
Abstract
Zoologists have adduced morphological convergence among embryonic stages of closely related taxa, which has been called the phylotypic stage of embryogenesis. Transcriptomic analyzes reveal an hourglass pattern of gene expression during plant and animal embryogenesis, characterized by the accumulation of evolutionarily older and conserved transcripts during mid-embryogenesis, whereas younger less-conserved transcripts predominate at earlier and later embryonic stages. In contrast, comparisons of embryonic gene expression among different animal phyla describe an inverse hourglass pattern, where expression is correlated during early and late stages but not during mid-embryo development. Here, multiplexed spatial-transcriptomic analyzes is used to investigate embryogenesis and homology in maize, which has grass-specific morphology. A set of shared, co-expressed genes is identified during initiation of maize embryonic organs, replete for ancient/conserved genes manifesting an hourglass pattern during mid-embryogenesis. Transcriptomic comparisons of maize and Arabidopsis embryogenesis with that of the moss Physcomitrium patens identify an inverse hourglass pattern across plant phyla, as in animals. The data suggest that the phylotypic stages in plants and animals are characterized by expression of ancient and conserved genes during histogenesis, organization of embryonic axes, and initial morphogenesis. We propose a mechanism for gene evolution during the innovation of morphological novelty.
Similar content being viewed by others


Introduction
The embryo is defined as the developmental stage spanning the period between fertilization and the appearance of primary tissues and organs. Post-fertilization, flowering plant embryogenesis consists of three conserved processes: (1) polarized and asymmetric cell divisions to create the embryo proper, (2) patterning of discrete histological tissue-layers, and (3) organogenesis of lateral organs and the formation of apical meristems. Persistent meristematic reservoirs comprising the shoot apical meristem (SAM) and root apical meristem enable plants to extend organogenesis beyond embryogenesis. The genes regulating these processes, however, are largely undescribed.
The embryos of the eudicot Arabidopsis thaliana and the monocot Zea mays ssp. mays involve asymmetric cell divisions that establish the embryo proper and the basal, haustorial suspensor that supplies nutrients to the embryo1,2,3 (Fig. 1a). Subsequent histological layering creates an outer epidermal layer surrounding internal tissues, and Arabidopsis embryos develop two embryonic leaves (cotyledons) and shoot and root apical meristems, whereupon organogenesis is suspended until seed germination1,4. The grass embryo differs from that of Arabidopsis2,3,5,6,7,8,9 in several important respects. The first lateral organ formed in the maize embryo is the scutellum (Fig. 1b), which at maturity forms a shield-shaped, haustorial organ that absorbs nutrients from the endosperm7,10. The coleoptile then emerges (Fig. 1c) to form a tubular-like sheath that protects the shoot during germination. Unlike Arabidopsis, grasses extend embryo organogenesis to develop several foliar leaves contained within the coleoptile before seed quiescence.

Differential interference contrast (DIC) microscopy of maize embryos at the Proembryo stage (a), Transition stage (b), Coleoptilar stage (c), and Stage 1 (d). All stages were analyzed in five replicates. Immunohistological analyzes of the Class I KNOX proteins mark the shoot apical meristem (SAM). ep, embryo proper; en, endosperm; su, suspensor; sc, scutellum; c, coleoptile; L, leaf; *, SAM. Scale bar: 50 μm (a and b) and 100 μm (c and d). e, Uniform Manifold Approximation and Projection (UMAP) of Stage 1 embryo single-cell transcriptomic profiling with tissue types assigned for each cell cluster.
In contrast, embryos of the moss Physcomitrium patens have no apical meristems sensu stricto, or lateral organs. After fertilization, a transverse zygotic cell division ultimately gives rise to a basal “foot” and a cell mass that develops into the seta and the spore capsule 11,12. The latter consists of a histologically layered sporangium, wherein meiosis results in thousands of haploid spores. Fundamental differences in embryonic growth polarity are also noted between non-vascular plant and vascular plants. Nonvascular plant (e.g. Physcomitrium) embryos develop exoscopically outward from the subtending gametophyte, whereas vascular plant (e.g. maize and Arabidopsis) embryos develop endoscopically into the gametophyte13. Indeed, embryo morphometric diversity is widespread across the land plants, although comparative transcriptomic analyzes of embryogenesis are few10,14,15.
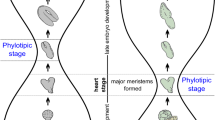
The diversity of land plant embryo development stands in contrast with that of animals, which appears to converge on to a period of maximum embryo morphological similarity among related vertebrate taxa16,17,18 during “mid-embryogenesis”. However, “earlier” and “later” stages of vertebrate embryogenesis are dissimilar morphologically. The result is an hourglass pattern. The convergent mid-embryo stage has been called the phylotypic stage19. Seminal analyzes of transcriptomic patterns in chordates revealed that the mid-embryonic stage in mouse is highly constrained20,21. Later transcriptomic analyzes of fruit fly (Drosophila), zebrafish (Danio), and Arabidopsis embryos found that gene expression during the predicted phylotypic stages showed a transcriptomic hourglass pattern, that featured enhanced accumulation of more ancient transcripts during mid-embryogenesis as compared to earlier and later ontogenetic stages22,23,24,25. In addition, the Arabidopsis study found that the transcripts accumulating during the predicted phylotypic stages are also sequence-conserved25. Cross-species comparative transcriptomic analyzes of several chordate embryos found that gene expression and morphological traits are correlated and conserved during mid-embryogenesis26,27. Plots of transcriptomic gene age conform to the “hourglass model” first described in animals16,17,18.
However, more widespread comparisons of ontogenetic gene expression across ten diverse animal phyla identify an inverse hourglass pattern, wherein the most prominent transcripts during early and later stages of development are more conserved among phyla as compared to genes expressed during the presumptive “phylotypic stage”28. An important issue in all embryological comparisons across diverse taxa involves temporal homology––what is meant by early-, mid-, and late-embryogenesis? This issue has added relevance when comparing metazoans and land plants, each of which is adduced to be monophyletic yet morphologically extremely diverse. This concern applies even to broad comparisons among land plant embryos, which manifest what appear to be three, distinct patterns of embryogenesis or proposed phylotypes: the nonvascular plants, the vascular seedless plants, and the vascular seed plants13. Notably, these proposed plant phylotypes reflect shared processes of embryo development, not morphological similarity13. Yet, another concern in this field of inquiry is the temporal delay between gene expression and embryological-morphogenetic response, which likely differs among genes within an individual organism and among phylogenetically diverse taxa. Neither of these concerns has been adequately addressed, and, as yet, a transcriptomic hourglass analysis is reported for only a single land plant genus (Arabidopsis)25. No such study has been reported within or across phyla for the morphologically diverse, land plants29.
Here we present multiplexed analyzes of gene expression during maize embryogenesis, a grass that evolved more than 150 million years after the appearance of flowering plants30. Single cell sequencing (scRNA-seq), spatial RNA-seq, laser-microdissection RNA-seq (LM-RNA-seq), and multiplexed RNA-targeting in situ hybridizations were utilized to characterize gene expression in developing maize embryos at high resolution. A set of genes co-expressed during the initiation of maize embryonic organs (i.e., scutellum, coleoptile, and leaf) is identified, indicating their likely collective homology. Within phylum comparisons of maize and Arabidopsis show conserved expression of relatively ancient and sequenced-conserved gene homologs in an hourglass pattern, peaking at mid-embryo stages. Cross-phylum comparisons of embryonic gene expression in the flowering plants maize and Arabidopsis to that of a nonvascular moss reveals inverse hourglass patterns, equivalent to those described in animals. The data indicate that the plant and animal phylotypic stages reflect shared ancestral morphometric processes of tissue histogenesis and meristematic growth of embryonic organs within each of the two kingdoms. An across kingdom model for the evolution of morphological novelty via the innovative expression of newer-evolved, sequence-conserved genes is presented.
Results
Multiplexed and single-cell profiling of the transcriptomic networks in the Stage 1 maize embryo
A combinatorial scRNA-seq approach was utilized for transcriptomic analyzes of the homology of maize embryonic organs31,32, and comparative analyzes of plant embryogenesis. Stage 1 maize embryos were ~1 mm long and contain a prominent, ovately-flattened scutellum, an emerged coleoptile that is beginning to surround the SAM, and the newly-initiated primordium of the first foliar leaf5 (Fig. 1d). Stage 1 embryos contained all the lateral organ types of the maize embryonic shoot and are thus an appropriate stage to analyze the organ-specific genes among grass embryonic organs. 10X Genomics Chromium™ was used to profile the single-cell transcriptomes of 11,459 protoplasts digested from over 400 Stage 1 embryos in two replicates (Fig. 1e). Cell clustering classified transcriptionally similar cells; twenty-two clusters were identified with median values of 10,592 UMI per cell and 4,029 genes per cell (Supplementary Fig. 1).
The first challenge encountered in analyzes of the scRNA-seq data was the identification of the tissue/organ-specific origins of each cell cluster. Prior RNA-seq analyzes in maize have focused on seedlings or inflorescences33, such that cell clusters derived from the SAM, embryonic stem, and vasculature34 can be readily identified, as is the residual starchy endosperm (Fig. 1e and clusters 6, 15 and 20 respectively). In contrast, relatively few transcriptomic analyzes have focused on the grass scutellum or coleoptile, such that the origins of several Stage 1 embryonic cell clusters were not immediately identifiable. To annotate all the cell clusters, a multiplexed transcriptomic approach was used, combining the scRNA-seq data with spatial-transcriptomics and laser-microdissection RNA-sequencing (LM-RNA-seq) to determine spatial information for all Stage 1 embryo cell clusters, as described below (Fig. 2 and Supplementary Fig. 2–3, Supplementary Data 1, 2).

