
Abstract
Sample relatedness is a major confounder in genome-wide association studies (GWAS), potentially leading to inflated type I error rates if not appropriately controlled. A common strategy is to incorporate a random effect related to genetic relatedness matrix (GRM) into regression models. However, this approach is challenging for large-scale GWAS of complex traits, such as longitudinal traits. Here we propose a scalable and accurate analysis framework, SPAGRM, which controls for sample relatedness via a precise approximation of the joint distribution of genotypes. SPAGRM can utilize GRM-free models and thus is applicable to various trait types and statistical methods, including linear mixed models and generalized estimation equations for longitudinal traits. A hybrid strategy incorporating saddlepoint approximation greatly increases the accuracy to analyze low-frequency and rare genetic variants, especially in unbalanced phenotypic distributions. We also introduce SPAGRM(CCT) to aggregate the results following different models via Cauchy combination test. Extensive simulations and real data analyses demonstrated that SPAGRM maintains well-controlled type I error rates and SPAGRM(CCT) can serve as a broadly effective method. Applying SPAGRM to 79 longitudinal traits extracted from UK Biobank primary care data, we identified 7,463 genetic loci, making a pioneering attempt to conduct GWAS for these traits as longitudinal traits.
Similar content being viewed by others
Introduction
Over the past decade, the emergence of biobanks containing hundreds of thousands of genotyped participants has spurred a rapid growth of large-scale genome-wide association studies (GWAS). Leveraging electronic health records (EHRs), the large-scale GWAS has been extended to complex traits with more intricate structures1,2,3,4,5,6,7,8. For example, quantitative traits with phenotypic values measured repeatedly over time are known as longitudinal traits9. The longitudinal trait can characterize the evolution of health status, and GWAS on longitudinal traits has contributed to novel findings which deepen our understanding of genetic architecture10,11,12,13.
As popular approaches for analyzing longitudinal traits, linear mixed models and generalized estimation equations can incorporate time-varying covariates and the correlation structure of repeated measures into analysis14,15. Numerous previous literatures have highlighted the advantages of these methods over heuristic strategies that convert longitudinal traits to cross-sectional traits, e.g., only a single time point (usually baseline) is considered16,17,18,19,20. Recently, Ko et al. proposed TrajGWAS in which a mixed-effects multiple location scale model21 was introduced in large-scale longitudinal trait GWAS22. The linear mixed-effects model allows TrajGWAS to identify genetic variants associated with within-subject (WS) variability, which is an important risk factor for complex diseases23,24,25,26. Although well-developed, TrajGWAS is only applicable to analyze unrelated subjects, leading to a substantial reduction in sample sizes and thus statistical power.
Sample relatedness is a major confounder in GWAS and could result in inflated type I error rates if not appropriately controlled. To address this issue, numerous methods have been proposed to incorporate a random effect with a variance-covariance matrix of genetic relationship matrix (GRM) into conventional regression models6,27,28,29,30,31,32,33,34. However, applying this strategy to complex traits with intricate structures, such as longitudinal traits, is challenging. A notable example is the demanding statistical task of accurate variance components estimation6,27,28,29,30,31,32,33,34,35. Furthermore, the scale of biobank data presents challenges in terms of both memory usage and computational efficiency3. REGENIE is a GRM-free method that uses ridge regression predictors to replace the random effect36. While appealing, this approach encounters difficulties in applying the ridge regression into complex statistical models, restricting its application to a longitudinal trait analysis.
Here, we propose SPAGRM, a saddlepoint approximation (SPA)37,38 implementation that leverages the GRM to effectively control for sample relatedness in large-scale GWAS. SPAGRM adjusts for the sample relatedness via a retrospective strategy in which the genotypes of related subjects are considered as a multivariate random variable39. The retrospective approaches are more robust to model misspecification than prospective analyses39,40,41,42,43. This advantage allows incorporating the GRM-related random effect in the null model fitting to be optional, rather than required. Thus, existing conventional statistical models and methods with or without incorporating GRM can either be used to fit a null model, which significantly extends the scope of its applicability. In this paper, we evaluated SPAGRM in longitudinal trait GWAS via simulation studies and real data analyses.
SPAGRM differs from existing retrospective approaches via employing the SPA to estimate the null distribution of score statistics. Unlike regular retrospective methods solely relying on the GRM, such as ROADTRIP40, MASTOR41, and L-GATOR42, SPAGRM calculates identity by descent (IBD)-sharing probabilities44 for each pair of related subjects and then employs the Chow-Liu algorithm45 to approximate the joint distribution of genotypes for families with more than two related subjects. In addition, incorporating SPA can ensure high accuracy for a wide range of genotypic distribution (common, low-frequency, and rare variants) and phenotypic distribution (balanced and unbalanced distribution).
Due to the wide applicability, SPAGRM can conduct valid longitudinal trait GWAS based on a wide variety of analytical approaches, including but not limited to the linear mixed model as in TrajGWAS. The complexity of longitudinal traits renders no models optimal in all scenarios. Leveraging Cauchy combination test (CCT)46,47, we propose SPAGRM(CCT) as a robust and powerful solution to aggregate the SPAGRM results from multiple models and underlying assumptions.
In this work, we conducted extensive simulation studies and real data analyses to evaluate SPAGRM-based approaches in longitudinal trait GWAS. SPAGRM maintained well-controlled type I error rates, and SPAGRM(CCT) proved to be a broadly effective method for longitudinal trait GWAS. We applied SPAGRM to analyze 79 health-related longitudinal traits including blood counts, blood and urine biochemistry, physical measures, functional tests for various organs, and more. The traits were extracted from UK Biobank primary care data via a semi-supervised algorithm48 and thorough manual reviews to address duplicated records, unit errors, and implausible values. The refined pipeline is crucial for the subsequent GWAS of longitudinal traits. In total, the genome-wide analyses identified 7463 genetic loci, which suggests a significant potential of SPAGRM in large-scale GWAS. To the best of our knowledge, this is a pioneering attempt to conduct GWAS for a majority of the 79 longitudinal traits, and the analysis results can benefit for the research community interested in metabolism, blood cells, and serum/urine biomarkers.
Results
An overview of SPAGRM framework
SPAGRM is an analysis framework for conducting GWAS in a large-scale study cohort including related subjects. SPAGRM contains two main steps (Fig. 1). In step 1, we fit a null model to adjust for the effects of covariates on phenotypes and calculate model residuals. The covariates can be age, sex, SNP-derived principal components (PCs), and leave-one-chromosome-out polygenic scores (LOCO-PGS)49,50,51. It is optional, rather than required, to incorporate a GRM-related random effect into null model fitting, which extends the applicability of SPAGRM to a broader range of traits. In the Methods section, we exemplify the regression models to fit longitudinal traits, along with the corresponding model residuals.

SPAGRM framework consists of two steps: (1) Fitting a null model to calculate model residuals; (2) Calculating p values following a hybrid strategy including normal distribution approximation and saddlepoint approximation for each genetic variant. To avoid redundant computations, the joint distribution of genotypes for related subjects is estimated in advance, as illustrated in step 0.
In step 2, SPAGRM associates the trait of interest to a single genetic variant by approximating the null distribution of score statistics \(S={\sum }_{i=1}^{n}{G}_{i}{R}_{i}\), where \(n\) is the number of individuals, and \({G}_{i}\) and \({R}_{i}\) are the genotype and model residual for subject \(i,\,i\le n\), respectively. In a retrospective context, SPAGRM treats the genotype vector \(G={\left({G}_{1},{G}_{2},\ldots,{G}_{n}\right)}^{T}\) as a multivariate random variable, while considering the model residuals as fixed coefficients. SPAGRM utilizes IBD-sharing probabilities and Chow-Liu algorithm to approximate the joint distribution of the genotype vector of related subjects. Subsequently, to calculate p values, normal distribution approximation and SPA are used to approximate the null distribution of score statistics. Further details are provided in the Methods and Supplementary Note.
To remain scalable for large-scale GWAS, SPAGRM employs several strategies to enhance computational efficiency. To avoid redundant computations in step 2, the joint distribution of the genotype vector is estimated in advance (illustrated as step 0 in Fig. 1). Since the genotype distribution complexity scales exponentially with family size, we limit the maximum family size to 5 by default, grouping pairs of subjects with genetic relatedness (i.e., the corresponding GRM element) > 0.05 into a family. For larger families, we use a greedy strategy to reduce family size while minimizing variance estimation bias. We also apply a variance ratio adjustment to ensure the variance from SPA is accurate. In step 2, SPAGRM first leverages GRM to calculate standardized score statistics for each genetic variant. If the standardized score statistics is close to 0, SPAGRM calculates p values following normal distribution. Otherwise, SPAGRM calibrates p values using SPA. The hybrid strategy combining normal distribution approximation and SPA can balance the computational efficiency and accuracy, as in previous studies5,6,22,28,30,38. Similar to fastSPA38, we employ a partial normal distribution approach to fast calculation of cumulant generating function (CGF), which is the most computationally intensive step in SPA. More details can be seen in the Methods section.
Simulation studies
We conducted extensive simulation studies to evaluate the performance of SPAGRM in terms of type I error rates and power for longitudinal trait analysis. We simulated data cohorts following the below three scenarios of family relatedness.
-
Small-family based dataset includes 25,000 unrelated subjects and 25,000 related subjects from 6,250 families, each of which includes 4 members (Supplementary Fig. 1a).
-
Large-family based dataset includes 25,000 unrelated subjects and 25,000 related subjects from 2,500 families, each of which includes 10 members (Supplementary Fig. 1b).
-
Unrelated dataset includes 50,000 unrelated subjects.
To mimic the genotype distribution in real data, we simulated genotype data using real genotype data of White British subjects in UK Biobank by performing gene-dropping simulations52. We simulated 100,000 common variants (minor allele frequency (MAF) > 5%) and rare variants (MAF < 5% and minor allele counts (MAC) > 20) from genotype calls (field ID: 22418) and sequencing data (field ID: 23155), respectively. MAF spectrums of the simulated variants were displayed in Supplementary Fig. 2. More details about the simulation can be found in the Data simulation subsection of the Methods section.
In simulation studies, we compared five methods including TrajGWAS, SPAGRM, NormGRM, SPAGRM(INT), and SPAGRM(CCT). In step 1, SPAGRM and NormGRM employed the same null model fitting as in TrajGWAS to calculate model residuals. In step 2, SPAGRM utilized a hybrid strategy including both normal distribution approximation and SPA, while NormGRM calculated p values using normal distribution approximation only. SPAGRM(INT) was similar to SPAGRM, with the only exception that model residuals were updated using a rank-based inverse normal transformation (INT)53. SPAGRM(CCT) combined p values from SPAGRM and SPAGRM(INT) via CCT46,47. More details can be seen in the Methods section.
Type I error rates
In each scenario, we conducted \(1\times {10}^{9}\) tests and evaluated empirical type I error rates at significance levels \(\alpha=5\times {10}^{-5}\) and \(5\times {10}^{-8}\) (Table 1 and Supplementary Fig. 3). Across all scenarios, SPAGRM, SPAGRM(INT), and SPAGRM(CCT) can well control type I error rates. Meanwhile, NormGRM cannot control type I error rates when testing rare variants. The result is consistent with previous studies, further affirming the importance of SPA. Since TrajGWAS cannot adjust for relatedness, the type I error rates were significantly inflated when the study cohort included related subjects. Meanwhile, when related subjects were removed, TrajGWAS can still effectively control the type I error rates (Supplementary Fig. 4). In addition, even in cohorts without related subjects, TrajGWAS produced inflation when testing rare variants in terms of \({\tau }_{g}\). The inflation stemmed from the ultra-rare variants with MAF less than 0.002 (Supplementary Fig. 5), which was not fully evaluated by Ko et al.22.
