
Abstract
As a global transcription activator or repressor, the representative OmpR/PhoB family response regulator PhoP plays a crucial role in regulating bacterial pathogenicity and stress adaptation. However, the molecular mechanisms underlying the transcriptional regulation that define its differential functions remain largely unclear. In the present study, we determine three cryo-EM structures of Mycobacterium tuberculosis (Mtb) PhoP-dependent transcription activation complexes (PhoP-TACs) and build one preliminary cryo-EM structure model of Mtb PhoP-dependent transcription repression complex (PhoP-TRC). In PhoP-TACs, tandem PhoP dimers cooperatively recognize various types of promoters through conserved PhoP-PHO box interactions, which displace the canonical interactions between the -35 element and σAR4 of RNA polymerase (RNAP), unraveling complex transcription activation mechanisms of PhoP. In PhoP-TRC, one PhoP dimer binds and significantly distorts the upstream PHO box of the promoter cross-talked with the global nitrogen regulator GlnR through the PhoP-PHO box, PhoP-GlnR and αCTD-DNA interactions. This unique binding of PhoP creates steric hindrances that prevent additional GlnR binding, positioning PhoP within a unique ‘competitive occluding model’, as supported by prior biochemical observations. Collectively, these findings reveal the dual molecular mechanisms of PhoP-dependent transcription regulation, and offer valuable insights for further exploration of the enormous PhoP-like OmpR/PhoB family response regulators.
Similar content being viewed by others

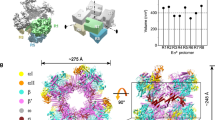

Introduction
Transcription initiation, finely orchestrated by the highly conserved multi-subunit RNA polymerase (RNAP, α2ββ‘ωσ) and various transcription factors, serves as a critical pivot for the precise regulation of gene expression in bacteria in response to changing signals. Regarding host-keeping genes, the principal σ factor specifically recognizes the promoter consensus -10 and -35 elements through its conserved domains, σR2 and σR4, respectively1. These interactions are further enhanced by the conserved σR2-β‘ clamp helices (β‘ CH), σR4-β flap interactions, and potential αCTD-DNA or/and αCTD-σR4 interactions, which collectively promote the formation of an active RNAP-promoter open complex (RPo) to initiate transcription2,3,4,5,6,7. In the context of promoters containing suboptimal elements, the formation of RPo is facilitated by specific transcription activators that recruit RNAP or bend DNA to compensate for the weaker σ-DNA interactions compared to those occurred in one canonical RPo. Consequently, this activator-containing RPo is referred to as a transcription activation complex (TAC)8,9. As recently reviewed, transcription activators from the σ70 family can be classified into four categories based on the positioning of their binding sites relative to the –35 element: Type I (upstream site), Type II (downstream site), Type III (no DNA binding), and Type IV (both upstream and downstream sites)10. Conversely, transcription initiation can be inhibited by transcription repressors that utilize mechanisms such as occlusion, DNA looping, or locking of RNAP to impede the function of either RNAP or the activators9,11. The complex formed in this scenario is termed a transcription repression complex (TRC). Notably, many transcription factors have been identified as either activators or repressors at different promoters, underscoring the intricate nature of finely tuned regulatory mechanisms.
As being identified, a majority of OmpR/PhoB family response regulators (RRs), characterized by distinct winged-helix DNA binding motifs, function as dual functional transcription regulators12,13. Typically, RRs, in conjunction with their associated membrane-bound sensor kinases, form integral signaling cascades—known as two-component systems (TCSs). These systems facilitate the transient regulation of stress-responsive genes that enhance bacterial survival and pathogenicity14. Numerous studies have shown that the OmpR/PhoB family RRs are widely distributed among bacteria, particularly in notable pathogens such as Mycobacterium tuberculosis (Mtb), Vibrio cholerae (Vch), Pseudomonas aeruginosa (Pae), Escherichia coli (Eco) or the model bacteria Streptomyces coelicolor (Sco)15,16,17,18,19,20. As a representative member of the enormous OmpR/PhoB family, Mtb PhoP remodels the transcriptional network of Mtb by regulating over 100 genes involved in virulence, lipid biosynthesis, carbon metabolism, low pH, hypoxia, and stress response, functioning as both a global transcriptional activator and repressor16,21,22,23,24. Notably, the complex regulatory mechanisms of PhoP in response to phosphate starvation and its interplay with nitrogen availability have been documented25,26: PhoP not only activates the transcription of phosphate assimilation genes, including those for high-affinity phosphate-specific transport systems genes (pstSCAB) and the global phosphate starvation stress response modulator gene (phoU)27,28,29, but also represses the transcription of relevant dissimilation or other metabolic genes. For example, PhoP represses the nitrogen metabolism gene amtB through cross-talk with the global nitrogen metabolism regulator GlnR30,31,32. However, what determines PhoP to deferentially act as a dual transcriptional regulator remain inadequately understood.
Structural based sequence alignment shows that Mtb PhoP shares high structural similarity with the other homologous members (Supplementary Fig. 1). It comprises a conserved N-terminal receiver domain (REC) and a C-terminal effective DNA binding domain (DBD). Biochemical assays indicate that phosphorylation of the REC by its cognate sensor kinase PhoR or by acetyl phosphate (AcP) enhances the DNA binding affinity of DBD21,22,33,34. The specific direct repeat units (DRus) PhoP binds are referred as PhoP binding boxes (PHO boxes), each of which carries two conserved PhoP binding sites (site a and site b) separated by several nucleotides (Fig. 1)34,35,36. The co-crystal structure of PhoP bound to a PHO box-containing promoter DNA reveals that a PhoP dimer interacts with the PHO box through conserved winged helices, akin to other transcription regulators in the OmpR/PhoB family12,22,37,38,39,40. These interactions facilitate a compact arrangement of the PhoP_RECs and PhoP_DBDs, which restricts a 4-bp spacer between the two binding sites41. Notably, PhoP is capable of regulating multiple PHO promoters that consists of two or more PHO boxes, a similar characteristic to its homolog PhoB20,27,29,35,42,43. The molecular details on how these PhoP-like OmpR/PhoB family regulators efficiently control such complex promoters remain to be elucidated.

a DNA scaffold used in structure determination of Mtb 2PhoP-TAC (top panel) and two views of the cryo-EM density map of Mtb 2PhoP-TAC (bottom panel). b DNA scaffold used in structure determination of Mtb 4PhoP-TAC (top panel) and two views of the cryo-EM density map of Mtb 4PhoP-TAC (bottom panel). c DNA scaffold used in structure determination of Mtb 6PhoP-TAC (top panel) and two views of the cryo-EM density map of Mtb 6PhoP-TAC (bottom panel). According to the structures, the PhoP binding box, −35 element, -10 element is boxed with yellow, violet, and brown colors, respectively. a1 site, b1 site, a2 site, b2 site, a3 site and b3 site of the PhoP binding box are shaded with light orange, dark cyan, brown, cyan, purple, and dark green, respectively. The EM density maps are colored as indicated in the color key. NT, non-template-strand promoter DNA; T, template-strand promoter DNA.
Here, we determine three cryo-EM structures of the PhoP-dependent transcription activation complexes (2PhoP-TAC, 4PhoP-TAC, 6PhoP-TAC) consisting of Mtb RNA polymerase (RNAP), Mtb PhoP, and the well-characterized consensus promoters including various numbers of PHO boxes, respectively. Structural comparisons of PhoP-TACs and the obtained Mtb RPo demonstrate that PhoP specifically recognizes promoter PHO boxes as either dimer or distinct tandem dimers (tetramer/hexamer). These downstream PhoP-DNA interactions displace the canonical interactions between the -35 element and σAR4 of Mtb RNAP. Additionally, PhoP engages in substantial protein-protein interactions with the conserved domains (β flap and σAR4) of RNAP. These indicate PhoP functions through diverse and complex transcription activation mechanisms. Based on the prior competitive identification of PhoP on the PhoP-GlnR cross-talked nitrogen assimilation promoter, the DNA binding characteristics of PhoP, and the cryo-EM map of PhoP-TRC obtained, we were able to build one preliminary cryo-EM structural model for the PhoP-dependent transcription repression complex (PhoP-TRC), which comprises Mtb RNAP, PhoP, GlnR, and their cross-talked nitrogen assimilation promoter DNA. Notably, PhoP is restrained in a unique repression mode within PhoP-TRC: competitive binding of the PhoP dimer to the upstream PHO box significantly distorts the DNA double-strands at the following spacer. This distortion creates substantial steric hinderances to impede the binding of a third GlnR dimer, thereby repressing GlnR-dependent transcription activation. Collectively, these structural and biochemical data shed new light on the dual molecular mechanisms underlying PhoP-dependent transcription regulation, and provide valuable insights for exploring the PhoP-like OmpR/PhoB family transcription regulators and target development.
