
Abstract
Toxic protein aggregates are associated with various neurodegenerative diseases, including Huntington’s disease (HD). Since no current treatment delays the progression of HD, we develop a mechanistic approach to prevent mutant huntingtin (mHttex1) aggregation. Here, we engineer the ATP-independent cytosolic chaperone PEX19, which targets peroxisomal membrane proteins to peroxisomes, to remove mHttex1 aggregates. Using yeast toxicity-based screening with a random mutant library, we identify two yeast PEX19 variants and engineer equivalent mutations into human PEX19 (hsPEX19). These variants effectively delay mHttex1 aggregation in vitro and in cellular HD models. The mutated hydrophobic residue in the α4 helix of hsPEX19 variants binds to the N17 domain of mHttex1, thereby inhibiting the initial aggregation process. Overexpression of the hsPEX19-FV variant rescues HD-associated phenotypes in primary striatal neurons and in Drosophila. Overall, our data reveal that engineering ATP-independent membrane protein chaperones is a promising therapeutic approach for rational targeting of mHttex1 aggregation in HD.
Similar content being viewed by others

Introduction
Maintaining proper protein homeostasis is essential for healthy cells. However, the cell is under continuous risk from newly synthesized proteins that might expose hydrophobic surfaces in the crowded cellular environment, leading to protein misfolding and aggregation1,2,3,4. To overcome these problems, cells invest in a sophisticated integrative chaperone network that supports accurate de novo protein folding, facilitates refolding of misfolded proteins, and prevents protein aggregation1,2,5,6. However, environmental stresses, genetic mutations, and aging can reduce the overall capacity of molecular chaperones, resulting in the accumulation of toxic aggregates and misfolded proteins in cells7,8,9. Such aggregates eventually lead to various diseases, including neurodegenerative diseases and type 2 diabetes10,11.
Huntington’s disease (HD) is the most common dominantly inherited neurodegenerative disorder and is caused by the abnormal expansion of CAG (polyQ) repeats in exon 1 of the huntingtin gene (Httex1)12,13. The length of polyQ repeats in the mutant Httex1 (mHttex1 with > 36 repeats) positively correlates with an increasing propensity to form aggregates and correlates inversely with the age of disease onset14,15. Aggregation of the polyQ repeat domain is also mediated by its flanking domains: the N-terminal conserved N17 domain and the C-terminal proline-rich domain (PRD). The N17 domain stimulates mHttex1 aggregation, whereas the PRD inhibits it16,17,18,19. Accumulation of mHttex1 aggregates in the nucleus and cytoplasm impairs the proteostasis network and disrupts cellular endomembranes, thus leading to dysregulation of diverse cellular processes including transcription, mitochondrial respiration, ER homeostasis, vesicular trafficking, and axonal transport20,21,22,23.
One suggested approach to correcting protein misfolding and removing pathological aggregates involves engineering a molecular chaperone to increase chaperone capacity in affected cells24,25,26. Indeed, the yeast AAA + protein disaggregase, Hsp104, has been engineered to rescue the proteotoxicity of TDP43, FUS, and α-synuclein for amyotrophic lateral sclerosis (ALS) and Parkinson’s disease24,27,28. However, most chaperones, including Hsp104, require subunit assembly, oligomerization, co-chaperones, or cofactors, such as ATP and metal ions, for optimal activity or substrate specificity5,29. Thus, engineered chaperones that rely on cellular ATP concentrations and the expression levels of their subunits and co-chaperones30 may complicate therapeutic applications. Furthermore, several ATP-independent human disaggregases such as DAXX, Karyopherin-β2, and high-temperature requirement A1 (HTRA1) are known to reverse the formation of various pathological aggregates31,32,33. These studies suggest that ATP-independent chaperones are promising therapeutic targets for treating neurodegenerative diseases.
Peroxisomal dysfunction in peroxisome biogenesis disorders is linked to neurodegenerative diseases34,35,36,37. Intriguingly, a previous study showed that PEX19-/- adult flies exhibit a defect in climbing ability, suggesting the potential functional relevance of PEX19 to neurodegeneration38. PEX19, an ATP-independent cytosolic chaperone, mediates the targeting of peroxisomal membrane proteins (PMPs) during peroxisome biogenesis39,40,41. Importantly, PEX19 does not require any co-chaperones, cofactors, or complex assembly steps for its chaperone activity. Therefore, we hypothesize that PEX19 could be readily engineered to provide a robust approach for mitigating mHttex1 proteotoxicity.
Here, using yeast toxicity-based screening42 with a random mutant library, we isolate yeast PEX19 (scPEX19) variants that suppress the proteotoxicity of mHttex1 aggregates. Using this information, we engineer the equivalent human PEX19 (hsPEX19) variants and show that they also potently suppress toxic mHttex1 aggregates. The isolated hsPEX19 variants directly bind the hydrophobic side of the amphipathic helix at the N17 domain of mHttex1, thereby effectively delaying the kinetics of mHttex1 aggregation. Overexpression of the hsPEX19 variant further rescues mHttex1-induced neurite degeneration in mouse striatal neurons and improves both the climbing ability and lifespan of flies expressing mHttex1-93Q. Altogether, our study suggests that fine-tuning the sequences of ATP-independent membrane protein chaperones could be a feasible approach to designing therapeutic chaperones for HD and potentially other diseases linked to protein aggregation.
Results
Engineered scPEX19 variants suppress the toxicity of mHttex1 in yeast
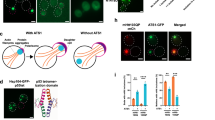
To isolate a scPEX19 mutant gene that suppresses the cellular toxicity of mHttex1 protein, we used the yeast toxicity-based screening method42 (Fig. 1a). Deletion of PRD in Httex1-97Q enhances its polyQ-induced toxicity in yeast19. This mutant is more optimal for screening since it results in a larger difference in cell viability compared to expressing non-toxic Httex1-25Q. To this end, we generated yeast strains carrying chromosomally integrated Httex1 genes (Httex1-25QΔP and Httex1-97QΔP), which encode an N-terminal FLAG tag, the first 17 amino acids of Httex1 (N17 domain), 25 or 97 repeats of glutamine, and a C-terminal GFP gene under the control of the galactose-inducible promoter (Fig. 1a). Expression of the wild-type scPEX19 did not alter the cellular toxicity of Httex1-97QΔP when compared with the empty vector control (Supplementary Fig. 1a). We randomly mutated the entire scPEX19 gene and screened the scPEX19 plasmid library (~ 2.5 × 105 library size) against Httex1-97QΔP toxicity. Among approximately 90,000 transformants, 21 colonies were able to grow on galactose plates. After assessing the cell viability of those colonies, we found that two colonies containing scPEX19 variants, m1 and m2, effectively suppressed the cellular toxicity of Httex1-97QΔP in yeast (Fig. 1b). Sequencing analysis based on colony PCR showed that these two scPEX19 variants displayed distinct single chromatogram peaks at the mutation sites (Supplementary Fig. 2). Thus, although yeast cells often harbor multiple plasmids, the selected colonies are highly likely to contain a single plasmid with the identified multiple mutations listed in Fig. 1a.

a Yeast toxicity-based screening to identify mHttex1 suppressors. scPEX19 plasmid library generated by random mutagenesis was transformed into Httex1-97QΔP-GFP-integrated yeast cells. Both scPEX19 variants and Httex1-97QΔP-GFP are under the control of the GAL1 promoter. The sequences of identified scPEX19 mutants, m1, and m2, are shown, and the common mutation sites are highlighted in red. Created in BioRender. Hyunju, C. (2025) https://BioRender.com/o38y171. b, c Growth test of Httex1-25QΔP-GFP- and Httex1-97QΔP-GFP-integrated yeast cells expressing scPEX19-WT and its scPEX19 variants. Five-fold serial dilutions of cells were spotted on galactose plates to coexpress Httex1-25QΔP or Httex1-97QΔP and the indicated scPEX19 proteins (Right) or on glucose plates as loading controls (Left). Representative images from three biological replicates (n = 3). d, e (d) Confocal microscopy images of Httex1-25QΔP-GFP and Httex1-97QΔP-GFP cells upon coexpression of scPEX19-WT, scPEX1.9-FV, and scPEX19-FI. Scale bar: 10 µm. The percentage of cells containing 97QΔP aggregates was quantified in (e). f, g (f) Representative image of Western blot monitoring SDS-insoluble and SDS-soluble Httex1-97QΔP-GFP proteins and (g) quantification of SDS-insoluble protein in (f). Yeast cells were lysed using glass beads, and then the total cell lysates were analyzed using Western blot. N-terminally FLAG-tagged Httex1-97QΔP-GFP was probed using FLAG antibody. PGK1 serves as a loading control. Data in (e), (g) are shown as mean ± SD, with three biological replicates (n = 3). Pairwise comparisons are shown as indicated, where ****p < 0.0001 by the ordinary one-way ANOVA with Tukey post-hoc test. Source data are provided as a Source Data file.
The isolated scPEX19 variants share two common mutation sites, L288F and E292V (Fig. 1a). Therefore, we hypothesized that these two mutation sites account for the ability of scPEX19 variants to rescue Httex1-97QΔP-induced toxicity in yeast. To test this hypothesis, we generated a double mutant scPEX19-L288F/E292V. The results of the spotting assay showed that scPEX19-L288F/E292V is sufficient to suppress the cellular toxicity of Httex1-97QΔP (Fig. 1b). In contrast, coexpression of the single mutants of scPEX19-L288F or scPEX19-E292V with Httex1-97QΔP did not restore cell viability (Supplementary Fig. 1b), suggesting that scPEX19-L288F/E292V is a minimally mutated suppressor of polyQ-induced toxicity in yeast. In addition, we substituted E292 with other hydrophobic amino acids on the scPEX19 variant. We found that only scPEX19-L288F/E292I suppressed Httex1-97QΔP-induced toxicity to the same degree as scPEX19-L288F/E292V (Fig. 1c), possibly due to the structural similarity between the valine and isoleucine side chains. Moreover, overexpression of scPEX19-L288F/E292V or scPEX19-L288F/E292I alone did not cause cellular toxicity at either 30 °C or 37 °C (Supplementary Fig. 1c). Therefore, we identified two scPEX19 variants, scPEX19-L288F/E292V (scPEX19-FV) and scPEX19-L288F/E292I (scPEX19-FI), that potently suppress mHttex1 toxicity in yeast.
Consistent with the results obtained with the spotting assay, both microscopy and Western blot analyses showed that scPEX19-FV and scPEX19-FI significantly reduced the aggregation of Httex1-97QΔP proteins compared to scPEX19-WT (Fig. 1d–g). Over 50% of Httex1-97QΔP was found in SDS-insoluble aggregates in scPEX19-WT expressing cells (Fig. 1f, g). In contrast, overexpression of scPEX19-FV and scPEX19-FI drastically reduced the relative amount of SDS-insoluble 97Q aggregate and simultaneously increased SDS-soluble 97Q protein levels (Fig. 1f, g, and Supplementary Fig. 1d–f). This enhancement of Httex1-97QΔP solubility by scPEX19-FV and scPEX19-FI is not due to different expression levels of PEX19 in the cells (Supplementary Fig. 1d, g).
