
Abstract
The role of non-coding regulatory elements and how they might contribute to tissue type specificity of disease phenotypes is poorly understood. Autosomal Dominant Leukodystrophy (ADLD) is a fatal, adult-onset, neurological disorder that is characterized by extensive CNS demyelination. Most cases of ADLD are caused by tandem genomic duplications involving the lamin B1 gene (LMNB1) while a small subset are caused by genomic deletions upstream of the gene. Utilizing data from recently identified families that carry LMNB1 gene duplications but do not exhibit demyelination, ADLD patient tissues, CRISPR edited cell lines and mouse models, we have identified a silencer element that is lost in ADLD patients and that specifically targets expression to oligodendrocytes. This element consists of CTCF binding sites that mediate three-dimensional chromatin looping involving LMNB1 and the recruitment of the PRC2 transcriptional repressor complex. Loss of the silencer element in ADLD identifies a role for non-coding regulatory elements in tissue specificity and disease causation.
Similar content being viewed by others

Introduction
Many neurological disorders are caused by mutations in genes that are expressed in multiple cell types. Why the CNS is preferentially impacted in these diseases has been a longstanding puzzle. The fatal, adult onset, progressive neurological disorder Autosomal Dominant Leukodystrophy (ADLD, OMIM# 169500) is one such disease. Symptoms begin around the fifth or sixth decade of life with the primary pathology being widespread CNS demyelination with patients rarely living beyond their mid 70s1,2,3,4.
Most cases of ADLD are caused by tandem genomic duplications (ADLD-Dup) involving the lamin B1 gene (LMNB1) while a smaller subset is caused by genomic deletions (ADLD-Del) upstream of the gene1,4,5,6,7. In both cases, the pathological variants are completely penetrant and increased expression of LMNB1 is thought to underlie the disease phenotype3,6,7. Lamin B1 is an intermediate filament protein that is an integral part of the nuclear lamina, a fibrous meshwork found adjacent to the inner nuclear membrane in most mammalian cells8,9. It is critical for the proper maintenance of nuclear architecture and multiple other cellular processes8,9. Lamin B1 is widely expressed in many different cell types and tissues10,11 and why duplications or upstream deletions involving this gene cause a specific CNS demyelinating phenotype is a mystery4,12. In addition, whether duplications and upstream deletions share a common mechanism to cause lamin B1 overexpression is unknown4.
Also unclear are the specific cell types that are involved in the disease process. We have previously demonstrated that oligodendrocyte (OLs), the cell type that produces myelin in the CNS, are critical to the disease process using a transgenic mouse where we targeted human FLAG-tagged LMNB1 overexpression to OLs using a Plp1 promoter construct13, a gene that is highly expressed in mature oligodendrocytes14,15. These PLP-LMNB1 transgenic (TG) mice exhibited severe vacuolar degeneration of the white matter resulting in late age onset motor dysfunction, muscle wasting, paralysis and did not survive beyond 13 months14. The vacuolar degeneration was very similar to the pathological changes observed in human ADLD brain sections while the age-dependent motor dysfunction was reminiscent of symptoms in ADLD patients16,17. However, targeting LMNB1 overexpression to astrocytes or neurons in transgenic mice did not produce a pathological phenotype15. Whether or how other cell types such as astrocytes, microglia or neurons contribute to the demyelination phenotype is still unclear.
In this report, we present the identification of three independent families with multiple members segregating tandem duplications involving the LMNB1 gene that, strikingly, do not demonstrate any demyelination phenotype, even at an advanced age. These data suggest that merely having an extra copy of LMNB1 does not result in ADLD and implicates other mechanisms regulating the overexpression. Using data derived from disease and non-disease-causing LMNB1 structural variants, ADLD patient tissues, CRISPR editing of diverse cell lines and mouse models, we have identified a silencer element that regulates the expression of lamin B1 specifically in oligodendrocytes. The loss of this silencer element in ADLD provides a parsimonious mechanism explaining tissue specificity and how both duplication and upstream deletion can lead to lamin B1 overexpression and furthers our understanding of the role of non-coding regulatory elements in disease causation.
Results
Large tandem genomic duplications involving LMNB1 do not cause ADLD
Genomic duplications of chr5q23.2, encompassing LMNB1, were initially identified in three independent multi-generational families (families F1-F3, Supplementary Fig. 1a–c) by whole genome wide array comparative genomic hybridization (aCGH) studies. A detailed description of the ages and clinical phenotypes of individuals with the duplications are provided in Supplementary clinical data and Table 1. Adult subjects from families F1-F3 had mean and median ages of 62 and 65 years, respectively (range—48–84 years). Magnetic resonance imaging (MRI), a key tool for the diagnosis of leukodystrophies, revealed that none of these individuals exhibited leukodystrophic changes (Fig. 1a–c). MRI abnormalities have been shown to precede neurological symptoms in ADLD (Fig. 1e) and can be identified as early as 29 years of age18. The mean and median ages at onset for ADLD patients from previous studies were estimated to be 47 and 48 years, respectively (range—40–58 years), while the mean and median ages at death were 66 and 68 years, respectively (range—56–75 years)18. The fact that the older subjects from F1-F3 with the duplications did not exhibit any MRI abnormalities or significant disability even in their mid-70s (Fig. 1a, b), an age at which no ADLD patients live beyond, further supports that they do not suffer from ADLD. Interestingly, although individuals did not exhibit the demyelination phenotype, some of them exhibited symptoms such as intermittent bowel or bladder dysfunction or orthostatic hypotension which can both be part of the early autonomic symptoms in ADLD (Table 1, Supplementary clinical data).

Images I-V are axial slices of the brain and images VI parts of the middle sagittal slices. Subjects of the families F1, F2, and F3 did not exhibit leukodystrophic changes (a–c), but the oldest subjects had age-related hyperintensities, best seen in F1-2 periventricularly (a.IV, arrows) and as small foci in deep white matter (a.V, one marked with an arrow). Subject F4-1 showed a pathologic high signal intensity in the pyramids of the medulla oblongata (d.I, arrow), in the pons and middle cerebellar peduncles (d.II), mesencephalon (d.III), and in all cerebral lobes (d.III-V). A less affected periventricular rim in T2w SE images (d.IV, arrows) is characteristic for LMNB1-related leukodystrophy. The corpus callosum is thin (d.VI, arrow). For comparison, images of two subjects from other families with a canonical ADLD-causing LMNB1 duplication are presented (e, f). The subject at the age 35 was still asymptomatic but had a mild T2 signal intensity increase in the pyramids (e.I, arrow), in the middle and upper cerebellar peduncles (e.II-III, white arrows), and in the corticospinal tracts both in the mesencephalic (e.III, black arrow) and uppermost parts (e.V, arrow). The other subject was 65 years old and had a clinical disease. She exhibited similar MR abnormalities to those in F4-1. A pathologic high signal intensity in the pyramids of the medulla oblongata (f.I, arrow), in the pons and cerebellar peduncles (f.II), mesencephalon (f.III), and in all cerebral lobes (f.III-V). A less affected periventricular rim is seen in T2w SE images (f.IV, arrows) and the corpus callosum is thin (f.VI, arrow). Abbreviations: T2w T2-weighted, T1w T1-weighted, SE spin echo sequence, FLAIR fluid attenuated inversion recovery sequence, GRE gradient echo sequence, TFE turbo field echo sequence.
Subjects with the LMNB1 duplications were subsequently analyzed with a custom high resolution CGH array that we had previously designed for the LMNB1 region5 which allowed us to identify precise duplication junction boundaries (Supplementary Fig. 2, Fig. 2a, Supplementary table 1). Sequencing PCR products using primers that flanked the duplication junctions revealed that they were in a tandem head-to-tail configuration (Supplementary Fig. 3a, Fig. 2a), similar to the ADLD-causing duplications5.

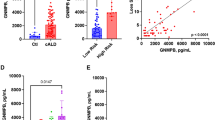
a Schematic of duplications in LN-Dup (F1-F3, green) and F4 (yellow) families. For the canonical disease-causing duplication (blue), duplications were composites based on previous reports5 where precise duplication boundaries were identified. ADLD-Dup majority (dark blue) represents the maximal extent of all disease-causing duplications from multiple families5, the centromeric end of which extends to chr5:126683196. In one family, A85 (dark blue), the centromeric end of the duplication extended beyond the ADLD-Dup majority (chr5:126667592) and is shown separately. ADLD-Del Min (red) is a composite of multiple patients with deletions and represents the smallest deleted region causing ADLD6,7. ADLD-Crit 1 and ADLD-Crit 2 represent the smallest critical regions that are included in the ADLD-Del Min and LN-Dup but not in ADLD-Dup Majority or A8, respectively. Inset box shows the orientation of the various structural variants, with LMNB1 depicted as a black arrow. Both ADLD Dup and LN-Dup (F1-F3) have duplications with a similar head-to-tail tandem configuration, but LN-Dups are larger and contain more of the LMNB1 upstream regulatory region, towards the centromeric end. The F4 duplication is inverted and inserted into the original duplicated segment as previously described5. b Fibroblasts from both LN-Dup and ADLD-Dup patients show significantly increased LMNB1 expression compared to controls, as measured by real time PCR. No difference is observed in LMNB1 expression between ADLD-Del2 and controls. Control n = 5; ADLD-Dup1 n = 3; ADLD-Dup3 n = 3; LN-Dup F2 n = 5; LN-Dup F3 n = 6; ADLD-Dup F4 n = 3; ADLD-Del2 n = 3. c ADLD-Dup and Del patients show significantly higher expression of LMNB1 in white matter vs. grey matter in comparison to control brain samples, as measured by real time PCR. Note that we did not have grey matter sample for the ADLD-Del-1 patient. *p < 0.05, **p < 0.01, ***p < 0.001. WM—Control n = 6; ADLD-Dup1 n = 5; ADLD-Dup2 n = 3; ADLD-Del1 n = 3. GM—Control n = 6; ADLD-Dup1 n = 5; ADLD-Dup2 n = 3. ADLD-Dup 1-3 and ADLD-Del 1-2 have been described previously5,6. For (b, c), all comparisons are between controls and LN-Dup or ADLD samples using one-way ANOVA. All samples are biological replicates and error bars are S.E.M.
