
Abstract
The multi-enzyme pyruvate dehydrogenase complex (PDHc) links glycolysis to the citric acid cycle and plays vital roles in metabolism, energy production, and cellular signaling. Although all components have been individually characterized, the intact PDHc structure remains unclear, hampering our understanding of its composition and dynamical catalytic mechanisms. Here, we report the in-situ architecture of intact mammalian PDHc by cryo-electron tomography. The organization of peripheral E1 and E3 components varies substantially among the observed PDHcs, with an average of 21 E1 surrounding each PDHc core, and up to 12 E3 locating primarily along the pentagonal openings. In addition, we observed dynamic interactions of the substrate translocating lipoyl domains (LDs) with both E1 and E2, and the interaction interfaces were further analyzed by molecular dynamics simulations. By revealing intrinsic dynamics of PDHc peripheral compositions, our findings indicate a distinctive activity regulation mechanism, through which the number of E1, E3 and functional LDs may be coordinated to meet constantly changing demands of metabolism.
Similar content being viewed by others

Introduction
The pyruvate dehydrogenase complex (PDHc) is a multi-enzyme complex that plays pivotal roles in cellular energy production, biosynthetic metabolism and cellular signaling. By converting pyruvate into acetyl-coenzyme A (acetyl-CoA)1,2,3,4, PDHc links glycolysis to the citric acid cycle, promotes energy production, and catalyzes the biosynthesis of fatty acids and steroids. Recent researches have shown that PDHc has the ability to translocate from the mitochondrion to the cell nucleus, which provides a pathway for nuclear acetyl-CoA synthesis required for histone acetylation and epigenetic regulation5,6,7. PDHc also involves in cell proliferation and viability by influencing cellular signaling pathways8,9,10. Due to its central roles in maintaining cellular energy homeostasis, metabolic regulation, and cellular signaling, PDHc is associated with many major diseases, such as neurodegenerative disorders, metabolic acidosis, diabetes, and cancer11,12,13,14.
The PDHc, along with the 2-oxoglutarate dehydrogenase complex (OGDHc, also known as α-ketoglutarate dehydrogenase complex) and the branched-chain α-keto acid dehydrogenase complex (BCKDHc), belongs to the α-keto acid/2-oxo-acid dehydrogenase (OADH) family, the largest cellular enzyme system with the molecular weight ranging from 4 to 10 million Da (MDa)15. The PDHc is composed of multiple copies of three catalytic enzymes: pyruvate dehydrogenase (E1; EC1.2.4.1), dihydrolipoyl transacetylase (E2; EC 2.3.1.12) and dihydrolipoyl dehydrogenase (E3; EC 1.8.1.4)16. E1 is a α2 dimer in Gram-negative bacteria and a α2β2 heterotetramer in Gram-positive bacteria and eukaryotes. E2 is a multidomain protein consisting of 1-3 tandem lipoyl domains (LDs) separated by flexible linkers, a peripheral subunit-binding domain (PSBD) and an inner catalytic (IC) domain17,18,19 (Fig. 1a). E3 is a homodimer20,21 and is shared among different members of the OADH family. E2 enzymes fold into a cube (24-mer, for gram negative bacteria) or pentagonal icosahedron (60-mer, for gram-positive bacteria and eukaryotes) from trimers22,23,24,25, serving as a scaffold for the assembly of peripheral E1 and E3 enzymes. Compared to the prokaryotic counterpart, the eukaryotic PDHc has additional dihydrolipoamide dehydrogenase-binding protein (E3BP)26,27,28, which is structurally very similar to E2 (Fig. 1a) and localizes at the PDHc core. The catalytic reaction of PDHc consists of three steps (Fig. 1b). First, E1 catalyzes the oxidative decarboxylation reaction and transfers an acyl group to lipoate in the mobile LD of an E2 enzyme. Subsequently, the activated LD localizes to the IC core and transports the lipoyl moiety to CoA, forming acetyl-CoA. Finally, the LD is reoxidized for another catalytic cycle by an E3 enzyme. The E3 enzyme facilitates the transfer of protons to NAD+, resulting in the formation of NADH3. In the catalytic reaction cycle, the PDHc core-linked LDs function as flexible “swinging arms” to transport intermediate substrates among the active sites of different enzymes29,30 (Fig. 1f).

a Organization of E2 and E3BP of mammalian PDHc. E2 or E3BP is composed of one or two lipoyl domains (LDs) followed by a peripheral subunit-binding domain (PSBD) and an inner catalytic (IC) domain that are connected by flexible linkers. “S” represents PSBD. E1 or E3 binds on the PSBD of E2 or E3BP, respectively. b Scheme of the multi-step reaction catalyzed by the individual components of PDHc. c A representative cryo-electron tomogram slice showing intact PDHc particles in a mitochondrion. The insertion shows the zoomed in view of one PDHc particle. Similar features were observed in more than 10 tomograms. d Three-dimensional surface view of the tomogram shown in (c). e A representative model of mammalian PDHc, with 21 E1 and 4 E3 surrounding the 60-mer E2/E3BP core (green: E2/E3BP, light coral: E1, blue: E3, lime: LD, teal: S). See also Supplementary Movie. Notice that due to the heterogeneity of E1 and E3, this is not a structure generated by sub-tomogram averaging. Instead, it is a model by manually placing the E1 and E3 at the most likely positions based on our statistical analysis. f A putative scheme (based on Arjunan et al. 30) of the substrate channeling mechanism in the catalytic cycle of PDHc. Only one copy of each enzyme is shown for simplicity. From left to right: the pre-catalytic state of PDHc, the first, second and third reaction step of the PDHc catalytic cycle, respectively. During the catalytic cycle, the LD interacts with each enzyme sequentially to transport the substrate.
X-ray crystallography has made significant contributions to reveal the structure of PDHc, characterizing its individual components of E1, E2, and E320,21,22,23,24,25,31,32,33,34,35,36,37,38,39,40,41,42,43,44, as well as the subcomplexes involving the PSBD of the PDHc core binding to the E145 and E320,39, which provide crucial structural information about PDHc components and their interaction loci. However, due to its large size and structural flexibility, resolving the intact structure of PDHc is challenging for crystallography. Recently, structures of endogenous PDHc from bacteria, fungi, and bovine were obtained by cryo-electron microscopy single-particle analysis (cryo-EM SPA)41,46,47,48,49,50, which offer further insights into the PDHc core assembly and its interaction with the LD. In fungal PDHc, E3BP was found to reside within the core as four separate complexes46,47,50. While in mammals, E3BP substitutes for E2 in the core scaffold33 at possible stoichiometric ratios of 48:1251,52,53 or 40:2020,54 E2:E3BP in an unknown arrangement. The interaction interface of LD and E2 IC domain was revealed from the resting state E2, which was obtained by growing Escherichia coli in a minimal medium supplemented with succinate instead of glucose48. The peripheral E1 and E3 were supposed to be ~5–10 nm away from the PDHc core36,52. Nonetheless, recent cryo-EM SPA studies41,47 exhibited limited resolutions for the peripheral densities and were unable to distinguish E1 or E3, hampering our understanding of the overall PDHc structure.
Characterizing structures of the OADH family complex in its intact state through crystallization or purification remains challenging due to the complexity of its assembly and the highly mobile E2-linked lipoyl “swinging arms”, which play a central role in the multi-step catalytic mechanism. Moreover, previous biochemical analyses have suggested that PDHcs also exhibit stoichiometry variations of the E1 and E3 components47,55, but the roles of such variation of stoichiometry and subunit organization41,46,56,57 remains elusive. Unraveling the molecular architecture of intact PDHcs is indispensable for a comprehensive understanding of its catalytic mechanisms.
In the current work, we determine the in-situ structure of an intact PDHc from the mitochondria of porcine hearts by cryo-electron tomography (cryo-ET). Our results show highly heterogeneous composition and spatial distribution of E1 and E3, indicating collaborative mechanisms among multiple components within PDHc. Furthermore, we observe structural snapshots of the LD transfer pathway. We then analyze the interactions of E1-LD and E2-LD by molecular dynamics (MD) simulations, which illustrate how the swinging arms bind to the active sites of E1 and E2 to deliver the substrate-carrying prosthetic group. Overall, our work provides further understanding on the regulation, assembly and collaborative catalytic reactions of multi-enzyme OADH family complexes.
Results
In-situ structure of intact PDHc
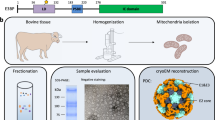
To characterize PDHc in its native environment, we reconstructed the in-situ structure of PDHc within mitochondria of porcine heart using cryo-ET. The mitochondrial membrane, cristae and ATPase are clearly visible in the cryo-electron tomograms (Fig. 1c, d). The cristae exhibit a conventional lamella shape, indicating the investigated regions retain the microenvironment of mitochondria. PDHc particles were readily seen in the reconstructed tomograms (Fig. 1c). To get an idea about the abundance of PDHc in mitochondria, we measured the volume of the examined mitochondria in our tomograms and calculated the density of the PDHc in native mitochondrial lumen. Based on our calculation, there is only one PDHc particle per 2.7\(\times\)10−3 μm3 of mitochondrial lumen volume. Considering that the volume of mitochondrion ranges from 0.04–0.08 μm3 58, the average number of PDHc is approximately 15–30 per mitochondrion in heart cells.
The PDHc core is found to be surrounded by a shell of protein densities, which lacks apparent symmetry, does not show any obvious local cluster organizations, and is speculated to be E1 or E3 (Fig. 1c insertion). To solve the in-situ structure of the PDHc at high resolution, we picked PDHc particles from hundreds of tomograms and carried out sub-tomogram averaging analysis (Supplementary Fig. 1 and Table 2). In the average structure, E2/E3BP folds into an icosahedral core (dark green in Fig. 1e), which agrees well with the reported mammalian PDHc core structure24,41. The surrounding shell densities were classified, refined, and then distinguished as E1 or E3 by comparing with the reported crystal structures and the atomic models predicted by AlphaFold259 (Supplementary Fig. 1). Moreover, local refinement and classification help us identified the LD and PSBD binding on the E1 or E3 (Fig. 1e, details will be shown in following sections). The numbers and locations of E1 and E3 vary across the PDHc particles, which will be presented in details in the next section. On average, each PDHc has twenty-one E1 and four E3 surrounding the 60-mer E2/E3BP core (Fig. 1e and Supplementary Movie).
