organocatalysis in reactions like acyl transfer, conjugate additions, and umpolung reactivity. Their tunable electronic and steric properties of NHCs allow chemists to modify their structure for specific catalytic applications, making them highly adaptable for a wide range of reactions. Their effectiveness in controlling reaction selectivity, both regio- and stereoselectivity, has led to widespread use in modern organic synthesis. Our groups have developed programmable selective acylation of saccharides using the combination of carbene and boronic acid35. In this study, we established an NHC catalyzed acylation system to selectively target individual hydroxyl groups within monosaccharides (Fig. 1c). Our approach relied on the chirality match/mismatch to achieve specific transformations of hydroxyl groups, facilitated by inter- and intramolecular hydrogen bonding network in the monosaccharides to increase the nucleophilicity at the corresponding site. Particularly, we successfully accomplished 3-O/2-O selective acylation on glycosides through π-π interactions, and the interaction between acyl azolium and the hydrogen bonding network. This was supported by both density functional theory (DFT) calculations and NMR analysis. The efficacy of our strategy was further demonstrated through selective synthesis of chaenomeloidin, Gal/Glu differentiation, α/β identification of glucosides, and orthogonal reactions. The carbene-catalyzed site-selective acylation offers valuable insights into the relationship between chirality and regioselectivity, which is particularly useful in the synthesis of complex glyco-structures and could have far-reaching implications in stereoselective organic synthesis.
Results and discussion
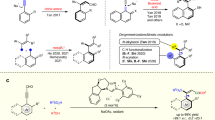
We initiated the establishment of site-selective acylation using methyl 6-O-trityl-α-D-glucopyranosides 1, which possess three secondary hydroxyl groups forming a common unit of hydrogen bonding network within six-membered sugars. This represents a common challenge encountered in carbohydrate synthesis. carbene-catalyzed oxidative reactions were employed to achieve selective acylation. Preliminary screening highlighted the challenges involved and revealed varying acylation ratios across the three secondary hydroxyl groups (Supplementary Fig. S4). However, NHC A catalyzed benzoylation of 1 in CH2Cl2 did not exhibit preference for either the 2-O or 3-O position in the presence of benzaldehyde, DQ (3,3’,5,5’-tetra-tert-butyldiphenoquinone), and K2CO3 (entry 1). 4-OH was successfully deactivated under such circumstance. In contrast, the use of achiral NHC B resulted in the predominant formation of the 3-O acylated product with a regioselectivity of 1:5 and a yield of 68% (entry 2). Upon employing chiral aminoindane-based NHC C, the site selectivity improved to 1:11 with an 80% yield (entry 3). However, replacing the -Mes groups of NHC with phenyl or pentafluorophenyl groups (NHC D, E) resulted in decreased yield and selectivity (entries 4-5). Subsequently, utilizing chiral morpholine-based NHCs F, G, and H led to the exclusive formation of the 3-O acylated product with yields ranging from 79% to 90% (entry 6–8). Notably, NHC F(entry 6), brominated NHC G (entry 7)and nitrated NHC H (entry 8, reaction time 4 h) all exhibited the excellent selectivity with 85%, 90% yield and 79% yield. This finding suggests that electronic tuning of N-heterocyclic carbene (NHC) catalysts can significantly impact both reaction rate and equilibrium. Introducing stronger electron-withdrawing groups may accelerate the reaction rate, although this could push the equilibrium toward less favorable conditions. This aligns with Burke’s notable research on electronic tuning of acyl donors and their counterions, where similar strategies of electron manipulation are employed to control reactivity and selectivity in catalytic processes53. Further optimization based on the conditions outlined in entry 7 resulted in the quantitative formation of the 3-O-benzoylated product (entry 9). Of important note, elongation of reaction time to 48 h did not change the regioselectivity ratios and yields. Additionally, various bases and solvents (entry 9–11, and see Supplementary Table S1) were examined for the selective acylation of the 3-OH position. We observed that weaker bases, NaHCO3, yielded only trace amounts of the product (entry 10), whereas stronger organic bases such as DBU and DABCO led to suppressed yields and diminished selectivity (entry 11-12).
