
Abstract
In eukaryotes, structural maintenance of chromosomes (SMC) complexes form topologically associating domains (TADs) by extruding DNA loops and being stalled by roadblock proteins. It remains unclear whether a similar mechanism of domain formation exists in prokaryotes. Using high-resolution chromosome conformation capture sequencing, we show that an archaeal homolog of the bacterial Smc-ScpAB complex organizes the genome of Thermococcus kodakarensis into TAD-like domains. We find that TrmBL2, a nucleoid-associated protein that forms a stiff nucleoprotein filament, stalls the T. kodakarensis SMC complex and establishes a boundary at the site-specific recombination site dif. TrmBL2 stalls the SMC complex at tens of additional non-boundary loci with lower efficiency. Intriguingly, the stalling efficiency is correlated with structural properties of underlying DNA sequences. Our study illuminates a eukaryotic-like mechanism of domain formation in archaea and a role of intrinsic DNA structure in large-scale genome organization.
Similar content being viewed by others
Introduction
In both eukaryotes and prokaryotes, structural maintenance of chromosomes (SMC) complexes play critical roles in regulating the 3D structure and function of genomes1,2,3. At the core of the SMC complex are two SMC proteins featured by a ~50-nm-long antiparallel coiled coil with the hinge dimerization domain at one end and the ATPase head domain at the other. The head domains of the dimer sandwich ATPs and hydrolyze them to drive conformational changes in the complex. The two SMC protomers are bridged by a Kleisin subunit to form a common tripartite ring structure. An intervening central domain of Kleisin further interacts with either Kleisin-interacting tandem winged-helix elements (Kites) or HEAT proteins associated with Kleisins (Hawks)4,5.
Single-molecule and chromosome conformation capture (3C) studies have provided a growing body of evidence that SMC complexes function as motors that progressively extrude DNA loops6,7. In eukaryotes, SMC-mediated loop extrusion is postulated to fold genomes into arrays of self-interacting domains. These domain structures, often called topologically associating domains (TADs) or loop domains, are believed to play versatile regulatory roles6,8,9. In most cases, eukaryotic chromosomal domains are formed by the SMC complex cohesin and a number of DNA-binding proteins (CTCF, RNA polymerases, etc.) that presumably stall cohesin-mediated loop extrusion at domain boundaries6,10,11,12,13,14,15. The CTCF roadblock is the most studied example and can nicely explain the formation of multiple boundaries and boundary-associated Hi-C features such as stripes and boundary-to-boundary loops10,16,17. Another eukaryotic SMC complex condensin also mediates domain formation in certain species, although it is less clear whether and how loop extrusion and boundary proteins are involved in this process18,19. As with eukaryotes, bacteria fold their genomes into arrays of self-interacting domains called chromosomal interaction domains (CIDs)20,21,22. However, two major classes of bacterial SMC complexes, Smc-ScpAB and MukBEF, do not play a role in CID formation, and they instead presumably use the loop extrusion activity to resolve replicated DNA molecules for chromosome segregation20,22,23,24,25,26,27,28,29,30. CIDs are formed by high levels of transcription occurring at their boundaries20,21,22. These findings have led to the prevailing view that domain formation driven by loop extrusion of SMC complexes is specific to eukaryotes.
Current evidence suggests the origin of eukaryotes within the prokaryotic domain Archaea31,32. Most archaea, with the notable exception of Crenarchaeota, possess homologs of the bacterial Smc-ScpAB subunits, namely the Smc ATPase, the Kleisin protein ScpA, and the Kite protein ScpB33,34. Recent in vitro experiments failed to detect a physical interaction between archaeal ScpA and ScpB, whereas their bacterial homologs form a stable subcomplex33,35,36. The roles of archaeal Smc, ScpA, and ScpB in vivo are poorly understood, although a recent study has provided an important clue about their functions (see below).
Our knowledge of archaeal 3D genome organization was considerably limited for a long time, partly due to a relatively low number of cultivated archaeal species, their small size, and the extreme growth conditions of most model archaea37,38. We and others have recently succeeded in applying genome-wide 3C techniques (Hi-C and 3C-seq) to diverse archaeal species, identifying a number of structural entities including self-interacting domains39,40,41,42,43,44,45. As is the case in bacteria, most of these archaeal chromosomal domains are demarcated by active transcription39,41. Intriguingly, formation of certain domain boundaries in the halophilic archaeon Halofarax volcanii depends on Smc rather than transcription41. This raises the possibility that SMC-mediated loop extrusion is a key driver of domain formation not only in eukaryotes but also in archaea. However, the underlying molecular mechanism is largely unknown, especially regarding how Smc-dependent boundaries are formed at specific loci.
To address this question, we set out to characterize the Smc-ScpAB homolog in the hyperthermophilic anaerobic archaeon Thermococcus kodakarensis. Like many other archaea, T. kodakarensis has a genome encoded in a single circular chromosome. Whereas H. volcanii lacks ScpB, T. kodakarensis possesses homologs of Smc (TK1017), ScpA (TK1018), and ScpB (TK1962). In this study, we provide evidence that these proteins act in concert with a roadblock nucleoid-associated protein (NAP) and sequence-dependent structural features of DNA to sculpt chromosomal domains.
Results
Domain formation in the genome of T. kodakarensis
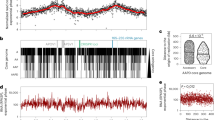
To investigate the role of Smc, ScpA, and ScpB for archaeal genome organization, we developed a high-resolution 3C-seq protocol for T. kodakarensis, starting from the Hi-C procedure for prokaryotes published recently41. In this 3C-seq protocol, we used two crosslinking reagents (formaldehyde and disuccinimidyl glutarate) to capture fine-scale structures46. Crosslinked DNA was digested with two four-base blunt-end cutters (AluI and HaeIII) for high resolution and uniform digestion. We applied the method to our laboratory strain KU216, a uracil-auxotrophic strain (∆pyrF) constructed from the wild-type T. kodakarensis strain KOD147. Cells were grown in nutrient-rich medium (ASW-YT-m1-S0) until mid-to-late log phase and immediately fixed for the experiment. An obtained contact map, binned at 5-kb resolution, was largely similar to that of KOD1 published previously (Fig. 1a and Supplementary Fig. 1a)41. Our 3C-seq data also generated a high-coverage contact map even at 500-bp resolution, whereas the published Hi-C data generated a highly sparse contact map at this resolution (Fig. 1b and Supplementary Fig. 1b). Since previous Hi-C/3C-seq analyses of archaea were conducted at resolutions ranging from 1 to 30 kb39,40,41,42,43,44,45, this study has provided the highest-resolution view of archaeal genome conformation.

a 3C-seq analysis on the genome of the T. kodakarensis strain KU216. The contact map was generated at 5-kb resolution. Two visible boundaries (boundary 1 and boundary 2) are indicated by arrows. Bins removed due to low coverage (see “Methods” for more detail) are shaded in gray throughout the paper. b Left panel: a magnified view of a showing a butterfly pattern indicative of a genomic inversion (500-bp resolution). Two proviral regions (TKV2 and TKV3) are indicated by cyan and green rectangles, respectively. Homologous sequences within TKV2 and TKV3 (TKV2HR and TKV3HR, respectively) are indicated by blue triangles. Right panel: a 3C-seq contact map was generated as in the left panel, except that it was generated for a virtual genome sequence in which a DNA segment flanked by the expected inversion breakpoints was flipped. In the two panels, the orientation of the inverted segment is indicated by white arrows. c Models for the suggested co-existence of chromosome copies with and without the inversion. d, e Magnified views of a showing stripes at 2-kb resolution. Boundaries 1, 2, and part of the inverted segment are indicated by a black triangle, a white triangle, and a white box with one side open, respectively. f Insulation score profile of the KU216 genome. Note that the genomic inversion has caused an artificial drop in the contact insulation at the breakpoints, manifested as deep valleys in the profile. Source data are provided as a Source Data file.
Of note, the 3C-seq contact map of KU216 displayed a butterfly-like pattern indicative of genomic inversion (Fig. 1b, left panel)48. Expected inversion breakpoints were located in highly homologous ~9-kb sequences (sequence identity: 96%) in the two proviral regions TKV2 and TKV349. These sequences, here denoted as TKV2HR and TKV3HR respectively, are oriented in opposite directions on the chromosome, which probably caused the inversion via intra-chromosomal crossover. As T. kodakarensis contains 7–19 copies of the chromosome per cell50, we wondered whether the inversion exists in all copies of the chromosome in the KU216 cell. To test this, we re-generated a 3C-seq contact map of KU216 using a virtual reference genome sequence in which the intervening segment between TKV2HR and TKV3HR was inverted. This manipulation did not eliminate the butterfly pattern (Fig. 1b, right panel), suggesting that chromosome copies with and without the inversion co-exist at the single-cell level or the population level (Fig. 1c). Published Hi-C data of the wild-type strain KOD141 also generated a weaker but visible butterfly signal on the original reference genome (Supplementary Fig. 1b). Thus, the genomic heterogeneity is not specific to our laboratory strain, and chromosome copies harboring the inversion can exist at a variable ratio.
The 3C-seq contact map of KU216 displayed two visible boundaries across which genomic contacts were relatively depleted (Fig. 1a). These boundaries, denoted as boundary 1 and boundary 2 respectively, can also be seen in the published Hi-C contact map of KOD1 (Supplementary Fig. 1a)41. Boundaries 1 and 2 also formed weak stripe patterns reminiscent of those formed by eukaryotic CTCF sites17, although the intensity of the stripe at boundary 1 must be carefully interpreted since it goes across the inversion breakpoints (Fig. 1d, e). To determine the precise locations of the boundaries, we used the metric called insulation score19, which represents the relative frequency of local contacts across a locus and thereby reflects contact insulation strength of the region. By determining local minima of insulation scores at 1-kb resolution, we determined the positions of boundaries 1 and 2 as 483–484 kb and 1561–1562 kb, respectively (Fig. 1f). Boundaries were also called using the published tool HiCDB51, which identified boundaries 1 and 2 essentially at the same positions (Supplementary Data 1). The insulation of local contacts was much weaker in the rest of the genome (Fig. 1f), leading us to conclude that boundaries 1 and 2 are the major boundaries on the T. kodakarensis genome. However, given the ensemble nature of the 3C-seq contact matrix, it remains to be determined how frequently these boundaries are formed and how stably they are maintained across individual chromosome copies and within individual cells.
A recent high-resolution Hi-C study on Escherichia coli identified transcription-induced domains (TIDs), which are featured by high levels of transcription and bundle-like patterns of short-range interactions52. We found that both boundaries 1 and 2 in T. kodakarensis were transcriptionally inactive (Supplementary Fig. 2), suggesting that they are structural entities that differ from TIDs. Boundary 1 also did not exhibit TID-like enrichment of short-range interactions, whereas boundary 2 was adjacent to a ~5-kb segment that was slightly enriched for local contacts (Supplementary Fig. 2).
The euryarchaeon H. volcanii and members of the Crenarchaeota form tens of DNA loops39,41,42. To search for DNA loops in T. kodakarensis, we analyzed the 3C-seq data of KU216 using Chromosight53. The interactions involving the inverted segment were excluded from the analysis. Chromosight detected ~10 loops from each of three biological replicates, but none of them were reproducible in all the replicates (Supplementary Table 1). Loop anchors might be obscured by the combined effect of the polyploidy and thermal motion of the chromosome under the high growth temperature of T. kodakarensis.
Smc, ScpA, and ScpB are all required to form chromosomal domains
To investigate whether Smc, ScpA, and ScpB contribute to boundary formation in T. kodakarensis, we constructed five deletion mutants lacking one or two of these proteins (Δsmc, ΔscpA, ΔscpB, Δsmc ΔscpA, and Δsmc ΔscpB) using KU216 as a parental strain. 3C-seq uncovered that all deletions tested resulted in loss of contact insulation at boundaries 1 and 2, especially at the former, as well as a slight increase in short-range interactions up to ~150 kb (Fig. 2a–d and Supplementary Fig. 3). The loss of these boundaries was also confirmed by HiCDB (Supplementary Data 1) and manifested as an increase in the insulation score for both loci (Fig. 2e). The insulation score was increased very similarly among the five mutants. From these results, we conclude that Smc, ScpA, and ScpB act in the same pathway to organize the T. kodakarensis genome into the domain structures. Our unpublished results also suggest that these three proteins form a ternary complex (see “Discussion”). According to these findings, we will refer to the T. kodakarensis counterpart of bacterial Smc-ScpAB simply as Smc-ScpAB.

a Comparison of 3C-seq contact maps (5-kb resolution) from KU216 and Δsmc cells (upper right and lower left triangles, respectively). b Differential contact map showing log2 ratios of contact frequencies between KU216 and Δsmc cells at 5-kb resolution. The genomic contacts between the inverted region and the other loci (shaded in gray) were omitted from the analysis. The locations of boundaries 1 and 2 in KU216 are indicated by arrows. Note that the deletion of the long smc gene (3570 bp) has caused artifacts in the map (indicated by an asterisk). c, d 3C-seq contact maps of genomic regions surrounding boundary 1 (c) and boundary 2 (d). KU216 and derivative strains lacking one or two of the Smc-ScpAB subunit homologs were used for the analysis. e Insulation score profiles of boundary 1 (upper panel) and boundary 2 (lower panel) are shown for KU216 (black solid line) and the deletion strains (dashed lines, Δsmc: red, ΔscpA: green, ΔscpB: blue, Δsmc ΔscpA: orange, Δsmc ΔscpB: cyan). Note that the graphs in the upper panel cover an inversion breakpoint that has caused artificial drops in the score. Source data are provided as a Source Data file.