The duplications in the three families ranged from 4.37 Mb (F1) and 1.03 Mb (F2) to 466 kb (F3) and did not share any common junction sequences suggesting that they were non-recurrent and arose independent of each other (Supplementary Fig. 3b, Supplementary Table 1). We have termed these duplications ‘large normal duplications (LN-Dups)’, as they were not associated with a leukodystrophy phenotype. While these LN-Dups were also tandem, they were larger and extended more centromeric (Fig. 2a) than the canonical ADLD-Dups which ranged from 128 kb to 324 kb5.
The exception to this range was the F4-1 patient that we previously identified5 (Supplementary Fig. 1d, denoted as BR-1 in that publication) with a duplication of ~475 kb involving LMNB1 but whose clinical features had not been previously described. This patient did not have a head-to-tail tandem duplication but rather an inverted duplication (ADLD-Inv-Dup) that was inserted centromeric to the LMNB1 gene (Fig. 2a)5. F4-1 exhibited, both clinically and radiologically, a more severe and accelerated form of ADLD with MRI features at age 34 (Fig. 1d, Supplementary clinical data) resembling ADLD patients in their 60s with advanced stages of the disease (Fig. 1f) and was wheelchair-bound at age 38. Furthermore, both his mother and grandmother were reported to have suffered from a similar neurological disorder and died in their mid 30s (Supplementary Fig. 1, Supplementary clinical data).
These findings demonstrate that simply possessing an extra copy of the LMNB1 is insufficient to cause the ADLD disease phenotype and that additional factors such as size and orientation of the duplication need to be considered when predicting the pathogenicity of LMNB1 duplications.
LMNB1 is differentially expressed in ADLD patient tissues
We analyzed LMNB1 expression from ADLD patients from whom we had fibroblast and brain samples (Fig. 2b, c, Supplementary table 2). Two brain samples were obtained from ADLD patients with duplications and one from an ADLD patient with a deletion. The duplications in brain samples 1 and 2 were 169 kb and 203 kb, respectively, while brain sample 3 was from a patient with a deletion that encompassed a 608 kb region upstream of the LMNB1 gene5,16 (Supplementary table 2). All three patients had typical ADLD presentations, and the duplications and deletion have been described previously5,6 (Supplementary table 2).
RNA expression analysis from ADLD-Dup fibroblasts demonstrated an increase of ~1.5 to 2-fold LMNB1 expression, consistent with these cells having 3 vs. 2 copies of the LMNB1 gene and similar to our previous results from other ADLD fibroblasts5 (Fig. 2b). However, the fibroblast line from the ADLD deletion patient did not exhibit any increase in LMNB1 expression (Fig. 2b). Interestingly, fibroblasts derived from LN-Dup individuals also exhibited similar LMNB1 overexpression comparable to ADLD-dup patients (Fig. 2b). Fibroblasts derived from the F4-1 patient exhibited the highest level of LMNB1 expression (Fig. 2b). These results were consistent with western blot analysis measuring LMNB1 protein abundance in cultured primary fibroblasts (Supplementary Fig. 4a, b).
LMNB1 expression was then analyzed from ADLD brain samples compared to age matched controls (Fig. 2c). We isolated RNA from the frontal grey matter, which is not affected in the disease and frontal white matter, which is a site of demyelination in ADLD. Intriguingly, while LMNB1 expression in the grey matter did not exhibit statistically significant increases, expression in the white matter was increased from ~4 to 7-fold in ADLD patients as compared to age and sex matched controls (Fig. 2c). A similar pattern was seen using western blot analysis of protein isolated from grey and white matter in brain tissue from unaffected controls and ADLD-Dup patients (Supplementary Fig. 4c, d). This increase in the white matter expression of LMNB1 is much higher than what would be expected from just an extra copy of the gene. As these are autopsy brain tissue it is possible that this increase of LMNB1 expression could be due to alterations in other cell-types secondary to the demyelination phenotype. However, it may also suggest a distinct transcriptional regulatory mechanism that directs the overexpression specifically to the white matter where OLs are one of the predominant cell types.
Oligodendrocytes are not uniquely susceptible to the pernicious effects of LMNB1 overexpression
As OLs clearly drive a significant portion of the pathology in our mouse model14, two possibilities may exist to explain their preferential involvement in ADLD patients and the specificity of the demyelination phenotype. One possibility is that these cell-types are uniquely susceptible to the effects of LMNB1 overexpression i.e., all cell types are exposed to similar LMNB1 overexpression but only OLs suffer deleterious consequences. Another possibility is that the genomic rearrangements (ADLD-Dup and ADLD-Del) that underlie the disease result in a mis-expression of LMNB1 that specifically targets OLs (and potentially other glial cells in the white matter) for maximal overexpression.
In addition to OLs in the CNS, the Plp1 promoter used in our TG mice also drives expression in Schwann cells13 that are responsible for peripheral myelination. We confirmed that the FLAG-tagged human LMNB1 (hLMNB1) was also expressed in the sciatic nerve, part of the peripheral nervous system (Fig. 3a–c). If OLs are uniquely susceptible to the deleterious effects of LMNB1, we would expect peripheral myelination to be unaffected in our TG mice despite the overexpression of LMNB1 in Schwann cells. We therefore studied the impact of LMNB1 overexpression on myelination in sciatic nerves and discovered that the TG mice exhibited significant age dependent demyelination, as evidenced by reduction of Luxol Fast Blue (LFB) staining, a myelin specific stain (Fig. 3d, e), loss of myelinated axons analyzed by electron microscopy analysis (Fig. 3f–h), and nerve conduction defects (Fig. 3i, j) when compared to wild type mice. These results indicate that overexpression of LMNB1 in Schwann cells can also have deleterious effects and that OLs are not unique in their susceptibility to increased LMNB1 levels. However, in ADLD patients, no peripheral nerve conduction defects have been described1,18 suggesting that peripheral myelination is unaffected. Combining the data from the previous two sections suggests that in ADLD patients, specific OL involvement is more likely due to targeted overexpression to the CNS.

a Real time PCR analysis from sciatic nerve samples from PLP-LMNB1 transgenic (TG) and WT mice demonstrate expression of the exogenous human LMNB1 (hLMNB1) only in TG samples. Mouse Lmnb1 (mLmnb1) expression is not altered between WT and TG samples. n = 3 independent mice for each group, ***p < 0.001, two-tailed t-test. b Representative western blot of sciatic nerves from WT and TG mice probed for endogenous mouse LMNB1, Flag-tagged exogenous hLMNB1 (arrows), and GAPDH as a loading control. MW—molecular weight markers in kDa. c Quantification of immunoblot demonstrating that total LMNB1protein (mLMNB1 and hLMNB1) is overexpressed in sciatic nerves from TG mice. n = 3 independent mice for each group, *p < 0.05, two-tailed t-tests. d Representative brightfield images of 20μm thick longitudinal sections of sciatic nerves from 9-month-old mice stained with luxol fast blue (LFB), scale bars = 100 μm. e Quantification of the LFB staining shows less LFB staining in nerves from 9-month TG mice when compared to WT controls. n = 3 independent mice samples for each group *p < 0.05, one-way ANOVA. f Representative TEM montages of semi-thin transverse sections of sciatic nerves from 9-month-old WT and TG mice stained with toluidine blue. g Representative zoomed in images of the sections in (f), scale bars = 50 μm. h Quantification of the number of myelinated axons. Sciatic nerves from 9-month-old TG mice have fewer myelinated axons than those from WT. n = 3 independent mice for each group, *p < 0.05, two-tailed t-tests. i Representative traces of recorded compound action potential (CAP) of Aβ large diameter fibers from sciatic nerves from 9-month-old WT (black line) and TG (red line) mice. j Quantification of conduction velocity in sciatic nerves from 9-month-old mice demonstrates slower conduction velocity in Aβ fibers from TG mice. n = 6 independent mice for each group, *p < 0.05, two-tailed t-tests. All data are presented as mean values +/- SEM.
Analysis of LMNB1 structural variants suggests the presence of a putative tissue-specific silencer element that down-regulates LMNB1 expression specifically in OL lineage cells
How does one explain the absence of the leukodystrophy phenotype in LN-Dups while the ADLD-dup, ADLD-del and ADLD-Inv-Dup result in demyelination? In light of the finding that LMNB1 overexpression in ADLD may be targeted to specific CNS tissues or cell types, one possible mechanism is that LMNB1 is under the control of a silencer element that maintains low levels of expression in OLs (Fig. 4a). Gene expression analysis studies of mouse OL lineage cells have revealed that Lmnb1 expression is dramatically reduced during the maturation of oligodendrocyte precursor cells (OPCs) into mature OLs19 (Supplementary Fig. 5a, b).

a Model 1—A silencer element acts to maintain low LMNB1 expression in oligodendrocytes (OLs). The silencer-LMNB1 interaction is disrupted in ADLD-causing variants but not in LN-Dup cases. Model 2—Based on a previous study of an ADLD-Del patient, proposes that the upstream deletion causes loss of a TAD boundary bringing an enhancer closer to LMNB1 leading to overexpression7. However, it is unclear how this would explain why there is no disease due to LN-Dups. b UCSC genome browser view showing the syntenic ADLD-Crit 1 region in the mouse genome (134kb-Del, grey bar) deleted by CRISPR/Cas9. c Dual guide RNAs (G1 and G2) were cloned into a CRISPR/Cas9 plasmid followed by transfection and FACS sorting for GFP+ single cells. Deletion positive clones were identified by PCR screening using primers that amplify across the deletion junction. Only clones with deletions (+) will show a PCR product. d Representative DNA copy number analysis from positive Oli-neu clone with deletion using real time PCR demonstrates reduced copy number compared to control clone using primers within deleted region (red). n = 3 technical replicates for each clone, ***p < 0.001, two-tailed t-tests. Primers outside deleted region (blue and green) show copy number similar to control cells. e Sequencing deletion junctions using the junction PCR primers shown in (b) reveals each Oli-neu clone has a unique sequence due to imperfect repair after CRISPR/Cas9 mediated deletions. Protospacer Adjacent Motif (PAM) sites are highlighted in blue. Note that PAM sites can be located on the reverse strand, with their orientation indicated by arrows. f Lmnb1 mRNA expression as measured by real time PCR relative to βActin (Actb) is significantly higher in Oli-neu cells with the deletion but is not significantly altered in N2A cells and reduced in 3T3 cells, compared to control cell lines. Oli-neu, control n = 3, del n = 4; N2A, control & del n = 3; 3T3, control n = 5, del n = 4, *p < 0.05, two-tailed t-tests. In all cases, independent clones were used. For all graphs, data are presented as mean values +/− SEM.