Following the successful differentiation of the 3-OH group, we endeavored to tackle the challenge of achieving selective acylation at the 2-OH position (see Supplementary Table S2 for more information). Aware of that bases may play the important role in the regioselective acylation54, we were pleased to observe an increased selectivity of 4:1 when employing NHC ent-G in CH2Cl2 in the presence of DABCO. Ether solvents such as THF (entry 14, 10%), Et2O (entry 15, 86%), and iPr2O (entry 16, 90%) resulted in enhanced 2-OH selectivity with ratios of 5:1, 9:1, and 10:1, respectively. This suggests that solvents affect the acylation of the 2-OH group, and ether solvents are favorable for the site selectivity. Subsequently, by increasing the loading of NHC ent-G (20 mol %) and DABCO (20 mol %) in a dilute solvent (iPr2O, 8 mL), excellent identification of the 2-OH group was achieved at 0 °C with a yield of 93% and a regioselectivity rate of 20:1 (entry 17). During the screening. No acyl migration was observed under the reaction conditions. Additionally, we investigated the substitution of NHC G and ent-G in the respective standard conditions (entry 18 and entry 19), which both resulted in decreased yields and regioselectivities.
With two optimized conditions established, exceptional site selectivity and reaction generality were initially demonstrated during the substrate scope extension of 3-OH acylation (Fig. 2). A wide array of aromatic and aliphatic aldehydes proved suitable in the catalytic system without compromising regioselectivity (3a-3x), including substituted benzaldehydes (F, Cl, Br, I, CN, CH3, OCH3, tBu, Ph, COOCH3, etc.) at various positions (para, meta, ortho), heterocyclic aldehydes (3t-3v), and aliphatic aldehydes (3w-3ad). Notably, only single isomers were observed in each case (3aa-3ad) without the formation of over-acylated products. However, conjugated π-planar systems (3f, 3s, 3x) and ortho-substituents (3q) may slightly impact the acylation process, resulting in slightly reduced yields (72%-80%) in these instances. Aliphatic aldehydes also led to lower yields probably due to their less reactivity. Alterations to the trityl group, anomeric configuration, and modification (3ae-3ai) did not compromise selectivity or yields, suggesting their interactions with the catalyst are distant from the key interactions governing selectivity. The examination of methyl N-acetyl-D-glucosamine revealed well-preserved selectivity and yield (3aj). Finally, we extend our method to the mannoside. The conversion rate was very low, and the product 3ak was obtained regioselectively in 36% yield in presence of NHC ent-G. Overall, the selectivity of this transformation is exceptional coupled with satisfactory yields.

The ratios referred to acylation on 2-OH:3-OH and were determined by 1H NMR. aReactions were performed at 0 °C. bReactions were performed at r.t. cIt was also synthesized by other methods to verify our results (see Supplementary Information 4.2).
Differing from the selective transformation of 3-OH, acylation of 2-OH proved sensitive to both the electronic and steric nature of substrates (Fig. 2). In the presence of NHC ent-G, deviations from the benzene ring, whether introducing electron-donating or electron-withdrawing substituent, resulted in decreased but still good yields and selectivity (4a-4ab). This underscores the critical importance of electronic property matching between chiral acyl azolium and carbohydrate substrates. Heterocycles (4w-4y) were also well tolerated with good yields and selectivity. Although aliphatic substrates were less suited to this reaction, conversion into phenol esters addressed this limitation. Marketed drugs were effectively adorned with glucosides using this approach without compromising selectivity (4z-4aa). Further exploration of glucoside alterations revealed the indispensable role of the α-anomeric configuration in selectivity (4ab-4ai). This method is not applicable to acylation of 2-OH of β-pyranoside substrates. Replacement of the anomeric -CH3 group with an allyl group or phenyl group resulted in products 4ac and 4ad with slightly increased yields (98% or 96%, respectively) and exclusive selectivity. Using p-NO2-phenyl glucoside with a strong electron-withdrawing anomeric group (4ae) and/or thioglycoside (4af), the exceptional selectivity was maintained with yields of 78% and 74%, respectively. Additionally, xyloside (4ag, 8:1 85%) and galactoside (4ah,89%) also exhibited good suitability in this transformation, likely due to structural alterations being distant from the site for chirality match / mismatch. Finally, examination of more complex glucosides with a bridged ring, such as ertugliflozin, yielded a sole product 4ai albeit with congested anomeric steric hindrance, providing further evidence of the key role of the anomeric unit in the selective acylation process.