Heterogeneous organization of E1 and E3
E1 and E3 components were distinguished by three-dimensional (3D) refinement, classification and comparison with atomic models (refer to Methods and Supplementary Fig. 1 for details), making it possible to analyze their spatial organization. The number of E1 in the periphery of each PDHc in mitochondria (hereafter called mitochondrial PDHc) is not constant, but varies from 1 to 46, with an average of 21 E1 per PDHc (Fig. 2a). The distribution of E1 number fits with two normal distributions (Fig. 2a). The number of E3 in each mitochondrial PDHc varies between 0 and 12, with an average of 4 E3 per PDHc (Fig. 2b). It is worth noting that each PDHc has at least one E1, whereas a subset of PDHc particles has no E3 at all (Fig. 2b). We then further conducted statistical analysis on the quantities of E1 and E3 in the same mitochondrial PDHc. The result shows a linear variation in the numbers of E1 and E3 (Fig. 2c). Specifically, for each mitochondrial PDHc, there was an additional assembly of E3 along with five more E1. In addition, the distributions of E1 and E3 in the same PDHc also exhibited two major proportions (Fig. 2c), with the centers locating around [7E1, 1E3] and [32E1, 6E3], which is consistent with the bimodal distribution of E1 (Fig. 2a). Our results show that native PDHcs do not have a specific subunit stoichiometry, consistent with previous reports47,52,54,55. The stoichiometry of E1:E2/E3BP:E3 in porcine PDHc varies from 1:60:0 to 46:60:12, with an average ratio of 21:60:4. Accordingly, the molecular weight of porcine PDHc varies from 4 to 11 MDa, with an average weight of ~7.5 MDa.

a Distribution of the number of E1 in each mitochondrial PDHc. SD: standard deviation. The dashed blue line shows the overall curve fitting of the E1 number distribution. The red lines show the two Gaussian fitting curves of the E1 number distribution. b Distribution of the number of E3 in each mitochondrial PDHc. c Distribution of the number of E1 and E3 in the same PDHc in mitochondria. As indicated by the grey line, the number of E3 has a linear relationship with the number of E1 (E3 = 0.2 * E1–0.4). d Statistics of the distance between the E1 center and the PDHc core center. e Statistics of the distance between the E3 center and the PDHc core center. f, g Orientation distribution plots of E1 and E3 relative to the PDHc core. The view of the core was shown in the left panel. Each point on the spheres (middle and right panels) represents an individual E1 or E3. Different colors of the sphere surface display the density of E1 (or E3) around each point. Refer to Methods for the generation of these plots.
The distances between E1/E3 centers and the icosahedral PDHc core were then measured to analyze the diameter of PDHc. The distance between the E1 center and the PDHc core center varies between 130 Å and 270 Å, with an average value and standard deviation of ~200 Å and ~24 Å, respectively (Fig. 2d). While the average distance between E3 center and the PDHc core center is ~193 Å, with the standard deviation ~30 Å (Fig. 2e). Therefore, taking into account of the size of E1 and E3, the diameter of PDHc typically ranges from 40–60 nm, with an average value of 50 nm.
The icosahedral PDHc core has two, three, and fivefold symmetries. We then analyzed the spatial distributions of E1 and E3 along different symmetry axes. The E1 scatters around the core, with minor preference locating along the twofold and threefold axes than the fivefold axis (Fig. 2f). On the other hand, the E3 primarily locates above the pentagonal opening (Fig. 2g).
Previous cryo-EM SPA studies using PDHc purified from endogenous sources suggest that E1 and E3 may form clusters surrounding the E2 core41,47,48, which is different from the current cryo-ET analysis of the mitochondrial PDHcs. To address the difference, we carried out both cryo-EM SPA and cryo-ET analysis of the PDHcs purified from the mitochondria of porcine hearts (hereafter called purified PDHcs). The SDS-PAGE, mass spectrometry and enzyme activity analysis show that the purified PDHcs maintain all necessary components and are catalytically active (Supplementary Figs. 2 and 3 and Supplementary Table 1).
Although the PDHc core structure was resolved to 3.2 Å resolution (Supplementary Fig. 4b–d), a simple asymmetric reconstruction of the peripheral densities did not provide informative insights into the structures of E1 or E3 (Supplementary Fig. 4e). Considering that the E1/E3 like densities were observed in the raw SPA images (Supplementary Fig. 4a), we hypothesized that collecting 3D tomograms of the PDHc particles may help us pick the E1/E3 like densities from the raw data and resolve the E1/E3 structures. Therefore, we carried out cryo-ET investigations of the purified PDHc. The tomograms show that the purified PDHcs encountered sample compression, resulting in fragmentation and the presence of scattered E1 and E3 like densities in the background (Supplementary Fig. 4f–h). Some PDHc particles locate around the tomogram center and possess E2 core with clear icosahedral features (Supplementary Fig. 4g). However, those particles which are closest to the graphene surfaces, which look perfect along the z-axis, are broken and have incomplete cores and periphery densities (Supplementary Fig. 4h). The complete and compressed particles can be easily distinguished in 3D tomograms, but not in the projection images for SPA. We thus speculate that the mixture of the compressed particles with the complete ones increases the difficulty for the subsequent distinguishment of E1 and E3.
To further analyze the differences between the purified PDHcs and mitochondrial PDHcs, we manually picked 1209 complete particles from 256 tomograms of the purified PDHc and carried out similar sub-tomogram averaging and statistical analysis as the mitochondrial PDHc. The quantity of E1 and E3 in each purified PDHc varies between 1–42 and 0–11, respectively, with the average value of 22 E1 and 3 E3 (Supplementary Fig. 5a, b). The distance between E1 center, E3 center and the PDHc core is ~200 Å and ~190 Å, respectively (Supplementary Fig. 5d, e). These features are similar to those native PDHcs observed within the mitochondrial lumen. However, it is notable that the number distribution of E1 in purified PDHc fits with one normal distribution (Supplementary Fig. 5c), rather than the bimodal distribution of mitochondrial PDHc. This difference could be attributed to the PDHc extraction, which may lead to sample homogenization and the loss of the natural distribution observed in the mitochondrial PDHc. Moreover, it further confirms that the mitochondria we observed retain their microenvironment to some extent. Together, our structural analysis here reveals the heterogeneous organization of E1 and E3, illustrating the intrinsic flexibilities of the composition and assembly of the PDHc (Supplementary Movie).
Binding of E1 and E3 on PSBD and the E1-LD interactions
The PSBD subunits of the PDHc core provide binding sites for E1 and E3, and the E2-linked LDs serve as swinging arms to transfer substrates among different enzymes3,29,60 (Fig. 3a). We carried out 3D classifications to analyze these interactions of E1 and E3 with PSBD and LD. The tomograms of the purified PDHc and mitochondrial PDHc were combined together to increase the particle number and improve the achievable resolution (Supplementary Fig. 1). The E1 were classified into two main classes (Fig. 3b, c). The previously reported mammalian E1 structure (PDB: 6cfo) fit well with the main architecture of both class averages, with all E1 possessing an additional density close to the C-terminal region (Fig. 3b, c) and ~82% E1 having additional density close to the N-terminal region (Fig. 3b). It was previously reported that the PSBD binds on the C terminus of E1β, while the N terminus of E1α interacts with LD45,61,62. We thus docked the PSBD-E1-LD and PSBD-E1 models (predicted by AlphaFold2) into the corresponding class averages, which agree well with our structures (Fig. 3b, c). Due to the resolution limitation, PSBD can be fitted in two different ways with opposite orientations (Fig. 3b, c and Supplementary Fig. 7d). Nevertheless, since E1 is a α2β2 heterotetramer, these two fitting strategies are thus structurally identical.

a The putative scheme of the substrate channeling mechanism in the catalytic cycle of PDHc, as shown in Fig. 1f. The diagram highlights the pre-catalytic state and the E1-LD interactions in the first step of catalytic cycle. b, c Class averages of the sub-tomogram averaging analysis of E1. b Class average showing the interaction of E1 with PSBD and LD. LD locates near the E1 N-terminal region and PSBD locates under the E1 C-terminal region. c Class averages showing the binding of E1 on PSBD. The docking models in (b, c) are predicted by AlphaFold2 (Supplementary Fig. 6a, b). d Enlarged view of the interaction interface shown in (b), highlighting the acidic amino acids of LD in the electrostatic potential region. e Electrostatic potential of E1 at the LD-E1 interaction interface, indicating the positions of basic amino acids. f The results of MD simulations showing the positions of hydrophilic (magenta) and hydrophobic (blue-purple) amino acids surrounding the substrate ThDP in E1 and the oxidized lipoic acid bound to K259 of LD. g Average structure showing the binding of E3 on the PSBD with the rigid fitting model predicted by AlphaFold2 (Supplementary Fig. 6d). LD: lipoyl domain, S: PSBD.
On the basis of our PSBD-E1-LD structure and the corresponding model, we carried out MD simulations to further analyze the interactions between E1 and LD (Supplementary Fig. 7a–c and Supplementary Fig. 8). The simulations show that LD interacts directly with the catalytic channel formed by E1α and E1β subunits (Fig. 3b). The interaction is stabilized by electrostatic interactions (Fig. 3d, e), which is notably supported by interactions involving the negatively charged residues E239, E248, D250, E254, D258, K259, E265, E268, E269 in LD (Fig. 3d), and the positively charged residues K77, K83, K85, H92, R235, H292, R304, R311 in E1α, and R37, R72, R59 in E1β (Fig. 3e). Additionally, we analyzed the amino acids in E1 that exhibit high interaction frequencies with the oxidized lipoic acid carried by K259 of LD (Supplementary Fig. 7c), which primarily consist of hydrophilic (H92, H292, Y118 on E1α, and R37, Y64, K69, H159 on E1β) and hydrophobic residues (F90, M229, M294 on E1α, and A67, M112, F116 on E1β). This suggests that the hydrophobic interactions play an important role in the E1-LD interaction and may also influence the proper positioning of the lipoic acid moiety or the kinetics of LD binding (Fig. 3f).