We also evaluated type I error rates of SPAGRM in presence of cryptic sample relatedness by selecting 50,000 White British individuals. The individuals were purposely sampled from related UKB participants to achieve a higher proportion of relatives compared to the overall UKB cohort (Methods and Supplementary Fig. 6). Using real genotypes, we simulated phenotypes to mimic the effect of cryptic relatedness and then conducted association tests. Similarly, SPAGRM-based methods demonstrated well-controlled type I error rates in this context (Supplementary Fig. 7). Additionally, we assessed the impact of pedigree cuts on SPAGRM under conditions of cryptic relatedness. SPAGRM maintained well controlled type I error rates across various maximum family size settings (Supplementary Fig. 8). Furthermore, using a real phenotype, we demonstrated that pedigree cuts had minimal impact on SPAGRM in the UKB analysis, though they did affect run time (see Supplementary Figs. 9, 10 and Supplementary Note). Overall, the default maximum family size setting in SPAGRM is accurate enough in UKB analyses while remaining computational efficiency.
Empirical power
To evaluate empirical power, we simulated longitudinal traits following three types of alternative models, denoted as WSalt/BSalt (\({\tau }_{g}=1.5,\,{\beta }_{g}=1\)), WSalt/BSnull (\({\tau }_{g}=1.5,\,{\beta }_{g}=0\)), and WSnull/BSalt (\({\tau }_{g}=0,\,{\beta }_{g}=1\)). More details about the simulation can be seen in the Methods section. NormGRM was not evaluated as it cannot control type I error rates. Since TrajGWAS cannot account for sample relatedness, the analyses were restricted to a maximal set of unrelated subjects. For a family with 4 members, the two founders (i.e. 50%) were retained in the maximal set, while the two offspring were excluded. Similarly, for a family with 10 members, the four founders (i.e. 40%) were retained in the maximal set, while the other six subjects were excluded (Supplementary Fig. 1). To evaluate the power under varying extent of sample relatedness, we simulated study cohorts following the below five scenarios.
-
Dataset A includes 25,000 unrelated subjects from the previous Unrelated dataset, all of which are included in TrajGWAS analysis.
-
Dataset B includes 25,000 related subjects from the previous Small-family based dataset, of which 12,500 (50%) subjects are the maximal set of unrelated subjects and included in TrajGWAS analysis.
-
Dataset C includes 25,000 related subjects from the previous Large-family based dataset, of which 10,000 (40%) subjects are the maximal set of unrelated subjects and included in TrajGWAS analysis.
-
Dataset D is the combination of Dataset A and Dataset B, of which 37,500 (75%, 25,000 from Dataset A and 12,500 from Dataset B) subjects are the maximal set of unrelated subjects and included in TrajGWAS analysis.
-
Dataset E is the combination of Dataset A and Dataset C, of which 35,000 (70%, 25,000 from Dataset A and 10,000 from Dataset B) subjects are the maximal set of unrelated subjects and included in TrajGWAS analysis.
The empirical distributions of the chi-square statistics derived from p values under WSalt/BSalt are demonstrated in Fig. 2. Mean chi-square statistics and empirical power estimates are displayed in Supplementary Tables 1 and 2, respectively. If the study cohort only includes unrelated subjects (i.e., Dataset A), TrajGWAS and SPAGRM were almost the same powerful when testing \({\beta }_{g}=0\). If the study cohort includes related subjects, SPAGRM was more powerful than TrajGWAS in terms of both \({\beta }_{g}\) and \({\tau }_{g}\), regardless of common or rare variants. This is expected as TrajGWAS requires removing related subjects to avoid inflated type I error rates. When testing \({\tau }_{g}=0\), SPAGRM was still more powerful than TrajGWAS even if the study cohort only includes unrelated subjects. To clarify this, we used an empirical method to construct the empirical cumulative distribution functions (CDFs) of the score statistics, by randomly resampling model residuals or genotypes. The empirical p values derived from these CDFs were compared to those from TrajGWAS and SPAGRM methods (see Supplementary Note). Through resampling, we showed that TrajGWAS produced conservative p values when model residuals were extremely unbalanced, whereas SPAGRM maintained well-calibrated p values close to the empirical p values (Supplementary Fig. 11). This can also be validated in real data analyses, as shown in Supplementary Fig. 12.

Subplots a and b correspond to the empirical power to test \({\beta }_{g}=0\) and \({\tau }_{g}=0\), respectively. From left to right, the plots considered five relatedness scenarios of Dataset A: 25,000 unrelated subjects (all were unrelated and used in TrajGWAS analysis); Dataset B: 25,000 related subjects from 4-member families (50% were unrelated and used in TrajGWAS analysis); Dataset C: 25,000 related subjects from 10-member families (40% were unrelated and used in TrajGWAS analysis); Dataset D: a combination of Dataset A and B (75% were unrelated and used in TrajGWAS analysis); Dataset E: a combination of Dataset A and C (70% were unrelated and used in TrajGWAS analysis). Genetic effect sizes were set to \(-\log 10\left({{\rm{MAF}}}\right)\times 0.1\) for common variants and \(-\log 10\left({{\rm{MAF}}}\right)\times 0.02\) for rare variants, with an additional multiplier of 1 for \({\beta }_{g}\) and 1.5 for \({\tau }_{g}\). The points with chi-square statistics less than 3.84 (i.e., p values > 0.05) were filtered out. The black dots in the box plot represent the mean, while the box indicates the interquartile range (IQR). The whiskers extend to 1.5 times the IQR from the quartiles. The black dashed line represents the mean chi-squared values of TrajGWAS analysis results, and the grey dashed line represents the chi-squared statistic corresponding to the p value of \(5\times {10}^{-8}\). Genetic variants were grouped to common variants with MAF in (0.05, 0.5) and rare variants with MAF in (2e-4, 0.05). MAF, minor allele frequency. 80 and 30 are breakpoints for the different scales of the y-axis in subplots a and b, respectively. Mean chi-square statistics are displayed in Supplementary Table 1, and empirical power estimates are displayed in Supplementary Table 2. P value is calculated using two-sided score tests.
The empirical distributions of the chi-square statistics derived from p values under WSalt/BSnull and WSnull/BSalt are demonstrated in Supplementary Figs. 13 and 14, respectively. Notably, when analyzing common variants, both SPAGRM and TrajGWAS can accurately distinguish the mean profile and WS variability via score statistics \({S}_{{\beta }_{g}}\) and \({S}_{{\tau }_{g}}\). For example, when testing for common variants under WSalt/BSnull (i.e., \({\tau }_{g}\ne 0\) and \({\beta }_{g}=0\)), SPAGRM was powerful to test the null hypothesis H0: \({\tau }_{g}=0\) while controlling type I error rates to test the null hypothesis H0: \({\beta }_{g}=0\). The excellent performance also holds for common variants under WSnull/BSalt. Nevertheless, given a large effect size \({\tau }_{g}\) (e.g., \({\tau }_{g}=1.5\)), the performance does not always hold when analyzing rare variants under WSalt/BSnull. If the effect size \({\tau }_{g}\) is moderate (e.g., \({\tau }_{g}=0.5\)), the type I error rates can also be well controlled when testing H0:\({\beta }_{g}=0\) (Supplementary Fig. 15). The inflation is expected to remain under control in real data analysis, as the number of genetic variants with \({\tau }_{g}\gg 0\) and \({\beta }_{g}=0\) is anticipated to be limited.
In addition to the settings mentioned above, we also simulated longitudinal traits using alternative configurations. To mimic a real-world situation where few genetic variants influence the phenotypic variance, we simulated longitudinal traits without random effects on the WS variability. SPAGRM-based methods showed well-calibrated type I error rates and outperformed TrajGWAS across all scenarios (Supplementary Figs. 16 and 17). We also used an alternative WS variability model instead of model (4) to simulate longitudinal traits, and the conclusions remained consistent (Methods and Supplementary Figs. 18 and 19). These results indicate that SPAGRM-based methods are robust to model misspecification.
SPAGRM(CCT) can serve as an optimal unified approach
SPAGRM follows the null model fitting as in TrajGWAS to calculate model residuals and score statistics. Thus, SPAGRM is powerful, particularly when the longitudinal trait can be characterized by a linear mixed model, as exemplified in TrajGWAS. However, the complexity of the longitudinal trait renders no models optimal in all scenarios. SPAGRM framework is a retrospective approach in which model residuals are treated as fixed coefficients. The feature extends its applicability to allow for different models or data transformation to calculate or update model residuals. In this section, SPAGRM(INT) applies a rank-based INT to update model residuals obtained from TrajGWAS. Then, SPAGRM(INT) uses the updated model residuals to construct score statistics and perform association tests.
Compared to TrajGWAS and SPAGRM, SPAGRM(INT) preserves the rank of model residuals for each subject, while effectively addressing outliers. When testing for \({\beta }_{g}=0\), the number of model residual outliers from TrajGWAS is limited and the original SPAGRM is slightly more powerful than SPAGRM(INT). Meanwhile, when testing for \({\tau }_{g}=0\), TrajGWAS obtains a substantial number of model residual outliers, which greatly reduces the statistical power of TrajGWAS and the original SPAGRM. In this case, SPAGRM(INT) were more powerful, with a significant improvement in terms of chi-square statistics: 6-12 folds for common variants and 2-3 folds for rare variants (Supplementary Table 1). SPAGRM(CCT) combines p value results from SPAGRM and SPAGRM(INT) via CCT and can serve as a broadly effective approach. Across the simulation settings, SPAGRM(CCT) was always close to the most powerful method. In addition to the SPAGRM(INT), other approaches such as generalized estimation equations, can also be applied for longitudinal trait analyses. The wide applicability of SPAGRM framework and CCT allows SPAGRM(CCT) to leverage the p values from distinct models to calculate a single p value.
Application of SPAGRM to 79 longitudinal traits in the UK Biobank
We applied SPAGRM and TrajGWAS to analyze the 79 longitudinal traits extracted from the UKB primary care data (\(n=230,000\)). For each trait, we utilized a semi-supervised algorithm48 and conducted thorough manual reviews to identify the corresponding Read v2 and CTV3 code terms (see the Methods section). The code terms for each trait are available in Supplementary Table 3. These traits encompass a wide range of health-related data, including blood counts, blood and urine biochemistry, physical measures, functional tests for various organs, and more. The sample sizes vary across traits, ranging from 181,248 for systolic blood pressure (SBP) to 6,163 for cancer antigen 125 (CA125). Notably, for approximately 60 traits, more than three times as many events were recorded compared to the sample size, indicating that longitudinal traits convey richer information compared to cross-sectional traits. Table 2 presents basic summary information of 31 selected longitudinal traits and more detailed information for all 79 traits can be seen in Supplementary Table 4.
For each longitudinal trait, we analyzed approximately 23 million genetic variants imputed using the Haplotype Reference Consortium panel54. The analyses were restricted to variants with an imputation INFO score \(\ge 0.6\), MAF \(\ge 2\times {10}^{-4}\) and Hardy-Weinberg equilibrium (HWE) p value > \(1\times {10}^{-6}\). A total of 227,437 linkage disequilibrium (LD) pruned (\({r}^{2} < 0.2\)), high-quality genetic variants (MAF \(\ge 1\%\) and missing rate \(\le 5\%\)) were selected to calculate GRM and IBD-sharing probabilities. The pairs of subjects were considered as unrelated to each other if their genetic relatedness (i.e., the corresponding GRM element) <0.05. In the SPAGRM analysis, all 191,305 White British subjects were included. Meanwhile, in the TrajGWAS analysis, approximately 12.9% (mean platelet volume, MPV) to 17.9% (oxygen saturation at periphery, SpO2) of related subjects, up to a third degree4,55, were excluded. The exclusion was necessary because TrajGWAS cannot adjust for sample relatedness, as demonstrated previously. Similar to the simulation sections, we also assessed NormGRM. NormGRM relies solely on normal distribution approximation to calculate p values and is expected to perform similarly as the regular retrospective approaches, such as ROADTRIP40, MASTOR41, and L-GATOR42. For more details in covariates correction, medication adjustment, and the derivation of independent loci, please refer to the Genome-wide association analysis subsection in the Methods section.