Based on our hypothesis, in the canonical ADLD-Dups, the LMNB1 gene is duplicated while the silencer regulatory element is not. As a result, the duplicated copy of the LMNB1 gene is no longer under the repressive control of the silencer element and overexpression occurs. In the case of the ADLD-Del, this silencer element is lost due to the upstream genomic deletion, leading to a similar overexpression (Fig. 4a, left panel). This mechanism would explain why LN-Dups are not associated with demyelination as, in this case, the duplications also include the putative silencer element. The duplicated LMNB1 gene is thus still repressed and would not exhibit a specific overexpression in OLs.
This model also explains the paradoxical case of the F4 inverted duplication that results in ADLD, even though this duplication has a comparable size to the LN-Dups. Although it includes the putative silencer element, it is not arranged in a tandem orientation. Rather, the duplication is inverted and inserted between the silencer element and the non-duplicated copy of the LMNB1 gene and could potentially result in the disruption of the spatial relationship between them (Fig. 4a). Why the F4 inverted duplication results is a more severe phenotype can also be explained by our silencer model and is discussed in detail below.
An alternative model (Fig. 4a, right panel), that was proposed when the first ADLD-del family was identified, posited that the deletion resulted in the adoption of an alternative enhancer that acted on LMNB17. However, this model assumed that the extra copy of LMNB1 was sufficient to cause disease, which we now know is not the case. It cannot explain the lack of a disease phenotype in the LN-Dup families and why the F4 inverted duplication results in a more severe form of ADLD.
Using the silencer model to explain the differential pathogenic effects of the LMNB1 structural variants (SV), and by comparing the maximal centromeric extents of the ADLD-causing duplications and telomeric extents of ADLD-causing deletions to that of the centromeric extents of the nonpathogenic LMNB1 duplications, we were able to delineate a minimal critical genomic region that would be predicted to include this putative LMNB1 suppressor element (Fig. 2a). We only utilized LMNB1 ADLD-causing duplications whose exact duplication junctions were identified5. We noted that with the exception of one case, the centromeric extent of the majority of the ADLD duplications extended to genomic coordinates (hg38) chr5:126683196. The duplication in one family, A8, extended to genomic coordinates chr5:1266675925. These two boundaries allowed us to identify the ADLD critical regions 1 and 2, with sizes of ~145 kb (ADLD Crit-1) and ~160 kb (ADLD Crit-2), respectively (Fig. 2a). An analysis of this region in the mouse and human genomes revealed synteny with almost complete conservation of gene order, suggesting that intergenic elements such as the putative silencer might also be conserved (Supplementary Fig. 6a, b).
To test whether the ADLD critical region influences lamin B1 expression, we carried out CRISPR/Cas9 mediated genomic deletions of a syntenic genomic segment in the mouse genome. This region in the mouse genome was identified using the ‘LiftOver’ function in the UCSC genome browser and corresponded to ~134 kb (mm10, chr18: 56504521-56638837) on mouse chromosome 18 (Fig. 4b). Genomic deletions were generated in Oli-neu, neuro2A (N2A) and NIH-3T3 (3T3) mouse cell lines that represented oligodendroglial, neuronal, and fibroblast lineages, respectively. Undifferentiated Oli-neu cells are considered similar to OPCs while differentiated Oli-neu cells are thought to recapitulate OLs in early stages of maturity20. They have been extensively utilized in multiple studies as surrogates for early stage OLs, especially in analyzing epigenetic modifications and regulatory pathways in the OL lineage21,22,23,24,25,26,27. Relevant to our work, Oli-neu cells also exhibit a significant reduction in Lmnb1 expression levels upon differentiation similar to OLs, suggesting that the regulatory mechanisms that control Lmnb1 expression are similar between primary OLs and Oli-neu cells (Supplementary Fig. 5b).
At least three independent clones with a genomic deletion of ~134 kb corresponding to the syntenic ADLD critical region were generated for each of the cell types. In all cases, clones were grown from single cells and presence of the deletions was confirmed by real-time PCR to measure DNA copy number and sequencing of junction PCR products across the deletion breakpoints (Fig. 4c–e, Supplementary Fig. 7a, b). Sequencing deletion junctions also confirmed that each clone was unique as non-homologous end joining (NHEJ) repair of the breakpoints resulted in sequence “scars” for each clone (Fig. 4e, Supplementary Fig. 7b). Strikingly, we observed a significant increase in Lmnb1 expression levels in differentiated Oli-neu cells with the 134 kb deletion but not in N2A or 3T3 cells (Fig. 4f) or undifferentiated Oli-neu cells (Supplementary Fig. 7e). These results suggest that the ADLD critical region does indeed contain a regulatory element that can potentially act as a silencer in an oligodendrocyte specific manner. However, as the 134 kb deletion results in the disruption of the centromeric boundary of the Topologically Associated Domain (TAD) boundary containing lamin B1 (Fig. 5a), the overexpression could still result from the action of an enhancer, as has been suggested previously7.

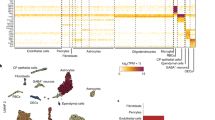
a Micro-C chromatin interaction map27 (hg38) from human Embryonic Stem Cells (hESC) reveals distinct long-range interactions between the LMNB1 promoter and upstream elements (dashed lines, oval). This 19 kb region (red bar) wholly overlaps ADLD Crit 1 and partially with ADLD Crit 2. The transcriptionally associated domain (TAD) containing LMNB1 is also depicted. b PLAC-Seq maps from human OLs30 confirm the interaction between the LMNB1 promoter and the 19 kb putative regulatory element. c ATAC seq data of human OLs30 identifies regions of open chromatin in the 19 kb region. d ChIP Seq data of human OLs30 reveals no enrichment of H3K27Ac in this region. Black bars above traces represent called peaks in both cases. e Single cell Hi-C data from human OLs31 reveals a similar interaction (oval) between LMNB1 promoter (arrow) and the 19 kb element (bar) as seen in (a). f The interaction (oval) between LMNB1 promoter (arrow) and the 19 kb regulatory element (bar) is recapitulated using the Orca simulation based on sequence data. Boxed region is the genomic segment depicted in (a). All coordinates are from the hg38 human genome build.
Examination of the Lamin B1 topologically associated domain identifies a candidate regulatory region
To confirm the silencer hypothesis and narrow down this putative element we analyzed the 3D chromatin organization using previously published chromatin conformation capture data from human embryonic stem cells (hESC)28 (Fig. 5a). Given that long-range chromatin interactions preferentially occur within TADs29,30, we reasoned that the putative silencer would reside within the TAD encompassing the LMNB1 gene. Two genomic regions located ~121 kb centromeric to the LMNB1 transcription and that were ~19 kb apart were identified to interact with the human LMNB1 gene (Fig. 5a). We also confirmed that the 3D genome organization was similar in mouse and humans and that this interacting region was found in both genomes (Supplementary Fig. 6b). This 19 kb region is completely included in ADLD Crit 1 and partially included in ADLD Crit2, towards their telomeric ends, and was thus an attractive candidate for the LMNB1 regulatory element (Fig. 5a). In addition, analysis of previously published PLAC-Seq data31 from human brain-derived OLs identified genomic regions that interacted with the LMNB1 promoter corresponding to the 19 kb candidate region we identified (Fig. 5b). This interaction was also present in human OLs based on single-cell Hi-C experiments (Fig. 5e)32. Analysis of previous ATAC-Seq data from human OLs revealed chromatin accessibility peaks in the 19 kb region (Fig. 5c)31. However, these regions were not enriched in H3K27ac marks that are usually associated with enhancer elements (Fig. 5d, Supplementary Fig. 8a). A comparison of the OL specific transcription factor gene myelin regulatory factor (MYRF)21 demonstrates the expected H3K27ac enrichment for known enhancer regions (Supplementary Fig. 8b). This would suggest that while this 19 kb region serves as a LMNB1 regulatory element, it is unlikely to be an enhancer.
Interactions with the putative 19 kb regulatory element are perturbed by disease-causing structural variants
To determine how the LMNB1 SVs altered 3D genome organization and impacted the interaction between the 19 kb regulatory element and LMNB1, we used a powerful, recently developed, sequence-based deep-learning approach, known as Orca (Figs. 5f, 6)33. Orca can predict, from sequence data, the 3D genome structure from kilobase-scale up to whole-chromosome-scale33. Remarkably, all ADLD disease-causing SVs (ADLD-Dup, ADLD-Del and ADLD-Inv-Dup) reduced interactions between the 19 kb critical region and one of the copies of the LMNB1 gene (Fig. 6a–e). The direction and magnitude of genomic interaction change predicted by Orca was quantified by the delta score (Δ), defined as the logarithmic fold change of the interaction score between the promoter region of the LMNB1 gene and the 19 kb critical region (log Mutant/WT, see Methods section for details). Specifically, the majority of the ADLD-causing duplications (ADLD-Dup majority) resulted in a duplicated LMNB1 promoter that had a dramatically reduced interaction with the critical 19 kb region, compared to the original promoter (Δ2 = −1.79, Fig. 6b). In the case of A8, only a portion of the 19 kb region was duplicated and the duplicated LMNB1 gene interacted only with partial regulatory element (Δ2 = −0.56, Fig. 6c). ADLD-del removed the critical region completely and therefore the interaction with the critical region was lost (Fig. 6d). Interestingly, for ADLD-Del, Orca was able to predict an interaction between an upstream element representing an ectopic enhancer in ADLD-del patients that had been previously identified7, further confirming the accuracy of the Orca model (Fig. 6d).