During protein targeting to peroxisome membranes, farnesylation of the C-terminal cysteine residue in the PEX19-CaaX motif increases the binding affinity of PMPs, effectively preventing PMP aggregation in the cytosol43. In addition, the PMP-bound PEX19 is recruited to the peroxisomal membrane by PEX3, which interacts with an N-terminal αa helix in PEX1944,45,46. Noticeably, the levels of farnesylated PEX19 were significantly reduced in scPEX19-FV and scPEX19-FI overexpressing cells (Supplementary Fig. 1d, h). Therefore, we tested whether two major aspects of PEX19-mediated PMP targeting, farnesylation and interaction with PEX343,44,45,47, are crucial for ameliorating polyQ-induced cellular toxicity in yeast. To this end, we introduced two further mutations, a farnesylation-defective mutation scPEX19-C339S and a PEX3 binding-defective mutation scPEX19-ΔN, into scPEX19-WT and the toxicity-reducing variants (Supplementary Fig. 3a). The results of a spotting assay showed that coexpression of scPEX19-FV/C339S, scPEX19-FV/ΔN, scPEX19-FI/C339S, and scPEX19-FI/ΔN with Httex1-97QΔP did not alter cell growth compared to scPEX19-FV and scPEX19-FI (Supplementary Fig. 3b, c). Therefore, both farnesylation and recruitment of PEX19 to the peroxisomal membrane by binding to PEX3 are dispensable for scPEX19-FV and scPEX19-FI to suppress the cellular toxicity of Httex1-97QΔP in yeast.
hsPEX19 variants suppress mHttex1 aggregation
Due to the immunogenic potential of yeast gene-derived proteins, we used the scPEX19 variants to create equivalent human PEX19 (hsPEX19) variants. Sequence alignment analysis showed that both mutated residues (L288F/E292V and L288F/E292I) are located in the α4 helix of the PEX19 protein (Fig. 2a, green highlighted boxes). These residues are highly conserved from Human (M255/Q259) to Arabidopsis (M202/Q206) (Fig. 2a). In addition, the M255 residue of hsPEX19 directly interacts with the farnesyl group near its C-terminal end43 (Fig. 2b), suggesting that this residue could be important for substrate recognition. Due to their highly homologous sequences, we hypothesized that introducing identical mutations (M255F/Q259V or M255F/Q259I) into hsPEX19 could also enhance the suppression of mHttex1 aggregation.

a Multiple sequence alignment of the α4 helix sequences of PEX19 across various species. The alignment was performed using the Clustal Omega and displayed with ESPript 3101,102. Conserved sequences of scPEX19-L288 and scPEX19-E292 are highlighted as green boxes. b NMR structure of hsPEX19-CTD (161–299 aa) (PDB 5LNF)43. Two conserved residues, M255 and Q259, are located in the α4 helix of hsPEX19 and are shown in magenta. The M255 residue of hsPEX19 is known to bind its C-terminally modified farnesyl group (cyan)43. c, d In vitro aggregation assay of Httex1-51Q in the absence and presence of hsPEX19 proteins. 3 μM of GST-TEV-Httex1-51Q-Stag and 1.5 μM (0.5×) or 3 μM (1×) of PEX19 proteins were incubated at 30 °C, and after the addition of TEV protease, samples were quenched at the indicated time points. SDS-insoluble Httex1-51Q aggregates in (c) and their replicates were quantified and shown in (d) (n = 3, mean ± SD). e ThioflavinT fluorescence assay to measure fibril formation of Httex1-51Q. The fluorescence intensity was measured every 15 min. Data are shown as mean ± SD with n = 3 (technical replicates). f Negatively stained transmission electron micrograph (TEM) images of Httex1-51Q in the absence and presence of hsPEX19 proteins. Scale bar: 500 nm g, h (g) Confocal microscopy images of HEK293T cells coexpressing Httex1-19Q-GFP or Httex1-134Q-GFP and hsPEX19. Empty vector control (denoted as vector control) was used as a negative control. Scale bar: 50 µm. h The percentage of cells containing 134Q aggregates was quantified. i (top) Representative image of the filter trap assays monitoring the SDS-insoluble Httex1-134Q-GFP aggregates in HEK293T cells upon coexpression of hsPEX19 proteins. (bottom) Quantification of the images and their replicates. Data in (h, i) are shown as mean ± SD, with three biological replicates (n = 3). Pairwise comparisons are shown as indicated, where **p < 0.01, ***p < 0.001, ****p < 0.0001 by the ordinary one-way ANOVA with Tukey post-hoc test. Source data are provided as a Source Data file.
To test whether both purified scPEX19 and hsPEX19 variants directly prevent Httex1-51Q aggregation in vitro, we used the well-established filter trap assay that detects heat-stable, SDS-insoluble aggregates16,48. In this assay, the N-terminal Httex1-51Q can be exposed by cleaving off a GST-tag using TEV protease, thus initiating polyQ aggregation (Supplementary Fig. 4a). In the absence of a chaperone, Httex1-51Q readily formed SDS-insoluble aggregates at 3 h (Fig. 2c, d and Supplementary Fig. 4b, c). In contrast, equimolar addition of scPEX19-FV and scPEX19-FI completely suppressed aggregation of the purified Httex1-51Q protein, while scPEX19-WT was insufficient to prevent Httex1-51Q aggregation (Supplementary Fig. 4b, c). Similar to scPEX19 variants, hsPEX19 variants effectively delayed aggregation of Httex1-51Q protein in vitro although hsPEX19-FI exhibited weaker chaperone activity for Httex1-51Q than hsPEX19-FV (Fig. 2c, d). This enhanced chaperone activity of hsPEX19 variants was not due to different TEV cleavage efficiency caused by their mutations (Supplementary Fig. 4d). Furthermore, hsPEX19-FV suppressed aggregation of Httex1-51Q more effectively than the known Httex1 aggregation suppressor, nascent polypeptide-associated complex (NAC)49 (Supplementary Fig. 4e–g).
Both the ThioflavinT assay and negatively stained transmission electron micrograph (TEM) analysis showed that hsPEX19-FV effectively suppressed the formation of Httex1-51Q fibrils (Fig. 2e, f, and Supplementary Fig. 5a, b). In contrast, hsPEX19-WT did not prevent Httex1-51Q fibril formation (Fig. 2e, f, and Supplementary Fig. 5a, b). Consistent with the results of the filter trap assay (Fig. 2c, d, and Supplementary Fig. 5c, d), hsPEX19-FI also delayed fibril formation of Httex1-51Q (Fig. 2e), albeit less effectively than hsPEX19-FV. TEM analysis of hsPEX19-FI revealed both larger Httex1-51Q aggregates and small fibrils at 15 h and 24 h, while hsPEX19-FV exhibited a complete suppression of Httex1-51Q fibril formation at the tested time points (Fig. 2f and Supplementary Fig. 5a, b). Collectively, hsPEX19-FV suppressed Httex1-51Q aggregation and fibril formation more effectively than hsPEX19-FI.
Similar to other amyloid diseases, external seeds are known to promote aggregation of mHttex1 in vitro50,51. Furthermore, the mHttex1 seeding activities detected in cerebrospinal fluid and brain tissues of HD mice and patients have been suggested to positively correlate with disease progression50,52,53. Therefore, we tested whether hsPEX19 variants inhibit the seed-catalyzed aggregation of mHttex1 (Supplementary Fig. 6a, b). In the presence of 100 nM of seed (~ 3% of total Httex1-51Q), fibril formation of Httex1-51Q was significantly increased compared to the non-seeded sample at 36 h (Supplementary Fig. 6a). Both hsPEX19-FV and hsPEX19-FI exhibited a complete suppression of Httex1-51Q fibril formation at 36 h, even in the presence of 100 nM seed (Supplementary Fig. 6b), suggesting that the PEX19 variants exhibit the potential to reduce the seeded aggregation of mHttex1. In contrast, hsPEX19 variants were unable to redissolve preformed Httex1-51Q aggregates when added at 3 h, suggesting that they do not have disaggregase activity (Supplementary Fig. 6c, d). Therefore, we conclude that hsPEX19 variants function as a holdase that prevents the initial aggregation process of Httex1-51Q.
To test whether hsPEX19 variants are also effective in reducing mHttex1 aggregation in a mammalian HD model, we coexpressed hsPEX19 variants with Httex1-19Q-GFP or Httex1-134Q-GFP in HEK293T cells54. Overexpression of hsPEX19-FV and hsPEX19-FI at ~ 3-fold over endogenous PEX19 levels strongly prevented the aggregation of Httex1-134Q, as demonstrated by both fluorescence microscopy analysis and the filter trap assay (Fig. 2g–i and Supplementary Fig. 7a–d). In contrast, overexpression of hsPEX19-WT reduced the Httex1-134Q aggregates by ~ 50% on average, suggesting that hsPEX19-WT itself exhibits a mild chaperone activity toward mHttex1 proteins, as also supported by the in vitro aggregation assays (Fig. 2g–i). The difference in rescuing effects observed in hsPEX19 variants relative to their wild-type protein was not due to different expression levels of exogenous PEX19 or Httex1-134Q (Supplementary Fig. 7a–c). In contrast to scPEX19 variants, over 95% of both hsPEX19-WT and its variants were farnesylated in HEK293T cells (Supplementary Fig. 7d). Consistent with this, overexpression of hsPEX19 variants did not perturb the peroxisomal localization of the peroxisomal membrane protein PMP70, suggesting that this approach is unlikely to interfere with peroxisome biogenesis (Supplementary Fig. 7e, f). Therefore, these data demonstrate that the substitution of two conserved residues on the α4 helix of hsPEX19 significantly increases its chaperone activity toward mHttex1.
PEX19 variants bind the N17 domain of mHttex1
The N17 domain of Httex1 has an amphipathic helical property, which contributes to the initiation and acceleration of mHttex1 aggregation16,55 (Fig. 3a). Furthermore, a recent study suggested that structural coupling between the N17 and polyQ repeat domains stabilizes the helical content of Httex1 and accelerates aggregation56. Deletion of the N17 domain of Httex1-51Q (Httex1-51Q-ΔN) delays the kinetics of Httex1-51Q aggregation16. Given that hsPEX19 variants generate a more hydrophobic environment at their C-terminal domain (CTD) than hsPEX19-WT, we hypothesized that they bind to the hydrophobic amino acids in mHttex1, possibly at the N17 domain of mHttex1. Thus, we tested whether hsPEX19 variants also suppress Httex1-51Q-ΔN aggregation in vitro. In contrast to Httex1-51Q-WT, hsPEX19 variants were insufficient to suppress the aggregation of Httex1-51Q-ΔN (Fig. 3b, c). Consistent with this, GST-Httex1-51Q-WT readily bound to hsPEX19-FV, whereas no binding was observed with GST-Httex1-51Q-ΔN (Supplementary Fig. 8a, b). Furthermore, hsPEX19 variants did not suppress the aggregation of another polyQ repeat protein, Ataxin3 (Fig. 3d–f). In contrast, both hsPEX19-WT and its variants reduced aggregation of the N-terminally N17-fused Ataxin3-78Q (N17-ATX3-78Q) to a similar extent (Fig. 3e, g). Since the N17 domain in N17-Ataxin3-78Q is distally located from the polyQ repeat domain (Fig. 3d), it is plausible that hsPEX19 variants more effectively suppress aggregation of the polyQ proteins when N17 is coupled to the polyQ repeat domain. Taken together, these results suggest that the N17 domain could be the primary recognition site of hsPEX19 variants within the Httex1-51Q protein.