Longitudinal cryosections of Stage 1 embryos embedded in optimal cutting temperature compound (OCT; (a–b) two replicates) or in paraffin (c–d) six replicates, for use in10x VisiumTM spatial transcriptomics analysis of embryo sections. The Toluidine Blue-stained sections (a, c) are overlayed with the10x VisiumTM spatial map (b, d); the various color-coded circles correspond to the capture spots within which spatial cDNA libraries were generated. Scale bars = 100 μm. e Spatial clustering of VisiumTM spatial transcriptomic analysis of the Stage 1 embryo. The spatial embryonic origins of each cluster are indicated by the color-coded embryo cartoon; numbers correspond to the cluster designations on the UMAP. f Longitudinal sections of paraffin-embedded Stage 1 embryo for use in LM-RNA-seq. g Enlarged view of a Stage 1 embryo showing regions microdissected for LM-RNA-seq; all LM-RNA-seq analyzes included three replicates. Figure labels: scu, scutellum; uc, upper coleoptile; lc, lower coleoptile. Scale bars =100 μm. h Heatmap illustrating the correlation between the single-cell (sc) clusters (y-axis) and VisiumTM spatial clusters (x-axis). The significance level is marked by −log10p. The p values were calculated based on the cumulative distribution function (CDF) of the one-tailed hypergeometric distribution (for exact p values, please refer to Source Data). i Expression of candidate genes identified in microdissected embryonic organs. Cell clusters exhibiting elevated candidate gene expression of the specific markers are indicated; expression values are log2 transformed, i.e., log2(TPM + 1). sc_cluster, single-cell cluster; V, VisiumTM spatial cluster; scu/Scu, scutellum; epi, epidermis; hypo, hypocotyl; ab, abaxial; ad; adaxial; sus, suspensor; col, coleoptile; emb_endo, embryo-endosperm boundary; UC, upper coleoptile; LC; lower coleoptile.
To further validate the identification of Stage 1 embryo single cell clusters, eight individual Stage 1 embryo sections were processed using the 10X Genomics VisiumTM spatial transcriptomics pipeline (Fig. 2a–d and Supplementary Data 3). Each spatially resolved spot on the VisiumTM slides was sorted into a spatial cluster, based upon shared gene expression. Nine spatial clusters were identified and categorized into four supergroups: (1) shoot-root axis, (2) scutellum epidermis, (3) internal scutellum, and (4) endosperm/embryo boundary (Fig. 2e). A hypergeometric test was used to analyze the overlap of cluster marker genes between the single-cell and spatial transcriptomic data, revealing that at least 7 of the 22 cell clusters (0-4 and 6-7) are derived from the scutellum (Fig. 2h). In cases where cell cluster identity was not solved by the hypergeometric test (i.e. smaller clusters such as 8, 10, 11, and 15–18), candidate marker genes differentially -expressed in individual cell clusters were identified in the VisiumTM spatial map. This strategy suggested that clusters 8 and 10 are associated with scutellum internal tissue, clusters 11 and 16 are comprise boundary areas, cluster 15 is vasculature tissue, cluster 17 comprised the scutellum tip, and cluster 18 is associated with the scutellum and suspensor area (Figs. 1e and Supplementary Fig. 2a, i, j, n–q). Comparisons of transcript localizations of known marker genes within the scRNA-seq UMAP and the spatial transcriptomic spots indicated that the resolution of VisiumTM technology comprises 5–10 embryonic cells. Thus, the 5–10 cell resolution is not sufficient to reliable distinguish transcripts specifically expressed in the diminutive coleoptile, SAM, or leaf, which contain very few cells. To increase transcriptomic resolution, LM-RNA-seq used to microdissect gene expression in the SAM, adaxial scutellum, upper/lower coleoptile, and leaf primordium of Stage 1 embryos (Fig. 2f, g). Principal component analyzes (PCA) showed that the transcriptomic profiles of these five, maize embryonic organ/tissues are distinct (Supplementary Fig. 3a). In particular, 9913 genes were identified as differentially-expressed in at least one embryonic structure, and that 89, 1058, 246, 84, and 53 genes were preferentially expressed in these same five embryonic organ/tissues (Supplementary Figs. 3b–f and Supplementary Data 4–6). Thus, the multiplexed transcriptomic approach enabled the identification of the tissue/organ origin of each cell cluster in the Stage 1 maize embryonic UMAP (Figs. 1e, 2i).
Spatiotemporal analyzes of maize embryogenesis and homology via multiplexed RNA-targeting
By definition, homologous organs express homologous genes during developmental patterning31,35. With an emphasis on regulatory genes differentially expressed in our transcriptomic analyzes of Stage 1 maize embryos, a total of100 transcripts were targeted for spatiotemporal analyzes of multiplexed, in-situ hybridization of transcript localization during embryogenesis, using the 10X XeniumTM protocols (Supplementary Data 7). Samples included longitudinal sections of (i) transition-staged embryos just forming a SAM and scutellum (Fig. 1b), (ii) Coleoptilar-staged embryos (Fig. 1c), and (iii) Stage 1 embryos initiating the first foliar leaf (Fig. 1d). Correlations of the scRNA-seq data, spatial-transcriptomic plots, and XeniumTM in situ RNA-targeting data were examined via comparisons of 16 genes preferentially expressed in one-or-two cell clusters (Supplementary Fig. 2). In all cases, the in situ hybridization patterns were consistent with the spatial expression data.
In light of the comparative consistencies of the multiplexed in situ hybridization data, the dynamic co-localization patterns of some key regulators of maize shoot development during serial stages in grass embryogenesis were examined. Consistent with prior studies36,37, the spatial-temporal expression of the indeterminacy marker KNOTTED1 (KN1; Fig. 3a–d) was first detected during SAM initiation at the maize Transition stage (Fig. 1b). At later stages, KN1 was observed to accumulate in the SAM and subtending embryonic stem, but was downregulated in lateral organs36,37. NARROW SHEATH1 (NS1) and WUSCHEL-RELATED HOMEOBOX 3 A (WOX3A), which encode homeodomain transcription factors required for mediolateral expansion of maize lateral organs22, were co-expressed at the edges of the developing coleoptile and initiating leaf primordia as described previously7,31,38,39 (Fig. 3b, c, f, g). Undetected in prior studies of grass embryogenesis, Fig. 3a reveals that NS1 transcripts accumulate in the initiating scutellum at the Transition stage. Moreover, transcripts of CUP-SHAPED COTYLEDON2 (CUC2), which mark developmental boundaries throughout maize ontogeny40, were detected between the expression domains of KN1 and NS1 during scutellum initiation (Fig. 3a), and in the boundaries between the SAM and later-staged embryonic lateral organs (Fig. 3b, c,e). DROOPING LEAF1 (DRL1), which regulates leaf architecture and is expressed in incipient and developing foliar leaf primordia41, was not detected in the coleoptile or scutellum at any embryo stage examined (Fig. 3c, h). These combinatorial in situ RNA-targeting analyzes enabled the identification of previously undescribed combinatorial patterns of dynamic, gene expression during maize embryo development.

a–c Signal point map of KN1, CUC2, NS1, WOX3A and DRL1 at the Transition stage (a), Coleoptilar stage (b) and Stage 1 (c). d–h Expression density map of KN1 (d), CUC2 (e), NS1 (f), WOX3A (g) and DRL1 (h) at Stage 1. Relative expression levels are color-coded. i–k, l–n, o–q and u–w, Signal point map of BOPa (i–k), YAB14 (l–n), IG1 (o–q), and ARF3 (u–w) at the Transition stage, Coleoptilar stage and Stage 1. r–t and x–z, Expression density map of IG1 (r–t) and ARF3 (x–z) at the corresponding stages. Figure labels: T, Transition stage; C, Coleoptilar stage; L: Stage 1. The point colors are arbitrarily determined. Yellow asterisks mark the adaxial expression of IG1 and ARF3, and red asterisks mark the abaxial expression of ARF3. The green asterisk in panel (k) marks the expression maximum of BOPa at the emerging leaf sheath. The yellow asterisks in panel r-t and z mark the expression maxima of IG1 at the adaxial side of the scutellum (r), the coleoptile (s), the coleoptile and the leaf (t), as well as the expression maxima of ARF3 at the adaxial side of the scutellum (z), respectively. The red asterisks in panel (x–z) mark the abaxial expression of ARF3 at the scutellum (x, y), the coleoptile (y, z) and the leaf (z). Scale bar for all panels = 50 μm.
Other candidate genes examined using XeniumTM displayed patterns of transcript accumulation that offer insights into the developmental homologies of maize embryonic lateral organs. For example, BLADE-ON-PETIOLEa (BOPa) accumulated throughout the developing coleoptile and in the proximal region of the initiating leaf at Stage 1 (Fig. 3i–k), consistent with the proposed function of BOP homologs as leaf proximal-identity genes42. Notably, BOPa was undetected throughout scutellum ontogeny, in a manner similar to the accumulation patterns noted for the BOPa paralog TASSELS REPLACE UPPER EARS1 (TRU1) (Supplementary Fig. 4). Transcripts of the DRL1-paralog YABBY14 (YAB14) densely accumulated in the apical tip of the scutellum at the Transition stage and in the elongating scutellum and newly emerged coleoptile at the Coleoptilar stage. During Stage 1, a new YAB14 expression maximum was organized at the primordial leaf tip (Fig. 3l–n). Moreover, the pleiotropic gene INDETERMINATE GAMETOPHYTE1 (IG1), reported to function during leaf polarity and patterning of maize leaves43,44, accumulated on the dorsal (i.e. adaxial) surfaces of initiating leaf primordia during Stage 1, and also in the adaxial layers of the emerging coleoptile and in the upper-adaxial region of the Transition-staged scutellum (Fig. 3o–t). No significant IG1 expression was observed during later stages of embryonic scutellum development (Fig. 3p, q, s, t). Another leaf polarity regulator, AUXIN RESPONSE FACTOR 3 (ARF3)45, was expressed on the abaxial region of the Transition-staged embryo during scutellum initiation, and in the abaxial domains of the newly-emerged coleoptile and leaf during later embryo stages (Fig. 3u–z). Interestingly, the expression of ARF3 in the scutellum moved from the ventral (i.e., abaxial) region in the Transition stage to adaxial region at Stage 1 (Fig. 3z).