SPAGRM outperforms TrajGWAS and NormGRM in longitudinal data analyses
Manhattan plots and Quantile-quantile (QQ) plots demonstrated that SPAGRM identified numerous significant associations while controlling type I error rates well (Figs. 3–4 and Supplementary Fig. 20). SPAGRM implicitly assumes hard-called genotypes, and we cannot theoretically justify its applicability to imputed data due to the complexity of imputation. Note that UK Biobank data analysis used non-discrete imputation dosage data and did not result in inflation or deflation, indicating SPAGRM’s robustness. At a significance level of \(5\times {10}^{-8}\), SPAGRM identified 7,463 and 362 genetic loci significantly associated with the mean and WS variability of the 79 longitudinal traits, respectively. Among these, 4,845 and 221 loci pass the significance level of \(5\times {10}^{-8}/79\) for the mean profile and WS variability, respectively. A complete list of the 7,825 (i.e., 7,463 + 362) loci is displayed in Supplementary Data 1 and 2.

a Mirror Manhattan plots of GWAS results of SPAGRM, SPAGRM(CCT), NormGRM, and TrajGWAS. b QQ plots in which genetic variants are grouped based on minor allele frequency (MAF): common variants with MAF in (0.05, 0.5), low-frequency variants with MAF in (0.01, 0.05), rare variants with MAF in (0.002, 0.01), and ultra-rare variants with MAF in (2e-4, 0.002). c Scatterplots comparing SPAGRM and SPAGRM(CCT) with TrajGWAS on common variants (i.e., MAF > 0.05). Across all subplots, upper and lower panels are results for testing mean profile (\({\beta }_{g}=0\)) and WS variability (\({\tau }_{g}=0\)), respectively. The red dashed line represents p value of \(5\times {10}^{-8}\), and the grey dashed line represents the breakpoint \({10}^{-15}\) for the different scales of the y-axis. For eGFR, 165,305 subjects were used for NormGRM, SPAGRM, and SPAGRM(CCT) analysis, of which 138,634 (83.9%) were used for TrajGWAS. An average of 8.30 eGFRs were measured per subject. P value is calculated using two-sided score tests.

a Mirror Manhattan plots of GWAS results of SPAGRM, SPAGRM(CCT), NormGRM, and TrajGWAS. b QQ plots in which genetic variants are grouped based on minor allele frequency (MAF): common variants with MAF in (0.05, 0.5), low-frequency variants with MAF in (0.01, 0.05), rare variants with MAF in (0.002, 0.01), and ultra-rare variants with MAF in (2e-4, 0.002). c, Scatterplots comparing SPAGRM and SPAGRM(CCT) with TrajGWAS on common variants (i.e., MAF > 0.05). Across all subplots, upper and lower panels are results for testing for mean profile (\({\beta }_{g}=0\)) and WS variability (\({\tau }_{g}=0\)), respectively. The red dashed line represents p value of \(5\times {10}^{-8}\), and the grey dashed line represents the breakpoint \({10}^{-15}\) for the different scales of the y-axis. For serum ferritin, 66,729 subjects were used for NormGRM, SPAGRM, and SPAGRM(CCT) analysis, of which 55,683 (83.4%) were used for TrajGWAS. An average of 2.26 values were measured for one subject. P value is calculated using two-sided score tests.
Traditionally, GWAS mainly focus on identifying loci that influence averaged trait levels. However, investigating trait variability can reveal loci that impact the stability of a trait, which might be overlooked. Most findings related to WS variability were concentrated in thyrotropin- and lipid-related phenotypes, suggesting that genetic effects may contribute to the volatility of these traits over time (Supplementary Fig. 21). For 79 longitudinal traits, 86.5% (313 out of 362) of the loci that affected WS variability also influenced mean levels. These could be caused by gene-environment interaction, selection, or epistasis22. Additionally, we observed some loci that affected WS variability without altering the mean, mostly in thyrotropin-related phenotypes.
Due to the inclusion of related subjects, SPAGRM successfully identified more significant loci compared to TrajGWAS. For the mean profile, of the 7,463 loci identified by SPAGRM, 1,338 (1,338/7,463 = 17.9%) loci were missed in TrajGWAS analysis. On the contrary, TrajGWAS analysis only exclusively identified 312 loci, of which 11 loci passed the significance level of \(5\times {10}^{-8}/79\). The detailed information of the 312 loci can also be found in Supplementary Data 1. For the WS variability, TrajGWAS identified a large number of spurious findings when testing low-frequency and rare variants. NormGRM performed nearly identically to SPAGRM when testing common variants, but it cannot control type one errors when testing low-frequency and rare variants. This finding is consistent with previous studies and the simulation studies, which indicates the necessary of SPA to calibrate p values.
To exemplify the advantages of SPAGRM framework, we selected three longitudinal traits including estimated glomerular filtration rate, serum ferritin, and serum thyroid stimulating hormone (see Supplementary Note). We also evaluated SPAGRM(CCT) in which p values of SPAGRM analyses with and without applying INT for model residuals were combined.
Genome-wide association studies of estimated glomerular filtration rate
Estimated glomerular filtration rate (eGFR) is widely accepted to evaluate the kidney function. In this study, we derived eGFR values (~8.3 measures per subject) from serum creatinine test using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation56. For SPAGRM, SPAGRM(CCT), and NormGRM, a total of 165,305 subjects were included in GWAS. And for TrajGWAS, 138,634 (83.9%) unrelated subjects were included.
Figure 3 displays the results via Manhattan plots and QQ plots. When testing for the mean level (i.e., \({\beta }_{g}\)), SPAGRM, SPAGRM(CCT), NormGRM, and TrajGWAS each identified hundreds of significant associations, with a substantial overlap among them. At a significance level of \(5\times {10}^{-8}\), SPAGRM identified 158 loci, of which 43 loci were missed in TrajGWAS analysis. In contrast, TrajGWAS analysis exclusively identified only 8 loci (8/43 = 18.6%). SPAGRM(CCT) showed minimal differences compared to SPAGRM, with 5 loci exclusively identified (Supplementary Data 3). In terms of the WS variability (i.e., \({\tau }_{g}\)), few loci were identified by SPAGRM, SPAGRM(CCT) and TrajGWAS. Meanwhile, a considerable number of rare variants were identified by NormGRM, which was highly spurious. This was primarily due to the highly skewed distribution of residuals after fitting a linear mixed model. To clarify this, we shuffled the order of the residuals and re-applied these methods, and NormGRM still produced a large number of false-positive rare variants (Supplementary Fig. 22).
Recently, Stanzick et al. conducted a large-scale GWAS with exceeding 1.2 million participants and identified 424 eGFR-associated SNPs57. The 424 eGFR-associated SNPs and the loci identified in SPAGRM analyses were highly consistent. For example, of the 43 loci detected by SPAGRM but missed in TrajGWAS analysis, 33 signals lay within 500 kb of at least one of these 424 eGFR-associated SNPs. In addition to the reported SNPs, SPAGRM identified several loci located more than 500 kb away from those identified by Stanzick et al.57. For example, SNP rs2729940 (SPAGRM p value = 2.55e-8) is an intronic variant in BLK gene, which encodes Tyrosine-protein kinase Blk. BLK was mainly identified as a susceptibility gene for systemic lupus erythematosus (SLE) in previous GWAS58. Di et al. showed that the polymorphisms of BLK were associated with renal disorder in patients with SLE59. In addition, Zhou et al. found that SNP markers in BLK gene were also associated with IgAN in a Chinese population60,61. SNP rs17663700 (SPAGRM p value = 2.08e-8) is located upstream of the ATP6V1B1 gene. A case report found that mutations in ATP6V1B1 would cause distal renal tubular acidosis62.
Genome-wide association studies of serum ferritin
Ferritin, a major iron storage protein, is predominantly utilized as a serum biomarker of total body iron stores. For SPAGRM, SPAGRM(CCT), and NormGRM, a total of 66,729 subjects were included in GWAS. And for TrajGWAS, 55,683 (83.4%) unrelated subjects were included. An average of 2.26 values were measured per subject.
Figure 4 displays the results via Manhattan plots and QQ plots. For mean profile, SPAGRM and SPAGRM(CCT) identified more peaks of association compared to TrajGWAS. Meanwhile, NormGRM identified numerous spurious associations when testing low-frequency and rare variants. For WS variability, SPAGRM identified a significant peak of association while effectively controlling false positive rates. SPAGRM(CCT) produced more significant p values compared to SPAGRM at known peaks. However, TrajGWAS and NormGRM appeared to inflate type I error rates when testing low-frequency and rare variants (Fig. 4b and Supplementary Fig. 22). In general, SPAGRM(CCT) and SPAGRM yielded more notable p values compared to TrajGWAS in terms of both mean profile and WS variability (Fig. 4c).
SPAGRM and SPAGRM(CCT) detected 39 (28 + 11) loci associated with mean levels and 9 (4 + 5) loci linked to WS variability (loci identified by SPAGRM + loci additionally identified by SPAGRM(CCT)). Of these loci, SNPs rs1800562 (nearest gene HFE) and rs855791 (TMPRSS6) have been previously associated with iron homeostasis biomarkers63,64. The exonic variant rs1800562 showed noteworthy associations with both the mean (SPAGRM p value = 2.83e-84; SPAGRM(CCT) p value = 5.66e-84) and the WS variability (SPAGRM p value = 1.38e-16; SPAGRM(CCT) p value = 2.99e-37) of serum ferritin, statistically affirming the relationship between HFE gene and iron homeostasis. The SNP rs855791 located in gene TMPRSS6 was found to be associated with the mean of serum ferritin (SPAGRM p value = 2.56e-16), which is consistent with previous findings63,64. Notably, there was evidence that TMPRSS6 was also associated with the ferritin variability (rs855791, SPAGRM(CCT) p value = 1.29e-10), while this association was overlooked without INT (SPAGRM p value = 0.70; TrajGWAS p value = 0.85).
Generalized estimation equations can contribute to greater power
Generalized estimation equations (GEE) can characterize longitudinal data through a user-specified correlation structure for multiple measures of a subject65. Unlike linear mixed-effects models (LMM), GEE is flexible in adopting various correlation structures. In this section, we focused the comparison on the longitudinal mean, as GEE is a marginal model and does not account for WS variabilities. We fitted GEE models with exchangeable and autoregressive working correlation structures to calculate model residuals, and then passed the residuals to SPAGRM framework to conduct GWAS (denoted as SPAGRM(GEEexc) and SPAGRM(GEEar1)). In addition, we applied CCT46,47 to combine p values of SPAGRM(GEEexc) and SPAGRM(GEEar1) (denoted as SPAGRM(CCT)). These methods were compared against SPAGRM using a null model fitting by LMM (denoted as SPAGRM(LMM)). We conducted a series of simulation studies and real data analyses to evaluate the performance of these methods. We simulated longitudinal traits under three generative models of linear mixed model and GEE models with exchangeable and autoregressive correlation structures. More details can be seen in Supplementary Note.
SPAGRM-based methods demonstrated well-controlled type I error rates across all scenarios, indicating that SPAGRM is robust against correlation structure misspecification (Supplementary Fig. 23). The empirical power of these methods varied depending on the data generating mechanism (Supplementary Table 5). Since no method was uniformly most powerful, SPAGRM(CCT) can serve as a broadly effective method across all scenarios. We also applied these methods to real data analyses. Supplementary Fig. 24 displayed that, in some cases, SPAGRM methods based on GEE model can produce more significant p values in the tail compared to SPAGRM(LMM). For example, SPAGRM(GEEar1) resulted in lower p values than SPAGRM(LMM) when analyzing eGFR, while SPAGRM(GEEexc) outperformed SPAGRM(LMM) in the analysis of TSH. Overall, no method was omnibus, indicating that different longitudinal traits could correspond to different architectures. Strikingly, SPAGRM(CCT) achieved p values on par with the most effective method across traits.