a Orca plot of reference human genome demonstrating interaction (horizontal oval) of the LMNB1 promoter (blue arrow) with 19 kb regulatory element (yellow bar). b Plot of ADLD-Dup majority demonstrating duplication of the LMNB1 gene but not the silencer element. ∆1 represents the strength of interaction of the 19 kb element and the non-duplicated LMNB1 promoter (horizontal oval) which is unchanged, while ∆2 represents the strength of interaction of 19 kb element with the duplicated LMNB1 gene (vertical oval) which is reduced. This reduction is represented in the Orca plots by a reduction in color intensity. The duplicated region is represented by grey and black bars on the left side of the plot. c The A8 ADLD-causing duplication contains only part of the putative silencer element. This SV also results in unchanged ∆1 (horizontal oval) and reduced ∆2 (vertical oval). In both (b, c), no other genomic regions are predicted to interact with either of the copies of the LMNB1 gene. d ADLD-Del results in loss of the putative silencer element (∆ cannot be calculated). This deletion results in the interaction (green oval) of an exogenous enhancer (green arrowhead) with LMNB1. Open arrowhead indicates the site of the junction subsequent to the deletion. e The ADLD-causing inverted duplication is inserted between the original copy of the LMNB1 gene and its cognate silencer element, resulting in the loss of interaction (reduced ∆1, horizontal oval). The interaction between the duplicated silencer and duplicated LMNB1 is unchanged (∆2, vertical oval). A detailed schematic of the Orca prediction for this SV is presented in Supplementary Fig. 7. f LN-Dup SVs duplicate both LMNB1 and the putative silencer element, and there is no alteration in the interaction between these two elements (∆1 and ∆2) in either of the duplicated copies (horizontal and vertical ovals). Note that no other potential regulatory genomic regions interact with either copy of the LMNB1 gene. g Deletion of the 19 kb putative silencer element does not result in new genomic interactions with LMNB1.
Although the ADLD inverted duplication (F4) includes the 19 kb region and is similar in size to one of the LN-Dups, it paradoxically results in the most severe disease phenotype. This finding can now be explained in the context of the silencer model of LMNB1 regulation (Figs. 2a, 6e, Supplementary Fig. 9). The inverted, duplicated copy of the LMNB1 gene together with its 19 kb regulatory element is inserted between the original LMNB1 gene and its cognate 19 kb region. The Orca-prediction demonstrated that this inverted and duplicated 19 kb element forms an interaction with the original 19 kb element effectively sequestering the latter element and preventing its interaction with the non-duplicated LMNB1 gene (Fig. 6e, Supplementary Fig. 9). As a result, the interaction of the original 19 kb element and the non-duplicated LMNB1 gene and the Δ score are reduced to an even greater extent (Δ1 = −2.49) than when compared the canonical ADLD duplications. One can expect that this rearrangement would lead to even higher LMNB1 overexpression and thus lead to increased disease severity as observed in the F4-1 patient.
In contrast, the LN-Dup SVs duplicated both the LMNB1 promoter and 19 kb critical region, and the interactions between these two regions were preserved in both original and duplicated gene-silencer pairs, as evidenced by a minimal alteration in the delta score (Δ1 = 0.04, Δ2 = −0.08, Fig. 6f), consistent with the lack of an ADLD phenotype associated with this SV. For both the ADLD-Dup and LN-Dup cases, Orca also predicted that no new long-range regulatory elements (either enhancers or silencers) interact with the original or duplicated copies of the LMNB1 promoter and that the loss of interactions with the 19 kb regulatory region in the ADLD-Dup is the only difference between these two SVs (Fig. 6b, c, f). Orca also predicted that deletion of the 19 kb regulatory element alone did not result in the formation of any new 3D chromatin interaction with LMNB1 (Fig. 6g). If the 19 kb region is a silencer element, as we predict, this loss of interaction in the ADLD-causing SVs should lead to an increase in LMNB1 gene expression.
Deletion of 19 kb candidate silencer region results in oligodendrocyte-specific overexpression of Lmnb1
To determine if the 19 kb genomic segment indeed acts as a silencer element, we deleted this region in the three mouse cell types described above and observed an increase in Lmnb1 expression only in Oli-neu cells but not in either the N2A or 3T3 cells (Fig. 7a, b, Supplementary Fig. 10a). The fold increase in Lmnb1 expression was similar to the deletion of the 134 kb ADLD critical region and suggested that silencing capacity of the ADLD critical region resided within this 19 kb element. To confirm that the increase in Lmnb1 expression was not an artifact of the CRISPR-Cas9 genomic editing process, we also made a similar-sized control deletion (~16 kb) outside the Lmnb1 TAD and did not observe any differences in Lmnb1 expression between CRISPR treated and control Oli-neu, N2A, and 3T3 cells (Supplementary Fig. 11). Given that the 19 kb region does not disrupt the TAD boundary containing Lmnb1 and that the magnitude of overexpression was similar between the 134 and 19 kb deletions, these analyses also suggest that the cause of the overexpression is unlikely due to the interaction with an enhancer element from outside the Lmnb1 TAD, as has been previously suggested7. The 19 kb deletion did not alter the expression of other genes in the nearby vicinity (Gramd3, Aldh7a1, and Phax), confirming that this regulatory element was specific for Lmnb1 (Supplementary Fig. 10d).

a UCSC genome browser view of genomic segment encompassing mouse Lmnb1 showing the 134 kb and 19 kb deleted regions. b Real-time PCR analysis of Lmnb1 mRNA expression in Oli-neu, N2A and 3T3 cells with the 19 kb deletion. Lmnb1 expression is significantly higher in Oli-neu cells with the deletion but lowered in N2A and 3T3 cells, relative to their respective controls. Lmnb1 expression is normalized to β actin (Actb). Oli-neu, control n = 6, Del n = 4; N2A, control n = 6, Del n = 4; 3T3, control n = 5, Del n = 3. In all cases, independent clones were used. *p < 0.05, **p < 0.01, two-tailed t-test. c Real-time PCR analysis of Lmnb1 mRNA expression in primary oligodendrocyte progenitor cells (OPC), oligodendrocytes (OL), astrocytes (AS), and ear fibroblasts (Fib) isolated from Lmnb1-Del19 (Del-19) and control mice. For OPCs, OLs and AS, n = 3 for both control and Del-19. For Fib, n = 4 for both control and Del-19. ***p < 0.001, two-tailed t-test. Lmnb1 is significantly increased in OLs but reduced in astrocytes and unchanged in OPCs and fibroblasts. Data are presented as mean values +/− SEM for all graphs. d Representative epifluorescence images of cultured primary differentiated OLs from Del-19 mice and WT controls stained with antibodies against LMNB1 (green) and the OL-specific marker CNP (red). Scale bar = 50 μm. e LMNB1 staining of OL nuclei from WT and Del-19 mice demonstrate increased presence of misshapen nuclei in the latter (arrows). Scale bar = 50 μm. Violin plot quantifications of (f) LMNB1 intensity (n = 50 cells for both genotypes), g ratio of misshapen nuclei (n = 15 fields for both genotypes), and (h) circularity reveal (n = 77 cells for Control and 90 for Del-19) increased LMNB1 intensity and ratio of misshapen nuclei and decreased nuclear circularity in OLs from the Del19 mice relative to control cells. n. *p < 0.05. ***p < 0.001, two-tailed Mann-Whitney test.
To further confirm that the 19 kb region silences Lmnb1 expression specifically in oligodendrocytes, we generated a mouse line (Lmnb1-Del-19) with a germline deletion of this genomic segment. The deletion was generated using the same gRNAs used to generate the deletion in mouse Oli-neu cells (Supplementary Fig. 12). Primary OLs isolated from these mice exhibited a significant increase of ~2 fold in Lmnb1 levels compared to wild type controls, similar in magnitude to what we have observed in Oli-neu cells (Fig. 7b, c). Such an increase was not observed in ear fibroblasts or primary astrocytes, representing non-OL cell types (Fig. 7c).
One of the most recognizable consequences of lamin B1 overexpression across multiple cell types is an alteration of nuclear structure, and we used this as a functional readout of lamin B1 overexpression3,34,35. We identified an increase in the frequency of misshapen nuclei and reduction in nuclear circularity in OL nuclei derived from Lmnb1-Del-19 mice when compared to wild type (Fig. 7d–h), consistent with previous reports of impact of lamin B1 overexpression on nuclear architecture34,35. Such nuclear abnormalities were not observed in astrocytes from the Lmnb1-Del 19 mice (Supplementary Fig. 13).
The results from the mouse model further strengthen our OL-specific silencer-mediated Lmnb1 overexpression hypothesis and confirm the results we obtained in the Oli-neu cell lines.
CTCF binding sites within the 19 kb region mediate Lmnb1 silencing
To elucidate the molecular mechanisms underlying the silencer element, we carried out a bioinformatic analysis of transcription factor binding sites in the two regions within the 19 kb element that interact with the LMNB1 promoter in both mouse and human genomes. In humans, we observed that both regions were enriched for binding sites for the transcription factor CCCTC binding factor (CTCF) that we named CTCF1 (chr5: 126656590-126656600) and CTCF2 (chr5:126676270-126676280) (Supplementary Fig. 14a). Based on the Hi-C data (Fig. 5a), these CTCF1&2 motifs interact with CTCF binding sites CTCF3 (chr5:126780430-126780440) and CTCF4 (hg38—chr5:126676270-126676280) in the first and third introns of the LMNB1 gene. The CTCF1 (mm10-56608876-56608895), 2 (mm10-56622463-56622482) and 3 (mm10-56711264—56711282) sites were also conserved in the mouse genome while CTCF4 was not (Supplementary Fig. 14b). Within TADs, one of the main functions of CTCF is to target regulatory elements to their cognate promoters by forming chromatin loops and loop forming CTCF sites need to be in opposite orientations36,37,38. We observed that CTCF1 and CTCF2 were in the forward orientation while CTCF3 and CTCF4 were in the reverse orientation (Supplementary Fig. 14c), indicating their ability to form a chromatin loop as indicated by the Hi-C and Orca maps. (Fig. 5a, f).
We confirmed that the CTCF 1, 2 and 3 predicted binding sites were indeed bound by the CTCF protein by carrying out genome wide analysis of CTCF binding in primary human fibroblasts and mouse embryonic stem (ES) cells and primary OLs using the Cleavage Under Targets and Release Using Nuclease (CUT&RUN) technique39,40 (Fig. 8a–c, Supplementary Fig. 15). The conservation of CTCF binding across cell types and species would indicate that they have a functionally important role in LMNB1 regulation.

