
Abstract
Designing efficient Ruthenium-based catalysts as practical anodes is of critical importance in proton exchange membrane water electrolysis. Here, we develop a self-assembly technique to synthesize 1 nm-thick rutile-structured high-entropy oxides (RuIrFeCoCrO2) from naked metal ions assembly and oxidation at air-molten salt interface. The RuIrFeCoCrO2 requires an overpotential of 185 mV at 10 m A cm−2 and maintains the high activity for over 1000 h in an acidic electrolyte via the adsorption evolution mechanism. We discuss the role of each element in the RuIrFeCoCrO2 and find that the Cr, Co, and Ir sites contribute to the catalytic activity, while the Cr atoms weaken the Ru-O bond covalency and improves the catalyst stability. The assembled proton exchange membrane electrolyzer operates stably for more than 600 h at a large current of 1 A cm−2. The naked ion assembly demonstrated in this work may provide an effective pathway for the controlled synthesis of a diversity of high-entropy materials.
Similar content being viewed by others


Introduction
Proton-exchange membrane water electrolyzer (PEMWE) has been considered as one of the most potential devices for green hydrogen production due to the high efficiency, fast response, and safety1,2. However, the rapid structural deterioration of anode catalysts under acidic and oxidative condition has severely hindered the industrial application3,4. In terms of cost, abundance and activity, ruthenium oxide (RuO2)-based catalysts are regarded the ideal candidate for anodic oxygen evolution reaction (OER)5,6. However, the high covalency of Ru-O bonds triggers the lattice oxygen oxidation mechanism (LOM), leading to oxygen vacancy and leaching of Ru species (soluble RuO4 species), which ultimately accelerates the crystal degradation5,7. Doping can enhance the Ru-O bond covalency and improves the activity at the sacrifice of stability. On the contrary, weakening the Ru-O bond covalency will induce the adsorption evolution mechanism (AEM) and improve the stability of Ru-based oxides8, which has been much less explored. Moreover, the doped metal elements are subject to leaching due to their thermodynamic instability and thus induce accelerated corrosion of catalysts9,10,11. Because of these factors, the operation durability of PEMWE cells often fall below 100 h under the industrial current level of 1 A cm−2.
High-entropy materials (HEM) exhibit unique features induced by entropy increase, which is conducive to enhancing the crystal stability and catalytic active sites12,13,14,15,16. However, a deep understanding of the role of each element in electrocatalysis is still a challenge17. The synthesis process of HEM usually requires heating temperatures above 1000 °C to achieve high mixing entropy, followed by quenching to preserve the high-entropy state18,19,20,21. Recently, it has been reported that liquid metals can be used to synthesize HEM by negative mixing enthalpy under mild conditions22,23. The obtained materials are mostly in form of irregular spherical particles, which corresponds to reduced atomic utilization and limited catalysis performance. In contrast, atomic-thin nanosheets have a larger specific surface area, which would expose more active atoms. However, controlled synthesis of such thin HEM nanosheets remains unexplored.
The air-liquid interface self-assembly is a process where freely moving units in a solution are driven to arrange into well-ordered superstructures24,25. As mentioned above, liquid metal induced HEM indicates that the liquid state is an ideal dynamic mixing environment for synthesizing multicomponent materials. One idea comes to us: can we overcome the size limit of the assembly units by reducing micro/nano particles to metal atoms under liquid condition? This would create an advanced synthesis strategy to realize complex multicomponent atomic-thin superstructures. However, metal ions typically exist in water as hydrated ions, which impedes their desolvation and directional movement.
Here, we develop a naked metal ions self-assembly strategy to realize bottom-up synthesis of atomic-thin HEM, exemplified by rutile-structured RuIrFeCoCrO2 high-entropy oxides (HEO). This bottom-up assembly technique deploys molten salts to desolvate metal ions from water molecules coordination, thereby forming freely moving naked ions. The vaporization of free water molecules drives the naked metal ions to directionally move towards the air-liquid interface, followed by rapid oxidation. Thus formed RuIrFeCoCrO2 HEO features an ~1 nm thickness and hundreds of micrometers size. The contribution of each atom to OER has been revealed. The decreased Ru-O bond covalency suppresses the leaching of Ru species and contribute to enhanced stability by AEM pathway, evidenced by in-situ experiments and computations; while the Cr, Co, Ir atoms are responsible for the activity. As a result, the RuIrFeCoCrO2 electrode delivers a low overpotential of 185 mV at 10 mAcm−2 and stable operation over 1000 h in an acidic electrolyte. The assembled PEMWE device (countered with Pt/C electrode) outputs a cell voltage of 1.68 V at a high current of 1 A cm−2 for over 600 h, highlighting the potential in industrial green hydrogen production.
Results
Air-liquid interface self-assembly requires the assembly units to be freely moving relative to the solution, allowing directional movement and assembly by a driving force. Due to the extremely small radius, atomic species are usually subjected to high resistance from water. The relationship between the desolvation coefficient (D) and temperature can be described as follow:
where Ea, kb and T are the desolvation energy, Boltzmann constant, and temperature, respectively26. The density functional tight binding methods with highest-level variant (GFN3-xTB) indicate that the D in water is around 1\(\times\)10−20 with high desolvation energy, making self-desolvation process for metal ions highly challenging and unfavorable for assembly (Fig. 1a and Supplementary Figs. 1–2). However, in molten salt (potassium nitrate, KNO3), the corresponding D (1\(\times\)10−2) for metal ions is higher by several orders of magnitude compared to that in water, which allows metal ions to become freely moving naked ions (Fig. 1a and c). The small amount of water (wt. 17%) remained in the metal precursors quickly vaporizes at high temperature of 360 °C (Supplementary Fig. 3). This process propels the naked ions rapidly towards the air-liquid interface for assembly, followed by oxidation and then forming HEO (Fig. 1d). By applying Ru, Ir, Fe, Co, Cr metal ions into this synthesis, large-scale 2D RuIrFeCoCrO2 were fabricated.