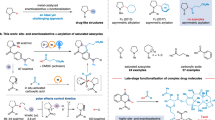
Chaenomeloidin and salicin belong to a large family of biologically active phenolic glycosides. In comparison to salicin, chaenomeloidin features a benzoyl group installed at the 3-OH position. Reported strategies have required four steps to convert salicin to chaenomeloidin, yielding less than 10%. Therefore, we endeavored to efficiently convert salicin into chaenomeloidin using our methodology. By simply masking the primary alcohols, gram scale synthesis of chaenomeloidin 6 in a site-selective manner was achieved with 71% yield (Fig. 3a).

a Selective synthesis of chaenomeloidin from Salicin. b Dual fluorescent labeling, c fluorescent labeling on glucosides over galactosides and α/β identification. d Orthogonal acylation on minimally protected saccharides.
Fluorescent labeling is a valuable technology for substance identification and biological tests. Selective fluorescent labeling of saccharides may offer fundamental tools for interpreting complicated glycan’s sequences55,56,57. To this end, we replaced the trityl group of the model substrate with a fluorescent 7-diethylaminocoumarin (7). Another fluorescent azobenzene 8 was selectively installed at the 3-OH position, resulting in the synthesis of a dual fluorescent-labeled glucoside 9 with excitation at 644 nm and emission at 646 nm (Fig. 3b). Additionally, we demonstrated the ability to label glucosides over galactosides in a competitive manner (Fig. 3c). At a concentration of 0.1 mM, the glucoside was identified by azobenzene 8 in 92% isolated yield, while the galactoside was recovered in 97% isolated yield. At a concentration of 6.25 mM, the α-glucoside 1α + 1β was converted into 4a with a regioselectivity of 20:1 and a yield of 93%, while the β-glucosides 1β were quantitatively recovered. These techniques may facilitate the development of chemical reporters and biomimetic receptors for monosaccharides and glycans.
Furthermore, we showcased orthogonal acylation on minimally protected sugars as another notable feature of our strategy (Fig. 3d). Product 12 was synthesized from 6-O-trityl-α-D-galactopyranosides using our 2-OH selective transformation. It underwent subsequent 3-OH selective acylation to afford the single product 13 with two minimally protected monosaccharide residues in 95% yield. These synthetic utilities proceeded smoothly with minimal protection, offering promising prospects in synthetic chemistry and biological applications.