a N-terminal GST-tagged Httex1-51Q-WT or Httex1-51Q-ΔN were used to monitor Htt51Q aggregation or interaction between hsPEX19 and Httex1-51Q proteins in vitro. The helical wheel illustrates the distribution of hydrophobic amino acids in the amphipathic helix of the N17 domain of Httex1-51Q. The sequences of hydrophobic amino acids in the N17 domain are also highlighted in red. “*” represents the Bpa incorporation site on Httex1-51Q. b, c In vitro aggregation assay of Httex1-51Q-ΔN in the absence and presence of hsPEX19 variants. SDS-insoluble aggregates of Httex1-51Q in (b) and their replicates were quantified and shown in (c) (n = 3, mean ± SD). d Schematic illustration of Ataxin3-78Q-WT and N17-Ataxin3-78Q. Ataxin3 consists of an N-terminal Josephin domain, several ubiquitin-interacting motifs (UIMs), and a polyQ repeat domain. The N17 domain of Httex1 was fused to the N-terminus of Ataxin3-78Q. e–g In vitro aggregation assay of Ataxin3-78Q-WT and N17-Ataxin3-78Q in the absence and presence of hsPEX19 variants. 30 µM of Ataxin3-78Q was incubated with 30 µM of hsPEX19 proteins at 37 °C. SDS-insoluble aggregates of Ataxin3-78Q-WT and N17-Ataxin3-78Q in (e) and their replicates were quantified and shown in (f) and (g), respectively (n = 3, mean ± SD). h, i Bpa crosslinking assay to monitor the direct association of hsPEX19Bpa with Httex1-51Q-WT or Httex1-51Q-ΔN. 3 μM of hsPEX19-FVBpa or hsPEX19-FIBpa was incubated with an equimolar concentration of Httex1-51Q-WT or Httex1-51Q-ΔN for 3 h at 30 °C. Crosslinked samples were analyzed using Western blots probed with PEX19 (h) and S-tag (51Q) (i) antibodies. j, k Bpa crosslinking assay to monitor the intermolecular interaction of Httex1-51Q-F11Bpa with hsPEX19-WT and its hsPEX19 variants. 3 μM of Httex1-51Q-F11Bpa was incubated with 1.5 μM of hsPEX19 proteins for 3 h at 30 °C. Crosslinked samples were subjected to Western blot analysis against PEX19 (j) and S-tag (51Q) (k) antibodies. All Bpa crosslinking assays in (h–k) were performed twice or three times independently (n = 3 for (h and i) or 2 for (j and k)). Source data are provided as a Source Data file.
To check whether the mutated hydrophobic residues in the hsPEX19 variants directly interact with Httex1-51Q, we used the Bpa crosslinking assay that employs a photocrosslinker, p-benzoyl-l-phenylalanine (Bpa) (Supplementary Fig. 9a). We site-specifically incorporated Bpa into the F255 residue of the hsPEX19 variants using amber suppression57. The addition of equimolar concentration of hsPEX19-FVBpa and hsPEX19-FIBpa to Httex1-51Q suppressed Httex1-51Q aggregation at 3 and 6 h (Supplementary Fig. 9b, c). At 3 h incubation, hsPEX19-FV×Httex1-51Q or hsPEX19-FI×Httex1-51Q crosslink at ~ 70 kDa was readily detectable, whereas there was no observed crosslinked band in the presence of Httex1-51Q-ΔN (Fig. 3h, i). Therefore, these results indicate that the F255 residue in the hsPEX19 variants specifically binds to the N17 domain of Httex1-51Q.
We further tested whether the hydrophobic amino acid residues in the N-terminal amphipathic helix of mHttex1 also bind the hsPEX19 variants (Fig. 3a). To minimize structural perturbation, we incorporated Bpa at the F11 residue of Httex1-51Q among seven hydrophobic amino acid residues (Fig. 3a). Similar to Httex1-51Q-WT, both hsPEX19-FV and hsPEX19-FI suppressed the aggregation of Httex1-51Q-F11Bpa more efficiently than hsPEX19-WT (Supplementary Fig. 9d, e). In the presence of hsPEX19 variants, two distinct hsPEX19×Httex1-51Q crosslinked bands at ~ 70 and ~ 80 kDa were observed (Fig. 3j, k), suggesting that Httex1-51Q -F11Bpa binds hsPEX19 variants, possibly with two different conformations. In contrast, hsPEX19-WT resulted in a distinct crosslinked band at ~ 80 kDa and a weak diffuse band at ~ 70 kDa (Fig. 3j, k). Consistent with its mild chaperone activity, hsPEX19-WT also binds to Httex1-51Q-F11Bpa, but likely with one dominant conformation (Fig. 3j, k). These observed differences in the aggregation and Bpa crosslinking assays are not due to different TEV cleavage efficiency (Supplementary Fig. 9f). Therefore, our data demonstrate that the F11 hydrophobic residue on Httex1-51Q directly interacts with hsPEX19 and consistent with the results in Fig. 2d, its variants have stronger chaperone activities for mHttex1.
The α4 helix of hsPEX19 variants serves as a specific binding site for the N17 domain of mHttex1
PEX19 binds to the moderately hydrophobic transmembrane domains (TMDs) of peroxisomal and mitochondrial membrane proteins39,46,58,59 (Fig. 4a). In addition, PEX19 interacts with TMDs located in diverse topologies of membrane proteins, multi-spanning PMPs, tail-anchored membrane proteins (TAs), and N-terminal signal-anchored membrane proteins (Fig. 4a). Since hsPEX19 binds to these moderately hydrophobic TMDs, we hypothesized that hsPEX19 variants might also interact with the isolated N17 domain of Httex1 (Fig. 4a). To test this, we fused the N17 domain of Httex1 to the N-terminus of the Maltose binding protein (MBP) (Fig. 4b). The hsPEX19-FVBpa and hsPEX19-FIBpa proteins readily crosslinked to the N17-MBP protein, whereas no crosslinked band appeared in the presence of wild-type MBP protein (Fig. 4c). These results suggest that the N17 domain of Httex1 is a minimum recognition motif for hsPEX19 variants that allows suppression of the mHttex1 aggregation.

a Sequences of the N17 domain of Httex1-51Q and TMDs of peroxisomal and mitochondrial membrane proteins and their Grand Average of Hydropathy (GRAVY) scores103. All listed membrane proteins are known to interact with hsPEX19 during their targeting to peroxisome or mitochondria39,46,58,59. PEX26, PEX11B, and ACBD5 are peroxisomal tail-anchored membrane proteins (TAs), whereas Fis1 is a dually localized TA in mitochondria and peroxisomes58,59. PEX13 and PMP34 are multi-spanning peroxisomal membrane proteins (PMPs)39,59. ATAD1 is an N-terminal signal-anchored membrane protein localized in both mitochondria and peroxisome46. b Schematic representation of Httex1-51Q, N17-MBP, MBP-WT, and PEX26. The isolated N17 sequence was N-terminally fused to MBP (maltose binding protein). The recombinant PEX26 protein contains the N-terminal 2 × Strep-tagged SUMO domain and the PEX26 targeting sequences (237–305 aa) encompassing the TMD and C-terminal charged tail of PEX2663. c, d Bpa crosslinking assay of hsPEX19Bpa with MBP-WT, N17-MBP, N17-MBP variants. e, f In vitro aggregation assay to monitor the chaperone activity of hsPEX19-FV and SGTA toward Httex1-51Q. SDS-insoluble Httex1-51Q aggregates in (e) and their replicates were quantified and shown in (f) (n = 3, mean ± SD). g, h Representative images of His6-PEX19 pulldown assay with PEX26 in (g) and their quantification in (h). I, FT, and E denote total input, flowthrough, and elution, respectively. The amounts of PEX26 bound to hsPEX19 were analyzed by western blot against Strep (PEX26) and PEX19 antibodies. Data in (h) are shown as mean ± SD, with n = 3 (technical replicates). i–k Bpa crosslinking assay of hsPEX19Bpa with either Httex1-51Q or PEX26. The Bpa crosslinking assays with Httex1-51Q were carried out under the same conditions as Fig. 3h and i. Prior to the Bpa crosslinking assay, 0.75 µM of PEX26 was incubated with 3 µM of hsPEX19Bpa at room temperature for 5 min. “*” represents the SDS-resistant PEX26 dimers in (k). All Bpa crosslinking assays were performed three times independently (n = 3). Source data are provided as a Source Data file.
To test the substrate specificity of hsPEX19 variants, we generated three mutants of the N17-MBP protein: N17-sc-MBP, N17-5AG-MBP, and N17-2I-MBP. N17-sc-MBP contains a scrambled sequence of five hydrophobic residues in the N17 domain, thus having no alterations in the hydrophobicity of the N17 domain (Fig. 4a). Replacement of hydrophobic residues in the N17 domain with five alanine/glycines reduces the hydrophobicity of the N17 domain (N17-5AG-MBP), whereas substituting two isoleucine residues for the lysine residues of N17 domain (N17-2I-MBP) generates a moderately hydrophobic PMP-TMD-like N17 domain (Fig. 4a). Among the three N17-MBP mutants, only N17-sc-MBP displayed a crosslinked band with hsPEX19-FVBpa, suggesting that the hydrophobicity of the N17 domain is crucial for the interaction with the α4 helix of hsPEX19 (Fig. 4d). In contrast to hsPEX19-FV, SGTA, a cytosolic co-chaperone that binds highly hydrophobic TMDs of ER TAs60,61, was unable to suppress the aggregation of Httex1-51Q (Fig. 4e, f). Collectively, our results suggest that mutations on the α4 helix of hsPEX19 enable binding to the relatively low hydrophobic N17 domain of mHttex1.
Several studies suggested that the α1 helix of PEX19-CTD serves as a binding site of PMPs41,43,62 (Supplementary Fig. 10a). We also previously showed that the M179 residue in the α1 helix of PEX19-CTD interacts with PEX26, the peroxisomal TA63 (Supplementary Fig. 10a). Given that Httex1-51Q binds to the α4 helix of hsPEX19 variants, we checked whether the hsPEX19 variants also interact with a bona fide PEX19 substrate, PEX2640. At an approximately 3-fold excess concentration of the endogenous hsPEX1964, the amounts of PEX26 loaded onto hsPEX19 variants were comparable to hsPEX19-WT, indicating that these mutations on the α4 helix of hsPEX19 do not largely alter the overall binding capacity of PEX26 (Fig. 4g, h). Consistent with the results of the His-PEX19 pulldown assay, the hsPEX19-M179Bpa-FV and -FI variants readily crosslinked with PEX26 (Supplementary Fig. 10b–d). In contrast to hsPEX19-M179Bpa variants, both hsPEX19-FVBpa and hsPEX19-FIBpa did not crosslink to PEX26 (Fig. 4i, k), suggesting that the α1 helix of PEX19 is a primary binding site for PEX26. Conversely, in contrast to hsPEX19-FVBpa and hsPEX19-FIBpa (Fig. 4i, j), hsPEX19-M179Bpa variants did not show a distinct crosslinked band with Httex1-51Q (Supplementary Fig. 10e, f). Taken together, these results suggest that F255 and V259/I259 mutations on hsPEX19 can create a specific binding site for the N17 domain of Httex1-51Q, eventually resulting in robust suppression activity for Httex1-51Q aggregation.
We tested whether hsPEX19-FV also prevents aggregation of a non-polyQ protein, TDP43, which is associated with another neurodegenerative disease, ALS. To this end, we performed an established in vitro aggregation assay using the purified TDP43-TEV-MBP-His6 protein65. The addition of TEV protease enables the initiation of TDP43 aggregation (Supplementary Fig. 11a, black). In contrast to Httex1-51Q aggregation in Fig. 2d, incubation with hsPEX19-WT or hsPEX19-FV exhibited only a minor delay in TDP43 aggregation kinetics (Supplementary Fig. 11a, blue and red). To further monitor TDP43 aggregation in cells, we generated a stable HEK293 cell line (TDP43-BiFC) that expresses both TDP43-VN and TDP43-VC. Given that phosphorylation and acetylation on TDP43 promote its aggregation66,67,68,69, we used Forskolin as a phosphorylation activator and Apicidin as an acetylation-inducing agent for TDP4370,71. Treatment with either Forskolin or Apicidin significantly increased the fluorescence intensities of TDP43-BiFC in the cytosol (Supplementary Fig. 11b, c). Overexpression of hsPEX19-WT or hsPEX19-FV showed at most a minor rescue of Forskolin or Apicidin-induced cytosolic TDP43 aggregation in HEK293 cells (Supplementary Fig. 11d–g). Together with Fig. 2, we conclude that hsPEX19-FV selectively suppresses the aggregation of mHttex1 in vitro and in mammalian cells.
hsPEX19-FV rescues HD-associated phenotypes
Since the in vitro data in Fig. 2 indicated that hsPEX19-FV has a higher chaperone activity for mHttex1 than hsPEX19-FI, we next assessed whether hsPEX19-FV can rescue the proteotoxicity of mHttex1 in primary neurons and Drosophila HD models. To test whether hsPEX19-FV protects striatal neurons from mHttex1 proteotoxicity, we coexpressed Httex1-134Q-GFP with hsPEX19-WT or hsPEX19-FV at 7 days in vitro (DIV) in primary striatal neurons (Fig. 5a). In contrast to Httex1-19Q-GFP- and vector control-coexpressing striatal neurons, at 48 h post-transfection, we observed largely fragmented neurites in the striatal neurons when coexpressed with Httex1-134Q-GFP and vector control, suggesting that mHttex1 induces neuritic degeneration72,73 (Fig. 5a, b). Over 80% of striatal neurons coexpressing Httex1-134Q-GFP and hsPEX19-FV exhibited unfragmented healthy neurites, while various degrees of fragmented neurites were observed in the Httex1-134Q-GFP-and hsPEX19-WT-coexpressing neurons (Fig. 5a, b). Furthermore, the results of the TUNEL cell death assay showed that hsPEX19-FV significantly reduced apoptotic DNA fragmentation in Httex1-134Q-GFP expressing neurons (Supplementary Fig. 12a, b). Therefore, these results suggest that hsPEX19-FV effectively protects against mHttex1-induced neurotoxicity in mouse striatal neurons.

