
Abstract
Interleukin-36 receptor (IL-36R), belonging to the IL-1 receptor family, is crucial for host defense and tissue repair. Targeting cytokine receptors with low molecular weight (LMW) compounds remains challenging due to their interaction with the large surface area of cytokine. In this study, two encoded library technologies are used to identify LMW molecules binding to IL-36R’s extracellular domain. The mRNA-based display technique identifies 36R-P138, a macrocyclic peptide blocking IL-36R signaling. Importantly, its optimized analog (36R-P192) also effectively suppresses expression of marker genes induced by IL-36 in human skin biopsies. DNA encoded libraries (DEL) screening delivers 36R-D481, a high affinity LMW IL-36R binder, effectively inhibiting IL-36 signaling. X-ray crystallography analysis reveals that both the cyclic peptide and DEL-compound bind to the IL-36R’s D1 domain, potentially disrupting IL-36 cytokine binding. This study demonstrates that it is possible to target a cytokine receptor within the IL-1 receptor family using a small molecule ( < 1000 Da).
Similar content being viewed by others


Introduction
The group of IL-36 cytokines belongs to the IL-1 superfamily and consists of three agonistic isoforms IL-36α, β and γ, and one antagonist named IL-36Ra1,2. The IL-36 receptor (also termed IL1RL2) plays a pivotal role in innate immunity as it is expressed mainly in epithelial barrier tissues such as skin, lung, kidney and colon where the IL-36 cytokines are involved in tissue homeostasis3,4,5. However, under pathological conditions, the aberrant activation of IL-36R can lead to acute and chronic tissue inflammation and damage1,2,3,4,5. A prominent role of IL-36 signaling in skin inflammatory diseases has been well documented6,7,8. Activated keratinocytes were shown to be one of the main sources of IL-36 pro-inflammatory cytokines in the inflamed skin tissue9. Due to the expression of IL-36R by keratinocytes themselves, the IL-36 pathway activation in these cells can lead to an uncontrolled skin inflammation characterized by the establishment of an autocrine amplification of IL-36 signaling in the skin10,11. In addition, IL-36 can activate immune and non-immune cells in the skin, resulting in up-regulation of CXC chemokines and release of IL-23 from dendritic and Langerhans cells2,9,12,13,14. The key-role of IL-36 signaling in skin diseases is also clinically proven, through individuals with mutations in the natural antagonist (IL-36Ra) who develop generalized pustular psoriasis (GPP), a severe subtype of psoriasis15,16,17. Based on the identified carriers of IL-36R loss of function mutations, presenting no increased risk of opportunistic infections (including candida or tuberculosis), inhibition of IL-36R signaling appears to be well tolerated in humans11. Therefore, inhibition of IL-36R signaling represents an attractive approach for the treatment of various inflammatory skin diseases. Recently, a low molecular weight inhibitor of IL-36γ, discovered through high-throughput screening, has been disclosed18. The described Ambrisentan analog A-552 was found to bind selectively to the γ-isoform which may potentially limit its efficacy, since under pathological conditions more than one IL-36 cytokine isoform is over-expressed11,19. In order to maximize the effect of an IL-36 pathway inhibitor in disease settings, our hit finding strategy was focused on the discovery of compounds binding to the IL-36 receptor. Further support for this strategy comes from two clinically tested antibodies (spesolimab and imsidolimab), both binding to IL-36R. In particular, spesolimab has already been approved for the treatment of flares of GPP patients20,21. Our focus was on developing orally bioavailable low molecular weight inhibitors, since therapeutic antibodies might suffer from limitations as they have to be administered either by intravenous infusions or by subcutaneous injections. Another limitation is the potential development of anti-drug antibodies against therapeutic antibodies, which can reduce their efficacy over time. In a clinical trial for GPP with spesolimab, anti-drug antibodies have been observed in 23 out of 50 patients who had received at least one dose of spesolimab22. In addition, systemically administered antibodies typically have lower tissue penetration compared to low molecular weight compounds, which could impact their efficacy23.
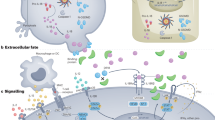
The IL-36R is a type I transmembrane protein consisting of N-terminal signal peptide, three Ig-like domains in the extracellular domain (ECD), named D1, D2 and D3, a single spanning transmembrane domain and a Toll-like and IL-1R (TIR) domain in the cytoplasmic sequence (Fig. 1). Although no crystal structure of the IL-36R ECD with any of the IL-36 ligands could be solved to date, structural modeling based on the known structures of other IL-1R family members suggests that each of the three ECD domains is involved in the recognition of IL-36 cytokines24. Kinetic studies proved that binding of IL-36 agonists to the IL-36R ECD is required for association of the IL-1 receptor accessory protein (IL-1RAcP)25. Formation of the ternary (IL-36/IL-36R/IL-1RAcP) complex is essential for receptor signaling24,25 as it allows for the IL-36R and IL-1RAcP containing TIR domains in the cytoplasm to heterodimerize (Fig. 1). This process is required for the recruitment of the adaptor protein myeloid differentiation primary response protein 88 (MyD88) that leads to the activation of mitogen activated protein kinases (MAPK’s) and the transcription factor NF-κB26.

Signaling through IL-36R relies on the formation of the IL-36 ternary complex, which occurs in two steps. In the first step, one of the agonistic IL-36 cytokines binds to IL-36R. The formed binary complex then associates with IL-1RAcP, resulting in the proximity of the TIR domains on both IL-36R and IL-1RAcP that is essential for downstream signaling through MyD88. Binding of IL-36 agonists and signaling through IL-36R is blocked by the natural antagonist (IL-36Ra) or by IL-36 inhibitors described in this paper.
In the search for IL-36R antagonists, the use of encoded libraries technologies (ELT) was envisioned because they enable screening of up to trillions of chemical entities, thereby significantly enlarging the investigated chemical space compared to the conventional high-throughput methods. Additionally, these technologies have the ability to identify ligands in a function agnostic manner which allows for probing of diverse binding sites and respective mechanism of actions. At Novartis, two ELT-based screening platforms are available, mRNA-encoded cyclic peptide libraries (Peptide Discovery Platform = PDP) and DNA-encoded chemical libraries (DEL)27. PDP mostly focuses on cyclic 10-14mer peptides containing natural and non-natural amino acids. The resulting molecules thus frequently serve as drug discovery tools in challenging drug discovery projects28,29. In contrast, DELs are primarily geared towards the conventional low molecular weight space and readily identify medicinal chemistry starting points30. For both technologies, there is a compound enrichment process, driven by binding of compounds to the protein of interest (POI). The identity of thereby enriched molecules is subsequently determined by sequencing the associated oligonucleotide tag. The respective ligands are then chemically resynthesized and tested for binding and function in follow-up assays.
In this work, we describe the use of encoded library technologies for the discovery of low molecular weight IL-36R antagonists. The X-ray crystallography of the identified IL-36R binders reveals a common binding site on IL-36R, interacting mainly with its D1 domain. To our knowledge, this study presents an example of inhibitor with molecular weight below 1 kDa, that is capable of binding to a member of the IL-1R family and effectively blocking its signaling.