UCSC genome browser tracks of CUT&RUN analysis of CTCF in (a) human fibroblast, (b) mouse ES cells, (c) mouse primary oligodendrocytes (OL), and (d) SUZ12 in mouse primary OLs confirm bioinformatically predicted CTCF binding sites within 19 kb silencer element, and that SUZ12 associates with the CTCF2 region. Arrows point to the conserved CTCF 1 and CTCF 2 sites. Black bars above traces represent called peaks. e Schematic showing deletions of CTCF 1 and 2 sites. Real-time PCR analysis of Lmnb1 mRNA expression in Oli-neu, N2A and 3T3 cells with CRISPR-mediated deletions of (f) CTCF1 and (g) CTCF2. Lmnb1 expression is significantly higher in Oli-neu cells with CTCF1 deleted but lowered in N2A and 3T3 cells, relative to their respective controls. Lmnb1 expression is increased in Oli-neu and N2A cells with CTCF2 deleted but lowered in 3T3 cells. Lmnb1 is normalized to β Actin (Actb). Graphs are mean ± SEM. For CTCF1 - Oli-neu, control & del n = 3; N2A, control n = 5, del n = 3; 3T3, control n = 4, del n = 5. For CTCF2 - Oli-neu, control n = 6, del n = 4; N2A, control n = 6, del n = 4; 3T3, control n = 5, del n = 4. In all cases, independent clones were used. *p < 0.05, **p < 0.01, two-tailed tailed t-tests.
To experimentally test whether the individual disruption of either site impacted Lmnb1 expression, we generated CRISPR deletions that removed each CTCF site independently in Oli-neu, N2A and 3T3 cell lines (Fig. 8e–g, Supplementary Figs. 16, 17). Deletions of the CTCF1 site resulted in an increase in Lmnb1 expression in Oli-neu cells but not in the other two cell types, while deletion of the CTCF2 site resulted in increased Lmnb1 expression in Oli-neu and N2A cells but not in 3T3 cells (Fig. 8f, g). The magnitude of the increase was similar to the deletion of the 19 kb critical region, indicating that these sites were responsible for the silencer function of this region. However, it is unclear why Lmnb1 expression is also increased in N2A cells with the deletion of only the CTCF2 deletion. It is possible that this region has complex roles with respect to the cell type specific regulation of Lmnb1 that need to be investigated further.
To further explore the role of CTCF in mediating Lmnb1 silencing in OLs, we carried out RNAi-mediated knockdown of CTCF in all three cell types and observed increased expression of Lmnb1 only in Oli-neu cells but not in N2A or 3T3 cells (Fig. 9b, Supplementary Fig. 18a). Consistent with our model, a similar experiment using Oli-neu cells with the 19 kb deletion did not exhibit any further increase in Lmnb1 expression when treated with CTCF RNAi (Fig. 9b), confirming that CTCF binding specifically in the 19 kb critical region was responsible for transcriptional downregulation of Lmnb1.

a Genome tracks of H3K27me3 CUT&RUN analysis of mouse OLs (upper panel). Black bars above traces represent called peaks. CUT&TAG analysis of mouse OLs, neurons and astrocytes (lower three panels). Enrichment of H3K27me3 in the 19 kb silencer (red bar and grey shaded region) element is observed specifically in OLs. CUT&TAG analysis is from previously published data25. b Ctcf and Eed siRNA treatment of Oli-neu, N2A and 3T3 cell lines reveal increased Lmnb1 expression only in Oli-neu cells, relative to respective untreated cell-specific controls. Scrambled siRNA treatment has no effect on Lmnb1 expression. No increase in Lmnb1 expression is observed in Oli-neu cells with the 19 kb deletion (Oli-neu Δ19) relative to untreated D19 cells. Graphs are mean ± SEM. n = 3 biological replicates per cell type and treatment. ***p < 0.001, using two-way ANOVA and Dunnett’s multiple comparisons test.
To further investigate the roles of individual CTCF sites in the regulation of LMNB1 expression, luciferase assays were carried out with luciferase constructs containing ~1 kb of genomic sequence surrounding CTCF1, CTCF2, CTCF2 in the reverse orientation, or a region located within the 16 kb control sequence that is devoid of CTCF motifs (Supplementary Fig. 19a), and luciferase activity was quantified and compared to plasmid containing only the promoter sequence. We observed that the presence of the CTCF2 sequence in both orientations significantly reduced luciferase activity only in Oli-neu cells, signifying silencer activity that is specific for oligodendrocyte-lineage cells (Supplementary Fig. 19b). We did not observe silencing with the CTCF1 site.
Epigenetic analysis of the 19 kb region identifies a role for the PRC2 complex in lamin B1 silencing
One mechanism by which CTCF mediates transcriptional silencing is by its association with the polycomb repressive complex 2 (PRC2) group of proteins41,42. PRC2 is a large multimeric protein complex and has been shown to function as a repressor protein complex in the establishment of long-range chromatin interactions43,44. The PRC2 complex is comprised of the core subunits SUZ12, EED, and the methyltransferase EZH2 or its closely related homolog EZH1 and are all required for the catalytic activity of the complex45.
We tested whether SUZ12, one of the components of the PRC2 complex exhibited any binding in the 19 kb silencer region in OLs using CUT&RUN and observed SUZ12 binding to the same location as CTCF2 but not at CTCF1 (Fig. 8d, Supplementary Fig. 20). This is consistent with the results from our luciferase assays (Supplementary Fig. 19) and explains why only the CTCF2 sequence exhibited reduced luciferase activity as this is the region bound by SUZ12 within the silencer element. These results greatly strengthen our model for the role of the PRC2 complex in oligodendrocyte-specific Lmnb1 silencing.
Chromatin regions that are silenced by the PRC2 complex are enriched in H3K27 tri-methylation (H3K27me3)44,46, and we analyzed previously published data on H3K27me3 binding within the Lmnb1 TAD from various mouse CNS cell types26 and also carried out H3K27me3 CUT&RUN analysis on primary OLs (Fig. 9a, Supplementary Fig. 21). Strikingly, we observed a significant enrichment of H3K27me3 binding within the 19 kb silencer region only in OLs and not in other CNS cell types such as astrocytes and neurons (Fig. 9a).
Similar to CTCF, we carried out RNAi-mediated knockdown of one of the PRC2 components, Eed, in all three cell types, and observed increased expression of Lmnb1 only in Oli-neu cells but not in N2A or 3T3 cells (Fig. 9b. Supplementary Fig. 18b). As predicted, Eed knockdown in the 19 kb del Oli-neu cells did not exhibit any further increase of Lmnb1 expression (Fig. 9b). Supporting our findings, an examination of previously published RNA-Seq data revealed that Lmnb1 expression was significantly increased by ~1.5 fold compared to controls in mouse OLs where the Eed was conditionally knocked out47.
Discussion
Our results provide insights into the tissue specificity of ADLD and provide a unifying mechanism to explain how duplications and upstream deletions can cause disease. These data indicate that ADLD is caused by a transcriptional targeting of lamin B1 overexpression specifically to OLs, mediated by the loss of an interaction with an OL-specific silencer element, rather than just the presence of an extra copy of the LMNB1 gene.
These results also establish a framework for the interpretation of the pathogenic potential of LMNB1 SVs, and other pathogenic SVs in general, that have significant implications for genetic testing and provide insights into how the disruption of long-range regulatory elements can lead to disease. Our findings demonstrate that not all LMNB1 duplications will result in ADLD, and we demonstrate that the size, orientation, location, and insertion site of the LMNB1 duplications are all essential for predicting disease onset (Supplementary Figs. 22, 23). Loss of interactions with the silencer element appears to be the critical determinant of pathogenicity of the LMNB1 SVs. In the case of ADLD-Del, it is yet unclear if interaction with the ectopic enhancer is also required for disease causation or whether deletion of the silencer element alone is sufficient (Supplementary Fig. 23). While our results favor the latter hypothesis, loss of the silencer element would still be required to facilitate the interaction of the ectopic enhancer with the LMNB1 promoter and would thus be a critical first step in ADLD pathogenesis.
Once the exact location, size, and orientation of the SVs are known, interaction with the silencer element can be predicted using bioinformatic tools such as Orca. While this is straightforward for deletions that remove the silencer element, this can be more complicated for duplications or inversions and a case-by-case review is required to predict pathogenicity. For simple tandem duplications that encompass the silencer element, we would predict that ADLD would not develop. However, if the duplications are not tandem, it is critical to identify their exact insertion site as they may be inserted in a manner that disrupts the lamin B1-silencer interaction, as has occurred in F4-1. While patients with the large duplications do not exhibit symptoms of ADLD, some do exhibit more subtle clinical phenotypes including those of autonomic dysfunction, and the exact role of LMNB1 duplications in these cases needs to be explored further.
When predicting the pathogenic impact of SVs, conventional wisdom has dictated that the larger the variant and the more genes involved, the greater likelihood that the variant will be deleterious48. Paradoxically, our results demonstrate that the larger duplications have a more benign impact and provide a rationale for explaining this phenomenon that may also be applicable to other disease-causing SVs. There have been very few reports implicating silencer elements in disease pathogenesis, especially those that silence target gene expression in specific cell types and none for Mendelian diseases49. To the best of our knowledge, we have also not encountered previous reports of duplications leading to loss of interactions with a silencer element as a disease mechanism. These represent unique consequences of genomic alterations that should be borne in mind with predicting the pathogenic potential of other SVs. While this manuscript was in revision another report was published that identified a family with a large duplication involving LMNB1 that also did not cause ADLD50. Consistent with our results and a previous publication51 they observed LMNB1 duplications that did not cause ADLD were larger and included the regulatory element. However, they did not determine the nature of this element that we have now identified to be the OL-specific silencer.
Our results indicate that OLs are the primary cell type that are targets of the lamin B1 overexpression and potentially the primary cell type that drives the ADLD disease process. This finding is consistent with our previous findings that transgenic mice where LMNB1 overexpression is targeted to OLs exhibit ADLD-like phenotypes while targeting LMNB1 overexpression to astrocytes or neurons does not result in demyelination14,15. Histopathological analysis of ADLD brain post-mortem tissue revealed no alteration in OL number but has identified abnormally shaped astrocytes as a hallmark of the disease16,17,52. Given that we did not identify overexpression of lamin B1 in astrocytes from the Lmnb1-Del-19 mice, this would indicate that astrocyte dysmorphology is likely a secondary, cell non-autonomous consequence of OL dysfunction. However, the role of other CNS cell types in ADLD cannot be discounted, especially since in-vitro studies have suggested that exogenous overexpression of LMNB1 in an astrocyte cell line resulted in cellular dysfunction53. Our finding that OLs may be the primary driver for the ADLD disease process has important implications for designing therapeutic approaches, especially for those that utilize lamin B1 reduction as a strategy. These approaches can now be targeted specifically to OLs and can reduce the potential side effects of a more indiscriminate reduction of lamin B1 levels in other cell types such as astrocytes or neurons.