a Confocal microscopy images of mouse striatal neurons coexpressing Httex1-19Q-GFP and Httex1-134Q-GFP with vector control, hsPEX19-WT, or hsPEX19-FV. Tuj1 (neuron marker) was stained using Tuj1 antibody. Scale bar: 20 µm. b The degrees of fragmentation in a primary neuron are classified as four fragmentation scores: score 1 (fragmented areas in a single neuron are less than 5%), score 2 (5% < fragmented areas < 50%), score 3 (50% < fragmented areas < 90%), score 4 (fragmented areas > 90%). The heatmap shows the population of fragmentation scores for each condition (from left to right, n = 23, 19, 25, 27 neurons). c Climbing ability of 12-day-old adult flies (W1118, Httex1-20Q, and Httex1-93Q) expressing vector control, hsPEX19-WT, and hsPEX19-FV in motor neurons. The data in (c) are shown as violin plots with mean and quartiles (from left to right, n = 102, 120, 119, 105, 103, 110, 101, 112, 105 adult flies). Climbing index (5 cm/5 sec). Statistical significance was evaluated using the two-way ANOVA with Tukey post-hoc test. **p < 0.01, ****p < 0.0001, ns = not significant. d Climbing ability of 10-day-old adult flies (W1118, Httex1-20Q, and Httex1-93Q) expressing vector control, hsPEX19-WT, and hsPEX19-FV in pan-neurons. Climbing index (5 cm/5 sec). The data in (d) are shown as violin plots with mean and quartiles (from left to right, n = 109, 102, 91, 251, 205, 152, 125, 103, 130 adult flies). Statistical significance was evaluated using two-way ANOVA with Tukey post-hoc test. **p < 0.01, ****p < 0.0001, ns = not significant. e–g Lifespan analysis of W1118, Httex1-20Q, and Httex1-93Q Drosophila expressing vector control, hsPEX19-WT, and hsPEX19-FV in pan-neurons. Lifespan data were plotted as Kaplan-Meier survival curves, and p-values were determined using the Log-rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test. Vector control, hsPEX19-WT, hsPEX19-FV; (e) n = 140, 148, 167, (f) n = 159, 114, 117, (g) n = 164, 151, 121 adult flies. *p < 0.05, **p < 0.01, ****p < 0.0001, ns = not significant. Genotypes of Drosophila used in (c–g) are listed in Supplementary Table 1. Source data are provided as a Source Data file.
We next tested whether the hsPEX19-FV variant could rescue HD-associated phenotypes in Drosophila HD models. To this end, we generated transgenic fly lines expressing pACU2 empty vector (vector control), hsPEX19-WT, or hsPEX19-FV and coexpressed Httex1-20Q or Httex1-93Q under the control of Elav-GAL4 (pan-neurons) or D42-GAL4 (motor neurons) drivers (Supplementary Table 1). As a negative control, we used the W1118 fly line which does not carry a Httex1 transgene. Compared to W1118/vector control and Httex1-20Q/vector control flies, motor- or pan-neuronal Httex1-93Q overexpression in Httex1-93Q/vector control flies led to a significant defect in their locomotion capacities (Fig. 5c, d). In contrast to hsPEX19-WT, hsPEX19-FV expression partially restored the impaired climbing ability of flies overexpressing Httex1-93Q in motor- and pan-neurons (Fig. 5c, d). Overexpression of hsPEX19-FV marginally increased the lifespan of W1118 flies only at later time points despite no significant effect observed in Httex1-20Q flies (Fig. 5e, f). In contrast, hsPEX19-FV significantly increased the lifespan of flies expressing Httex1-93Q (Fig. 5g). hsPEX19-WT exhibited a modest increase in survival rates in both Httex1-20Q and Httex1-93Q flies (Fig. 5f, g). Overall, these results indicated that hsPEX19-FV rescued HD-relevant behavioral deficits and improved the survival of HD flies.
We further addressed the question of whether hsPEX19-FV has a protective role in the mHttex1 aggregation-associated neurotoxicity in HD flies. The results of immunohistochemistry showed that the numbers of Httex1-93Q-positive puncta in Httex1-93Q/hsPEX19-FV flies were significantly reduced in both motor- and pan-neurons compared to Httex1-93Q/vector control flies (Supplementary Fig. 13a–h). Furthermore, despite the exclusive cytosolic localization of hsPEX19-FV, Httex1-93Q/hsPEX19-FV flies displayed nuclear-localized soluble Httex1-93Q in both motor- and pan-neurons (Supplementary Fig. 13g, h). Consistent with these results, overexpression of hsPEX19-FV in Httex1-93Q flies at 15 days of age revealed a notable enhancement in their locomotion and a significant decrease in mHttex1 aggregation levels in the HD fly brains (Supplementary Fig. 14a–c). In contrast, hsPEX19-WT exhibited no significant differences in Httex1-93Q aggregation despite a mild improvement in the climbing ability of HD flies (Supplementary Fig. 14a–c). More importantly, hsPEX19-FV ameliorated mHttex1 aggregation-associated neurotoxicity more effectively than hsDNAJB1, a known mHttex1 aggregation suppressor74,75 (Supplementary Fig. 15a–c). Taken together, hsPEX19-FV effectively reduced mHttex1 aggregation-induced neurotoxicity, thereby leading to neuroprotection in mouse striatal neurons and improved survival and locomotion in HD flies.
Discussion
Here, we used the yeast toxicity-based screening method to identify two yeast PEX19 variants, scPEX19-FV (L288F/E292V) and scPEX19-FI (L288F/E292I), that rescue the toxicity of mHttex1 in yeast. Since the sites of these mutations in the α4 helix of PEX19 are highly conserved, we further generated the human variants hsPEX19-FV (M255F/Q259V) and hsPEX19-FI (M255F/Q259I). We confirmed that hsPEX19 variants effectively suppress mHttex1 aggregation in vitro and in mammalian cells. The mutated phenylalanine residue in the α4 helix of hsPEX19 variants directly interacts with the N17 domain of mHttex1, thereby significantly delaying the aggregation of mHttex1. Finally, our results demonstrate that hsPEX19-FV rescues mHttex1-induced neuritic degeneration in primary striatal neurons and HD-associated behavioral deficits and lifespan in the Drosophila HD models.
Several chaperones have been identified as mHttex1 aggregation suppressors, which target different domains of mHttex1. Previous studies showed that the TRiC chaperonin and Hsc70 chaperone bind to the N17 domain of mHttex1, thereby preventing mHttex1 aggregation16,76. In addition, two J-domain proteins (JDPs), DNAJB6 and DNAJB8, and the β subunit of the nascent polypeptide-associated complex (NAC) directly interact with the PolyQ repeat domain, thereby suppressing polyQ-mediated aggregation49,77,78,79. Furthermore, a recent study showed that another JDP, DNAJB1, together with Hsc70 and Apg2, binds to the PRD of mHttex1, and the trimeric chaperone system prevents and redissolves mHttex1 aggregates74. The S/T-rich region of DNAJB6 was suggested to form hydrogen bonds with the polyQ residues77, whereas the positively charged N terminus of βNAC is involved in interactions with the polyQ repeat domain49. These polyQ-binding domains in DNAJB6 and βNAC are low complexity linkers located between the JD and C-terminal substrate binding domain and N-terminal unstructured small domain (~ 40 aa), respectively49,77,80. In this study, we showed that the hydrophobic interactions between the α4 helix of hsPEX19 variants and the N17 domain of mHttex1 enable the suppression of mHttex1 aggregation. In contrast to DNAJB6 and βNAC that form electrostatic interactions or hydrogen bonds with mHttex1, the amphipathic N17 domain appears to dock into the hydrophobic farnesyl group-binding groove of hsPEX19-CTD43. The hydrophobic residues in the N17 domain are likely to be protected from aqueous cytosolic environments, thereby inhibiting the self-assembly of mHttex1. Together with these previous studies, our results further suggest that, depending on their mHttex1-binding domains, chaperones can employ different molecular mechanisms to prevent mHttex1 aggregation.
Membrane protein chaperones recognize their cargo membrane proteins primarily based on the hydrophobicity and location of TMDs81,82. TMDs are typically between 15 and 30 amino acids long with widely variable hydrophobicity. Regardless of TMD location, PEX19 generally recognizes moderately hydrophobic TMDs in peroxisomal and mitochondrial membrane proteins39,41,46,58,59 (Fig. 4a). In contrast, SGTA (Sgt2 in yeast) and TRC40 (Get3 in yeast) preferentially bind to more hydrophobic TMDs located near the C-terminus in the ER TAs41,60,82,83,84. Consistent with the low hydrophobicity and N-terminal localization of the N17 domain of mHttex1 (Fig. 4a), hsPEX19-WT exhibits a mild chaperone activity toward Httex1-51Q (Fig. 2d), whereas SGTA was not able to suppress the aggregation of Httex1-51Q (Fig. 4f). The hsPEX19 variants suppressed the formation of both SDS-insoluble larger aggregates and fibrils more efficiently than hsPEX19-WT (Fig. 2c–f). Despite the unaltered overall binding capacity of hsPEX19 variants to PEX26, a hydrophobic residue, F255, in the α4 helix of hsPEX19 variants did not interact with PEX26 (Fig. 4g–k). In contrast, the α1 helix of hsPEX19, the primary binding site of PMPs41,43,62, did not bind to the N17 domain (Supplementary Fig. 10e, f), suggesting that the α4 helix of hsPEX19 variants is a specific binding site for the N17 domain of mHttex1. Alternatively, the α4 helix of hsPEX19 could be a unique structural feature that discriminates membrane proteins with low hydrophobicity of TMDs. Nevertheless, further structural analysis of hsPEX19 variants would explain how hsPEX19 variants efficiently suppress mHttex1 aggregation.
Accumulation of mHtt aggregates in the cytoplasm sequesters a variety of cytosolic proteins, thereby interfering with diverse cellular functions and endomembrane structures20,21,23,85,86. Several studies showed that cytoplasmic mHtt aggregates impair nucleocytoplasmic transport of proteins and mRNAs by sequestering nuclear-shutting factors to the aggregates85,86. Furthermore, cytoplasmic mHtt aggregates trap with the cytoskeletal transport system as well as other polyQ proteins in the cytosol, thus further disrupting axonal transport in a Drosophila HD model87. Despite the predominant nuclear localization of mHttex1 aggregates in Httex1-93Q expressing Drosophila, our results showed that overexpressing the hsPEX19-FV variant in the cytosol significantly reduces the nuclear aggregation of mHttex1 in both motor- and pan-neurons (Supplementary Fig. 13). Prior to the nuclear import of mHttex1, hsPEX19-FV could prevent the aggregation of mHttex1 in the cytosol (Supplementary Fig. 7e, f). Together with previous studies85,86,87, our results suggest that maintaining a soluble form of mHttex1 assisted by molecular chaperones in the cytosol could also modulate the conformational quality of nuclear mHttex1.