a Calculated comparison of the motor behaviors of metal ions in molten salt and water. b Metal ions coordinated by six water molecules show confined movement and sluggish desolvation in water. The arrows in (b) indicate the movement direction of coordinated water molecule. c Water coordinated metal ions are completely desolvated and free to move in molten salt. The background green (b) and gray (c) colors indicate water and molten salt solvents, respectively. d Naked metal ions in molten salt move rapidly toward the air-liquid interface driven by water evaporation for self-assembly, and then react with oxygen to form atomic-thin HEO. The comet tails (c, d) indicate the movement direction of metal ions in molten salt.
The obtained RuIrFeCoCrO2 catalyst is characterized by a large surface over hundreds of micrometers square (Fig. 2a). X-ray diffraction (XRD) pattern proves that the main diffraction peaks can be assigned to rutile RuO2 crystals (Fig. 2b). The severe-lattice-distortion in RuIrFeCoCrO2 is evidenced by the positive shifted and broadened diffraction peaks. Atomic force microscopy (AFM) image further displays the two-dimensional structure with an average thickness of ~1 nm, corresponding to ~5 atomic layers (Fig. 2c). Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) confirms the two-dimensional structures are connected by amounts of HEO clusters with average size of 2 nm. The corresponding selected area electron diffraction (SAED) pattern shows the polycrystalline of RuIrFeCoCrO2. A closer inspection of aberration-corrected HAADF-STEM images display the characteristics of severe lattice distortion, amorphous-crystal interfaces, and twinning boundaries (Fig. 2e). In such a HAADF image, the heavy and light elements present high and low imaging contrast based on atomic numbers, respectively. Furthermore, the atomic line profiles analysis clearly shows the coexistence of Ru, Ir, Fe, Co, and Cr elements, which is confirmed by various signal intensities (Fig. 2f). To investigate the unique atomic arrangement in HEO, the geometric phase analysis (GPA) is conducted to detect the surficial lattice strain (Fig. 2g and Supplementary Figs. 4–5). The obvious color change in mapping indicates the presence of large lattice strain, which can be ascribed to the large radius differences between different metal atoms. HAADF-STEM and the corresponding energy dispersive spectrum (EDS) mapping images show the uniform distribution of Ru, Ir, Fe, Co, Cr, and O elements (Fig. 2i). The molar content of Ru, Ir, Fe, Co, and Cr for RuIrFeCoCrO2 is around 23.8, 18.9, 21.5, 18.7, and 17.1 %, respectively, which was determined by inductively coupled plasma mass spectrometry (ICP-MS). These results verify an atomic-thin superstructure. For comparison, RuO2, IrO2, RuIrO2 and RuIrFeCoO2 were synthesized with similar method, and commercial RuO2 (C-RuO2) and IrO2 (C-IrO2) catalysts were prepared for characterization and electrochemical tests (Supplementary Figs. 6–13).

TEM (a), XRD (b), and AFM images (c) of RuIrFeCoCrO2. d, e Aberration-corrected HAADF-STEM images. The inset in (d) is the corresponding SAED image. The red dots in (e) indicate the metal atoms. f Line-scanning intensity profiles obtained from the atomic arrangements highlighted with yellow and blue boxes. g GPA analysis for strain distribution. The red and blue areas present tensile and compressive strain (εxx), respectively, along the x direction. h, i HAADF-STEM image and elemental mappings.
The electronic structure and chemical environment of RuIrFeCoCrO2 and control catalysts with varying elements were investigated by X-ray analytical techniques. As presented in the O 1 s spectra from X-ray photoelectron spectroscopy (XPS) in Fig. 3a, the peaks located at about 529.2, 531.1, and 532.8 eV can be ascribed to lattice oxygen (OL), oxygen vacancy (OV), and hydroxyl groups (OOH), respectively27. The integrated area ratio of OV/OL increases from 0.62, 0.65 to 0.74 in C-RuO2, RuIrFeCoO2, and RuIrFeCoOCrO2, respectively, indicating more oxygen vacancies generated by Cr atoms. Moreover, the peak of lattice oxygen from RuIrFeCoO2 positively shifted toward a higher binding energy, while that of Ru shifted to a lower binding energy, when compared to those of C-RuO2 and RuIrFeCoO2 (Fig. 3b). The positive shift of O 1 s and negative shift of Ru 3p binding energies present sufficient evidence for the decreased Ru-O bond covalency by high-entropy effects, indicating the improved structural stability8,28,29. For more detailed analysis, the ratio of Ru4+/Ru3+ species decreases from 3.8 in C-RuO2, 2.88 in RuIrFeCoO2 to 2.19 in RuIrFeCoCrO2. The multiple doping of Ir, Fe, and Co elements lead to a higher oxidation state of Ru in RuIrFeCoO2 and that in RuO2. This may result in structural collapse during acidic OER due to the formation of unstable high valanced Ru species30. It should be noted that the introduction of Cr greatly reduces the oxidation state of Ru (Ru<4+) in RuIrFeCoCrO2, which is even lower than that of C-RuO2 (Ru4+). Atomic Cr species thus could induce electron pumping onto Ru sites. The Ir 4 f XPS spectra display the peaks located at about 61.1 and 64.1 eV, which can be assigned to Ir 4 f7/2 and Ir 4f5/2 of Ir4+ (Fig. 3c). Moreover, the Ir 4f peak in RuIrFeCoCrO2 shows a positive shift compared to those in C-IrO2 and RuIrFeCoO2 references. The oxidation state of Ir (Ir>4+) was enhanced by Cr atoms, which is generally considered a high activity center during OER31. Therefore, we propose that Cr in RuIrFeCoCrO2 enhances the structural stability of Ru-O configuration while simultaneously increasing the catalytic activity of Ir. The Ar+ sputtering-assisted XPS depth profiling spectra indicates that all the metal elements in RuIrFeCoCrO2 maintain the same oxidation state, verifying the uniform crystal structures (Fig. 3d and Supplementary Fig. 14).