Our findings are consistent with a recent report that has characterized a class of silencer elements known as H3K27me3-rich regions or methylation rich regions (MRRs)46. As we have demonstrated for LMNB1, these MRRs are located at sites of long-range chromatin interactions and are thought to function through chromatin looping. They also answer an important question of whether the silencing effects are directly mediated by PRC2, or whether PRC2 is an effector of CTCF–cohesin-mediated chromatin looping49. Our results suggest the latter as the chromatin loops linking the silencer element are conserved across cell types while H3K7me3 modifications are only observed in OLs. Furthermore, the luciferase assay results demonstrating silencing specifically in Oli-neu with the presence of the CTCF2 site, regardless of orientation, is consistent with the results that CTCF2 is bound by the PRC2 complex (Suz12) and is enriched in H3K27me3. In the context of the whole genome, it is possible that the CTCF1 site is required for chromatin looping, bringing the PRC2-bound CTCF2 region in close proximity to the LMNB1 promoter. This would explain why deletion of CTCF1 also results in LMNB1 overexpression in Oli-neu cells, despite its inability to reduce expression in the luciferase assay. This suggests that the PRC2 complex is specifically recruited to the silencer element only in OLs, indicating that PRC2 is not required for the formation of the CTCF-mediated chromatin loops.
A yet unanswered question that arises from our findings is the mechanism underlying the OL specificity of the silencer we have identified. How this occurs is unclear and would likely involve the interaction of an OL specific transcription factor with PRC2. While enhancers in OLs have been previously reported21,22, this report presents the identification of an OL-specific silencer element. Our results can help identify additional OL specific silencer elements and determine whether CTCF-PRC2 interactions are a common mechanism for the downregulation of other genes in maturing OLs. They also provide insights into how long-range non-coding regulatory elements can modulate gene expression and identify a hitherto unknown role for silencer elements in tissue specificity and disease causation.
Methods
This research was carried out in compliance with all relevant ethical regulations and approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC), protocol # 24054947 and the University of Pittsburgh Institutional Review Board (IRB), protocol # 19100151.
Clinical and MRI examination and collection of tissue samples
Clinical and magnetic resonance imaging (MRI) examinations of all subjects were carried out at the respective clinical centers after obtaining appropriate informed consent and Institutional Review Board approval (University of Pittsburgh protocol 19100151) to participate in this study which included publication of non-identifiable clinical details including age and sex and brain imaging and complies with all relevant ethical regulations. Fibroblast samples were obtained from skin punches and cultured as described below. Brain tissue was collected from ADLD patients at autopsy and samples were flash-frozen. In all cases, affected frontal white matter and unaffected frontal grey matter were used for RNA and protein isolation. Control brain samples were obtained from same brain region from age and sex-matched individuals. Fibroblasts and brain samples were obtained after informed consent (University of Pittsburgh IRB protocol 19100151 and University of Pittsburgh CORID protocol 709, respectively). Participants were not compensated for this study.
Genomic DNA isolation and array-based comparative genomic hybridization (Array CGH)
Genomic DNA from whole blood, saliva, primary fibroblasts and brain tissue was isolated using the Gentra Puregene kit (Qiagen) according to the manufacturer’s instructions. Array CGH was performed at the University of Pittsburgh on genomic DNA from clinical samples, hybridized on a custom 8x15K HD-CGH microarray previously described5, scanned in an G2565CA Agilent microarray scanner, and analyzed using Agilent CGH Analytics software (Agilent Technologies). Human genome assembly GRCh38/hg38 was used for all genome coordinates. PCR primers were designed to specifically amplify the tandem duplication junctions for each family using Longamp Taq Polymerase (New England Biolabs), then Sanger sequenced (Eurofins Genomics) after treatment with ExoSAP-IT reagent (Applied Biosystems). The duplication junction primers sequences are listed in Supplementary Data 1.
CRISPR guide RNA design, generation and validation of CRISPR clones
“Left” and “right” CRISPR guide RNA (gRNA) sequences for either end of the genomic deletions were selected using the CRISPOR online tool54, selecting for high specificity and efficiency, adjacent to S. pyogenes Cas9 Protospacer Adjacent Motif (PAM) sites (NGG), encompassing the regions of interest in the mouse genome (mm10) on chromosome 18. The genomic targets of the guides were Sanger sequenced to ensure there were no known variants in the binding sequences or PAM. Both guides were cloned into pDG458 (Addgene) via golden gate cloning, as previously described55, then purified using EndoFree Plasmid Maxi Kit (Qiagen). The gRNA spacer sequences for each of the deletions are listed in Supplementary Data 1.
20 µg of purified pDG458 plasmids containing both CRISPR guides were transfected into Oli-neu, N2A, and 3T3 cells grown to 90% confluence in 10 cm dishes using Lipofectamine 3000 (ThermoFisher) according to manufacturer’s instructions and incubated for 24 h post-transfection. Cells were then detached into a single-cell suspension using Accutase (Millipore Sigma), Fluorescence Activated Cell Sorting (FACS) was then used to sort GFP-positive single cells into 96-well plates to establish clonal populations. Cells were separated from debris by FACS using a FSC vs SSC gate then doublet cells were excluded from single cells and finally GFP+ cells were separated from GFP - cells using a 488 nm laser with a 530/30 bandpass filter. Clones that grew were transferred into 24-well plates, then genotyped using DirectPCR Cell Lysis buffer + 0.2 mg/mL proteinase K (Viagen) and Taq polymerase (New England Biolabs) using primers that spanned the deletion junction (Fig. 4c). Cells that were positive for deletions were expanded for validation and testing.
For clone validation, genomic DNA was isolated from cells using Gentra Puregene Kit (Qiagen) and CRISPR-induced deletions were confirmed via PCR using primers adjacent to each end of the deletion. The primers to sequence deletion junctions are listed in Supplementary Data 1. Amplicons were Sanger sequenced (Azenta) to verify the coordinates and uniqueness of deletions. Clones with deletions were used for expression analysis via quantitative RT-PCR. Cell lines were also tested to ensure they did not contain genomic inversion via PCR using a three-primer strategy as described in Supplementary Fig. 7c.
Cell culture
For fibroblast isolation, patient and control skin biopsies were minced to ~1.0 mm3 pieces then incubated with 0.05% Trypsin-EDTA (Sigma) for 3 h at 37 °C with gentle mixing. Pieces were then plated on a 10 cm tissue culture dish (ThermoFisher) with a minimal amount of DMEM complete (high glucose DMEM (Corning) supplemented with 10% FBS (Fisher), 2 mM L-glutamine (Millipore), and 1% penicillin-streptomycin (Hyclone)) until they adhered to the culture dish. Primary fibroblasts were trypsinized and re-plated into new dishes as they exited the explant. Details of patient fibroblasts are provided in Supplementary table 2.
Neuro2A (ATCC# CCL-131) and NIH-3T3 (ATCC# CRL-1658) cell lines were cultured in DMEM complete medium. Oli-neu cells were provided by Dr. Franca Cambi, University of Pittsburgh and were cultured as previously described56,57. Primary astrocytes and OPCs were isolated by immunopanning from 6–7 day-old mouse pups (cells from both sexes were mixed together) and OPCs were differentiated into OLs for 4 days according to previously-established protocols58. All cells were cultured at 37 °C and 5% CO2 in a humidified chamber.
RNAi treatment
For transfection of RNAi constructs, cells were seeded at a density of ~1 × 105 cells per well of a six-well plate for 16–18 h prior to transfection. 50–75 pmol of pre-designed MISSION® esiRNA (Sigma) using Lipofectamine™ RNAiMAX Transfection Reagent (Thermo Fisher) were transfected in each well. After 24 h the medium was replaced with fresh proliferation medium and cultured another 24 h. In the case of Oli-neu cells, they were then switched to differentiation medium for 4 days.
Mouse husbandry and generation of Lmnb1-Del-19 mice
All mouse studies were approved by the University of Pittsburgh IACUC. Mice were housed under conditions of 12 h cycles of light and dark at an ambient temperature between 68 °F and 76 °F and a relative humidity between 30% RH and 70% RH. The C57BL/6 mouse strain was utilized, and animals of both sexes were used.
Lmnb1-Del-19 mice mice were generated using CRISPR/Cas9 technology as previously described59. Briefly: guide RNAs used to generate the 19 kb deletion and cloned in the pDG458 plasmid were in-vitro transcribed using the TranscriptAID T7 High Yield Transcription Kit (ThermoFisher) and purified using GeneJET RNA purification kit (ThermoFisher). Guide RNAs (200 ng/μl) together with the Cas9 protein (100 ng/μl, Alt.R S.p. Cas9) Nuclease 3NLS, IDT) were electroporated into C57BL/6 mouse zygotes and transferred the following day as two-cell stage embryos into the oviducts of pseudo-pregnant females. Founder lines were screened using primers designed to amplify across the 19 kb deletion listed in Supplementary Data 1.
RNA isolation, cDNA synthesis, and real-time PCR
RNA was isolated from cells, sciatic nerves and brain tissue using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. Details of ADLD brain tissue are provided in Supplementary table 2. cDNA was synthesized from 1 μg of RNA using qScript cDNA Synthesis Kit (Quanta Bio). Real-time PCR was performed using PerfeCTa SYBR Green SuperMix with ROX (Quanta Bio) on an ABI QuantStudio 12 K Flex (Applied Biosystems). Gene expression was analyzed using the ΔΔCT60 method using Actb mRNA as an endogenous control. Primer efficiencies were validated using 6-fold serial dilutions of cDNA. Primers used for real time PCR analyses are listed in Supplementary Data 1.
Real-time copy number analysis
Purified genomic DNA samples from control and CRISPR-edited cells were diluted to 5 ng/µL and real-time PCR was performed using PerfeCTa SYBR Green SuperMix with ROX (Quanta Bio) on an ABI QuantStudio 12 K Flex (Applied Biosystems). Copy numbers were calculated using the ΔΔCT method60 normalized against Actb genomic DNA and plotted as relative to undeleted control lines across three regions: within the deletion region and on each side (centromeric or telomeric) of the deleted region. Cell lines without a significant reduction of deleted region DNA abundance compared to undeleted control were omitted from analysis. Copy number analysis real-time primer sequences are listed in Supplementary Data 1.