DFT calculations were conducted to elucidate the mechanism of site-divergent O-acylation catalyzed by different NHC enantiomers, specifically focusing on entries 9 and 17 of Table 1, which demonstrated selectivity towards O3- and O2-acylation, respectively (see SI Section 7). We examined the key transition state (TS) structures for the acylation step, allowing attacks from either the (Re)-face or the (Si)-face of the acyl azolium intermediate by the 2-OH and 3-OH groups. Thorough conformational samplings ensure that the most stable TS for each possibility is used for comparison (see SI section 7.3 on conformational considerations). For NHC G (entry 9), the calculated TS barriers for the model reaction indicate that both the 2-OH and 3-OH groups most favorably attack at the carbonyl carbon of the acyl azolium intermediate from the (Re)-face (Fig. 4a). However, the calculated TS barriers suggest that 3-OH acylation via TS_b1_G_O3_Re has a barrier that is 3.7 kcal/mol lower than 2-OH acylation via TS_b1_G_O2_Re. This predicts that O3-acylation is kinetically most favorable, resulting in an estimated regioselectivity ratio of > 516:1 over O2-acylation, consistent with the experimentally observed exclusive 3-OH acylated product. The lower energy barrier of O3-acylation TS, TS_b1_G_O3_Re, over O2-acylation TS, TS_b1_G_O2_Re, may be attributed to the late transition state and more favorable non-covalent interactions (NCIs) observed in the former. In TS_b1_G_O3_Re, the O–C bond formed between hydroxyl group of sugar and carbonyl carbon of acyl azolium intermediate is shorter (2.18 Å) than in TS_b1_G_O2_Re (2.48 Å, Supplementary Fig. S6); in addition, the π-π interaction is presumably stronger in the former (3.64 Å) than the latter (3.68 Å, Supplementary Fig. S6). Additionally, the NCI plots suggests that TS_b1_G_O3_Re may benefit from more attractive interactions than in TS_b1_G_O2_Re (Supplementary Fig. S7), thereby greatly stabilizing the transition state structure. For NHC ent-G (entry 17), our results indicate that the 2-OH group attack at the carbonyl carbon of the acyl azolium intermediate most favorably from the (Si)-face (Fig. 4b), while the 3-OH group attacks more favorably from the (Re)-face (Supplementary Fig. S8). Overall, the calculated TS barriers for the NHC ent-G catalyzed model reaction suggest that 2-OH acylation via the most stable TS_b2_ent-G_O2_Si is lower by 1.8 kcal/mol compared to 3-OH acylation via TS_b2_ent-G_O3_Re. This predicts that O2-acylation is kinetically most favorable, translating to a O2: O3 acylation ratio of 28:1, agrees qualitatively well with our experimentally observed 2-OH acylated product selectivity (20:1). This lower barrier for O2-acylation TS, TS_b2_ent-G_O2_Si, can be attributed to the π-π interaction between the trityl group of sugar and the bromo-phenyl group of the acyl azolium intermediate (Supplementary Figs. S8 and S9). In contrast, TS_b2_ent-G_O3_Re experiences some steric hindrance between the trityl group and the acyl azolium intermediate (Supplementary Fig. S9). Upon close examination, acylation was found to occur via interaction among hydrogen-bonding network and the carbonyl group of the acyl azolium species. We hypothesized that an intermolecular hydrogen bonding network may play a role in stabilizing the most stable transition states for both 2-OH and / or 3-OH acylation. To investigate this further, we conducted NMR analysis with the stable Breslow intermediate 13 and acyl azolium 14 (Fig. 4c). The NMR results indicated that Breslow intermediate 13 showed no obvious interaction with the hydrogen bonding network of glucoside 1, while acyl azolium 14 made all the hydroxyl groups more acidic (evident by broad peaks). This suggests that ketone unit within the acyl azolium may interact with the hydrogen bonding network in some way, contributing to the stabilization of transition states. Finally, we proposed a reaction pathway in Fig. 4d. The distinct interactions between acyl azoliums (IIa and IIb) and the substrate indicate that the inter- and intramolecular hydrogen bonding network plays a significant role in modulating the reactivity and selectivity of hydroxyl group functionalization. The OH groups of the saccharides then engage with the acyl azoliums in a chirality match/mismatch manner, yielding regioselective outcomes.

a and b DFT optimized transition state structures for the most favored TSs for each reaction using either NHC G for O3-selective acylation or NHC ent-G for O2-acylation; c NMR analysis; d Proposed mechanism.
Here, we unveil carbene-controlled site-divergent acylation on monosaccharides, effectively differentiating the reactivity of secondary OH groups via chirality match/mismatch between acyl and saccharides assisted by a hydrogen bonding network. The site-divergent acylation occurred via steric hindrance and non-covalent interaction including π−π interaction and hydrogen bonding. Thus, the adjacent microenvironment of OH groups played a crucial role in the identification process, while distal alterations had minimal or no steric effect on selectivity. These findings were supported by experimental observations and DFT calculations. Furthermore, we demonstrated the efficient late-stage modification of salicin into chaenomeloidin. Additionally, we showcased the potential utility of this methodology in fluorescent labeling and the development of biomimetic receptors through competing reactions and orthogonal acylation.