Results
Overall structures of PhoP-TAC and PhoP-TRC
To gain deeper structural insights into PhoP-dependent transcription regulation, we reconstituted the PhoP-TACs using purified Mtb RNAP, Mtb PhoP protein, and previously characterized phosphate metabolism promoters that contain various numbers of PHO boxes (Fig. 1 and Supplementary Fig. 2)27,29. The high-affinity phosphate-specific transporter promoter pstS comprises one PHO box, with the a1 and b1 sites overlapping the -35 element (Fig. 1a). In contrast, the promoter for the global phosphate starvation response modulator, phoU4, contains two PHO boxes, featuring upstream a2-b2 sites and downstream a1-b1 sites (Fig. 1b). An extension of the phoU4 scaffold by 27 base pairs (bp) results in a longer phoU6 scaffold, which consists of three PHO boxes corresponding to the upstream a3-b3 sites, the middle a2-b2 sites, and the downstream a1-b1 sites (Fig. 1c). Similarly, the PhoP-TRC was assembled by incubating Mtb RNAP, PhoP, GlnR, and the identified PhoP-GlnR cross-talked nitrogen assimilation promoter amtB, which is repressed by PhoP30,32. Furthermore, the downstream region of each promoter was annealed to include a consensus –10 element, a 13-nucleotide transcription bubble, and an 11-bp downstream double-stranded DNA segment, as previously reported37,44,45,46. To enhance the DNA binding affinity, the PhoP proteins used for assembling the PhoP-TACs or PhoP-TRC were phosphorylated with AcP prior to their incubation with each DNA scaffold and the RNAP holoenzyme21,34.
Following the purification process, each complex achieved to a high degree of homogeneity. As anticipated, SDS-PAGE analysis of the complexes purified through size-exclusion chromatography confirmed the presence of all assembled components (Supplementary Fig. 3a–d). Consistently, in vitro transcription assays utilizing the PhoP target promoter fused with a fragment of the mango sequence showed enhanced transcriptional activity of PhoP on the pstS promoter (which contains one PHO box), the phoU promoter (which contains three PHO boxes), and the pks2 promoter which is predicted to contain PHO boxes (Supplementary Fig. 3e). The inclusion of phosphorylated PhoP in electrophoretic mobility shift assays (EMSA) resulted in the formation of a specific larger band of the PhoP-TAC compared to the PhoP-DNA complex in the presence of heparin, a competitive inhibitor. In contrast, substituting PhoP with the homologous OmpR/PhoB family protein Mtb GlnR, or replacing the PhoP specific promoter with a heterologous negative control promoter, prevented the formation of similar discernible larger bands, indicating that the PhoP-TAC was specifically assembled (Supplementary Fig. 3f). The purified PhoP-dependent transcription activation complexes present promising profiles for subsequent cryo-EM analysis. After multiple attempts to optimize the samples and data collection parameters, we successfully obtained a dataset of Mtb RPo that exclusively contains Mtb RNAP and promoter DNA, and three datasets of Mtb PhoP-TAC featuring various numbers of PhoP molecules (Figs. 1–3, Tables 1–2, and Supplementary Figs. 4–8). Additionally, SDS-PAGE analysis also showed an assembled complex with varied ratio of PhoP and GlnR proteins on the cross-talked promoter amtB (Fig. 4a), and presence of PhoP clearly reduced the corresponding GlnR-dependent transcriptional activities (Fig. 4b). These findings demonstrate that all purified proteins are enzymatically active and that PhoP exerts dual transcriptional regulatory roles on the phosphate-nitrogen metabolism promoters, as previously verified25,28,32. As anticipated, one cryo-EM dataset of Mtb PhoP-TRC that includes Mtb RNAP, promoter DNA engaged by both PhoP and GlnR proteins was finally obtained (Fig. 4c–e, Tables 1–2, and Supplementary Fig. 9).
Structural analysis show that each cryo-EM map reveals distinct densities for the major and minor grooves of the downstream promoter DNA, as well as for each subunit of RNAP, thereby allowing for the fitting of the reported model of Mtb RPo (PDB ID: 6VVY)47 (Supplementary Figs. 10–13). The RNAP core enzyme exhibits a local resolution of approximately 3.0–4.5 Å, however, the peripheral maps of the αCTD and PhoP molecules that interact with the upstream promoter DNA appear more flexible. Notably, our recently resolved model of Mtb GlnR-TAC (PDB ID: 8HIH) containing two GlnR dimers fits well into the downstream densities of PhoP-TRC37. Based on the structural characteristics of protein-DNA and protein-protein interactions within the PhoP-TACs, GlnR-TAC, and previous competition verification of PhoP on the PhoP-GlnR cross-talked promoter10,30,37, a preliminary structural model of PhoP-TRC were feasible to be built for docking analysis. In PhoP-TRC, the electron densities engaging the upstream DNA suggest the presence of a PhoP dimer comprising PhoPV_DBD and PhoPVI_DBD. Structural analysis reveals that binding of the PhoP dimer greatly contributes to significant distortion of the upstream DNA (Fig. 4e and Supplementary Figs. 9, 13). These findings suggest that PhoP acts through a unique mechanism of transcription repression. Although the electron densities for PhoP_RECs and GlnR-RECs are incomplete in the structures, those adjacent to RNAP αNTD offer new insights into their potential regulatory roles in stabilizing PhoP-TRC.
In comparison to the Mtb RPo that lacks the PhoP protein, we designate the PhoP-TACs containing two, four, and six PhoP molecules as 2PhoP-TAC, 4PhoP-TAC, and 6PhoP-TAC, respectively. The high-resolution structure of 6PhoP-TAC not only exhibits similar protein-DNA and protein-protein interactions as those observed in 2PhoP-TAC and 4PhoP-TAC, but it also reveals the engagement of αCTD and an additional PhoP dimer. Therefore, we select 6PhoP-TAC to elucidate the distinct structural basis of PhoP-dependent transcription activation (Figs. 2, 3 and Supplementary Figs. 5–7, 10–12, 14), while employing PhoP-TRC to investigate the structural basis of PhoP-dependent transcription repression (Fig. 4 and Supplementary Figs. 9, 13, 14).
Tandem PhoP dimer cooperatively binds to promoter PHO box
Structural analyses of both PhoP-TACs and PhoP-TRC reveal a notable feature: two PhoP molecules form a tandem dimer that cooperatively binds to each PHO box (Figs. 1, 2a, and Supplementary Figs. 10c, 11c, 11e, 12a, 12c, 12e, 13h). These findings are consistent with previously reported structures of Mtb PhoP-DNA and other members of the OmpR/PhoB family regulators12,13,37,38,39,41,48. In the three structures of PhoP-TACs, the downstream PhoPII_DBD and PhoPI_DBD form a head-to-tail dimer, and interact with the a1 and b1 sites that overlap with the −35 element, respectively (Fig. 2b, c and Supplementary Figs. 10c, 11c, 12a). The long helix α7 of each PhoP_DBD inserts into the DNA major groove, allowing for specific recognition of the DNA bases. Meanwhile, the C-terminal β hairpin extends into the adjacent DNA minor groove, and helix α5 interacts with the DNA backbone at its terminus (Fig. 2b, c). Furthermore, the dimer is stabilized by polar and hydrophobic interactions between PhoPI_DBD and PhoPII_DBD (Fig. 2d). By virtue of similar PhoP-DNA and dimeric PhoP-PhoP interactions, two or three tandem PhoP dimers collaboratively interact with the corresponding PHO boxes of promoter DNA, thereby facilitating the formation of 4PhoP-TAC and 6PhoP-TAC, respectively (Figs. 1, 2, and Supplementary Figs. 11c, 11e, 12a, 12c, 12e). Consistently, mutations in the key residues of PhoP (E161A, E164A, P176A, N212A, E215A, S219L, and R237A) involved in these interfaces impair PhoP-dependent transcription activities (Fig. 2e), underscoring the importance of these residues in both dimerization and DNA binding activity of PhoP.