Computational efficiency
We evaluated the computational efficiency of SPAGRM in analyzing longitudinal traits. All analyses were conduct on a CPU model of Intel(R) Xeon(R) Gold 6342 CPU 2.80 GHz. SPAGRM requires GRM and IBD-sharing probabilities as input, which only need to be calculated once for a specific study cohort. GRM can be efficiently computed using tools like GCTA66. For example, constructing a sparse GRM for 408,961 UKB white British participants using 227,437 LD-pruned SNPs required 240 CPU hours, which can be divided into 250 separate tasks, each taking under one hour. Since SPAGRM only needs IBD-sharing probabilities for related pairs (e.g., when the GRM element is greater than 0.05), computing these probabilities using our implemented function took only two CPU hours (see Code availability). If users want, other tools can also be used to calculate GRM and IBD-sharing probabilities67, with format easily convertible for SPAGRM analysis.
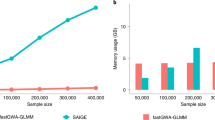
We used longitudinal BMI data to evaluate the computational performance of SPAGRM, SPAGRM(CCT), and TrajGWAS, where SPAGRM(CCT) is the combination of SPAGRM and SPAGRM(INT). We randomly sampled 10%, 25%, 50%, 75%, and 100% of individuals with BMI measurements (\(n={\mathrm{175,443}}\)) and performed GWAS on each subset for 23 million imputed variants. For SPAGRM, the estimation of the joint distribution of genotypes is completed in step 0, which generally takes a few hours (Supplementary Fig. 25). The null model fitting in step 1, for both LMM and GEE models, is computationally efficient. Therefore, we focused on the comparison of association tests in step 2, as it accounts for most of the run time. SPAGRM took 302 CPU hours to analyze 23 million imputed variants on 175,443 subjects (Fig. 5). Meanwhile, TrajGWAS took approximately 188 CPU hours to accomplish the same task, which was approximately 1.6-fold faster than SPAGRM. SPAGRM(CCT) was the most time-consuming, taking up to 408 hours. For memory costs, SPAGRM required a peak memory usage of 2.7 GB in step 2, which was more resource-efficient than TrajGWAS. We also evaluated SPAGRM’s computational performance with GEE models fitting the null model and found that it performed similarly to the LMM model (Supplementary Fig. 26).

a Runtime. The x axis represents the sample size and the y axis represents the run time in hourly units. b Memory usage. The x axis represents the sample size and the y axis represents the memory usage in GB units. We compared three methods: SPAGRM, SPAGRM(CCT), and TrajGWAS, where SPAGRM(CCT) is the combination of SPAGRM and SPAGRM(INT). Analysis was performed on 10%, 25%, 50%, 75%, and 100% of 175,443 individuals with longitudinal BMI measurements. The association tests were conducted on 23 million imputed variants. All analyses were conduct on a CPU model of Intel(R) Xeon(R) Gold 6342 CPU 2.80 GHz.
Discussion
In this paper, we propose a scalable and accurate analysis framework, SPAGRM, to adjust for sample relatedness in a large-scale GWAS involving hundreds of thousands of subjects. SPAGRM treats genotypes as random variables and employs IBD and Chow-Liu algorithm to approximate the joint distribution of genotypes for related subjects. Because it is not required to incorporate a random effect to characterize sample relatedness, SPAGRM is widely applicable to complex traits with intrinsic structures. A hybrid strategy including SPA ensures the accuracy to analyze low-frequency and rare variants, even if the phenotypic distribution is unbalanced. Additional strategies, such as partial normal distribution approximation, greatly reduce the computational burden and thus SPAGRM is scalable to analyze hundreds of thousands of subjects.
TrajGWAS is not valid to analyze a study cohort including related subjects. Meanwhile, SPAGRM can well control type I error rates and is more powerful than TrajGWAS when analyzing related subjects. Due to the wide applicability, SPAGRM allows for more flexible analytical approaches to calculate or update model residuals. For example, applying a rank-based inverse normal transformation to update model residuals or fitting null model based on GEE could result in greater power. Real data analyses validated that no method was uniformly most powerful, which indicates that different longitudinal traits correspond to different architectures. Thus, we propose SPAGRM(CCT) to combine p values obtained from applying SPAGRM framework to distinct models. SPAGRM(CCT) can serve as a broadly efficient approach that controls type I error and is nearly as powerful as the most effective of the component methods.
We applied SPAGRM to analyze 79 longitudinal traits extracted from UK Biobank primary care data. The analyses identified 7,463 and 362 loci for mean profile and WS variability of the longitudinal trajectories, respectively. The 79 longitudinal traits encompassed blood counts, blood and urine biochemistry, physical measures, and functional tests of multiple organs, most of which were analyzed as a longitudinal trait in GWAS for the first time.
SPAGRM can further gain statistical power through incorporating PGS as covariates with fixed effects. Recent reports have shown that adjusting for PGS can account for polygenic effects and increase statistical power49,50,51. This strategy can complement the reduced power for sparse-GRM-related methods like fastGWA32,34. In Supplementary Note, we employed this idea to implement a two-stage strategy, SPAGRM-PGS. In stage 1, we conduct the first round of GWAS via SPAGRM and then calculate the LOCO-PGS49,50,51 based on the summary statistics. In stage 2, the LOCO-PGS is included as an additional covariate for a second round of GWAS via SPAGRM. Using several longitudinal traits, we demonstrated SPAGRM-PRS had superior performance compared to the original SPAGRM.
As a universal analysis framework, SPAGRM is not limited to longitudinal analyses. In Supplementary Note, we also evaluated the performance of SPAGRM when analyzing quantitative and binary traits. SPAGRM achieved comparable performance to existing sparse-GRM-based methods32,34, and showed solid consistency with dense-GRM-based methods29,30 and REGENIE29,30,36 after PGS adjustment. The comparison demonstrated that SPAGRM after adjusting for PRS performs similarly as the original frameworks explicitly incorporate random effects in the null model fitting. We do not intend to replace the current method but to give some insights for phenotypes that no existing method is available. For example, if a new type of phenotype is needed, researchers can feel free to use SPAGRM and do not need to propose a new mixed-effect method to address relatedness.
There are several limitations in SPAGRM and the real data analysis in UK Biobank. First, SPAGRM assumes that genotype marginally follows a binomial distribution. Thus, HWE test is required to exclude genetic variants whose genotypic distribution deviate significantly. As expected, most of genetic variants have passed HWE p value of 1e-6 in real data analyses (Supplementary Fig. 27). Although SPAGRM does not exhibit type I error rates inflation at a looser HWE p value threshold of 1e-15 (Supplementary Fig. 28), we still recommend using 1e-6 as the threshold because extremely significant HWE test p value may also indicate potential quality control issues. Second, SPAGRM is based on score test and does not fit a full model. If genetic effect size is required for the follow-up analysis, SPAGRM can serve as a screening process to prioritize variants to fit a full model. Third, in UK Biobank data analyses, we only focused on the mean and WS variability of a longitudinal trajectory. In the future, we plan to extend GWAS to other patterns harbored in the longitudinal trajectory. A notable example is the dynamic process (upward or downward) of complex traits after specific medical treatments or surgical procedures. Finally, the current version of SPAGRM only supports analyzing autosomes.
The current framework assumes that, for each individual, genotypes marginally follow the same binomial distribution, i.e., the allele frequency is the same. However, for individuals from different ancestries, the allele frequencies for a single SNP could be different, which violate the assumption and the results. We plan to incorporate global genetic PCs and local ancestry information to allow for ancestry-specific allele frequencies.
Currently, there is a growing trend toward utilizing complex traits with intricate structures in GWAS. For most of the traits, conventional regression models have been developed by statisticians but there is still a substantial gap to apply them in GWAS. One challenge is to efficiently adjust for sample relatedness in large-scale biobanks. SPAGRM can serve as a universal analysis framework to address this issue. For any trait of interest, users only need to select conventional regression models as the null model and then calculate the first derivative of likelihood as the model residuals, then SPAGRM can handle follow-up processes including 1) reading in genotype from BGEN or PLINK, 2) adjusting for sample relatedness, and 3) remaining statistical powerful while controlling type I error rates for both common and rare variants. We believe that SPAGRM framework can bridge the gap between statistics and GWAS to expedite the implementation of GWAS using complex and precise statistical regression models.
Methods
Ethics statement
The research reported herein was conducted in compliance with all ethical requirements. This UK Biobank project was conducted under the application number 78795. Ethical approval for the UK Biobank resource was granted by the North West Centre for Research Ethics Committees (reference number 11/NW/0382). The UK Biobank study was carried out in accordance with the principles outlined in the Declaration of Helsinki, with participants providing informed written consent to participate and to be followed up through national record linkage.
Linear mixed effect model for longitudinal traits
We let \(n\) denote the number of subjects, \({m}_{i}\) denote the number of measurements for subject \(i\), and \(m={\sum }_{i=1}^{n}{m}_{i}\) denote the total number of measurements for all subjects. For the j-th measurement of subject \(i\), Ko et al.22 proposed a linear mixed effect model (LMM) to characterize mean profile and within-subject (WS) variability as below.
where \({g}_{i}\) is the genotype of a single variant, \({{{\boldsymbol{x}}}}_{{ij}}\,(p\times 1)\) and \({{{\boldsymbol{z}}}}_{{ij}}\,(q\times 1)\) are two vectors of covariates with fixed coefficient \({{\boldsymbol{\beta }}}\) and random coefficient \({{{\boldsymbol{\gamma }}}}_{i}\), respectively, and \({y}_{{ij}}\) is the trait. Random term \({\varepsilon }_{{ij}}\) follows a normal distribution with a mean of zero and a standard error of \({\sigma }_{{\varepsilon }_{{ij}}}\). The standard error \({\sigma }_{{\varepsilon }_{{ij}}}\) is determined by covariate vector \({{{\boldsymbol{w}}}}_{{ij}}\,(l\times 1)\), genotype \({g}_{i}\), and a random intercept \({\omega }_{i}\). Covariates \({{{\boldsymbol{x}}}}_{{ij}}\), \({{{\boldsymbol{z}}}}_{{ij}}\), and \({{{\boldsymbol{w}}}}_{{ij}}\) can be time-invariant (e.g. sex) or time-varying (e.g. age during measurement). The genetic effects on mean and WS variability are \({\beta }_{g}\) and \({\tau }_{g}\), respectively. Random effects \({({{{{\boldsymbol{\gamma }}}}_{i}}^{T},{\omega }_{i})}^{T}\) follow a multivariate normal distribution with a mean of zero and a variance-covariance matrix of
Suppose that the study cohort includes genetically related subjects and the genetic relationship matrix (GRM) is denoted as \(\varPhi\). We can incorporate random effects, \({b}_{i}\) and \({\widetilde{b}}_{i}\), into models (1) and (2) to characterize the sample relatedness.
Here, \({b}_{i}\) and \({\widetilde{b}}_{i}\) are assumed to follow multivariate normal distributions with variance-covariance matrices of \({\sigma }^{2}\varPhi\) and \({\widetilde{\sigma }}^{2}\varPhi\). The assumption is to mimic the polygenic effects, i.e., the sum of additive effects of a large number of genetic variants on the phenotypic mean and WS variability, respectively29,30,36. Given evidence that genetic loci influencing phenotypic variance are fewer than those influencing phenotypic mean, the assumption in terms of \({\widetilde{b}}_{i}\) may not always hold24,26,68. Therefore, we conducted a series of simulations to evaluate the performance of our purposed methods under more realistic conditions (see the Data simulation subsection below).