Methods
Experimental procedure A for 3-O-benzoate formation
To a 10 mL screwtop test tube was added monosaccharide (0.05 mmol, 1.0 equiv.), aldehyde (2.0 equiv.), NHC G catalyst (15 mol %), K2CO3 (15 mol %) and DQ (1.1 equiv.). Then, CH2Cl2 (2 mL) was added to the mixture. The reaction was allowed to stir vigorously for 12 h at room temperature. The reaction mixture was concentrated under reduced pressure and was directly purified by flash column chromatography on silica with an appropriate solvent (Hexane/EtOAc = 3:1) to afford the product.
Experimental Procedure B for 2-O-benzoate formation
To a 10 mL screwtop test tube was added monosaccharide (0.05 mmol, 1.0 equiv.), aldehyde (2.0 equiv.), NHC ent-G catalyst (20 mol %), DABCO (20 mol %) and DQ (1.1 equiv.). Then, iPr2O (8 mL) was added to the mixture. The reaction was allowed to stir vigorously for 24 h at room temperature or 10 °C. The reaction mixture was concentrated under reduced pressure and was directly purified by flash column chromatography on silica with an appropriate solvent (Hexane/EtOAc = 3:1) to afford the product.
Data availability
The data generated in this study are provided in the Supplementary Information file. For the experimental procedures and data of NMR analysis, see Supplementary Information. All data are available from the corresponding author upon request. The X-ray crystallographic coordinates for the structure reported in this study has been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CDCC 2339260 for NHC G and 2338074 for NHC ent-G. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.Geometries of all optimized structures are included in a separate folder named DFT optimized structures with an associated readme.txt file. All these data have been deposited with this Supplementary Information and uploaded to https://zenodo.org/records/13925754 (DOI: 10.5281/zenodo.13925754). Source data are provided with this paper.
References
Haas, B. C. et al. Enantioselective Sulfonimidamide Acylation via a Cinchona Alkaloid-Catalyzed Desymmetrization: Scope, Data Science, and Mechanistic Investigation. J. Am. Chem. Soc. 146, 8536–8546 (2024).
Fang, L. L., Xiao, L., Jun, Y. W., Onishi, Y. & Kool, E. T. Reversible 2′-OH acylation enhances RNA stability. Nat. Chem. 15, 1296–1305 (2023).
Zhou, Y. et al. Nε-Fatty acylation of Rho GTPases by a MARTX toxin effector. Science 358, 528–530 (2017).
Liu, Y. et al. Mechanisms and inhibition of Porcupine-mediated Wnt acylation. Nature 607, 816 (2022).
Xu, Y. et al. A light-driven enzymatic enantioselective radical acylation. Nature 625, 74–78 (2024).
Shang, S., Liu, J. & Hua, F. Protein acylation: mechanisms, biological functions and therapeutic targets. Signal Transduct. Target Ther. 7, 396 (2022).
Mikami, T., Majima, S., Song, H. & Bode, J. W. Biocompatible Lysine Protecting Groups for the Chemoenzymatic Synthesis of K48/K63 Heterotypic and Branched Ubiquitin Chains. ACS Cent. Sci. 9, 1633–1641 (2023).
Weng, J. K., Philippe, R. N. & Noel, J. P. The Rise of Chemodiversity in Plants. Science 336, 1667–1670 (2012).
Wu, H. et al. Chemical Synthesis and Biological Evaluations of Adiponectin Collagenous Domain Glycoforms. J. Am. Chem. Soc. 143, 7808–7818 (2021).
Wang, J. et al. Total Synthesis of Mannopeptimycin β via β-Hydroxyenduracididine. Ligation. J. Am. Chem. Soc. 143, 12784–12790 (2021).
Strieth-Kalthoff, F. et al. Artificial Intelligence for Retrosynthetic Planning Needs Both Data and Expert Knowledge. J. Am. Chem. Soc. 146, 11005–11017 (2024).
Wang, H. et al. Radical thioesterification via nickel-catalysed sensitized electron transfer. Nat. Synth. 2, 1116–1126 (2023).