The initiation of maize embryonic lateral organs utilizes a shared gene expression network
By Stage 1, the three grass embryonic lateral organs are reported to initiate morphological divergence and continue to do so before seed quiescence5. Therefore, the determination of organ homology32 and of a transcriptomic hourglass in maize requires the examination of gene expression throughout embryogenesis. Toward this end, the LM-RNA-seq data from Stage 1 embryonic organs were combined with the LM-RNA-seq data from serial stages of maize embryonic development34 to generate a more comprehensive sampling throughout embryogenesis. Specifically, these prior analyzes generated transcriptomic data precisely from the newly initiated embryonic organ and the adjacent SAM, which were serially sampled at the Transition stage (scutellum +SAM), the Coleoptilar stage (coleoptile +SAM) and Stage 1 (leaf 1 + SAM). Figure 2i shows that the expression levels of several leaf developmental genes were highest in Stage 1 leaves (L) and steadily decreased in the lower coleoptile (LC), upper coleoptile (UC), and scutellum (S). Notably, the expression of these genes during the Transition stage and the Coleoptilar stage was similar to that of leaf 1 during Stage 1 (Supplementary Data 4)34. These data provide evidence that the gene expression networks correlated with foliar leaf initiation at Stage 1 are also associated with the initiation of the scutellum and coleoptile at successively earlier stages in embryogenesis.
To further investigate this hypothesis, a weighted gene co-expression network analysis (WGCNA)46,47 was performed on the Stage 1 embryonic LM-RNA-seq data to identify co-expression modules correlated with the expression pattern L > LC > UC > S. A total of 5797 gene transcripts with high expression variability were selected. These transcripts were grouped into twelve co-expression modules (ME1 through ME12; Supplementary Fig. 5a). Two co-expression modules (ME2 and ME3) comprising 963 genes emerged as correlated with the L > LC > UC > S pattern. (Supplementary Fig. 5a and Supplementary Data 8). Intersection of these 963 genes with transcripts identified by LM-RNA-seq in the initiating scutellum, coleoptile and leaf 134 (excluding putative SAM-specific genes) identified 130 genes that are co-expressed during initiation of the first three embryonic organs of maize (Fig. 4a and Supplementary Fig. 5b and Supplementary Data 9). Co-expression of these genes was significantly higher during initiation of these embryonic organs as compared to later morphometric stages (Fig. 4b, Supplementary Fig. 5b-e).

a A Venn diagram of embryonic transcriptomics identifies 130 genes co-expressed in initiating embryonic organs. b Normalized expression of these 130 genes at the Proembryo stage (PE), Transition stage (T), Coleoptilar stage (C), Stage 1 scutellum (Scu), Stage 1 upper coleoptile (UC), Stage 1 lower coleoptile (LC), Stage 1 leaf (L), as well as P0 and P1 of the 14 day-after-germination seedlings. The letters above the boxplots represent significance levels (non-overlapping letters indicate significant differences at p < 0.05 using the Tukey-Kramer HSD test (for exact p values, please refer to Source Data). The middle line reflects the median value; the box shows the 25th and 75th percentiles and the whiskers reflect 1.5 times the interquartile range. c and d WGCNA (c) and hdWGCNA test (d) hierarchical cluster tree graph of transcriptomic similarities among tissue-specific co-expression modules, respectively. The height (y-axis) scales the length of tissue-specific branches, indicating the transcriptomic distances among modules. The hierarchical cluster trees were constructed based on topological overlap dissimilarity, derived from a signed (one-sided) adjacency matrix calculated using the Pearson correlation. In d the significance levels of coexpression module-sc cluster correlations are marked by color-coded circles (significance levels were calculated via −log10p; red or blue colors represent relatively strong or weak correlations, respectively); colored branches indicate correlated structures (red branches represent Scutellum-Coleoptile super-cluster; and blue branches represent the SAM&hypocotyl-Leaf super-cluster). The p values (see Supplementary Fig. 7) were calculated based on the cumulative distribution function (CDF) of the one-tailed hypergeometric distribution. e Heatmap illustrating expression correlations of the gene set in Maize (y-axis) and Arabidopsis (x-axis) during multiple embryonic stages. Heatmap colors are determined by correlation coefficient (r) via Pearson correlation test with significance level p < 0.05 (*) or p < 0.01 (**) (for exact p values, please refer to Source Data). Scu, Stage 1 scutellum; Col, Stage 1 coleoptile; epi: epidermis; hypoc, hypocotyl; M, co-expression modules; sc_cluster, single-cell cluster; PE, Proembryo; T, Transition stage; C, Coleoptilar stage; L: Stage 1 leaf.
In light of the controversy regarding the homology of the grass cotyledon (i.e., whether the coleoptile is a foliar leaf8, the entire cotyledon9, or the proximal component of a single grass cotyledon3,39), a second WGNCA analysis was performed on the LM-RNA-seq data to identify co-expression modules among developing embryonic organs and seedlings. Batch effects were removed (Supplementary Fig. 6a, b), and the merged dataset was filtered to select genes with variability in transcript accumulation levels across embryonic organs. In the analysis, 5128 out of 5137 genes were grouped into 15 co-expression modules (LM1, LM2, …, LM15); the remaining transcripts did not resolve to a module (Supplementary Data 10). Transcriptomic relationships among the co-expression modules are shown in a tree graph (Fig. 4c), in which the y-axis (height) scales the length of organ-specific branches, indicating transcriptomic distances between these modules (Supplementary Figs. 6a, b). Pearson correlation tests between module eigengenes and organ-specific transcriptomes identified associations between specific co-expression modules and the corresponding embryonic organs (Fig. 4d and Supplementary Fig. 6c). Two major branches comprise (1) the Stage 1 leaf, SAM, P0 and P1, and (2) the coleoptile and the scutellum.
To further compare the transcriptomic similarities among Stage 1 maize embryonic lateral organs at single-cell resolution, high-dimensional WGCNA (hdWGCNA49) was performed on the scRNA-seq data (Fig. 4d). A total of 4762 genes transcripts were grouped into 34 co-expression modules (M1, M2, …, M34; Supplementary Data 11). Overlap between genes in co-expression modules and genes in single-cell clusters was analyzed using a hypergeometric test (Supplementary Fig. 7). The transcriptomic similarities between co-expression modules were evaluated by the module eigengene tree (Fig. 4d), which showed that the modules associated with the stage 1 coleoptile group closer to the scutellum whereas the stage 1 leaf is closer to the SAM, consistent with the LM-RNA-seq WGCNA data (Fig. 4c).
The WGCNA results linking the scutellum and coleoptile (Fig, 4c, d) were supported by genetic analysis. LONELY GUY7 (LOG7), which encodes a cytokinin-activating required for maize SAM maintenance48, was expressed at the SAM tip in Stage 1 embryos (Supplementary Figs. 8a–c). Null mutations in log7 resulted in SAM termination48. Histological examination of log7 mutant Stage 1 embryos revealed the absence of a detectable SAM or foliar leaf, although the scutellum and coleoptile were morphologically normal (Supplementary Figs. 8d–h). These data expose morphogenetic differences between leaves, and the scutellum and coleoptile.
Maize embryonic gene expression is conserved in Arabidopsis embryos
To investigate the transcriptomic similarities of embryonic lateral organs of maize and those of the eudicot model Arabidopsis, comparative genomic toolkits [GRAMENE (https://www.gramene.org/), TAIR (https://www.arabidopsis.org/), BLAST tools (https://blast.ncbi.nlm.nih.gov/Blast.cgi)] were used to select Arabidopsis orthologs of the genes expressed during maize embryogenesis, and to investigate the gene expression atlas of Arabidopsis embryo development50. Embryonic nomenclature differs between eudicots and grasses1. In Arabidopsis, histological layering and SAM initiation occur during the globular stage, cotyledon initiation and emergence defines the heart stages, whereas the cotyledons elongate during the torpedo and bent embryo stages.
Out of 130 genes co-expressed in the initiating scutellum, coleoptile, and leaf 1 of maize embryos, forty-nine had corresponding Arabidopsis orthologs expressed during embryo development, commencing during cotyledon initiation at the early heart stage and persisting until the late cotyledon stage (Supplementary Data 12). Notably, the stage-specific accumulation patterns of these homologous maize transcripts in the emerging scutellum (Transition stage), coleoptile (Coleoptilar stage), and foliar leaf (Stage 1) were significantly correlated with the reported expression of the corresponding Arabidopsis orthologs during specific stages in cotyledon initiation and development in this eudicot embryo50 (Fig. 4e, Supplementary Fig. 9). In addition, transcripts accumulating in the expanding Stage 1 scutellum correlated with expression of Arabidopsis orthologs during the later stages of cotyledon growth (i.e. late-torpedo stage50). Likewise, maize transcripts accumulating in the elongating Stage 1 coleoptile were significantly correlated with expression of orthologous Arabidopsis genes during earlier stages of cotyledon expansion (i.e., the early heart stage50; Fig. 4e). In contrast, the genes expressed in the maize Proembryo and Stage 2 were not significantly correlated with any stages of Arabidopsis (Fig. 4e). These data were interpreted to indicate that despite the morphological dissimilarities of these grass and eudicot “mid-staged” embryos, the development of the maize and Arabidopsis embryonic lateral organs share a homologous regulatory genes, whereas in earlier and later-stages embryogenesis, this homology is not obvious13.