Our analysis of the E3 illustrates that one PSBD binds to the dimerization interface of E3 (Fig. 3g), which agrees with previously reported PSBD-E3 structures20,39. The structures show that both the E1 and E3 bind on one PDHc core linked PSBD. The E1 roughly faces the PDHc core with its twofold axis, while the twofold axis of E3 tilts ~60˚ relative to the pentagonal opening of the PDHc core (Fig. 1e and Supplementary Fig. 7e). Overall, our structures capture snapshots of PSBD-E1-LD, PSBD-E1 and PSBD-E3 interactions, illustrating the binding of E1 and E3 to the E2/E3BP core and the substrate transfer between E1 and LD.
Interactions between the LD and the PDHc core
PDHc transfers substrates between different enzymes by the LDs22, which are connected to the catalytic domains of the PDHc core through flexible linker regions (Fig. 4a). Analysis of the interactions between LD and different enzymes of the PDHc is essential for understanding the catalytic reactions. The in-situ structure of the PDHc core was resolved by sub-tomogram averaging to analyze the interactions between LD and the PDHc core. The PDHc core structure was resolved to a resolution of 4.3 Å (Fig. 4b). The overall structure shows the expected 60-meric central core with icosahedral symmetry and the density matches well with our cryo-EM SPA resolved core structure of porcine PDHc (PDB: 9j1w) (Fig. 4b). Noticeably, if we lower the threshold of the core structure, significant protruding densities can be found at the bridges of E2 subunits (arrow indicated in Fig. 4b), which are supposed to be the LDs.

a The putative scheme of the substrate channeling mechanism in the catalytic cycle of PDHc, as shown in Fig. 1f. The diagram highlights the second step of the catalytic cycle, emphasizing the interactions between the LD and the PDHc core in the second step of PDHc catalytic cycle. b 4.3 Å resolution sub-tomogram average structure of the E2/E3BP core (grey) fitting with our PDHc core atomic structure (green, PDB: 9j1w). The transparent density shows one E2/E3BP trimer with a lower threshold. The additional density indicated by the red arrow represents the extension from the PDHc core. The extended densities were classified into two classes shown in (c, d). c One class average show E2 trimer density, which has no significant extended density and fits well with the E2 trimer (PDB: 9j1w). d The other class average with extended densities on the E2 trimer, which fits well with the E2-LD model predicted by AlphaFold2 (Supplementary Fig. 6c). e The interaction interface between the LD and E2 trimer, corresponding to the structure shown in (d) with a rotation to better visualize the active site region. f Enlarged view of the dash framed region in (e), which shows the interaction interface between the LD and E2 trimer. The dihydrolipoamide (DHLA) was placed in the active site channel based on our cryo-EM SPA resolved PDHc core structure (Supplementary Fig. 4b–d). g The electrostatic surface potential map of the E2-LD interaction interface. The key amino acids involved in the interaction was analyzed by MD simulations and shown in different colors. h Frequency distribution of the interactions between K259 of LD and residues of E2. Residues where the frequency exceeds 40% in all repetitions of MD simulations were marked with red stars, while those exceeding 40% in part of repetitions were marked with black stars. The names of residues and bars are colored according to their polarity. LD: lipoyl domain, S: PSBD.
To better visualize the protruding densities, we performed asymmetric reconstruction of the E2 subunits. All 60 E2 subunits were extracted from each icosahedral PDHc core, aligned together and then 3D classified into two main classes. One class average only consists of the density of E2 trimer (Fig. 4c and Supplementary Fig. 10b and e), while the other class average revealed significant protruding densities (Fig. 4d and Supplementary Fig. 10c and f), which fit well with the AlphaFold2 predicted E2-LD model (Fig. 4d). Due to the resolution limitation, we did not observe the substrate density in the PDHc core structure resolved by cryo-ET (Fig. 4b–e). Fortunately, in our cryo-EM SPA resolved PDHc core structure, a cylindrical density extending thinly through the active site channel was observed near the catalytic residues S556 and H’620 of E2, accommodating a dihydrolipoamide (DHLA) model (PDB: 1eae) (Supplementary Fig. 4c, d and Supplementary Fig. 10l). This is consistent with the reported position of DHLA in the SPA study of bovine PDHc41. Since the E2 active site channel is located at the interface between two catalytic domains48. Our structures thus illustrate that the LD binds above the E2 catalytic pocket and inserts the lipoyl moiety attached lysine residue (K259) to a funnel-shaped opening that leads to the E2 active site channel (Fig. 4e, f).
MD simulations were performed to further analyze the interactions between LD and E2 (Supplementary Fig. 9). Results show that the electrostatic and hydrophobic interactions play crucial roles in the E2-LD interactions (Fig.4g). In detail, the E2-LD interaction involves residues K552-H556, F558-G560 of E2 and L226-M230, T257-D258, A260-I262 of LD (Fig. 4g). Additionally, we tallied the amino acids in E2 that interact frequently with K259 of LD in MD simulations (Fig. 4h). The majority of these amino acids are hydrophobic, highlighting the contribution of hydrophobic interactions to the E2-LD interaction. In brief, we captured the previously indistinct interactions between E2 and LD in mammalian PDHc, providing insights into the transacetylase reaction taking place within mitochondria.
Discussion
In this study, we purified mitochondria from porcine hearts and utilized cryo-ET and related approaches to elucidate the in-situ structure of mammalian PDHc. Our results show the intrinsic flexibilities of the PDHc on both diameter and peripheral organizations. In mitochondrial PDHc, E1 scatters around the icosahedral core, while E3 primarily locates above the pentagonal faces (Fig. 2f, g). In addition, two normal distributions of the number of E1 and E3 in each mitochondrial PDHc were observed (Fig. 2c), suggesting that there may be distinct subpopulations or heterogeneity within the PDHc population and an additional regulation to respond to different cellular states or nutrition conditions. Another possible reason for the observation of the biomodal distribution of E1 is the potential cell environment change, including temperature, osmotic pressure, oxidative stress level and others, for those mitochondria during the purification, which may regulate the PDHc composition63. Future efforts to characterize the PDHc within cells or tissues utilizing cryoFocused Ion Beam milling may enable us to visualize this multi-enzyme complex in its truly native environment (please refer to the Supplementary Information for more detailed discussions).
As determined by biochemical analysis, E3BP substitutes for a corresponding E2 in the PDHc core scaffold in mammals33,47,54 instead of being an additional subunit as in fungus46,47,50. No significant densities were found in our porcine PDHc core structure (Fig. 4b and Supplementary Fig. 4), thus our results agree with the substitution model of mammalian PDHc core. The number of E2 and E3BP constituting the 60-mer core of mammalian PDHc is still under debate. Recent studies favor 48:1233,51,52,53 or 40:2054,64 for the ratio of mammalian E2:E3BP. Due to the resolution limitation and high similarity between E2 and E3BP, we are unable to discriminate the densities of E3BP and E2 in our PDHc core structure. However, considering E3 specifically binds on the E3BP-linked PSBD27,28, our cryo-ET analysis reveals up to 12 E3 in each mitochondrial PDHc, which indicates that the porcine PDHc core scaffold is likely composed of 48 E2 and 12 E3BP. Additionally, the distinct location of E3, found exclusively along the fivefold axis of the PDHc core, agrees well with the mammalian PDHc core model proposed by Zdanowicz et al. 53, where each of the 12 E3BP locates at one pentagonal face of the icosahedral core. On the basis of 48:12 E2:E3BP ratio, each PDHc core could bind up to 48 E1 and 12 E3, the actual numbers of E1 and E3 we found in mitochondrial PDHcs are typically far from saturation (Fig. 2c), similar to the scenarios that were previously proposed by M. Smolle et al.65 Such structural characteristics might also apply to prokaryotic PDHcs, which could function effectively without fully occupying all the binding sites of E1 and E331, and potentially allow various substitutions to emerge during evolution.
The dynamic movement of the N-terminally-located, E2-linked LDs plays a pivotal role in the catalytic process of PDHc. These LDs, functioning as swinging arms, facilitate the transferring of their prosthetic groups to the active sites of all three enzymes throughout the catalytic cycle. Here, we captured the previously indistinct dynamic interactions of LDs with both E1 (Fig. 3) and E2 (Fig. 4) in mammalian PDHc, and analyzed their interaction details by MD simulations.
As for the E1-LD interaction, it is worth noting that although the α2β2 heterotetrameric E1 has two active sites (Supplementary Fig. 7a), only one active site was observed to interact with LD in our results (Fig. 3b). Biochemistry, kinetics, and spectroscopy studies have shown that E1 functions through an alternating active-site mechanism, known as the flip-flop mechanism, in which, while one active site is involved in the decarboxylation of pyruvate, the other simultaneously catalyzes the reductive acetylation of an E2-lipoamide molecule66,67,68,69. Our results provide structural evidence supporting this alternating active-site mechanism. Additionaly, our class averages show that the number of E1 enzymes binding with LD is ~4.5 times of that E1 without LD binding (Supplementary Table 2). This suggests that E1-LD interaction is likely stable rather than transient, or the binding/unbinding ratio may be related with the oxidative or metabolic states in purified mitochondria.
Regarding the E2-LD interaction, our MD simulations show that the interaction primarily relies on electrostatic interactions (Fig. 3d, e and Fig. 4g), which is consistent with previously reported E2-LD interactions in both prokaryotic48 and fungal49 PDHcs. This implies that a general electrostatic-based interaction between LD and E2 may exist across various species. Furthermore, the sequence alignments of the LDs in prokaryotes, fungi, and mammals revealed that there are 12 to 15 acidic amino acids with conserved electronic properties across these species (Supplementary Fig. 10i). We thus propose that the acidic patches on the LD (Supplementary Fig. 10g) serve as a general binding site for its interactions with different components of PDHc. In addition, the reported LD position relative to E2 in the resting state of PDHc in E. coli differs from our observations in porcine PDHc (Supplementary Fig. 10j, k), whereas the position of the DHLA carried by the lysine residue barely changes (Supplementary Fig. 10j–l). The difference in LD positioning could be attributed to species differences. Alternatively, it might be because the E2-LD interactions are highly dynamic, allowing LD to deliver the substrate-carrying prosthetic group into the E2 active site from various directions.