Wang, Y. et al. A Desilylative Approach to Alkyl Substituted C(1)-Ammonium Enolates: Application in Enantioselective [2+2] Cycloadditions. Angew. Chem. Int. Ed. 61, e202208800 (2022).
Shivatare, S. S., Shivatare, V. S. & Wong, C. H. Glycoconjugates: Synthesis, Functional Studies, and Therapeutic Developments. Chem. Rev. 122, 15603–15671 (2022).
Thomford, N. E. et al. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 19, 1578 (2018).
Lou, Y. R. et al. It happened again: Convergent evolution of acylglucose specialized metabolism in black nightshade and wild tomato. Sci. Adv. 7, eabj8726 (2021).
Neta, N. S., Teixeira, J. A. & Rodrigues, L. R. Sugar Ester Surfactants: Enzymatic Synthesis and Applications in Food Industry. Crit. Rev. Food Sci. Nutr. 55, 595–610 (2015).
Choi, J., Yoon, K. D. & Kim, J. Chemical constituents from and their α-glucosidase inhibitory activities. Bioorg. Med. Chem. Lett. 28, 476–481 (2018).
Bernal, C. A. et al. Peruvioses A to F, sucrose esters from the exudate of fruit as α-amylase inhibitors. Carbohydr. Res. 461, 4–10 (2018).
Quan, H. F., Zang, L. L., Ma, M., Dong, L. & Fu, X. Y. Chemical Constituents from Populus tomentosa Leaves and Their Anti-Cancer Activity. Chem. Nat. Compd. 59, 920–925 (2023).
Hilgers, L. A. T. et al. Carbohydrate fatty acid monosulphate esters are safe and effective adjuvants for humoral responses. Vaccine 35, 3249–3255 (2017).
Ashibe, S. et al. Non-Enzymatic Oxidation of a Pentagalloylglucose Analogue into Members of the Ellagitannin Family. Angew. Chem. Int. Ed. 56, 15402–15406 (2017).
Takeuchi, H. et al. Total Synthesis of Ellagitannins through Regioselective Sequential Functionalization of Unprotected Glucose. Angew. Chem. Int. Ed. 54, 6177–6180 (2015).
Richieu, A., Peixoto, P. A., Pouységu, L., Deffieux, D. & Quideau, S. Bioinspired Total Synthesis of (−)-Vescalin: A Nonahydroxytriphenoylated C-Glucosidic Ellagitannin. Angew. Chem. Int. Ed. 56, 13833–13837 (2017).
Dimakos, V. & Taylor, M. S. Site-Selective Functionalization of Hydroxyl Groups in Carbohydrate Derivatives. Chem. Rev. 118, 11457–11517 (2018).
Blaszczyk, S. A. et al. S-Adamantyl Group Directed Site-Selective Acylation: Applications in Streamlined Assembly of Oligosaccharides. Angew. Chem. Int. Ed. 58, 9542–9546 (2019).
Cramer, D. L., Bera, S. & Studer, A. Exploring Cooperative Effects in Oxidative NHC Catalysis: Regioselective Acylation of Carbohydrates. Chem.-Eur. J. 22, 7403–7407 (2016).
Blaszczyk, S. A., Homan, T. C. & Tang, W. P. Recent advances in site-selective functionalization of carbohydrates mediated by organocatalysts. Carbohydr. Res. 471, 64–77 (2019).
Wu, J. C. et al. Site-Selective and Stereoselective O-Alkylation of Glycosides by Rh(II)-Catalyzed Carbenoid Insertion. J. Am. Chem. Soc. 141, 19902–19910 (2019).
Li, J. Q., Grosslight, S., Miller, S. J., Sigman, M. S. & Toste, F. D. Site-Selective Acylation of Natural Products with BINOL-Derived Phosphoric Acids. ACS Catal. 9, 9794–9799 (2019).
Wang, S. et al. Studies of Catalyst-Controlled Regioselective Acetalization and Its Application to Single-Pot Synthesis of Differentially Protected Saccharides. J. Am. Chem. Soc. 143, 18592–18604 (2021).