Genes expressed during maize embryogenesis comprises a transcriptomic hourglass
The data reported in this study support the hypothesis that genes co-expressed in initiating maize embryonic organs are conserved in Arabidopsis thaliana embryos; strongest correlations were found with transcriptomic signatures of the eudicot heart and torpedo stage embryos. Prior studies reported that during mid-embryogenesis, old and conserved genes are preferentially expressed over younger and divergent genes, marking the transcriptomic hourglass of Arabidopsis embryogenesis25. To investigate if these maize genes co-expressed during initiation of all maize embryonic organs expression also comprises a transcriptomic hourglass in grass embryogenesis, the age index and sequence divergence of genes comprising this maize embryo co-expression network were examined.
To evaluate gene age, a phylostratigraphic analyzes obtained 13 phylostrata (PS), PS1-PS13 (Fig. 5a and Supplementary Data 13) using statistical genetic methods to identify homologous founder genes across lineages (including 350 species) and date their phylogenetic origin (Supplementary Data 14)51. Significantly, 102 of the 130 transcripts enriched in initiating maize embryonic organs appear in one of three phylostrata: either phylostratum PS1 (29; unicellular eukaryotes and prokaryotes), PS2 (36; all eukaryotes), or PS4 (37; Bryophyta). Less than 10 percent of this embryonic gene set comprises newer transcripts (from PS5-PS13) that appear to have arisen later in plant evolution (Supplementary Figs. 9a–c). Notably, the majority of these genes appear within the Bryophyte phylostratum PS4, which marks the first appearance of the plant embryo30. In contrast, just 12 embryonic transcripts (9.23%) are identified in the algal phylostratum (PS3), which comprises plants lacking embryos. In contrast, genes expressed during the Proembryo stage are dominated by later evolved genes from PS9-PS11 (i.e., the Poaceae or grasses, the panicoid grasses, and the genus Zea; Fig. 5a), whereas transcripts from Stage 2 embryos and seedling leaves are enriched for newly evolved genes from PS12 and PS13 and the genus Zea (Supplementary Data 15).

a Phylostratigraphic map of maize. All maize protein-coding transcripts were assigned to thirteen phylostrata (PS1-PS13). Parentheses designate the number of genes per phylostratum; taxonomic groupings are marked above the corresponding PS. b–c The transcriptome age index (TAI; b) and transcriptome divergence index profiles (TDI; c) during maize embryogenesis and formation of seedling leaves after germination. Plots were rendered using Python package Matplotlib; embryo images created in Adobe Photoshop™ version 25.7.0. Figure labels: PE, Proembryo stage; T, transition stage; C, Coleoptilar stage; L, Stage 1 leaf; Scu, Stage 1 scutellum; Col, Stage 1 coleoptile; P0 and P1, Plastochron stages of the 15th and 14th leaves in the 14-day after planted seedlings. The gray lines represent the standard deviation estimated by bootstrap analysis as described22. The overall patterns of TAI and TDI profiles are significant, as measured by the flat line test (PTAI = 3.56 × 10−6, PTDI = 4.80 × 10−4), reductive hourglass test (PTAI = 1.49 × 10−4, PTDI = 1.53 × 10−3), and reductive early conservation test (PTAI = 1, PTDI = 0.998).
The transcriptome age index (TAI) measures gene age adjusted by gene expression level for each embryogenic stage (Supplementary Data 16). As shown in Fig. 5b and Supplementary Fig. 10c, the TAI displays a “classic” hourglass pattern featuring a significant decrease beginning at the Proembryo stage that reaches a minimum value during Stage 1, followed by an increased TAI at Stage 2 that continues in the newly initiated leaves of later, germinated maize seedlings. The transcriptome divergence index (TDI) measures the average sequence divergence of genes (Ka/Ks ratios), weighted by transcript accumulation level, at different developmental stages (Supplementary Data 17). Consistent with the TAI results, the TDI also showed an hourglass pattern that commences at the Proembryo stage and extends to Stage 1, followed by a notable increase at Stage 2 and in germinated seedling leaf primordia (Figs. 5c and Supplementary Fig. 10d–e). Thus, measures of both transcriptional gene age and gene sequence divergence reveal a mid-embryogenic, “hourglass” pattern during maize embryogenesis. The data also reveal that homologous genes are expressed during mid-embryogenesis in maize and Arabidopsis (Fig. 4e), as is predicted for two seed plants from the same proposed, morphological phylotype13,28.
Cross-phyla transcriptomic comparisons of plant embryogenesis reveal an inverse hourglass pattern
A comprehensive transcriptomic survey of embryonic/sporophytic development in the non-vascular plant Physcomitrium patens is available for cross-phylum analyzes of gene expression52. Ortiz-Ramirez et al.52 sampled gene expression in the zygote and young embryo housed within the gametophytic archegonium, and from four subsequent ontogenetic increments in moss embryo development (labeled S1-3, and SM). As described in Janzen (1929)11 and modeled in Fig. 6a, key morphogenic events in P. patens sporophyte development include the establishment of a multicellular embryo during S1 [5-6 days after fertilization (AF)] and subsequent proliferative growth of the seta and spore capsule during both the S2 (9-11 days AF) and S3 stages (18-20 days AF) (Fig. 6a). Formation of the sporangial epidermal layers occurs during S3, followed by meiosis in the mature sporangium during the SM stage (28-30 days AF). It is important to note that the archegonium samples consist primarily of gametophytic, and not embryonic/sporophytic tissues52.

a Development of embryo/sporophyte in the moss P. patens. b Phylostratigraphic map of P. patens. All P. patens protein-encoding transcripts were assigned to one of five phylostrata (PS1-PS5). Parentheses refer to the number of genes per phylostratum; taxonomic groupings are noted. c The transcriptome age index (TAI) and transcriptome divergence index (TDI) profiles of P. patens archegonia; sporophyte stages S1, S2, S3 and SM. Colored bars illustrate the duration and timing of different S stages along the 30-day time scale. d Comparative expression levels of stage-specific embryonic genes between Arabidopsis and P. patens, and maize and P. patens (for exact correlation coefficients r and p values, please refer to Source Data). Figure labels: (in moss) AR, archegonium; S1, sporophyte 1 stage; S2, sporophyte 2 stage; S3, sporophyte 3 stage; SM, mature sporophyte stage; (in maize) PE, Proembryo stage; T, Transition stage; C, Coleoptilar stage; L, Stage 1 leaf; Scu, Stage 1 scutellum; Col, Stage 1 coleoptile; S2, Stage 2. The gray lines represent the standard deviation estimated by bootstrap analysis as described22. The overall patterns of TAI and TDI profiles are significant, as measured by the flat line test (PTAI = 0.0131, PTDI = 4.73 × 10−3), reductive hourglass test (PTAI = 0.0332, PTDI = 0.0249), and reductive early conservation test (PTAI = 0.998, PTDI = 0.973). Images in (a) and (c) are modified from Ortiz-Ramirez et al.46.
Five phylostrata were used to determine the ages of genes enriched in P. patens embryos, whereas their sequence divergence was calculated by comparing gene homologs in Physcomitrellopsis africana (Fig. 6b and Supplementary Data 18-20). TAI and TDI were rendered for transcripts enriched during each of the five developmental increments: archegonia, and sporophyte stages S1, S2, S3, and SM. The data indicate an increasing TAI and TDI pattern from the archegonium stage to S1, whereas older embryonic transcripts increase markedly during S2, reaching a peak at S3, followed by increasing TAI and TDI at SM reflecting the use of relatively younger genes in the mature sporophyte (Fig. 6c). Thus, embryos of this moss also manifest an hourglass pattern of gene expression at mid-embryogenesis.
To test if embryos of P. patens utilize similar genes as the seed plants Arabidopsis and maize, correlations of embryonic gene expression were made across phyla. The data reveal an inverse hourglass pattern (Fig. 6d), with little correlation between mid-embryonic transcripts in moss (S2-S3) and maize or Arabidopsis. In contrast, early (S1) and later-staged (SM) embryonic-expressed genes in moss (i.e., the bulb regions of the transcriptomic hourglass) are more correlated with genes expressed during early and late stages in maize and Arabidopsis embryogenesis. Thus, as described in animal embryos, cross-phylum comparisons of plant embryonic gene expression yield an inverse hourglass pattern characterized by the expression of distinct gene sets.
Discussion
Multiplexed transcriptomic approaches were applied to investigate embryogenesis in a monocot grass plant, which has evolved novel embryonic structures with previously undefined gene expression networks. Spatial transcriptomic technologies complement the ambiguities encountered in cell cluster identification that can otherwise complicate scRNA-seq analyzes, whereas the precision of LM-RNA-seq can delineate the tissue/organ-specific transcriptomes of embryonic organs that are unresolved using spatial transcriptomics alone (Fig. 2). Moreover, high-throughput, multiplexed in situ RNA-targeting technologies enable localizations of multiple transcripts simultaneously and combinatorically, throughout embryogenesis. The RNA-targeting analyzes presented in this study support the expression of a gene network that functions during the initiation of the grass scutellum, coleoptile, and foliar leaf (Fig. 3). Thus, the combinatorial application of multivariate transcriptomic approaches enables a high-throughput and spatially-informed transcriptomic profile of grass embryo development at cellular resolution (Fig. 1).