Earlier studies63,70 indicate that the catalytic activity of PDHc increases as more E1 and E3 assembled to the core. Dave et al.’s work71 showed that reducing the number of tandem LDs in each E2 chain of E. coli PDHc adversely affects the cell growth, suggesting that the activity of PDHc also correlates with the number of functional LDs. Together with the dynamic architecture of PDHc we observed in current work, we speculate that PDHc can regulate its catalytic activity through the variation in quantity of E1, E3 and functional LDs. Following the speculation, we proposed a regulation model for the catalytic performance of mammalian PDHc in response to different metabolic demands, which may be influenced by the cell energy demands, oxidative stress levels and other physiological conditions. In a low metabolic demand state, several E1 and E3 enzymes are sufficient to sustain catalytic reactions (Fig. 5a). Although each E2 chain in the core possesses two tandem LDs, only one LD is required for substrate transfer since there is a limited number of E1/E3 active sites (Fig. 5b–d). Initially, E1 captures and decarboxylates a pyruvate and then transfers it to a mobile LD (Fig. 5b). Subsequently, the substrate is conveyed by the LD to an active site on E2 to produce acetyl-CoA (Fig. 5c). Finally, the lipoyl moiety is shipped to a catalytic site of E3 by the LD, where it is reoxidized to complete one reaction (Fig. 5d).

a–d Schematic diagram of PDHc reaction with low catalytic performance. In low metabolic demand, limited amount of E1 and E3 bind to the PDHc core. Each E2 has two tandem LDs, but only one of the LDs functions as swing arm to carry out substrate transfer among E1, E2, and E3. a E1 catalyzes the decarboxylation of pyruvate. b E1 participates in the subsequent oxidation reaction. c E2 is involved in generating acetyl-CoA. d E3 catalyzes the reduction and dehydrogenation processes. e–h Schematic of PDHc reaction with high catalytic performance. With increasing metabolic demand, more E1 and E3 bind to the PDHc core and provide more active sites. More LDs are required to function as swing arms for substrate transfer to increase the catalytic performance. e E1 enzymes participate in pyruvate decarboxylation. f E1 enzymes engage in oxidation, while both tandem LDs of some E2 chains may interact with E1 enzymes to convert the acetyl group into S-acetyldihydrolipoamide. The question marker indicates the lack of evidenceregarding whether two consecutive LDs belonging to the same E2 chain can simultaneously bind to distinct E1 subunits. g The two LDs transfer the acetyl group to coenzyme A, producing acetyl-CoA. h E3 reoxidizes the lipoyl moiety, allowing the cycle to repeat and produce NADH. The question marker indicates the undertatinty regarding whether a single E3 dimer can interact with two LDs simultaneously. In this diagram, all E1 and E3 bind on the PSBD and connect with the PDHc core by flexible linkers. For simplicity, only one E2 trimer of the core and only the linkers of two E2 chains are shown. Other linkers are omitted. LD: lipoyl domain, S: PSBD.
In a state of high metabolic demand, more E1 and E3 would be assembled on the PDHc core to provide more active sites. Meanwhile, more LDs are required to involve in the catalytic reactions and both tandem LDs of a single E2 chain may engage concurrently to achieve more efficient transfer of substrates (Fig. 5e–h). We speculate that both LDs of some E2 chains will interact with different E1 subunits simultaneously (Fig. 5f) and transfer acetyl groups, facilitating reactions at two catalytic sites on the E2 core (Fig. 5g). Subsequently, the reduced lipoyl moiety is conveyed to the catalytic sites on E3 (Fig. 5h). Although there are significantly fewer E3 enzymes (no more than 12) in the PDHc compared to LD, it is currently unclear whether a single E3 dimer can interact with two LDs simultaneously or not (indicated by the question mark in Fig. 5h). Nevertheless, considering that E1 catalyzes the rate-limiting step of the overall PDHc reaction72, this coordinated process enables simultaneous completion of multiple reactions, hence improving the overall catalytic efficiency. This high-performance catalytic process continues until the energy demand of the mitochondrion decreases. Then a portion of the E1 and E3 dissociate from the E2 core, and the PDHc reverts to the state where only one LD is involved in the catalytic reaction. In summary, the canonical substrate channeling mechanism in the PDHc may be regulated by modular binding of E1 and E3, and multiple LDs may be used at high turnover, potentially even simultaneously.
Previous studies have postulated two different mechanisms to explain the higher-order interaction of the components of OADH complexes: the “multiple random coupling” (MRC) mechanism73,74 and the “division-of-labor” (DOL) mechanism54,75. Both the dynamic architecture of mammalian PDHc we observed and the model we proposed support the MRC mechanism, in which all LDs have the freedom to visit various active sites within the complex and the overall activity of the PDHc is influenced by redundancies and random processes. Given the similarity among members of the OADH family, the regulation mechanism proposed here is likely applicable to the entire OADH family.
Regulation of the mammalian PDHc is crucial for maintaining glucose homeostasis in feeding and fasting states. It has been reported that the catalytic reaction of PDHc is regulated through the end-product inhibition by acetyl-CoA and NADH76,77, as well as through the reversible phosphorylation and dephosphorylation of the E1 subunit mediated by PDK and PDP3,78,79. Acetyl-CoA/CoA and NADH/NAD+ ratios act as effectors in feedback inhibition by influencing the redox and acetylation status of the lipoamide cofactors. While PDK and PDP inhibit PDHc activity by phosphorylation of the serine residues on E1. Given that the (de-)phosphorylation regulation directly relates with the enzyme activity of E1, we suspect such (de-)phosphorylation of the E1 subunit might affect the (dis-)assembly of E1 and E3, thereby regulating the catalytic performance of PDHc. To verify this hypothesis, it would be helpful to perform structural analysis for samples at periods of high and low expression level of PDP and PDK.
In summary, we obtained the in-situ structure of mammalian PDHc which indicates the dynamic substrate transporting by the LDs between different enzymes. We structurally characterized the location and quantity of peripheral enzymes as well as the interface of their interactions with the PDHc core. Overall, our results provide further insights into the structure and regulation of OADH family complexes.
Methods
Ethics statement
The pig hearts were purchased from a local slaughterhouse, and the subsequent handling procedures were performed at Zhejiang Agriculture and Forestry University, following the protocols established by the institution. The study was approved by the Experimental Animal Ethics Committee of Zhejiang A&F University (EAEC-ZAFU).
Isolation of mitochondria from porcine heart
All the following procedures were carried out on ice or at 4 °C. Fresh porcine heart was obtained from the slaughterhouse and transported to the laboratory as soon as possible. The heart muscles were washed twice with Milli-Q water. 720 × g of heart muscles were isolated and cut into small cubes and suspended in 1000 mL of buffer A (0.25 M sucrose, 10 mM potassium phosphate, 0.1 mM EDTA, pH 7.6). These tissues were homogenized in a large-capacity blender for 5 min. The homogenate was centrifuged in l-liter bottles at 2000 × g for 10 min. The supernatant fluid was strained through eight layers of cheesecloth. The mitochondrial fraction was collected by centrifugation at 22,000 × g for 17 min. The crude mitochondria were washed twice with 200 mL 0.02 M potassium phosphate, pH 7.0 and paste was collected by centrifugation at 22,000 × g for 10 min. The crude extracted mitochondria were prepared for further purification of PDHc or preparation of cryo-ET samples.
PDHc purification
The crude mitochondria were suspended in 60 mL 0.02 M potassium phosphate (pH 7.0). 5 M NaCl and Cooktail were added into the suspension at a ratio of 1:100, and the pH was adjusted to 6.4. The suspension was disrupted twice at 276 bar with a cell disruptor (JNBIO) equipped with a cooling system. The mitochondria lysate was centrifuged at 30,000 × g for 30 min, and the supernatant was added 10 mM MgCl2 and PEG precipitated in 5% (w/v) PEG 6000 for 10 min. The precipitate was collected by centrifugation at 30,000 × g for 20 and resuspended in 25 mL buffer B (50 mM MOPS-KOH buffer, pH 7.0, 0.02 mM thiamin pyrophosphate, 1 mM MgCl2, 2 mM DTT). The suspension was stored 4 h in a refrigerator and then centrifuged at 40,000 × g for 20 min before discarding the precipitates. To the clear supernatant fluid was added 2 mM EGTA. After 20 min the solution was loaded onto 10 mL 35% (w/v) sucrose cushions centrifuged at 105,000 × g for 3.5 h in a Beckman type SW32 rotor. The precipitate is resuspended with buffer B supplemented with 2 mM EGTA. After 20 min, the solution was added to a 10% to 40% sucrose gradient configured by buffer C (50 mM potassium phosphate buffer, pH 7.0, 0.02 mM thiamin pyrophosphate,1 mM MgCl2, 1 mM NAD+, 0.1 mM EDTA, 0.4 mM DTT)) and centrifuged at 144,000 × g for 2.5 h using a SW41Ti rotor with a Beckman Coulter Optima XPN-100 centrifuge. The gradients were fractionated and assessed by SDS-PAGE and negative staining, PDHc around the sucrose density of 15%. The samples were concentrated to 0.4 mg mL−1 by using a membrane concentrator with a 100 kDa cut-off for biochemical and cryo-ET analyses.
Biochemical analysis of PDHc
After preparing a 12% polyacrylamide gel and assemble the electrophoresis tank, we loaded a 20 μL aliquot of PDHc protein sample and 5 μL of protein marker onto the gel for polyacrylamide gel electrophoresis (SDS-PAGE).