a Relative locations of Mtb PhoPI_DBD, PhoPII_DBD, PhoPIII_DBD, PhoPIV_DBD, PhoPV_DBD, PhoPVI_DBD located at the upstream double-stranded DNA. b, The relative locations between PhoPI_DBD and PhoPII_DBD bound to the promoter DNA. The secondary structural elements involved in PhoPI_DBD and PhoPII_DBD are labeled, respectively. c Detailed interactions between Mtb PhoPI_DBD, PhoPII_DBD, and their corresponding PhoP binding sites. The key residues involved are shown as green spheres. d The detailed interactions between PhoPI_DBD and PhoPII_DBD. The key residues involved in PhoPI_DBD and PhoPII_DBD are shown as wheat and blue spheres, respectively. e Substitutions of PhoP residues involved in promoter engagement and dimeric interface reduce in vitro transcription activities. Data for in vitro transcription assays are means of 3 technical replicates. Error bars represent mean ± SEM of n = 3 experiments. Source data are provided as a Source Data file. f Relative locations of PhoPII_DBD, σAR4, and the upstream double-stranded DNA in Mtb 6PhoP-TAC. g Relative locations of σAR4 and the upstream typical −35 element DNA in Mtb RPo (PDB ID: 6VVY). Colors are shown as in Fig. 1c.
In addition, structural comparisons between the PhoP-TACs and Mtb RPo reveal a significant distinction: PhoPII_DBD inserts into the DNA major groove overlaps with the -35 element and largely elevates the DNA strands (Fig. 2f, g). This unique modification disrupts the direct interactions between σAR4 and the -35 element, which are typically observed in most bacterial RPo, thereby enabling PhoP to drive RNAP for the specific transcription of PhoP target genes.
Cryo-EM analysis of different PhoP-TACs reveals the structural basis for PhoP functioning as a global transcription activator
By comparing different PhoP-TACs with Mtb RPo, we can clearly delineate the characteristics of PhoP-dependent transcription activation—PhoP specifically recognizes promoter PHO boxes by forming distinct assemblies of tandem dimer, tetramer, and hexamer, and recruits RNAP through extensive interactions with its conserved domains, including the N-terminal domain of RNAP α subunit (αNTD), β flap, σAR4, and αCTD (Figs. 1–3 and Supplementary Figs. 10–14).

a Relative locations of Mtb PhoPI_DBD and RNAP β flap (Left and Middle). PhoPI_DBD and RNAP β flap are shown in surface style (Left). Residues involved between PhoPI_DBD and RNAP β flap are shown as slate (PhoPI_DBD) and cyan (RNAP β flap) spheres (Right). b Relative locations of Mtb PhoP_DBDs and RNAP σAR4 (Left and Middle). PhoPI_DBD, PhoPII_DBD and RNAP σAR4 are shown in surface (Left). Residues involved in interactions between PhoP_DBDs and RNAP σAR4 are shown as blue (PhoPI_DBD), wheat (PhoPII_DBD) and yellow (RNAP σAR4) spheres (Middle and Right). RNAP β flap is colored in cyan, and RNAP σAR4 is colored in yellow. c Conformational comparisons of αCTD and DNA from 6PhoP-TAC and GlnR-TAC. gray, PhoP or GlnR; pink, αCTD; red, DNA from 6PhoP-TAC; gray, DNA from GlnR-TAC. d Substitutions of PhoP residues involved in PhoP-β flap, PhoP-σAR4 interfaces compromise in vitro transcription activities. Data for in vitro transcription assays are means of 3 technical replicates. Error bars represent mean ± SEM of n = 3 experiments. Source data are provided as a Source Data file. The other colors are shown as in Fig. 1.
Although the structure of 4PhoP-TAC closely resembles our recently reported cryo-EM structure of Mtb GlnR-TAC (PDB ID: 8HIH)37, the αCTD is not visualized beneath the upstream DNA in 4PhoP-TAC (Figs. 1, 3c and Supplementary Fig. 14). Notably, three tandem PhoP dimers interact specifically with the three typical PHO boxes (namely, a1-b1, a2-b2, and a3-b3 sites) in 6PhoP-TAC, collectively inducing significant distortion of the upstream DNA in conjunction with the PhoP_RECs adjacent to the αNTDs (Figs. 1c, 2, 3 and Supplementary Figs. 12a, 12c, 12e). Though the densities of PhoP_RECs are not particularly unambiguous, the buried surface areas between PhoP_RECs and αNTD are approximately 350 Å2 and 120 Å2, respectively. This observation suggests a positive role of PhoP_RECs in cooperatively bridging the upstream PhoP_DBDs and the main body of RNAP, thus stabilizing 6PhoP-TAC through assembling into compact architectures41.
Besides, all PhoP-TACs are also stabilized by substantial protein-protein interactions between PhoP and the conserved domains (β flap, σAR4, and αCTD) of RNAP (Fig. 3 and Supplementary Figs. 10b, 10d, 11b, 11d, 12b, 12d), underscoring the general significance of these interactions in maintaining the stability of PhoP-TACs. Helix α6 of the PhoP I_DBD forms both polar and hydrophobic contacts with the loop of the RNAP β flap (Fig. 3a). Furthermore, the C-terminal loop of helix α6 in PhoPI_DBD is likely to engage in electrostatic interactions with the long loop from σAR4, while helix α6 from the PhoPII_DBD interacts with the conserved C-terminal helix of σAR4 (Fig. 3b). Notably, the αCTD, which is observed in 6PhoP-TAC but absent in 2PhoP-TAC and 4PhoP-TAC, is positioned approximately 2 nm (equivalent to one helix diameter) upstream compared to its location in GlnR-TAC (Fig. 3c and Supplementary Fig. 14). As suggests a favorable role for αCTD in stabilizing the structure of 6PhoP-TAC. Consistent with this observation, site-directed mutagenesis of the involved residues (T191A, K197A, H201A, D206A, and F207A) or truncation of the REC domain (DBD) significantly diminishes PhoP-dependent transcriptional activities, particularly for K197A, F207A, and DBD, highlighting the critical role of these interactions in promoting the transcriptional activity of PhoP (Fig. 3d).
Cryo-EM analysis of PhoP-TRC reveals unique structural mechanism for PhoP acting as a transcription repressor
Both in vitro and in vivo investigations demonstrate that six GlnR binding sites (a1-b1 sites, a2-b2 sites, and a3-b3 sites) within the amtB promoter are required for efficient transcription activation of nitrogen metabolism genes in response to nitrogen starvation30,49. PhoP, in turn, competitively represses GlnR-dependent transcription at the upstream sites of the PhoP-GlnR cross-talked promoter amtB, thereby maintaining a balance between the environmental phosphate and nitrogen availability50. Based on the above competition verification of PhoP and the structural characteristics of protein-DNA, protein-protein interactions in PhoP-TACs and GlnR-TAC, we were able to build a preliminary cryo-EM structural model of PhoP-TRC for docking analysis, providing deep insights into the molecular mechanism of PhoP-dependent transcription repression (Fig. 4 and Supplementary Figs. 9, 13). Additionally, a model of 6GlnR-TAC targeting the amtB promoter can also be constructed according to its high structural similarities with 6PhoP-TAC (Supplementary Fig. 14b).