Although questions about the evolution and homology of the cotyledon remain unresolved, genetic and morphological models indicate that the likely cotyledon evolved via the modification of the first embryonic leaf1,4,53. In support of these models, our transcriptomic comparisons of initiating, maize embryonic organs identified a conserved gene co-expression network (Fig. 4a), consistent with the proposition that the scutellum, coleoptile, and leaf 1 are indeed homologous organs. Likewise, our co-expression analyzes, RNA-targeting assays, and genetic analyzes of meristem mutants collectively support the hypothesis that the cotyledon is a bipartite organ comprising an apical, haustorial scutellum fused to a basal, emergent and sheathing coleoptile3,39. The scutellum and coleoptile group together in WGCNA analyzes (Fig. 4c, d) and distant from foliar leaves, reflecting the expression of genes that are more closely-shared within these first-elaborated embryonic organs as compared to leaves. Genetic analyzes also reveal that Zmlog7 mutants fail to make a SAM or leaves, although the scutellum and coleoptile are unaffected, likewise consistent with morphogenetic differences between leaves and both the scutellum and coleoptile (Supplementary Fig. 8). Similar phenotypes are reported for null mutations of the meristem maintenance gene KNOTTED1, wherein the SAM and leaf are absent, but the coleoptile and scutellum are unperturbed by loss of leaf and meristem integrity36. Furthermore, expression of the proximal, sheath marker genes BOPa and TRU140,44,54 is seen in the Stage 1 coleoptile but not in the scutellum (Fig. 3i–k and Supplementary Fig. 4), in support of models where the coleoptile is purported to be homologous to the proximal, leaf sheath and the scutellum comprises the highly modified distal component of a fused, bipartite, grass cotyledon3,39.
Our multiplexed transcriptomic analyzes of maize embryogenesis identify a set of 130 genes co-expressed during initiation of embryonic organs (Fig. 5b, c); these genes predominate in the descending regions of the embryonic, transcriptomic hourglass. As previously reported in the eudicot Arabidopsis and in the embryos of fruit flies (Drosophila melanogaster) and zebrafish (Danio rerio)23,24,25, the peak of the transcriptomic hourglass is defined by the preferential expression of ancient and sequence-conserved genes during maize embryogenesis. We note this period in maize embryogenesis commences during the late Proembryo stage and forms an extended hourglass shape corresponding to successive initiations of the scutellum (at Transition stage), coleoptile (at Coleoptilar stage), and leaf 1 (at Stage 1). Significant decreases in both transcript age and conservation occur during later stages of maize embryogenesis (Stage 2), and upon the initiation of seedling leaves after germination (Fig. 5b, c). Within phylum comparisons of this maize embryo organ gene set to homologous transcripts accumulating during Arabidopsis embryogenesis suggests that the maize and Arabidopsis transcriptomic hourglasses are phylotypic, although we note that just two species within this plant phylum are compared.
As in the seed plants maize and Arabidopsis, embryos of the non-vascular moss Physcomitrium patens display transcriptomic hourglass patterns that peak mid-embryonically (Fig. 6c). In this context, it is worth noting that the genes expressed in this P. patens hourglass comprise transcripts that are markedly distinct from the genes expressed during mid-embryogenesis in Arabidopsis and maize. Indeed, cross-phylum transcriptomic comparisons display an inverse hourglass pattern, where the least homologous embryonic genes are expressed at mid-embryonic stages, whereas early and later stages of moss sporophyte/embryo development show increased co-expression with maize and Arabidopsis genes (Fig. 6d). These findings are consistent with cross-phylum comparisons in animals28, and likewise support predictions that non-vascular plants and seed plants comprise distinct, embryonic phylotypes13,29.
Historically, the morphological and transcriptomic phylotypic embryonic stages in animals are mid-embryogenic20,21,22,23,24. Studies in plants so far suggest that the transcriptomic hourglass may also peak during mid-embryogeneis25 (Fig. 5b, c; Fig. 6c), although such chronological staging is especially problematic in plants where embryo-morphogenic outcomes can vary dramatically among taxa. Nonetheless, the question arises as to why older, sequence-conserved genes are preferentially utilized during mid-embryogenesis in both plants and animals? What morphogenetic characteristics embody this mid-embryogenic stage, to favor the utilization of ancient and sequence-conserved genes?
One concern when answering this question is the temporal delay between gene expression curves and the developmental phenomenology that expression evokes. The maize transcriptomic hourglass begins its steep decline in TAI at the Proembryo stage when the inner-and-outer embryonic layers are formed, and culminates during mid-embryogenic Stage 1 when the scutellum and coleoptile are expanding primordia and the first foliar leaf is just initiating5 (Fig. 1d). Similarly in Arabidopsis, the transcriptomic hourglass begins its decent during histological patterning at the globular stage, and peaks during the torpedo stage when the two cotyledon primordia exhibit expansive growth25. In addition, the expression level of the 130 genes differentially expressed in initiating embryonic organs also exhibits an hourglass pattern from the Transition stage to the Stage 1 leaf (Fig. 4b). Similar findings are reported in animal studies21,22,24. Moreover, the hourglass peak of the P. patens embryo begins during S2 and peaks during S3, stages comprising expansive growth of the spore capsule (from a central meristematic layer and histogenesis of the sporangium) (Fig. 6). Thus, although no lateral organs are formed in the moss embryo, the transcriptomic hourglass in P. patens correlates with expansive embryonic growth and histogenesis.
Likewise in Drosophila embryogenesis23,24, gene age (TAI) peaks during germband elongation23, at the onset of embryo segmentation and initiation of the organogenic imaginal disks55. Notably, the trend toward the usage of older gene transcripts in the fruit fly begins earlier in embryogenesis, during gastrulation and formation of tissue layers 23. A similar pattern is observed in zebrafish (Danio rerio), wherein the hourglass begins its descent toward older gene transcripts during gastrulation (histogenesis) and then peaks in the subsequent late segmentation/early pharyngula stages, when organ primordia initiate and grow23. A later reduction in relative transcriptomic gene age is noted during the zebrafish larval stage, which is accompanied by additional morphogenesis. Taken together, these data suggest that the mid-embryogenic period in plants and animals correlates with tissue layer organization, specification of embryonic axes, and expansive growth of embryonic organs, corresponding to the phylotypic stages of embryogenesis.
Despite their evolutionary distances, we demonstrate that in maize and a moss, as in Arabidopsis, mouse, fruit flies, and fish, the processes of embryonic tissue-layer histogenesis and primordial growth utilize old and conserved genes, comprising anciently evolved, developmental genes proposed to initiate the fundamental body plan23,24,25. Importantly, these genes expressed during the mid-embryogenesis across phyla are not homologous, but instead comprise disparate, old and conserved genes that are predicted to generate the variable, mid-embryo stage morphologies seen among different phylotypes of plants or animals13,28. At the end of this embryonic phylotypic period, plant and animal organs express newer and divergent gene sets in species-specific ways, to generate the vast morphological diversity found in nature.
Our transcriptomic analyzes of the maize embryo suggest an evolutionary mechanism for the innovation of morphological novelty. The maize embryo hourglass period displays significantly higher TAI values (expression of relatively newer genes) at the Transition and Coleoptilar stages than during Stage 1 (Fig. 5b). Notably, no such deviation in the shape of the transcriptomic hourglass is detected during cotyledon initiation in Arabidopsis embryogenesis25. These data likely reflect the relatively recent evolution of the bipartite grass cotyledon (the scutellum and coleoptile), which arose long after the cotyledon evolved from an ancestral embryonic leaf 13,4 and whose evolution involved relatively newer genes. At the same time, the initiating scutellum expresses genes highly sequence-conserved, marking the TDI minimum of maize embryogenesis (Fig. 5c). These data indicate that after the scutellum first appeared, the expression of highly-sequence-conserved genes during scutellum initiation ensured that this morphologically novel, embryonic organ was retained in subsequent generations. Relatedly speaking, the slight decrease in the use of sequence-conserved genes (TDI value) during the Coleoptilar stage may be attributable to morphogenetic differences in the initiating coleoptile, ensuring that the bipartite components of the single maize cotyledon (scutellum and coleoptile) are morphologically distinct. We interpret these data to suggest a testable, conserved mechanism for genome evolution, involving opposite trends in TAI and TDI values during the innovation of evolutionary novelty in plants and animals.
In the study, we utilized both TAI/TDI and cross-species comparison of transcriptomes to evaluate patterns during embryogenesis. Whereas TAI and TDI were used to analyze transcript age and divergence within single species during embryogenesis, these methods have limitations in cross-species comparisons. Thus, we also performed within-and-between-phyla comparisons of homologous gene expression and abundance. The integration of these two approaches provides support for the hourglass model in plant embryogenesis.
Methods
Plant Materials and growth conditions
B73 inbred lines and lines segregating log7 mutants (obtained from the Maize Genetics COOP Stock Center: UFMu-02863 mu1030680::Mu) were grown in the Gutermann greenhouse facility, Cornell University (Ithaca, NY). Transition staged, Coleoptilar staged, Leaf 1-staged, and Leaf 2-staged embryos were collected at 8–9 DAP, 10–11 DAP, 12–13 DAP, 14–15 DAP, respectively.