For mass spectrometry, purified PDHc obtained by purification method was concentrated to 30 μL using a 100 kDa concentrator. Protein precipitation was performed using TCA precipitation method (n = 1). The precipitated protein was resolubilized by adding a resolubilization solution (8 M urea,100 mM Tris-HCl, pH 8.5), and the protein concentration was determined using the BCA method. Equal amounts of protein were taken and adjusted to the same volume. TCEP (tris(2-carboxyethyl)phosphine) and CAA (chloroacetamide) were added for reduction and alkylation reaction at 37 °C for 1 h. The reduced and alkylated samples were diluted with 100 mM Tris-HCl solution to reduce the Urea concentration to below 2 M. Trypsin was added at a 1:50 ratio of enzyme to protein and incubated overnight at 37 °C with shaking for enzymatic digestion. The next day, the digestion reaction was terminated by adding TFA, and the supernatant was subjected to desalting using an SDB-RPS desalting column. After vacuum drying, the samples were stored at −20 °C. Mass spectrometry analysis was performed using an ion mobility-quadrupole-time-of-flight mass spectrometer (timsTOF Pro) from Bruker. Sample injection and separation were performed using an UltiMate 3000 RSLCnano liquid chromatography system coupled online with the mass spectrometer. Peptide samples were injected and trapped on a Trap column (75 μm × 20 mm, 2 μm particle size, 100 Å pore size, Thermo), followed by elution onto an analytical column (75 μm × 250 mm, 1.6 μm particle size, 100 Å pore size, ionopticks) for separation. The analysis gradient was established using two mobile phases (mobile phase A: 0.1% formic acid in H2O and mobile phase B: 0.1% formic acid in ACN). The flow rate of the liquid phase was set to 300 nL/min. Peptides were introduced into the mass spectrometer via CaptiveSpray nanospray ion source for DDA scanning. TIMS function was enabled, and PASEF scanning mode was used. Each scan cycle consisted of 1.1 s, including one MS1 scan and 10 PASEF MS/MS scans, with each PASEF MS/MS scan generating 12 MS/MS spectra. The mass spectrometry data were analyzed using MaxQuant (V2.0.1) software with the Andromeda database search algorithm. The protein reference database used for the search was the Sus_scrofa protein reference database from Uniprot (2023-01-09, containing 46,139 protein sequences). The main search parameters were as follows: variable modifications—Oxidation (M), Acetyl (Protein N-term); fixed modification—Carbamidomethyl (C); enzyme—Trypsin/P; primary mass tolerance - 20 ppm in the first search and 4.5 ppm in the main search; secondary mass tolerance - 20 ppm. The search results were filtered at a 1% FDR (false discovery rate) threshold at the protein and peptide levels. A total of 236 proteins were identified, of which 232 proteins have quantitative information. The average molecular weight of the amino acid residues in the proteins is approximately 110 Da, so most of the peptides detected by mass spectrometry are located within 727 amino acids. Most proteins do not have missed cleavage sites, while a small number of peptides may have 1 or 2 missed cleavage sites. Protein entries corresponding to reverse sequences, contaminating proteins, and peptides with a single modification were removed, and the remaining identification information was used for further analysis.
PDH activity assay
The Pyruvate Dehydrogenase Activity Assay was performed using the Kinetic Activity Assay Kit (Catalog Number ab287837, Abcam). Experiments were conducted following the manufacturer’s protocol. Briefly, the assay involves a coupled enzyme reaction that produces a colorimetric product detectable at 450 nm, with the intensity correlating to enzymatic activity.
Samples were prepared according to detailed experimental procedures provided in Supplementary Fig. 3. Absorbance was measured at 450 nm in kinetic mode over 20 min at 37 °C. Background readings were subtracted from all measurements, and a standard curve for NADH was generated to quantify the amount of NADH produced during the reaction time (∆T = T2–T1). The pyruvate dehydrogenase activity of each sample was calculated using the formula: B/(∆T × V) × D = nmol/min/mL = mU/mL, where B represents the amount of NADH, ∆T is the reaction time, V is the sample volume, and D is the dilution factor. One unit of pyruvate dehydrogenase activity is defined as the amount of enzyme that generates 1.0 µmol of NADH per minute under standard conditions (pH 7.5, 37 °C).
Cryo-EM SPA sample preparation and data collection
An aliquot of a 3 μL purified PDHc sample was applied to a holey carbon grid covered with graphene-oxide (Quantifoil R1.2/1.3, Au, 300 mesh). After waiting for 60 s, the grids were blotted for 3.5 s at a humidity of 100% and 4 °C and plunge-frozen in liquid ethane using a Vitrobot (Thermo Fisher Scientific).
Purified PDHc samples were detected by a Titan Krios cryogenic electron microscope (Thermo Fisher) operated at 300 kV and equipped with a Selectris energy filter with a slit width of 10 eV and a Falcon4 camera (Thermo Fisher), and cryo-EM images were acquired using EPU in the counting mode at a nominal magnification of ×105,000, corresponding to a calibrated physical pixel size of 1.2 Å. The defocus range was set between −1.3 and −2.0 μm. Images were recorded with a 5.85 s exposure to give a total dose of 51 e−/Å2.
Cryo-EM SPA image processing
The beam-induced motion correction of image stacks was carried out using MotionCor280 to generate average dose-weighted micrographs. The contrast transfer function parameters of these average micrographs were calculated by Gctf 81. Other procedures of data processing were performed in cryoSPARC82. For the data processing procedures of purified PDHc, 413,042 particles were automatically selected, and then 2D and 3D classifications were carried out to select consistent particle classes. Finally, 68,914 particles were selected for 3D refinement. The PDHc core structure was refined with icosahedral symmetry, resulting in a map with resolution of 3.2 Å. Asymmetric reconstruction was applied to the peripheral part, with a mask focusing on the peripheral densities surrounding the PDHc core. The data processing details are summarized in Supplementary Fig. 11.
E2 model building and refinement
The models of E2 trimers predicted by AlphaFold2 were first fitted into the cryo-EM density map using Chimera83. Subsequently, based on the map’s symmetry, they were arranged into a dodecahedron structure, comprising 60 E2 chains, and then adjusted in Coot84. The models of the E2 core were manually built in Coot. The entire structure was further refined through manual adjustments and using phenix.real_space_refine85. Cryo-EM collection, refinement and validation statistics are summarized in Supplementary Table 3.
Preparation of cryo-ET samples
Preparation of purified PDHc cryo-ET samples followed the procedure described for cryo-EM SPA sample preparation. For the mitochondrial PDHc samples, the crude extracted mitochondria was rinsed gently with 1 mL of 0.02 M potassium phosphate. An aliquot of a 5 μL sample was applied to a glow-discharged holey carbon grid (Quantifoil R2/2, Au, 400 mesh). After waiting for 60 s, the grids were blotted for 7 s at a humidity of 100% and 22 °C and plunge-frozen in liquid ethane using a Vitrobot.
Cryo-ET data collection and tomogram reconstruction
The frozen-hydrated samples were transferred to a 300 kV Titan Krios cryogenic transmission electron microscope (Thermo Fisher Scientific) equipped with a Selectris energy filter and a Falcon 4 direct electron detector (Thermo Fisher Scientific). All images were recorded at ×81,000 magnification with a pixel size of 1.5 Å at the specimen level. PaceTomo script86 was used with the SerialEM software to collect tilt series at −2 to −4 µm defocus (dose-symmetric collection theme, start from 0˚, group size 2), with accumulative dose of ~90–100 e−/Å2 distributed over 35 images and covering angles from −51˚ to 51˚, with a tilt step of 3˚.
All recorded images were first motion-corrected by Relion-4.087 or Warp88 and then stacked by IMOD89 and aligned by AreTomo90. For posterior processing using RELION-4.0 and Protomo-i391,92, Gctf was used to determine the defocus of each tilt image. For sub-tomogram averaging using Protomo-i3, the ‘ctfphaseflip’ function in IMOD was used to do the contrast transfer function (CTF) correction for the tilt images. For other processing, Warp was used for the defocus estimation and CTF correction. Tomograms were reconstructed by weighted back-projection (WBP) or simultaneous iterative reconstruction (SIRT) methods using IMOD or tomo3D with the CTF-corrected aligned stacks.
Sub-tomogram averaging and corresponding analysis
All PDHc core particles were first manually picked from 6\(\times\) binned SIRT reconstructed tomograms. Then sub-tomograms of PDHc particles were cut from the 4× binned WBP reconstructed tomograms and aligned by Protomo-i391,92.
Refinement of the PDHc core structure: the sub-tomogram averaging pipeline in RELION-4.0 was used for high-resolution structure determination. The coordinates and Euler angles of each PDHc particle generated from the previous Protomo-i3 alignment step were first transferred to RELION-4.0 by a homemade script package. 3D alignment and classification were generated under binning 4× and 2×, followed by local refinement and 2 times of CTF and frame refinement for unbinned data, resulting in the 4.3 Å resolution PDHc core structure.
Local refinement and classification of E2: each PDHc core consists of 60 E2 subunits. After the alignment of PDHc core, the position of each E2 subunit was calculated using the “symmetry expansion” and “shift center” functions in RELION-4.0 (60 E2 subunits corresponding to each PDHc core). Then the corresponding sub-tomograms under binning 2× were reconstructed by Warp, and RELION-3.1 was used to do 3D classifications for all E2 subunits with fixed alignments. A spherical mask including the region adjacent to the E2 subunit was applied for classification. Those particles contain obvious LD density were sorted out and local refined to generate the E2-LD structure. Other particles were merged to generate the E2 trimer structure. Afterwards, three iterations of the geometry refinement (including image warp, particle poses, stage angles and volume warp) and three iterations of CTF refinement (including defocus, odd and even Zernike orders) by the software M93 were applied to generate the classification results shown in Fig. 4c, d.