SMC-mediated genome organization is critical for cell viability in the bacterial model organisms E. coli and Bacillus subtilis23,24. An early study reported that loss of smc causes a discernible growth defect in the archaeon Methanococcus voltae, whereas Cockram et al. more recently revealed that deletion of smc has no apparent impact on the cellular fitness of H. volcanii41,54. To explore whether the SMC-mediated domain formation has phenotypic consequences in T. kodakarensis, we first measured the growth of the five deletion mutants in nutrient-rich medium. All of them proliferated at similar rates as the parental strain KU216 during exponential phase, although the mutants exhibited a slight decrease in growth rate immediately before the transition to stationary phase (Supplementary Fig. 4a, b). We next compared morphological and cellular features of the Δsmc, ΔscpA, and ΔscpB strains with those of KU216. The three mutants were slightly larger than KU216 but displayed a similar nucleoid distribution in which the nucleoid occupied most part of the cell as in KU216 (Supplementary Fig. 4c, d). No anucleate cells were found among the 1000–1700 cells analyzed for each strain. Finally, RNA-seq analysis using the Δsmc, ΔscpA, and ΔscpB strains grown to mid-to-late exponential phase revealed only minor differences in their transcriptomes compared to that in KU216 (Supplementary Fig. 4e). These results suggest that the domain structures shaped by Smc-ScpAB have a very small impact on the cellular fitness of T. kodakarensis, at least under the growth conditions tested here.
Boundary formation at dif-surrounding sequences in diverse euryarchaea
Boundary 1 was located in a large intergenic region (~1.9 kb) between the convergently oriented genes TK0561 and TK0562. We found that this intergenic region also contains a previously reported putative dif sequence composed of imperfect inverted repeats of 11 bp separated by a 6-bp spacer (Fig. 3a and Supplementary Fig. 5a)55,56. dif is known as a target site of Xer site-specific recombinases (XerC and XerD in bacteria and XerA or Xer in archaea), which resolve chromosome dimers arising from an odd number of crossover events between circular sister chromosomes57,58. Chromatin immunoprecipitation sequencing (ChIP-seq) on the sole homolog of XerA (TK0777) in T. kodakarensis showed that the dif sequence was the bona fide binding site for XerA (Fig. 3b). We did not find any discernible DNA motif around boundary 2.

a Genomic position of the dif sequence in T. kodakarensis. The coordinate of the first base of the dif sequence is shown in parentheses. The position of boundary 1 (determined at 1-kb resolution) is indicated by a black capped line. Neighboring genes and their orientations are indicated by gray pentagons. b ChIP-seq tracks of XerA are shown for KU216 and ΔxerA cells of T. kodakarensis. The genomic positions of boundaries 1 and 2 in KU216 are indicated by black triangles. The genomic position of dif is indicated by an arrow. The coordinate of the first base of the dif sequence is shown in parentheses. Asterisks indicate a non-specific peak. c The genomic positions of dif1 and dif2 in Thermoplasma acidophilum are shown as in (a). dif1 is denoted in brackets to highlight that it is not functional59. d Left panel: 3C-seq analysis on the whole genome of T. acidophilum at 5-kb resolution. The genomic positions of dif1 and dif2 are indicated by black triangles. Right panel: a magnified view of the left panel showing the colocalization of dif2 with a boundary structure (indicated by an arrow) at 1-kb resolution. e Insulation score profile of the T. acidophilum genome. The genomic positions of dif1 and dif2 are indicated by red arrows. Source data are provided as a Source Data file. f Left panel: the genomic position of a putative dif sequence in Haloferax volcanii are indicated as in (a). Cyan pentagons represent an operon containing a homolog of xerA. Right panel: published Hi-C data41 were used to generate contact maps (5-kb resolution) around the putative dif sequence (indicated by an arrow) in wild-type and Δsmc cells of H. volcanii (upper right and lower left triangles, respectively). g Left panel: the genomic position of a putative dif sequence in Halobacterium salinarum is shown as in (f). Right panel: published Hi-C data41 were used to generate a contact map (5-kb resolution) around the putative dif sequence (indicated by an arrow) in wild-type H. salinarum.
A previous study identified two XerA-binding sites (dif1 and dif2) in Thermoplasma acidophilum, a euryarchaeon possessing homologs of Smc, ScpA, and ScpB (Fig. 3c). Of these two sites, only dif2 can serve as a substrate for XerA-mediated recombination in vitro59. By applying 3C-seq to T. acidophilum, we identified a boundary-like structure near dif2 but not dif1 (Fig. 3d, e). To further explore the generality of dif-associated boundaries, we searched for dif-like sequences around the previously reported Smc-dependent boundaries in the multipartite genome of H. volcanii41. We found that two of the nine Smc-dependent boundaries, one of which resides on the main chromosome and the other on the megaplasmid pHV3, are adjacent to putative dif sequences (Fig. 3f and Supplementary Fig. 5b). The dif-like element on the main chromosome is located upstream of an operon containing a xerA homolog (Fig. 3f, left panel). We also inspected a published Hi-C dataset of Halobacterium salinarum, another euryarchaeon with a multipartite genome41. In this organism, a dif-like sequence is found upstream of the same xerA-containing operon and located close to a boundary structure (Fig. 3g). It remains unknown whether this boundary is formed by an SMC complex41. Taken together, the colocalization of a boundary structure and dif is conserved among a wide range of euryarchaeal lineages.
A recent study has shown that bacterial XerD also serves as an unloader of Smc-ScpAB at the dif-like sequences named XDS in the replication terminus (ter)60. Presumably due to this unloading function, an artificial array of XDS inserted on a bacterial chromosome inhibits the translocation of Smc-ScpAB and forces the complex to form a boundary-like structure60. These findings led us to hypothesize that archaeal XerA is responsible for specifying the dif-associated boundary. To test this possibility, we tried to construct T. kodakarensis strains lacking either XerA or the whole 28-bp sequence of dif using KU216 as a parental strain. Consistent with the successful deletion of Xer in the crenarchaeon Saccharolobus solfataricus57, we obtained both mutants of T. kodakarensis. To our surprise, 3C-seq revealed that boundary 1 was unchanged in these mutants (Fig. 4). In addition, conformational changes in other genomic regions were not observed in ΔxerA or Δdif. Altogether, despite the conserved colocalization of a boundary and archaeal dif, the Xer/dif system is not essential for the boundary formation in T. kodakarensis.

a Comparison of 3C-seq contact maps (5-kb resolution) from KU216 and deletion strains (upper right and lower left triangles, respectively). b Differential contact maps displaying log2 ratios of contact frequencies between KU216 and deletion strains are shown as in Fig. 2b. c 3C-seq contact maps (1-kb resolution) of a genomic region surrounding boundary 1 are shown for T. kodakarensis strains lacking either xerA (left panel) or dif (right panel). d Insulation score profiles of boundary 1 from KU216 (black solid line), ΔxerA (red dashed line), and Δdif (orange dashed line) strains of T. kodakarensis. Note that the graphs cover an inversion breakpoint that has caused artificial drops in the score. Source data are provided as a Source Data file.
To investigate whether the formation of boundary 1 is dictated by an underlying DNA sequence other than dif, we deleted the entire part of the TK0561-0562 intergenic region that contains boundary 1 and dif (referred to as the boundary1 locus). 3C-seq revealed that deletion of boundary1 resulted in a complete loss of contact insulation at the corresponding region (Fig. 5a, b). To verify this finding, we next constructed a DNA sequence that contains the whole intergenic region between TK0561 and TK0562 but lacks the 28-bp dif sequence. This construct, denoted as boundary1Δdif, was introduced into an ectopic locus outside the inverted segment in KU216 and the Δboundary1 strain. 3C-seq showed that the insert formed a boundary structure in both strains (Fig. 5a, b). Since the Δboundary1 strain carrying the boundary1Δdif insert has only one copy of the dif-surrounding sequence, this strain allowed us to see in detail how the insert interacted with other loci. First, the insert generated a stripe that was not contaminated by the inversion, corroborating the presence of the stripe at the endogenous boundary1 locus (Figs. 1d, 5c). Second, the insert formed a weak but reproducible loop on the 3C-seq contact map by interacting with boundary 2 (Fig. 5c and Supplementary Fig. 6a). The looping interaction was also confirmed by virtual 4C plots generated from the 3C-seq data (Fig. 5d and Supplementary Fig. 6b). These results support the role of the dif-surrounding sequence in directing the formation of boundary 1 and additionally highlight its structural properties shared with eukaryotic CTCF sites.

a, b Comparison of 3C-seq contact maps (5-kb resolution) between KU216 and strains lacking the boundary1 locus and/or possessing the boundary1Δdif sequence at an ectopic location. The contact maps were generated at 5-kb (a) and 1-kb (b) resolutions. The endogenous positions of boundaries 1 and 2 are indicated by cyan ellipses and cyan rectangles, respectively. The deletion of boundary1 is represented by X. The insertion site of boundary1Δdif is indicated by magenta ellipses. The inverted segment is indicated by white rectangles (with or without one side open). In (b), the genomic contacts between the inverted segment and other loci are shaded in gray. c Left panel: a 3C-seq contact map was generated at 2-kb resolution to visualize a stripe extending from the ectopic boundary1Δdif sequence that had been introduced into the Δboundary1 strain. The positions of boundary 2 and the boundary1Δdif insert are indicated as in (a, b). Right panel: a magnified view of the left panel focusing on a loop anchored by the ectopic boundary1Δdif sequence and boundary 2. d Virtual 4C plot confirming the looping interaction shown in (c). The ectopic boundary1Δdif sequence was selected as a viewpoint (white triangle). The position of boundary 2 is indicated by a black triangle. The inverted segment was omitted from the analysis (shaded in gray).
The NAP TrmBL2 is required for the formation of boundary 1
To identify a protein that specifies boundary 1, we performed DNA affinity purification of proteins that bind to the intergenic region encompassing boundary 1. We prepared two biotinylated DNA probes, denoted as difL and difR respectively, that cover the intergenic region (Fig. 6a). The kanamycin resistance gene kanR was used as a control probe. These probes were conjugated to streptavidin beads and incubated with cell extracts of KU216 in the presence of 150 mM NaCl. After washing with 150 mM NaCl, bound proteins were eluted in a single step by adding SDS (“total bound proteins” in Fig. 6b). The difL and difR probes yielded two common specific bands (bands 1 and 2) as well as many other non-specific bands (Fig. 6c). We reduced these non-specific proteins by eluting the bound material with increasing concentrations of NaCl before adding SDS. Bands 1 and 2 were isolated from two eluates (eluates 1 and 2) and analyzed by mass spectrometry (Fig. 6b, c). Band 1 was identified as TrmBL2 (TK0471), a NAP known to form a stiff nucleoprotein filament and repress more than a hundred of genes61,62. Band 2 was identified as a protein encoded by TK0795, which shares weak homology with the modified cytosine restriction protein McrB but is functionally uncharacterized49. Although both difL and difR contained dif, we did not see a clear specific band corresponding to XerA (33 kDa) under our experimental conditions.

a Genomic regions covered by the two probes (difL and difR) for DNA affinity purification are indicated by black arrows. The other features are depicted as in Fig. 3a. b Workflow of DNA affinity purification. c Proteins from the DNA affinity purification experiment were separated by SDS-PAGE and visualized by silver staining. 0.1% of the lysate was run as input. Positions of common specific bands for the difR and difL probes are indicated by white triangles (bands 1 and 2). Bands indicated by black rectangles were subjected to mass spectrometry (MS). Source data are provided as a Source Data file. d Comparison of 3C-seq contact maps (5-kb resolution) from KU216 (upper left triangles) and derivative strains lacking either trmBL2 or TK0795 (lower left triangles). e Differential contact maps displaying log2 ratios of contact frequencies between KU216 and deletion strains are shown as in Fig. 2b. f Contact maps of genomic regions surrounding boundary 1 (upper panels) and boundary 2 (lower panels) were generated using the 3C-seq data from ΔtrmBL2 and ΔTK0795 cells. g Insulation score profiles of boundary 1 (top panel) and boundary 2 (bottom panel) are shown for KU216 (black solid line) and deletion strains (dashed lines, ΔtrmBL2: red, ΔTK0795: orange). Note that the graphs on the top panel cover an inversion breakpoint that has caused artificial drops in the score. Source data are provided as a Source Data file.
To investigate whether the identified proteins are required to build boundary 1, we constructed deletion mutants of trmBL2 and TK0795 using KU216 as a parental strain. 3C-seq demonstrated that the trmBL2 deletion caused a complete loss of boundary 1, while the TK0795 deletion rather slightly enhanced the contact insulation at boundary 1 (Fig. 6d–g). The loss of boundary 1 in the ΔtrmBL2 mutant was also confirmed by HiCDB (Supplementary Data 1). The insulation of contacts was largely maintained at boundary 2 in both mutants. Except for the loss of boundary 1 in the ΔtrmBL2 mutant, we did not see a clear structural change in either of the mutants (Fig. 6d, e). Taken together, TrmBL2 is responsible for specifying boundary 1, while another, unknown factor defines boundary 2.
TrmBL2 localizes Smc-ScpAB to boundary 1 and tens of other loci
We wondered whether TrmBL2 positions Smc-ScpAB at boundary 1 as eukaryotic CTCF does for cohesin to form TAD boundaries6,10. To test this possibility, we carried out ChIP-seq of Smc and TrmBL2 using antisera against these proteins. The specificity of the antisera was confirmed by performing ChIP-seq on Δsmc and ΔtrmBL2 cells (Fig. 7a, b). On the genome of KU216, Smc was most highly enriched at boundary 1 (Fig. 7a). At a finer scale, boundary 1 was nestled by two sharp peaks of Smc, each of which overlapped with a distinct TrmBL2 peak (Fig. 7c). Furthermore, these Smc peaks were almost completely lost in ΔtrmBL2 cells. These results support the role of TrmBL2 in localizing Smc-ScpAB to boundary 1.

a ChIP-seq tracks of Smc for the whole genomic regions of KU216, ΔtrmBL2, and Δsmc strains. Enrichment of immunoprecipitated versus input DNA (IP/input) is shown at 1-kb resolution. The genomic positions of boundaries 1 and 2 in KU216 are indicated by gray dashed lines. b ChIP-seq tracks of TrmBL2 for the whole genomic regions of KU216 and ΔtrmBL2 strains are shown as in (a). ChIP-seq tracks of Smc (KU216: blue solid line, ΔtrmBL2: cyan dashed line) and TrmBL2 (KU216: orange solid line) are shown for boundary 1 (c), boundary 2 (d), and non-boundary loci (e, f) in T. kodakarensis. Enrichment of immunoprecipitated versus input DNA (IP/input) is shown at 50-bp resolution. The locations of genes, boundaries, and dif are indicated as in Fig. 3a. g Venn diagram showing the overlap of Smc and TrmBL2 peaks detected by ChIP-seq analysis of KU216. Statistical significance of the overlap was determined by two-sided permutation test. h MEME-ChIP122 was performed to search for DNA motifs enriched in the three peak groups in (g). Statistical significance of the enrichment was evaluated using E-values. Only the most significant motif is shown for each group. i Smc occupancy was calculated for non-boundary loci forming Smc peaks in KU216. Occupancy was defined as the ChIP-seq IP/input ratio of a 200-bp region centered at the peak summit. Statistical significance of the difference was determined by two-sided Wilcoxon rank sum test. Source data are provided as a Source Data file. j Occupancies of TrmBL2 and Smc were plotted for their common binding sites shown in (g). The binding sites adjacent to boundary 1 are highlighted in red. The Spearman rank correlation coefficient (r) and corresponding two-sided p-value (p) are also shown. Source data are provided as a Source Data file.