Single-cell transcriptomic analysis
Two batches of B73 Leaf1 staged embryos were collected in summer 2021 and winter 2022. For each batch, 200 embryos were dissected from developing seeds for protoplast isolation as previously described56. Protoplast (1000-2000 cells per µL) were loaded into the 10X Genomics Chromium platform to generate a Gel Bead-In-Emulsion (GEM). The GEM-captured RNA was reverse transcribed and amplified to make 3′ cDNA libraries, followed by the high throughput RNA sequencing on the Illumina NextSeq 2000 sequencer at the Cornell University Biotechnology Resource Center.
The resulting raw base call (BCL) files were demultiplexed to generate FASTQ files via CellRanger mkfastq v6.0 (10x Genomics, Pleasanton, CA). FASTQ reads were trimmed, aligned (to B73 reference genome V357) and assigned to cell barcodes to generate expression count matrices via CellRanger count with default settings. The expression count matrices were then analyzed using Seurat v4.4058,59,60. In detail, the matrices were filtered to remove low quality cells (number of 1500 < genes < 30,000; 6000 < UMI < 100,000). The filtered matrices were normalized and log transformed via SCTransform function. The normalized matrices were merged, and batch effects were removed via R package Harmony v1.2.061. The cell-cycle associated genes were scored and regressed via CellCycleScoring and ScaleData function. Matrix dimension was performed using RunPCA. The cell clusters and the Uniform Manifold Approximation and Projection (UMAP) plot were generated with the setting (dim = 1:40, resolution = 0.95, n.neighbors = 40, min.dist = 0.01, spread = 3). The differential expressed genes and cell cluster markers were identified via FindAllMarkers function with the log2 fold change threshold = 0.2.
Spatial transcriptomic analysis
For use in spatial transcriptomics, B73 Leaf1 staged embryos were processed for either paraffin-embedding and microtome sectioning, or cryoembedding and cryosectioning. For paraffin -embedding, two embryos were fixed in Farmer’s Fixative (Ethanol:Acetic acid = 3:1), followed by dehydration in an ethanol concentration gradient (75%, 85%, 95%, 100%) and clearing with Ethanol:Xylene gradient of 3:1, 1:1, 1:3, and 100% Xylene at room temperature. Cleared embryos were embedded in Paraplast Plus® (Leica Biosystems, Deer Park, IL). The embedded blocks were microtome-sectioned at 10 μm at room temperature; sections were subsequently mounted onto 10x VisiumTM Spatial Gene Expression Slides (10x Genomics, Pleasanton, CA). Slides were deparaffinized with two changes of 100% Xylene and 95% Ethanol for 10 min and 2 min, respectively. For cryoembedding, two kernels were dissected longitudinally to remove left and right margin of the kernel; the central region containing the embryo was embedded in Tissue-Tek® Optimal Cutting Temperature (O.C.T., Sakura Finetek, Torrance, CA), and snap-frozen in the dry ice. Frozen tissue blocks were cryosectioned at10 µm, and the sections were mounted on 10x VisiumTM Spatial Gene Expression Slide (10x Genomics, Pleasanton, CA).
The 10x VisiumTM Spatial Gene Expression Slides were processed for spatial library construction following the manufacturer instructions. Briefly, the sections were baked at 37 oC for 1 min followed by re-fixation in 100% methanol at −20 oC for 30 min. Subsequently, the sections were stained with Toluidine Blue for histological imaging. Tissue permeabilization was performed at 37 oC for 6 min to release RNA from the fixed tissue, followed by first strand cDNA synthesis. Subsequently, second strand cDNA was synthesized and amplified for library construction and sequencing. High-throughput RNA sequencing was conducted on an Illumina NextSeq 2000 sequencer at the Cornell University Biotechnology Resource Center.
Raw reads were demultiplexed, mapped to the Toluidine Blue stained images, and aligned to the B73 reference genome V357 to generate spatial expression count matrices using SpaceRanger v2.0 (10x Genomics, Pleasanton, CA). The matrices were analyzed using Seurat v458,59,60. In detail, the matrices were normalized and log transformed via SCTransform function. The normalized matrices were merged followed by dimensionality reduction. Cell clusters and the UMAP were generated with the following settings (dim = 1:40; resolution = 1.5; neighbors = 10; minimum distance = 0.01). The spatial data were visualized with DimPlot and SpatialPlot; differential expressed genes and cell cluster markers were identified via FindAllMarkers function with the log2 fold change threshold = 0.1.
Laser-microdissection RNA-seq analysis
Sixty maize Leaf1 staged kernels in two replicates were dissected longitudinally to remove the left and right margins of the kernels. This embryo-containing central region was fixed in Farmer’s Fixative (Ethanol:Acetic acid= 3:1). After fixation, the samples were dehydrated, cleared, embedded and sectioned as described previously62,63. Tissue sections were mounted on PEN membrane slides (Leica Microsystems, Deer Park, IL) pre-treated at 180 oC for 4 hours followed by UV irradiation at 254 nm for 30 minutes. The sections were deparaffinized with two changes of 100% Xylene and 100% Ethanol for 10 min and 2 min, respectively. Laser microdissection was performed using a PALM Laser Microbeam System (Zeiss Group, Oberkochen, Germany). Adaxial scutellum, SAM, leaf, as well as upper and lower coleoptiles were microdissected (total area for each captured organ is over 0.3 mm2) and RNA extraction was performed using the ArcturusTM PicoPureTM RNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA). The extracted RNA was amplified using ArcturusTM RiboAmpTM HS PLUS RNA Amplification Kit (Thermo Fisher Scientific, Waltham, MA). RNA quality control, library construction and sequencing were performed at the Cornell University Biotechnology Resource Center. The raw reads quality control was conducted via FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Raw reads were mapped to the. B73 reference genome V3 by HISAT2 (v2.1.0)64,65. Read counts and the normalized number of transcripts (transcript per million reads, TPM) were calculated as previously described66. DEseq2 (v1.26.0)67 was used to identify differentially expressed genes (DEGs) with the cutoff criteria of log2 fold change > 1 or < 1 with the false discovery rate (FDR) < 0.05.
Multiplex in situ hybridization
Transition staged, Coleoptilar staged and Leaf1 staged kernels (two replicate for each stage) were dissected longitudinally to remove the left and right kernel margins. The central region containing the embryo was fixed in 4% paraformaldehyde (PFA); fixed samples were dehydrated, cleared, embedded and sectioned as described previously68,69. Sections were mounted on 10x Genomics XeniumTM slides, deparaffinized and de-crosslinked following the manufacturer’s instructions. The Xenium custom gene expression panel containing probes of 99 maize genes were applied on each section to perform hybridization, ligation and rolling circle amplification, followed by cycles of fluorescent probe hybridization, imaging and decoding. The data was visualized by Xenium Explorer v1.3. The XeniumTM multiplex in situ hybridization assays were conducted at Memorial Sloan Kettering Cancer Center (New York, NY).
Histology and immunohistolocalizations
Developing kernels from the Proembryo stage, Transition stage, Coleoptilar stage, and the Stage 1 were fixed in FAA, dehydrated, cleared, embedded in paraplast, sectioned and stained as described previously69. Immunohistolocalizations were performed using an antibody to maize Class I KNOX proteins, KNOTTED1 and ROUGH SHEATH1, as described70.
WGCNA and hdWGCNA
For WGCNA to pursue to initiation network, log2-transformed TPM data from LM-RNA-seq (SAM data was excluded) were filtered following these criteria: row mean > 0.2 and coefficient of variation > 0.4. The general pipeline was performed as per R package instructions46 with minor modifications as follows. The network type was set to “signed”. The soft power, the minimum module size, and the merging threshold were set to 13, 20, and 0.3, respectively. For the analysis to pursue transcriptomic relationship between organs, the LM-RNA-seq data in this study was merged with the LM-RNA-seq data of the 14-day seedling P0 and P1 data48, which were integrated as a reference for later leaf ontogeny. The data was normalized and batched effect removed as described, and filtered following the same criteria as the WGCNA for the initiation network. The network type was set to “signed”. The soft power, the minimum module size, and the merging threshold were set to 16, 20, and 0.4, respectively.
For hdWGCNA, the scRNA-seq data was used to perform the analysis. The data was SCTransformed and normalized via Seurat v4.4058,59,60. The general pipeline was conducted as the R package hdWGCNA (V0.3.03) instructions49 with the following minor modifications. The fraction of cells that a gene needs to be expressed in order to be included was set to 0.001. The network type was set to “signed”, and the soft power and minimum module size were set to 10 and 30, respectively. Hypergeometric tests were performed to analyze the correlation between the hdWGCNA modules and the single-cell clusters following the previously described procedures71. The module eigengene tree representing the module similarity was generated via hclust function incorporated in the hdWGCNA package.
Statistical analysis
Hypergeometric tests were performed to analyze the correlation between single-cell clusters and VisiumTM spatial clusters, as well as between the single-cell clusters and hdWGCNA modules. We followed previously algorithms71 to statistically evaluate if the two groups of data were significantly correlated by calculating the number of overlapping gene relative to the total number of protein-coding genes in the maize B73 reference genome V357. The p values were transformed by –log10p, and visualized as heatmap built by the R package gplots (https://github.com/cran/gplots).
LM-RNA-seq read count data were normalized to fit the generalized linear model, and Empirical Bater shrinkage estimation was used for calculating dispersions, expression fold changes and adjust p values. In addition, the Pearson correlations and correlation tests were performed between (1) WGCNA module eigengenes and corresponding cell types or gene expression patterns, (2) all the embryonic genes of maize and Arabidopsis, (3) the 130 genes comprising the maize embryo organ initiation network, (4) the genes comprising the embryo organ initiation network of maize and Arabidopsis, (5) the embryonic genes of maize and Physcomitrium patens, and (6) the embryonic genes of Arabidopsis and Physcomitrium patens.