a Gel filtration maps and SDS-PAGE analysis of Mtb PhoP-TRC. Source data are provided as Source Data files. b Relative transcription activity of Mtb PhoP on the PhoP-GlnR co-regulated amtB promoter and the amtB_mut promoter determined by in vitro transcription assays. Mutations of PhoP (truncation of the PhoP_REC domain) or the upstream PhoP binding sites (amtB_mut promoter) cause defects on the transcription repression activity. Data for in vitro transcription assays are means of 3 technical replicates. Error bars represent mean ± SEM of n = 3 experiments. Source data are provided as a Source Data file. c DNA scaffold used in structure determination of Mtb PhoP-TRC. GlnR binding box (a1-b1 sites, a2-b2 sites, and a3-b3 sites) and PhoP binding box (a3’-b3’ sites) are framed in green and yellow color, respectively. The a1 site, b1 site, a2 site, b2 site, a3 site, b3 site, a3’ site, b3’ site are shaded in light orange, dark cyan, brown, cyan, khaki, light green, purple, and dark green, respectively. d Two views of the cryo-EM density map of Mtb PhoP-TRC. The EM density map is colored as indicated in the color key. e The promoter DNA bends toward the main body of RNAP in the preliminary structure model of PhoP-TRC compared with the modeled structure of Mtb 6GlnR-TAC. Mtb 6GlnR-TAC is modeled based on the structural similarities of 6PhoP-TAC. The other colors are shown as in Fig. 1.
In the structural model of PhoP-TRC, four GlnR molecules bind collaboratively to the a1, b1, a2, and b2 sites of the promoter DNA, simultaneously engaging in various protein-protein interactions with the conserved domains of RNAP (β flap, σAR4, αCTD, and αNTD) (Fig. 4c, d and Supplementary Fig. 13). These interactions closely resemble those observed in the Mtb GlnR-TAC structure (Supplementary Fig. 14). Strikingly, one PhoP dimer (PhoPV and PhoPVI) competitively interacts with the upstream PhoP binding box (a3’-b3’ sites) which overlaps with the a3 site, thereby occluding similar binding by the third GlnR dimer as observed in 6GlnR-TAC (Fig. 4c, Supplementary Figs. 13h, 14b). Structural analysis reveals the unique spacer between the b3’ and a2 sites is much longer ( ~ 15 bp) than those in 6PhoP-TAC or 6GlnR-TAC ( ~ 6 bp), which enables PhoPV_DBD and the dimeric PhoP_RECs to interact with the adjacent GlnR_RECs, presenting buried surface areas of approximately 95 Å2 and 168 Å2, respectively (Fig. 4e, middle panel). This finding underscores the importance of the long spacer in facilitating PhoP-dependent transcription repression. Consistently, the DBD mutant, which is a truncation of the PhoP_REC domain, or mutation at the upstream a3’-b3’ sites supresses PhoP-dependent repression activities (Fig. 4b), emphasizing the necessity of the PhoP_REC domain and the spacer for enabling PhoP-dependent transcription repression.
In addition, the RNAP αCTD shifts downstream through a helical movement, establishing interactions with both DNA and GlnR_DBDIV in PhoP-TRC (Fig. 4d, e). These interactions may further enhance the stability of the upstream DNA and facilitate the engagement of the PhoP dimer. In contrast to the DNA bending configuration observed in 6GlnR-TAC, the PhoP-mediated protein-DNA and protein-protein interactions in PhoP-TRC collectively induce a significant DNA distortion, approximately 45° towards the RNAP main body, particularly at the long spacer (Fig. 4d right panel and Supplementary Fig. 14b). This distortion markedly alters the DNA curvature at the a3-b3 sites, creating substantial steric hindrances that impede the accommodation of the third GlnR dimer. Consequently, it is likely that PhoP acts through a “competitive occluding model” to repress efficient GlnR-dependent transcription initiation.
Discussion
Throughout evolution, bacteria has adeptly developed intricate transcription regulatory networks to tightly control differential expression of stress genes, a process that is essential for bacterial viability and pathogenicity25. The dual regulation of global transcription regulators, such as the widely distributed OmpR/PhoB family RRs, has been established as one of the most effective strategies which function through extensive protein-DNA and protein-protein interactions to drive RNAP37,38,51,12,39. By using the global transcription regulator PhoP as a model for the large OmpR/PhoB family, we elucidate the cryo-EM structures of Mtb PhoP-TACs and Mtb PhoP-TRC (Figs. 1–4), and provide complex structural insights into the dual regulatory mechanisms governing PhoP-dependent transcription activation and PhoP-dependent transcription repression.
As reviewed recently, bacterial transcription activators exhibit a remarkable diversity in the mechanisms of transcription activation, and are succinctly classified into four categories based on the key structural features of the reported TACs10. In the three structures of PhoP-TACs, the downstream PhoP dimer not only specifically binds to the a1-b1 sites that overlap promoter -35 element through the PhoP-PHO box interactions, but also engages with the conserved β flap and σAR4 via extensive PhoP-RNAP interactions (Figs. 1–3). Collectively, these interactions stabilize the displacement of σAR4. Such characteristics support PhoP as a complex type IV σ70 family-dependent transcription activator, which activates transcription by utilizing a sigma adaptation mechanism that compensates for the canonical strong interactions between σAR4 and the -35 element in canonical RPo (Supplementary Fig. 8). This mechanism is reminiscent of other reported transcription activators, such as MotA-AsiA, Rob, and the DNA damage factor PafBC10,52. Intriguingly, the second and third PhoP dimers join tandemly and cooperatively to recognize multiple PHO promoters in distinct tetrameric and hexametric configurations, thereby stabilizing the structures of 4PhoP-TAC and 6PhoP-TAC, respectively (Figs. 1, 2, 5a). It is noteworthy that RNAP αCTD and the oligomeric PhoP_RECs also contribute to the stabilization of 6PhoP-TAC and RNAP engagement (Figs. 1, 3, 4). These arrangements contrast sharply with the reported TACs carrying monomeric or dimeric transcription activators10,45,46,52,53,54,55. Considering these features, PhoP can be recognized as a distinct type IV transcription activator characterized by finely tuned complex regulatory mechanisms10. The observation of similar dimeric and tetrameric arrangements in PmrA-TAC and GlnR-TAC suggests that this tandem architecture may represent a generalized transcription activation mechanism for the OmpR/PhoB family regulators37,38. Regarding the physiological significance of these diverse regulatory modes on the multiple PHO promoters mediated by tandem PhoP dimers, we propose that this mechanism has evolved to recognize a broad range of promoter targets. This capability enables PhoP to initiate transcription of various target regulons in a precise and sequential manner in response to different stresses, thereby significantly enhancing bacterial survival in challenging environments.

a The canonical RPo and three types of PhoP-dependent transcription activation in which PhoP exists as a tandem dimer, tetramer, and hexamer, respectively. b Classic transcription repression models by occluding RNAP binding sites at a promoter (upper panel) or locking RNAP on a promoter (lower panel). c A proposed “competitive occluding model” for PhoP-dependent transcription repression. In the absence of the upstream PhoP dimer, six GlnR molecules cooperatively and efficiently activate transcription of the target promoter. However, when the PhoP dimer interacts competitively with the a3’-b3’ sites and engages the adjacent GlnR molecules, these interactions cause significant distortion of the upstream DNA in the long spacer region, creating steric hindrances that impede the binding of a third GlnR dimer. Consequently, the formation and stabilization of a competent 6GlnR-TAC is disrupted, resulting in the repression of efficient GlnR-dependent transcription initiation. The repressor and spacer are represented as a five-pointed star and a rectangle, respectively, both colored in red.