Mitigating any potential off-target effects caused by the artificial mutations on a target chaperone would be critical for further therapeutic applications. The identified hsPEX19 variants appear to be specific to HD, relative to proteins linked to other neurodegenerative diseases, potentially due to both the highly conserved sequences88 and the amphipathic helix property of N17 domain of Httex1. hsPEX19-WT and its variants were unable to suppress the aggregation of Ataxin3-78Q (Fig. 3e, f). In addition, hsPEX19-FV displayed only a very modest chaperone activity in TDP43 in vitro aggregation assays (Supplementary Fig. 11a). Substituting the E292 residue into the α4 helix of scPEX19 variants with various hydrophobic amino acid residues led to different capacities for ameliorating mHttex1-induced toxicity in yeast (Fig. 1c). These results further suggest that direct modulation of the amino acid sequences on the N17 domain-binding site of PEX19 variants could generate higher substrate specificity for HD. Therefore, further tuning other amino acid sequences in the α4 helix of hsPEX19-FV would help to eliminate unidentified side effects caused by hsPEX19 variants for HD.
The rescuing effects of hsPEX19-FV observed in mHttex1-expressing flies might not be entirely due to the increased chaperone action of hsPEX19-FV on the N17-mediated mHttex1 aggregates. We note that the PEX26 binding capacity of hsPEX19-FV was not drastically altered compared to hsPEX19-WT (Fig. 4g, h), suggesting that the variant can act on peroxisomal membrane proteins as well as mHttex1. Indeed, given that a variety of cytosolic chaperones are sequestered to mHttex1 aggregates20,21, as also observed with hsPEX19-WT in Httex1-134Q expressing HEK293T cells (Supplementary Fig. 7f), overexpression of hsPEX19-FV could assist PMP targeting to the peroxisome, thereby maintaining proper peroxisome biogenesis in mHttex1-expressing flies.
Overall, our study demonstrates that engineering an ATP-independent membrane chaperone is a feasible approach to reducing N17-mediated mHttex1 aggregates in HD. Several tetratricopeptide repeat (TPR) domain-containing chaperones, i.e., ATP-independent chaperones involved in ER and mitochondrial membrane protein biogenesis, are known to have decreased expression levels in HD patients7. Furthermore, a recent study identified the TPR domain-containing chaperones, TTC1 and TOMM70A, as mitochondrial membrane protein biogenesis factors89. Thus, our approach could be readily applicable for designing other ATP-independent membrane protein chaperones that specifically reduce mHttex1 toxicity and maintain proper membrane protein biogenesis for other organelles. Furthermore, given that amphipathic helix-mediated aggregation was previously observed in α-Synuclein, islet amyloid polypeptide, and apolipoprotein C-II90,91,92,93, this approach may not be limited to HD but could also be used for other diseases such as Parkinson’s disease, diabetes, and cardiac amyloidosis. Therefore, ATP-independent membrane protein chaperones could serve as a design platform for therapeutic development that targets various diseases associated with misfolding and aggregation of amphipathic helix.
Methods
Ethical statement
All experiments involving animal procedures were approved by the Institutional Animal Care and Use Committees (IACUC) of IBS (Deajeon, Korea). No ethical approval is required for experiments with Drosophila melanogaster.
Plasmids
To generate yeast integration plasmids for pRS306-Gal-FLAG-Httex1-25QΔP-GFP and pRS306-Gal-FLAG-Httex1-97QΔP-GFP, the insert genes for FLAG-Httex1-25QΔP-GFP and FLAG-Httex1-25QΔP-GFP were amplified from pYES2-FLAG-Httex1-25QΔP-GFP and pYES2-FLAG-Httex1-97QΔP-GFP (gift from F. Ulrich Hartl Lab)19, respectively. The vector backbone for the pRS306-Gal gene was amplified from pRS306-Gal-NDC80-RFP-MPS1 (gift from Won-ki Huh Lab)94. Gibson assembly was performed to incorporate the insert genes into the pRS306-Gal vector. For pRS413-Gal-scPEX19, the scPEX19 gene was amplified from the isolated yeast genomic DNA and further incorporated into pRS413-Gal vector using Gibson assembly. For His6-scPEX19 and His6-hsPEX19, the yeast PEX19 gene and the human PEX19 gene were cloned into pET-33b, respectively.
Yeast strains
Yeast strain used in this study is W303a (MATa, can1-100, his3-11,15, leu2-3,112, trp1-1, ura3-1, ade2-1; gift from Won-ki Huh Lab). To generate FLAG-Httex1-25QΔP-GFP and FLAG-Httex1-97QΔP-GFP integrated-W303a strains, the linearized pRS306-Gal-FLAG-Httex1-25QΔP-GFP and pRS306-Gal-FLAG-Httex1-97QΔP-GFP by NcoI were transformed into W303a strain and selected on SD media lacking Ura.
Spotting assay
Yeast cells were grown to mid-log phase in the selective media containing 2% raffinose and 0.1% glucose. Overnight cultured cells were diluted to OD600 of 0.1. The diluted cells were spotted onto 3% galactose and 1% raffinose-containing plates in serial 5-fold dilutions. Equal spotting was confirmed by spotting the same diluted cells on plates containing 2% glucose. After 2-3 days of incubation at 30 °C, the images were acquired using the iBrightTM FL 1000 imaging system (Thermo Fisher Scientific).
Yeast PEX19 library generation
A random mutagenesis library was generated by error-prone PCR using a GeneMorph II mutagenesis kit (Agilent, #200552). The cloned wild-type scPEX19 gene was used as a template, and the reactions were performed according to the manufacturer’s protocol. To generate diverse mutations, two different amounts of template (5 ng and 50 ng) were used in PCR reactions for 34 cycles, followed by two more sequential PCR reactions. The resulting PCR products in each reaction were re-amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs, #M0491L) and gel-purified using a QIAquick Gel Extraction Kit (Qiagen, #28704). The gel-purified PCR products were inserted into the pRS413 vector using Gibson assembly. The Gibson assembly mixture was desalted and then transformed into DH5α competent cells using electroporation. The transformed colonies were pooled and purified using a Midi prep kit (MACHEREY-NAGEL, # 740410.10).
Yeast toxicity-based screening
The scPEX19 plasmid library was transformed into Httex1-97QΔP strain using the LiAc transformation method95, and the cells were spread onto a 2% glucose-containing SD-His-Ura plate. Approximately 90,000 colonies were pooled and further cultured in SR-His-Ura media supplemented with 2% raffinose and 0.1% glucose overnight at 30 °C. The culture cells were adjusted OD600 to 0.004 and spread onto an SG-His-Ura plate (3% galactose and 1% raffinose). We used the scPEX19-WT-transformed Httex1-97QΔP and Httex1-25QΔP cells as negative and positive controls, respectively. The selected colonies from the SG-His-Ura plate were further confirmed using the spotting assay. The scPEX19 variants were amplified using colony PCR, and then mutation sites were identified with sequencing. The selective scPEX19 variants were generated by Quick Change mutagenesis using pRS413-Gal-scPEX19-WT as a template. Those plasmids were transformed into Httex1-97QΔP and Httex1-25QΔP cells and further confirmed their suppression activity using the spotting assay.
Preparation of yeast cell extracts
For total yeast cell extract, the cell pellets were resuspended in 100 µL of 0.3 M NaOH and incubated for 3 min at room temperature. After washing the cells with water, the cell pellets were resuspended in 100 µL of lysis buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 2% SDS) supplemented with protease inhibitor cocktail (cOmplete mini, EDTA-free protease inhibitor cocktail, Roche, # 11836170001) and then incubated at 95 °C for 5 min. After centrifugation at 15,000 × g for 5 min, the clarified lysate was subjected to Western blot analysis.
To monitor SDS-insoluble 97Q aggregates in yeast, the cell extracts were prepared as described with minor modifications96. The cells were induced at OD600 of 0.1 with 1% galactose and 3% raffinose for 4 h. The cell pellets were resuspended in 200 µL of lysis buffer (25 mM Tris (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 5% glycerol) supplemented with protease inhibitor cocktail and benzonase, and then lysed by Distruptor Genie (Scientific industries) with glass beads. The extracts were clarified by centrifugation of 500 × g for 5 min at 4 °C.
Protein expression and purification
Expression and purification of GST-Httex1-51Q-WT and GST-Httex1-51Q-ΔN proteins were performed as described18. GST-Httex1-51Q-WT and GST-Httex1-51Q-ΔN were expressed BL21 StarTM (DE3) (Invitrogen, #C601003) with 1 mM IPTG at 18 °C for 2.5 h. Cells were resuspended in PBS supplemented with 150 mM NaCl, 1 mM EDTA, and cOmpleteTM EDTA-free protease inhibitor cocktail (Roche, #11836170001). After sonication, the lysate was centrifuged at 20,000 × g for 20 min at 4 °C. The supernatant was incubated with glutathione agarose resin (Thermo Scientific, #16100) for 1 h at 4 °C. The resin was washed with PBS containing 500 mM NaCl, 5 mM MgCl2, 2 mM ATP, and then the protein was eluted with 15 mM glutathione dissolved in PBS. After dialysis in the buffer (50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5% glycerol), the purified GST-Httex1-51Q proteins were concentrated with Amicon® Ultra 30,000 MWCO centrifugal filters and 0.22 μm-filtered through prior to storing at − 80 °C.
His6-hsPEX19 or His6-scPEX19 proteins were expressed in BL21 StarTM (DE3) with 0.5 mM IPTG at 18 °C overnight. His6-SGTA, MBP-His6, and N17-MBP-His6 proteins were expressed in BL21 StarTM (DE3) with 0.1 mM IPTG for 3 h at 37 °C. Cells were resuspended in Buffer A (20 mM Tris-HCl (pH 8.0), 300 mM NaCl, 5 mM imidazole, 2 mM 2-mercaptoethanol, 10% glycerol) supplemented with cOmpleteTM EDTA-free protease inhibitor cocktail and lysed using sonication. The clarified lysate was incubated with Ni-NTA agarose resin (Qiagen, #3023), and the proteins were eluted with 300 mM imidazole dissolved in Buffer A. After dialysis in Buffer B (20 mM HEPES (pH 7.5), 150 mM NaCl, 2 mM 2-mercaptoethanol, 10% glycerol), the purified proteins were stored at − 80 °C.
To site-specifically incorporate p-benzoyl-L-phenylalanine (Bpa) into His6-hsPEX19 or GST-Httex1-51Q proteins, the coding sequences for the residue F255 in the His6-hsPEX19 variants or the residue F11 in GST-Httex1-51Q were replaced with an amber codon (TAG) using npfu-special polymerase (Enzynomics, #P100S) according to the manufacturer’s introduction. Expression plasmids for His6-hsPEX19-FVamb or His6-hsPEX19-FIamb, or GST-Httex1-51Q-F11amb and tRNACUAOpt synthetase57 were co-transformed into BL21 StarTM (DE3) cells. The expression of tRNA synthetase was induced with 0.2% arabinose at OD600 of 0.3. At OD600 of 0.6, proteins were induced with 0.5 mM IPTG and 1 mM Bpa (Bachem, #4017646) at 18 °C overnight. Bpa incorporation into the proteins was confirmed by SDS-PAGE analysis. His6-hsPEX19-FVBpa, His6-hsPEX19-FIBpa, and GST-Httex1-51Q-F11Bpa were purified in the same way as their non-Bpa proteins.