Immunofluorescence (IF)
IF was performed as described previously11. Primary antibodies used and dilutions are listed in Supplementary Table 3. AlexaFluor 488 or Cy3-conjugated secondary antibodies were used (Jackson ImmunoResearch) After antibody staining, cells were mounted onto glass microscope slides with Vectashield antifade mounting medium containing DAPI (Vector Laboratories) and imaged using a Leica CTR5000 fluorescence microscope using identical exposure settings. Investigators were blinded to comparison groups.
Protein isolation and Western blotting
Sciatic nerves were harvested from WT and PLP-LMNB1 (TG) mice and stored at –80 °C until protein isolation. To isolate protein, nerves were incubated in T-PER supplemented with 1x protease inhibitor cocktail (Thermo Scientific) and homogenized. Protein from brain tissue was isolated using RIPA buffer using the same protocol as above. Protein isolation and Western blotting were carried out as described previously with 50 µg protein loaded per well on a 10% gel11. The blot was imaged and quantified using the LI-COR Odyssey CLx infrared scanner and Image Studio software (LI-COR Biosciences). Antibodies used are listed in Supplementary Table 3. Images of uncropped blots are provided in Source Data and in Supplementary information.
Myelin visualization and electron microscopy analysis
To assess myelin in the periphery, sciatic nerves from 9-month-old WT and PLP-LMNB1 mice were stained with Luxol fast blue (LFB) as previously described61. Sections were examined and images captured using a Leica CTR5000. Staining was quantified from five representative areas per nerve using ImageJ and an average mean intensity was measured for each nerve.
For transmission electron microscopy (TEM) analysis, mice were perfused with cold PBS and 4% PFA, sciatic nerves from WT and PLP-LMNB1 mice were post-fixed in cold 2.5% glutaraldehyde in 0.01 M PBS (Fisher, Pittsburgh, PA), pH 7.3 and processed as previously described14. Sections were examined on a JEOL 1011 transmission electron microscope (JEOL Peabody, MA) or JEOL 1400 transmission electron microscope with a side mount AMT 2k digital camera (Advanced Microscopy Techniques, Danvers, MA). Images of transverse sciatic nerve sections were captured at 5000× magnification. Counts of myelinated axons were calculated from calibrated TEM images using ImageJ. Investigators were blinded to comparison groups.
Sciatic nerve conduction velocity
Compound action potential (CAP) recordings were performed as previously described62. Briefly, Sciatic nerves were harvested from 9-month-old WT and TG mice and immediately placed in oxygenated Krebs solution. CAPs were measured at room temperature with oxygenated Krebs solution perfused by gravity into the recording chamber containing the nerves. Current was delivered through a stimulating electrode suctioned to one end of the nerve while recordings were taken from a recording electrode suctioned to the other end of the nerve. Conduction velocity was calculated by diving the length of the nerve (distance from one electrode to the other) by the latency between the initiation of stimulus artifact and peak of CAP.
Predicting ADLD structural variant effects on 3D genome with Orca sequence models, identification of CTCF sites and synteny analysis
We generated 3D genome interaction map predictions with Orca33 for all SVs including ADLD-dup, LN-Dup, ADLD-del, ADLD-inv-dup. As the model output values represents log fold over distanced-based expectation, we converted the prediction to log balanced count matrix scale by multiplying the model output by distance-based expectation matrix. The 3D genome predictions for both the wildtype and mutated sequences were visualized as heatmaps.
After we made the prediction for the WT and mutant sequences, we compared the interaction strength between LMNB1 promoter and 19 kb critical region in the mutant and WT samples. In the case of duplication events, we compared the interaction between duplicated critical region and the LMNB1 gene promoter with interaction strength between original copies of these two regions in the WT samples. The effects of the SV on the 3D genome interaction changes were quantified based on Orca predicted log-fold change (Δ) in interaction scores (for H1-ESC cell, Orca-32Mb predictions at 2 Mb scale). When the genomic region(s) of interest were duplicated by the SV, interaction log-fold change scores for each duplicate were computed separately. CTCF sites in the 19 kb critical region and in the lamin B1 gene vicinity were identified using the Find Individual Motif Occurrences (FIMO) online tool63. Synteny was analyzed using the JAX Synteny Browser64 (https://syntenybrowser.jax.org/browser) and the Cinteny tool65 (https://cinteny.cchmc.org).
CUT&RUN analysis
CUT&RUN was performed as previously described under native conditions, using recombinant Protein A-MNase (pA-MNase)66,67. After separating released fragments through centrifugation, fragments isolated were used as input for a library build consisting of end repair and adenylation, NEBNext stem-loop adapter ligation, and subsequent purification with AMPure XP beads (Beckman Coulter). Barcoded fragments were then amplified by 17 cycles of high-fidelity PCR and purified using AMPure XP beads. Libraries were pooled and sequenced on an Illumina NextSeq2000 to a depth of ~10 million mapped reads. Antibodies used for CUT&RUN are listed in Supplementary Table 3.
CUT&RUN data was analyzed as previously described66,67. Paired-end FASTQ files were mapped to the mm10 genome with bowtie2 (options -q -N 1 -X 1000 --very-sensitive-local)68. Mapped reads were filtered for PCR duplicates using Picard tools ([http://broadinstitute.github.io/picard/) and filtered for MAPQ ≥ 10 using SAMtools69. Fragment distribution plots were generated using Picard. Fragment classes corresponding to TF footprints (<120 bp) were generated using a custom awk script and SAMTools. UCSC files were generated from size classed sam files using HOMER70. Reads were converted to bigWig files using deepTools with RPGC read normalization71. Heatmaps were generated using deepTools computeMatrix (options -a 2000 -b 2000 -bs 20 --missingDataAsZero) and plotHeatmap. Peaks were called using MACS272.
Luciferase reporter assay
1 kb regions surrounding the CTCF1 site, the CTCF2 site in the forward and reverse direction, or a control region devoid of CTCF binding sites were amplified using primers designed to incorporate complementary MluI and XmaI overhangs (Supplementary Data 1) using the bacterial artificial chromosome (BAC) RP23-383H12 serving as the DNA template (BACPAC Resource Center, Children’s Hospital Oakland Research Institute, California, USA). All PCR products were subsequently cloned into the pGL3.0 firefly luciferase Promoter Vector (Promega) using MluI and XmaI restriction sites, resulting in the creation of the CTCF1, CTCF2, Reverse CTCF2, and control constructs.
Cells were transiently co-transfected with 5 μg of the different constructs cloned in pGL3.0 firefly along with 0.5 μg of the wild-type Tk-Renilla luciferase control reporter using electroporation with the Amaxa system. Following a 24-hour incubation period, the culture medium was aspirated and replaced with fresh medium. Subsequently, luciferase activity was quantified using the Dual-Luciferase Reporter Assay System (Promega). Promoter activity was assessed in relative units of luciferase (RUL), comparing firefly to Renilla luciferase activities, and normalized to the control.
Statistical analysis
For qPCR/qRT-PCR, western blot, and lamin B1 immunofluorescence intensity data, Student’s t-test was used to compare experimental groups with controls. For multiple comparisons, Dunnett’s test was performed to compare multiple treatment groups with controls in each cohort. Two-tailed Mann-Whitney tests were performed to compare proportions of misshapen nuclei and nuclear circularities in cells from Del-19 mice compared to WT controls. Statistical analysis was performed using Graphpad Prism software.
List of online resources utilized
UCSC genome browser (hg38 version) - https://www.genome.ucsc.edu/
CRISPOR - http://crispor.tefor.net/
HiGlass Hi-C visualization tool - https://higlass.io/
FIMO motif finding tool - https://meme-suite.org/meme/tools/fimo
JAX Synteny Browser (https://syntenybrowser.jax.org/browser
Cinteny tool (https://cinteny.cchmc.org).
Picard tools - http://broadinstitute.github.io/picard/
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genomic data and array data have been uploaded to GEO Omnibus under accession numbers GSE238093 and GSE285491. Additional data is provided in the Supplementary information. Source data are provided with this paper.
Code availability
All Orca-based analyses of structural variant effects on 3D genome organization can be reproduced with code provided in the open-source repository https://github.com/jzhoulab/orca or the webserver https://orca.zhoulab.io/.
References
Raininko, R., Gosky, M.& Padiath, Q. S. LMNB1-related autosomal dominant leukodystrophy. In: GeneReviews((R)) (eds Adam MP, et al.) (2021).
Padiath, Q. S. & Fu, Y. H. Autosomal dominant leukodystrophy caused by lamin B1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol. 98, 337–357 (2010).
Padiath, Q. S. et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet 38, 1114–1123 (2006).
Padiath, Q. S. Autosomal dominant leukodystrophy: a disease of the nuclear lamina. Front Cell Dev. Biol. 7, 41 (2019).
Giorgio, E. et al. Analysis of LMNB1 duplications in autosomal dominant leukodystrophy provides insights into duplication mechanisms and allele-specific expression. Hum. Mutat. 34, 1160–1171 (2013).
Nmezi, B. et al. Genomic deletions upstream of lamin B1 lead to atypical autosomal dominant leukodystrophy. Neurol. Genet 5, e305 (2019).
Giorgio, E. et al. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD). Hum. Mol. Genet 24, 3143–3154 (2015).
Wong, X., Melendez-Perez, A. J. & Reddy, K. L. The Nuclear Lamina. Cold Spring Harb. Perspect. Biol. 14, a040113 (2022).
Gerace, L. & Huber, M. D. Nuclear lamina at the crossroads of the cytoplasm and nucleus. J. Struct. Biol. 177, 24–31 (2012).
Dauer, W. T. & Worman, H. J. The nuclear envelope as a signaling node in development and disease. Dev. Cell 17, 626–638 (2009).
Nmezi, B. et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl Acad. Sci. USA 116, 4307–4315 (2019).
Padiath, Q. S. Lamin B1 mediated demyelination: linking lamins, lipids and leukodystrophies. Nucleus 7, 547–553 (2016).
Fuss, B. et al. Purification and analysis of in vivo-differentiated oligodendrocytes expressing the green fluorescent protein. Dev. Biol. 218, 259–274 (2000).
Rolyan, H. et al. Defects of lipid synthesis are linked to the age-dependent demyelination caused by lamin B1 overexpression. J. Neurosci. 35, 12002–12017 (2015).