XPS spectra of O 1 s (a), Ru 3p (b), and Ir 4 f (c) for C-RuO2, C-IrO2, RuIrFeCoO2 and RuIrFeCoCrO2. The OOH, OV, and OL in (a) are ascribed to hydroxyl groups, oxygen vacancy, and lattice oxygen, respectively. The sat. in (c) is the abbreviation of satellite peak. d Ar+ sputtering-assisted XPS depth profiling spectra for Ru. e Normalized Ru K-edge XANES spectra of samples. f Fourier transforms EXAFS spectra. g Normalized Ir L3-edge XANES spectra. h Average oxidation states and d-band holes of Ir calculated from the XANES spectra. i Fourier-transform EXAFS spectra of Ir foil, C-IrO2, and RuIrFeCoCrO2.
X-ray absorption near-edge spectroscopy (XANES) was conducted for the RuIrFeCoCrO2 at the Ru K-edge and Ir-L3 to investigate intrinsic electronic structures. The absorption edges of Ru in RuIrFeCoO2, C-RuO2, and RuIrFeCoCrO2 samples show the gradually negative shift, which is consistent with XPS results. This implies that increased oxidation state of Ru by incorporation of Ir, Fe, and Co, while reduced Ru valance by the introduction of Cr atoms. The extended X-ray adsorption fine structure (EXAFS) and fitting results suggest that the band distance of Ru-O appeared at 1.95 Å in C-RuO2, and that is slightly contracted to 1.93 Å for RuIrFeCoCrO2 (Fig. 3f, Supplementary Fig. 15, and Supplementary Table 1), suggesting the modified to Ru-O bond length. The changed band distance was also observed in the two-O and single-O bridged Ru-Ru scattering paths. Moreover, the coordination number of Ru-O in RuIrFeCoCrO2 is 5.8, which is lower than those in C-RuO2 (6), RuIrFeCoO2 (5.9). This means more oxygen vacancies induced by Cr doping in RuIrFeCoCrO2. Compared to Ir foil and C-IrO2, the white-line intensity of Ir L3-edge in RuIrFeCoCrO2 gradually increases (Fig. 3g). The oxidation states of Ir can be estimated to be 0 for Ir foil, + 4 for IrO2, and + 5.1 for RuIrFeCoCrO2, by using Ir foil (5d7) and IrO2 (5d3) as references (Fig. 3h). The corresponding d-band hole number for Ir foil, C-IrO2, and RuIrFeCoCrO2 was evaluated to be 3, 7, and 8.1, respectively, indicating the presence of varied unoccupied Ir 5d-orbitals. The tunable d vacancy would tailor the adsorption energy barrier of intermediates, modifying the reaction performance32. The corresponding EXAFS fitting result suggests the Ir-O in RuIrFeCoCrO2 exhibits a lower coordination number (5.8) and enlarged Ir-O distance (2.00 Å) than those in C-IrO2 (6.0 and 1.98 Å) due to the lattice distortion (Fig. 3i, Supplementary Fig. 16, and Supplementary Table 2). Based on the above fine structure analysis, the RuIrFeCoCrO2 shows decreased Ru-O bond covalency and high-valance Ir.
To investigate the acidic OER performance of RuIrFeCoCrO2, a three-electrode cell in 0.5 M H2SO4 aqueous electrolyte was used. From the linear sweep voltammograms (LSV) (Fig. 4a), RuIrFeCoCrO2 displays the highest OER activity, as verified by the lowest overpotentials of 185 and 226 mV to reach the currents of 10 and 100 mA cm−2, respectively, comparable to RuIrO2 (225 and 280 mV), RuIrFeCoO2 (230 and 301 mV), RuO2 (300 and 390 mV), IrO2 (320 and 420 mV), and C-IrO2 (341 and 436 mV), as compared in Fig. 4b. The corresponding LSV curves without iR compensation were also displayed in Supplementary Fig. 17. The corresponding Tafel slope for RuIrFeCoCrO2 (41 mV dec−1) is much lower than those of RuIrO2 (58 mV dec−1), RuIrFeCoO2 (66 mV dec−1), RuO2 (72 mV dec−1), IrO2 (79 mV dec−1), and C-IrO2 (98 mV dec−1), suggesting the boosted reaction kinetics (Fig. 4c). The double-layer capacitance (Cdl) measurement was performed to evaluate the electrochemically active surface areas (ECSAs) of various catalysts (Fig. 4d and Supplementary Fig. 18). When normalized to their corresponding ECSA values (Supplementary Fig. 19), RuIrFeCoCrO2 displays the highest OER activity at a potential of 1.5 V vs. RHE (0.087 mA cm−2), which is over 40, 62, and 124 times that of C-IrO2 (0.002 mA cm−2), RuO2 (0.0015 mA cm−2), IrO2 (0.0007 mA cm−2), respectively (Fig. 4e). Moreover, the turnover frequency (TOF) values were calculated based on the loading amounts of metal atoms to evaluate the intrinsic OER activity. Notably, the TOF value at η = 300 mV for RuIrFeCoCrO2 is 0.2 s−1, which is 10 times that of RuIrFeCoO2 (0.02 s−1), showing the positive contribution to activity by Cr doping (Fig. 4e and Supplementary Table 3). In addition to enhanced mass transfer, the charge transfer process in the RuIrFeCoCrO2 electrode is also improved, as suggested by the lower charge-transfer resistance (Rct) in electrochemical impedance spectroscopy (EIS) measurement (Fig. 4f). In particular, the introduction of Cr in RuIrFeCoO2 ignites a sharply drop of Rct, probably due to its role in tuning electronic configuration. Moreover, a series of working electrodes with different catalyst loading (200, 150, 100, 75, and 50 μL for 1, 0.75, 0.5, 0.375, and 0.25 mg cm−2, respectively) were further prepared for comparison. When the catalyst mass loading decreases to 0.75 or 0.5 mg cm−2, the OER activity is very close to the working electrode with a mass loading of 1 mg cm−2 (Supplementary Fig. 20). The negligible change in overpotential indicates the working electrode with mass loading of 0.5 mg cm−2 is still comparable to the reported catalysts.