Results
PDP identifies peptides binding to the IL-36R
Due to overexpression of all three IL-36 cytokine isoforms under pathological conditions, our hit finding strategy was focused on identification of binders to IL-36R rather than to a single IL-36 agonist. Along with several constructs of the full-length extracellular domain of the IL-36 receptor, a shorter D1-D2 domain (20–220) construct was prepared as well. Interestingly, the shorter construct behaved better than the entire extracellular domain, in terms of expression, purification, and aggregation, most likely due to the absence of the highly flexible linker between the D2 and D3 domains31. Inspection of the IL-36R structural model24 revealed that the only cavity for binding of low molecular weight compounds is at the concave side of D1-D2 domain. In addition, the published co-crystal structure of the spesolimab-Fab bound to IL-36R D1-D2 demonstrated that this antibody binds exclusively to the D1-D2 domain of the receptor31. Therefore, we have decided to focus on the shorter, D1-D2 domain protein for hit finding using mRNA display technology25 (Fig. 2a). We anticipated that the high diversity of mRNA encoded cyclic peptide libraries, in combination with the RaPID selection process, would generate suitable macrocyclic binders32,33,34,35. Our mRNA libraries encode for macrocycles containing 10-14 amino acids with a fixed C-terminal cysteine. The initiation methionine was replaced with N-chloroacetyl L-Phe for spontaneous macrocyclization after translation. Further, we introduced N-methyl L-Phe, N-methyl L-Gly, N-methyl L-norleucine (NMe-Nle) and N-methyl L-Ala into our codon schemes through flexizyme-mediated tRNA acylations. Two independent selection campaigns were performed with six standard rounds followed by three rounds with increased selection pressure in which we reduced the amount of immobilized target protein and extended the washing times. Those rounds correspondingly aimed towards increasing the off-rates of the retained ligands (koff rounds). In the first selection, we panned the in vitro translated peptides against IL-36R D1-D2. In the second iteration, we included the spesolimab-Fab in selected rounds to discriminate binders to the described binding site of spesolimab31 from those binding other sites of the receptor. For both selections, we observed increasing amounts of recovered cDNA over the course of the selection process with a temporary decrease in the koff rounds. The recovered cDNAs from all the rounds were submitted for Next Generation Sequencing (NGS) to analyze the enriched peptide sequences. We observed a significant enrichment for the cyclic peptide 36R-P138 and related chemotypes (Fig. 2b). 36R-P138 was subsequently chemically synthesized through Fmoc-solid phase peptide synthesis (SPPS) on Rink Amide resin for further characterization. 36R-P138 showed strong binding to IL-36R D1-D2 in SPR with a dissociation constant (Kd) of 1.1 µM (Fig. 2c). As expected, same affinity was measured for its binding towards the entire ECD (D1-D3) of IL-36R (Kd of 1.1 µM, Fig. 2c). In the absence of structural information, we explored the influence on binding of each individual amino acid through an Ala-scan and found that Trp4, Tyr5, Val10 and Trp11 were crucial for binding of the peptide to IL36R D1-D2. After several rounds of optimization, four beneficial amino acid replacements were identified (Table SI2) and their combination led to the discovery of 36R-P192 (Fig. 2b). This optimized peptide demonstrated a significantly improved binding affinity as measured in SPR (Kd of 0.57 nM for D1-D2 and Kd of 0.66 nM for D1-D3, Fig. 2c). The increased binding affinity was mainly driven by slowing down the off-rate by 3 log units (0.98–1.29 × 10−4 s−1 compared to 1.26–1.45 × 10−1 s−1 for 36R-P138, Fig. 2c, d). To get an insight whether 36R-P192 could also interfere with IL-36 agonist binding, IL-36α binding to IL-36R D1-D3 was assessed by SPR, in the absence and presence of 36 R-P192 at 0-300 nM concentration (Fig. 2e). The full-length IL-36R ECD (D1-D3) was required for this purpose as no binding of the IL-36 cytokines to D1-D2 could be determined even at 50 µM concentration. In this experiment, we evaluated the binding of IL-36α at a concentration of 0.6 μM, which is approximately the dissociation constant for IL-36α binding to IL-36R. Conducting the measurements in the presence of different IL36R-P192 concentrations revealed that 36 R-P192 competes with IL-36α for binding to IL-36R, with EC50 of 2.7 nM (Fig. 2e), indicating that the peptide may be an orthosteric inhibitor of the IL-36R.

a Illustrative mRNA display selection cycle used to identify macrocyclic peptides against IL-36R. Selections were performed with and without addition of the spesolimab-Fab. b Chemical structures of peptides 36R-P138 and 36R-P192, amino acid replacements shown in red. c Affinities of 36R-138 and 36R-P192 to IL-36R measured by SPR. d Single cycle kinetics shown for 36R-P192/IL-36R D1-D3 with 36R-P192 concentrations from 0.4 to 100 nM; experimental data in black, fit in red. e SPR measurements of IL-36α/IL-36R D1-D3 with IL-36α concentrations from 0.02 to 2.5 µM (left panel), binding of IL-36α at 0.6 µM to IL-36R D1-D3 in the presence of 36R-P192 (middle panel), quantification of competition of IL-36α binding IL36R D1-D3 at 0.6 µM with 36R-P192 (right panel).
PDP peptides are selective orthosteric inhibitors of the IL-36R in cell-based assays and inhibit IL-36R signaling in ex vivo IL-36 treated skin biopsies
To prove the functionality of the peptides, two orthogonal cell-based assay formats were used. In the first assay, HekBlueTM Null1 cells (InvivoGen) were stably transfected with cDNA, encoding the sequence of the human IL-36R (further named HekBlue_IL-36R). These cells express a secreted embryonic alkaline phosphatase (SEAP) reporter construct under the control of the interferon-β minimal promotor fused to NF-κB and AP-1 binding sites and respond to IL-36 agonists due to co-expression of recombinant IL-36R and endogenously expressed IL-1RAcP. The second cellular assay was based on A431 cells, a human squamous carcinoma cell line, with endogenous expression of IL-36R and IL-1RAcP. Stimulation of these A431 cells with IL-36 agonists leads to the secretion of high amounts of the chemokine IL-8, as quantified by ELISA. Both assays were run under low serum conditions (0.1% FCS) to avoid the influence of plasma protein binding of the measured compounds (Fig. 3a).

a Left panel, HekBlue_IL-36R cells had been stimulated with either 0.03 ng/mL IL-36α or 5 ng/mL IL-1β in the absence or presence of 36R-P138 and 36R-P192, respectively and SEAP was measured after 6 h; right panel, A431 cells had been stimulated with either 10 ng/mL IL-36α or 5 ng/mL IL-1β in the absence or presence of 36R-P138 and 36R-P192, respectively and released IL-8 was measured in cell supernatants after 18 h of incubation. Representative experiment is shown and mean IC50’s with stdv from three independent experiments are indicated in the boxes. b Effect of 36R-P192 in dose response on SEAP expression of HekBlue_IL-36R cells upon stimulation with various concentrations of IL-36α (upper panel), IL-36β (middle panel) and IL-36γ (lower panel). Experiment was done in duplicates and a representative experiment is shown. c Skin biopsies from 4-6 different donors had been pre-incubated for 30 min with either 200 nM spesolimab or various concentrations of 36R-P192 and stimulated with 50 ng/mL IL-36α for 24 h; mRNA was extracted, and marker gene expression was quantified by qPCR and calculated as fold induction compared to unstimulated controls. Statistical analysis was conducted using ordinary one-way ANOVA (multiple comparison) and error bars indicate the standard deviation; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Source data are provided as a Source Data file.
Initial peptide characterization was carried out using the HekBlue_IL-36R SEAP reporter assay and IL-36α as a stimulus. When tested at IL-36α concentrations roughly equivalent to ED50 of the assay, 36R-P138 showed a decent inhibition with an IC50 of around 14 µM, whilst the optimized peptide 36R-P192 displayed a more than 100-fold boost in potency (IC50 of 0.068 µM, Fig. 3a). Since HekBlue_IL-36R cells also express endogenous IL-1 receptor (IL-1R1), these cells do respond also to IL-1 agonists. To demonstrate the specificity of the discovered peptides towards IL-36R, their potential interference with IL-1R1 was tested by measuring the IL-1β induced SEAP activity. Both peptides showed no inhibition of the IL-1β driven activity up to the highest (100 µM) concentration tested (Fig. 3a).
To further confirm their inhibitory function, both peptides were tested also in A431 cells, which express IL-36R, IL-1RAcP and IL-1R1 endogenously. Again, both peptides blocked dose-dependently IL-36R signaling and inhibited IL-36α induced IL-8 release from these cells. Similar to the data obtained from the HekBlue_IL-36R reporter assay, 36R-P138 showed 6.3 µM potency whereas an increased potency was observed for 36R-P192 (IC50 of 83 nM), when tested at roughly ED50 for IL-36α. The specificity towards IL-36R was confirmed again by measuring IL-1R1 signaling, as no inhibition of IL-1β induced IL-8 release assay in the presence of peptides was observed (Fig. 3a). 36R-P192 also effectively inhibited IL-36R signaling even when IL-36β and IL-36γ were used to induce IL-8 release from A431 cells (Fig. SI6).