The peak summits of TrmBL2 at boundary 1 were reproducibly located ~100 bp inward from the cognate peak summits of Smc (Fig. 7c and Supplementary Fig. 7). This distribution could be explained if TrmBL2, akin to CTCF, serves as an asymmetric barrier for the Smc-ScpAB complex that diffuses along DNA or actively extrudes a loop16,63. However, this scenario does not fit well with the crystal structure of DNA-bound Pyrococcus furiosus TrmBL2, in which TrmBL2 forms a dimer-of-dimer whose overall structure is symmetric64. Another possibility is that each of the two closely located TrmBL2-binding sites at boundary 1 stalls Smc-ScpAB with high efficiency, possibly ~100%. In this case, as loading of Smc-ScpAB onto the intervening region should be a relatively rare event, the left and right binding sites for TrmBL2 will encounter Smc-ScpAB mostly from the left and right, respectively. This directionally biased encounter and subsequent stalling will generate the observed ChIP-seq profile of Smc.
In KU216, a high ChIP-seq peak of Smc was also found near boundary 2 (Fig. 7a, d). In contrast to boundary 1, however, this region was not enriched for TrmBL2. Moreover, deletion of trmBL2 did not reduce the Smc binding to boundary 2 (Fig. 7d). These results are consistent with a TrmBL2-independent formation of boundary 2 (Fig. 6d–g).
Our ChIP-seq identified a total of 90 Smc peaks in KU216, among which 66 overlapped with TrmBL2 peaks in a statistically significant manner (Fig. 7c, e–g). Common and TrmBL2-specific binding sites were enriched for very similar AT-rich motifs, whereas Smc-specific binding sites were characterized by a dissimilar AT-rich motif (Fig. 7h). As observed for boundary 1, many non-boundary regions that were co-occupied by Smc and TrmBL2 in KU216 showed decreased binding of Smc in ΔtrmBL2 cells (Fig. 7i). This was not due to global depletion of Smc from the chromosome, because the loss of TrmBL2 did not reduce Smc occupancy at Smc-specific binding sites (Fig. 7d, i). Taken together, TrmBL2 is required to position Smc-ScpAB at many non-boundary regions as well as boundary 1.
To explore the potential difference between boundary 1 and other colocalization sites of TrmBL2 and Smc-ScpAB, we plotted the occupancies of TrmBL2 and Smc at their common binding sites (Fig. 7j). The two values showed a high level of positive correlation on average (r = 0.67). However, Smc was disproportionally enriched at a number of loci despite their modest enrichment for TrmBL2, and this disproportionality was most evident at boundary 1. The variability is also exemplified by the ChIP-seq profiles in Fig. 7e, f, which display different heights of Smc peaks relative to the cognate TrmBL2 peaks. These results suggest that TrmBL2 positions Smc-ScpAB very effectively at boundary 1 (possibly with ~100% efficiency as described above) and much less, but still significantly, at non-boundary sites. Importantly, this difference cannot simply be explained by the level of TrmBL2 occupancy.
DNA sequences correlate with the efficiency of TrmBL2-dependent positioning of Smc-ScpAB
How does TrmBL2 position Smc-ScpAB with variable efficiency? We noticed that the intergenic region encompassing boundary 1 exhibited unusually high AT content (Fig. 8a). The percentage was especially high around the peaks of Smc and TrmBL2, reaching a maximum of over 75% at 200-bp scale, while the overall AT content of the T. kodakarensis genome is 48%. AT content was also high at other loci co-occupied by TrmBL2 and Smc (Supplementary Fig. 8a, b). These observations led us to hypothesize that AT-rich sequences affect how efficiently TrmBL2 positions Smc-ScpAB.

a AT-content track of boundary 1. AT content was calculated with sliding window size of 200 bp and step size of 100 bp. The peak positions of Smc and TrmBL2 are indicated by black and white triangles, respectively. The other features are depicted as in Fig. 3a. b AT content and Smc positioning efficiency were plotted for 57 TrmBL2-binding sites that showed TrmBL2-dependent localization of Smc. The Spearman rank correlation coefficient (r) and corresponding two-sided p-value (p) are also shown. The two sites associated with boundary 1 are highlighted in red. Source data are provided as a Source Data file. c Spearman correlation coefficients between DNA tetranucleotide frequencies and Smc positioning efficiency were calculated for the 57 colocalization sites. Only the sequences with high statistical significance (two-sided p-value adjusted for multiple comparisons <0.05) are shown with the adjusted p-values. Source data are provided as a Source Data file. d The Spearman correlation coefficient and corresponding two-sided p-value between the frequency of all types of A-tract tetranucleotides and Smc positioning efficiency were calculated for the 57 colocalization sites. The partial Spearman correlation coefficient and corresponding two-sided p-value were also calculated to examine their relationship without the contribution of AT content. Source data are provided as a Source Data file. e Partial Spearman correlation coefficients between Smc positioning efficiency and structural properties of the 57 colocalization sites were calculated by removing the contribution of AT content. Two-sided p-values adjusted for multiple comparisons are also shown. Source data are provided as a Source Data file. f Model for the TrmBL2-mediated regulation of Smc-ScpAB dynamics in T. kodakarensis. The diagram assumes that the complex functions as a dimer as suggested for bacterial Smc-ScpAB25. ScpB probably forms a complex with Smc (green) and ScpA (magenta) but is not included in the diagram for simplicity. See “Discussion” for more detail.
To explore this possibility, we defined a simple metric named Smc positioning efficiency. When a given TrmBL2-binding site overlapped with an Smc-binding site (each defined as a 200-bp region centered at the peak summit), their occupancy ratio log2(Smc/TrmBL2) was defined as the Smc positioning efficiency of the TrmBL2-binding site. If TrmBL2 positions Smc-ScpAB more efficiently at a given locus than another, the value of Smc positioning efficiency is expected to be higher at the former. We calculated Smc positioning efficiency for the 57 TrmBL2-binding sites that overlapped with TrmBL2-dependent Smc-binding sites (see “Methods” for more detail). Comparison of Smc positioning efficiency with the AT content of the TrmBL2-bound 200-bp region revealed that the two metrics were well correlated with each other (Fig. 8b). In contrast, AT content showed a weaker correlation with Smc occupancy and no significant correlation with TrmBL2 occupancy (Supplementary Fig. 8c, d). Thus, AT content is strongly linked to the positioning efficiency of Smc-ScpAB but not to the DNA binding affinities of Smc-ScpAB and TrmBL2.
We further explored what feature pertinent to AT content could affect Smc positioning efficiency. The high growth temperature (85 °C) of T. kodakarensis raises the possibility that AT-rich sequences affect Smc positioning via DNA melting. To test this, we predicted the melting temperatures (Tm) of the TrmBL2-bound 200-bp regions used for the calculation of Smc positioning efficiency (see “Methods” for more detail). The predicted Tm value and Smc positioning efficiency showed a strong inverse relationship with a Spearman correlation coefficient of –0.63 (Supplementary Fig. 8e). However, the predicted Tm value and AT content also had a very strong negative correlation with a Spearman correlation coefficient of –0.99, making it difficult to infer which of the two factors is more directly related to Smc positioning. We next considered whether Smc positioning efficiency is associated with a specific form of AT-rich sequences. To test this, we counted occurrence of each of all possible DNA tetranucleotides in the TrmBL2-bound 200-bp regions. The Spearman correlation coefficient was calculated between the tetranucleotide frequency and Smc positioning efficiency. This analysis identified all types of so-called “A-tract” tetranucleotides (AAAA/TTTT, AAAT/ATTT, and AATT/AATT) as the most positively correlated sequences (Fig. 8c). A-tracts are four or more consecutive A:T base pairs without a TpA step65,66,67. The total number of the A-tract tetranucleotides showed a significant correlation with Smc positioning efficiency even if the contribution of AT content was removed by partial correlation analysis (Fig. 8d). This indicates a potential role of A-tracts in localizing Smc-ScpAB to TrmBL2-binding sites.
A-tracts display unique structural features in multiple ways. For example, long poly(A) sequences (often 10-20 bp or even longer) disfavor nucleosome wrapping in eukaryotes, while A-tracts induce DNA curvature when in phase with the helical pitch of DNA67,68,69. A-tracts also form narrow and negatively charged minor grooves70. The observed correlation between A-tract frequency and Smc positioning efficiency raises the possibility that TrmBL2-dependent localization of Smc-ScpAB is affected by structural properties of the underlying sequence. To test this, we quantified structural features associated with A-tracts for colocalization sites solely by using their sequence information. We first focused on persistence length (PL), within which a polymer segment can be seen as a straight rod. Although the PL of DNA is often regarded as its stiffness to bending deformation, the metric can actually be decomposed into dynamic and static PLs, which reflect bona fide stiffness and intrinsic shape of the DNA chain, respectively71,72. These two types of PLs have recently been estimated for all possible DNA tetranucleotides using all-atom molecular dynamics (MD) simulation data73. We used these values to calculate the mean dynamic and static PLs of all tetranucleotide steps in each stalling site, seeing the values as the local PLs of the site. We also used the recently developed software Deep DNAshape74 to predict the width and electrostatic potential of the minor groove averaged over the bases of each sequence. Partial correlation analysis controlling the effect of AT content uncovered that, among these four structural features, the local static PL and the minor-groove electrostatic potential showed significant positive and negative correlations with Smc positioning efficiency, respectively (Fig. 8e). Thus, these two features may be causally related to the positioning of Smc-ScpAB.
Current models posit that DNA bending is prerequisite for loop-extruding SMC complexes to reel in DNA for translocation7,75,76. Given this, it is somewhat surprising that the local dynamic PL of DNA did not significantly contribute to TrmBL2-dependent positioning of Smc. Experimental evidence suggests that SMC complexes capture a bent DNA segment of ~200 bp to carry out a loop extrusion cycle77,78,79,80. The effect of DNA stiffness may need to be evaluated at this length scale. This prompted us to estimate the dynamic PLs of entire 200-bp segments (here referred to as gross dynamic PL) of colocalization sites by performing coarse-grained MD simulations81. To save the computational cost for the modeling, we selected and analyzed the top five and bottom five colocalization sites in terms of Smc positioning efficiency. The directional correlation decay of the modeled DNA polymer was used to calculate gross dynamic PL. The obtained value actually showed a high positive correlation with Smc positioning efficiency (r = 0.85) (Supplementary Fig. 8f). However, the gross dynamic PL varied only from 64.8 to 68.1 nm among the ten sequences, whereas the local static PL varied in a much larger range of 88.0 to 211 nm among the 57 colocalization sites (Supplementary Fig. 8g). At this stage, it remains unclear whether the small differences in the gross dynamic PLs contribute to the variability in Smc positioning efficiency. It is possible that our MD simulation analysis has underestimated the gross dynamic PL.
Discussion
In this study, we have demonstrated that Smc, ScpA, and ScpB are all required for the domain formation in T. kodakarensis (Fig. 2). This finding raises the possibility that these proteins function, at least in some archaeal lineages, as a ternary complex akin to bacterial Smc-ScpAB. Indeed, our analyses using yeast two-hybrid assays and modeling by AlphaFold282 suggest that archaeal ScpA and ScpB can form a complex when Smc triggers a conformational change of ScpA (Takemata, Yamauchi, Takada, and Atomi, manuscript in preparation).
In T. kodakarensis, TrmBL2 is required to localize Smc-ScpAB to tens of loci, most strikingly at boundary 1 (Fig. 7). We propose that TrmBL2, analogous to eukaryotic CTCF6,10,63, stalls loop-extruding Smc-ScpAB and that the stalling occurs most frequently at the dif-proximal TrmBL2-binding sites, resulting in the formation of boundary 1 (Fig. 8f). Assuming high stalling efficiencies (possibly close to 100%) for the TrmBL2-enriched regions at boundary 1, this model can explain the distribution pattern of Smc observed at boundary 1 (Fig. 7c and Supplementary Fig. 7). A potential alternative explanation for the colocalization of Smc and TrmBL2 is that TrmBL2 serves as a loader of Smc-ScpAB. A simulation study showed that, if a loop-extruding factor shapes a boundary at its loading site as postulated above, it must perform asymmetric loop extrusion combined with one-dimensional diffusion along DNA83. In contrast to this requirement, the ability of the ectopic boundary1Δdif sequence to serve as an anchor for loop- and stripe-type contacts supports the idea that this sequence can stably retain Smc-ScpAB (Fig. 5c). In addition, although the loop extrusion of Smc-ScpAB has not been directly observed in vitro, Hi-C studies suggested that the complex forms a dimer that extrudes a DNA loop symmetrically25. In vitro single-molecule experiments also demonstrated that the Kite-containing eukaryotic SMC complex SMC5/6 and the bacterial MukBEF-like complex Wadjet form dimeric supercomplexes that extrude DNA loops symmetrically84,85. Taken together, it is more plausible that T. kodakarensis Smc-ScpAB is also a symmetric loop extruder and is stalled by TrmBL2 for the boundary formation.
We note a number of differences in the Smc-mediated genome organization between archaea and bacteria. First, the above model places T. kodakarensis Smc-ScpAB as a key player for a eukaryotic-like mechanism of domain formation, whereas bacterial Smc-ScpAB is not involved in sculpting domains20,21. The chromosomal loading of bacterial Smc-ScpAB is mediated by the DNA-binding CTPase ParB, which binds to parS centromere-like sequences scattered around the origin27,28,86,87,88. Notably, the T. kodakarensis homolog of ParB functions as an ADP-dependent serine kinase involved in cysteine biogenesis, and thus this enzyme does not appear to play a role in Smc-ScpAB loading89.