a OER polarization curves of C-IrO2, IrO2, RuO2, RuIrO2, RuIrFeCoO2, and RuIrFeCoCrO2 in 0.5 M H2SO4. The voltage is corrected by an automatic 90% of iR compensation. Comparison of overpotentials at 10 and 100 mA cm−2 (b) and Tafel plots (c). d Half of current density differences (Ja-Jc) plotted against scan rates at non-Faradaic current region. e Comparison of TOF at an overpotential of 300 mV and activity normalized by ECSA at 1.5 V vs. RHE. The solid and sparse columns indicate TOF and intrinsic activity values, respectively. f EIS Nyquist plots of the catalysts acquired in the frequency range of 0.01-100 kHz at an A.C. amplitude of 5 mV. g Chronopotentiometric test of RuIrFeCoCrO2 at the current density of 10 mA cm−2 over 1000 h. The inset is the comparation between of C-RuO2, RuIrO2, RuIrFeCoO2, and RuIrFeCoCrO2 at 100 mA cm−2.
During the long-term test in chronopotentiometric mode, the RuIrFeCoCrO2 electrode shows only a marginal potential drop (1.2%) at the current of 10 mA cm−2 after 1000 h (Fig. 4g). At the higher current of 100 mA cm−2, the potential increases only by 0.02 V after operation for 100 h, much lower than those of RuIrFeCoO2 (0.09 V for 50 h) and RuIrO2 (0.08 V for 60 h). To compare, the RuIrFeCoCrO2 outperforms the advanced RuIr-based electrocatalysts for acidic OER in terms of mass loading, overpotential, Tafel slope, and structure stability (Fig. 4h and Supplementary Table 4). Moreover, the accelerated durability test-cyclic voltammetry (ADT-CV) test for compared samples were conducted (Supplementary Fig. 21). The C-RuO2 shows a sharp drop in current densities from the 1st, 1000th to 5000th cycle and those of RuO2, RuIrO2, and RuIrFeCoO2 show slightly decrease. However, the RuIrFeCoCrO2 shows no obvious change in activity, indicating the advanced durability. After stability measurement, the C-RuO2 particles shows seriously aggregation with unstable crystal structure. However, the molar content of Ru, Ir, Fe, Co, and Cr for RuIrFeCoCrO2 is 20.1, 23.7, 18.4, 16.3, and 21.5 %, respectively (Supplementary Table 5), nearly the same as their initial values. The composition, nanosheet morphology, and crystalline phase of the RuIrFeCoCrO2 are well retained, as revealed by XPS, TEM image and XRD characterization, highlighting the high stability (Supplementary Figs. 22–28).
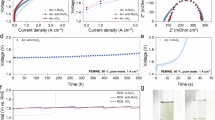
To verify the feasibility of electrocatalysts under industrial conditions, RuIrFeCoCrO2 and C-RuO2 were used as the anode in PEMWE cell tested in 0.5 M H2SO4 electrolytes at 80 °C (Fig. 5a, b). The cathode is commercial 20% Pt/C and the PEM is Nafion membrane (Nafion 117). Comparing the polarization curves (Fig. 5c) shows that the RuIrFeCoCrO2 || Pt/C cell delivers a consistently lower cell voltage than that of C-RuO2 || Pt/C cell. As for the stability, the RuIrFeCoCrO2 || Pt/C in the PEMWE can stably run for >600 h with only marginal increase of cell voltage, while the cell voltage of C-RuO2 || Pt/C undergoes sharp increase within only 20 h (Fig. 5d), which is comparable to the reported PEMWE cells (Supplementary Table 6). Hence, the advanced structural stability and activity of the RuIrFeCoCrO2 electrode renders its potential in industrial green hydrogen production.

a, b Photograph (a) and the corresponding schematic structure (b) of PEMEW device. c Polarization curves of the PEMWE no iR-correction. d Durability test of the single-cell PEMWE measured with a current density of 1 A cm−2 in 0.5 M H2SO4 aqueous solution at 80 °C. The blue arrows in (b) indicate the transfer direction of molecules.
To gain insight to the structural stability and reaction mechanism, we conducted in-situ Raman spectroscopy, differential electrochemical mass spectrometry (DEMS), and theoretical calculations. The chemical states of Ru in RuO2 and RuIrFeCoCrO2 were monitored by in-situ Raman under the continuous potential change from the open-circuit potential to 1.6 V vs. RHE (Fig. 6a, b). The three peaks located at ~ 910, 989, and 1060 cm−1 are due to the asymmetric stretching vibration of HSO4−, the total symmetric stretching vibration of SO42-, and the symmetric stretching of HSO4− ions from H2SO4, respectively33. Moreover, there are two major Raman peaks for rutile phase of RuO2 located at ~ 432 and 598 cm−1, which are assigned to Ru4+-O and Ru3+-O bonds, respectively34. The atomic ratio of Ru3+/Ru4+ indicates the structural stability. When the high-valence Ru species leach from the catalyst, it will cause structural collapse and catalytic deactivation. As shown in Fig. 6c, the intensity ratio (\({{{\rm{I}}}}_{{{{\rm{Ru}}}}^{3+}}\)/\({{{\rm{I}}}}_{{{{\rm{Ru}}}}^{4+}}\)) of RuIrFeCoCrO2 is much higher than that of RuO2. The more Ru3+ species on the high-entropy surface would weaken the bonding between Ru atoms and lattice oxygen, resulting in enhanced structural stability in the harsh acidic OER environment. The in-situ DEMS measurements were further carried out to disclose the reaction mechanisms for C-RuO2, RuIrFeCoO2, and RuIrFeCoCrO2 by using the 18O isotope. The catalysts were labeled with 18O in 0.5 M H2SO4 of H218O electrolyte, following by rinsed with H216O. The labeled catalysts were then tested in 0.5 M H2SO4 of H216O solution to produce oxygen bubbles, which were analyzed by DEMS in real time during OER (Fig. 6d and Supplementary Fig. 29). The presence of both 34O2 (18O in the lattice and 16O in electrolyte) and 32O2 indicates C-RuO2 and RuIrFeCoO2 follow the LOM pathway (Fig. 6e and Supplementary Fig. 30). However, there is only 32O2 signal detected during the OER for RuIrFeCoCrO2 without obvious 34O2, which reveals the AEM pathway (Fig. 6f). The switch of reaction mechanism from LOM for RuO2 and RuIrFeCoO2 to AEM for RuIrFeCoCrO2 also gives strong support for structural stability. The result comparison of RuIrFeCoO2 and RuIrFeCoCrO2 also verifies the positive contribution of Cr atoms to the mechanistic transition.