Since initial SPR experiments suggested, that 36R-P192 is an orthosteric inhibitor of the IL-36R, we tested the peptide in a dose-response experiment using the HekBlue_IL-36R reporter assay with various concentrations of IL-36α, IL-36β and IL-36γ (Fig. 3b). In this assay, 36R-P192 was found to dose-dependently inhibit the activity of all three IL-36 agonists. As expected, the measured potency shifted with increasing amounts of IL-36 agonists, which further supports 36R-P192 to be an orthosteric inhibitor, competing with IL-36 cytokines for binding to the IL-36R (Fig. 3b). Interestingly, the HekBlue_IL-36R reporter assay and IL-8 release from A431 cells showed a remarkable difference in their responsiveness towards IL-36 agonists. Presumably, due to the recombinant overexpression of IL-36R, the SEAP reporter in HekBlue_IL-36R cells responded in a concentration range of 0.001–1 ng/mL to IL-36, whereas higher concentrations (1–100 ng/mL) of IL-36 were required for triggering IL-8 release from A431 cells. It might be, that in the recombinant HekBlue_IL-36R cells due to high receptor expression in combination with an optimized reporter construct, less functional signaling competent IL-36 R complexes are needed to evoke an IL-36 response compared to the more physiological setting in the A431 cells with endogenous receptor expression levels. Despite its competitive behavior and lower concentrations of IL-36 used in the HekBlue_IL-36R reporter assay, 36R-P192 showed similar potency when tested at roughly ED50 for IL-36 in both assays (Figs. 3a and SI 5) demonstrating that 36R-P192 is a potent IL-36R inhibitor under physiological IL-36R expression levels.
Inhibition of IL-36R signaling by 36R-P192 could be demonstrated also in a more complex setting, i.e. in human skin punch biopsies from healthy donors being stimulated with IL-36α (Fig. 3c). IL-36α was shown to drive the expression of a large number of genes in the skin explants. More than 100-fold increase in il36a mRNA expression was measured which confirms the autocrine amplification of IL-36 signaling also in the skin samples. To test the efficacy of 36R-P192 under these conditions, the skin biopsies were pre-incubated with 100, 33, 11 and 3.7 µM of 36R-P192 or 200 nM of the IL-36R blocking antibody spesolimab (positive control) followed by stimulation with 50 ng/mL of IL‑36α. In this experiment, 36R-P192 dose-dependently silenced mRNA expression of marker genes, including expression of il36a, il23 and serpin6 mRNA (Fig. 3c). For a significant silencing of gene expression though, higher concentrations of 36R-P192 were required. The observed decrease in potency, compared to cellular assays, may potentially be caused by high plasma protein binding of the peptide.
DEL screen identifies a high affinity low molecular weight IL-36R binder demonstrating functional inhibition of IL-36R signaling
After successful identification of peptides blocking the interaction between the IL-36R and the IL-36 cytokines, we have also performed a screen based on the DEL technology using the same IL-36R D1-D2 (20-220) construct as for PDP-screen. In contrast to PDP, DEL produces pools of diverse small molecules and accordingly, the identified ligands are more suitable starting points for development of orally bioavailable inhibitors.
To screen the Novartis DEL collections, comprising of 27 individual libraries and 1.9 billion diverse chemical structures, D1-D2 along with the full ECD (D1-D3) were employed. Since the binding site of spesolimab to D1-D2 is known31, the complex of D1-D2 with the spesolimab-Fab was included in the screening setup, to potentially inform on the binding site of the enriched ligands. Sequencing of the selection output, after two rounds of affinity panning, revealed multiple families of related molecules exhibiting good enrichment against the D1-D2 constructs and, to lesser extent, against the D1-D3 construct. The selections against the D1-D2 construct in complex with the spesolimab-Fab yielded an even larger number of highly enriched ligands that were absent in the D1-D2 selection. Therefore, this selection was excluded from further analysis, because it indicated that those molecules possibly bind to the Fab itself or to a pocket induced by the Fab.
Eight clusters enriched against the D1-D2 domain were selected for resynthesis, whereas the PEG-based DNA linker was replaced by a morpholinoethyl group. One cluster including 36R-D481 (Fig. 4a) could be validated by SPR against both, the truncated and the full ectodomain construct. This substance exhibited a high binding affinity for both, IL-36 R D1-D2 (Kd of 77 nM) and D1-D3 (Kd of 105 nM) (Fig. 4a). Importantly, 36R-D481 also demonstrated inhibition of IL-36R signaling in IL-36α and IL-36γ induced SEAP-release assay (IC50 of 1.42 and 6.78 µM, Fig. 4b). However, due to unknown reasons, this compound was unable to block the effect of IL-36β (Fig. 4b). This observation was consistent with the results obtained when measuring IL-36-induced IL-8 release from A431 cells (Fig. SI6), where 36R-D481 effectively inhibited IL-36α and IL-36γ but not IL-36β induced IL-8 release. As for the peptides, 36R-D481 appears to be an orthosteric inhibitor since it demonstrated a competitive inhibition when tested in a dose-response at various IL-36α concentrations (Fig. 4c).

a Chemical structure of 36R-D481 and SPR measurements of 36R-D481 binding to IL-36R; calculated Kd’s for binding either to IL-36R D1-D2 or IL-36R D1-D3 are indicated in the table. b HekBlue_IL-36 R cells had been stimulated with either 0.01 ng/mL IL-36α, 0.01 ng/mL IL-36γ, 0.003 ng/mL IL-36β or 5 ng/mL IL-1β in the absence or presence of 36R-D481 and SEAP was measured after 6 h; representative experiment is shown and mean IC50’s with stdv from three independent experiments are shown in the table. c Effect of 36R-D481 measured in dose response on HekBlue_IL-36R cells in the presence and of various concentrations of IL-36α. Experiment was done in duplicates and a representative experiment is shown. Source data are provided as a Source Data file.
Co-Crystal structure of IL-36R D1-D2 with cyclic peptide 36R-P192
In order to understand the binding mode of 36R-P192, we explored co-crystallization with the IL-36R. Recently, an X-ray structure of BI-655130 (spesolimab) Fab-domain, bound to the D1-D2 IL-36R (20-215) was disclosed (PDB: 6U6U)31. Beside revealing the binding site and mechanism how spesolimab blocks the IL-36R, this study also highlighted the difficulty of obtaining an X-ray structure of the full length ECD IL-36 R, no matter whether the Fab was used for stabilization of the protein or not. Surprisingly, it was found to be challenging to get an X-ray structure of even the more rigid D1-D2 construct without using the Fab. Since the original peptide 36R-P138 was shown to bind to D1-D2 domain with a similar affinity, with and without Fab (KD of 0.36 and 1.1 µM, respectively), the system based on spesolimab-Fab:D1-D2 domain (20–220) was investigated for the co-crystallization with 36R-P192. Crystallization screening and optimization was performed by the vapor diffusion method in sitting drops with a 4-fold molar excess of peptide over the protein-Fab complex. X-ray analysis of the obtained crystals provided a 2.3 Å resolution structure of the ternary complex of 36R-P192, D1-D2 IL-36R and spesolimab-Fab (PDB: 9ETH; Fig. 5a). While the spesolimab-Fab binds to the convex site of the IL-36R31, 36R-P192 occupies its concave, putative cytokine binding site24. The peptide 36R-P192 binds rather on the surface of the protein (Fig. 5b), mainly interacting with the D1-domain. The cyclic peptide itself forms a hairpin-like structure with five internal hydrogen bonds (Fig. 5c). Its conformation is further stabilized by an intramolecular ionic interaction between 3-Py-6-NH2-A5 and E3. An additional intramolecular π-π interaction between 3-Py-6-NH2-A5 and Y8 helps to make the ligand’s conformation even more rigid (Fig. 5c). The contact of 36R-P192 to IL-36R is mediated by extensive hydrogen bond interactions including a bidentate interaction between N29-amide residue and the ligand’s 5-Cl-W4 backbone atoms (Fig. 5d). Interestingly, the 3-Py-6-NH2-A5 not only contributes to the stabilization of the ligand’s conformation, but it is also involved in an ionic interaction with D130 of IL-36R, located at the end of a loop between D1 and D2 domain (Fig. 5d). A second, water-mediated interaction with this loop, is formed by the backbone-carbonyl of 5-Cl-W4 and IL-36R E131-carboxylate (Fig. 5d). The only interaction of the ligand with the D2 domain (H149) is mediated by a π-π stacking using ligand’s W6 (Fig. 5e). Apart from these interactions, the ligand also makes a number of van der Waals contacts to the receptor, especially using its two valines (V2 and V9) as well as the three different tryptophans. Surprisingly, the cyclohexylglycine in position 10 is oriented away from the receptor and as such it is not involved in any lipophilic contact to the receptor despite its profound influence on binding affinity (15-fold increase vs. valine in the same position, Fig. 2b). This indicates that the bulky nature of the cyclohexyl residue may play a role in stabilizing the necessary conformation36 of the peptide inhibitor allowing it to better bind to the IL-36R by slowing down its dissociation (Table SI2).