In addition to these differences, our study suggests a common feature of the SMC flux in diverse prokaryotes—disfavoring SMC traversal across the chromosome dimer resolution site dif. T. kodakarensis Smc-ScpAB is likely stalled on both sides of dif to form boundary 1, whereas the bacterial Smc-ScpAB complex traveling from the replication origin is unloaded by XerD before the complex reaches the dif site in ter60. E. coli MukBEF is also kept away from the dif-containing ter region by the unloader protein MatP, whose binding sites are scattered in ter26,90,91. These similarities may indicate a common role of prokaryotic SMC complexes in dif regions (see below).
dif and dif-like sequences are found proximal to boundary structures in T. acidophilum, H. volcanii, and H. salinarum (Fig. 3). All of these organisms possess both Smc proteins and TrmBL2-like proteins33,41,61,92, suggesting that the mechanism and function of the dif-associated boundary are conserved in multiple euryarchaeal lineages. Despite this presumed conservation, loss of the dif boundary does not cause any discernible phenotype in T. kodakarensis. What are the physiological consequences of the dif-associated boundary in archaea? Here, we describe two potential roles, presuming that the boundary formation involves loop extrusion by Smc-ScpAB.
-
(1)
The dif-boundary formation conceivably promotes chromosome dimer resolution, because two dif copies on the chromosome dimer will be brought into proximity by loop-extrusion-mediated formation of a loop array between the dif boundaries. In polyploid euryarchaeal cells, loop extrusion will also be useful to selectively drive intra-molecular pairing of dif sequences over inter-molecular pairing. The Smc-dependent synapsis of dif sites and their recombination by XerA might become crucial when chromosome dimers are frequently formed due to increased DNA damage.
-
(2)
A recent Hi-C study has proposed that the MukBEF-free dif/ter region in E. coli serves as a hub to which DNA catenanes are brought to be resolved by the type IIA topoisomerase Topo IV93. In accordance with this localized decatenation activity, ter possesses a major Topo IV-binding site where Topo IV is targeted by XerC in E. coli94,95. A similar decatenation hub may be installed in the archaeal dif boundary region, and loop-extruding Smc may bring catenation links to this hub. In line with this idea, loop extrusion has been suggested to direct DNA entanglements to the decatenation hub in E. coli and to CTCF boundaries in mammalian cells93,96. Although it remains unclear whether a decatenation hub exists in bacteria possessing Smc-ScpAB, this complex has been implicated in minimizing sister DNA interconnections—either DNA catenanes or protein-mediated bridges—through loop extrusion to ensure efficient chromosome segregation25,27,28,29,30. It is tempting to speculate that archaeal Smc-ScpAB, bacterial Smc-ScpAB, and MukBEF all contribute to sister chromosome resolution through loop extrusion, despite their different outcomes in overall chromosome folding. Such an active mechanism for sister chromosome resolution might be crucial to euryarchaeal cells when, for instance, their chromosome copy numbers are reduced in response to nutrient availability50,97.
In any case, it will be important to further explore the physiological function of the dif boundary in euryarchaea, especially in the context of their polyploid nature.
According to current models, the loop extrusion cycle of SMC complexes requires a DNA bend to reel a DNA segment through the lumen of the SMC ring7,75,76. We propose that TrmBL2, a NAP that forms a filamentous structure along DNA, serves as an obstacle that blocks loop extrusion by slowing or prohibiting DNA bending (Fig. 8f). A previous biophysical study demonstrated that TrmBL2 increases the persistence length of bound DNA in a concentration-dependent manner62. The persistence length can be increased even above 90 nm, approximately twice as large as the size of the SMC ring. In line with our model, a recent study observed that the loop extrusion of eukaryotic condensin is stalled by a linear array of Rap1, a telomeric protein that can induce local DNA stiffening98,99.
To our surprise, we found that TrmBL2-dependent positioning of Smc-ScpAB is associated with the AT richness of the underlying sequence (Fig. 8b). Given the high growth temperature of T. kodakarensis, one could imagine that AT-rich sequences interfere with loop extrusion by being denatured into single-stranded DNA (ssDNA), even though our correlation analysis failed to address this possibility. ssDNA has been suggested to serve as a high-affinity substrate for Smc-ScpAB binding and thereby may trap the complex100. However, due to the fragility of ssDNA that threatens genome integrity, hyperthermophiles are more likely to suppress heat-induced denaturation of genomic DNA. For this suppression, all hyperthermophiles appear to leverage reverse gyrase, a type IA topoisomerase that generates positive DNA supercoils and is critical for the thermal adaptation of hyperthermophiles101,102,103. Hyperthermophilic archaea also protect their genomes from thermal denaturation by coating genomic DNA with NAPs more extensively than mesophilic archaea do104. We prefer the idea that physical properties of AT-rich sequences other than their proneness to denaturation facilitate TrmBL2-mediated stalling of Smc-ScpAB. This notion is supported by the positive correlation of Smc positioning efficiency with A-tracts, a specific type of AT-rich sequences that displays unique structural features (Fig. 8c, d). A-tracts are characterized by their intrinsically straight conformation and enhanced negative electrostatic potential in the minor groove67,70,73. It is plausible that these two features of A-tracts help TrmBL2 to impede Smc-ScpAB translocation, which is consistent with their correlations with Smc positioning efficiency (Fig. 8e). We also note that the DNA motifs found at TrmBL2- and Smc-binding sites contain A-tract-like sequences (Fig. 7h). The structural properties of A-tracts may affect their interaction with TrmBL2 or Smc-ScpAB, thereby influencing the stalling competency. In this case, the effect of A-tracts on DNA binding of TrmBL2 or Smc-ScpAB will be qualitative rather than quantitative, given that AT richness is not markedly correlated with the binding level of TrmBL2 or Smc (Supplementary Fig. 8c, d). The electrostatic potential of DNA minor groove can indeed affect protein-DNA interaction via so-called shape readout mechanisms70,105.
In summary, our study has not only uncovered a eukaryotic-like mechanism of chromosomal domain formation in archaea, but also shed light on the potential role of intrinsic DNA structure in defining higher-order genome organization. It will be interesting to see whether a similar interplay of SMC complexes, boundary proteins, and structural features of boundary sequences shapes eukaryotic 3D genomes.
Methods
T. kodakarensis strains
The uracil-auxotrophic strain KU216 (ΔpyrF)47 was used as a parental strain for strain construction. For all experiments, cells were grown anaerobically at 85 °C. Unless otherwise stated, cells were cultivated as follows. Cells were inoculated into the ASW-YT-m1-S0 rich medium106 and pre-cultured overnight. The pre-culture was inoculated into fresh ASW-YT-m1-S0 rich medium to an OD660 of 0.015. The culture was grown until mid-to-late log phase (OD660: 0.2-0.25) and used for experiments. ASW-YT-m1-S0 was used with the redox indicator resazurin added to a concentration of 0.5 mg/L.
T. acidophilum strain
The T. acidophilum wild-type strain DSM 1728 was grown as described previously59 to an OD600 of ~0.2.
E. coli strain
The BL21-CodonPlus(DE3)-RIL strain (Agilent, 230245) was used for protein expression. Unless otherwise stated, cells harboring an expression plasmid were grown in LB medium containing 100 μg/mL of ampicillin and 30 μg/mL of chloramphenicol.
Construction of T. kodakarensis mutants
Except xerA (TK0777), all genes were deleted using a pop-in/pop-out approach targeting the entire (or almost entire) coding region47. For xerA deletion, its coding sequence was replaced with a marker gene via double crossover107. For dif deletion, the entire 28-bp sequence of dif (TTTTGATATAATGTACCTTATATGACAA) was deleted using the pop-in/pop-out method. For ectopic insertion of boundary1Δdif into the genome of the Δboundary1 strain, boundary1Δdif was integrated into the intergenic region between TK0712 and TK0713 via the pop-in/pop-out method. For ectopic insertion of boundary1Δdif into the genome of KU216, boundary1Δdif was integrated into the TK0712-TK0713 intergenic region together with a marker via double crossover.
Deletion/insertion constructs
The primers used for the construction are listed in Supplementary Data 2. For deletions of smc (TK1017), scpA (TK1018), and scpB (TK1962), each target sequence was PCR amplified together with upstream and downstream ~1-kb regions and cloned into the pUD3 plasmid, which contains the pyrF marker108. pUD3 was digested at the PstI and SalI sites for the cloning. The target sequence was then removed by inverse PCR to generate the deletion plasmids pUD3-Δsmc, pUD3-ΔscpA, and pUD3-ΔscpB. For double deletion of smc and scpA, constituting a part of an operon, pUD3-∆smc was amplified by inverse PCR to remove the downstream homology arm. The obtained PCR fragment was fused with a downstream ~1-kb region of scpA using In-Fusion HD Cloning Plus reagents (Takara Clontech, 638910) to generate pUD3-ΔsmcΔscpA. For dif deletion, upstream and downstream ~1-kb regions of the dif sequence were PCR amplified separately and cloned together into pUD3 (digested at BamHI and HindIII sites) using In-Fusion HD Cloning Plus reagents. The obtained plasmid was named pUD3-Δdif. For TK0795 deletion, upstream and downstream ~1-kb regions of the target sequence were PCR amplified separately and cloned together into the EcoRI site of pUD3 using In-Fusion HD Cloning Plus reagents. The obtained plasmid was named pUD3-ΔTK0795. For trmBL2 (TK0471) deletion, upstream and downstream ~1-kb regions of the target sequence were PCR amplified as a single fragment using genomic DNA of the KCP1 strain55 (ΔpyrF ΔtrpE ΔtrmBL2) as a template. The KCP1 strain was kindly provided by Hugo Maruyama. The fragment was cloned into the EcoRI site of pUD3 to generate the deletion plasmid pUD3-ΔtrmBL2. For xerA deletion, a PCR fragment containing the pyrF marker and upstream and downstream ~1-kb regions of the target sequence were PCR amplified separately. The three fragments were cloned together into the EcoRI site of pUC118 using In-Fusion HD Cloning Plus reagents. The obtained deletion plasmid was named pUC118-ΔxerA. For boundary1 deletion, upstream and downstream ~1-kb regions of the entire intergenic region between the ORFs of TK0561 and TK0562 were PCR amplified separately and cloned together into the EcoRI site of pUD3 using In-Fusion HD Cloning Plus reagents. The obtained plasmid was named pUD3-Δboundary1.
For ectopic insertion of boundary1Δdif into the genome of the Δboundary1 strain, upstream and downstream ~1-kb regions of the insertion site (located in the intergenic region between TK0712 and TK0713) were PCR amplified and cloned into the EcoRI site of pUD3 using In-Fusion HD Cloning Plus reagents. The obtained plasmid, named pUD3-TK0712, was linearized at the TK0712-TK0713 intergenic region by inverse PCR. We also PCR amplified a boundary1Δdif fragment using the genome of the Δdif strain as a template. The two PCR fragments were assembled using In-Fusion HD Cloning Plus reagents to generate the pUD3-TK0712-boundary1Δdif plasmid. For ectopic insertion of boundary1Δdif into the genome of KU216, the insertion site was PCR amplified and cloned into the EcoRI site of pUC118 in the same way as for the construction of pUD3-TK0712. The obtained plasmid, named pUC118-TK0712, was linearized by inverse PCR in the same way as for the construction of pUD3-TK0712-boundary1Δdif. This fragment was assembled with the boundary1Δdif fragment (the same sequence as used for the construction of pUD3-TK0712-boundary1Δdif) and the pyrF marker using In-Fusion HD Cloning Plus reagents to generate the pUC118-TK0712-pyrF-boundary1Δdif plasmid.
Transformation
For gene disruption or sequence insertion via pop-in/pop-out recombination, transformation with deletion or insertion constructs described above was performed as described previously106. The synthetic medium ASW-AA-m1-S0 was made according to Su et al. 109. For selection of ∆trmBL2 cells, cells were spread onto solid ASW-AA-m1-S0 medium containing 7.5 g/L 5-fluoroorotic acid (FUJIFILM Wako, 064-03664), 44 mM NaOH, and 5 mg/L uracil. The ∆smc ∆scpA strain was constructed by transforming KU216 with pUD3-∆smc∆scpA. The ∆smc ∆scpB strain was constructed by disrupting scpB in the ∆smc mutant.
Double-crossover deletion of xerA was conducted as follows. KU216 cells from 20 mL of an overnight culture were pelleted and resuspended in 200 µL of 0.8 × ASW-m1, which is composed of 0.8 × artificial seawater (ASW)110 supplemented with 20 μM KI, 20 μM H3BO3, and 10 μM NiCl2. The cell suspension was mixed with 3 μg of pUC118-ΔxerA and incubated on ice for 5 min. The cells were heat shocked for 45 s at 85 °C and cooled on ice for 5 min. This heating-chilling cycle was repeated five times. The cell suspension was inoculated into 20 mL of the uracil-free ASW-AA-m1-S0 medium and grown for 3 days. 400 µL of the culture were inoculated into fresh ASW-AA-m1-S0 medium and further grown for 3 days. For single colony isolation, 50 µL of the culture were spread onto ASW-YT-m1 medium solidified with 10 g/L Gelrite (FUJIFILM Wako, 075-05655) and grown for 1 day. Deletion of xerA was confirmed by colony PCR and DNA sequencing.
Ectopic introduction of boundary1Δdif into the genome of KU216 was conducted as follows. A fragment containing the upstream and downstream homology arms, boundary1Δdif, and pyrF was PCR amplified using pUC118-TK0712-pyrF-boundary1Δdif as a template. The fragment was integrated into the genome of KU216 via double crossover. Transformation was carried out as described for ∆xerA. Insertion was confirmed by colony PCR and DNA sequencing.
Growth measurement of T. kodakarensis
Cells from an overnight pre-culture were inoculated into three vials containing fresh ASW-YT-m1-S0. These three cultures were grown at 85 °C and used separately for OD660 measurement.