His6-Ataxin3-78Q (gift from Sheena Radford lab) and N17-His6-Ataxin3-78Q were expressed in BL21 StarTM (DE3) with 0.5 mM IPTG at 30 °C for 3 h. His6-Ataxin3-78Q and N17-His6-Ataxin3-78Q were purified using Ni-NTA, the same procedure used for His6-PEX19 proteins. The eluted proteins were loaded onto a superdexTM 200 increase 10/300 column (Cytiva, #28990944), and the monomer fractions were collected and further concentrated with Amicon® Ultra 50,000 MWCO centrifugal filter. The purified Ataxin3-78Q proteins were snap-frozen and used for the filter trap assay.
TDP43-TEV-MBP-His6 (Addgene plasmid #104480) was expressed BL21 StarTM (DE3) with 0.1 mM IPTG overnight at 18 °C. The purification of TDP43-TEV-MBP-His6 was carried out as described65 with minor modifications. Briefly, the cells were resuspended in Buffer C (20 mM Tris-HCl (pH 8.0), 1 M NaCl, 5 mM imidazole, 2 mM 2-mercaptoethanol, 10% glycerol) and then sonicated. The bound TDP43-TEV-MBP-His6 protein onto Ni-NTA resin was eluted with 300 mM imidazole dissolved in Buffer C. The eluted proteins were loaded onto a superdexTM 200 increase 10/300 column and further purified in Buffer D (20 mM Tris-HCl (pH 8.0), 300 mM NaCl, 1 mM DTT). The purified TDP43-TEV-MBP-His6 protein was concentrated using Amicon® Ultra 50,000 MWCO centrifugal filter and stored at − 80 °C.
Expression and purification of 2 × Strep-SUMO-PEX26 (237-305aa) were carried out as described in the previous study63. Briefly, the protein was induced with 0.1 mM IPTG in BL21 StarTM (DE3) at 37 °C for 1 h. Cells were lysed by incubating with 0.5% N,N-Dimethyl-1-Dodecanamine-N-Oxide (LDAO, Anatrace, #D360), and 1 × CelLyticTM B Cell Lysis Reagent (Sigma) for 40 min at room temperature. The clarified lysate was then diluted 3-fold with Buffer A and loaded onto a Strep-Tactin Sepharose column (IBA Lifesciences, #2-1201-025). The proteins were eluted with 15 mM d-Desthiobiotin (Sigma, #D1411) dissolved in the buffer (20 mM Tris-HCl (pH 8.0), 300 mM NaCl, 2 mM 2-Mercaptoethanol, 0.05% LDAO, 10% glycerol) and further dialyzed in the buffer (20 mM HEPES (pH 7.5), 200 mM NaCl, 10% glycerol, 0.05% LDAO).
The human NAC heterodimer consisting of NACα with an N-terminal 6×His tag and NACβ were expressed and purified as described previously97, with minor modifications. The cell lysate was loaded onto Ni-NTA resin, and the NAC protein was eluted with 300 mM imidazole. The eluted proteins were further purified using HiTrap Q HP anion exchange chromatography column (Cytiva, #29051325). Elution fractions containing the NAC heterodimer were pooled out and concentrated using an Amicon® Ultra 10,000 MWCO centrifugal filter.
In vitro aggregation assay – Filter trap assay
3 µM of GST-TEV-Httex1-51Q-Stag proteins were mixed with 1.5 µM or 3 µM of hsPEX19 or scPEX19 proteins in the 1X TEV reaction buffer (Invitrogen, #12575015). The polyQ aggregation reaction was initiated by adding 0.05 Units/µL of AcTEV protease (Invitrogen, #12575015) and further incubated at 30 °C. For the in vitro aggregation of Ataxin3-78Q, 30 µM of His6-Ataxin3-78Q were incubated with the equimolar concentration of hsPEX19 protein in the reaction buffer (20 mM HEPES (pH 7.5), 25 mM NaCl, 2 mM DTT, 5% glycerol) at 37 °C. Samples were quenched at the indicated time points by mixing an equal volume of the quench buffer (4% (w/v) SDS, 0.1 M DTT) and then boiling at 95 °C for 10 min. The quenched samples were filtered through a 0.22 µm cellulose acetate membrane (Hyundai Micro, #CA020090A) and then washed with 0.1% SDS. The membrane was probed using an S-tag antibody (1:3000 dilution, Invitrogen, #MA1-981) or a His antibody (1:3000 dilution, Genscripts, #A00186) and the secondary mouse antibody IRDye800 (1:15,000 dilution, LiCor, #926-32210). The membrane-trapped polyQ aggregates were detected using the iBrightTM FL1000 imaging system.
In vitro aggregation assay – Turbidity assay
7.5 µM of TDP43-MBP-His6 protein was mixed with either 7.5 µM of hsPEX19-WT or hsPEX19-FV proteins in the reaction buffer (20 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM DTT). After the addition of AcTEV protease, the optical density values at 395 nm were recorded using a BioTek Epoch 2 plate reader (Agilent).
ThT (Thioflavin T) fluorescence assay
3 µM of GST-TEV-Httex1-51Q-Stag proteins were mixed with 3 µM of hsPEX19 proteins. After supplementing the mixture of 12.5 µM Thioflavin T and 0.05 Units/µL of AcTEV protease, the samples were loaded on a clear, flat bottom 96-well black plate (Corning, #CLS3904). The fluorescence intensity values at an excitation wavelength of 440 nm and emission wavelength of 480 nm were recorded every 15 min using Infinite M Plex microplate (Tecan).
Negative-stain TEM analysis
3 µM of GST-TEV-Htt51Q-Stag proteins were mixed with 3 µM of hsPEX19 proteins in the 1X TEV reaction buffer supplemented with AcTEV protease and incubated at 30 °C for 15 h and 24 h. A copper grid coated with a continuous carbon film (Electron Microscopy Sciences, #CF300-Cu) was negatively glow-discharged at 15 mA for 30 sec. 3 µL of protein samples were applied to a glow-discharged grid, incubated at room temperature for 3 min, and washed twice with distilled water and 0.75% uranyl formate once. The samples were negatively stained with 0.75% uranyl formate for 1 min with gentle shaking. The negatively stained specimens were examined under a FEI TecnaiTM G2 spirit microscope operated at 120 kV. Micrographs were collected using an FEI Eagle 4 K x 4 K CCD camera at a nominal magnification of 15,000X with an electron dose of ~ 30 e-/A2.
Bpa crosslinking assay
In vitro, aggregation assay with hsPEX19-FVBpa/hsPEX19-FIBpa or Httex1-51Q-F11Bpa was performed as described in the section of Filter trap assay. The reaction was stopped by freezing samples at the indicated time points, and frozen reaction aliquots were crosslinked on dry ice ∼ 4 cm away from a UVP B-100AP lamp (Analytik Jena) for 10 min. Crosslinked and uncrosslinked hsPEX19 or Httex1-51Q proteins were resolved on SDS-PAGE and probed with PEX19 (1:3000 dilution, Novus Biologicals, #NBP2-43757) and S-tag (1:3000 dilution, Invitrogen, #MA1-981) antibodies, respectively.
Biolayer interferometry (BLI)
The BLI experiments were performed on an Octet R8 Protein Analysis System (Sartorius) at 30 °C. His-hsPEX19-FV protein was immobilized using a Ni-NTA biosensor (Sartorius, #18-5101). The biosensor was subsequently incubated in the assay buffer (20 mM HEPES (pH 7.5), 150 mM KoAc) containing various concentrations (ranging from 250 nM to 15.6 nM) of GST-Httex1-51Q proteins for 300 s to allow association. Dissociation was then monitored for 600 s in the assay buffer. All data were fitted with the 1:1 binding model using the Octet Analysis Studio 12 software (Sartorius) to estimate kon, koff, and KD values.
HEK293 cell culture and plasmid transfection
HEK293T cells (ATCC CRL-3216) were cultured in Dulbecco’s Modified Eagle Medium, GlutaMAXTM (DMEM, Gibco, #10569044) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco, #10082147), 100 U/ml streptomycin, and 100 µ/ml penicillin and incubated in a humidified chamber with 5% CO2 at 37 °C. For coexpression of Httex1-134Q-GFP and hsPEX19 proteins, 3 × 105 cells per well were seeded on a 6-well plate one day prior to the transfection. Transient transfections of plasmids (each 1.25 µg) were carried out with Polyethylenimine (Polysciences, #23966) according to the manufacturer’s instructions.
Filter trap assay using HEK293T cell lysates
HEK293T cell lysates were prepared for filter trap assay as described previously98 with minor modifications. Briefly, cells were harvested with PBS supplemented with 1% Triton X-100 and 1x protease inhibitor cocktail (Thermo Scientific, #87785) and further incubated on ice for 30 min. The cell lysates were bath-sonicated in ice water for 5 min. After supplementing with 1% SDS and 50 mM DTT, the cell lysates were heated at 95 °C for 10 min and stored at − 80 °C for filter trap assay and Western blot analysis. Filter trap assays were carried out as described above in the in vitro aggregation assay section. SDS-insoluble Httex1-134Q-GFP aggregates were probed with anti-GFP antibody (1:3000 dilution, Sigma, #SAB4301138) and quantified using iBright Analysis Software (Thermo Scientific). To check protein expression levels, the cell lysates were loaded onto 10% Tris-glycine gels, and then Httex1-134Q-GFP, PEX19, and actin were probed in immunoblots with GFP (1:3000 dilution, Sigma, #SAB4301138), HA (1:3000 dilution, Genscript, #26183), and actin (1:5000 dilution, Invitrogen, #MA5-15452) antibodies, respectively.
Live cell imaging
HEK293T cells (ATCC CRL-3216) were cultured and cotransfected with Httex1-134Q-GFP and hsPEX19-WT or its variants in a confocal 6-well plate (SPL Life Sciences, #230206). At 48 h post-transfection, the plate was inserted in an inverted Eclipse Ti-E microscope (Nikon) equipped with a stage-top incubator (37 °C, 5% CO2). Live cell images were acquired with a 20× 0.5 NA objective and an automated perfect focus system (PFS).
Primary neuronal cell culture and transfection
Mice were housed under a 12 h light/dark cycle (light cycle: 7 AM–7 PM) at 20–26 °C with 40–60% humidity. Striata were from mouse pups of ICR mice aged postnatal day 1 (P1). Dissected tissues were digested in an enzyme solution containing 0.1% w/v Papain, 100 µg/mL DNase I, and 1 mM HEPES in Earle’s Balanced Salt Solution (EBSS) (Sigma, #E7510-500) at 37 °C for 30 min. After incubation, the enzyme solution was carefully aspirated, and dissected tissues were rinsed with a Neurobasal A medium containing 20% FBS. The tissues were dissociated by mechanical trituration, and the isolated cells were resuspended in the neuro culture medium containing 2% v/v B-27 Supplement, 1 mM L-Glutamine, 1% Penicillin/Streptomycin in Neurobasal A medium (Gibco, #10888-022). 3 × 105 cells were plated on glass coverslips precoated with 0.1 mg/mL Poly-D-Lysine (Gibco, #A38904-01) and 5 µg/mL Laminin (Gibco, #23017-015) in a 24-well plate. Striatal neurons were cultivated at 37 °C with 5% CO2 in a humidified incubator and used for experiments at 7 days in vitro (DIV). The cells were cotransfected with 1 µg of each plasmid using Lipofectamine™ LTX Reagent with PLUS™ Reagent (Invitrogen, #15338030).