Mensah, E., Camasso, N., Kaplan, W. & Nagorny, P. Chiral Phosphoric Acid Directed Regioselective Acetalization of Carbohydrate-Derived 1,2-Diols. Angew. Chem. Int. Ed. 52, 12932–12936 (2013).
Yamatsugu, K. & Kanai, M. Catalytic Approaches to Chemo- and Site-Selective Transformation of Carbohydrates. Chem. Rev. 123, 6793–6838 (2023).
Sun, X., Lee, H., Lee, S. & Tan, K. L. Catalyst recognition of cis-1,2-diols enables site-selective functionalization of complex molecules. Nat. Chem. 5, 790–795 (2013).
Lv, W. X. et al. Programmable selective acylation of saccharides mediated by carbene and boronic acid. Chem 8, 1518–1534 (2022).
Shibayama, H., Ueda, Y., Tanaka, T. & Kawabata, T. Seven-Step Stereodivergent Total Syntheses of Punicafolin and Macaranganin. J. Am. Chem. Soc. 143, 1428–1434 (2021).
Li, R. Z. et al. Site-Divergent Delivery of Terminal Propargyls to Carbohydrates by Synergistic Catalysis. Chem 3, 834–845 (2017).
Rao, V. U. B. et al. A synergistic Rh(I)/organoboron-catalysed site-selective carbohydrate functionalization that involves multiple stereocontrol. Nat. Chem. 15, 424–435 (2023).
Dimakos, V., Garrett, G. E. & Taylor, M. S. Site-Selective, Copper-Mediated O-Arylation of Carbohydrate Derivatives. J. Am. Chem. Soc. 139, 15515–15521 (2017).
Lee, D., Williamson, C. L., Chan, L. & Taylor, M. S. Regioselective, Borinic Acid-Catalyzed Monoacylation, Sulfonylation and Alkylation of Diols and Carbohydrates: Expansion of Substrate Scope and Mechanistic Studies. J. Am. Chem. Soc. 134, 8260–8267 (2012).
Gouliaras, C., Lee, D., Chan, L. & Taylor, M. S. Regioselective Activation of Glycosyl Acceptors by a Diarylborinic Acid-Derived Catalyst. J. Am. Chem. Soc. 133, 13926–13929 (2011).
Allen, C. L. & Miller, S. J. Chiral Copper(II) Complex-Catalyzed Reactions of Partially Protected Carbohydrates. Org. Lett. 15, 6178–6181 (2013).
Shang, W. D. et al. Site-Selective O-Arylation of Glycosides. Angew. Chem. Int. Ed. 57, 314–318 (2018).
Lewis, C. A. & Miller, S. J. Site-Selective Derivatization and Remodeling of Erythromycin A by Using Simple Peptide-Based Chiral Catalysts. Angew. Chem. Int. Ed. 45, 5616–5619 (2006).
Yoganathan, S. & Miller, S. J. Structure Diversification of Vancomycin through Peptide-Catalyzed, Site-Selective Lipidation: A Catalysis-Based Approach To Combat Glycopeptide-Resistant Pathogens. J. Med. Chem. 58, 2367–2377 (2015).
Griswold, K. S. & Miller, S. J. A peptide-based catalyst approach to regioselective functionalization of carbohydrates. Tetrahedron 59, 8869–8875 (2003).
Wang, H. et al. Chemical Cell-Surface Receptor Engineering Using Affinity-Guided, Multivalent Organocatalysts. J. Am. Chem. Soc. 133, 12220–12228 (2011).
Dey, K. & Jayaraman, N. Trivalent dialkylaminopyridine-catalyzed site-selective mono-O-acylation of partially-protected pyranosides. Org. Biomol. Chem. 22, 5134–5149 (2024).
Huber, F. & Kirsch, S. F. Site-Selective Acylations with Tailor-Made Catalysts. Chem. Eur. J. 22, 5914–5918 (2016).
Kawabata, T., Muramatsu, W., Nishio, T., Shibata, T. & Schedel, H. A catalytic one-step process for the chemo- and regioselective acylation of Monosaccharides. J. Am. Chem. Soc. 129, 12890–12895 (2007).