Microscopy
Microscopic observation of T. kodakarensis cells was performed according to a previous study111. For each strain, 500 μL of hot cell culture were dispensed into each of two 1.5-mL tubes containing 500 μL of pre-chilled 0.8 × ASW-m1. The tubes were cooled in ice-cold water for 5 min. After centrifugation (10,000 × g, 4 °C, 5 min), the two pellets were resuspended together in 200 μL of pre-chilled 0.8 × ASW-m1. The cells were centrifuged (10,000 × g, 4 °C, 5 min), resuspended in 100 μL of pre-chilled 0.8 × ASW-m1 containing 1 μg/mL Hoechst33342 (FUJIFILM Wako, 346-07951), and incubated for 10 min on ice in the dark. 3 µL of the cell suspension were mounted on a glass slide covered with a thin layer of 1% agarose containing 0.8 × ASW-m1. Bright field and fluorescence images were obtained at room temperature using Axio Imager.M2 equipped with a Plan-Apochromat 100×/1.4 NA oil immersion objective, an Axiocam 305 mono camera, and ZEN Pro software (Carl Zeiss). Hoechst33342-stained DNA was visualized using a 385-nm laser from the Colibri 7 LED illumination system (excitation filter: 385/30 nm, emission filter: 450/40 nm) (Carl Zeiss). Cell detection and quantitative analysis were performed using MicrobeJ112.
3C-seq
Cell fixation
T. kodakarensis cells were crosslinked by dispensing 8 mL of hot cell culture into each of eight 50-mL tubes containing 32 mL of fixative solution 1 (4.3 mL of 37% formaldehyde [Nacalai Tesque, 16223-55], 6.4 mL of 4 × ASW-m1, and 21.3 mL of MilliQ water). The mixtures were incubated for 30 min at 25 °C with agitation at 75 rpm. Crosslinking was quenched by adding 10 mL of 2.5 M glycine to each tube and incubating the mixtures for 10 min at room temperature. The cells were collected by centrifugation (8000 × g, 15 min, 25 °C). Supernatant was removed with ~2 mL per tube left for resuspension. The cell suspensions were combined and dispensed into four 5-mL tubes. The cells were centrifuged (10,000 × g, 5 min, 25 °C), and two each of the pellets were resuspended in 3 mL of fixative solution 2 (30 µL of 300 mM DSG [Thermo Fisher Scientific, 20593] dissolved in DMSO [FUJIFILM Wako, 046-21981], 600 µL of 4 × ASW-m1, and 2370 µL of MilliQ water) for additional crosslinking. The two cell suspensions were incubated for 40 min at 25 °C with agitation at 75 rpm. Crosslinking was quenched by adding 750 µL of 2.5 M glycine and incubating for 5 min at room temperature. The cells were collected by centrifugation (10,000 × g, 5 min, 4 °C). The two pellets were resuspended together in 1 mL of 1 × PBS. The cells were centrifuged (10,000 × g, 5 min, 4 °C), resuspended in 1 mL of 1 × PBS, and centrifuged again (10,000 × g, 5 min, 4 °C). The pellet was stored at –80 °C until use.
For crosslinking of T. acidophilum, 50 mL of cell culture were pelleted (8000 × g, 5 min, room temperature) and resuspended in a mixture of 3.35 mL 1 × PBS and 0.65 mL 37% formaldehyde (final 6%). The reaction was incubated for 30 min at 25 °C with agitation at 100 rpm. Crosslinking was quenched by adding 680 µL of 2.5 M glycine and incubating the mixture for 10 min at room temperature. The cells were pelleted by centrifugation (10,000 × g, 3 min, 4 °C). The centrifugation was repeated with the opposite side of the tube outward. The cells were washed by resuspending the pellet in 1 mL of 1 × PBS and centrifuging the suspension (10,000 × g, 2 min, 4 °C). This step was repeated once. The pellet was stored at –80 °C until use.
Restriction digestion
A frozen pellet of T. kodakarensis was resuspended in wash buffer (10 mM Tris-HCl [pH 8.0], 50 mM NaCl, 10 mM MgCl2) to an OD660 of 3. 1.2 mL of the cell suspension were used for the experiment. A frozen pellet of T. acidophilum was resuspended in 1 mL of wash buffer, and the whole cell suspension was used for the experiment. The cells were centrifuged (10,000 × g, 10 min, 4 °C) and resuspended in 50 μL of 1 × NEBuffer 2 (New England Biolabs, B7002S). 50 μL of the suspension were transferred to an 1.5-mL tube, mixed with 5.55 μL of 10% SDS, and incubated for 15 min at 65 °C with agitation at 600 rpm. The suspension was immediately cooled on ice for 90 s and placed at room temperature until use. 12.5 µL of the lysate were stored for purification of undigested control DNA. For DNA digestion, 37.5 µL of the lysate were mixed with 232.5 µL of premixed buffer composed of 26.3 µL of 10 × NEBuffer 2, 60 µL of 10% v/v Triton X-100, and 146.2 μL of MilliQ water. This was further mixed with 15 μL each of 10 U/μL HaeIII (New England Biolabs, R0108L) and 10 U/μL AluI (New England Biolabs, R0137L) and incubated for 3 h at 37 °C with agitation at 600 rpm. The tube was also inverted every 30 min for mixing. After the incubation was completed, the reaction was mixed with 33.3 μL of 10% SDS and incubated for 10 min at room temperature to inactivate the restriction enzymes.
Proximity ligation
A previous study demonstrated that, for prokaryotic Hi-C, using only the insoluble material for proximity ligation significantly improves the data quality41. According to this strategy, the insoluble fraction was collected by centrifugation (16,000 × g, 10 min, 25 °C), resuspended in 500 μL of wash buffer, and centrifuged again (16,000 × g, 10 min, 25 °C). The pellet was resuspended in 1185 μL of 1.01 × T4 DNA Ligase Reaction Buffer (New England Biolabs, B0202S). 395 µL of the sample were mixed with 5 µL of MilliQ water and stored for purification of un-ligated control DNA. For ligation, 790 µL of the sample were mixed with 10 μL of 400 U/μL T4 DNA Ligase (New England Biolabs, M0202L) and incubated for 3 h at 16 °C with agitation at 600 rpm. The reaction was also mixed every 30 min by inverting the tube.
Reverse crosslinking
The insoluble fraction was collected by centrifugation (16,000 × g, 10 min, 25 °C) and resuspended in 115 μL (for T. kodakarensis) or 230 µL (for T. acidophilum) of decrosslinking buffer (100 µL of TE buffer [pH 8], 10 µL of 10% SDS, and 5 µL of 0.5 M EDTA [pH 8]). The suspension was mixed with 3 μL (for T. kodakarensis) or 6 µL (for T. acidophilum) of 800 U/mL proteinase K (New England Biolabs, P8107). For T. kodakarensis, decrosslinking was performed by incubating the sample overnight at 37 °C with agitation at 600 rpm. After that, 3 μL of 800 U/mL proteinase K were additionally added, and the sample was incubated for 4 h at 37 °C with agitation at 600 rpm. For T. acidophilum, decrosslinking was performed by incubating the sample for 6 h at 65 °C with agitation at 600 rpm, which was followed by overnight incubation at 30 °C with agitation at 600 rpm. After that, 6 µL of 800 U/mL proteinase K were additionally added, and the sample was incubated for 4 h at 30 °C with agitation at 600 rpm.
Sonication and purification of DNA
90 μL of the sample were used for DNA sonication, while the remainder of the sample was stored for purification of unsonicated control DNA. The 90-µL aliquot was transferred to a microTUBE AFA Fiber Pre-Slit Snap-Cap 6 × 16 mm (Covaris, 520045) and sonicated for 200 s at 7 ° C using an M220 Focused-ultrasonicator (Covaris) with the peak power set to 50, the duty factor set to 20, and the cycles/burst set to 200. DNA was then extracted twice with phenol:chloroform:isoamyl alcohol (Sigma Aldrich, 77618-100ML) and ethanol-precipitated together with 2 μL of 20 mg/mL glycogen (Thermo Fisher Scientific, R0561). The DNA was dissolved in 30 μL of 10 mM Tris-HCl (pH 8.0) containing 0.05 mg/mL RNase A (Thermo Scientific, EN0531).
Library construction
The sheared DNA was used for library construction with NEBNext Ultra II DNA Library Prep with Sample Purification Beads (New England Biolabs, E7103S). Reaction was performed according to the manufacturer’s instructions with size selection for a 300–400 bp insert.
RNA-seq
RNA extraction
10 mL of hot cell culture were dispensed into a 50-mL tube containing 20 mL of pre-chilled 0.8 × ASW-m1 and further chilled in iced water for 5 min. The cells were centrifuged (10,000 × g, 4 °C, 10 min), resuspended in 1 mL of pre-chilled 0.8 × ASW-m1, and centrifuged again (10,000 × g, 4 °C, 5 min). The pellet was resuspended in 750 µL of lysis buffer (75 µL of 10% SDS, 25 μL of 3 M sodium acetate [pH 5.2], and 650 µL of RNase-free water). The RNA was extracted twice with acid phenol (NIPPON GENE, 315-90291) and isopropanol-precipitated. The pellet was dissolved in 51 µL of RNase-free water. The solution was mixed with 6 µL of 10 × DNase I Reaction Buffer (New England Biolabs, M0303S) and 3 µL of 2000 U/mL DNase I (New England Biolabs, M0303S). After 30 min of incubation at 37 °C, the RNA was extracted with acid phenol and ethanol-precipitated. The pellet was dissolved in 25 µL of RNase-free water.
Ribosomal RNA depletion
Ribosomal RNAs (rRNAs) were removed according to a previous study using NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat) (New England Biolabs, E7400L) and a 85 μM DNA oligo mixture containing 85 DNA oligos complementary to either 23S rRNA, 16S rRNA, or 5S rRNA113. 1 µg of the RNA was diluted in RNase-free water for a total volume of 11 µL. The diluted RNA was mixed with 2 µL of NEBNext Probe Hybridization Buffer (New England Biolabs, E7400L) and 2 µL of the DNA oligo mixture. Annealing was performed by heating the sample for 2 min at 95 °C and cooled down to 22 °C (0.1 °C/s) in a thermocycler. After additional 5-min incubation at 22 °C, the sample was mixed with 2 µL of RNase H Reaction Buffer (New England Biolabs, E7400L), 2 µL of NEBNext Thermostable RNase H (New England Biolabs, E7400L), and 1 µL of nuclease-free water. rRNA digestion was performed by incubating the sample for 30 min at 50 °C in a thermocycler with the lid temperature set to 55 °C. The reaction was quickly chilled on ice and mixed with 5 µL of DNase I Reaction Buffer (New England Biolabs, E7400L), 2.5 µL of NEBNext RNase-free DNase I (New England Biolabs, E7400L), and 22.5 µL of nuclease-free water. This was followed by incubation for 30 min at 37 °C in a thermocycler with the lid temperature set to 40 °C. The RNA was purified using Monarch RNA Cleanup Kit (New England Biolabs, 32855).
Library construction
The cleaned-up RNA was used for library construction with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, E7765L). Reaction was performed according to the manufacturer’s instructions.
ChIP-seq
ChIP-seq was carried out as described previously with modifications108.
Cell fixation and lysate preparation
For crosslinking, 80 mL of hot cell culture were mixed with 2.5 mL of 37% formaldehyde (final 1%). The mixture was immediately cooled in iced water with occasional agitation. After 5 min, the mixture was incubated for 20 min at room temperature with agitation at 75 rpm. Crosslinking was quenched by adding 4.5 mL of 2.5 M glycine (final 0.13 M). After 20 min of incubation at room temperature, the mixture was transferred to 50-mL tubes while removing as much powder of elemental sulfur as possible. The cells were collected by centrifugation (16,000 × g, 10 min, 4 °C) and resuspended in 1 mL of 0.8 × ASW–m1. The cell suspension was transferred to an 1.5-mL tube and centrifuged (10,000 × g, 5 min, 4 °C). The pellet was resuspended in 1 mL of TBS-TT (20 mM Tris-HCl [pH 8], 150 mM NaCl, 0.1% v/v Triton X-100, and 0.1% v/v Tween 20) and transferred to a milliTUBE 1 mL AFA Fiber (Covaris, 520135) for DNA fragmentation using an M220 Focused-ultrasonicator (Covaris). The fragmentation was performed for 12 min at 7 °C with the peak power set to 75, the duty factor set to 26, and the cycles/burst set to 200. The lysate was transferred to an 1.5-mL tube and centrifuged (20,400 × g, 30 min, 4 °C). The supernatant was transferred to a new 1.5-mL tube, flash frozen in liquid nitrogen, and stored at –80 °C until use.
Immunoprecipitation
500 μL of the lysate were mixed with 10 µL of antiserum (IP) or TBS-TT and incubated for 2.5 h at 4 °C. 50 μL of Dynabeads Protein G (Thermo Fisher Scientific, 10004D) were washed with TBS-TT and added to the IP sample. The lysate-bead mixture was incubated for 1 h at 4 °C. The beads were washed five times with TBS-TT, once with TBS-TT containing 0.5 M NaCl, and once with TBS-TT containing 0.5% v/v Triton X-100 and 0.5% v/v Tween 20. Protein-DNA complexes were eluted in 200 μL of elution buffer (20 mM Tris-HCl [pH 7.5], 10 mM EDTA [pH 8], and 0.5% SDS) for 30 min at 65 °C with agitation at 1400 rpm. 186 µL of the input sample were mixed with 4 µL of 0.5 M EDTA (pH 8) and 10 µL of 10% SDS and heated in the same way. Each of the IP and input samples was incubated with 3 μL of 800 U/mL proteinase K overnight at 37 °C. The DNA was extracted with phenol:chloroform:isoamyl alcohol and ethanol-precipitated together with 2 μL of 20 mg/mL glycogen. The DNA was dissolved in 55 μL of 10 mM Tris-HCl (pH 8.0).
Library construction
The DNA was used for library construction with NEBNext Ultra II DNA Library Prep with Sample Purification Beads. Reaction was performed according to the manufacturer’s instructions.