Immunofluorescence
For HEK293T cells, transfection was carried out under the same conditions for live cell imaging. At 48 h post-transfection, cells were washed with PBS and fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature. After washing with PBS twice, cells were permeabilized with 0.1% Triton X-100 for 15 min and then blocked with 2% BSA for 1 h at room temperature. Cells were incubated with HA-Tag antibody (1:100 dilution, Invitrogen, #26183) for hsPEX19-WT and its variants proteins and PMP70 antibody (1:200 dilution, Invitrogen, #PA1-650) in 0.1% BSA solution at 4 °C for two overnights. After washing with PBS, the cells were incubated with Alexa Fluor 647 secondary antibody (1:1000 dilution, Invitrogen, #A-21235) for hsPEX19 proteins and Alexa Fluor 568 secondary antibody (1:1000 dilution, Invitrogen, #A-11011) for PMP70 for 1 h at room temperature. To visualize nuclei, cells were additionally stained with 300 nM of DAPI (Invitrogen, #D1306). Images were acquired with an inverted Eclipse Ti-E microscope (Nikon) with a 60× 1.4 NA oil objective.
At 48 h post-transfection, primary neuronal cells were washed with PBS and fixed for 15 min in 4% PFA at room temperature. After washing with PBS twice, the cells were blocked with 10% Normal Donkey Serum (Jackson immunoresearch, #017-000-121) in PBS containing 0.1% Triton X-100 for 1 h at room temperature. Cells were incubated in the blocking solution anti-Tuj1 antibody (1:500 dilution, Abcam, #ab18207) overnight at 4 °C. After washing with PBS, the cells were incubated with Cy3 secondary antibody (1:1000 dilution, Jackson immunoresearch, #706-166-148) for 1 h at room temperature. Nuclei were stained with DAPI.
TUNEL assay
DNA fragmentation-associated cell death was measured by TUNEL assay21. At 48 post-transfection, primary striatal neurons were washed and fixed in 4% PFA for 15 min at room temperature. Neurons were permeabilized in 0.1% Triton X-100 and then further incubated with terminal deoxynucleotide transferase and TMR red dUTP (In Situ Cell Death Detection kit, Roche, #12156792910) for 1 h at 37 °C. Tuj1 and Nuclei were stained using an anti-Tuj1 antibody (1:500 dilution, Abcam, #ab18207) and DAPI, respectively. TUNEL-positive neurons were counted for each condition.
TDP43-BiFC measurements
HEK293 TDP43-BiFC cells were cultured in the same HEK293T cell media supplemented with 100 μg/mL Geneticin (G418). All cells were maintained in a humidified chamber with 5% CO2 at 37 °C. HEK293 TDP43-BiFC cells were plated on a 96-well plate with an Opti-MEM medium (Gibco, #31985070). After 12 h, the cells were transfected with 0.1 μg of hsPEX19-WT or hsPEX19-FV plasmid using Lipofectamine®2000 reagent (Invitrogen, #11668027). At 13 h post-transfection, TDP43-BiFC cells were treated with Forskolin (30 μM) or Apicidine (1 μM). After 36 h, nuclei were counterstained with Hoechst 33342 (Invitrogen, #H1399). Fluorescence images were automatically acquired using Operetta CLS (PerkinElmer) with a 20× water immersion objective (TDP43-BiFC; λex = 460–490 nm and λem = 500–550 nm, Hoechst; λex = 355–385 nm and λem = 430–500 nm). The fluorescence intensities of TDP43-BiFC were quantified using Harmony v4.9 software (PerkinElmer). Data for each replicate were collected from 20 fields of view per well in the 96-well plate.
Drosophila melanogaster stocks
The fly lines W1118 (stock #5905), UAS-Httex1-20Q (stock #68412), UAS-Httex1-93Q (stock #68418), UAS-hsDNAJB1 (stock #82244), UAS-hsHSPA1A (stock #97467), Elav-Gal4 (stock #8765), and D42-Gal4 (stock #8816) were obtained from the Bloomington Drosophila Stock Center (USA). All flies were maintained at 27 °C.
To generate transgenic fly lines, N-terminally HA-tagged hsPEX19-WT and hsPEX19-FV genes were subcloned into the pACU2 vector (gift from Chun Han, Cornell University). The pACU2 vector lacking the HA-hsPEX19 gene was used as a negative control. The UAS-pACU2 vector, UAS-HA-hsPEX19-WT, and UAS-HA-hsPEX19-FV transgenic fly lines were generated by BestGene, Inc. These hsPEX19 transgenic fly lines were crossed with Httex1 transgenic fly lines, and the genotypes of generated transgenic fly lines used in this study are listed in Supplementary Table 1.
Climbing assay
Ten to fifteen male flies were collected and transferred into an acrylic cylinder (3 cm diameter, 18 cm height) without the use of any CO2 anesthesia but with cotton-sealed. Prior to the climbing assay, the collected flies were transferred into a new food vial within 24 h. The flies were acclimatized for 20 min in the cylinder. The climbing ability was measured by tapping the cylinder against a table, which was recorded for 1 minute. A climbing index is the proportion of flies climbing > 5 cm from the bottom of the cylinder within 5 or 6 s. Seven technical trials were conducted for each individual experiment, and the average of these trials was considered as one biological replicate.
Lifespan assay
A maximum of 15 male flies were collected within 24 h after pupal eclosion (APE) in a food vial and transferred to a new vial every two days. The number of dead flies was counted daily until all flies died. Lifespan data were plotted as Kaplan-Meier survival curves and statistical analyses were performed using both the log-rank (Mantel-Cox) test and the Gehan-Breslow-Wilcoxon test. The log-rank and Gehan-Breslow-Wilcoxon are more sensitive to distributional differences in the survival curves at a later time and an early time99, respectively.
Immunohistochemistry
Male fly heads were dissected to extract brains in Schneider’s insect medium (Sigma, #S0146). The brains were fixed with 3.7% formaldehyde for 20 min at room temperature. After washing with the washing buffer (0.3% Triton X-100 in PBS) for 10 min (a total of six washes), the brain samples were incubated in the blocking buffer (5% Normal Donkey Serum in PBS containing 0.3% Triton X-100) for 1 h at room temperature with gentle shaking. To stain Httex1-93Q and HA-hsPEX19 proteins, the samples were incubated with Huntingtin (mEM48) mouse (1:200 dilution, Sigma-Aldrich, #MAB5374) and HA rabbit (1:200 dilution, Cell Signaling, # 3724S) primary antibodies at 4 °C overnight, respectively. After washing with the washing buffer, the samples were further incubated with rabbit Alexa Fluor 555 (1:200 dilution, Thermo Scientific, #A-21428) and mouse Alexa Fluor 488 (1: 200 dilution, Invitrogen, #A-11001) secondary antibodies for 2 h at room temperature. The stained samples were mounted onto slides using Antifade Mounting Medium with DAPI (VectorLabs, #H-1200-10). All stained brain samples were taken at 400× magnification using a 40× water immersion objective, acquired by Zeiss LSM700 confocal microscopy. Confocal microscopy images were set to a threshold to eliminate non-puncta fluorescence signals using Zeiss ZEN software. The same threshold settings were applied to all the images of the experiment. The total number of puncta in the region of interest was calculated using Fiji software.
Preparation of Drosophila head lysates
Drosophila head lysates were prepared for the filter trap assay as described in the previous study with minor modifications100. 30 male fly heads were ground for 1 min in homogenization buffer (2% SDS, 1× Protease Inhibitor Cocktail, 1 µg/mL DNase). Lysates were centrifuged at 6900 × g for 10 min at 4 °C. 75 µg of the clarified lysate was used for the filter trap assay and the Httex1-93Q aggregates were probed using MW8 antibody (1:1000 dilution, DSHB, #concentrate 0.1 mL).
Statistics & reproducibility
Statistical analysis was conducted using GraphPad Prism Software. As indicated in Figure legends, we used the Log-rank (Mantel-Cox) test, Gehan-Breslow-Wilcoxon test, one-way ANOVA with Tukey post-hoc test, and two-way ANOVA with Tukey post-hoc test. All data were shown as mean ± SD or SEM and the exact numbers of samples were shown in the figure legends. p-values were represented with asterisks: *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. No statistical method was used to predetermine the sample size, and no data were excluded from the analyses. For Drosophila experiments, flies of the same genotype were randomly selected for each experiment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated in this study are provided in the manuscript’s main text (Figs. 1–5), Supplementary information (Supplementary Figs. 1–15), and Source Data file. A previously published structure of PEX19 used to show both mutation sites and Bpa incorporation positions is available under PDB code 5LNF. Source data are provided in this paper.
References
Hartl, F. U., Bracher, A. & Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011).
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M. & Hartl, F. U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 (2013).
Ellis, R. J. & Minton, A. P. Protein aggregation in crowded environments. Biol. Chem. 387, 485–497 (2006).
Yu, I. et al. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife 5, e19274 (2016).
Balchin, D., Hayer-Hartl, M. & Hartl, F. U. In vivo aspects of protein folding and quality control. Science 353, aac4354 (2016).
Nollen, E. A. A. et al. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 101, 6403–6408 (2004).
Brehme, M. et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150 (2014).
Shemesh, N. et al. The landscape of molecular chaperones across human tissues reveals a layered architecture of core and variable chaperones. Nat.Commun. 12, 2180 (2021).
Walther, D. M. et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932 (2015).
Iadanza, M. G., Jackson, M. P., Hewitt, E. W., Ranson, N. A. & Radford, S. E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 19, 755–773 (2018).
Hipp, M. S., Kasturi, P. & Hartl, F. U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435 (2019).
A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983 (1993).
DiFiglia, M. et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 (1997).
Scherzinger, E. et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 (1997).
Gusella, J. F. & MacDonald, M. E. Molecular genetics: Unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 1, 109–115 (2000).
Tam, S. et al. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285 (2009).
Crick, S. L., Ruff, K. M., Garai, K., Frieden, C. & Pappu, R. V. Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 110, 20075–20080 (2013).
Shen, K. et al. Control of the structural landscape and neuronal proteotoxicity of mutant Huntingtin by domains flanking the polyQ tract. Elife 5, e18065 (2016).
Gruber, A. et al. Molecular and structural architecture of polyQ aggregates in yeast. Proc. Natl. Acad. Sci. USA 115, E3446–E3453 (2018).
Kim, Y. E. et al. Soluble oligomers of polyQ-expanded huntingtin target a multiplicity of key cellular factors. Mol. Cell 63, 951–964 (2016).
Riguet, N. et al. Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties. Nat. Commun. 12, 6579 (2021).
Gidalevitz, T., Ben-Zvi, A., Ho, K. H., Brignull, H. R. & Morimoto, R. I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311, 1471–1474 (2006).
Bäuerlein, F. J. B. et al. In situ architecture and cellular interactions of polyQ inclusions. Cell 171, 179–187.e10 (2017).
Jackrel, M. E. et al. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 156, 170–182 (2014).
Wang, J. D., Herman, C., Tipton, K. A., Gross, C. A. & Weissman, J. S. Directed evolution of substrate-optimized GroEL/S chaperonins. Cell 111, 1027–1039 (2002).
Quan, S. et al. Genetic selection designed to stabilize proteins uncovers a chaperone called Spy. Nat. Struct. Mol. Biol. 18, 262–269 (2011).
Mack, K. L. et al. Tuning Hsp104 specificity to selectively detoxify α-synuclein. Mol. Cell 83, 3314–3332. (2023).
Tariq, A. et al. Mining disaggregase sequence space to safely counter TDP-43, FUS, and α-synuclein proteotoxicity. Cell Rep. 28, 2080–2095 (2019).
Kampinga, H. H. & Craig, E. A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 (2010).
Sathyanarayanan, U. et al. ATP hydrolysis by yeast Hsp104 determines protein aggregate dissolution and size in vivo. Nat. Commun. 11, 5226 (2020).
Huang, L. et al. DAXX represents a new type of protein-folding enabler. Nature 597, 132–137 (2021).
Guo, L. et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173, 677–692 (2018).
Chen, S. et al. HTRA1 disaggregates α-synuclein amyloid fibrils and converts them into non-toxic and seeding incompetent species. Nat. Commun. 15, 2436 (2024).
Jo, D. S., Park, N. Y. & Cho, D.-H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 52, 1486–1495 (2020).