a 36R-P192 (moccasin) shown as solvent accessible surface bound to IL-36R D1-D2 (blue ribbon) and spesolimab-Fab (ribbon; HC in magenta, LC in pink) complex (PDB: 9ETH). b 36R-P192 (mocassin sticks) binding to IL-36R D1-D2 shown as solvent accessible surface. c Detailed intramolecular interactions within the cyclic peptide structure of 36R-P192. d, e Interactions of 36R-P192 with IL-36R D1-D2. f IL-36R D1-D2 (blue ribbon) with 36R-P192 (moccasin, skin representation) and IL-36γ (green ribbon) (PDB: 4IZE) superimposed to ternary IL1β/IL-1R1/IL-1RAcP (PDB: 4DEP, not shown, only used for the overlay). The modelled structure demonstrates possible clash between 36R-P192 and IL-36γ, thus competing for the same binding site on the concave D1-D2 site of the IL-36 receptor.
Co-Crystal structure of IL-36R D1-D2 with 36R-D481
Crystal structure analysis was also used for gaining insights on binding of the DEL-compound 36R-D481 to IL-36R. Co-crystallization of the compound with D1-D2 (20-220) and spesolimab-Fab provided an X-ray structure with 2.4 Å resolution (PDB: 9ETI). The structure revealed 36R-D481 to bind to the same, concave, cytokine binding site as the peptide 36R-P192 (Fig. 6a). It binds exclusively to the D1-domain, except for a single van der Waals contact of its morpholinoethyl linker to H149 (Fig. 6b). Despite the presence of a deep pocket between the D1-D2 domains (as shown in blue in Fig. 6a), both 36R-D481 and peptide 36R-P192 bind to the receptor surface rather than within the pocket. A notable difference is that 36R-D481 utilizes its Br-phenyl group to bind to a deep, lipophilic pocket as depicted in Fig. 6a. This pocket is absent in the structures of apo-IL-36R or IL-36R bound 36R-P192 (Fig. SI1c). Apart from van der Waals contacts, the phenyl ring in 36R-D481 is engaged in a T-shaped edge-to-face interaction37 with the F44-residue. The bromine atom on this substituent is deeply buried and engaged in a unique, chalcogene interaction to sulfur38,39 of C42, that is involved in a disulfide bridge with C95. The morpholine ring occupies another, albeit very small pocket, while making a hydrogen bond interaction to the N41-backbone-NH (Fig. 6a, b). This interaction was unexpected and potentially of low significance for binding, since it was not present during the DEL-screen. The morpholine-ring binds at the start of a larger/deeper pocket identified between D1-D2 domains (Fig. 6a) which could be used for further scaffold optimization. Besides the morpholine-mediated interaction with the receptor’s β3 sheet, there are two more hydrogen bond interactions with β3-4 loop formed by the ligand’s amide-NH and pyrazole (Fig. 6b). In addition, the ligand’s carboxylate makes an interaction to the β1-2 loop by a hydrogen bond to the backbone-NH of K28. Compared to the ligand-free structure, the β3-4 loop had to move in order to accommodate 36R-D481, resulting in a shift of the F44 residue to allow for a π-π interaction with the pyrazole-ring in 36R-D481 (Figs. 6b and SI1a).

a 36R-D481 (magenta) bound to IL-36R D1-D2 (water accessible surface shown in skin representation, PDB: 9ETI). Pocket between D1 and D2 shown in blue. b Key interactions between 36R-D481 bound to IL36R (blue). c Possible interaction of IL-36R D1-D2 (binding 36R-D481) with IL-36γ (green, PDB: 4IZE) modelled by superimposition of IL36R D1-D2 with the D1-D2 domains of IL-1R1 and IL-36γ superimposed with IL1β in the ternary IL1β/IL-1R1/IL-1RAcP complex (PDB: 4DEP). The modelled complex structure demonstrates a possible clash between 36R-D481 (magenta) and IL-36γ (green).
A comparison of the IL-36R D1-D2 domains between the two new complex structures and the previously published structure (PDB: 6U6U)31, using structural superposition, revealed RMSD values of 1.000 Å for the 36R-P192 complex (177 Cα-atoms) and 0.335 Å for the 36R-D481 complex (167 Cα-atoms). The largest structural differences were observed for the β3-4 loop (Fig. SI1a). Whereas this loop was only marginally shifted in the structure of 36R-D481 compared to the ligand-free structure (PDB: 6U6U), a larger movement was seen in the structure when 36R-P192 was bound (Fig. SI1a). F44 in this loop demonstrated a remarkable flexibility when comparing the three structures. In the 36R-P192 structure, the side chain of F44 is situated at the site where the Br-phenyl substituent of 36R-D481 is located, and as such, the pocket occupied by this substituent is non-existing in the peptide 36R-P192 bound structure. In order to access this pocket, also the side chain of M27 (β1-2 loop) needs to move away. Interestingly, in the ligand-free structure (Fig. SI1a), the side chain of M27 also blocks access to this pocket and therefore, binding of 36R-D481 seems to induce this cryptic pocket. Since the F44 in the 36R-P192 structure already occupies this pocket, the M27 also in this structure is shifted as compared to the ligand-free structure. Besides, the side chain of N29 adopts a different rotamer in the 36R-P192 bound structure, since it is forming a bidentate hydrogen bond to the main chain of the peptide. Finally, the comparison of the buried surface area by these two ligands shows a larger area being covered by the peptide 36R-P192 (864.8 Å2) compared to 36R-D481 (616.9 Å2, Fig. SI1b). This difference may explain the 100-fold slower dissociation (koff) measured for 36R-P192 (0.98-1.29 × 10−4 s−1), compared to 36 R-D481 (6.0-9.0 × 10−2 s−1), whereas the on-rate (kon) was comparable for the two compounds (Figs. 2c and 4a).