Unlike most transcription repressors that inhibit RNAP by occupying promoter elements or locking RNAP to prevent transcription initiation (Fig. 5b)9, the global transcription regulator PhoP has been shown to cooperatively repress transcription of the PhoP-GlnR cross-talked metabolic promoters, as well as in conjunction with other regulators25,56. Consistent with these genetic and biochemical findings, structural analysis of our preliminary structural model of the PhoP-TRC elucidates the molecular determinants of PhoP-dependent transcription repression, urging us to propose a unique “competitive occluding model” (Fig. 5c). In the absence of PhoP, six GlnR molecules effectively activate the transcription of target promoter, similar to PhoP in 6PhoP-TAC. However, when phosphorated PhoP is present, the tandem PhoP dimer competitively binds to the upstream a3’-b3’ sites through conserved PhoP-PHO box interactions and contacts the adjacent GlnR_RECs via its PhoPV_DBD and dimeric PhoP_RECs. Additionally, the RNAP αCTD also shifts downstream through a helical movement, potentially contributing to the stabilization of the PhoP-TRC. These collaborative interactions significantly distort the upstream promoter DNA at the long spacer (Fig. 4e right panel and Supplementary Fig. 14b), creating steric hindrances that prevent the accommodation of a third GlnR dimer, thereby repressing the formation of an effective transcription-competent 6GlnR-TAC. This finely tuned coordination mechanism aligns well with the hypothesis that the upstream binding sites may be evolved as a transcription strategy for stringent regulation of cellular nutrient starvation and essential phosphate–nitrogen metabolic homeostasis. This may serve as a model for PhoP in relation to other cross- regulated regulons. Furthermore, this repression model bears resemblance to the previously described “RNAP locking repression model” associated with the phage Ф29 regulatory protein P4, which locks RNAP on the A2c promoter through interactions between P4 and αCTD57,58 (Fig. 5b, lower panel). In contrast, PhoP primarily represses transcription via competitive binding and the resulting DNA distortion, which is maintained through complex interactions with GlnR molecules and RNAP αNTD.
Mtb, the primary causative agent of tuberculosis (TB), continues to infect nearly one-third of the global population and results in approximately two million deaths annually. Nowadays, there is an urgent need to investigate the molecular mechanisms underlying Mtb infections and to identify new therapeutic targets. In comparison to the virulent Mtb H37Rv strain, the conserved mutation S219L of the key virulence regulator PhoP, where serine at position 219 is substituted with leucine, has been shown to contribute to virulence attenuation in the avirulent Mtb H37Ra strain. This S219L mutation disrupts the synthesis of essential virulent cell wall lipids and alters bacterial morphology59,60,61. Consistent with its physiological significance, the S219 residue, located in the conserved α7 helix of PhoP_DBD, specifically interacts with the bases of the template DNA strand in both PhoP-TACs and PhoP-TRC structures (Fig. 2b and Supplementary Figs. 10c, 11c, 11e, 12a, 12c, 12e, 13h). Notably, the PhoP S219L mutant displays significantly reduced transcriptional activity compared to the wild-type PhoP (Fig. 2e). This interaction underscores the critical importance of this highly conserved residue and clarifies the functional states of these PhoP-dependent transcription regulation complexes. These findings provide substantial structural evidence supporting the potential of PhoP as a promising target for anti-tuberculosis strategies.
In addition to the crucial roles of PhoP-mediated protein-DNA and protein-protein interactions, the variability in promoter characteristics within PhoP-TACs and PhoP-TRC—such as the number of PHO boxes, consensus nucleotides, and intervening spacer lengths—holds evolutionary significance in determining whether PhoP functions as an activator or a repressor. This variability finely tunes the transcription initiation of different stress genes. These findings elucidate the dual regulatory mechanisms of the PhoP-like OmpR/PhoB family regulators, and significantly enhance our understanding of the intricate transcription network that governs transcriptional plasticity and stress adaptation. Given that the high conservation of the global virulence factor PhoP among Mtb and other prominent pathogens, our findings may guide the development of innovative drugs or interventions targeting PhoP to combat the concerning bacterial infections.
Methods
Plasmids and DNA
To construct the expression plasmid pET28a-phop, the gene encoding Mtb PhoP, which is fused with an N-terminal His×6 tag and regulated by the T7 promoter, was synthesized into pET28a by Sangon Biotech, Inc. Recombinant plasmids containing site-directed Mtb PhoP mutants were generated following the instructions provided in the QuikChange Site-Directed Mutagenesis Kit, Agilent, Inc. To assess the transcriptional activities of PhoP on promoters containing various numbers of PhoP binding sites (2, 4, and 6 sites), the PhoP target promoters pstS, phoU, and proposed promoter pks2, each of which followed by an RNA aptamer coding sequence (Mango III) were amplified and purified using the QIAquick PCR Purification Kit (Qiagen, Inc.). One heterogeneous control promoter which isn’t a PhoP target regulon is used as a negative control promoter. The purified DNA fragments were designated as pstS, phoU, pks2, and control promoters, respectively. Nucleic acid scaffolds for assembling Mtb RPo, Mtb PhoP-TACs, and Mtb PhoP-TRC were generated by annealing synthetic template-strand DNA (T) and non-template-strand DNA (NT). This annealing process involved heating the mixture at 95 °C for 5 min, followed by gradual cooling to 25 °C in 2 °C increments, all conducted in the annealing buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl). The primers utilized in this study are detailed in Supplementary Table S1.
Mtb PhoP and Mtb PhoR
Plasmids pET28a-phop and its derivatives were initially transformed into BL21(DE3) (Invitrogen, Inc.). Subsequently, single colonies of positive transformants were inoculated and expanded in 5 L of LB broth supplemented with 100 μg/mL kanamycin, incubating at 37 °C with shaking. Once OD600 of the cultures reached approximately 0.8, PhoP expression was induced by the addition of 0.5 mM IPTG, followed by overnight incubation at 20 °C. Following centrifugation at 4,800 g for 15 min at 4 °C, the cell pellets were resuspended in 100 mL of buffer A (20 mM Tris–HCl, pH 7.9, 300 mM NaCl, and 5% glycerol) and lysed using an ATS AH−10013 cell disrupter (ATS, Inc.). After centrifugation at 13,000 g for 30 min at 4 °C, the supernatant was loaded onto a 5-mL Ni-NTA agarose column (Qiagen, Inc.) that had been pre-equilibrated with buffer A. The column was washed with 10 mL of buffer A containing 40 mM imidazole followed by elution with 30 mL of buffer A containing 200 mM imidazole. The elutes were then concentrated and further purified using a 120-mL HiLoad 16/600 Superdex 75 column (GE Healthcare, Inc.) with buffer B (20 mM Tris–HCl, pH 7.9, 300 mM NaCl, 5 mM MgCl2, and 1 mM DTT). After verification by SDS-PAGE, the target elutions containing PhoP were pooled and stored at –80 °C. The yield was approximately 1.5 mg/L, with a purity exceeding 90%. To enhance the relative DNA binding affinity, the purified PhoP proteins were phosphorylated by incubating them with 50 mM AcP at 30 °C for 90 min. Following centrifugation at 13,000 g for 30 min at 4 °C, the sample was loaded onto a 25-mL Hitrap desalting column (Cytiva, Inc.) and eluted with buffer B to remove excess AcP. Finally, the phosphorylated PhoP proteins were then pooled and stored as previously described. Similarly, the phosphorylated PhoP derivatives were prepared. The Mtb PhoR coding gene phoR, which contains the kinase domain was constructed into pGEX 4T-1 and transformed into BL21(DE3). The recombinant proteins were purified through a glutathione magarose column (Smart-Life Sciences) in buffer C (20 mM Tris–HCl, pH 7.9, 200 mM NaCl, and 5% glycerol) and eluted in buffer C supplemented with 10 mM Glutathione. Then the elutes were further purified by using a HiLoad 16/600 Superdex 75 column (GE Healthcare, Inc.) and concentrated as PhoP. The purified PhoP proteins were also phosphorylated by incubating them with Mtb PhoR at 25 °C for 15 min as described in the in vitro transcription assay and electrophoretic mobility shift assay (EMSA). For clarity, the phosphorylated PhoP and its derivatives are designated as PhoP and PhoP derivatives, respectively.