Protein purification and antibody production
Purification of XerA
The coding region of T. kodakarensis XerA was cloned into the pET21a(+) plasmid (Merck Millipore, 69740-3CN) and expressed without any additional sequence. E. coli cells harboring the expression plasmid were grown to an OD600 of ~0.8 in LB at 37 °C. Protein expression was induced with 0.1 mM IPTG for 15 h at 28 °C. The cells were pelleted by centrifugation, resuspended in 1 × PBS, and pelleted again. The cells were resuspended in 50 mM Tris-HCl (pH 7.5) containing 1 mM EDTA and disrupted by sonication. The lysate was cleared by centrifugation and heated for 20 min at 70 °C. The heat-denatured proteins were removed by centrifugation. The supernatant was concentrated using an Amicon Ultra Centrifugal Filter 10 kDa MWCO (Merck Millipore, UFC901096) and loaded onto a HiTrap Heparin HP affinity column (Cytiva, 17040701) and eluted with a linear gradient of NaCl (0–1 M). The main peak fractions were pooled and fractionated through a Superdex-200 10/300 GL column (Cytiva, 17517501) using 50 mM Tris-HCl (pH 7.5) containing 150 mM NaCl as a mobile phase. The main peak fractions were pooled and concentrated using an Amicon Ultra Centrifugal Filter 10 kDa MWCO. The sample was stored in the presence of 650–800 mM NaCl to avoid precipitation.
Purification of Smc
The coding sequence of T. kodakarensis Smc with a 5’ extension of ATGTGG was cloned into pET21a(+) and expressed without any additional sequence. E. coli cells harboring the expression plasmid were cultured in LB medium containing 50 μg/mL of ampicillin and 34 μg/mL of chloramphenicol. The culture was grown to an OD600 of ~0.4 in LB at 37 °C. Expression of the smc gene was induced with 1 mM IPTG for 4 h at 37 °C. The cells were pelleted by centrifugation and resuspended in 50 mM Tris-HCl (pH 8) containing 1 mM EDTA and 500 mM NaCl. After cell disruption by sonication, the cell extract was obtained by centrifugation followed by heating for 20 min at 80 °C. The heat-denatured proteins were removed by centrifugation. The supernatant was mixed with polyethylenimine (final 0.15%) and incubated for 10 min on ice. Precipitated nucleic acids were removed by centrifugation (23,708 × g, 10 min, 4 °C). Ammonium sulfate was added to the supernatant to 70% saturation, and the soluble proteins were precipitated by overnight incubation at 4 °C. The proteins were recovered by centrifugation (23,708 × g, 20 min, 4 °C). The precipitate was dissolved in 50 mM Tris-HCl (pH 8) containing 1 mM EDTA and 1 M ammonium sulfate. Debris were removed from the solution through a 0.45-μm Minisart Filter (Sartorius, S6555—FMOSK). The sample was loaded onto a HiTrap Phenyl HP column (Cytiva, 17135101), and the chromatography was developed with a linear gradient of ammonium sulfate (1–0 M). The main peak fractions were pooled and dialyzed overnight against 50 mM Tris-HCl (pH 8) containing 1 mM EDTA and 100 mM NaCl. The dialyzed sample was loaded onto a HiTrap Heparin HP affinity column. Elution was performed with a linear gradient of NaCl (0–1 M). The main peak fractions were pooled and diluted in 50 mM Tris-HCl (pH 8) containing 1 mM EDTA so that the NaCl concentration was adjusted to 100 mM. Lastly, the Smc protein was purified through a HiTrap Q HP column (Cytiva, 17115401) with a linear gradient of NaCl (0.1–1 M).
Purification of TrmBL2
The coding sequence of T. kodakarensis TrmBL2 was cloned into pET21a(+) and expressed without any additional sequence. The expression plasmid was kindly provided by Hugo Maruyama61. Protein expression was induced as described previously61. A heat-stable protein extract was prepared and concentrated as described for XerA purification, except that heating was performed at 80 °C. The extract was loaded onto a HiTrap Heparin HP affinity column and eluted with a linear gradient of NaCl (0–2 M). A pool of the main peak fractions was loaded onto a ResourceQ anion exchange column (Cytiva, 17117901) and eluted with a linear gradient of NaCl (0–1 M). The main peak fractions were pooled and concentrated using an Amicon Ultra Centrifugal Filter 10 kDa MWCO. The sample was stored in 50 mM Tris-HCl (pH 8.0) containing 240 mM NaCl.
Antibody production
The purified recombinant proteins were used to raise rabbit polyclonal antibodies (Eurofins Genomics).
DNA affinity purification
DNA probes
PCR was performed to amplify difL and difR (using genomic DNA of T. kodakarensis as a template) and kanR (using the pGBKT7 plasmid as a template) and clone them separately into the pUC19 plasmid (digested at the BamHI and SphI sites) using In-Fusion HD Cloning Plus reagents. The primer sets used for the cloning are shown in Supplementary Data 2. The inserted fragments were PCR amplified to generate biotinylated DNA probes (see Supplementary Data 2 for oligonucleotide sequences). The probes were purified using FastGene Gel/PCR Extraction Kit (Nippon Genetics, FG-91302).
Cell extract preparation and affinity purification
80 mL of hot cell culture of KU216 were dispensed into two 50-mL tubes, chilled in iced water for 10 min, and centrifuged (10,000 × g, 10 min, 4 °C). The two pellets were resuspended together in 3.5 mL of buffer H (20 mM Tris-HCl [pH 7.5], 500 mM NaCl) and sonicated. The lysate was cleared by centrifugation (12,000 × g, 10 min, 4 °C). 3 mL of the supernatant were mixed with 7 mL of 20 mM Tris-HCl (pH 7.5) containing 0.714% v/v Tween-20. The diluted lysate corresponding to 0.9 mg protein was mixed with 45 µL of 2 mg/mL Poly(dI-dC) (Sigma Aldrich, 81349-500UG). The sample was mixed with Dynabeads MyOne Streptavidin T1 (Thermo Fisher Scientific, 65602) pre-coupled to 9 µg of a biotinylated probe. The mixture was incubated for 30 min at 25 °C with agitation at 600 rpm. The beads were recovered and resuspended in 1.5 mL of buffer MT (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% v/v Tween-20). Beads were recovered from 500 µL of the sample for total bound proteins and from 1000 µL for eluates 1 and 2 in Fig. 6.
The beads for total bound proteins were washed with buffer MT and resuspended in 30 µL of buffer W600 (20 mM Tris-HCl [pH 7.5], 600 mM NaCl, 0.5% v/v Tween-20). The sample was mixed with 15 µL of 3 × Blue Protein Loading Dye (New England Biolabs, B7703S) and incubated for 10 min at 37 °C with agitation at 600 rpm to elute the bound proteins. The supernatant was recovered, mixed with 1.5 µL of 1.25 M DTT (New England Biolabs, B7703S), and heated for 5 min at 95 °C. The beads for eluates 1 and 2 were first washed with buffer MT. Proteins were then sequentially eluted from the beads with (1) 56.2 µL of buffer W300 (20 mM Tris-HCl [pH 7.5], 300 mM NaCl, 0.5% v/v Tween-20), (2) 58.1 µL of buffer W450 (20 mM Tris-HCl [pH 7.5], 450 mM NaCl, 0.5% v/v Tween-20), (3) 60 µL of buffer W600, (4) 48 µL of buffer W750 (20 mM Tris-HCl [pH 7.5], 750 mM NaCl, 0.5% v/v Tween-20), (5) 36 µL of buffer W1000 (20 mM Tris-HCl [pH 7.5], 1 M NaCl, 0.5% v/v Tween-20), and (6) 90 µL of SDS sample buffer (buffer W600 containing 1 × Blue Protein Loading Dye). Each elution was performed by incubating the bead suspension for 10 min at 37 °C with agitation at 600 rpm and recovering the supernatant. The supernatant from (5), used as eluate 1, was mixed with 12 μL of 20 mM Tris-HCl (pH 7.5) and 30 µL of 3 × Blue Protein Loading Dye. The supernatant from (7) was used as eluate 2. Eluates 1 and 2 were each mixed with 3 µL of 1.25 M DTT and heated for 5 min at 95 °C.
Detection of bound proteins
20 µL of the DNA affinity purification sample per lane were subjected to SDS-PAGE using e-PAGEL HR 5–20% gradient gel (ATTO, 2331970). The gel was stained with Pierce Silver Stain for Mass Spectrometry (Thermo Fisher Scientific, 24600). Indicated gel bands were excised, digested with trypsin, and subjected to label-free quantification analysis by NanoLC-MS/MS using a Q Exactive mass spectrometer (Thermo Fisher Scientific) and an Ultimate 3000 nanoLC pump (AMR). Peptides and proteins were identified by means of automated database searching using Sequest HT (Thermo Fisher Scientific) against the T. kodakarensis database (Taxon ID 311400, UniProtKB/SWISS-PROT, release 2018-01) with a precursor mass tolerance of 10 p.p.m., a fragment ion mass tolerance of 0.02 Da, and trypsin specificity that allows for up to two missed cleavages. A reversed decoy database search was conducted with Percolator node, setting the false discovery rate (FDR) threshold to 5% at the peptide level.
Identification of putative dif sequences near domain boundaries
Prior work reported nine Smc-dependent boundaries and their closest genes in H. volcanii41. We first retrieved the sequences of these genes together with their upstream and downstream 4-kb regions. To identify potential boundary-associated dif sequences, we searched these nine regions for imperfect inverted repeats containing TAA(N)nTTA (n = 6–8). We selected this motif since it is the most highly conserved feature among the previously reported dif sequences in archaea (Supplementary Fig. 5a). We identified three sequences containing the motif, one of which is located upstream of the xerA-containing operon as illustrated in Fig. 4E. Another hit is located upstream of HVO_B0249 as shown in Supplementary Fig. 5b. The third hit (AATCGAGAGTAAGGAGAGTATTACTAACCGGC) is located ~200 bp away from the second one. The third sequence was removed from the candidates due to a smaller number of base matches than that in the second one. The dif-like sequence in H. salinarum was identified by searching for the intergenic region upstream of the same xerA-containing operon.
Illumina DNA sequencing and read mapping
The 3C-seq, RNA-seq, and ChIP-seq libraries generated in this study were paired-end sequenced on either of the Illumina HiSeq X Ten, NovaSeq 6000, NextSeq 550, and NextSeq 500 platforms. The sequencing was performed by Macrogen, Single-cell Genome Information Analysis Core at Kyoto University, and the NGS core facility of the Graduate Schools of Biostudies at Kyoto University. Sequence data were mapped to the reference genome of T. kodakarensis KOD1 (GCA_000009965.1), T. acidophilum DSM 1728 (GCA_000195915.1), H. volcanii DS2 (GCA_000025685.1) or H. salinarum NRC-1 (GCA_000006805.1). For analysis of the T. kodakarensis strains lacking boundary1, the deletion was also introduced into the reference genome sequence. For analysis of the T. kodakarensis strains possessing the ectopic boundary1Δdif sequence, the insert was also introduced into the reference genome sequence.
3C-seq data analysis
Generation of 3C-seq contact maps
3C-seq contact maps were generated using HiC-Pro (version 3.0.0)114. The data were iteratively corrected with the MAX_ITER parameter of 500. The obtained matrices were normalized so that the sum of interaction scores is equal to 1000 for each row and column. For T. kodakarensis, genomic bins with extremely low coverage were filtered out by setting the FILTER_LOW_COUNT_PER parameter to 0.006. 3C-seq contact maps reflecting the genomic inversion in T. kodakarensis were generated by flipping the sequence corresponding to the genomic coordinates 327001-520000.
Insulation score analysis
Insulation score was determined as described previously at 1-kb resolution39. The size of the sliding square was set to 40 kb. The insulation score profile from KU216 formed two major valleys with the local minima at 483–484 kb and 1561–1562 kb, which was defined as the positions of boundaries 1 and 2 respectively. We also used the boundary caller RHiCDB (version 1.0), an R-packaged version of HiCDB51, to verify the presence of boundaries 1 and 2 in KU216. RHiCDB was also applied to the 3C-seq data of the Δsmc and ΔtrmBL2 strains to test the loss of the two boundaries. RHiCDB was used with the following parameters: resolution = 1000, wd = 20, and wdsize = 4.
Detection of loop structures
DNA loops in the T. kodakarensis strain KU216 were searched for using Chromosight (version 1.5.0)53 as described previously39. 3C-seq contact maps at 1-kb resolution were used for the analysis. A genomic segment containing the inversion (the genomic coordinates 321001–525000) were omitted due to the uncertainty resulting from the genome heterogeneity caused by the inversion. The max-dist parameter was set to 1044000.
Virtual 4C analysis
3C-seq contact maps at 2-kb resolution were used for the analysis. The genomic bin 616001–618000 was used as a viewpoint, and its contact frequencies with the other bins were plotted. The genomic interval 362001–524000, containing TKV2HR, TKV3HR, and the inverted segment, were omitted.
RNA-seq data analysis
Differential gene expression analysis was performed using the GFF annotation file of the T. kodakarensis reference genome. The 23S, 16S, and 5S rRNA genes were omitted from the analysis. The orientation of the 7S (SRP) RNA was inverted, because the original orientation in the genome annotation is probably incorrect as reported previously110. RNA levels were first quantified by Salmon (version 1.9.0)115 and then analyzed using edgeR (version 3.40.2)116 as follows. The read count was normalized using the TMM normalization method. Common, trended, and tagwise dispersions were estimated using the GLM method. Statistical significance of the gene expression difference was tested using the glmFit function.
ChIP-seq data analysis
Mapping
Reads were mapped using Bowtie 2 (version 2.3.5.1)117. Low-quality alignments (MAPQ < 30) were removed using SAMtools (version 1.9)118.
Generation of ChIP-seq tracks
Generated BAM files were processed using the bamCoverage function (version 3.5.1) of deepTools119 to calculate Reads Per Kilobase region per Million mapped reads (RPKM) for genomic bins of 50 bp and 1 kb. RPKM ratios of immunoprecipitated versus input DNA were plotted as IP/input.
Peak analysis
Mapped data were processed for peak calling using MACS (version 3.0.0a6)120 with the following setting: --fe-cutoff 3, --keep-dup all, --call-summits, -m 1 50. To identify common binding sites for Smc and TrmBL2, overlaps of ±100-bp regions from the identified peaks were examined using the numOverlaps function of regioneR (version 1.30.0)121. Statistical significance of the overlaps was evaluated using the overlapPermuTest function of regioneR (ntimes = 1000). DNA motifs enriched at identified peaks were determined using MEME-ChIP (version 5.5.5)122 with the following setting: -ccut 100, -order 2, -minw 4, -maxw 15, -meme-mod zoops. To calculate protein occupancy at the identified peak, the read coverage of the ±100-bp region from the peak summit was calculated using the multicov function of BEDtools (version 2.26.0)123. The coverage was normalized to the total read number, and the IP/input ratio of the normalized coverage values was used as the occupancy of the protein at the peak.