Discussion
The IL-36R is expressed on a variety of immune and non-immune cells, explaining the pleiotropic effects of IL-36 agonists and their implications in inflammatory diseases1,2,3,4,5. Since aberrant IL-36 pathway activation plays a particularly important role in skin diseases, IL-36R antibodies were developed for treatment of such diseases and are being tested in the clinic20,22,40. With the aim of identifying orally bioavailable inhibitors of IL-36R signaling, we explored the possibility of inhibiting the IL-36R with a low molecular weight compound. To this end, we first performed a mRNA display selection for IL-36R binders using large libraries of cyclic peptides (PDP). Although peptides are generally considered as less suited for an oral application, new strategies are evolving to develop orally bioavailable peptidic therapeutics by enhancing peptide stability and permeability, or by using permeation enhancers41,42, leading to a steady growth of the list of FDA approved peptidic drugs43. The selections using the truncated IL36R (D1-D2) variant provided 36 R-P138 which demonstrated a moderate binding affinity for the D1-D2 variant as well as for full length IL-36R extracellular domain (Fig. 2c). Gratifyingly, this peptide also efficiently inhibited the IL-36R signaling as assessed using two different cellular assays formats (Fig. 3a). Several rounds of optimization led to the discovery of 36R-P192, which binds to both, the full ECD of IL-36R as well as to its shorter D1-D2 variant, with an excellent affinity (KD of 0.57-0.66 nM, Fig. 2c). SPR as well as cellular experiments suggest that 36R-P192 is an orthosteric inhibitor of the IL-36R, competing with all three IL-36 agonists for binding to its receptor (Figs. 2e and 3b). This finding was further supported by X-ray crystallography, demonstrating that 36R-P192 binds mainly to the IL-36R D1 domain at a site overlapping with the putative binding site of IL-36 cytokines (Fig. 5f). Unfortunately, as described by others31, we did not succeed in the crystallization of the entire IL-36R ECD (alone or in complex with any of the IL-36 cytokines) nor the apo-structure of IL-36R D1-D2, i.e. with no ligand and spesolimab-Fab. Thus, our interpretations are mainly based on structural modeling of IL-36 R/IL-36 complexes using published IL-1R1 structures44,45,46. In this model, the bound peptide was found to clash with the assumed contact site of IL-36 cytokines with IL-36R24,47 (Fig. 5f). It is worth noting that the spesolimab-binding site is distinct to both the assumed cytokine and the co-receptor IL-1RAcP binding sites31. In the absence of the apo-structure of IL-36R D1-D2, i.e. structural data for the protein construct without the Fab, it is difficult to judge whether possible conformational changes are induced by the Fab. However, since 36R-P138 binds simultaneously with the spesolimab-Fab to IL36R D1-D2, their mode of actions might be different. Interestingly, an antagonistic IL-1R1-binding peptide (AF10847), discovered by phage-display technology, was described more than two decades ago48. The X-ray crystal structure of this linear peptide bound to IL-1R1 has been solved49 (PDB: 1G0Y), demonstrating that AF10847 binds to a “closed” conformation of the receptor’s ECD in which the N-terminal D1 domain comes into a close proximity and makes contacts to the C-terminal D3 domain49 (Fig. SI2a). For signaling, cytokines of the IL-1-family receptors bind into an “open” conformation which is required for formation of the ternary signaling complex50. Therefore, stabilization of such a unique “closed” conformation appears to be an attractive approach for inhibiting the IL-1R1, as it makes it impossible for the cytokines to bind into such a conformation. In the case of 36R-P192 however, it is rather unlikely that this cyclic peptide could induce a similar structural rearrangement of the IL-36R ECD. The presence of the cyclic peptide within the D1 domain significantly reduces the likelihood of the D1 domain attracting the D3 domain to achieve this type of “closed” conformation (Fig. SI2b). However, it cannot be definitively ruled out that the cyclic peptide may still contribute to stabilizing a closed receptor conformation by interacting with the D3 domain.
Based on the encouraging results from the PDP screen, our in-house DEL platform was used to screen for low molecular weight compounds binding to the IL-36R. Surprisingly, only a single compound (36R-D481) could be confirmed as a hit despite screening more than a billion of compounds. This molecule turned out to bind the IL-36R full ECD (D1-D3) as well as D1-D2 with high affinity (Fig. 4a). More importantly, it also showed a functional inhibition of IL-36R signaling in the HekBlue_IL-36R SEAP assay (Fig. 4b) and A431 cells (Fig. S1.6). We were pleased with the impressive potency exhibited by the low molecular weight hit, particularly considering that 36R-D481 has not undergone any optimization, apart from the substitution of the DNA-linker with a morpholinoethyl group. As for the peptide 36R-P192, this compound demonstrated a competitive behavior in the HekBlue_IL-36R SEAP reporter assay, indicating that this compound is also an orthosteric inhibitor of the IL-36R (Fig. 4c). The co-crystal structure of 36R-D481 bound to Fab-complexed IL-36R D1-D2 revealed a highly overlapping binding site on D1-D2 with 36R-P192 (Fig. SI1b), whilst binding of 36R-D481 was found to be restricted to the D1 domain. Compound 36R-D481 exhibits a distinct binding mode, where its Br-phenyl substituent occupies a pocket that is absent in both the 36R-P192 structure and the published structure of IL-36R D1-D2 with spesolimab-Fab alone (PDB: 6U6U)31, indicating an induced-fit conformation. Using a model of the IL-36γ/IL-36R complex, compound 36R-D481 was shown to occupy a part of the binding site on IL-36R used by IL-36 cytokines (Fig. 6c), which may explain its antagonistic effect. However, the precise mechanism of this antagonism remains unclear due to the lack of detailed structural information regarding the binding of IL-36 cytokines to the IL-36R. For example, while 36R-D481 was found to be able to block the IL-36α and IL-36γ induced IL-36R signaling with low micromolar potency, no inhibition up to 100 µM concentration was observed when IL-36β was used. In contrast, the peptide 36R-P192 inhibits the action of all isoforms with a similar potency. While IL-36β displays the highest affinity for the IL-36 R/IL-1RAcP complex51 compared to IL-36α and IL-36γ, this marginal difference may not fully explain the observed discrepancies. However, structural modeling suggests that IL-36α and IL-36γ interact similarly with the D1 domain, whereas the positioning of IL-36β is distinct24. Consequently, displacing IL-36β with 36R-D481 might prove to be more challenging in comparison to the larger peptide 36R-P192 that increases the likelihood of potential interference with the binding of IL-36β to the D1 domain of IL-36R. To our knowledge, the work presented here describes the discovery of an orthosteric IL-36 R inhibitors that compete for binding with IL-36 agonists. In particular, 36R-D481 represents a unique example of a compound with low molecular weight (< 1000 Da) targeting a cytokine receptor of the IL-1R family. The recent advances in hit finding technologies enable the rapid project progress from peptidic tools to low molecular weight starting point by employing complementary encoded library technologies in a subsequent fashion. First, the peptide ligands confirmed that binding to IL36R D1-D2 is sufficient to obtain orthosteric inhibition. Second, the DEL derived molecules translate this learning into a chemical space commonly suited for orally bioavailable drugs. In summary, this work demonstrates that it is possible to identify small molecules blocking a cytokine-receptor protein-protein interaction using state-of-the-art hit finding strategies.
Methods
Ethics statement
Human abdominal skin discards from plastic surgery were either obtained from the local University Hospital Basel or from ALPHENYX. Ethical approvals were provided by Comité de Protection des Personnes Sud Méditerranée for Alphenyx (# AC-2019-3567) and Ethikkommission Nordwest und Zentralschweiz, Basel, Switzerland (EKNZ # 2019-02188). All samples were obtained with written informed consent from the participants in accordance with the Declaration of Helsinki principles and the Swiss Human Research Act.
Protein expression and purification
Human IL-36α (aa 6–158/Q9UHA7), IL-36β (aa 5-157/Q9NZH7-2) and IL-36γ (aa 18-169/Q9NZH8) were all expressed in E.coli with an N-terminal His6-GS-Sumo tag. E. coli (BL21;DE3) expressions were induced overnight at 20 oC with 0.1 mM IPTG and the cell pellets from 1 L culture were resuspended in 8 volumes of 50 mM NaH2PO4, containing 300 mM NaCl and 20 mM imidazole (buffer A). EDTA-free protease inhibitor tablets were added (cOmpleteTM Mini, Roche, 1 tablet/50 mL) and the suspension lysed by 3 passes through a microfluidizer at 20,000 psi. The lysates were centrifuged at 16,000 g for 45 min and the supernatants subsequently filtered (0.45 mM) before application to a 5 mL His-Trap column (Cytiva), which had been equilibrated with 4 column volumes of buffer A. The column was subsequently washed with 10 column volumes of buffer C and then eluted with a 0–100% gradient of buffer B (buffer A containing 0.5 M imidazole) over 15 column volumes. The eluted proteins were pooled following analysis by SDS-PAGE, ULP-1 protease was added (1:60 ratio) before dialysis against 50 volumes of buffer A. Following filtration (0.45 mM), the pools were reapplied to the His-Trap column under the same conditions. This time the unbound fraction was collected, containing the mature processed IL-36 proteins. The non-cleaved proteins and the His-Sumo tags remained on the column and were eluted later. The mature IL-36 proteins were concentrated to ~10 mL using a 3000MWCO centrifugal concentrator for loading onto an XK26/600 column of Superdex 30 (Cytiva), which had been equilibrated with PBS pH 7.4. Eluted proteins were again analyzed by SDS-PAGE and the main peaks, containing monomeric proteins were pooled, aliquoted, and frozen at −80 oC.