To statistically compare the expression of the initiation network between the Transition stage, Coleoptilar stage and the Stage 1, the TPM data was log2 transformed [log2(TPM + 1)] and compared using Tukey-Kramer HSD test by JMP Pro 17 (SAS Institute, Cary, NC) with default settings.
For the correlation test, the Proembryo stage, Transition stage, Coleoptilar stage, Stage 1, as well as Stage 2 TPM data was double log2 transformed, i.e. log2[log2(TPM + 1) + 1], to fix the data skewness caused by the outliers. The Arabidopsis TPM data was log2 transformed. The Pearson correlation was performed in R v4.3.2 with the significance cutoff value p < 0.05.
Calculating TAI and TDI for embryo transcriptomic data
Irie & Sehara-Fujisawa (2007) determined the ancestor index of genes expressed throughout embryo development in multiple chordates. The procedures of constructing a phylostratigraphic map, calculating TAI and TDI, and statistical tests, have been presented previously23,25,51,72. For each of the thirteen phylostrata for maize (Fig. 5a), the amino acid sequences of all 350 species (Supplementary Dataset 15) with completely sequenced genomes were extracted from NCBI, Ensembl genomes, Treegene (https://treegenesdb.org), Fernbase (https://fernbase.org), or Maizegdb (https://maizegdb.org). An equivalent strategy was applied for the five phylostrata to P. patens (Fig. 6b), encompassing 174 species (Supplementary Dataset 17). Each of the 63,031 amino acid sequences in Zea mays ssp. mays (v3) and 24, 002 sequences in P. patens (v1.6) with minimum length of 30 amino acids was analyzed by BLASTp (BLAST version 2.15.0) with an E-value cut-off of 10-5. Each such gene of Zea mays ssp. mays was assigned to the phylogenetically most distant (oldest) phylostratum containing at least one species with at least one blast hit. In circumstances where no blast hit was identified in older phylostrata, the Zea mays ssp. mays gene was assigned to phylostratum 13 (PS13; Fig. 5a). In this way, each gene of P. patens was likewise assigned to one P. patens phylostratum, from PS1 to PS5 (Fig. 6b).
To evaluate gene sequence divergence, the Ka/Ks ratio was calculated between maize and its sister species Zea diploperennis or Zea Mexicana, between Physcomitrium patens and the closely-related species Physcomitrellopsis africana73. Ka and Ks refer to the number of non-synonymous and synonymous substitutions, respectively, between each orthologous pair. Orthologous gene pairs were determined with the method of best hits using BLASTp. Amino acid sequence alignments of each pair generated with MAFFT74 (L-INS-i option) were used for codon alignments generated with PAL2NAL75 to compute sequence divergence levels (Ka/Ks) with KaKs Calculator (version 3.0)76. Gene pairs with Ka < 0.5, Ks < 5 and Ka/Ks ratios < 2 were retained.
Transcriptomic data for maize embryonic developmental stages were collected from four data sources 1) LM-RNA-seq data of Proembryo stage34, 2) organ initiation network (this study), 3) RNA-seq data of Stage 2 embryo77 and 4) LM-RNA-seq data of 14-day after germination seedling P0 and P1 leaves48. The datasets were processed with batch effect removal and normalization as described above. The TAI and the TDI are weighted means of evolutionary age and sequence divergence, respectively, and defined previously23,25. The transcriptome age index TAIs and the transcriptome divergence index TDIs of developmental stage s (s = Proembryo, Transition, Coleoptilar, Stage 1 leaf, Stage 1 scutellum, Stage 1 coleoptile, seedling leaves P0 and seedling leaves P1) are the weighted mean of the evolutionary age (phylostratum) weighted by the expression level of each gene at developmental stage s, and the weighted mean of the Ka/Ks ratio of each gene weighted by the expression level of each gene at developmental stage s, respectively. Transcriptomic data for embryo/sporophyte stages in P. patens were from Ortiz-Ramirez et al.52. Low/high PS values correspond to evolutionarily old/young genes, so low/high TAI values correspond to evolutionarily old/young transcriptomes. Low/high Ka/Ks ratios correspond to conserved/divergent genes, so low/ high TDI values correspond to conserved/divergent transcriptomes. For gene models with multiple predicted peptides, TAI was calculated with the most conserved phylostratum assigned to that locus and TDI was calculated with the conserved codon divergence strata assigned to that locus.
To determine the statistical significance of the TAI and TDI profiles, the flat line test25, reductive hourglass test and reductive early conservation test were performed via myTAI package (v1.0.1)73. The flat line test is a permutation test based on the variance of the TAI or TDI values of a given TAI or TDI profile. For the reductive hourglass test in maize, the embryonic stages were partitioned into three modules, early (Proembryo stage), mid (Transition stage, Coleoptilar stage, Stage 1 leaf, Stage 1 scutellum, and Stage 1 coleoptile), and late (Stage 2 and seedling leaves), and a permutation test was performed for the minimum differences of the mean TAI or TDI values between early and mid-modules, and between late and mid modules. For the reductive early conservation test in maize, a permutation test was performed for the minimum differences of the mean TAI or TDI values between mid and early modules, and between late and early modules. For the reductive hourglass test and reductive early conservation tests on P. patens, the three modules comprised the archegonium and S1 stage (early), S2 and S3 (middle), and SM (late). Detailed procedures are described72. The TAI and TDI curves are plot by Python package matplotlib78.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq, VisiumTM spatial transcriptomics, and LM-RNA-seq data generated in this study are deposited in the NCBI Sequence Read Archive (SRA) with the BioProject ID: PRJNA1045558. The XeniumTM multiplex in situ hybridization data generated in this study was deposited in Science Data Bank with the link: https://www.scidb.cn/s/F7ZbEz, which could be visualized by Xenium Explorer v1.3. LM-RNA-seq data for Proembryo-staged, Transition-staged, Coleoptilar-staged, and Stage 1 maize embryos is available as described in Takacs et al.34; LM-RNA-seq data for P1-staged P0-staged data are from Knauer et al.48; RNA-seq data from Stage 2 embryos is from Chen et al.77; raw RNA-seq data for Arabidopsis embryogenesis is presented in Hofmann et al.50; microarray expression data for Physcomitrium patens embryos is provided in Ortiz-Ramírez et al.52. All processed transcriptomic data generated in this study are provided in the Supplementary Data files. Source data are provided in this paper.
Code availability
The code for TAI and TDI calculations was available on GitHub (https://github.com/ruqiangzhang/Maize_hourglass; https://doi.org/10.5281/zenodo.14497921)79.
References
Kaplan, D. R. & Cooke, T. J. Fundamental concepts in the embryogenesis of dicotyledons: a morphological interpretation of embryo mutants. Plant Cell 9, 1903–1919 (1997).
Randolph, L. F. Developmental morphology of the caryopsis in maize. J. Agric Res 53, 881–916 (1936).
Kaplan, D. R., & Specht, C. D. Kaplan’s Principles of Plant Morphology. New York, NY: CRC Press (2022).
Chandler, J. W. Cotyledon organogenesis. J. Exp. Bot. 59, 2917–2931 (2008).
Abbe, E. C. & Stein, O. L. The growth of the shoot apex in maize: embryogeny. Am. J. Bot. 41, 285–293 (1954).
Bommert, P. & Werr, W. Gene expression patterns in the maize caryopsis: clues to decisions in embryo and endosperm development. Gene 271, 131–142 (2001).
Nardmann, J., & Werr, W. Patterning of the maize embryo and the perspective of evolutionary developmental biology. In: Handbook of Maize: Its Biology (eds Jeff L. Bennetzen, Hake SC). Springer (2009).
Weatherwax, P. Position of scutellum and homology of coleoptile in maize. Botanical Gaz. 69, 179–182 (1920).
Boyd, L. Evolution in the monocotyledonous seedling: a new interpretation of the morphology of the grass embryo. Trans. Botanical Soc. Edinb. 30, 286–303 (1931).
Chen, M. et al. Comparative analysis of embryo proper and suspensor transcriptomes in plant embryos with different morphologies. Proc Natl Acad Sci USA 118 (2021).
Janzen, P. Die Entwicklung und Biologie von Funaria hygrometrica. In: L. Loeske, ed., Die Laubmoose Europas. II. Funariaceae. pp, 72 -111. Verlag Max Lande, Berlin-Schoneberg (1929).
Reski, R. Development, genetics and molecular biology of mosses. Botanica Acta 111, 1–15 (1998).
Niklas, K. J. et al. Haeckel’s biogenetic law and the land plant phylotypic stage. BioScience 66, 510–519 (2016).
Dresselhaus, T. & Jürgens, G. Comparative embryogenesis in angiosperms: activation and patterning of embryonic cell lineages. Annu. Rev. Plant Biol. 72, 641–676 (2021).
Frank, M. H. & Scanlon, M. J. Transcriptomic evidence for the evolution of shoot meristem function in sporophyte-dominant land plants through concerted selection of ancestral gametophytic and sporophytic genetic programs. Mol. Biol. Evolution 32, 355–367 (2015).
Haeckel, E. Generelle Morphologie der Organismen. Allgemeine Grundzüge der Organischen Formen-Wissenschaft, Mechanisch begründet durch die von Charles Darwin reformierte Descendenz-Theorie, vols I and II. Verlag Georg Reimer, Berlin (1866).