a, b In-situ Raman spectra for RuO2 and RuIrFeCoCrO2 in 0.5 M H2SO4 conducted at continuously changed potentials from the open-circuit potential (OCP) to 1.6 V vs. RHE. c Intensity ratio of Ru3+/Ru4+ (\({{{\rm{I}}}}_{{{{\rm{Ru}}}}^{3+}}\)/\({{{\rm{I}}}}_{{{{\rm{Ru}}}}^{4+}}\)) in (a, b) as a function of applied potential. d schematic of the in-situ DEMS (PTFE, polytetrafluoroethylene). e, f DEMS measurements of 32O and 34O signals from the reaction products for 18O-labeled RuIrFeCoO2 (e) and RuIrFeCoCrO2 (f) in 0.5 M H2SO4 in H216O. g Crystal orbital Hamilton population (COHP) analysis for Ru-O bonds in RuO2 (top) and RuIrFeCoCrO2 (bottom). h Density-of-states (DOS) analysis. i, j The OER reaction pathway on Ru site for RuO2 and Co site for RuIrFeCoCrO2. k Calculated energy barrier diagrams of RuO2 and RuIrFeCoCrO2. l Theoretical overpotential volcano plot for RuO2 and RuIrFeCoCrO2, calculated from the scaling relationship of ∆GOOH = 0.79∆GOH + 3.46 eV. The O, OH and OOH present the corresponding adsorption intermediates. The Co_11, Ir_5, and Ir_19 indicates different atomic sites. The color change from red to blue indicates an increase in overpotentials for OER. The η in (k) and (l) presents the overpotential of OER. The LOM (i) and AEM (j) stand for lattice oxygen mechanism and adsorption oxygen mechanism, respectively.
In the density functional theory calculations, we establish and screen the optimized slabs for RuO2 and RuIrFeCoCrO2 to investigate the adsorption of intermediates on catalysts during the four-electron OER process (Supplementary Figs. 31–33). To understand the structural stability of RuIrFeCoCrO2, the crystal orbital Hamilton population (COHP) analysis was performed, which is a partitioning of the band-structure energy in terms of orbital-pair contributions (Fig. 6g)35. The value of integrated COHP (iCOHP) for Ru-O in RuIrFeCoCrO2 (3.67 eV) is lower than that in RuO2 (4.16 eV), indicating weakened Ru-O bonds in the HEO. In addition, the downward shift of p-band center (εp) of RuO2 suggests charge redistribution, which will regulate the adsorptions of oxygen intermediates (Fig. 6h and Supplementary Fig. 34). The charge redistribution is also reflected by the d-band center (εd) shift, in accordance to the valence change of Ru36. Because of the reduction in Ru valence state and the weakening of Ru-O interaction, the lattice oxygen reaction process could be inhibited and the dissolution of high-valence Ru species is effectively suppressed28,29. These two effects jointly improve the structural durability. To gain insights to the enhanced OER activity and reaction mechanism, we calculated the reaction pathways with Gibbs free energy of the reaction intermediates for RuO2 and RuIrFeCoCrO2 (Fig. 6i, j, Supplementary Figs. 35–38). As shown in Fig. 6k, the rate-determining-step (RDS) changes from the formation of *OOH on RuO2 to *O on RuIrFeCoCrO2. Moreover, the free energy barrier for RDS of RuO2 is 1.95 eV (U = 0 V), corresponding to the overpotential of 0.72 V (U = 1.23 V). These two values drop to 1.70 eV and 0.35 V for RuIrFeCoCrO2. The overpotentials (∆GOOH-∆GO, RDS) for possible metal active sites were calculated versus ∆GOH and ∆GO-∆GOH by using the scaling relationship of ∆GOOH = 0.79∆GOH + 3.46 eV (Supplementary Figs. 39–40). As shown in the volcano plot in Fig. 6l, the calculated overpotential during acidic OER for RuO2 electrode is 0.78 V, while overpotentials of the high active sites in RuIrFeCoCrO2 drop to 0.47 V (Co site), 0.46 V (Cr site), and 0.52 V (Ir site), illustrating the boosted activity. In addition, the overpotential of RuO2 for LOM is lower than that for AEM, validating the unstable crystal structure. Our calculation also agrees with experiments that the reaction mechanism changes from LOM in RuO2 to AEM in RuIrFeCoCrO2.
Discussion
We have synthesized atomically thin high-entropy oxide, RuIrFeCoCrO2, and demonstrated its high activity and stability for PEMWE. The fabrication is realized by high mobility of naked metal ions in molten slat (in contrast to solvated ions with confinement movement in water) and their self-assembly at the liquid-air interface. When applied as the acidic OER catalyst, the RuIrFeCoCrO2 delivers a low overpotential of 185 mV at 10 mA cm−2 and stable operation over 1000 h. The assembled PEMWE device outputs a cell voltage as low as 1.68 V at an industrial current level of 1 A cm−2 for >600 h, suggesting potential for industrial production. The contribution of each atom in the RuIrFeCoCrO2 to the electrocatalysis performance has been scrutinized. Specifically, all the atoms contribute to the entropy increasing effect. The weakened Ru-O bond covalency by Cr prohibits the leaching of Ru atoms and thus benefits the crystal stability. Finally, according to calculations, the Cr, Co, Ir atoms are responsible for the boosted OER activity. Overall, the OER reaction mechanism changes from LOM pathway on RuO2 to AEM on RuIrFeCoCrO2, improving the crystal stability and activity. This work enhances our understanding the contribution by individual elements in high-entropy oxide electrode to the high catalytic performance. Meanwhile, the proposed naked metal ions self-assemble strategy may guide the controlled mild-condition synthesis of complex functional superstructures.