Generalized estimation equations for longitudinal traits
Generalized estimating equations (GEE) are also commonly used approaches for modeling repeated measures of outcomes15. Consider a linear regression,
where genetic effect on mean trajectories is \({\beta }_{g}\). Unlike LMM, GEE is a semi-parametric method that requires assumptions about the conditional distribution of error term \({\varepsilon }_{{ij}}\) with a user-specified correlation structure. In Supplementary Note, we clarify various working correlation structures, as well as null model fitting and model residuals calculation for GEE.
Null model fitting and model residuals calculation (step 1)
While the strategy of incorporating the GRM has been widely used to model various types of traits, it still presents substantial technical challenges in null model fitting. For example, fitting models (3) and (4) is not a simple extension of fitting models (1) and (2). SPAGRM method employs a retrospective framework, in which incorporating the GRM is optional, rather than required, when fitting null model.
In longitudinal data analysis, SPAGRM follows Ko et al.22 to employ Julia package WiSER69 to fit models (1) and (2) under null hypothesis \({H}_{0}:{\beta }_{g}={\tau }_{g}=0\). Given the estimated parameters \(\hat{{{\boldsymbol{\beta }}}}\), \(\hat{{{\boldsymbol{\tau }}}}\), and\(\,{\hat{{{\boldsymbol{\Sigma }}}}}_{{{\boldsymbol{\gamma }}}}\), score statistics \({S}_{{\beta }_{g}}={R}_{{\beta }_{g}}^{T}G\) and \({S}_{{\tau }_{g}}={R}_{{\tau }_{g}}^{T}G\) are to test genetic mean profile (\({\beta }_{g}=0\)) and WS variability (\({\tau }_{g}=0\)), where \(G={\left({g}_{1},{g}_{2},...,{g}_{n}\right)}^{T}\) is genotype vector, \({R}_{{\beta }_{g}}\) and \({R}_{{\tau }_{g}}\) are model residual vectors. More details about the score statistics and residuals can be found in previous study22. Note that score statistics have the consistent format of \(S={R}^{T}\cdot G\), regardless of testing the mean or WS variance effects. Thus, our discussion will pertain to the consistent format of \(S={R}^{T}\cdot G\).
Score testing with normal distribution approximation (step 2)
Similar to regular retrospective methods40,41,42, we assume that under Hardy-Weinberg equilibrium (HWE), genotype \({G}_{i}\) follows a Binomial distribution \(B(2,\,\mu )\) where \(\mu\) is the MAF of this variant. The mean and variance of the score statistics \(S={R}^{T}\cdot G\) are as below
where \({\sigma }_{g}=\sqrt{2\mu \left(1-\mu \right)}\) is the standard error of the genotype \({G}_{i}\) and \({\rho }_{{ij}}\) is the correlation between \({G}_{i}\) and \({G}_{j}\) (i.e., the corresponding element of GRM matrix). Suppose that GRM is \(\varPhi\), the mean and variance
where \({{{\boldsymbol{1}}}}_{n}\) is an n-dimensional vector of ones. Note that if the sum of the model residuals is zero, then the mean of score statistics is also zero. Since the calculation of \({R}^{T}\cdot \varPhi \cdot R\) is independent of genotype \(G\), it only needs to be computed once for a genome-wide analysis, making normal distribution approximation computationally efficient. Under the null hypothesis, the probability \(\Pr \left(S < {s|R}\right)=F\left\{\left(s-E\left(S\right)\right)/\sqrt{{Var}(S)}\right\}\) where \(F\left\{\cdot \right\}\) is the cumulative distribution function (CDF) of a standard normal distribution.
Score testing with saddlepoint approximation (step 2)
The normal distribution approximation is not accurate if the distribution of the residuals \(R\) is highly skewed or the MAF of the genetic variant is close to 0. To address this issue, we use SPA to more accurately calibrate p values.
Moment generating function of score statistics
Drawing upon a sparse GRM and a predetermined threshold (e.g. 0.05), we classify all subjects into \(q\) families, ensuring that the corresponding GRM elements between individuals in distinct families is below the specified cutoff. Suppose that family \(i\le q\) includes \({n}_{i}\) related subjects, then the score statistics can be decomposed as
where \({S}_{i}={\sum }_{j=1}^{{n}_{i}}{R}_{{ij}}{G}_{{ij}}\) is the score statistics for family \(i\le q\), \({R}_{{ij}}\) and \({G}_{{ij}}\) are the model residual and genotype of the j-th subject in family \(i\), respectively. The moment generating function (MGF) of \(S\) is
where \({M}_{{S}_{i}}\left(t\right)\) is the MGF of score statistics \({S}_{i}={\sum }_{j=1}^{{n}_{i}}{R}_{{ij}}{G}_{{ij}}\). If the family \(i\) only includes one subject, that is, \({n}_{i}=1\), then the MGF \({M}_{{S}_{i}}\left(t\right)={\left[1-\mu+\mu {e}^{t\cdot {R}_{i1}}\right]}^{2}\). Otherwise, the MGF of \({S}_{i}\) are estimated as below.
MGF estimation for families with two related subjects
We consider families with two related subjects and calculate MGF of score statistics \({S}_{i}={R}_{{i}_{1}}{G}_{{i}_{1}}+{R}_{{i}_{2}}{G}_{{i}_{2}}\). IBD-sharing probabilities have been widely used to characterize the relatedness of two subjects44. For two related subjects, we let non-negative values \({\delta }^{(0)}\), \({\delta }^{(1)}\), and \({\delta }^{(2)}\) denote the IBD-sharing probabilities that there are 0, 1, and 2 IBD-sharing alleles, and the corresponding MGFs of \({S}_{i}\) are
respectively (more details can be seen in Supplementary Note). The MGF estimation of \({S}_{i}\) is
Given IBD-sharing probabilities \({\delta }^{(1)}\) and \({\delta }^{(2)}\), kinship coefficient is \({\delta }^{(1)}/4+{\delta }^{(2)}/2\). However, if kinship coefficients for two pairs of related subjects are equal, it does not necessarily imply that the IBD-sharing probabilities and the corresponding MGFs are also equal. For example, the kinship coefficient between a pair of full siblings and a pair of parent-offspring are both 0.25. However, the corresponding MGFs are not the same. For a pair of full-siblings, \({\delta }^{(0)}\), \({\delta }^{(1)}\), and \({\delta }^{(2)}\) are 0.25, 0.5, and 0.25, respectively, and the MGF is \({M}_{{S}_{i}}\left(t\right)=0.25\cdot {M}_{{S}_{i}}^{(0)}\left(t\right)+0.5\cdot {M}_{{S}_{i}}^{(1)}\left(t\right)+0.25\cdot {M}_{{S}_{i}}^{(2)}\left(t\right)\). For a pair of parent-offspring, \({\delta }^{(0)}\), \({\delta }^{(1)}\), and \({\delta }^{(2)}\) are 0, 1, and 0, respectively, and the MGF is \({M}_{{S}_{i}}\left(t\right)={M}_{{S}_{i}}^{(1)}\left(t\right)\).
For each pair of related individuals, we estimate probabilities \({\delta }^{(0)}\), \({\delta }^{(1)}\), and \({\delta }^{(2)}\) using raw genotype data. We define two metrics \({\rho }_{1}\) and \({\rho }_{2}\) as below:
where \({\rho }_{1}\) is 2 times of kinship coefficient which can be obtained from the GRM, and \({\rho }_{2}\) is a method of moments (MOM) estimator for zero-IBD-sharing probabilities. Note that \({\rho }_{2}\) is equivalent to the definition implemented in previous study44. Since the metrics \({\rho }_{1}\) and \({\rho }_{2}\) do not depend on \(\mu\), we use genotype of \(s\) common variants (e.g. MAF > 0.05) to empirically estimate them as below
where \({G}_{{i}_{1}k}\) and \({G}_{{i}_{2}k}\) are genotypes of variant \(k\) of the two related subjects indexed by \({i}_{1}\) and \({i}_{2}\), \({\hat{\mu }}_{k}\) is the empirical allele frequency, and weight
is to characterize the contribution of genetic variant \(k\). Given \({\hat{\rho }}_{1}\) and \({\hat{\rho }}_{2}\), IBD-sharing probabilities \({\delta }^{(0)}\), \({\delta }^{(1)}\), and \({\delta }^{(2)}\) were estimated by solving a linear model:
MGF estimation for families including more than two related subjects
Suppose that family \(i\le q\) includes \({n}_{i} > 2\) related individuals, we use Chow-Liu algorithm45 to approximate the discrete joint distribution of \({G}_{{i}_{1}},{G}_{{i}_{2}},\ldots,{G}_{{i}_{{n}_{i}}}\) and then estimate the MGF of \({S}_{i}={\sum }_{j=1}^{{n}_{i}}{R}_{{i}_{j}}{G}_{{i}_{j}}\). We let \({{{\boldsymbol{G}}}}_{i}\) denote vector \({\left({G}_{{i}_{1}},{G}_{{i}_{2}},\ldots,{G}_{{i}_{{n}_{i}}}\right)}^{T}\) and \(\Pr ({{{\boldsymbol{G}}}}_{i})\) its joint distribution. Chow-Liu algorithm approximated the distribution \(\Pr ({{{\boldsymbol{G}}}}_{i})\) as
where \({m}_{k},1\le k\le {n}_{i}\) is a rearrangement of integers ranging from \(1\) to \({n}_{i}\), and the mapping \({m}_{h(k)}\) is called the dependence tree of the distribution. Define that \(P({G}_{{i}_{{m}_{k}}}|{G}_{{i}_{0}})=P({G}_{{i}_{{m}_{k}}})\). For example, a possible approximation of \(\Pr ({{{\boldsymbol{G}}}}_{i})\) for a 3-member family can be \(\hat{\Pr }({{{\boldsymbol{G}}}}_{i})={{\rm{P}}}({G}_{{i}_{1}}){{\rm{P}}}({G}_{{i}_{2}}|{G}_{{i}_{1}}){{\rm{P}}}({G}_{{i}_{3}}|{G}_{{i}_{1}})\).
The selection of second-order terms \(P({G}_{{i}_{{m}_{k}}}|{G}_{{i}_{{m}_{h(k)}}})\) is the most critical step for the approximation. Chow-Liu algorithm used a mutual information \(I({G}_{{i}_{{j}_{1}}},{G}_{{i}_{{j}_{2}}}),\,1\le {j}_{1} < {j}_{2}\le {n}_{i}\) between \({G}_{{i}_{{j}_{1}}}\) and \({G}_{{i}_{{j}_{2}}}\) and constructed a maximum-weight tree by adding the maximum mutual information pair to the tree. They further showed that the maximum-weight tree is the maximum-likelihood estimate of the distribution. More detailed derivations can be seen in Supplementary Note.
Given the joint distribution estimation \(\hat{\Pr }({{{\boldsymbol{G}}}}_{i})\), the MGF of \({S}_{i}={\sum }_{j=1}^{{n}_{i}}{R}_{{i}_{j}}{G}_{{i}_{j}}\) is estimated as below
In Supplementary Note, we used pedigree data to evaluate the accuracy of MGF estimation via the Chow-Liu algorithm, as the theoretical MGF is accessible given a known family structure. Compared to relying solely on the normal distribution approximation, Chow-Liu algorithm can estimate MGF more accurately, regardless of genotype and phenotype distributions (Supplementary Fig. 29 and Supplementary Note). Estimating MGF for families with more than two related individuals using Chow-Liu algorithm is crucial for SPAGRM and represents a key distinction from NormGRM, which performs poorly when genotype or phenotype distributions are high unbalanced. Note that the MGF estimation is based on empirical IBD estimation only, instead of family structure.