Human IL-36R (ILRL2/Q9HB29) proteins
IL-36R (aa 1-333)-His6-Avi and IL-36R (aa 1-220)-His6-Avi were expressed transiently using a pACM vector in HEK-ExpiGnti- cells. 24 h post-transfection, sodium butyrate was added to 2 mM, and the cultures incubated for a further 3 days, the cell suspensions containing either the mature IL-36R (20-333)-His6-Avi or the mature IL-36R (20-220)-His6-Avi were centrifuged for 20 min at 3,500 rpm to remove cells and the resultant supernatants filtered through a 0.45 mm Sartobran capsule (Sartorius). Purification was carried out using a 5 mL HisTrap crude column (Cytiva) which had been equilibrated with 50 mM NaH2PO4 pH 8.0 containing 300 mM NaCl, 20 mM imidazole and 10 % v/v glycerol (buffer C). The unbound proteins were washed out with 6 column volumes of buffer C and the bound protein eluted with a gradient of 0-100 % buffer D (buffer C containing 300 mM imidazole) over 8 column volumes. Fractions were analyzed by SDS-PAGE and the appropriate fractions pooled, concentrated to approximately 4 mL and loaded onto a Superdex75 XK16/600 column (Cytiva) equilibrated with 50 mM Tris pH 8.0, 50 mM NaCl, 10% (v/v) glycerol. Concentrated IL-36R proteins were in-vitro biotinylated overnight at 4 oC by addition of BirA to 1/50 (w/w), MgCl2 and ATP each to 10 mM and biotin to 300 µM. The separate pools were then concentrated to ~5 mL using a 10,000 MWCO centrifugal concentrator and applied to an XK16/600 column of Superdex 200 (Cytiva), run and equilibrated with 50 mM Tris-HCl, 50 mM NaCl pH 8.0. Peak fractions were analyzed by SDS-PAGE, pooled, aliquoted, and frozen at −80 oC.
The VH/VL regions of spesolimab were cloned into CVM-promoter-driven expression plasmids already featuring the constant regions of human IgG1 and kappa, respectively. The antibody was produced by transient transfection in the HEK293-6E cell line. Purification was done by affinity chromatography on Protein A Sepharose followed by a polishing step by size exclusion chromatography. To generate the Fab, a papain digestion of the full IgG was carried out and followed by another size exclusion step to remove the Fc-part. Both molecules were characterized for integrity and homogeneity by SDS-PAGE and mass spectrometry analysis.
Surface plasmon resonance (SPR)
IL-36R (20-220)-His6-Avi and IL-36R (20-333)-His6-Avi, both in vitro biotinylated, were immobilized on a streptavidin chip (Serie S sensor chip SA, Cytiva) by the affinity capture method. To immobilize these receptors on the chip, 50 µg/mL protein was injected at 10 µL/min for 300 s and immobilized until approximately 4000 RU. The immobilization and running buffer contain 10 mM HEPES (pH 7.5), 150 mM NaCl, 3 mM EDTA, and 0.05% Tween20. All SPR experiments were conducted at 25 °C. A Biacore T200 instrument (Cytiva) was used to characterize direct binding exhibiting fast kinetics using Standard Kinetics method. The peptides were diluted from 10 µM to 10 nM serial concentrations in running buffer with final 2% DMSO, and a volume of 166 µl was injected for 200 s at a rate of 50 µL/min over all four flow cells of the chip. 36R-D481 was diluted from 5 µM to 1 nM serial concentrations in running buffer with final 2% DMSO, and a volume of 100 µL was injected for 120 s at a rate of 50 µL/min over all four flow cells of the chip. A Biacore 8 K instrument (Cytiva) using the Single Cycle Kinetics method was employed to measure 36R-P192 due to its long off-rate. Nine increasing concentrations (0.4 to 100 nM) were injected at a rate of 50 µL/min successively for 240 s, followed by a final dissociation phase of 10,000 seconds. IL-36α binding to IL-36R (20-333) was assessed using ABA injection method (A: 36R-P192 and B: IL-36α). IL-36α concentration ranged from 0.02 to 2.5 µM and the concentration of IL-36R-P192 was kept at a fixed concentration for each channel at 300, 100, 30, 10, 3, 1 and 0 nM. ABA injection parameters are 336 s for pre-analyte, 60 s for post analyte and 60 s for injection B, at a rate of 50 µL/min. Sensorgrams were analyzed using the Biacore Evaluation Software 2.0 for T200 and Biacore Insight 5.0.18.22102 for 8 K (Cytiva). The kinetic rate constants (Kon and Koff) were determined using a 1:1 Langmuir binding model. Solvent correction and double referencing were applied to all data to ensure accurate determination of binding affinities. The data supporting the presented SPR data can be found in Source Data file as well as in the Supplementary Data 1 file.
PDP screen
The peptide coding region of the naïve mRNA template for the IL-36R selection campaigns contained a 10-13 NNU codon region to encode the random sequence which is flanked by the 5’ initiator AUG codon and a 3’ eight codon region to encode CGSGSGS followed by an amber stop codon (TAG). The in vitro translation system was reprogrammed through Flexizyme-mediated genetic code reprogramming as previously described32,34. Briefly, N-chloroacetyl L-Phe was used in place of the initiator methionine. For elongator positions, the natural amino acids Ser, Tyr, Pro, His, Arg, Asn, Val, Asp, Gly and Cys along with the non-natural elongators N-Methyl-Phe, N-Methyl-Gly, N-Methyl-Ala and N-Methyl-Nle were used. Applying this reprogrammed translation system to the aforementioned mRNA template affords a 12 to 15 amino acid thioether macrocyclic peptide library with each peptide containing a C-terminal Gly-Ser linker. Starting with an initial round containing >1013 unique cyclic peptides, IL-36R D1-D2-specific binders were enriched using His-avi-tagged IL-36R D1-D2 (20-220)-bound streptavidin conjugated magnetic beads and non-specific binders were removed using the streptavidin-conjugated magnetic beads only. A second selection was performed adding the spesolimab-Fab in addition to the IL-36R construct to the selected selection round. Library-IL-36R D1-D2 +/- Fab binding steps were performed in an iterative manner while the binding stringency was increased in latter rounds through temperature, incubation times and/or wash steps. All selection rounds were submitted for NGS to analyze enriched peptides sequences. Synthesis of 36R-P138 and 36R-P192 are described in the supplementary information.
DEL screen
DEL-selections were performed in analogy to Satz et al.52 20-40 µg of the biotinylated Avi-tagged target proteins were immobilized on 5 µL Streptavidin PhyTip columns (PhyNexus, Cat. No. PTR 92-05-05) in Hepes-buffered saline (HBS) pH 7.4, 0.05% Tween20, 1 mg/mL sheared Salmon sperm DNA (Thermo Fisher Scientific, Cat. No. 15632011). Residual protein was removed in several subsequent washes. The DEL pools were panned against the immobilized targets as two separate pools of a total of either 1 nmol (1.6 billion members) or 340 pmol (305 million members). The non-binding molecules were removed in several subsequent washes, the binding molecules were eluted by heat denaturation of the protein. The eluate was panned a second time against freshly immobilized target protein and the selection was repeated. The molecules in the eluate after the second round of selection were quantified by qPCR (108–109 molecules) and subjected to Illumina sequencing. Approximately 150 million reads were obtained per selection sample. Synthesis of 36R-D481 is described in the supplement information.
Cellular assays and skin biopsies
Stably transfected HekBlue_IL-36R cells were generated using the coding sequence of IL-36R (IL1RL2; Q9HB29) cloned into the vector backbone of pD2529 vector (Addgene). After transfection into HekBlue Null1TM cells (InvivoGen, Cat. No. hkb-null1), single cell clones were selected by cell sorting after puromycin selection. For the assay, 10.000 cells/well were seeded on poly-lysine coated 384-well plates in OptiMem medium without phenol red (ThermoFisher, Cat. No. 11058021) containing 0.1% FCS. Cells were cultured overnight. Next morning compounds were added 15 min prior to cell stimulation with either IL-36 or IL-1β. SEAP was measured 6 h later by adding QUANTI-Blue™ (InvivoGen, Cat. No. rep-qbs) and measuring OD at 635 nM.