Methods
Chemicals. Potassium nitrate (KNO3, ≥99 %), ruthenium chloride hydrate (RuCl3·3H2O, 99.9%), iridium chloride (IrCl3·xH2O, 99.9%), iron chloride hexahydrate (FeCl3·6H2O, ≥98%), cobalt chloride hexahydrate (CoCl2·6H2O, 98%), chromium chloride (CrCl3, 99%), absolute ethanol (C2H5OH), sulfuric acid (H2SO4, 98%), Nafion®117 solution (5%), hydrogen peroxide solution (H2O2, 3 wt.% in H2O), ruthenium oxide (RuO2, 99.9%), iridium oxide (IrO2, 99.9%), and platinum on graphitized carbon (Pt/C, 20 wt.%) were purchased from Sigma-Aldrich. The titanium (Ti) fiber felt with a thickness of 0.4 mm was obtained from Zhejiang Jiuyu Technology Co., Ltd. The Carbon fiber paper (CFP, TGP-H-060) a thickness of 0.2 mm was purchased from Thermo Fisher Scientific Inc. The deionized water with a resistivity of 18 MΩ was produced by a Merck Millipore SIMSVO1JP water generation device. All chemicals were used without further purification.
Synthesis of RuIrO2, RuIrFeCoO2, and RuIrFeCoCrO2. For the synthesis of RuIrFeCoCrO2, a 20 mL of CH3OH solution containing RuCl3·3H2O (0.13 g), IrCl3·xH2O (0.075 g), FeCl3·6H2O (0.068 g), CoCl2·6H2O (0.06 g), and CrCl3 (0.04 g) was vigorously stirred and then dried at 60 °C for 12 h in an oven to get mixed precursor powder. Then, the obtained precursor powder (~5 mg for each addition) was continuously added into molten KNO3 solution with a temperature of 360 °C. After reaction for 2 h, the temperature of mixed solution gradually decreased to room temperature. The obtained solid mass was added into 200 mL of DI water, followed by sonication treatment for 10 min. When the solid mass was totally dissolved into water, the RuIrFeCoCrO2 was collected by washing repeatedly with deionized water and centrifugated at 7104 ɡ and for further use. The RuIrO2 and RuIrFeCoO2 were synthesized by the same method except for removing the corresponding precursors.
Characterizations
The microstructures of the samples were characterized using TEM (FEI, Tecnai G2 F20) equipped with energy dispersive spectroscopy for elemental mapping. The operation voltage was 200 kV. Aberration-corrected HAADF-STEM images was obtained from JEM-ARM200F. XRD and XPS spectra were collected by using Miniflex-600 X-ray diffractometer and ESCALAB MK II X-ray photoelectron spectrometer, respectively. The inductively coupled plasma mass spectrometry (ICPMS-2030, Shimadzu) was used to determine the elemental contents. X-ray absorption spectroscopy measurements were carried out in transmission mode at Shanghai Synchrotron Radiation Facility. The corresponding XANES and EXAFS analysis was performed using Athena and Artemis software. In-situ Raman spectra was collected on the Ramantouch microspectrometer (Nanophoton Inc., Osaka, Japan) equipped with a laser of 532 nm. During the test, the laser was focus on samples by a 20× objective lens for 5 s. The Raman test cell includes carbon fiber working electrode loaded by electrocatalysts, saturated Ag/AgCl reference electrode, and platinum wire as the counter electrode. The acidic electrolyte was 0.5 M H2SO4 aqueous solution.
Electrochemical measurements
An electrochemical workstation equipped with a three-electrode cell setup (CHI 760e) was used to collect all the electrochemical data at 23 °C. The graphite rod (diameter: 6 mm) was the counter electrode and Ag/AgCl with a salt bridge was the reference electrode. To prepare the working electrode, a mixture solution of Nafion®117 (50 μL), DI water (500 μL), and ethanol (450 μL) was prepared. 5 mg RuIrFeCoCrO2 or control samples were then added into the above solution to obtain the electrocatalyst ink, with the assistance of sonication. After that, to obtain an electrode with mass loading of 1 mg cm−2, 200 μL electrocatalyst ink was uniformly dropped on the CFP electrode (1 cm × 1 cm), followed by infrared light irradiation. For the preparation of electrolyte, a typical volume-fixing method was used in a 1-liter volumetric flask. 26.65 mL H2SO4 (98%) was diluted to 1 liter by adding DI water to obtain 0.5 M H2SO4 electrolyte. 80 mL O2-saturated 0.5 M H2SO4 aqueous solution was served as electrolyte. For the EIS test, the frequency was varied from 0.01–100 kHz with an A.C. amplitude of 5 mV. LSV curves were measured at a scan rate of 2 mV s−1 with automatic ohmic potential drop (iR) correction of 90%. The electrochemical double layer capacitance (Cdl) was calculated from CV curves with the scan rate varying from 4–20 mV s−1 with an interval of 4 mV s−1. The electrochemical durability was tested by the constant current chronopotentiometry in three-electrode system at two currents of 10 and 100 mA cm−2. All the polarization data was calibrated by converting to the reversible hydrogen electrode (RHE). In this work, the reference electrode potential versus RHE was determined by:
The ECSA was obtained from the following equation:
where Cdl was determined by taking half the slope of the current differences (Δj = janodic - jcathodic) that were plotted as a function of the scan rate in a CV experiment. Cs and S are the general surface specific capacitance (0.06 mF cm−2) and geometric surface area of the working electrode (1 cm2), respectively.
The TOF was calculated from this following equation:
where J presents the current density collected at an overpotential of 300 mV; A is geometric surface area of working electrode (1 cm2); F is Faradic constant (96485 C mol-1), n is the mole number of metal atoms, and 4 refers to the number of electrons involved in OER.