Saddlepoint approximation to calibrate p values
The CGF of score statistics \(S\) is
and its first and second derivatives are
where \({M}_{S}(t)\) is the MGF. The variance derived from CGF is \({Va}{r}_{{CGF}}\left(S\right)={K}_{S}^{{\prime} {\prime} }\left(0\right)\), which is slightly different from \({Var}\left(S\right)\) due to family size reduction. We calculate adjusted score statistics \({s}_{{CGF}}=s\cdot \sqrt{{Va}{r}_{{CGF}}\left(S\right)/{Var}(S)}\) and \(\zeta\) such that \({K}_{S}^{{\prime} }\left(\zeta \right)={s}_{{CGF}}\). Then, we calculate \(\omega={\mathrm{sgn}}(\zeta )\sqrt{2(\zeta s-{K}_{S}\left(\zeta \right))}\) and \(\nu=\zeta \sqrt{{K}_{S}^{{\prime} {\prime} }\left(\zeta \right)}\). According to the Barndorff-Nielson method37,70, the cumulative distribution function of \(S\) at \(s\) is approximated by
where \(F\left\{\cdot \right\}\) is the CDF of a standard normal distribution.
Strategies to increase computational efficiency
To remain scalable for large-scale biobank-based GWAS, SPAGRM employs several strategies to enhance computational efficiency.
Pre-calculation of the joint distribution of genotypes
While Chow-Liu algorithm is computationally efficient, it is still not scalable to repeat it for millions of times. For a given family structure, the selection of second-order terms \(P({G}_{{i}_{{m}_{k}}}|{G}_{{i}_{{m}_{h(k)}}})\) and the corresponding \(\hat{\Pr }({{{\boldsymbol{G}}}}_{i})\) may also vary depending on MAF. Thus, we use a grid idea to divide the MAF region into 10 intervals given MAF cutoffs of 1e-4, 5e-4, 0.001, 0.005, 0.010, 0.050, 0.100, 0.200, 0.300, 0.400, and 0.500. We first calculate and store the discrete joint distribution of \(\hat{\Pr }({{{\boldsymbol{G}}}}_{i})\) at each MAF cutoff in step 0. Then, we use a linear interpolation to approximate the discrete joint distribution of \(\hat{\Pr }({{{\boldsymbol{G}}}}_{i})\) if the MAF falls within one of the specified intervals in step 2 when needed.
Family size reduction
Suppose that family \(i\) consists of \({n}_{i}\) subjects, the number of all possible outcomes for the discrete distribution \(\Pr ({{{\boldsymbol{G}}}}_{i})\) is \({3}^{{n}_{i}}\) as the genotype of each subject can take on one of three values 0, 1, or 2. Hence, the summation presented above consists of a total of \({3}^{{n}_{i}}\) elements, whose computational burden increases at an alarming rate with \({n}_{i}\). Thus, it is essential to restrict the family size below a pre-given cutoff (e.g. \(\le 5\)). We propose a heuristic greedy algorithm to divide large families into multiple families with more manageable sizes.
Large pedigree splitting has long been used in family-based linkage analysis, such as PedCut71 and PedStr72. Unlike many other splitting algorithms based solely on the kingship coefficient \({\rho }_{{i}_{{j}_{1}},{i}_{{j}_{2}}}\) to assess the degree of relatedness, we calculate \(|{\rho }_{{i}_{{j}_{1}},{i}_{{j}_{2}}}{R}_{{i}_{{j}_{1}}}{R}_{{i}_{{j}_{2}}}|,\,1\le {j}_{1} < {j}_{2}\le {n}_{i}\) for each pair of related subjects \({i}_{{j}_{1}}\) and \({i}_{{j}_{2}}\). First, we iteratively remove the relatedness pair in the increasing order of \(|{\rho }_{{i}_{{j}_{1}},{i}_{{j}_{2}}}{R}_{{i}_{{j}_{1}}}{R}_{{i}_{{j}_{2}}}|\) and re-evaluate the family structure until the largest family size is less than the pre-given cutoff. Then, we recovered the removed pairs in the decreasing order of \(|{\rho }_{{i}_{{j}_{1}},{i}_{{j}_{2}}}{R}_{{i}_{{j}_{1}}}{R}_{{i}_{{j}_{2}}}|\) only if the family size is less than the pre-given cutoff after recovering. The greedy strategy is to reduce the family size while remaining the largest variance of the original pedigree, and covariance-like metrics \(|{\rho }_{{i}_{{j}_{1}},{i}_{{j}_{2}}}{R}_{{i}_{{j}_{1}}}{R}_{{i}_{{j}_{2}}}|\) can achieve this aim better than the kingship coefficients.
Fast version of saddlepoint approximation
In SPAGRM, the most computationally demanding section is the calculation of the CGF \({K}_{S}\left(t\right)\) and its derivatives. To alleviate the computation burden, we adopt the basic idea of fastSPA38 to employ a partially normal distribution approximation. We use \({\alpha }_{0.25}\) and \({\alpha }_{0.75}\) to represent the 25th and 75th percentiles of the model residuals, respectively. Additionally, we define the interquartile range (IQR) as \({\alpha }_{0.75}-{\alpha }_{0.25}\). If a model residual falls outside the range between \({\alpha }_{0.75}-{{\rm{IQR}}}\cdot \gamma\) and \({\alpha }_{0.25}+{{\rm{IQR}}}\cdot \gamma\), it is considered as an outlier residual. In this paper, we use the default \(\gamma=1.5\).
Given the definition of the outlier residual, we reformulate the score statistics as
Of the first \({q}_{1}\) families, each includes at least one outlier residual, and the remaining \({q}_{2}\) families do not include any outlier residual. We let
For \({S}_{o}\), the calculation of the MGF and CGF are the same as in the previous sections. For \({S}_{{non}}\), we applied normal distribution approximation to calculate the MGF and CGF. The mean and the variance of \({S}_{{non}}\) under \({H}_{0}\) are
where \(\mu\) is the MAF of the genetic variant, \({R}_{{non}}\) and \({\varPhi }_{{non}}\) are the residuals and the GRM corresponding to the \({q}_{2}\) families without any outlier residual. Assume that \({S}_{{non}}\) follows a normal distribution, the CGF of \({S}_{{non}}\) can be approximated by
and the CGF of \(S={S}_{o}+{S}_{{non}}\) can be approximated by
Similar to the normal distribution approximation, we can pre-calculate common metrics of \({\sum }_{i=1}^{{q}_{2}}{\sum }_{j=1}^{{n}_{i}}{R}_{{i}_{j}}\) and \({R}_{{non}}^{T}\cdot {\varPhi }_{{non}}\cdot {R}_{{non}}\). When calculating \({K}_{{S}_{{non}}}\left(t\right)\) for each genetic variant, only an estimation of \(\mu\) is required. Consequently, the partially normal approximation significantly reduces the computational burden.
In this paper, we also use a hybrid strategy of normal distribution approximation and SPA to balance the high computational efficiency and accuracy. Given a pre-selected cutoff \(r\), if \(\left|s\right| < r\cdot \sqrt{\overline{{Var}}\left(S\right)}\), the normal distribution approximation is applied to calibrate p values, otherwise, more accurate SPA is used to calibrate p values. In this paper, we consider \(r=2\), following fastSPA38.
Variance ratio adjustment for saddlepoint approximation
Pre-calculation of the joint distribution of genotypes and family size reduction strategies may slightly compromise the accuracy of SPA. Thus, we apply a variance ratio adjustment for SPA to ensure the variance from SPA is accurate. The empirical variance of the score statistic is estimated as \({Var}\left(S\right)=2\mu \left(1-\mu \right)\cdot {R}^{T}\cdot \varPhi \cdot R\). Define the calculated variance from SPA as \({{Var}}_{{SPA}}\), and the variance ratio is \(r={Var}\left(S\right)/{{Var}}_{{SPA}}\). Then we use \(S/{sqrt}(r)\) as the observed score statistic for SPA to calibrate p values. Generally, the ratio r is very close to 1.
SPAGRM(INT) and SPAGRM(CCT) could increase statistical power
Suppose that model residuals obtained from a fitted null model are \(R={\left({R}_{1},{R}_{2},...,{R}_{n}\right)}^{T}\), SPAGRM(INT) applies a rank-based inverse normal transformation (INT)53 to update
where \({{\rm{rank}}}\left({R}_{i}\right)\) is the rank of \({R}_{i}\) in model residual vector \(R\), \({qnorm}()\) is the quantile function of a standard normal distribution. Here we use the conventional Blom offset of \(c=3/8\), as in previous study53. Then, SPAGRM(INT) passed the updated residuals \(\widetilde{R}\) to SPAGRM to construct score statistics \(S={\widetilde{R}}^{T}\cdot G\) and calculate p values.
We propose SPAGRM(CCT) to aggregate the results following different models via Cauchy combination test (CCT)46. For a genetic variant, suppose there are \(d\) distinct p values from different models or data transformation, then SPAGRM(CCT) p value is \(\frac{1}{2}-\frac{1}{\pi }\arctan \left({\sum }_{i=1}^{d}\frac{1}{d}\tan \left\{\left(0.5-{p}_{i}\right)\pi \right\}\right)\). For example, SPAGRM(CCT) can combine p values from SPAGRM and SPAGRM(INT). SPAGRM(CCT) can also combine p values from GEE with various correlation structures.
Data simulation
We carried out a series of simulations to evaluate the performance of SPAGRM in terms of type I error rates and power for longitudinal trait analysis. To mimic real genotypes, we used the real variants of unrelated white British subjects in UKB. We randomly selected hundreds of thousands of common variants (\({{\rm{MAF}}} > 0.05\)) and rare variants (\({{\rm{MAF}}} < 0.05\)) from genotype calls (field ID: 22418) and sequencing data (field ID: 23155), respectively. Then we performed the gene-dropping simulation52 using these variants as founder haplotypes that were propagated through the pedigrees of 4 family members and 10 family members shown in Supplementary Fig. 1. Variants with missing genotype rates \( > 0.05\) were excluded from our simulations. Finally, a total of 100,000 common variants (\({{\rm{MAF}}} > 0.05\)) and 100,000 rare variants (\({{\rm{MAF}}} < 0.05{{\rm{\&\; MAC}}}\ge 20\)) were chosen for each dataset. Note that we did not filter for Hardy-Weinberg equilibrium (HWE), so the distribution of HWE p values for the simulated genotypes resembled that of real data. Variants with HWE p values below 1e-6 were excluded from subsequent analyses, unless otherwise specified.
We simulated longitudinal traits following models (3) and (4). For subject \(i\), the number of measurements \({m}_{i}\) was simulated equally distributed ranging from 6 to 15. Three covariates were simulated as \({x}_{{ij}}\) and \({w}_{{ij}}\): the first one is time-invariant following a Bernoulli distribution with a probability of 0.5; the second one is time-invariant variable following the standard normal distribution, and the third one is time-varying with each measurement following an independent standard normal distribution. One time-varying covariate was simulated following an independent standard normal distribution as \({z}_{{ij}}\). In addition, covariates \({{{\boldsymbol{X}}}}_{i}\), \({{{\boldsymbol{W}}}}_{i}\) and \({{{\boldsymbol{Z}}}}_{i}\) have an intercept column of ones. We followed Ko et al.22 to set fixed coefficients \({{\boldsymbol{\beta }}}={\left(1,0.5,0.5,-0.3\right)}^{T},{{\boldsymbol{\tau }}}={\left(0.25,0.3,-0.15,0.1\right)}^{T}\), and variance components \({{{\boldsymbol{\Sigma }}}}_{{{\boldsymbol{\gamma }}}\omega }=\left(\begin{array}{ccc}2 & 0 & 0.2\\ 0 & 1.2 & 0.1\\ 0.2 & 0.1 & 1\end{array}\right)\). Unless otherwise specified, we set variance component parameters \(\sigma=\widetilde{\sigma }=1\) to simulate random effects \({b}_{i}\) and \({\widetilde{b}}_{i}\).