Smc positioning efficiency
Using ChIP-seq data from KU216, we first calculated Smc occupancy for the Smc peaks overlapping with those of TrmBL2. Smc occupancy in ∆trmBL2 was also calculated for the same regions. These values were used to calculate the Smc occupancy ratio between KU216 and ∆trmBL2. Loci with the ∆trmBL2/KU216 ratio of –0.5 or larger were filtered out. We then calculated TrmBL2 occupancy for the TrmBL2 peaks overlapping with the retained Smc peaks in KU216. These peak pairs were used to calculate the ratio of Smc versus TrmBL2 occupancies, which was defined as Smc positioning efficiency.
Analysis on DNA sequence features
AT-content tracks were generated using SeqKit (version 2.5.1)124 with sliding window size of 200 bp and step size of 100 bp. For analysis on colocalization sites of TrmBL2 and Smc, 200-bp sequences centered at TrmBL2 peaks were used. The melting temperatures of colocalization sites were predicted using Tm for Oligos Calculator (https://www.promega.jp/resources/tools/biomath/tm-calculator/), which is based on the nearest-neighbor method. The following parameters were used for the prediction: primer concentration: 200 nM, Na+/K+ concentration: 700 mM, Mg2+ concentration: 1 mM. Although the high concentration of Na+/K+ was adopted according to intracellular K+ concentrations in thermophilic methanogenic archaea125, this value did not significantly affect the result. Occurrence of DNA tetranucleotide sequences was counted for each stalling site using the countDnaKmers function of seqTools (version 1.32.0). For non-palindrome sequences, their occurrence frequencies plus those of the reverse complements were used for analyses. The occurrence frequency dataset was then used to identify the tetranucleotide sequences that were correlated with Smc positioning efficiency. The same dataset was used to calculate the average of 4-bp-scale persistence lengths for each stalling site. The persistence length values were according to a previous study73. The other DNA properties were predicted using the Deep DNAshape web server (https://deepdnashape.usc.edu/)74 with the Deep DNAshape layer set to 4. This server predicts a selected feature value for each base of the input sequence. The base average of the feature value was calculated for each stalling site and used for correlation analysis. Adjusted p-values were determined using the Benjamini-Hochberg method.
Coarse-grained MD simulations of TrmBL2/Smc colocalization sites
The coarse-grained MD simulations were conducted with CafeMol (version 3.2.1)126. The 3SPN.2C sequence-dependent coarse-grained DNA model was used81. 5’- and 3’ termini of stalling sites were capped with five CG repeats to insulate the analyzed sequences from end effects, but the CG caps were omitted when gross dynamic PLs were calculated from the obtained MD data. The temperature was set to 300 K. The monovalent ion concentration of Debye-Hückel model was set to 150 mM. Langevin dynamics simulations were performed with a step size of 0.2 in the CafeMol time unit for 2 × 108 steps per stalling site, including 5 × 107 steps for equilibration. The coordinates were stored every 5 × 103 steps.
The gross dynamic PL, \({l}_{{{\rm{d}}}}\), was estimated from the directional correlation decay of DNA polymer as \({C}_{{{\rm{d}}}}\left(s\right)\equiv {C}_{{{\rm{p}}}}(s)/{C}_{{{\rm{s}}}}(s)\approx \exp \left(-s/{l}_{{{\rm{d}}}}\right)\) as performed previously72. Here, \({C}_{{{\rm{p}}}}\left(s\right)\) is the autocorrelation function of the helical axis vectors \({{\boldsymbol{h}}}\) defined as \({C}_{{{\rm{p}}}}\left(s\right)=\, < {\left\{{{\boldsymbol{h}}}\left(l\right)\bullet {{\boldsymbol{h}}}\left(l+s\right)\right\}}_{l} > \,\approx \exp (-s/{l}_{{{\rm{p}}}})\), \({C}_{{{\rm{s}}}}(s)\) is that for minimum-energy ground state conformation defined as \({C}_{{{\rm{s}}}}\left(s\right)={\{{{{\boldsymbol{h}}}}^{\min }\left(l\right)\bullet {{{\boldsymbol{h}}}}^{\min }\left(l+s\right)\}}_{l}\approx \exp \left(-s/{l}_{{{\rm{s}}}}\right)\), \(s\) is the spacing between helical axis vectors, \(l\) is the position along the contour length, \({l}_{{{\rm{p}}}}\) is the PL, and \({l}_{{{\rm{s}}}}\) is the static PL. \(s\) is a multiple of 10 in order to remove any residual effects of DNA helicity. {・} represents an average over the position along the contour length and <・> represents a long-time average. The sequence-dependent ground state conformation was obtained using x3DNA (version 2.4)127.
Data visualization and statistical test
Unless otherwise stated, data were visualized using R software (http://www.r-project.org/).
Statistics and reproducibility
Statistical test was performed using R software. Reproducibility of 3C-seq results was confirmed using two (all strains except KU216) or three (KU216) biological replicates. Reproducibility of RNA-seq results were confirmed using three biological replicates. Reproducibility of ChIP-seq results was confirmed using two biological replicates. Unless otherwise stated, reads from the biological replicates were pooled for analysis. Growth measurement of T. kodakarensis cells were repeated twice independently with similar results. Microscopic observation of T. kodakarensis cells was performed for a single culture. In the microscopic analysis, 50 images were taken for each strain, all of which show similar results. The number of cells used for the image quantification is shown in Supplementary Fig. 4d. The specific bands observed in the DNA affinity purification in Fig. 6c were reproduced in three independent experiments. Samples obtained from one of the three experiments were analyzed by mass spectrometry in technical duplicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequence data generated in this study have been deposited in the NCBI’s Gene Expression Omnibus database under the GEO series accession codes https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE267297, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE267298, and https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE267299. The mass spectrometry data generated in this study have been deposited in the ProteomeXchange Consortium via the jPOST partner repository128 (http://jpostdb.org) with the PDX identifier https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD053073. The raw microscopic image data generated in this study have been deposited in FigShare (https://figshare.com/projects/Microscopic_images_of_Smc-ScpAB_mutants/231431). The published Hi-C data41 used in this study are available in the NCBI Sequence Read Archive (SRA) database under the following accession codes: T. kodakarensis: https://www.ncbi.nlm.nih.gov/sra/?term=SRR12717836 and https://www.ncbi.nlm.nih.gov/sra/?term=SRR12717848, H. volcanii DS2: https://www.ncbi.nlm.nih.gov/sra/?term=SRR11747722 and https://www.ncbi.nlm.nih.gov/sra/?term=SRR11747734, H. volcanii ∆smc: https://www.ncbi.nlm.nih.gov/sra/?term=SRR11747720 and https://www.ncbi.nlm.nih.gov/sra/?term=SRR11747731, H. salinarum: https://www.ncbi.nlm.nih.gov/sra/?term=SRR12717837 and https://www.ncbi.nlm.nih.gov/sra/?term=SRR12717838. The insulation score profiles generated from 3C-seq data are available in the Source Data file. The growth curve data and uncropped gel image data generated in this study are provided in the Source Data file. Source data are provided with this paper.
References
Yatskevich, S., Rhodes, J. & Nasmyth, K. Organization of chromosomal DNA by SMC complexes. Annu. Rev. Genet. 53, 445–482 (2019).
Uhlmann, F. SMC complexes: from DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 17, 399–412 (2016).
Hirano, T. Condensin-based chromosome organization from bacteria to vertebrates. Cell 164, 847–857 (2016).
Wells, J. N., Gligoris, T. G., Nasmyth, K. A. & Marsh, J. A. Evolution of condensin and cohesin complexes driven by replacement of Kite by Hawk proteins. Curr. Biol. 27, R17–R18 (2017).
Palecek, J. J. & Gruber, S. Kite proteins: a superfamily of SMC/Kleisin partners conserved across bacteria, archaea, and eukaryotes. Structure 23, 2183–2190 (2015).
Davidson, I. F. & Peters, J. M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 22, 445–464 (2021).
Kim, E., Barth, R. & Dekker, C. Looping the genome with SMC complexes. Annu. Rev. Biochem. 92, 15–41 (2023).
Nora, E. P. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Nora, E. P. et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944 e922 (2017).
Banigan, E. J. et al. Transcription shapes 3D chromatin organization by interacting with loop extrusion. Proc. Natl. Acad. Sci. USA 120, e2210480120 (2023).
Jeppsson, K. et al. Cohesin-dependent chromosome loop extrusion is limited by transcription and stalled replication forks. Sci. Adv. 8, eabn7063 (2022).
Rao, S. S. P. et al. Cohesin loss eliminates all loop domains. Cell 171, 305–320 e324 (2017).
Valton, A. L. et al. A cohesin traffic pattern genetically linked to gene regulation. Nat. Struct. Mol. Biol. 29, 1239–1251 (2022).
Costantino, L., Hsieh, T. S., Lamothe, R., Darzacq, X. & Koshland, D. Cohesin residency determines chromatin loop patterns. Elife 9, e59889 (2020).
Sanborn, A. L. et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 112, E6456–E6465 (2015).
Vian, L, et al. The energetics and physiological impact of cohesin extrusion. Cell 173, 1165–1178.e20 (2018).
Kakui, Y., Rabinowitz, A., Barry, D. J. & Uhlmann, F. Condensin-mediated remodeling of the mitotic chromatin landscape in fission yeast. Nat. Genet. 49, 1553–1557 (2017).
Crane, E. et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244 (2015).
Le, T. B., Imakaev, M. V., Mirny, L. A. & Laub, M. T. High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342, 731–734 (2013).
Wang, X. et al. Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev. 29, 1661–1675 (2015).
Lioy, V. S. et al. Multiscale structuring of the E. coli chromosome by nucleoid-associated and condensin proteins. Cell 172, 771–783 e718 (2018).
Britton, R. A., Lin, D. C. & Grossman, A. D. Characterization of a prokaryotic SMC protein involved in chromosome partitioning. Genes Dev. 12, 1254–1259 (1998).
Niki, H., Jaffe, A., Imamura, R., Ogura, T. & Hiraga, S. The new gene mukB codes for a 177 kd protein with coiled-coil domains involved in chromosome partitioning of E. coli. EMBO J. 10, 183–193 (1991).
Wang, X., Brandao, H. B., Le, T. B., Laub, M. T. & Rudner, D. Z. Bacillus subtilis SMC complexes juxtapose chromosome arms as they travel from origin to terminus. Science 355, 524–527 (2017).
Makela, J. & Sherratt, D. J. Organization of the Escherichia coli chromosome by a MukBEF axial core. Mol. Cell 78, 250–260.e5 (2020).
Gruber, S. & Errington, J. Recruitment of condensin to replication origin regions by ParB/SpoOJ promotes chromosome segregation in B. subtilis. Cell 137, 685–696 (2009).
Sullivan, N. L., Marquis, K. A. & Rudner, D. Z. Recruitment of SMC by ParB-parS organizes the origin region and promotes efficient chromosome segregation. Cell 137, 697–707 (2009).
Tran, N. T., Laub, M. T. & Le, T. B. K. SMC Progressively aligns chromosomal arms in Caulobacter crescentus but is antagonized by convergent transcription. Cell Rep. 20, 2057–2071 (2017).
Gruber, S. et al. Interlinked sister chromosomes arise in the absence of condensin during fast replication in B. subtilis. Curr. Biol. 24, 293–298 (2014).
Spang, A., Caceres, E. F. & Ettema, T. J. G. Genomic exploration of the diversity, ecology, and evolution of the archaeal domain of life. Science 357, eaaf388 (2017).
Imachi, H. et al. Isolation of an archaeon at the prokaryote-eukaryote interface. Nature 577, 519–525 (2020).
Jeon, J. H. et al. Evidence for binary Smc complexes lacking kite subunits in archaea. IUCrJ 7, 193–206 (2020).
Yoshinaga, M., Nakayama, T. & Inagaki, Y. A novel structural maintenance of chromosomes (SMC)-related protein family specific to Archaea. Front. Microbiol. 13, 913088 (2022).
Burmann, F. et al. An asymmetric SMC-kleisin bridge in prokaryotic condensin. Nat. Struct. Mol. Biol. 20, 371–379 (2013).
Kamada, K., Miyata, M. & Hirano, T. Molecular basis of SMC ATPase activation: role of internal structural changes of the regulatory subcomplex ScpAB. Structure 21, 581–594 (2013).
Takemata, N. & Bell, S. D. Emerging views of genome organization in Archaea. J. Cell Sci. 133, jcs243782 (2020).
Bell, S. D. Form and function of archaeal genomes. Biochem. Soc. Trans. 50, 1931–1939 (2022).
Takemata, N. & Bell, S. D. Multi-scale architecture of archaeal chromosomes. Mol. Cell 81, 473–487 e476 (2021).
Takemata, N., Samson, R. Y. & Bell, S. D. Physical and functional compartmentalization of archaeal chromosomes. Cell 179, 165–179.e118 (2019).
Cockram, C., Thierry, A., Gorlas, A., Lestini, R. & Koszul, R. Euryarchaeal genomes are folded into SMC-dependent loops and domains, but lack transcription-mediated compartmentalization. Mol. Cell 81, 459–472.e410 (2021).
Badel C. & Bell S. D. Chromosome architecture in an archaeal species naturally lacking structural maintenance of chromosomes proteins. Nat. Microbiol. 9, 263–273 (2023).
Badel, C., Samson, R. Y. & Bell, S. D. Chromosome organization affects genome evolution in Sulfolobus archaea. Nat. Microbiol. 7, 820–830 (2022).
Sobolev AS, M. et al. 3C-seq-captured chromosome conformation of the hyperthermophilic archaeon Thermofilum adornatum. bioRxiv, (2021).
Pilatowski-Herzing E. et al. Capturing chromosome conformation in Crenarchaea. Mol. Microbiol. https://doi.org/10.1111/mmi.15245 (2024).
Akgol Oksuz, B. et al. Systematic evaluation of chromosome conformation capture assays. Nat. Methods 18, 1046–1055 (2021).