In-situ DEMS measurement
The catalyst ink was loaded onto the porous Au disk electrode with a mass loading of 0.2 mg cm−2, which served as the working electrode. The Ag/AgCl electrode and carbon rod were the RE and CE, respectively. During the in-situ DEMS test, the catalysts were labeled with 18O isotope by CV tests (5 mV s−1) in 0.5 M H2SO4 electrolyte (H218O). The CV potential range for RuO2, RuIrFeCoO2, and RuIrFeCoCrO2 were set as 1.3–2.0 V (vs. Ag/AgCl), 1.1 -1.7 V (vs. Ag/AgCl), and 1.0 -1.5 V (vs. Ag/AgCl) to achieve the similar current density, respectively. After rinsed with H216O for several times, the electrodes were treated by CV in 0.5 M H2SO4 of H216O electrolyte. The mass spectrometry was conducted to analyze the products of O2 in real time by aQAS 100 device.
PEMWE test
The proton exchange membrane (PEM, N117, Dupont, 183 μm × 1.5 cm × 1.5 cm) was treated at 80 °C in a 5 wt.% H2O2 aqueous solution for 1 h to remove organic impurities. Then, the membrane was soaked in 0.5 M H2SO4 solution at 80 °C for 1 h, followed by thorough rinse by DI water at 80 °C for another 1 h. For the anode, 4 mg RuIrFeCoCrO2 was mixed with 400 μL DI water, 450 μL ethanol, and 150 μL Nafion®117 to obtain the electrocatalyst ink. After sufficient sonication in ice water for 1 h, 500 μL of the above ink solution was applied on Ti felt with a geometric surface area of 1 cm2 (mass loading: 2 mg cm−2) to obtain OER electrode. For the cathode, 5 mg commercial Pt/C powder was mixed with 400 μL DI water, 450 μL ethanol, and 150 μL Nafion®117 by sonication in ice water for 1 h to form the HER catalyst ink. 1 mL of the obtained ink was then coated onto Ti felt (1 cm2) with a mass loading of 5 mg cm−2. Then the Ti felt coated with HER and Ti felt coated with OER catalysts were carefully attached onto the two sides of PEM, followed by a hot-press process under 7 Mpa at 120 °C for 20 min. The PEMWE test was conducted at a temperature of 80 °C in 0.5 M H2SO4 aqueous solution. The 0.5 M H2SO4 aqueous solution was preheated at 80 °C of oil bath before pumping into the anode. The flowing rate was fixed at 60 mL min−1 by a peristaltic pump. The LSV curves of the PEMWE was collected at the cell voltage window of 1.2 - 2.0 V at 80 °C with a scan rate of 2 mV s−1.
Theoretical calculations
GFN3-xTB26 method was employed to calculate the desolvation energy barriers with the xTB program37. The potential energy surface for desolvation process were determined by scanning the cation-ligand bond length. Density Functional Theory (DFT) calculations38,39 were conducted by using the Vienna ab initio simulation package (VASP)40 with DFT + U method. The Projector Augmented Wave (PAW) method41 was employed. Moreover, the Perderw, Burke, and Ernzerhof (PBE) functional42, augmented with D3 van der Waals corrections43 were operated to describe exchange-correlation energies. The cut-off energy was set to 520 eV. The on-site interaction parameters (U – J) for Cr, Fe, Co, Ir, and Ru were set as 3.7 eV, 5.3 eV, 3.32 eV, 4.2 eV, and 3.8 eV, respectively. A three-layer nonstoichiometric RuO2 slab was constructed with (110) surface. And a vacuum layer >15 Å was used for all configurations. Geometry optimizations were performed by using the conjugate gradient method, with convergence thresholds set at 10−4 eV for energy and 0.05 eV Å−1 for force. To construct the RuIrFeCoCrO2, the Ru sites of RuO2 slab were randomly substituted by Cr, Ir, Co, and Fe atoms in specific ratios. 20 different configurations of oxides were optimized, and the one exhibiting the lowest energy was chosen as the RuIrFeCoCrO2 structure. Crystal orbital Hamilton populations (COHP) calculations were conducted using the lobster44 software package. For the assessment of free energies, including thermodynamic corrections, the VASPKIT45 software was employed.
Data availability
All relevant data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper. We have deposited the atomic coordinates theoretical calculations in MaterialsCloud (https://doi.org/10.24435/materialscloud:16-4a). Source data are provided with this paper.
References
Wang, Y. et al. Nano-metal diborides-supported anode catalyst with strongly coupled TaOx/IrO2 catalytic layer for low-iridium-loading proton exchange membrane electrolyzer. Nat. Commun. 14, 5119 (2023).
Wen, Y. et al. Stabilizing highly active Ru sites by suppressing lattice oxygen participation in acidic water oxidation. J. Am. Chem. Soc. 143, 6482–6490 (2021).
Pang, B. et al. Ru Doped Ir nanowires for high-efficient and durable proton exchange membrane water electrolyzers. Adv. Funct. Mater. 34, 2411062 (2024).
Zhao, G. et al. Metallic Ru─Ru interaction in ruthenium oxide enabling durable proton exchange membrane water electrolysis. Adv. Mater. 36, 2404213 (2024).
Pei, Z. et al. Electrocatalytic acidic oxygen evolution: from catalyst design to industrial applications. Matter 6, 4128–4144 (2023).
Sun, P. et al. Designing 3d transition metal cation-doped MRuOx as durable acidic oxygen evolution electrocatalysts for PEM water electrolyzers. J. Am. Chem. Soc. 146, 15515–15524 (2024).
Xue, Y. et al. Stabilizing ruthenium dioxide with cation-anchored sulfate for durable oxygen evolution in proton-exchange membrane water electrolyzers. Nat. Commun. 14, 8093 (2023).
Chen, D. et al. Bicontinuous RuO2 nanoreactors for acidic water oxidation. Nat. Commun. 15, 3928 (2024).
Lin, Y. et al. Chromium-ruthenium oxide solid solution electrocatalyst for highly efficient oxygen evolution reaction in acidic media. Nat. Commun. 10, 162 (2019).
Lee, K. et al. Modulating the valence electronic structure using earth-abundant aluminum for high-performance acidic oxygen evolution reaction. Chem 9, 3600–3612 (2023).
Jose, V. et al. Activating amorphous Ru metallenes through Co integration for enhanced water electrolysis. Adv. Energy Mater. 13, 2301119 (2023).