We randomly selected 5 common variants and 5 rare variants as causal variants, and let
where \({g}_{{ik}}\) and \({\widetilde{g}}_{{ik}}\) are standardized genotypes of the \(k\)-th common and rare variant, respectively, genetic effect sizes \({\theta }_{k}=-\log 10\left({{\rm{MAF}}}\right)\times 0.1,\,{\widetilde{\theta }}_{k}=-\log 10\left({{\rm{MAF}}}\right)\times 0.02\). To comprehensively evaluate type I error rates and power for mean and WS variabilities, we simulated four scenarios as below:
-
1.
WSnull/BSnull: \({\tau }_{g}=0,\,{\beta }_{g}=0\);
-
2.
WSalt/BSalt: \({\tau }_{g}=1.5,\,{\beta }_{g}=1\);
-
3.
WSalt/BSnull: \({\tau }_{g}=1.5,\,{\beta }_{g}=0\);
-
4.
WSnull/BSalt: \({\tau }_{g}=0,\,{\beta }_{g}=1\).
For scenario 1, 10,000 datasets including covariates and longitudinal traits were simulated to evaluate type I error rates. For scenarios 2-4, 200 datasets were simulated. In total, \(1\times {10}^{9}\) tests were conducted in scenario 1, and \(1\times {10}^{3}\) tests were conducted in scenarios 2-4.
To assess type I error rates of SPAGRM in presence of cryptic relatedness, we selected 50,000 UKB participants of white British ancestry. Rather than randomly sampling from the full set of 408,961 white British participants, we specifically sampled these individuals from 186,760 related samples, which excluded unrelated individuals, to enhance the proportion of related subjects (Supplementary Fig. 6). Under the null hypothesis of no genetic effects, we simulated longitudinal traits following scenario 1. WSnull/BSnull as mentioned above, except that polygenic effects \({b}_{i}={\sum }_{k=1}^{s}{g}_{{ik}}{\beta }_{k}\) and \({\widetilde{b}}_{i}={\sum }_{k=1}^{s}{g}_{{ik}}{\widetilde{\beta }}_{k}\) were simulated based on \(s=50,000\) randomly selected real genotypes from the odd chromosomes of these individuals, where \({g}_{{ik}}\) is centered genotype value for the \(k\)-th variant and genetic effects \({\beta }_{k},{\widetilde{\beta }}_{k} \sim N(0,1/s)\) for mean and WS variabilities, respectively. Each simulation was repeated 100 times. In total, 100,000 common variants (\({{\rm{MAF}}} > 0.05\)) and 100,000 rare variants (\({{\rm{MAF}}} < 0.05{{\rm{\&\; MAC}}}\ge 20\)) were chosen from the even chromosomes as null SNPs and \(1\times {10}^{7}\) association tests were conducted, respectively. (QQ plots of p values of SPAGRM-based methods in presence of cryptic sample relatedness are displayed in Supplementary Fig. 7. We also evaluated the impact of pedigree cuts on SPAGRM by setting the maximum family size to 3, 5, 7, and 10, and applied these settings to the aforementioned cohort with cryptic relatedness (Supplementary Fig. 8). We also assessed SPAGRM-based methods using a real phenotype (see Supplementary Note). The results demonstrated that pedigree cuts had minimal impact on SPAGRM in the UKB analysis, though they did affect run time (Supplementary Figs. 6c, 9, and 10).
We also used alternative settings to simulate longitudinal traits, distinct from those mentioned above. We first re-simulated scenario 1. WSnull/BSnull and 2. WSalt/BSalt with the variance component parameter \(\widetilde{\sigma }=0\), mimicking a real-world situation where only a few genetic variants influence phenotypic variance24,26,68. Scenario 1 was replicated 100 times with \(1\times {10}^{7}\) tests to evaluate type I error rates, while scenario 2 was replicated 200 times with \(1\times {10}^{3}\) tests to assess empirical power (Supplementary Figs. 16 and 17). We also re-simulated scenario 1. WSnull/BSnull and 2. WSalt/BSalt using an alternative model to investigate the robustness of our proposed method. Instead of the log-normal model from model (4), we simulated WS random effect \({\omega }_{i}\) from the natural logarithm of an inverse-gamma distribution, which is commonly used as a conjugate prior for variance in Bayesian statistics69. We used the setting Inv-Gamma(5, 10) without any special consideration. Supplementary Figs. 18 and 19 indicated that SPAGRM-based methods are robust to model misspecification.
Longitudinal traits extracted from UK Biobank primary care data
The UK Biobank (UKB) primary care data (Category ID: 3001) is derived from electronic health records (EHRs) maintained by General Practitioners (GPs) from multiple data providers in England, Wales, and Scotland (see Data availability). As of the latest release in September 2019, approximating 230,000 UKB participants have been linked to their corresponding primary care data. This dataset includes clinical event records (Field ID: 42040) spanning over 30 years, rich in information of diagnoses, history, symptoms, lab results, and procedures. Two controlled clinical terminologies, Read version 2 (Read v2) and Clinical Terms Version 3 (CTV3) are used to record these primary clinical events.
To generate clinical terms for analyzed longitudinal traits, we initially established mappings between the Read code look-ups (Resource 592) and the clinical event records (i.e., gp_clinical table). Subsequently, we followed a previously validated algorithm48 to created Read v2 and CTV3 clinical terms. All terms that appeared > ~10,000 times in the gp_clinical table underwent manual review to ensure no significant codes were inadvertently overlooked. And clinical terms with low frequency (<100 times), post-treatment, and exhibited ambiguity were manually excluded for each trait.
For each trait, we extracted the measurements from the gp_clinical table using the previous defined codes, and then compared the distributions with those obtained from the UKB assessment center (if available). Referring to the phenotype ranges as well as units provided through the UKB showcase, we excluded records containing implausible values and unit errors. For phenotypes not present in the UKB, we performed the quality control according to previous studies and/or expert knowledge. Taking prostate-specific antigen (PSA) as an example, we followed Hoffmann et al73. to omit PSA > 10 for low-risk prostate cancer PSA levels. Expanding upon the QC by Ko et al22., we also considered the presence of duplicated measurements and outlier values in the gp_clinical table. In general, if the differences among repeated observations were within a pre-given cutoff (usually the standard error of the phenotype), we took the mean of these data or selected the measurement closest to the individual’s overall mean as an approximation. Otherwise, repeated rows were excluded to avoid introducing implausible values. Furthermore, we inspected longitudinal values for each individual and removed outliers defined as values exceeding four times the standard deviation from the individual mean. Here we take the red blood cell (RBC) as an example to display the general pipeline of generating phenotypes used in SPAGRM and TrajGWAS analyses (Supplementary Figs. 30-31).
We particularly emphasize the importance of quality control (QC) to deal with the raw UKB primary care data. The raw clinical events often contain a large amount of duplicated records, unit errors, and implausible values, which should be addressed to ensure the credibility of the results. For example, when examining raw blood pressure data, we identified approximately 370,000 rows (4.6%) of duplicated records. Expanding upon the QC by Ko et al22., we implemented a refined and meticulous QC pipeline for each of the 79 longitudinal traits described above. We selected several longitudinal traits to demonstrate that the refined QC pipeline can help reduce spurious discoveries and increase statistical power (Supplementary Fig. 32 and Supplementary Note).
Genome-wide association studies of 79 longitudinal traits
For each trait, we generally adjust for age, age2, sex, age×sex, and BMI (except for longitudinal BMI) for both mean and WS variability. Age was treated as time-varying covariates; sex was not adjusted for sex-specific traits, PSA for male and CA125 for female; BMI at the enrollment visit was extracted from the UKB assessment center (field ID: 21001). All covariates (except for sex) and phenotypes were standardized with the mean of zero and variance of unity. We additionally adjusted for the first ten principal components (PCs) on the mean component. PCs were not adjusted for the WS variance because this makes no differences for the final results22. Furthermore, we included smoke status and genotyping array as additional covariates for pulmonary measures (FEV1, FVC, FEV1/FVC ratio, and PEF). Supplementary Table 6 gives detailed information about covariates correlation.
We also controlled the effects of medication and/or operation on certain traits. We follow similar framework for these biomarkers also evaluated in Ko et al.22. That is, adding 15 mmHg for SBP and 10 mmHg for DBP to the raw records for subjects taking blood-pressure-lowering medication74; adding 0.208 mmol/L for triglycerides, 1.347 mmol/L for total cholesterol, 1.290 mmol/L for LDL cholesterol, and subtracting 0.060 mmol/L for HDL cholesterol for subjects taking lipid-controlling treatments75. For glycemic measures (HbA1c, random and fasting glucose) and non-HDL cholesterol, a sensible adjustment factor was not available. Thus, we used an “indicator” method by treating medication as a covariate for both mean and WS components. Operation effects were hard to evaluate; thus, we excluded subjects who had ever taken surgical resections of the prostate for PSA, and subjects who had ever taken pituitary and thyroidectomy for TSH, free triiodothyronine (fT3), and free thyroxine (fT4).
We extracted genome-wide significant genetic variants with p values less than \(5\times {10}^{-8}\). We generally excluded variants with MAF \( < 0.002\) for TrajGWAS, due to its inability to control type I error rates for ultra-rare variants when testing WS variance (i.e., \({\tau }_{g}\)). Significant variants identified by SPAGRM, located \( > 200\) kb away from any significant variants detected through TrajGWAS, were clustered as one locus. These were defined as additionally discovered loci by the SPAGRM. Similarly, we also defined loci that were missed by the SPAGRM. Taken 200 kb as one locus, we identified distinct genome-wide significant variants discovered by SPAGRM, along with few markers additionally detected through TrajGWAS. Moreover, we used ANNOVAR76 to functionally annotate these variants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The individual-level genotype and phenotype data are available through formal application to the UK Biobank (http://www.ukbiobank.ac.uk). Description of UK Biobank primary care data is available at https://biobank.ndph.ox.ac.uk/ukb/label.cgi?id=3001. Results from the genome-wide association study analyses in this paper are available on Zenodo at https://doi.org/10.5281/zenodo.14633793 (ref. 77).
Code availability
SPAGRM is implemented as an open source R package available at https://hexupku.github.io/SPAGRM.github.io/. Code used to generate simulation results, real data analyses can be found on Zenodo at https://doi.org/10.5281/zenodo.14619471 and on Github at https://github.com/HeXuPKU/SPAGRM (ref. 78). TrajGWAS (version 0.4.6) can be found at https://github.com/OpenMendel/TrajGWAS.jl. GRAB (version 0.1.1) can be found at https://wenjianbi.github.io/grab.github.io/. GCTA (version 1.94.1) can be found at https://yanglab.westlake.edu.cn/software/gcta/#Overview.
References
Backman, J. D. et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634 (2021).
Beesley, L. J. et al. The emerging landscape of health research based on biobanks linked to electronic health records: Existing resources, statistical challenges, and potential opportunities. Statistics medicine 39, 773–800 (2020).
Bi, W. J. & Lee, S. Scalable and Robust Regression Methods for Phenome-Wide Association Analysis on Large-Scale Biobank Data. Front. Genet. 12, 682638 (2021).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Bi, W. J., Fritsche, L. G., Mukherjee, B., Kim, S. & Lee, S. A Fast and Accurate Method for Genome-Wide Time-to-Event Data Analysis and Its Application to UK Biobank. American Journal Human Genetics 107, 222–233 (2020).
Bi, W. J. et al. Efficient mixed model approach for large-scale genome-wide association studies of ordinal categorical phenotypes. Am. J. Hum. Genet 108, 825–839 (2021).
Meng, W. et al. A Meta-Analysis of the Genome-Wide Association Studies on Two Genetically Correlated Phenotypes Suggests Four New Risk Loci for Headaches. Phenomics 3, 64–76 (2023).