Mtb RNAP and Mtb GlnR
The Mtb RNAP holoenzyme, including σA, was induced from cultures of E. coli strain BL21(DE3) (Invitrogen, Inc.) that had been co-transformed with the plasmids pACYC Duet-rpoA-rpoD, pCDF-rpoZ, and pET Duet-rpoB-rpoC, and preliminarily purified by poly(ethyleneimine) precipitation to a ratio of 0.7% (m/v), and ammonium sulfate precipitation to a ratio of 30.0% (m/v)37. Then the precipitation pellets were resuspended and further purified through a Ni-NTA agarose column, a Mono Q 10/100 GL column (GE Healthcare, Inc.), and a HiLoad 16/600 Superdex 200 column (GE Healthcare, Inc.). The purified fractions containing Mtb RNAP were pooled and stored. In addition, Mtb GlnR was induced from cultures of E. coli strain BL21(DE3) transformed with the plasmid pET28a-glnR, and purified through a Ni-NTA agarose column and HiLoad 16/600 Superdex 75 column37 in similar methods to those of PhoP.
Assembly of PhoP-TAC and PhoP-TRC
The DNA scaffolds pstS containing two PhoP binding sites, phoU4 containing four PhoP binding sites, and phoU6 containing six PhoP binding sites were utilized to assemble the complexes 2PhoP-TAC, 4PhoP-TAC, and 6PhoP-TAC, respectively. In addition, the DNA scaffold amtB, which comprises two PhoP binding sites and four GlnR binding sites was selected to construct the repression complex PhoP-TRC. The assembly of the PhoP-TACs was initiated by incubating Mtb RNAP, the DNA scaffold, and PhoP in a molar ratio of 1: 1: 8 at 4 °C overnight. The resultant sample was subsequently applied to a 120-mL HiLoad 16/600 Superdex 200 column (GE Healthcare, Inc.) and eluted using buffer C (20 mM Tris–HCl, pH 7.9, 75 mM NaCl, 5 mM MgCl2, and 1 mM DTT). Following verification via SDS-PAGE, the fractions containing each component of the assembled PhoP-TAC were concentrated using Amicon Ultra centrifugal filters (10 KDa MWCO, Merck Millipore, Inc.). Furthermore, the PhoP-GlnR complex was assembled by incubating Mtb RNAP, amtB scaffold, PhoP and GlnR in a molar ratio of 1:1:8:16 at 4 °C overnight, followed by purification using a 120-mL HiLoad 16/600 Superdex 200 column and assessment through SDS-PAGE. The Oligonucleotides synthesized for the preparation of the DNA scaffolds are detailed in Supplementary Table S1.
Cryo-EM grid preparation
Initially, the Quantifoil grids (R1.2/1.3 Cu400 mesh; Quantifoil, Inc.) underwent glow_discharge for 120 seconds at a current of at 25 mA. Following this, 3 μl of each purified Mtb PhoP-dependent transcription regulation complex was applied to the grids after a 1-minute incubation with 8 mM CHAPSO (Hampton Research Inc.) at 25 °C. -The sample-loaded grids were then blotted using a Vitrobot Mark IV (FEI)and rapidly plunge-frozen in liquid ethane maintaining a chamber humidity of 95% at a temperature of 10 °C. Finally, grids exhibiting a moderate density and uniform distribution of single particles were selected for extensive cryo-EM data collection.
Cryo-EM data acquisition and processing
Cryo-EM data for each Mtb PhoP-dependent transcription regulation complex were acquired using a consistent set of parameters on a 300 kV Titan Krios (FEI, Inc.) equipped with a K3 Summit direct electron detector. The data were processed sequentially with the appropriate cryo-EM data analysis software including RELION 4.162 and CryoSPARC v4.263 (Tables 1, 2, and Supplementary Figs. 4–7, 9). A varying number of images were recorded using the EPU software in counting mode, featuring a pixel size of 1.2 Å, a dose rate of 10 e/pixel/s, and an electron exposure dose of 50 e/Å2. Movies were captured over a duration of 8.38 seconds with the defocus range varying from -2.0 μm to -1.0 μm. Subframes of individual movies were aligned using MotionCor264, While the contrast-transfer-function for each summed image was estimated using CTFFIND465. From the summed images, approximately 10,000 particles were manually picked and subjected to 2D classification in RELION. The resulting 2D classes, exhibiting diverse orientations were further selected, auto-picked, manually inspected, and subjected to iterative 2D classification. By eliminating poorly populated classes, the selected particles were then subjected to 3D classification in RELION using a map of Mtb RPo (PDB ID: 6VVY)47 (for PhoP-TACs and RPo) or Mtb GlnR-TAC (PDB ID: 8HIH)37(for PhoP-TRC), both low-pass filtered to 40 Å resolution as a reference. Subsequently, particles from the optimal class, which displayed clear density for RNAP, DNA, PhoP, and GlnR were re-processed through 3D auto-refinement, re-extracted, CTF-refined, Bayesian polishing, 3D auto-refinement and post-processing in RELION.
For the dataset of RPo, the polished particles obtained from RELION were subsequently imported into CryoSPARC and subjected to masked 3D classification targeting the upstream DNA region. The high-quality 3D classes generated were then selected for further analysis, which included homogeneous refinement, local resolution estimation, and local filtering, ultimately yielding the final map.
For the datasets of 2PhoP-TAC, 4PhoP-TAC, and 6PhoP-TAC, the well-aligned particles obtained from RELION were firstly imported into the CryoSPARC program. Subsequently, masked 3D classification was performed on the upstream PhoP binding regions, accompanied by homogeneous refinement, local resolution estimation, and local filtering to generate the final density maps. The final particles were further subjected to particle subtraction to preserve the signal from the upstream PhoP binding regions, followed by masked local refinements to improve the map quality and interpretability.
For the dataset of PhoP-TRC, particles that underwent 3D auto-refinement were sequentially subjected to post-processing, local resolution estimation and local filtering to generate the final map for PhoP-TRC. To enhance the map quality of the upstream PhoP and GlnR binding regions, a sequential analysis was performed using 3D multi-body refinement, masked local refinement, local resolution estimation, and local filtering in RELION.
The final mean map resolutions for each Mtb PhoP-dependent transcription regulation complex, as assessed using the Gold-standard Fourier-shell-correlation method, are as follows: 3.63 Å for RPo, 3.46 Å for 2PhoP-TAC, 3.70 Å for 4PhoP-TAC, 3.31 Å for 6PhoP-TAC, and 3.68 Å for PhoP-TRC (Table 1 and Supplementary Figs. 4–7, 9). Additionally, the resolution for the PhoP and GlnR regions in 2PhoP-TAC, 4PhoP-TAC, 6PhoP-TAC, and PhoP-TRC are 7.38 Å, 7.01 Å, 5.90 Å, and 5.90 Å, respectively (Table 2 and Supplementary Figs. 4–7, 9).
Cryo-EM model building and refinement
The model of RNAP and the downstream DNA, derived from the cryo-EM structure of Mtb RPo (PDB ID: 6VVY), as well as the co-crystal structure of PhoP and DNA were fitted into the cryo-EM density map of Mtb RPo and PhoP-TACs using Chimera66. Likewise, the model of Mtb RNAP, GlnR and DNA from the cryo-EM structure of Mtb GlnR-TAC (PDB ID: 8HIH) was fitted into the cryo-EM density map of Mtb PhoP-TRC to generate a preliminary structural model of Mtb PhoP-TRC for docking analysis. The model of the upstream nucleic acids was built manually using Coot67. The coordinates were subsequently calculated and validated through real-space refinement incorporating secondary structure restraints in Coot and Phenix (v1.19.2)68. Structures were analyzed using Chimera and PyMOL69. The Map versus Model FSCs of the four cryo-EM maps (RPo, 2PhoP-TAC, 4PhoP-TAC, 6PhoP-TAC) in this work were generated by Phenix. The statistics of cryo-EM refinement were summarized in Tables 1 and 2.