Liang, J., Cao, G., Zeng, M. & Fu, L. Controllable synthesis of high-entropy alloys. Chem. Soc. Rev. 53, 6021–6041 (2024).
Li, M. et al. High-entropy alloy electrocatalysts go to (sub-) nanoscale. Sci. Adv. 10, eadn2877 (2024).
Hsu, W. L. et al. Clarifying the four core effects of high-entropy materials. Nat. Rev. Chem. 8, 471–485 (2024).
Hao, J. et al. Unraveling the electronegativity-dominated intermediate adsorption on high-entropy alloy electrocatalysts. Nat. Commun. 13, 2662 (2022).
Zhu, H. et al. A high-entropy atomic environment converts inactive to active sites for electrocatalysis. Energy Environ. Sci. 16, 619–628 (2023).
Yao, Y. et al. High-entropy nanoparticles: synthesis-structure-property relationships and data-driven discovery. Science 376, eabn3103 (2022).
George, E. P. et al. High-entropy alloys. Nat. Rev. Mater. 4, 515–534 (2019).
Dey, G. R. et al. Chemical insights into the formation of colloidal high entropy alloy nanoparticles. ACS Nano 17, 5943–5955 (2023).
Xin, Y. et al. High-entropy alloys as a platform for catalysis: progress, challenges, and opportunities. ACS Catal 10, 11280–11306 (2020).
Zhu, H. et al. High-entropy alloy stabilized active Ir for highly efficient acidic oxygen evolution. Chem. Eng. J. 431, 133251 (2022).
Cao, G. et al. Liquid metal for high-entropy alloy nanoparticles synthesis. Nature 619, 73–77 (2023).
Liang, J. et al. High-entropy alloy array via liquid metal nanoreactor. Adv. Mater. 36, 2403865 (2024).
Liu, D. et al. Air-liquid interfacial self-assembly of two-dimensional periodic nanostructured arrays. ChemNanoMat 5, 1338–1360 (2019).
Zhang, T. et al. Periodic porous alloyed Au–Ag nanosphere arrays and their highly sensitive SERS performance with good reproducibility and high density of hotspots. ACS Appl. Mater. Interfaces 10, 9792–9801 (2018).
Bannwarth, C. et al. GFN2-xTB-an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theor. Comput. 15, 1652–1671 (2019).
Zhao, Z. et al. Engineering active and robust alloy-based electrocatalyst by rapid joule-heating toward ampere-level hydrogen evolution. Nat. Commun. 15, 7475 (2024).
Ping, X. et al. Locking the lattice oxygen in RuO2 to stabilize highly active Ru sites in acidic water oxidation. Nat. Commun. 15, 2501 (2024).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule 7, 558–573 (2023).
Stoerzinger, K. A. et al. The role of Ru redox in pH-dependent oxygen evolution on rutile ruthenium dioxide surfaces. Chem 2, 668–675 (2017).
Li, A. et al. Atomically dispersed hexavalent iridium oxide from MnO2 reduction for oxygen evolution catalysis. Science 384, 666–670 (2024).
Zhang, X. et al. Fast modulation of d-band holes quantity in the early reaction stages for boosting acidic oxygen evolution. Angew. Chem. Int. Ed. 62, e202308082 (2023).
Tomikawa, K. et al. Raman study of sulfuric acid at low temperatures. J. Phys. Chem. A 102, 6082–6088 (1998).
Zhang, D. et al. Construction of Zn-doped RuO2 nanowires for efficient and stable water oxidation in acidic media. Nat. Commun. 14, 2517 (2023).
Deringer, V. L. et al. Crystal orbital hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 115, 5461–5466 (2011).
He, J. et al. Regulating electron redistribution of intermetallic iridium oxide by incorporating Ru for efficient acidic water oxidation. Adv. Energy Mater. 11, 2102883 (2021).
Bannwarth, C. et al. Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci. 11, e1493 (2021).
Hohenberg, P. et al. Inhomogeneous electron gas. Phy. Rev. 136, B864–B871 (1964).
Kohn, W. et al. Self-consistent equations including exchange and correlation effects. Phy. Rev. 140, A1133–A1138 (1965).
Kresse, G. et al. Ab initio molecular dynamics for liquid metals. Phy. Rev. B 47, 558–561 (1993).
Blöchl, P. E. Projector augmented-wave method. Phy. Rev. B 50, 17953–17979 (1994).
Perdew, J. P. et al. Generalized gradient approximation made simple. Phy. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Maintz, S. et al. LOBSTER: a tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035 (2016).
Wang, V. et al. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phy. Commun. 267, 108033 (2021).
Acknowledgements
We acknowledge the financial support from the Singapore Ministry of Education by Tier 1 (RG80/22, RG125/21, H.J.F), the Singapore Ministry of Education by Tier 2 (MOE-T2EP50121-0006, H.J.F.), the University of Electronic Science and Technology of China for startup funding (A1098531023601467, B.Z.), and the support of computational resources provided by the High-performance Computing Platform of University of Electronic Science and Technology of China.
Author information
Authors and Affiliations
Contributions
T.Z. and H.J.F. supervised and conceived this project. T.Z. designed the experiments, conducted the characterization and electrical performance measurement, and analyzed the experimental and XAFS data. M.W. and N.W. collected the XAFS data. T.Z. and B.Z. performed and analyzed the theoretical calculations. Q.L. and H.B. provide some suggestions about Raman test. T.Z. and H.J.F. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Han Zhu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, T., Liu, Q., Bao, H. et al. Atomically thin high-entropy oxides via naked metal ion self-assembly for proton exchange membrane electrolysis. Nat Commun 16, 1037 (2025). https://doi.org/10.1038/s41467-025-56189-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56189-y
This article is cited by
-
Scalable metal–organic framework-based electrodes for efficient alkaline water electrolysis
Nature Chemical Engineering (2025)
-
Recent progress of green hydrogen production technology
Frontiers of Chemical Science and Engineering (2025)
-
Design strategies of lridium-based electrocatalysts for proton exchange membrane water electrolysis
Rare Metals (2025)