Heng, M. Y. et al. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J. Clin. Investig. 123, 2719–2729 (2013).
Alturkustani, M., Sharma, M., Hammond, R. & Ang, L. C. Adult-onset leukodystrophy: review of 3 clinicopathologic phenotypes and a proposed classification. J. Neuropathol. Exp. Neurol. 72, 1090–1103 (2013).
Melberg, A., Hallberg, L., Kalimo, H. & Raininko, R. MR characteristics and neuropathology in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. AJNR Am. J. Neuroradiol. 27, 904–911 (2006).
Finnsson, J., Sundblom, J., Dahl, N., Melberg, A. & Raininko, R. LMNB1-related autosomal-dominant leukodystrophy: clinical and radiological course. Ann. Neurol. 78, 412–425 (2015).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Jung, M. et al. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur. J. Neurosci. 7, 1245–1265 (1995).
Kim, D. et al. A principled strategy for mapping enhancers to genes. Sci. Rep. 9, 11043 (2019).
Kim, D., An, H., Fan, C. & Park, Y. Identifying oligodendrocyte enhancers governing Plp1 expression. Hum. Mol. Genet. 30, 2225–2239 (2021).
Winterstein, C., Trotter, J., & Kramer-Albers, E. M. Distinct endocytic recycling of myelin proteins promotes oligodendroglial membrane remodeling. J. Cell Sci. 121, 834–842 (2008).
Feldmann, A. et al. Transport of the major myelin proteolipid protein is directed by VAMP3 and VAMP7. J. Neurosci. 31, 5659–5672 (2011).
Gobert, R. P. et al. Convergent functional genomics of oligodendrocyte differentiation identifies multiple autoinhibitory signaling circuits. Mol. Cell Biol. 29, 1538–1553 (2009).
Bartosovic, M., Kabbe, M. & Castelo-Branco, G. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39, 825–835 (2021).
Fan, C., Kim, D., An, H. & Park, Y. Identifying an oligodendrocyte enhancer that regulates Olig2 expression. Hum. Mol. Genet. 32, 835–846 (2023).
Krietenstein, N. et al. Ultrastructural details of mammalian chromosome architecture. Mol. Cell 78, 554–565.e557 (2020).
Dixon, J. R., Gorkin, D. U. & Ren, B. Chromatin domains: the unit of chromosome organization. Mol. Cell 62, 668–680 (2016).
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Nott, A. et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019).
Lee, D. S. et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat. Methods 16, 999–1006 (2019).
Zhou, J. Sequence-based modeling of three-dimensional genome architecture from kilobase to chromosome scale. Nat. Genet 54, 725–734 (2022).
Nmezi, B. et al. Development and optimization of a high-content analysis platform to identify suppressors of lamin B1 overexpression as a therapeutic strategy for autosomal dominant leukodystrophy. SLAS Discov. 25, 939–949 (2020).
Ferrera, D. et al. Lamin B1 overexpression increases nuclear rigidity in autosomal dominant leukodystrophy fibroblasts. FASEB J. 28, 3906–3918 (2014).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Ren, G. & Zhao, K. CTCF and cellular heterogeneity. Cell Biosci. 9, 83 (2019).
Ren, G. et al. CTCF-mediated enhancer-promoter interaction is a critical regulator of cell-to-cell variation of gene expression. Mol. Cell 67, 1049–1058 e1046 (2017).
Skene, P. J., Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856 (2017).
Hainer, S. J. & Fazzio, T. G. High-resolution chromatin profiling using CUT&RUN. Curr. Protoc. Mol. Biol. 126, e85 (2019).
Xu, M. et al. CTCF controls HOXA cluster silencing and mediates PRC2-repressive higher-order chromatin structure in NT2/D1 cells. Mol. Cell Biol. 34, 3867–3879 (2014).
Wang, J. et al. CTCF-mediated chromatin looping in EGR2 regulation and SUZ12 recruitment critical for peripheral myelination and repair. Nat. Commun. 11, 4133 (2020).
Laugesen, A., Hojfeldt, J. W. & Helin, K. Role of the polycomb repressive complex 2 (PRC2) in transcriptional regulation and cancer. Cold Spring Harb. Perspect. Med. 6, a026575 (2016).
Ngan, C. Y. et al. Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat. Genet. 52, 264–272 (2020).
Laugesen, A., Hojfeldt, J. W. & Helin, K. Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol. Cell 74, 8–18 (2019).
Cai, Y. et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 12, 719 (2021).
Wang, J. et al. EED-mediated histone methylation is critical for CNS myelination and remyelination by inhibiting WNT, BMP, and senescence pathways. Sci. Adv. 6, eaaz6477 (2020).
Collins, R. L. et al. A structural variation reference for medical and population genetics. Nature 581, 444–451 (2020).
Pang, B., van Weerd, J. H., Hamoen, F. L. & Snyder, M. P. Identification of non-coding silencer elements and their regulation of gene expression. Nat. Rev. Mol. Cell Biol. 24, 383–395 (2023).
Dimartino, P. et al. Structural variants at the LMNB1 locus: deciphering pathomechanisms in autosomal dominant adult-onset demyelinating leukodystrophy. Ann. Neurol. 96, 855-870 (2024).
Fonseca, A. C. et al. The segregation of different submicroscopic imbalances underlying the clinical variability associated with a familial karyotypically balanced translocation. Mol. Cytogenet. 8, 106 (2015).
Coffeen, C. M. et al. Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum. Mol. Genet 9, 787–793 (2000).
Ratti, S. et al. Cell signaling pathways in autosomal-dominant leukodystrophy (ADLD): the intriguing role of the astrocytes. Cell Mol. Life Sci. 78, 2781–2795 (2021).
Concordet, J. P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46, W242–W245 (2018).
Adikusuma, F., Pfitzner, C. & Thomas, P. Q. Versatile single-step-assembly CRISPR/Cas9 vectors for dual gRNA expression. PLoS One 12, e0187236 (2017).
do Rosario, M. C. et al. Variants in the zinc transporter TMEM163 cause a hypomyelinating leukodystrophy. Brain 145, 4202–4209 (2022).
Bey, G. R. & Padiath, Q. S. Enhanced differentiation of the mouse Oli-neu oligodendroglial cell line using optimized culture conditions. BMC Res. Notes 16, 161 (2023).
Emery, B. & Dugas, J. C. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb. Protoc. 2013, 854–868 (2013).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Niemi, J. P. et al. A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J. Neurosci. 33, 16236–16248 (2013).
Labelle-Dumais, C. et al. COL4A1 mutations cause neuromuscular disease with tissue-specific mechanistic heterogeneity. Am. J. Hum. Genet 104, 847–860 (2019).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Kolishovski, G. et al. The JAX Synteny Browser for mouse-human comparative genomics. Mamm. Genome 30, 353–361 (2019).
Sinha, A. U. & Meller, J. Cinteny: flexible analysis and visualization of synteny and genome rearrangements in multiple organisms. BMC Bioinforma. 8, 82 (2007).
Patty, B. J. & Hainer, S. J. Transcription factor chromatin profiling genome-wide using uliCUT&RUN in single cells and individual blastocysts. Nat. Protoc. 16, 2633–2666 (2021).
Hainer, S. J., Boskovic, A., McCannell, K. N., Rando, O. J. & Fazzio, T. G. Profiling of pluripotency factors in single cells and early embryos. Cell 177, 1319–1329 e1311 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Acknowledgements
Q.S.P. would like to acknowledge the Padiath lab members, Dr. Svetlana Yatsenko and Dr. Rob Nicholls for helpful discussions and the ADLD patient families and the ADLD Center for their continuing support. The work was funded by NIH grants R01NS126193, R21NS131906, R33NS104384, R33NS106087, R01NS095884 and ADLD Center grant ADLD-23-001-02 to Q.S.P., Clinical Research Scholar Junior 1 Award from the Fonds de Recherche du Quebec-Santé the New Investigator Salary Award from the Canadian Institutes of Health Research and the Clinical Research Scholar Senior award from the FRQS to G.B. F.G.L. holds a tier 1 Canada Research Chair in tissue-engineering and in vitro brain disease modeling. K.R.S. and P.F.C. are supported by the MRC International Centre for Genomic Medicine in Neuromuscular Disease (MR/S005021/1). Jvd.A. is supported by a Wellcome Clinical Research Career Development Fellowship (219615/Z/19/Z). P.F.C. is a Wellcome Principal Research Fellow (212219/Z/18/Z), who receives support from a Wellcome Collaborative Award (224486/Z/21/Z) and an MRC research grant (MR/S035699/1). Jvd.A. and P.F.C. acknowledge core funding from the MRC to the MRC Mitochondrial Biology Unit (MC_UU_00028/8 and MC_UU_00028/7). This research was supported by the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). S.M.L. and S.J.H. were supported by R35GM133732. This project used the University of Pittsburgh HSCRF Genomics Research Core, RRID: SCR_018301 NextSeq2000, with special thanks to the assistant director, Will MacDonald. This research was supported in part by the University of Pittsburgh Center for Research Computing, RRID:SCR_022735, through the resources provided. Specifically, this work used the HTC cluster, which is supported by NIH award number S10OD028483. The views expressed are those of the author(s) and not necessarily thoseof theNIHRor theDepartment of Health andSocialCare. For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
B.N., G.R.B. and Q.S.P. conceived and designed the overall study with contributions from S.J.H., M.S.G. and J.Z. B.N., G.R.B., S.M.L., T.D.O., A.J., N.H., S.R. and E.L.A. carried out the experiments. K.D. and J.Z. carried our bioinformatic and Orca analyses. D.K. and Y.P. carried out bioinformatic analysis and provided conceptual input. J.R., E.K., N.G., K.S., Jvd.A., P.F.C., S.B.S., F.P., C.T., F.Pe.V., J.S., L.P. carried out patient clinical analysis and provided patient biological material. G.B. and R.R. carried out interpretation of clinical and MRI results. J.K., M.A., R.H. and F.G.-L. provided ADLD brain samples. Q.S.P. and B.N. wrote the paper with contributions from G.B., K.D., J.Z., S.H. and R.R.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks J. Robert Manak, Baoxu Pang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nmezi, B., Rodriguez Bey, G., Oranburg, T.D. et al. An oligodendrocyte silencer element underlies the pathogenic impact of lamin B1 structural variants. Nat Commun 16, 1373 (2025). https://doi.org/10.1038/s41467-025-56378-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56378-9