Meckel, J.F. Entwurf einer Darstellung der zwischen dem Embryozustande der höheren Tiere und dem permanenten der niederen stattfindenden Parallele: Beyträge zur vergleichenden Anatomie. Carl Heinrich Reclam, Leipzig (1811).
Serrés, E. R. A. Théorie des formations organiques, ou rescherches d’anatomie transcendante sur les lois de l’organogénie, appliquée à l’anatomie pathologique. Annales de. la Soc. des. Sci. Naturelles et. d. ’Archeologie de. Toulon et. du Var. 21, 5–49 (1827).
Sander, K. In Development and Evolution: the sixth Symposium of the British Society for Developmental Biology. Cambridge University Press. (1983).
Irie, N. & Sehara-Fujisawa, A. The vertebrate phylotypic stage and an early bilaterian-related stage in mouse embryogenesis defined by genomic information. BMC Biol. 5, 1 (2007).
Hazkani-Covo, E. et al. In search of the vertebrate phylotypic stage: a molecular examination of the developmental hourglass model and von Baer’s Third Law. J. Expert Zool. (Mol. Dev. Evol.) 304B, 150–158 (2005).
Cruickshank, T. & Wade, M. J. Microevolutionary support for a developmental hourglass: gene expression patterns shape sequence variation and divergence in Drosophila. Evol. Dev. 10, 583–590 (2008). 5.
Domazet-Loso, T. & Tautz, D. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468, 815–818 (2010).
Kalinka, A. T. et al. Gene expression divergence recapitulates the developmental hourglass model. Nature 468, 811–814 (2010).
Quint, M. et al. A transcriptomic hourglass in plant embryogenesis. Nature 490, 98–101 (2012).
Irie, N. & Kuratani, S. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat. Commun. 2, 248 (2011).
Hu, H. et al. Constrained vertebrate evolution by pleiotropic genes. Nat. Ecol. Evol. 1, 1722–1730 (2017).
Levin, M. et al. The mid-developmental transition and the evolution of animal body plans. Nature 531, 637–641 (2016).
Drost, H.-G. et al. Cross-kingdom comparison of the develomental hourglass. Curr. Opin. Genet Dev. 45, 69–75 (2017).
Soltis, P.S. et al. The diversification of flowering plants. In: Assembling the tree of life, (eds Cracraft J, Michael J D). Oxford University Press (2004).
Satterlee, J. W. et al. A Wox3-patterning module organizes planar growth in grass leaves and ligules. Nat. Plants 9, 720–732 (2023).
Kaplan, D. R. The concept of homology and its central role in the elucidation of plant systematic relationships. In: Cladistics: Perspectives on the Reconstruction of Evolutionary History (eds Duncan T, Stuessy TF). First Edition edn. Columbia University Press (1984).
Xu, X. et al. Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Dev. Cell 56, 557–568 e556 (2021).
Takacs, E. M. et al. Ontogeny of the maize shoot apical meristem. Plant Cell 24, 3219–3234 (2012).
Shubin, N. et al. Deep homology and the origins of evolutionary novelty. Nature 457, 818–823 (2009).
Smith, L. G. et al. Expression of knotted1 marks shoot meristem formation during maize embryogenesis. Developmental Genet. 16, 344–348 (1995).
Vollbrecht, E. et al. Shoot meristem size is dependent on inbred background and presence of the maize homeobox gene, knotted1. Development 127, 3161–3172 (2000).
Nardmann, J. et al. The maize duplicate genes narrow sheath1 and narrow sheath2 encode a conserved homeobox gene function in a lateral domain of shoot apical meristems. Development 131, 2827–2839 (2004).
Wu, H. et al. Genetic analyses of embryo homology and ontogeny in the model grass Zea mays L. subsp. mays. N. Phytol. 243, 1610–1619 (2024).
Johnston, R. et al. Transcriptomic analyses indicate that maize ligule development recapitulates gene expression patterns that occur during lateral organ initiation. Plant Cell 26, 4718–4732 (2014).
Strable, J. et al. Maize YABBY genes drooping leaf1 and drooping leaf2 regulate plant architecture. Plant Cell 29, 1622–1641 (2017).
Toriba, T. et al. BLADE-ON-PETIOLE genes temporally and developmentally regulate the sheath to blade ratio of rice leaves. Nat. Commun. 10, 619 (2019).
Evans, M. M. The indeterminate gametophyte1 gene of maize encodes a LOB domain protein required for embryo sac and leaf development. Plant Cell 19, 46–62 (2007).
Leiboff, S. et al. Network analyses identify a transcriptomic proximodistal prepattern in the maize leaf primordium. N. Phytol. 230, 218–227 (2021).
Fukushima, K. & Hasebe, M. Adaxial-abaxial polarity: the developmental basis of leaf shape diversity. Genesis 52, 1–18 (2014).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Langfelder, P. & Horvath, S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 1, 54 (2007).
Knauer, S. et al. A high-resolution gene expression atlas links dedicated meristem genes to key architectural traits. Genome Res 29, 1962–1973 (2019).
Morabito, S. et al. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep. Methods 3, 100498 (2023).
Hofmann, F. et al. The embryonic transcriptome of Arabidopsis thaliana. Plant Reprod. 32, 77–91 (2019).
Domazet-Loso, T. et al. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 23, 533–539 (2007).
Ortiz-Ramírez, C. et al. A transcriptome atlas of Physcomitrella patens provides insights into the evolution and development of land plants. Mol. Plant 9, 205–220 (2016).
Spurr, A. R. Histogenesis and organizationof the embryo in Pinus Strobus L. Am. J. Bot. 36, 629–641 (1949).
Richardson, A. E. et al. Evolution of the grass leaf by primordium extension and petiole-lamina remodeling. Science 374, 1377–1381 (2021).
Gilbert, S. F. Developmental Biology, 6th edn. Sunderland (MA): sinauer associates (2000).
Satterlee, J. W. & Scanlon, M. J. Protoplast isolation from undifferentiated maize seedling shoot tissue. Methods Mol. Biol. 2464, 123–130 (2022).
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115 (2009).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2023).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Scanlon, M. J. et al. Laser microdissection-mediated isolation and in vitro transcriptional amplification of plant RNA. Curr. Protoc. Mol. Biol. https://doi.org/10.1002/0471142727.mb25a03s87 (2009).
Zhang, S. et al. Laser-capture microdissection of maize kernel compartments for rna-seq-based expression analysis. Methods Mol. Biol. 1676, 153–163 (2018).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Kim, D. et al. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Wu, H. et al. NAKED ENDOSPERM1, NAKED ENDOSPERM2, and OPAQUE2 interact to regulate gene networks in maize endosperm development. Plant Cell 36, 19–39 (2023).
Love, M. I. et al. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Laureyns, R. et al. An in situ sequencing approach maps PLASTOCHRON1 at the boundary between indeterminate and determinate cells. Plant Physiol. 188, 782–794 (2022).
Jackson, D. In situ hybridization in plants. In: Molecular Plant Pathology: A Practical Approach, (eds D. J. Bowles, S. J. Gurr, McPherson M). Oxford University Press (1991).
Scanlon, M. J. et al. The maize mutant narrow sheath fails to establish leaf margin identity in a meristematic domain. Development 122, 1683–1691 (1996).
Zhan, J. et al. RNA sequencing of laser-capture microdissected compartments of the maize kernel identifies regulatory modules associated with endosperm cell differentiation. Plant Cell 27, 513–531 (2015).
Drost, H. G. et al. Evidence for active maintenance of phylotranscriptomic hourglass patterns in animal and plant embryogenesis. Mol. Biol. Evol. 32, 1221–1231 (2015).
Vuruputoor, V. S. et al. Crossroads of assembling a moss genome: navigating contaminants and horizontal gene transfer in the moss Physcomitrellopsis africana. G3 Genes|Genomes|Genetics,14, jkae104 (2024).
Katoh, K. et al. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33, 511–518 (2005).
Suyama, M. et al. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34, W609–W612 (2006).
Zhang, Z. KaKs_Calculator 3.0: calculating selective pressure on coding and non-coding sequences. Genomics Proteom. Bioinforma. 20, 536–540 (2022).
Chen, J. et al. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 166, 252–264 (2014).
Hunter, J. D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Hao, W. et al. Multiplexed transcriptomic analyses of the plant embryonic hourglass. Zenodo https://doi.org/10.5281/zenodo.14497921 (2024).
Acknowledgements
We thank L. Evans and R. Ragas for helpful discussions of the data and comments on the manuscript. Special thanks to Cornell Genomics Facility for performing single-cell and Visium™ sequencing, to the Cornell Transcriptional Regulation and Expression Facility for RNA sequencing, and the Cornell Guterman Bioclimatic Labs for our plants. Thanks to the Integrated Genomics Operation (IGO) of Memorial Sloan Kettering Cancer Center for performing Xenium assays. H. Wu was supported by a grant from the National Science Foundation (IOS-2016021). R. Zhang is supported by another grant from the National Science Foundation (IOS-2210259).
Author information
Authors and Affiliations
Contributions
M.J.S., H.W., R.Z., and K.J.N. conceived the study. H.W., M.J.S., and R.Z. generated and analyzed the data. M.J.S., H.W., R.Z., and K.J.N. interpreted the results. M.J.S., H.W., R.Z., and K.J.N. wrote and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks C. Stewart Gillmor and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, H., Zhang, R., Niklas, K.J. et al. Multiplexed transcriptomic analyzes of the plant embryonic hourglass. Nat Commun 16, 802 (2025). https://doi.org/10.1038/s41467-024-55803-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55803-9
This article is cited by
-
How toxicity drives evolution: past, present and future
Ecotoxicology (2025)