To measure IL-8 release from A431 cells (DSZM No ACC91), 30.000 cells/well were seeded on 96-well plates in DMEM containing 10% FCS. After cell adherence, medium was changed to DMEM containing 0.1% FCS and compounds were added 15 min prior to cell stimulation with either IL-36 or IL-1β. After 18 h of incubation, cell supernatants were collected and IL-8 was quantified using a commercial ELISA following the protocol of the provider (BioLegend, Cat. No. 431504). Curve fit and IC50 calculations were carried out using a non-linear curve fit with GraphPad Prism 9.5.1 software.
4 mm punch biopsies from human abdominal skin were placed in 48well plates containing IMDM medium (Life Technology, Cat. No. 21056023)/10% KnockOut Serum Replacement (Life Technology, Cat. No. 10828010) and 1% Pen-Strep (Life Technologies, Cat. No. 15140). Spesolimab (200 nM) or 36R-P192 were added 30 min prior to the addition of 50 ng/mL f.c. IL-36α. 24 h later biopsies were transferred into 48-well plates containing 0.4 mL RNAlater (Quiagen, Cat. No. 76106) and kept at −80 °C until RNA extraction. Tissue homogenization was done using a Precellys® homogenizer and Precellys® tissue homogenizing kit CKMix (Cat. No. P000918-LYSK0-A). RNA was extracted using a RNeasy miniKit (Quiagen, Cat. No. 74104) and RNA was quantified using a NanoDrop spectrophotometer. Reverse transcription was done according to the Applied Biosystems manual and by using the HighCapacity cDNA ReverseTranscription kit (Applied Biosystems, Cat. 4368813. qPCR was performed using Applied Biosystems QuantStudio 7 Pro real-time PCR systems (Thermo Fisher), its integrated design and analysis software and the TaqMan gene expression Mastermix (Applied Biosystems, Cat. 4369016). All FAM-MGB dye labelled TaqMan primers were obtained from Thermo Fisher: IL-36α (Cat. No. hs-00205367), IL-23 (Cat. No. hs-00900828), SERPINA6 (Cat. No. hs-01547823). mRNA levels were normalized to the housekeeper gene RPLP0 (hs-F99999902). Data were calculated as fold induction by setting the unstimulated control value to one and all other values were calculated according to this reference. Statistical analysis was done with GraphPad Prism 9.5.1 software using ordinary one-way ANOVA (multiple comparison).
X-ray structure and analysis
Complex preparation: IL-36R (20-220) was mixed with a 1.5 molar excess of anti-IL-36R Fab fragment (spesolimab) and incubated overnight at 4 oC. The sample was concentrated for size-exclusion chromatography as described above. An XK16/600 Superdex 200™ was equilibrated with PBS pH 7.2, containing 0.2 M sucrose, 5% v/v glycerol, 0.01% w/v CHAPS and 1 mM TCEP. The sample was run on the column as before and 2 mL fractions collected. Two major peaks were observed corresponding to complex, followed by excess Fab fragment. Fractions of the complex were pooled following SDS-PAGE analysis, resulting in 12.5 mL of complex, containing 30 mg of protein. 7.5 mL of this pool were concentrated to 0.5 mL to give a protein concentration of 23.4 mg/mL which was used for crystallization screening.
Crystallization, X-ray data collection and structure determination: All crystals were grown by using the sitting-drop vapor-diffusion method at 20 °C after mixing 200 nL protein complex solution with 200 nL reservoir solution (Mosquito robot dispenser, TTP LabTech). Compounds have been directly added to the purified Fab-protein complex in SEC buffer at 23.4 mg/mL in molar excess (8-fold for 36R-D481; 4-fold for 36R-P192) from stock solutions of 50 mM in DMSO-d6/D2O (9:1). Crystals for the 36 R-D481 complex were obtained in 1.6 M sodium citrate tribasic dihydrate pH 6.5. Prior to shock-cooling the crystals for data collection they were soaked in reservoir solution containing 10% glycerol and 1 mM 36R-D481 for 30 s. Crystals of the 36R-P192-complex were grown using 0.1 M citric acid pH 5 and 3.1 M ammonium sulfate as reservoir solution. These crystals were directly flash-cooled in liquid nitrogen for data collection. X-ray diffraction data were collected at the Swiss Light Source (SLS, beamline X10SA) using a Eiger pixel detector. The data were processed with XDS53 and autoPROC54 making use of the anisotropic data handling as implemented in STARANISO (https://staraniso.globalphasing.org/cgi-bin/staraniso.cgi, Global Pashing Ltd.). The structures were solved by molecular replacement with PHASER55 using as search model the coordinates of the published IL-36 R (20-215):Fab complex (PDB: 6U6U)31. The software programs COOT56 and BUSTER (BUSTER version 2.11.8, Global Phasing Ltd.) were used for iterative rounds of model building and structure refinement. PyMol (retrieved from http://www.pymol.org/pymol) and ICM-Pro (Molsoft L. L. C.) standard commands were used for structural alignments and solvent accessible surface calculations. MOE (2022.02; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022) was used for generation of interaction maps. The refined coordinates of the complex structure have been deposited in the Protein Data Bank (www.wwpdb.org) under accession code 9ETH and 9ETI.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The crystallographic coordinates of the complexes between IL-36R D1-D2/spesolimab-Fab and compounds 36R-P192 and 36R-D481 were deposited in the PDB database and can be accessed under accession codes 9ETH and 9ETI. Source data are provided with this paper.
References
Gabay, C. & Towne, J. E. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J. Leukoc. Biol. 97, 645–652 (2015).
Bassoy, E. Y., Towne, J. E. & Gabay, C. Regulation and function of interleukin-36 cytokines. Immunol. Rev. 281, 169–178 (2018).
Xu, P., Guan, H., Xiao, W. & Sun, L. The role of IL-36 subfamily in intestinal disease. Biochem Soc. Trans. 50, 223–230 (2022).
Buhl, A. L. & Wenzel, J. Interleukin-36 in Infectious and Inflammatory Skin Diseases. Front Immunol. 10, 1162 (2019).
Manzanares-Meza, L. D., Valle-Rios, R. & Medina-Contreras, O. Interleukin-1 Receptor-Like 2: One Receptor, Three Agonists, and Many Implications. J. Interferon Cytokine Res 42, 49–61 (2022).
Hovhannisyan, Z. et al. Enhanced IL-36R signaling promotes barrier impairment and inflammation in skin and intestine. Sci. Immunol. 5, eaax1686 (2020).
Iznardo, H. & Puig, L. IL-1 Family Cytokines in Inflammatory Dermatoses: Pathogenetic Role and Potential Therapeutic Implications. Int J. Mol. Sci. 23, 9479 (2022).
Hwang, J., Rick, J., Hsiao, J. & Shi, V. Y. A review of IL-36: an emerging therapeutic target for inflammatory dermatoses. J. Dermatol. Treat. 33, 2711–2722 (2022).
Hernandez-Santana, Y. E., Leon, G., St Leger, D., Fallon, P. G. & Walsh, P. T. Keratinocyte interleukin-36 receptor expression orchestrates psoriasiform inflammation in mice. Life Sci. Alliance 3, e201900586 (2020).
Elias, M. et al. IL-36 in chronic inflammation and fibrosis - bridging the gap? J. Clin. Invest 131, e144336 (2021).
Mahil, S. K. et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci. Transl. Med 9, eaar6600 (2017).
Goldstein, J. D. et al. IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci. Alliance 3, e202000688 (2020).
Blumberg, H. et al. IL-1RL2 and its ligands contribute to the cytokine network in psoriasis. J. Immunol. 185, 4354–4362 (2010).
Scholz, G. M., Heath, J. E., Walsh, K. A. & Reynolds, E. C. MEK-ERK signaling diametrically controls the stimulation of IL-23p19 and EBI3 expression in epithelial cells by IL-36gamma. Immunol. Cell Biol. 96, 646–655 (2018).
Onoufriadis, A. et al. Mutations in IL36RN/IL1F5 Are Associated with the Severe Episodic Inflammatory Skin Disease Known as Generalized Pustular Psoriasis. Am. J. Hum. Genet 89, 432–437 (2011).