Electrophoretic mobility shift assay
The electrophoretic mobility shift assay (EMSA) was performed using an EMSA buffer composed of 40 mM Tris–HCl, pH 7.9, 100 mM NaCl, 10 mM MgCl2, and 5 % glycerol. The total reaction mixture volume was 20 μl, containing the following components at their respective final concentrations: 30 μM Mtb PhoP (or its derivatives), 15 μM Mtb PhoR, 30 μM Mtb GlnR, 0.2 μM Mtb RNAP, and either 30 nM phoU promoter or a control promoter as specified. Initially, RNAP was incubated with DNA for 10 min at 37 °C, while PhoP was incubated with PhoR and ATP (1.5 mM) for 15 min at 25 °C. Subsequently, PhoP was mixed with GlnR and/or additional PhoP and incubated for 20 min at 37 °C. After a 2-minute incubation with 0.03 mg/mL heparin at 22 °C, the reaction mixtures were loaded onto 5% polyacrylamide slab gels (29:1 acrylamide/bisacrylamide) and subjected to electrophoresis in a buffer containing 90 mM Tris–borate (pH 8.0) and 0.2 mM EDTA. The gels were stained with 4S Red Plus Nucleic Acid Stain (Sangon Biotech, Inc.) according to the procedure of the manufacturer.
In vitro transcription assay
In vitro transcription assays were conducted in a transcription buffer consisting of 40 mM Tris–HCl, pH 7.9, 50 mM NaCl, 10 mM MgCl2, and 5% glycerol, utilizing 96-well microplates (Corning incorporated, USA). Each reaction mixture (80 μl) contained the following components: 0.15 μM Mtb RNAP, 30 nM mango-ended DNA (pstS, phoU, amtB, amtB_mut, pks2), 30 μM Mtb PhoP or its derivatives, 15 μM Mtb PhoR, 0.1 mM NTP mix (comprising ATP, UTP, GTP, and CTP), and 1 μM TO1-biotin. Mtb PhoP was phosphorylated by PhoR in the presence of 1.5 mM ATP for 15 min at 25 °C, after which it was purified according to previously established protocols. At first, Mtb RNAP and DNA were incubated together for 15 min at 37 °C. Subsequently, the phosphorylated PhoP or its combination with GlnR was introduced into the mixture and incubated for an additional 10 min at 37 °C. Following this, the NTP mix and TO1-biotin were added, and the mixture was incubated for another 30 min at 37 °C. Finally, fluorescence emission intensities were quantified using a multimode plate reader (EnVision, PerkinElmer Inc.; excitation wavelength = 510 nm; emission wavelength = 535 nm). The relative transcription activities of PhoP derivatives were calculated using the following Eq. (1).
where IWT and I represent the fluorescence intensities in the presence of PhoP and its derivatives, respectively. I0 denotes the fluorescence intensity in the absence of PhoP.
Statistics and Reproducibility
To analyze the data for in vitro transcription assays (Figs. 2e, 3d, and 4b), the mean values and the corresponding standard errors were analyzed from three independent measurements. Data for electrophoretic mobility shift assays (Supplementary Fig. 3) were repeated for three times. Source data are provided as Source Data files.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The accession codes for the atomic coordinates reported in this paper are deposited in the Protein Data Bank: 9JI2 for RPo [https://doi.org/10.2210/pdb9JI2/pdb], 9KET for 2PhoP-TAC [https://doi.org/10.2210/pdb9KET/pdb], 9KEU for 4PhoP-TAC [https://doi.org/10.2210/pdb9KEU/pdb], 9KEV for 6PhoP-TAC [https://doi.org/10.2210/pdb9KEV/pdb]. The accession codes for the 3D cryo-EM density maps are deposited in the Electron Microscopy Data Bank (EMDB): EMD-61492 for RPo [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-61492], EMD-62293 for 2PhoP-TAC [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-62293], EMD-62294 for 4PhoP-TAC [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-62294], EMD-62295 for 6PhoP-TAC [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-62295], EMD-62331 for PhoP-TRC [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-62331], EMD-61494 for the PhoP region in 2PhoP-TAC [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-61494], EMD-61496 for the PhoP region in 4PhoP-TAC [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-61496], EMD-61498 for the PhoP region in 6PhoP-TAC [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-61498], and EMD-61499 for the PhoP and GlnR regions in PhoP-TRC [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-61499]. The PDB entries 6VVY and 8HIH are used for structure comparison in this study. Source data are provided with this paper.
References
Decker, K. B. & Hinton, D. M. Transcription regulation at the core: similarities among bacterial, archaeal, and eukaryotic RNA polymerases. Annu Rev. Microbiol 67, 113–139 (2013).
Feklistov, A. & Darst, S. A. Structural basis for promoter-10 element recognition by the bacterial RNA polymerase sigma subunit. Cell 147, 1257–1269 (2011).
Feklistov, A., Sharon, B. D., Darst, S. A. & Gross, C. A. Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev. Microbiol 68, 357–376 (2014).
Saecker, R. M., Record, M. T. & Dehaseth, P. L. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 412, 754–771 (2011).
Bae B., Feklistov A., Lass-Napiorkowska A., Landick R., Darst S. A. Structure of a bacterial RNA polymerase holoenzyme open promoter complex. Elife 4, e08504 (2015).
Narayanan, A. et al. Cryo-EM structure of Escherichia coli sigma(70) RNA polymerase and promoter DNA complex revealed a role of sigma non-conserved region during the open complex formation. J. Biol. Chem. 293, 7367–7375 (2018).
Chen, J., Boyaci, H. & Campbell, E. A. Diverse and unified mechanisms of transcription initiation in bacteria. Nat. Rev. Microbiol 19, 95–109 (2021).
Lee, D. J., Minchin, S. D. & Busby, S. J. W. Activating transcription in bacteria. Annu. Rev. Microbiol., Vol. 66 66, 125–152 (2012).
Browning, D. F., Butala, M. & Busby, S. J. W. Bacterial transcription factors: regulation by pick “N” mix. J. Mol. Biol. 431, 4067–4077 (2019).
Kompaniiets, D., Wang, D., Yang, Y., Hu, Y. & Liu, B. Structure and molecular mechanism of bacterial transcription activation. Trends Microbiol 32, 379–397 (2024).
Lloyd, G., Landini, P. & Busby, S. Activation and repression of transcription initiation in bacteria. Essays Biochem 37, 17–31 (2001).
Kenney, L. J. Structure/function relationships in OmpR and other winged-helix transcription factors. Curr. Opin. Microbiol 5, 135–141 (2002).
Sadotra S., et al. Structural basis for promoter DNA recognition by the response regulator OmpR. J. Struct. Biol. 213, 107638 (2021).
Galperin, M. Y. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J. Bacteriol. 188, 4169–4182 (2006).
Groisman, E. A. The pleiotropic two-component regulatory system PhoP-PhoQ. J. Bacteriol. 183, 1835–1842 (2001).
Parish, T. Two-component regulatory systems of mycobacteria. Microbiol Spectr. 2, MGM2-0010-2013 (2014).
Tierney, A. R. P. & Rather, P. N. Roles of two-component regulatory systems in antibiotic resistance. Future Microbiol 14, 533–552 (2019).
De la Nieta, R. S., Santamaria, R. I. & Díaz, M. Two-Component Systems of Streptomyces coelicolor: An Intricate Network to Be Unraveled. Int J. Mol. Sci. 23, 15085 (2022).
Francis V. I., Stevenson E. C., Porter S. L. Two-component systems required for virulence in Pseudomonas aeruginosa. Fems. Microbiol. Lett. 364, fnx104 (2017).
Diniz, M. M. P. et al. Fine-tuning control of phoBR expression in vibrio cholerae by binding of PhoB to multiple Pho boxes. J. Bacteriol. 193, 6929–6938 (2011).
Goyal, R. et al. Phosphorylation of PhoP protein plays direct regulatory role in lipid biosynthesis of Mycobacterium tuberculosis. J. Biol. Chem. 286, 45197–45208 (2011).
Gupta, S., Pathak, A., Sinha, A. & Sarkar, D. Mycobacterium tuberculosis PhoP recognizes two adjacent direct-repeat sequences to form head-to-head dimers. J. Bacteriol. 191, 7466–7476 (2009).
Ryndak, M., Wang, S. S. & Smith, I. PhoP, a key player in mycobacterium tuberculosis virulence. Trends Microbiol. 16, 528–534 (2008).