Sato, T., Fukui, T., Atomi, H. & Imanaka, T. Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl. Environ. Microbiol. 71, 3889–3899 (2005).
Harewood, L. et al. Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol. 18, 125 (2017).
Fukui, T. et al. Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res. 15, 352–363 (2005).
Spaans, S. K., van der Oost, J. & Kengen, S. W. The chromosome copy number of the hyperthermophilic archaeon Thermococcus kodakarensis KOD1. Extremophiles 19, 741–750 (2015).
Chen, F., Li, G., Zhang, M. Q. & Chen, Y. HiCDB: a sensitive and robust method for detecting contact domain boundaries. Nucleic Acids Res. 46, 11239–11250 (2018).
Bignaud, A. et al. Transcription-induced domains form the elementary constraining building blocks of bacterial chromosomes. Nat. Struct. Mol. Biol. 31, 489–497 (2024).
Matthey-Doret, C. et al. Computer vision for pattern detection in chromosome contact maps. Nat. Commun. 11, 5795 (2020).
Long, S. W. & Faguy, D. M. Anucleate and titan cell phenotypes caused by insertional inactivation of the structural maintenance of chromosomes (SMC) gene in the archaeon Methanococcus voltae. Mol. Microbiol. 52, 1567–1577 (2004).
Cortez, D. et al. Evidence for a Xer/dif system for chromosome resolution in archaea. PLoS Genet. 6, e1001166 (2010).
Cossu, M., Da Cunha, V., Toffano-Nioche, C., Forterre, P. & Oberto, J. Comparative genomics reveals conserved positioning of essential genomic clusters in highly rearranged Thermococcales chromosomes. Biochimie 118, 313–321 (2015).
Duggin, I. G., Dubarry, N. & Bell, S. D. Replication termination and chromosome dimer resolution in the archaeon Sulfolobus solfataricus. EMBO J. 30, 145–153 (2011).
Midonet C., Barre F. X. Xer site-specific recombination: promoting vertical and horizontal transmission of genetic information. Microbiol Spectr 2, https://doi.org/10.1128/microbiolspec.MDNA3-0056-2014 (2014).
Jo, M., Murayama, Y., Tsutsui, Y. & Iwasaki, H. In vitro site-specific recombination mediated by the tyrosine recombinase XerA of Thermoplasma acidophilum. Genes Cells 22, 646–661 (2017).
Karaboja, X. et al. XerD unloads bacterial SMC complexes at the replication terminus. Mol. Cell 81, 756–766 e758 (2021).
Maruyama, H. et al. Histone and TK0471/TrmBL2 form a novel heterogeneous genome architecture in the hyperthermophilic archaeon Thermococcus kodakarensis. Mol. Biol. Cell 22, 386–398 (2011).
Efremov, A. K. et al. Transcriptional repressor TrmBL2 from Thermococcus kodakarensis forms filamentous nucleoprotein structures and competes with histones for DNA binding in a salt- and DNA supercoiling-dependent manner. J. Biol. Chem. 290, 15770–15784 (2015).
Li, Y. et al. The structural basis for cohesin-CTCF-anchored loops. Nature 578, 472–476 (2020).
Ahmad, M. U. D. et al. Structural insights into nonspecific binding of DNA by TrmBL2, an archaeal chromatin protein. J. Mol. Biol. 427, 3216–3229 (2015).
Haran, T. E. & Mohanty, U. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 42, 41–81 (2009).
Hagerman, P. J. Sequence-directed curvature of DNA. Annu. Rev. Biochem. 59, 755–781 (1990).
Marin-Gonzalez, A., Vilhena, J. G., Perez, R. & Moreno-Herrero, F. A molecular view of DNA flexibility. Q Rev. Biophys. 54, e8 (2021).
Segal, E. & Widom, J. Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr. Opin. Struct. Biol. 19, 65–71 (2009).
Marin-Gonzalez, A. et al. Understanding the paradoxical mechanical response of in-phase A-tracts at different force regimes. Nucleic Acids Res. 48, 5024–5036 (2020).
Rohs, R. et al. The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253 (2009).
Olson W. K. DNA Bending & Curvature (Adenine Press, 1988).
Mitchell, J. S., Glowacki, J., Grandchamp, A. E., Manning, R. S. & Maddocks, J. H. Sequence-dependent persistence lengths of DNA. J. Chem. Theory Comput. 13, 1539–1555 (2017).
Velasco-Berrelleza, V. et al. SerraNA: a program to determine nucleic acids elasticity from simulation data. Phys. Chem. Chem. Phys. 22, 19254–19266 (2020).
Li, J., Chiu, T. P. & Rohs, R. Predicting DNA structure using a deep learning method. Nat. Commun. 15, 1243 (2024).
Dekker, C., Haering, C. H., Peters, J. M. & Rowland, B. D. How do molecular motors fold the genome? Science 382, 646–648 (2023).
Marko, J. F., De Los Rios, P., Barducci, A. & Gruber, S. DNA-segment-capture model for loop extrusion by structural maintenance of chromosome (SMC) protein complexes. Nucleic Acids Res. 47, 6956–6972 (2019).
Strick, T. R., Kawaguchi, T. & Hirano, T. Real-time detection of single-molecule DNA compaction by condensin I. Curr. Biol. 14, 874–880 (2004).
Ryu, J. K. et al. Condensin extrudes DNA loops in steps up to hundreds of base pairs that are generated by ATP binding events. Nucleic Acids Res. 50, 820–832 (2022).
Cui, Y., Petrushenko, Z. M. & Rybenkov, V. V. MukB acts as a macromolecular clamp in DNA condensation. Nat. Struct. Mol. Biol. 15, 411–418 (2008).
Sun, M., Nishino, T. & Marko, J. F. The SMC1-SMC3 cohesin heterodimer structures DNA through supercoiling-dependent loop formation. Nucleic Acids Res. 41, 6149–6160 (2013).
Freeman, G. S., Hinckley, D. M., Lequieu, J. P., Whitmer, J. K. & de Pablo, J. J. Coarse-grained modeling of DNA curvature. J. Chem. Phys. 141, 165103 (2014).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Banigan, E. J., van den Berg, A. A., Brandao, H. B., Marko, J. F. & Mirny, L. A. Chromosome organization by one-sided and two-sided loop extrusion. Elife 9, e53558 (2020).
Pradhan, B. et al. The Smc5/6 complex is a DNA loop-extruding motor. Nature 616, 843–848 (2023).
Pradhan, B. et al. Loop extrusion-mediated plasmid DNA cleavage by the bacterial SMC Wadjet complex. Mol. Cell 85, 107–116 (2024).
Jalal A. S., Tran N. T. & Le T. B. ParB spreading on DNA requires cytidine triphosphate in vitro. Elife 9, e53515 (2020).
Soh, Y. M. et al. Self-organization of parS centromeres by the ParB CTP hydrolase. Science 366, 1129–1133 (2019).
Osorio-Valeriano, M. et al. ParB-type DNA segregation proteins Are CTP-dependent molecular switches. Cell 179, 1512–1524.e1515 (2019).
Makino, Y. et al. An archaeal ADP-dependent serine kinase involved in cysteine biosynthesis and serine metabolism. Nat. Commun. 7, 13446 (2016).
Mercier, R. et al. The MatP/matS site-specific system organizes the terminus region of the E. coli chromosome into a macrodomain. Cell 135, 475–485 (2008).
Nolivos, S. et al. MatP regulates the coordinated action of topoisomerase IV and MukBEF in chromosome segregation. Nat. Commun. 7, 10466 (2016).
Herrmann, U. & Soppa, J. Cell cycle-dependent expression of an essential SMC-like protein and dynamic chromosome localization in the archaeon Halobacterium salinarum. Mol. Microbiol. 46, 395–409 (2002).
Conin, B. et al. Extended sister-chromosome catenation leads to massive reorganization of the E. coli genome. Nucleic Acids Res. 50, 2635–2650 (2022).
El Sayyed, H. et al. Mapping topoisomerase IV binding and activity sites on the E. coli genome. PLoS Genet. 12, e1006025 (2016).
Sutormin, D., Galivondzhyan, A., Gafurov, A. & Severinov, K. Single-nucleotide resolution detection of Topo IV cleavage activity in the Escherichia coli genome with Topo-Seq. Front. Microbiol. 14, 1160736 (2023).
Canela, A. et al. Genome organization drives chromosome fragility. Cell 170, 507–521.e518 (2017).
Zerulla, K. et al. DNA as a phosphate storage polymer and the alternative advantages of polyploidy for growth or survival. PLoS ONE 9, e94819 (2014).
Le Bihan, Y. V. et al. Effect of Rap1 binding on DNA distortion and potassium permanganate hypersensitivity. Acta Crystallogr. D Biol. Crystallogr. 69, 409–419 (2013).
Analikwu B. T. et al. Telomere protein arrays stall DNA loop extrusion by condensin. bioRxiv, 2023.2010.2029.564563 (2023).
Yano, K., Noguchi, H. & Niki, H. Profiling a single-stranded DNA region within an rDNA segment that affects the loading of bacterial condensin. iScience 25, 105504 (2022).
Forterre, P. A hot story from comparative genomics: reverse gyrase is the only hyperthermophile-specific protein. Trends Genet. 18, 236–237 (2002).
Atomi, H., Matsumi, R. & Imanaka, T. Reverse gyrase is not a prerequisite for hyperthermophilic life. J. Bacteriol. 186, 4829–4833 (2004).
Lipscomb, G. L., Hahn, E. M., Crowley, A. T. & Adams, M. W. W. Reverse gyrase is essential for microbial growth at 95 degrees C. Extremophiles 21, 603–608 (2017).
Hocher, A. et al. Growth temperature and chromatinization in archaea. Nat. Microbiol. 7, 1932–1942 (2022).
Chiu, T. P., Rao, S., Mann, R. S., Honig, B. & Rohs, R. Genome-wide prediction of minor-groove electrostatic potential enables biophysical modeling of protein-DNA binding. Nucleic Acids Res. 45, 12565–12576 (2017).
Michimori, Y. et al. Removal of phosphoglycolate in hyperthermophilic archaea. Proc. Natl. Acad. Sci. USA 121, e2311390121 (2024).
Sato, T., Fukui, T., Atomi, H. & Imanaka, T. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 185, 210–220 (2003).
Samson, R. Y. et al. Specificity and function of archaeal DNA replication initiator proteins. Cell Rep. 3, 485–496 (2013).
Su, Y., Michimori, Y. & Atomi, H. Biochemical and genetic examination of two aminotransferases from the hyperthermophilic archaeon Thermococcus kodakarensis. Front. Microbiol. 14, 1126218 (2023).
Jager, D., Forstner, K. U., Sharma, C. M., Santangelo, T. J. & Reeve, J. N. Primary transcriptome map of the hyperthermophilic archaeon Thermococcus kodakarensis. BMC Genom. 15, 684 (2014).
Villain, P. et al. The hyperthermophilic archaeon Thermococcus kodakarensis is resistant to pervasive negative supercoiling activity of DNA gyrase. Nucleic Acids Res. 49, 12332–12347 (2021).
Ducret, A., Quardokus, E. M. & Brun, Y. V. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat. Microbiol. 1, 16077 (2016).
Sas-Chen, A. et al. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature 583, 638–643 (2020).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Gel, B. et al. regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 32, 289–291 (2016).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 11, e0163962 (2016).
Hensel, R. & König, H. Thermoadaptation of methanogenic bacteria by intracellular ion concentration. FEMS Microbiol. Lett. 49, 75–79 (1988).
Kenzaki, H. et al. CafeMol: A coarse-grained biomolecular simulator for simulating proteins at work. J. Chem. Theory Comput. 7, 1979–1989 (2011).
Lu, X. J. & Olson, W. K. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 3, 1213–1227 (2008).
Okuda, S. et al. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. 45, D1107–D1111 (2017).
Acknowledgements
We thank Single-Cell Genome Information Analysis Core (SignAC) at WPI-ASHBi at Kyoto University for Illumina DNA sequencing. We thank Takeshi Yamagami and Suzuka Kajikawa for their technical assistance to purify the Smc protein, Hugo Maruyama for his kind gifts of the T. kodakarensis KCP1 strain and the TrmBL2 expression plasmid, and Stephen Bell for helpful comments on the manuscript. The T. acidophilum strain DSM 1728 (JCM 9062) was provided by Japan Collection of Microorganisms at RIKEN BRC, which is participating in the National BioResource Project of the MEXT, Japan. The supercomputer of Academic Center for Computing and Media Studies (Kyoto University) was used for the MD simulations. K.Y. is supported by JST, the establishment of university fellowships towards the creation of science technology innovation (JPMJFS2123) and SPRING (JPMJSP2110). N.T. is funded by JST PRESTO (JPMJPR20K7), JST FOREST (JPMJFR224V), JSPS Grants-in-Aid for Young Scientists Start-up (JP21K20636), JSPS Grants-in-Aid for Transformative Research Areas (JP23H04281), Takeda Science Foundation, and the Uehara Memorial Foundation. S.T. is funded by JSPS Grants-in-Aid for Transformative Research Areas (JP20H05934). Y.I. is funded by JSPS Grant-in-Aid for Challenging Research (Exploratory) (JP19K22289).
Author information
Authors and Affiliations
Contributions
K.Y. and M.K. performed 3C-seq and strain construction for T. kodakarensis. K.Y. also performed growth measurement, RNA-seq, and ChIP-seq. N.T. performed 3C-seq on T. acidophilum and DNA affinity purification. T.T. and I.H. performed mass spectrometry analysis. K.Y. and S.I. performed protein purification to raise antibodies for ChIP-seq. K.Y., A.O., and N.T. performed microscopic analysis. N.T. performed data analysis on DNA sequencing data. M.Y. and S.T. performed MD simulations. N.T. and H.A. supervised the project. N.T., S.T., and Y.I. acquired funding. K.Y., N.T., A.O., M.Y., and T.T. wrote the manuscript. All co-authors critically read and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hironori Niki and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yamaura, K., Takemata, N., Kariya, M. et al. Chromosomal domain formation by archaeal SMC, a roadblock protein, and DNA structure. Nat Commun 16, 1312 (2025). https://doi.org/10.1038/s41467-025-56197-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56197-y