Hussain, S. et al. IL36RN mutations define a severe autoinflammatory phenotype of generalized pustular psoriasis. J. Allergy Clin. Immunol. 135, 1067–1070 e1069 (2015).
Tauber, M. et al. IL36RN Mutations Affect Protein Expression and Function: A Basis for Genotype-Phenotype Correlation in Pustular Diseases. J. Invest Dermatol 136, 1811–1819 (2016).
Todorovic, V. et al. Small Molecule IL-36gamma Antagonist as a Novel Therapeutic Approach for Plaque Psoriasis. Sci. Rep. 9, 9089 (2019).
Ding, L., Wang, X., Hong, X., Lu, L. & Liu, D. IL-36 cytokines in autoimmunity and inflammatory disease. Oncotarget 9, 2895–2901 (2018).
Blair, H. A. Spesolimab: First Approval. Drugs 82, 1681–1686 (2022).
Burden, A. D. Spesolimab, an interleukin-36 receptor monoclonal antibody, for the treatment of generalized pustular psoriasis. Expert Rev. Clin. Immunol. 19, 473–481 (2023).
Bachelez, H. et al. Trial of Spesolimab for Generalized Pustular Psoriasis. N. Engl. J. Med. 385, 2431–2440 (2021).
Shah, D. K. & Betts, A. M. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs 5, 297–305 (2013).
Yi, G. et al. Structural and Functional Attributes of the Interleukin-36 Receptor. J. Biol. Chem. 291, 16597–16609 (2016).
Zhou, L. et al. Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation. J. Biol. Chem. 293, 403–411 (2018).
Zhou, L. & Todorovic, V. Interleukin-36: Structure, Signaling and Function. Adv. Exp. Med Biol. 21, 191–210 (2021).
Ottl, J., Leder, L., Schaefer, J. V. & Dumelin, C. E. Encoded Library Technologies as Integrated Lead Finding Platforms for Drug Discovery. Molecules 24, 1629 (2019).
Hayashi, Y., Morimoto, J. & Suga, H. In vitro selection of anti-Akt2 thioether-macrocyclic peptides leading to isoform-selective inhibitors. ACS Chem. Biol. 7, 607–613 (2012).
Cremosnik, G. S. et al. mRNA Display Identifies Potent, Paralog-Selective Peptidic Ligands for ARID1B. ACS Chem. Biol. 19, 1142–1150 (2024).
Goodnow, R. A. Jr., Dumelin, C. E. & Keefe, A. D. DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov. 16, 131–147 (2017).
Larson, E. T. et al. X-ray crystal structure localizes the mechanism of inhibition of an IL-36R antagonist monoclonal antibody to interaction with Ig1 and Ig2 extra cellular domains. Protein Sci. 29, 1679–1686 (2020).
Goto, Y., Katoh, T. & Suga, H. Flexizymes for genetic code reprogramming. Nat. Protoc. 6, 779–790 (2011).
Goto, Y. & Suga, H. Flexizymes as a tRNA acylation tool facilitating genetic code reprogramming. Methods Mol. Biol. 848, 465–478 (2012).
Ishizawa, T., Kawakami, T., Reid, P. C. & Murakami, H. TRAP display: a high-speed selection method for the generation of functional polypeptides. J. Am. Chem. Soc. 135, 5433–5440 (2013).
Goto, Y. & Suga, H. The RaPID Platform for the Discovery of Pseudo-Natural Macrocyclic Peptides. Acc. Chem. Res 54, 3604–3617 (2021).
Jwad, R., Weissberger, D. & Hunter, L. Strategies for Fine-Tuning the Conformations of Cyclic Peptides. Chem. Rev. 120, 9743–9789 (2020).
Meyer, E. A., Castellano, R. K. & Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int Ed. Engl. 42, 1210–1250 (2003).
Wilcken, R. et al. Addressing Methionine in Molecular Design through Directed Sulfur-Halogen Bonds. J. Chem. Theory Comput 7, 2307–2315 (2011).
Ford, M. C., Saxton, M. & Ho, P. S. Sulfur as an Acceptor to Bromine in Biomolecular Halogen Bonds. J. Phys. Chem. Lett. 8, 4246–4252 (2017).
Warren, R. B. et al. Imsidolimab, an anti-interleukin-36 receptor monoclonal antibody, for the treatment of generalized pustular psoriasis: results from the phase II GALLOP trial. Br. J. Dermatol 189, 161–169 (2023).
Brayden, D. J., Hill, T. A., Fairlie, D. P., Maher, S. & Mrsny, R. J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 157, 2–36 (2020).
Zizzari, A. T., Pliatsika, D., Gall, F. M., Fischer, T. & Riedl, R. New perspectives in oral peptide delivery. Drug Discov. Today 26, 1097–1105 (2021).
Sharma, K., Sharma, K. K., Sharma, A. & Jain, R. Peptide-based drug discovery: Current status and recent advances. Drug Discov. Today 28, 103464 (2023).
Schreuder, H. et al. A new cytokine-receptor binding mode revealed by the crystal structure of the IL-1 receptor with an antagonist. Nature 386, 194–200 (1997).
Vigers, G. P., Anderson, L. J., Caffes, P. & Brandhuber, B. J. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1beta. Nature 386, 190–194 (1997).
Thomas, C., Bazan, J. F. & Garcia, K. C. Structure of the activating IL-1 receptor signaling complex. Nat. Struct. Mol. Biol. 19, 455–457 (2012).
Gunther, S. & Sundberg, E. J. Molecular determinants of agonist and antagonist signaling through the IL-36 receptor. J. Immunol. 193, 921–930 (2014).
Yanofsky, S. D. et al. High affinity type I interleukin 1 receptor antagonists discovered by screening recombinant peptide libraries. Proc. Natl Acad. Sci. USA 93, 7381–7386 (1996).
Vigers, G. P. et al. X-ray crystal structure of a small antagonist peptide bound to interleukin-1 receptor type 1. J. Biol. Chem. 275, 36927–36933 (2000).
Fields, J. K., Gunther, S. & Sundberg, E. J. Structural Basis of IL-1 Family Cytokine Signaling. Front Immunol. 10, 1412 (2019).
Towne, J. E. et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J. Biol. Chem. 286, 42594–42602 (2011).
Satz, A. L. et al. DNA-encoded chemical libraries. Nat. Rev. Methods Prim. 2, 3 (2022).
Kabsch, W. Xds. Acta Crystallogr D. Biol. Crystallogr 66, 125–132 (2010).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D. Biol. Crystallogr 67, 293–302 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl Crystallogr 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Acknowledgements
We would like to thank Sascha Mueller, Oleksiy Parkhomenko, Nicole Battaglia, Isabelle Adam, Sabine Rudolph, Felix Freuler and Andreas Boettcher for technical support. We would also like to thank Corinne Marx for HRMS and Thomas Lochmann for NMR measurements. We also acknowledge the project support by Christian Bruns, Rene Hersperger, Ulrich Hommel, Frédéric Berst and Andreas Marzinzik.
Author information
Authors and Affiliations
Contributions
J.V. conceived project and manuscript writing; G.C. conceived project, supervising experiments and manuscript writing; C.S. analysis of crystal structures, manuscript writing; P.M. synthesis of IL36R-D482; R.F. peptide synthesis; E.W. SPR and protein crystallization; P.R. supervised experiments; M.S. structural modeling; C.K. protein expression, purification and crystallization; S.L. protein crystallization; R.K. cellular assays; S.S. skin biopsies and mRNA expression; D.M-R. cellular assays; S.P., R.C. production of spesolimab and Fab thereof; P.L.,K.K., P.E., T.R.,J.G. conceived project; C.D. conceived project, supervised DEL screen and manuscript writing; G.M-B. conceived project, supervised experiments and manuscript writing
Corresponding authors
Ethics declarations
Competing interests
All authors are or were at the time of their involvement with the research employees of Novartis BioMedical Research and may hold stocks in Novartis. There are no more competing interests.
Peer review
Peer review information
Nature Communications thanks Sebastian Günther and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Velcicky, J., Cremosnik, G., Scheufler, C. et al. Discovery of selective low molecular weight interleukin-36 receptor antagonists by encoded library technologies. Nat Commun 16, 1669 (2025). https://doi.org/10.1038/s41467-025-56601-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56601-7