Cipolla, C. M. & Lodhi, I. J. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 28, 297–308 (2017).
Uzor, N.-E., McCullough, L. D. & Tsvetkov, A. S. Peroxisomal dysfunction in neurological diseases and brain aging. Front. Cell Neurosci. 14, 44 (2020).
Germain, K. et al. Upregulated pexophagy limits the capacity of selective autophagy. Nat. Commun. 15, 375 (2024).
Bülow, M. H. et al. Unbalanced lipolysis results in lipotoxicity and mitochondrial damage in peroxisome-deficient Pex19 mutants. Mol. Biol. Cell 29, 396–407 (2018).
Jones, J. M., Morrell, J. C. & Gould, S. J. PEX19 is a predominantly cytosolic chaperone and import receptor for class 1 peroxisomal membrane proteins. J. Cell Biol. 164, 57–67 (2004).
Yagita, Y., Hiromasa, T. & Fujiki, Y. Tail-anchored PEX26 targets peroxisomes via a PEX19-dependent and TRC40-independent class I pathway. J. Cell Biol. 200, 651–666 (2013).
Chen, Y. et al. Hydrophobic handoff for direct delivery of peroxisome tail-anchored proteins. Nat. Commun. 5, 5790 (2014).
Ripaud, L. et al. Overexpression of Q-rich prion-like proteins suppresses polyQ cytotoxicity and alters the polyQ interactome. Proc. Natl. Acad. Sci. USA 111, 18219–18224 (2014).
Emmanouilidis, L. et al. Allosteric modulation of peroxisomal membrane protein recognition by farnesylation of the peroxisomal import receptor PEX19. Nat. Commun. 8, 14635 (2017).
Sato, Y. et al. Structural basis for docking of peroxisomal membrane protein carrier Pex19p onto its receptor Pex3p. EMBO J. 29, 4083–4093 (2010).
Schmidt, F. et al. Insights into peroxisome function from the structure of PEX3 in complex with a soluble fragment of PEX19. J. Biol. Chem. 285, 25410–25417 (2010).
Liu, Y., Yagita, Y. & Fujiki, Y. Assembly of peroxisomal membrane proteins via the direct pex19p-pex3p pathway. Traffic 17, 433–455 (2016).
Rucktäschel, R. et al. Farnesylation of pex19p is required for its structural integrity and function in peroxisome biogenesis. J. Biol. Chem. 284, 20885–20896 (2009).
Tam, S., Geller, R., Spiess, C. & Frydman, J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 8, 1155–1162 (2006).
Shen, K. et al. Dual Role of Ribosome-Binding Domain of NAC as a Potent Suppressor of Protein Aggregation and Aging-Related Proteinopathies. Mol. Cell 74, 729–741 (2019).
Ast, A. et al. mHTT Seeding activity: A marker of disease progression and neurotoxicity in models of huntington’s disease. Mol. Cell 71, 675–688 (2018).
Pandey, N. K. et al. The 17-residue-long N terminus in huntingtin controls stepwise aggregation in solution and on membranes via different mechanisms. J. Biol. Chem. 293, 2597–2605 (2018).
Lee, C. Y. D. et al. Disease-related Huntingtin seeding activities in cerebrospinal fluids of Huntington’s disease patients. Sci. Rep. 10, 20295 (2020).
Tan, Z. et al. Huntington’s disease cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol. Psychiatry 20, 1286–1293 (2015).
Rozales, K. et al. Differential roles for DNAJ isoforms in HTT-polyQ and FUS aggregation modulation revealed by chaperone screens. Nat. Commun. 13, 516 (2022).
Sivanandam, V. N. et al. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566 (2011).
Elena-Real, C. A. et al. The structure of pathogenic huntingtin exon 1 defines the bases of its aggregation propensity. Nat. Struct. Mol. Biol. 30, 309–320 (2023).
Young, T. S., Ahmad, I., Yin, J. A. & Schultz, P. G. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374 (2010).
Costello, J. L. et al. Predicting the targeting of tail-anchored proteins to subcellular compartments in mammalian cells. J. Cell Sci. 130, 1675–1687 (2017).
Fransen, M., Wylin, T., Brees, C., Mannaerts, G. P. & Van Veldhoven, P. P. Human pex19p binds peroxisomal integral membrane proteins at regions distinct from their sorting sequences. Mol. Cell Biol. 21, 4413–4424 (2001).
Shao, S., Rodrigo-Brenni, M. C., Kivlen, M. H. & Hegde, R. S. Mechanistic basis for a molecular triage reaction. Science 355, 298–302 (2017).
Guna, A., Volkmar, N., Christianson, J. C. & Hegde, R. S. The ER membrane protein complex is a transmembrane domain insertase. Science 359, 470–473 (2018).
Schueller, N. et al. The peroxisomal receptor Pex19p forms a helical mPTS recognition domain. EMBO J. 29, 2491–2500 (2010).
Oh, J., Kim, D. K., Ahn, S. H., Kim, H. M. & Cho, H. A dual role of the conserved PEX19 helix in safeguarding peroxisomal membrane proteins. iScience 27, 109537 (2024).
Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N. & Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 (2014).
Hallegger, M. et al. TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell 184, 4680–4696 (2021).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
Brady, O. A., Meng, P., Zheng, Y., Mao, Y. & Hu, F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 116, 248–259 (2011).
Neumann, M. et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 117, 137–149 (2009).
Cohen, T. J. et al. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 6, 5845 (2015).
Lokireddy, S., Kukushkin, N. V. & Goldberg, A. L. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. USA 112, E7176–E7185 (2015).
Han, J. W. et al. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res. 60, 6068–6074 (2000).
Li, H., Li, S. H., Yu, Z. X., Shelbourne, P. & Li, X. J. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J. Neurosci. 21, 8473–8481 (2001).
Li, H., Li, S. H., Johnston, H., Shelbourne, P. F. & Li, X. J. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat. Genet. 25, 385–389 (2000).
Ayala Mariscal, S. M. et al. Identification of a HTT-specific binding motif in DNAJB1 essential for suppression and disaggregation of HTT. Nat. Commun. 13, 4692 (2022).
Rujano, M. A., Kampinga, H. H. & Salomons, F. A. Modulation of polyglutamine inclusion formation by the Hsp70 chaperone machine. Exp. Cell Res. 313, 3568–3578 (2007).
Monsellier, E., Redeker, V., Ruiz-Arlandis, G., Bousset, L. & Melki, R. Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 290, 2560–2576 (2015).
Kakkar, V. et al. The S/T-Rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol. Cell 62, 272–283 (2016).
Gillis, J. et al. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J. Biol. Chem. 288, 17225–17237 (2013).
Månsson, C. et al. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones 19, 227–239 (2014).
Karamanos, T. K., Tugarinov, V. & Clore, G. M. Unraveling the structure and dynamics of the human DNAJB6b chaperone by NMR reveals insights into Hsp40-mediated proteostasis. Proc. Natl. Acad. Sci. USA 116, 21529–21538 (2019).
Hegde, R. S. & Keenan, R. J. The mechanisms of integral membrane protein biogenesis. Nat. Rev. Mol. Cell Biol. 23, 107–124 (2022).
Chio, U. S., Cho, H. & Shan, S. Mechanisms of tail-anchored membrane protein targeting and insertion. Annu. Rev. Cell Dev. Biol. 33, 417–438 (2017).
Rao, M. et al. Multiple selection filters ensure accurate tail-anchored membrane protein targeting. Elife 5, e21301 (2016).
Cho, H. et al. Dynamic stability of Sgt2 enables selective and privileged client handover in a chaperone triad. Nat. Commun. 15, 134 (2024).
Woerner, A. C. et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351, 173–176 (2016).
Gasset-Rosa, F. et al. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 94, 48–57 (2017).
Lee, W.-C. M., Yoshihara, M. & Littleton, J. T. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 101, 3224–3229 (2004).
Tartari, M. et al. Phylogenetic comparison of huntingtin homologues reveals the appearance of a primitive polyQ in sea urchin. Mol. Biol. Evol. 25, 330–338 (2008).
Muthukumar, G. et al. Triaging of α-helical proteins to the mitochondrial outer membrane by distinct chaperone machinery based on substrate topology. Mol. Cell 84, 1101–1119 (2024).
Zhu, M. & Fink, A. L. Lipid binding inhibits alpha-synuclein fibril formation. J. Biol. Chem. 278, 16873–16877 (2003).
Burré, J., Sharma, M. & Südhof, T. C. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Neurosci. 35, 5221–5232 (2015).
MacRaild, C. A., Howlett, G. J. & Gooley, P. R. The structure and interactions of human apolipoprotein C-II in dodecyl phosphocholine. Biochemistry 43, 8084–8093 (2004).
Apostolidou, M., Jayasinghe, S. A. & Langen, R. Structure of α-Helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem. 283, 17205–17210 (2008).
Lim, G. & Huh, W.-K. Rad52 phosphorylation by Ipl1 and Mps1 contributes to Mps1 kinetochore localization and spindle assembly checkpoint regulation. Proc. Natl. Acad. Sci. USA 114, E9261–E9270 (2017).
Gietz, R. D. & Schiestl, R. H. Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 38–41 (2007).
Klaips, C. L., Gropp, M. H. M., Hipp, M. S. & Hartl, F. U. Sis1 potentiates the stress response to protein aggregation and elevated temperature. Nat. Commun. 11, 6271 (2020).
Lentzsch, A. M. et al. NAC guides a ribosomal multienzyme complex for nascent protein processing. Nature 633, 718–724 (2024).
Tashiro, E. et al. Prefoldin protects neuronal cells from polyglutamine toxicity by preventing aggregation formation. J. Biol. Chem. 288, 19958–19972 (2013).
Martinez, R. L. M. C. & Naranjo, J. D. A pretest for choosing between logrank and wilcoxon tests in the two-sample problem. METRON 68, 111–125 (2010).
Ast, A., Schindler, F., Buntru, A., Schnoegl, S. & Wanker, E. E. A filter retardation assay facilitates the detection and quantification of heat-stable, amyloidogenic mutant huntingtin aggregates in complex biosamples. Methods Mol. Biol. 1780, 31–40 (2018).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
Kyte, J. & Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 (1982).
Acknowledgements
We thank S.-h. Park, K. Shen, Y. Cho, and the members of Shan lab for valuable discussions on experimental methods. We thank the Research Solution Center and Korea Virus Research Institute at the Institute for Basic Science for the confocal microscopy facility and fluorescence plate reader, respectively. We thank W.-k. Huh, F.U. Hartl, J. Frydman, R. Shalgi, S.E. Radford, and S.-o. Shan for sharing the W303a yeast strain or plasmids. This work was supported by grants from the Institute for Basic Science (IBS-R030-Y1 to H.C. and IBS-R030-C1 to H.M.K.) and the National Research Foundation (NRF) grant funded by the Korea government (MSIT) (RS-2023-00261784 to Y.K.K. and 2022R1C1C100714612 to S.L.).
Author information
Authors and Affiliations
Contributions
H.C. conceived the study. H.C., S.B.L., and H.M.K. designed the study. J.O., C.C., E.S.K., K.W.M., H.C.J., H.K., M.K., S.H.A., N.L., M.G.J., H.S.B, and H.C. carried out all experiments. J.O., C.C., E.S.K., K.W.M., H.C.J., H.K., M.K., N.L., M.G.J., S.L., and H.C. performed data analysis. H.C., S.B.L., H.M.K., S.L., and Y.K.K. supervised the study. H.C., J.O., E.S.K., K.W.M., H.K., M.K., and S.L. drafted the manuscript. H.C. edited the manuscript with input from J.O., E.S.K., S.B.L., and H.M.K. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oh, J., Catherine, C., Kim, E.S. et al. Engineering a membrane protein chaperone to ameliorate the proteotoxicity of mutant huntingtin. Nat Commun 16, 737 (2025). https://doi.org/10.1038/s41467-025-56030-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56030-6