Refinement and classification of E1 and E3: (1) we speculate the densities surrounding the PDHc cores in raw tomograms (Fig. 1c and Supplementary Fig. 4g, h) are E1 and E3. Therefore, we manually picked ~10k particles of these densities from the 6x binned SIRT reconstructed tomograms. During the picking, we assigned the density of each particle to the nearby PDHc core. Since the core positions were already known from the previous alignment of the PDHc core, we thus got two positions for each E1/E3 like particle, one is the PDHc core position and another from the manual picking, which help us defined the initial Euler angles of the E1/E3 like particles. (2) The corresponding sub-tomograms were cut from the 4× binned WBP reconstructed tomograms, followed by 3D alignment and classification using Protomo-i3 to get an initial model. In the initial model, part of the icosahedral PDHc core is still visible, with strong densities roughly locating along its C2, C3, and C5 axis and the distance between the densities and PDHc core is ~20 nm. (3) To generate equally spaced and overlapping regions of interest for E1/E3 reconstructions, we did icosahedral symmetry expansion of the points at a distance of 20 nm from each aligned PDHc core center. We thus got 60 positions corresponding to each PDHc core. Those positions were used as particle centers to reconstruct sub-tomograms by Warp. The volume size was set to ~40 nm to make sure there are enough overlaps between different sub-tomograms, so that we did not miss any possible E1 or E3 particles. (4) The sub-tomograms were aligned to the initial model generated from Protomo-i3, followed by 3D classifications by RELION-3.1. Then the E1 particles and E3 particles were selected from the class averages by comparing the density shape with the reported E1, E3 structures and the models predicted by AlphaFold2, then merged and aligned separately to generate optimized references for E1/E3. Afterwards, the optimized E1/E3 references and randomly selected junk particle averages were used as new references to align and classify the sub-tomograms for another cycle. This was repeated for multiple iterations to make sure we classified the E1 and E3 particles with high confidence. (5) The selected E1 and E3 particles were separately aligned and classified by RELION-3.1. Resulting in the PSBD-E1, PSBD-E1-LD and PSBD-E3 structures. Afterwards, three iterations of the geometry refinement (including image warp, particle poses, stage angles and volume warp) were carried out in M. See also Supplementary Fig. 1 for ths sub-tomogram averaging workflow.
Orientation distribution analysis of E1 and E3
The coordinates of E1, E3, PDHc cores and the rotation matrices of PDHc cores, derived from the sub-tomogram averaging, were used to analyze the orientation distribution of E1 and E3. Specifically, the rotation matrix describes how each PDHc core can be rotated to align with a defined reference view (left panels in Fig. 2f, g). The vector formed by the coordinates of E1 (or E3) and the coordinates of the corresponding PDHc core was rotated according to the PDHc core’s rotation matrix. Subsequently, such vectors corresponding to all E1 (or E3) were shifted to a common origin, and their lengths were normalized to 1. As a result, the E1 (or E3) points were mapped onto the surface of a 3D sphere, with each point representing an individual E1 (or E3) (middle and right panels in Fig. 2f, g). The heatmaps (middle and right panels in Fig. 2f, g) display the density of E1 (or E3) around each point, quantified by the number of points observed within a 0.2 unit length of the reference point.
Mitochondrial volume size calculation
Using the IMOD software89, we manually outline the area of each mitochondrion in all tomograms and obtained the area size based on the IMOD contour information. We then rotate the tomogram along the Z-axis, measure the height of the mitochondria, and calculate the volume size of each mitochondrion by multiplying the area size with the height.
Model prediction by AlphaFold2
AlphaFold294,95 was utilized to predict the atomic models of the following complexes: PSBD-E1, PSBD-E1-LD, E2-LD, and PSBD-E3. The input sequences utilized for AlphaFold2 are detailed in Zenodo (https://doi.org/10.5281/zenodo.13225551). For all predictions, the top-ranked model was selected based on the confidence metrics such as pLDDT, PAE, pTM, and ipTM (Supplementary Fig. 6) for posterior analysis. The previously reported structure (PDB: 7b9k) was used as a guidance for the initial placement of LD relative to E2 for the prediction of E2-LD model.
Model building for MD simulations
In order to simulate the acetyl transfer processes in E1 and E2, the oxidized lipoic acid (LA) and S-acetyl-dihydrolipoamide (SA-DHLA) were covalently linked to K259 of LD in the PSBD-E1-LD and E2-LD models predicted by AlphaFold2, respectively. The flexible terminal loop region was excluded for docking and model refinement. For the model building of E2-LD system, our cryo-EM SPA resolved PDHc core structure (Supplementary Fig. 4) and the previously reported structure (PDB: 7b9k) was used as a guidance, then the SA-DHLA was docked into the E2 pocket by AutoDock Vina96, and subsequently connected to K259 of LD using PyMOL97 builder. Afterwards, the CoA molecule was docked to E2 based on the relative position of SA-DHLA and the vacuum electron density of the protein. For the model building of PSBD-E1-LD system, the TPP, HE-TPP molecules and four Mg2+ were docked into the corresponding pockets according to the reported structure (PDB: 3exe), with HE-TPP being positioned closer to K259 of LD. LA was docked into the space between the E1α and E1β monomers, and then connected to K259 using PyMOL.
Generation of topology and parameter files for ligands
The restrained electrostatic potential (RESP) charges are generated as follows: A methylamino group is added to the C-terminal, and an acetyl group is added to the N-terminal of lysine, where the N atom of the sidechain is connected with LA (K-LA), and SA-DHLA (K-SA-DHLA), respectively. All ligands (including K-LA, K-SA-DHLA, TPP, HE-TPP, CoA) are optimized in B3LYP/6-311g (d, p) using Gaussian 1698, with solvent effects corrected using the PCM method. Then, the RESP charges are calculated from the optimized geometry and wave function using Multiwfn99. The parameters of the Generated Amber force field (GAFF) for the ligands were obtained using Sobtop100.
MD simulations
Molecular dynamics (MD) simulations were conducted using GROMACS-v2023101 for the E1-LD (with TPP, HE-TPP, and K-LA) and E2-LD (with CoA and K-SA-DHLA) systems.
For the E2-LD system, topology and parameter files for CoA and K-SA-DHLA were incorporated into the force field of AMBER99SB-ILDN102, and the bonded information (including bond, angle, and dihedral angle) of K-SA-DHLA to surrounding residues was completed based on the existing topology. The system was solvated in a rectangular water box of TIP3 water, which extended 1.0 nm beyond the complex in any dimension, and neutralized with 150 mM NaCl. MD simulations were conducted in three stages, each imposing different degrees of restraint: (1) In the “First stage”, atoms of K-SA-DHLA, together with the two nearest residues (D258 and A260), water, and ions, were allowed to move without any restraints. (2) In the “Second stage”, only atoms of K-SA-DHLA and its two nearest residues, along with CoA, and the sidechain of protein residues within 8 Å radius around K-SA-DHLA and CoA, water, and ions, were permitted to move. (3) The “Third stage” involved restraining of only the backbone of E2, and releasing all residues of LD, ligands, and the water-ion environment. Since the complete PDHc core is excessively large for MD simulations. The simulations were only carried out for one E2 trimer interacting with one LD, and the position restraints applied on the E2 backbone were used to mimic the restraints imposed by the whole core and to make sure the trimer has a stable structure across the simulation. Before MD production, energy minimization was employed with the “first stage” restraint, using the steepest descent algorithm until the maximum force fell below 1000 kJ/mol/nm. This process utilized a cutoff distance of 1.2 nm for the neighbor list, Van der Waals interactions, and Coulomb interactions. During the equilibrium run, the “second stage” restraint was applied. This involved successive runs in a 100 ps NVT ensemble and a 200 ps NPT ensemble, utilizing velocity-rescale temperature coupling with a time constant of 0.1 ps. In the NPT ensemble, equilibrium was achieved using isotropic pressure coupling with the Berendsen algorithm103, applying a time constant of 2 ps at 1 bar pressure. The first cycle production run was conducted with the “third stage” restraint, carrying out simulations in the NPT ensemble at a pressure of 1 bar for a total of 47.6 ns. Temperature and pressure were coupled using the velocity-rescale method with a time constant of 1.0 ps and isotropic pressure coupling with the Parrinello-Rahman algorithm with a time constant of 5.0 ps, respectively. The second cycle’s 1000 ns production run mirrored the first cycle which employed the “third stage” restraint and was repeated for three times.
The simulation of the E1-LD system mirrored that of the E2-LD system, with the only difference being the equilibrium achieved in the NPT ensemble using isotropic pressure coupling with the Parrinello-Rahman algorithm for 100 ps and no restrictions in all processes. The 1000 ns production run was repeated for four times. Necessary files to repeat the simulations are deposited at Zenodo (https://doi.org/10.5281/zenodo.13225551).
MD analysis
The Root-Mean-Square-Deviation (RMSD) was analyzed using GROMACS-v2023. Contact maps were generated to illustrate the interaction frequency of specific residue pairs between LD and E1, as well as LD and E2. We define a contact to be formed if: (1) the distance between the closest atoms of residues is shorter than 4 Å, (2) the residues are positioned at the interface, with each residue in a pair originating from a different monomer, and (3) the contact is present for more than 30% of the simulation time, ensuring that we focus on positive contacts. The residues were identified using MDAnalysis104, and the minimum distance between residue pairs was calculated using PLUMED105.
Model fitting and visualization
Atomic models were rigidly fitted to the corresponding densities using the “Fit in Map” tool in UCSF chimera or ChimeraX106. EMAN2 was used for segmentation of the mitochondrial membranes and cristae. ArtiaX107 was used for mapping the PDHc complexes and ATP back to the raw tomograms. UCSF Chimera and ChimeraX were used for rendering the graphics.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
PDB accession codes of the atomic models used for model building or as initial models in this work: 6cfo, 1eae, 7b9k, and 3exe. Specific details are described in the corresponding sections of the article. The coordinates and structures of PDHc generated in this work were deposited in Electron Microscopy Data Bank108 under accession codes. Cryo-ET related maps: E2/E3BP core (Fig. 4b): EMD-38712, PSBD-E1-LD (Fig. 3b): EMD-61082, PSBD-E1 (Fig. 3c): EMD-61081, E2-LD (Fig. 4d): EMD-61084, E2 trimer (Fig. 4c): EMD-61083, E3-PSBD (Fig. 3g): EMD-38716. Cryo-EM SPA related maps: E2/E3BP core (Supplementary Fig. 4b): EMD-61080, PDB: 9j1w. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository109,110 with the dataset identifier PXD059530. Source Data are provided as a Source Data file. Source data are provided with this paper.
Code availability
The sequence files and MD simulation input, output and parameters files have been stored in Zenodo (https://doi.org/10.5281/zenodo.13225551).