Li, Q., Levi, S. M., Wagen, C. C., Wendlandt, A. E. & Jacobsen, E. N. Site-selective, stereocontrolled glycosylation of minimally protected sugars. Nature 608, 74–79 (2022).
Xiao, G. Z. et al. Catalytic Site-Selective Acylation of Carbohydrates Directed by Cation-n Interaction. J. Am. Chem. Soc. 139, 4346–4349 (2017).
Wilcock, B. C. et al. Electronic tuning of site-selectivity. Nat. Chem. 4, 996–1003 (2012).
Kattnig, E. & Albert, M. Counterion-Directed Regioselective Acetylation of Octyl β-D-Glucopyranoside. Org. Lett. 6, 945–948 (2004).
Marando, V. M. et al. Biosynthetic Glycan Labeling. J. Am. Chem. Soc. 143, 16337–16342 (2021).
Banahene, N., Kavunja, H. W. & Swarts, B. M. Chemical Reporters for Bacterial Glycans: Development and Applications. Chem. Rev. 122, 3336–3413 (2022).
Lee, S. Y. et al. Selective Glycan Labeling of Mannose-Containing Glycolipids in Mycobacteria. J. Am. Chem. Soc. 146, 377–385 (2023).
Acknowledgements
This work was supported by grants from Natural Science Foundation of China (No.22101266), Excellent Youth Program of Hennan Province (242300421119, Y.-G.L.) and International Postdoctoral Exchange Fellowship Program (Talent-Introduction Program, No. YJ20210304, Y.-G.L.) by the Office of China Postdoc Council, China Postdoctoral Science Foundation (No. 2022M722865, Y.-G.L.) and Zhengzhou University (2024ZDGGJS068, Y.-G.L.), Open Projects Fund of Shandong Key Laboratory of Carbohydrate Chemistry and Glycobiology, Shandong University(No. 2023CCG08, Y.-G.L.); We acknowledge funding support from the National Key Research and Development Program of China (2022YFD1700300 Y.R.C.), the National Natural Science Foundation of China (U23A20201, 22071036, Y.R.C.), the Frontiers Science Center for Asymmetric Synthesis and Medicinal Molecules,Department of Education, Guizhou Province [Qianjiaohe KY number (2020)004, Y.R.C.], the Natural Science Foundation of Guizhou University [Guida Tegang Hezi (2023)23, Y.R.C.], the Program of Introducing Talents of Discipline to Universities of China (111 Program, D20023, Y.R.C.) at Guizhou University, the Singapore National Research Foundation under its NRF Competitive Research Program (NRF-CRP22-2019-0002, Y.R.C.), the Singapore Ministry of Education under its MOE AcRF Tier 1 Award (RG70/21, RG84/22, Y.R.C.), MOE AcRF Tier 2 Award (MOE-T2EP10222-0006, Y.R.C.), and MOE AcRF Tier 3 Award (MOE2018-T3-1-003, Y.R.C.), a Chair Professorship Grant, and Nanyang Technological University. X.Z. acknowledges the funding support from the Chinese University of Hong Kong (CUHK) under the Vice-Chancellor Early Career Professorship Scheme Research Startup Fund (Project Code 4933634, X.Z.) and Research Startup Matching Support (Project Code 5501779, X.Z.).
Author information
Authors and Affiliations
Contributions
Z.Z. competed the acylation on 2-OH, Y.T. competed the acylation on 3-OH. H. W., X. P. and P.P. contributed in discussion, revision and experimentation. S.V.C.V. performed the DFT calculations under the supervision of X.Z. Y.R.C. and Y.-G.L. conceptualized and directed the project and drafted the manuscript with assistance from all co-authors. All authors contributed to the discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hai Dong and the other anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, YG., Zhong, Z., Tang, Y. et al. Carbene-catalyzed chirality-controlled site-selective acylation of saccharides. Nat Commun 16, 54 (2025). https://doi.org/10.1038/s41467-024-55282-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55282-y