Liu, Z. et al. Phenome-Wide Association Analysis Reveals Novel Links Between Genetically Determined Levels of Liver Enzymes and Disease Phenotypes. Phenomics 2, 295–311 (2022).
Yang, R., Tian, Q. & Xu, S. Mapping quantitative trait loci for longitudinal traits in line crosses. Genetics 173, 2339–2356 (2006).
Khera, A. V. et al. Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell 177, 587–596.e9 (2019).
Tanaka, T. et al. Plasma proteomic biomarker signature of age predicts health and life span. eLife 9, e61073 (2020).
Klarin, D. et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet 50, 1514–1523 (2018).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat Rev Genet 20, 467–484 (2019).
Laird, N. M. & Ware, J. H. Random-Effects Models for Longitudinal Data. Biometrics 38, 963 (1982).
Liang, K.-Y. & Zeger, S. L. Longitudinal data analysis using generalized linear models. Biometrika 73, 13–22 (1986).
Zeger, S. L. & Liang, K. Y. An overview of methods for the analysis of longitudinal data. Stat Med 11, 1825–1839 (1992).
Furlotte, N. A., Eskin, E. & Eyheramendy, S. Genome-wide association mapping with longitudinal data. Genet Epidemiol 36, 463–471 (2012).
Costanza, M. C., Beer-Borst, S., James, R. W., Gaspoz, J. M. & Morabia, A. Consistency between cross-sectional and longitudinal SNP: blood lipid associations. Eur J Epidemiol 27, 131–138 (2012).
Xu, Z., Shen, X. & Pan, W. & Alzheimer’s Disease Neuroimaging, I. Longitudinal analysis is more powerful than cross-sectional analysis in detecting genetic association with neuroimaging phenotypes. PLoS One 9, e102312 (2014).
Chiu, Y. F., Justice, A. E. & Melton, P. E. Longitudinal analytical approaches to genetic data. BMC Genet 17, 4 (2016).
Dzubur, E. et al. MixWILD: A program for examining the effects of variance and slope of time-varying variables in intensive longitudinal data. Behav Res Methods 52, 1403–1427 (2020).
Ko, S. et al. GWAS of longitudinal trajectories at biobank scale. Am. J. Hum. Genet 109, 433–445 (2022).
Rothwell, P. M. et al. Prognostic significance of visit-to-visit variability, maximum systolic blood pressure, and episodic hypertension. Lancet 375, 895–905 (2010).
Ivarsdottir, E. V. et al. Effect of sequence variants on variance in glucose levels predicts type 2 diabetes risk and accounts for heritability. Nat Genet 49, 1398–1402 (2017).
Zhou, J. J., Coleman, R., Holman, R. R. & Reaven, P. Long-term glucose variability and risk of nephropathy complication in UKPDS, ACCORD and VADT trials. Diabetologia 63, 2482–2485 (2020).
Kemper, K. E. et al. Genetic influence on within-person longitudinal change in anthropometric traits in the UK Biobank. Nat Commun 15, 3776 (2024).
Chen, H. et al. Control for Population Structure and Relatedness for Binary Traits in Genetic Association Studies via Logistic Mixed Models. Am J Hum Genet 98, 653–666 (2016).
Dey, R. et al. Efficient and accurate frailty model approach for genome-wide survival association analysis in large-scale biobanks. Nat Commun 13, 5437 (2022).
Loh, P.-R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nature Genetics 47, 284–290 (2015).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nature Genetics 50, 1335–1341 (2018).
He, L. & Kulminski, A. M. Fast Algorithms for Conducting Large-Scale GWAS of Age-at-Onset Traits Using Cox Mixed-Effects Models. Genetics 215, 41–58 (2020).
Jiang, L., Zheng, Z., Fang, H. & Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nature Genetics 53, 1616–1621 (2021).
Loh, P.-R., Kichaev, G., Gazal, S., Schoech, A. P. & Price, A. L. Mixed-model association for biobank-scale datasets. Nature Genetics 50, 906–908 (2018).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nature Genetics 51, 1749–1755 (2019).
Bi, W. et al. Scalable mixed model methods for set-based association studies on large-scale categorical data analysis and its application to exome-sequencing data in UK Biobank. American Journal Human Genetics 110, 762–773 (2023).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nature Genetics 53, 1097–1103 (2021).
Kuonen, D. Miscellanea Saddlepoint approximations for distributions of quadratic forms in normal variables. Biometrika 86, 929–935 (1999).
Dey, R., Schmidt, E. M., Abecasis, G. R. & Lee, S. A Fast and Accurate Algorithm to Test for Binary Phenotypes and Its Application to PheWAS. Am J Hum Genet 101, 37–49 (2017).
Jiang, D., Mbatchou, J. & McPeek, M. S. Retrospective Association Analysis of Binary Traits: Overcoming Some Limitations of the Additive Polygenic Model. Hum Hered 80, 187–195 (2015).
Thornton, T. & McPeek, M. S. ROADTRIPS: case-control association testing with partially or completely unknown population and pedigree structure. Am J Hum Genet 86, 172–184 (2010).
Jakobsdottir, J. & McPeek Mary S. MASTOR: Mixed-Model Association Mapping of Quantitative Traits in Samples with Related Individuals. American Journal Human Genetics 92, 652–666 (2013).
Wu, X. & McPeek, M. S. L-GATOR: Genetic Association Testing for a Longitudinally Measured Quantitative Trait in Samples with Related Individuals. Am J Hum Genet 102, 574–591 (2018).
Hayeck, T. J. et al. Mixed Model Association with Family-Biased Case-Control Ascertainment. Am J Hum Genet 100, 31–39 (2017).
Thornton, T. et al. Estimating Kinship in Admixed Populations. American Journal Human Genetics 91, 122–138 (2012).
Chow, C. & Liu, C. Approximating discrete probability distributions with dependence trees. IEEE Transactions Information Theory 14, 462–467 (1968).
Liu, Y. & Xie, J. Cauchy combination test: a powerful test with analytic p-value calculation under arbitrary dependency structures. J Am Stat Assoc 115, 393–402 (2020).
Liu, Y. et al. ACAT: A Fast and Powerful p Value Combination Method for Rare-Variant Analysis in Sequencing Studies. Am J Hum Genet 104, 410–421 (2019).
Denaxas, S. et al. A semi-supervised approach for rapidly creating clinical biomarker phenotypes in the UK Biobank using different primary care EHR and clinical terminology systems. JAMIA Open 3, 545–556 (2020).
Jurgens, S. J. et al. Adjusting for common variant polygenic scores improves yield in rare variant association analyses. Nat Genet 55, 544–548 (2023).
Campos, A. I. et al. Boosting the power of genome-wide association studies within and across ancestries by using polygenic scores. Nat Genet 55, 1769–1776 (2023).
Bennett, D., O’Shea, D., Ferguson, J., Morris, D. & Seoighe, C. Controlling for background genetic effects using polygenic scores improves the power of genome-wide association studies. Sci Rep 11, 19571 (2021).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30, 97–101 (2002).
McCaw, Z. R., Lane, J. M., Saxena, R., Redline, S. & Lin, X. Operating characteristics of the rank-based inverse normal transformation for quantitative trait analysis in genome-wide association studies. Biometrics 76, 1262–1272 (2020).
Loh, P. R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 48, 1443–1448 (2016).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015).
Levey, A. S. et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 150, 604–612 (2009).
Stanzick, K. J. et al. Discovery and prioritization of variants and genes for kidney function in >1.2 million individuals. Nat Commun 12, 4350 (2021).
Deng, Y. & Tsao, B. P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol 6, 683–692 (2010).
Di, D. et al. Polymorphisms of BLK are associated with renal disorder in patients with systemic lupus erythematosus. J Hum Genet 65, 675–681 (2020).
Zhou, X. J. et al. Association of systemic lupus erythematosus susceptibility genes with IgA nephropathy in a Chinese cohort. Clin J Am Soc Nephrol 9, 788–797 (2014).
Zhang, Y. M. et al. Shared genetic study gives insights into the shared and distinct pathogenic immunity components of IgA nephropathy and SLE. Mol Genet Genomics 296, 1017–1026 (2021).
Norgett, E. E. et al. A role for VAX2 in correct retinal function revealed by a novel genomic deletion at 2p13.3 causing distal Renal Tubular Acidosis: case report. BMC Med. Genet. 16, 38 (2015).
Moksnes, M. R. et al. Genome-wide meta-analysis of iron status biomarkers and the effect of iron on all-cause mortality in HUNT. Commun Biol 5, 591 (2022).
Bell, S. et al. A genome-wide meta-analysis yields 46 new loci associating with biomarkers of iron homeostasis. Commun Biol 4, 156 (2021).
Gardiner, J. C., Luo, Z. & Roman, L. A. Fixed effects, random effects and GEE: what are the differences? Stat Med 28, 221–239 (2009).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82 (2011).
Tang, K., Naseri, A., Wei, Y., Zhang, S. & Zhi, D. Open-source benchmarking of IBD segment detection methods for biobank-scale cohorts. Gigascience 11, giac111 (2022).
Yang, J. et al. FTO genotype is associated with phenotypic variability of body mass index. Nature 490, 267–272 (2012).
German, C. A., Sinsheimer, J. S., Zhou, J. & Zhou, H. WiSER: Robust and scalable estimation and inference of within‐subject variances from intensive longitudinal data. Biometrics 78, 1313–1327 (2021).
Barndorff-Nielsen, O. E. Approximate Interval Probabilities. Journal Royal Statistical Society: Series B (Methodological) 52, 485–496 (1990).
Liu, F., Kirichenko, A., Axenovich, T. I., van Duijn, C. M. & Aulchenko, Y. S. An approach for cutting large and complex pedigrees for linkage analysis. Eur J Hum Genet 16, 854–860 (2008).
Kirichenko, A. V., Belonogova, N. M., Aulchenko, Y. S. & Axenovich, T. I. PedStr software for cutting large pedigrees for haplotyping, IBD computation and multipoint linkage analysis. Ann Hum Genet 73, 527–531 (2009).
Kachuri, L. et al. Genetically adjusted PSA levels for prostate cancer screening. Nat Med 29, 1412–1423 (2023).
Giri, A. et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat Genet 51, 51–62 (2019).
Yusuf, S. et al. Cholesterol Lowering in Intermediate-Risk Persons without Cardiovascular Disease. N. Engl J Med 374, 2021–2031 (2016).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
He, X. et al. SPAGRM. Zenodo https://doi.org/10.5281/zenodo.14633793 (2025).
He, X. et al. SPAGRM. Zenodo https://doi.org/10.5281/zenodo.14619471 (2025).
Acknowledgements
This research was supported by National Natural Science Foundation of China (62273010, W.B.) and the Fundamental Research Funds for the Central Universities. UK Biobank data was accessed under the accession number 78795. This research is supported by high-performance computing platform of Peking University.
Author information
Authors and Affiliations
Contributions
H.X., P. Z., W.Y, and W.B designed the experiments. H.X. and W.B. performed the analyses. Y.M. gave valuable suggestions on the retrospective idea. Yufei Liu and Ying Li contributed to the code programming. H.X. and W.B. wrote the manuscript with the assistance of L.X., Yin Li, P.Z., X.Z, W.Z. and S.L. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, H., Ma, Y., Xu, Ll. et al. SPAGRM: effectively controlling for sample relatedness in large-scale genome-wide association studies of longitudinal traits. Nat Commun 16, 1413 (2025). https://doi.org/10.1038/s41467-025-56669-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56669-1
This article is cited by
-
Applying weighted Cox regression to genome-wide association studies of time-to-event phenotypes
Nature Computational Science (2025)
-
Boosting power for time-to-event GWAS analysis affected by case ascertainment
Nature Computational Science (2025)