References
Bisswanger, H. Substrate specificity of the pyruvate dehydrogenase complex from Escherichia coli. J. Biol. Chem. 256, 815–822 (1981).
Patel, M. S. & Roche, T. E. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 4, 3224–3233 (1990).
Patel, M. S., Nemeria, N. S., Furey, W. & Jordan, F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J. Biol. Chem. 289, 16615–16623 (2014).
Cox, L. J. Ra. D. J. Macromolecular organization of enzyme systems. Annu. Rev. Biochem. 35, 57–84 (1966).
de Boer, V. C. & Houten, S. M. A mitochondrial expatriate: nuclear pyruvate dehydrogenase. Cell 158, 9–10 (2014).
Sutendra, G. et al. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 158, 84–97 (2014).
Matsuda, S. et al. Nuclear pyruvate kinase M2 complex serves as a transcriptional coactivator of arylhydrocarbon receptor. Nucleic Acids Res. 44, 636–647 (2016).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Cerniglia, G. J. et al. The PI3K/Akt pathway regulates oxygen metabolism via pyruvate dehydrogenase (PDH)-E1α phosphorylation. Mol. Cancer Ther. 14, 1928–1938 (2015).
Zhang, L. et al. Mitochondrial STAT5A promotes metabolic remodeling and the Warburg effect by inactivating the pyruvate dehydrogenase complex. Cell Death Dis. 12, 634 (2021).
Saunier, E., Benelli, C. & Bortoli, S. The pyruvate dehydrogenase complex in cancer: an old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J. Cancer 138, 809–817 (2016).
Jeoung, N. H. Pyruvate dehydrogenase kinases: therapeutic targets for diabetes and cancers. Diab Metab. J. 39, 188–197, (2015).
Lee, I. K. The role of pyruvate dehydrogenase kinase in diabetes and obesity. Diab Metab. J. 38, 181–186, (2014).
Sun, W., Liu, Q., Leng, J., Zheng, Y. & Li, J. The role of Pyruvate Dehydrogenase Complex in cardiovascular diseases. Life Sci. 121, 97–103 (2015).
Reed, L. J. A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J. Biol. Chem. 276, 38329–38336 (2001).
Koike, M., Reed, L. J. & Carroll, W. R. α-Keto acid dehydrogenation complexes. J. Biol. Chem. 238, 30–39 (1963).
Green, J. D. F., Perham, R. N., Ullrich, S. J. & Appella, E. Conformational studies of the interdomain linker peptides in the dihydrolipoyl acetyltransferase component of the pyruvate-dehydrogenase multienzyme complex of Escherichia coli. J. Biol. Chem. 267, 23484–23488 (1992).
Perham, R. N., Duckworth, H. W. & Roberts, G. C. Mobility of polypeptide chain in the pyruvate dehydrogenase complex revealed by proton NMR. Nature 292, 474–477 (1981).
Stephens, P. E., Darlison, M. G., Lewis, H. M. & Guest, J. R. The pyruvate dehydrogenase complex of Escherichia coli K12. Nucleotide sequence encoding the dihydrolipoamide acetyltransferase component. Eur. J. Biochem. 133, 481–489 (1983).
Brautigam, C. A. et al. Structural insight into interactions between dihydrolipoamide dehydrogenase (E3) and E3 binding protein of human pyruvate dehydrogenase complex. Structure 14, 611–621 (2006).
Mattevi, A., Schierbeek, A. J. & Hol, W. G. J. Refined crystal-structure of lipoamide dehydrogenase from Azotobacter-Vinelandii at 2.2Å Resolution. A comparison with the structure of glutathione-reductase. J. Mol. Biol. 220, 975–994 (1991).
Reed, L. J. & Oliver, R. M. Structure-function-relationships in pyruvate and alpha-ketoglutarate dehydrogenase complexes. Adv. Exp. Med. Biol. 148, 231–241 (1982).
Milne, J. L. et al. Molecular architecture and mechanism of an icosahedral pyruvate dehydrogenase complex: a multifunctional catalytic machine. EMBO J. 21, 5587–5598 (2002).
Jiang, J. et al. Atomic structure of the E2 inner core of human pyruvate dehydrogenase complex. Biochemistry 57, 2325–2334 (2018).
Izard, T. et al. Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. USA 96, 1240–1245 (1999).
Demarcucci, O. & Lindsay, J. G. Component-X - an immunologically distinct polypeptide associated with mammalian pyruvate-dehydrogenase multi-enzyme complex. Eur. J. Biochem. 149, 641–648 (1985).
Powers-Greenwood, S. L., Rahmatullah, M., Radke, G. A. & Roche, T. E. Separation of protein X from the dihydrolipoyl transacetylase component of the mammalian pyruvate dehydrogenase complex and function of protein X. J. Biol. Chem. 264, 3655–3657 (1989).
Rahmatullah, M. et al. Subunit associations in the mammalian pyruvate dehydrogenase complex: structure and role of protein X and the pyruvate dehydrogenase component binding domain of the dihydrolipoyl transacetylase component. J. Biol. Chem. 264, 2221–2227 (1989).
Perham, R. N. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu. Rev. Biochem. 69, 961–1004 (2000).
Arjunan, P. et al. Novel binding motif and new flexibility revealed by structural analyses of a pyruvate dehydrogenase-dihydrolipoyl acetyltransferase subcomplex from the pyruvate dehydrogenase multienzyme complex. J. Biol. Chem. 289, 30161–30176 (2014).
Byron, O. & Lindsay, J. G. The pyruvate dehydrogenase complex and related assemblies in health and disease. Subcell. Biochem. 83, 523–550 (2017).
Ciszak, E. M., Korotchkina, L. G., Dominiak, P. M., Sidhu, S. & Patel, M. S. Structural basis for flip-flop action of thiamin pyrophosphate-dependent enzymes revealed by human pyruvate dehydrogenase. J. Biol. Chem. 278, 21240–21246 (2003).
Hiromasa, Y., Fujisawa, T., Aso, Y. & Roche, T. E. Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J. Biol. Chem. 279, 6921–6933 (2004).
Mattevi, A. et al. Atomic structure of the cubic core of the pyruvate dehydrogenase multienzyme complex. Science 255, 1544–1550 (1992).
Stoops, J. K. et al. On the unique structural organization of the Saccharomyces cerevisiae pyruvate dehydrogenase complex. J. Biol. Chem. 272, 5757–5764 (1997).
Zhou, Z. H., McCarthy, D. B., O’Connor, C. M., Reed, L. J. & Stoops, J. K. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. USA 98, 14802–14807 (2001).
Baker, T. S. et al. 3-Dimensional structure of the truncated core of yeast pyruvate-dehydrogenase complex. Faseb J. 6, A345–A345 (1992).
Stoops, J. K. et al. 3-Dimensional structure of the truncated core of the Saccharomyces-Cerevisiae pyruvate dehydrogenase complex determined from negative stain and cryoelectron microscopy images. J. Biol. Chem. 267, 24769–24775 (1992).
Mande, S. S., Sarfaty, S., Allen, M. D., Perham, R. N. & Hol, W. G. J. Protein-protein interactions in the pyruvate dehydrogenase multienzyme complex: Dihydrolipoamide dehydrogenase complexed with the binding domain of dihydrolipoamide acetyltransferase. Structure 4, 277–286 (1996).
Toyoda, T., Suzuki, K., Sekiguchi, T., Reed, L. J. & Takenaka, A. Crystal structure of eucaryotic E3, lipoamide dehydrogenase from yeast. J. Biochem. 123, 668–674 (1998).
Liu, S., Xia, X., Zhen, J., Li, Z. & Zhou, Z. H. Structures and comparison of endogenous 2-oxoglutarate and pyruvate dehydrogenase complexes from bovine kidney. Cell Discov. 8, 126 (2022).
Frank, R. A. W., Titman, C. M., Pratap, J. V., Luisi, B. F. & Perham, R. N. A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 306, 872–876 (2004).
Blackwell, N. C., Cullis, P. M., Cooper, R. A. & Izard, T. Rhombohedral crystals of 2-dehydro-3-deoxygalactarate aldolase from. Acta Crystallogr. Sect. D. -Biol. Crystallogr. 55, 1368–1369 (1999).
Arjunan, P. et al. Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 A resolution. Biochemistry 41, 5213–5221 (2002).
Frank, R. A. W., Pratap, J. V., Pei, X. Y., Perham, R. N. & Luisi, B. F. The molecular origins of specificity in the assembly of a multienzyme complex. Structure 13, 1119–1130 (2005).
Forsberg, B. O., Aibara, S., Howard, R. J., Mortezaei, N. & Lindahl, E. Arrangement and symmetry of the fungal E3BP-containing core of the pyruvate dehydrogenase complex. Nat. Commun. 11, 4667 (2020).
Kyrilis, F. L. et al. Integrative structure of a 10-megadalton eukaryotic pyruvate dehydrogenase complex from native cell extracts. Cell Rep. 34, 108727 (2021).
Skerlova, J., Berndtsson, J., Nolte, H., Ott, M. & Stenmark, P. Structure of the native pyruvate dehydrogenase complex reveals the mechanism of substrate insertion. Nat. Commun. 12, 108727 (2021).
Tuting, C. et al. Cryo-EM snapshots of a native lysate provide structural insights into a metabolon-embedded transacetylase reaction. Nat. Commun. 12, 6933 (2021).
Skalidis, I. et al. Cryo-EM and artificial intelligence visualize endogenous protein community members. Structure 30, 575–589.e576 (2022).
Hezaveh, S., Zeng, A.-P. & Jandt, U. Investigation of core structure and stability of human pyruvate dehydrogenase complex: a coarse-grained approach. ACS Omega 2, 1134–1145 (2017).
Hezaveh, S., Zeng, A. P. & Jandt, U. Full enzyme complex simulation: interactions in human pyruvate dehydrogenase complex. J. Chem. Inf. Model. 58, 362–369 (2018).
Zdanowicz, R. et al. Stoichiometry and architecture of the human pyruvate dehydrogenase complex. Sci. Adv. 10, eadn4582 (2024).
Prajapati, S. et al. Structural and functional analyses of the human PDH complex suggest a “Division-of-Labor” mechanism by local E1 and E3 clusters. Structure 27, 1124–1136 (2019).
Smolle, M. & Lindsay, J. G. Molecular architecture of the pyruvate dehydrogenase complex: bridging the gap. Biochem. Soc. Trans. 34, 815–818 (2006).
Jung, H. I., Bowden, S. J., Cooper, A. & Perham, R. N. Thermodynamic analysis of the binding of component enzymes in the assembly of the pyruvate dehydrogenase multienzyme complex of. Protein Sci. 11, 1091–1100 (2002).
Perham, R. N. Domains, motifs, and linkers in 2-Oxo acid dehydrogenase multienzyme complexes - a paradigm in the design of a multifunctional protein. Biochemistry 30, 8501–8512 (1991).
Kaasik, A., Safiulina, D., Zharkovsky, A. & Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 292, C157–163, (2007).
Skolnick, J., Gao, M., Zhou, H. & Singh, S. AlphaFold 2: why it works and its implications for understanding the relationships of protein sequence, structure, and function. J. Chem. Inf. Model. 61, 4827–4831 (2021).
Ali, S. T. & Guest, J. R. Isolation and characterization of lipoylated and unlipoylated domains of the E2p subunit of the pyruvate-dehydrogenase complex of Escherichia-Coli. Biochem. J. 271, 139–145 (1990).
Patel, M. S., Korotchkina, L. G. & Sidhu, S. Interaction of E1 and E3 components with the core proteins of the human pyruvate dehydrogenase complex. J. Mol. Catal. B-Enzymatic 61, 2–6 (2009).
Whitley, M. J. et al. Pyruvate dehydrogenase complex deficiency is linked to regulatory loop disorder in the αV138M variant of human pyruvate dehydrogenase. J. Biol. Chem. 293, 13204–13213 (2018).
Lee, J. et al. The plasticity of the pyruvate dehydrogenase complex confers a labile structure that is associated with its catalytic activity. PLoS ONE 15, e0243489 (2021).
Brautigam, C. A., Wynn, R. M., Chuang, J. L. & Chuang, D. T. Subunit and catalytic component stoichiometries of an in vitro reconstituted human pyruvate dehydrogenase complex. J. Biol. Chem. 284, 13086–13098 (2009).
Smolle, M. et al. A new level of architectural complexity in the human pyruvate dehydrogenase complex. J. Biol. Chem. 281, 19772–19780 (2006).
Yi, J. Z. et al. Effect of substitutions in the thiamin diphosphate-magnesium fold on the activation of the pyruvate dehydrogenase complex from Escherichia coli by cofactors and substrate. J. Biol. Chem. 271, 33192–33200 (1996).
Sergienko, E. A. et al. Spectroscopic detection of transient thiamin diphosphate-bound intermediates on benzoylformate decarboxylase. Biochemistry 39, 13862–13869 (2000).
Sergienko, E. A. & Jordan, F. New model for activation of yeast pyruvate decarboxylase by substrate consistent with the alternating sites mechanism: Demonstration of the existence of two active forms of the enzyme. Biochemistry 41, 3952–3967 (2002).
Kovina, M. V. & Kochetov, G. A. Cooperativity and flexibility of active sites in homodimeric transketolase. Febs Lett. 440, 81–84 (1998).
Domingo, G. J., Chauhan, H. J., Lessard, I. A. D., Fuller, C. & Perham, R. N. Self-assembly and catalytic activity of the pyruvate dehydrogenase multienzyme complex from Bacillus stearothermophilus. Eur. J. Biochem. 266, 1136–1146 (1999).
Dave, E., Guest, J. R. & Attwood, M. M. Metabolic engineering in Escherichia coli: lowering the lipoyl domain content of the pyruvate dehydrogenase complex adversely affects the growth rate and yield. Microbiology 141, 1839–1849 (1995).
Cate, R. L., Roche, T. E. & Davis, L. C. Rapid intersite transfer of acetyl groups and movement of pyruvate dehydrogenase component in the kidney pyruvate dehydrogenase complex. J. Biol. Chem. 255, 7556–7562 (1980).
Hackert, M. L., Oliver, R. M. & Reed, L. J. Evidence for a multiple random coupling mechanism in the alpha-ketoglutarate dehydrogenase multienzyme complex of Escherichia coli: a computer model analysis. Proc. Natl. Acad. Sci. USA 80, 2226–2230 (1983).
Hackert, M. L., Oliver, R. M. & Reed, L. J. A computer model analysis of the active-site coupling mechanism in the pyruvate dehydrogenase multienzyme complex of Escherichia coli. Proc. Natl. Acad. Sci. USA 80, 2907–2911 (1983).
Milne, J. L. et al. Molecular structure of a 9-MDa icosahedral pyruvate dehydrogenase subcomplex containing the E2 and E3 enzymes using cryoelectron microscopy. J. Biol. Chem. 281, 4364–4370 (2006).
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F. & Kroemer, G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 (2015).
Luengo, A. et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol. Cell 81, 691–707.e696 (2021).
Harris, R. A., Bowker-Kinley, M. M., Huang, B. L. & Wu, P. F. Regulation of the activity of the pyruvate dehydrogenase complex. Adv. Enzym. Regul., Vol. 42, Proc. 42, 249–259 (2002).
Patel, M. S. & Korotchkina, L. G. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 34, 217–222 (2006).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Eisenstein, F. et al. Parallel cryo electron tomography on in situ lamellae. Nat. Methods https://doi.org/10.1038/s41592-022-01690-1 (2022).
Zivanov, J. et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. eLife 11, e83724 (2022).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Kremer, J. R., Mastronarde, D. N. & McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996).
Zheng, S. et al. AreTomo: an integrated software package for automated marker-free, motion-corrected cryo-electron tomographic alignment and reconstruction. J. Struct. Biol. X 6, 100068 (2022).
Winkler, H. 3D reconstruction and processing of volumetric data in cryo-electron tomography. J. Struct. Biol. 157, 126–137 (2007).
Winkler, H. et al. Tomographic subvolume alignment and subvolume classification applied to myosin V and SIV envelope spikes. J. Struct. Biol. 165, 64–77 (2009).
Tegunov, D., Xue, L., Dienemann, C., Cramer, P. & Mahamid, J. Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 A in cells. Nat. Methods 18, 186–193 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at bioRxiv https://doi.org/10.1101/2021.10.04.463034 (2021).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput Chem. 31, 455–461 (2010).
Delano, W. PyMOL:an open source molecular graphics tool. CCP4. Newsl. Protein Crystallogr. 40, 82–92 (2002).
Frisch, M. J. et al. Gaussian 16, Revision C.01. https://gaussian.com (2016).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Sobtop, T. L. Version [Dev 3.1]. http://sobereva.com/soft/Sobtop, https://doi.org/10.1038/s41467-021-27287-4.
Pall, S. et al. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 153, 134110 (2020).
Lindorff-Larsen, K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010).
Berendsen, H. J. C., Postma, J. P. M., Vangunsteren, W. F., Dinola, A. & Haak, J. R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput Chem. 32, 2319–2327 (2011).
Tribello, G. A., Bonomi, M., Branduardi, D., Camilloni, C. & Bussi, G. PLUMED 2: new feathers for an old bird. Comput. Phys. Commun. 185, 604–613 (2014).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Ermel, U. H., Arghittu, S. M. & Frangakis, A. S. ArtiaX: an electron tomography toolbox for the interactive handling of sub-tomograms in UCSF ChimeraX. Protein Sci. 31, e4472 (2022).
Consortium, T. W. EMDB—the Electron Microscopy Data Bank. Nucleic Acids Res. D456–D465, https://doi.org/10.1093/nar/gkad1019.
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–D1527 (2022).
Acknowledgements
We thank Lingyun Wu, Chenyu Yang and Menghan Zhang in the Center of Cryo-Electron Microscopy (CCEM), Zhejiang University for their technical assistance on 200kv and 120kv cryo-electron microscopy. We thank Deyue Xu from the Laboratory Animal Center, Zhejiang University for providing fresh porcine heart in pre-experiment. We thank Shuaiqi (Phil) Guo and Donghyun (Raphael) Park for critical reading of the manuscript. This work was supported by the National Key Research and Development Program of China (2018YFA0507700 to X.Z.) and National Natural Science Foundation of China (32200980 to Y.C.).
Author information
Authors and Affiliations
Contributions
X.Z. initiated the project. X.Z., Y.C., and H.G. supervised the research. C.W., C.M., H.W., Y.W., S.A., and Q.H. performed the sample preparation and characterization. Y.C., C.W., and S.C. collected the cryo-ET data. Y.C. and C.W. processed the cryo-ET data and reconstructed the cryo-ET density map. Y.X. and X.C performed the MD simulations and related analysis. C.W., Y.X., and Y.C. prepared figures. C.M., C.W., Y.X., S.C., and J.C. performed the AlphaFold2 prediction and sequence alignment. C.Y., J.X, Y.J., and Z.J. assisted in the sample preparation and data analysis. Y.C., X.Z., C.W., C.M., and H.G. wrote the manuscript. All authors discussed and commented on the results and the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bjoern Forsberg and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, C., Ma, C., Xu, Y. et al. Dynamics of the mammalian pyruvate dehydrogenase complex revealed by in-situ structural analysis. Nat Commun 16, 917 (2025). https://doi.org/10.1038/s41467-025-56171-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56171-8
This article is cited by
-
FDX1-mediated cuproptosis promotes cholestatic liver injury exacerbated by taurocholic acid-enhanced copper accumulation
Cell Death Discovery (2026)
-
From maternal microbes to offspring development: gut microbiota-derived thiamine regulates the gut microbiota and drives the placental Notch pathway to coordinate angiogenesis and nutrient transport
Microbiome (2025)
-
Comparative transcriptomics reveals differential carbohydrate and lipid metabolism in roots and nodules of Rhizobia-Inoculated alfalfa (Medicago sativa L.)
BMC Plant Biology (2025)
-
Immunometabolism: crosstalk with tumor metabolism and implications for cancer immunotherapy
Molecular Cancer (2025)