Singh, P. R., Vijjamarri, A. K. & Sarkar, D. Metabolic switching of mycobacterium tuberculosis during hypoxia is controlled by the virulence regulator PhoP. J. Bacteriol. 202, e00705 (2020).
Martin, J. F., Rodriguez-Garcia, A. & Liras, P. The master regulator PhoP coordinates phosphate and nitrogen metabolism, respiration, cell differentiation and antibiotic biosynthesis: comparison in Streptomyces coelicolor and Streptomyces avermitilis. J. Antibiot. (Tokyo) 70, 534–541 (2017).
Martin, J. F. & Liras, P. The balance metabolism safety net: integration of stress signals by interacting transcriptional factors in streptomyces and related actinobacteria. Front Microbiol 10, 3120 (2020).
Sola-Landa, A., Rodriguez-Garcia, A., Franco-Dominguez, E. & Martin, J. F. Binding of PhoP to promoters of phosphate-regulated genes in Streptomyces coelicolor: identification of PHO boxes. Mol. Microbiol 56, 1373–1385 (2005).
Allenby, N. E., Laing, E., Bucca, G., Kierzek, A. M. & Smith, C. P. Diverse control of metabolism and other cellular processes in Streptomyces coelicolor by the PhoP transcription factor: genome-wide identification of in vivo targets. Nucleic Acids Res. 40, 9543–9556 (2012).
Santos-Beneit, F., Rodriguez-Garcia, A., Franco-Dominguez, E. & Martin, J. F. Phosphate-dependent regulation of the low- and high-affinity transport systems in the model actinomycete Streptomyces coelicolor. Microbiol. (Read.) 154, 2356–2370 (2008).
Sola-Landa, A., Rodriguez-Garcia, A., Amin, R., Wohlleben, W. & Martin, J. F. Competition between the GlnR and PhoP regulators for the glnA and amtB promoters in Streptomyces coelicolor. Nucleic Acids Res. 41, 1767–1782 (2013).
Martin, J. F. et al. Cross-talk of global nutritional regulators in the control of primary and secondary metabolism in Streptomyces. Micro Biotechnol. 4, 165–174 (2011).
Rodriguez-Garcia, A., Sola-Landa, A., Apel, K., Santos-Beneit, F. & Martin, J. F. Phosphate control over nitrogen metabolism in Streptomyces coelicolor: direct and indirect negative control of glnR, glnA, glnII and amtB expression by the response regulator PhoP. Nucleic Acids Res. 37, 3230–3242 (2009).
Pathak, A., Goyal, R., Sinha, A. & Sarkar, D. Domain structure of virulence-associated response regulator PhoP of Mycobacterium tuberculosis: role of the linker region in regulator-promoter interaction(s). J. Biol. Chem. 285, 34309–34318 (2010).
Sinha, A., Gupta, S., Bhutani, S., Pathak, A. & Sarkar, D. PhoP-PhoP interaction at adjacent PhoP binding sites is influenced by protein phosphorylation. J. Bacteriol. 190, 1317–1328 (2008).
He, X. & Wang, S. DNA consensus sequence motif for binding response regulator PhoP, a virulence regulator of Mycobacterium tuberculosis. Biochemistry 53, 8008–8020 (2014).
Sola-Landa, A., Rodriguez-Garcia, A., Apel, A. K. & Martin, J. F. Target genes and structure of the direct repeats in the DNA-binding sequences of the response regulator PhoP in Streptomyces coelicolor. Nucleic Acids Res. 36, 1358–1368 (2008).
Shi, J. et al. Structural insights into the transcription activation mechanism of the global regulator GlnR from actinobacteria. Proc. Natl Acad. Sci. USA 120, e2300282120 (2023).
Lou, Y. C. et al. Structural basis of transcriptional activation by the OmpR/PhoB-family response regulator PmrA. Nucleic Acids Res. 51, 10049–10058 (2023).
Mizuno, T. & Tanaka, I. Structure of the DNA-binding domain of the OmpR family of response regulators. Mol. Microbiol 24, 665–667 (1997).
Blanco, A. G., Sola, M., Gomis-Ruth, F. X., Coll, M. & Tandem, D. N. A. recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10, 701–713 (2002).
He, X. Y., Wang, L. Q. & Wang, S. S. Structural basis of DNA sequence recognition by the response regulator PhoP in Mycobacterium tuberculosis. Sci. Rep. -Uk 6, 24442 (2016).
Cimino M., et al. Identification of DNA binding motifs of the PhoP/PhoR two-component signal transduction system. Plos One 7, e42876 (2012).
Apel, A. K., Sola-Landa, A., Rodríguez-García, A. & Martín, J. F. Phosphate control of phoA, phoC and phoD gene expression in reveals significant differences in binding of PhoP to their promoter regions. Microbiol-Sgm 153, 3527–3537 (2007).
Shi, J. et al. Structural basis of Q-dependent transcription antitermination. Nat. Commun. 10, 2925 (2019).
Shi, J. et al. Structural basis of transcription activation by the global regulator Spx. Nucleic Acids Res. 49, 10756–10769 (2021).
Shi, J. et al. Structural basis of three different transcription activation strategies adopted by a single regulator SoxS. Nucleic Acids Res. 50, 11359–11373 (2022).
Lilic, M. et al. The antibiotic sorangicin A inhibits promoter DNA unwinding in a Mycobacterium tuberculosis rifampicin-resistant RNA polymerase. Proc. Natl Acad. Sci. USA 117, 30423–30432 (2020).
Friedland, N. et al. Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry 46, 6733–6743 (2007).
Li, Z. et al. Insight into the molecular mechanism of the transcriptional regulation of amtB operon in Streptomyces coelicolor. Front Microbiol 9, 264 (2018).
Wang, Y., Cen, X. F., Zhao, G. P. & Wang, J. Characterization of a new GlnR binding box in the promoter of amtB in Streptomyces coelicolor inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB. J. Bacteriol. 194, 5237–5244 (2012).
Blanco, A. G., Canals, A., Bernues, J., Sola, M. & Coll, M. The structure of a transcription activation subcomplex reveals how sigma(70) is recruited to PhoB promoters. EMBO J. 30, 3776–3785 (2011).
Shi, J. et al. Structural basis of transcription activation by Rob, a pleiotropic AraC/XylS family regulator. Nucleic Acids Res. 50, 5974–5987 (2022).
Feng, Y., Zhang, Y. & Ebright, R. H. Structural basis of transcription activation. Science 352, 1330–1333 (2016).
Liu, B., Hong, C., Huang, R. K., Yu, Z. & Steitz, T. A. Structural basis of bacterial transcription activation. Science 358, 947–951 (2017).
Shi, W., Jiang, Y. N., Deng, Y. B., Dong, Z. G. & Liu, B. Visualization of two architectures in class-II CAP-dependent transcription activation. Plos Biol. 18, e3000706 (2020).
Santos-Beneit, F., Rodriguez-Garcia, A., Sola-Landa, A. & Martin, J. F. Cross-talk between two global regulators in Streptomyces: PhoP and AfsR interact in the control of afsS, pstS and phoRP transcription. Mol. Microbiol 72, 53–68 (2009).
Maria, M. M. M., Fernando, R. & Margarita, S. Activation and repression of transcription at two different phage φ29 promoters are mediated by interaction of the same residues of regulatory protein p4 with RNA polymerase. Embo J. 15, 383–391 (1996).
MARIA, M. MM, MARGARITA S, AND F. Protein p4 represses phage Ф29 A2c promoter by interacting with the a subunit of Bacillus subtilis RNA polymerase. Proc. Natl Acad. Sci. USA 93, 8913–8918 (1996).
Lee, J. S. et al. Mutation in the transcriptional regulator PhoP contributes to avirulence of Mycobacterium tuberculosis H37Ra strain. Cell Host Microbe 3, 97–103 (2008).
Zheng, H. et al. Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv. PLoS One 3, e2375 (2008).
Chesne-Seck, M. L. et al. A point mutation in the two-component regulator PhoP-PhoR accounts for the absence of polyketide-derived acyltrehaloses but not that of phthiocerol dimycocerosates in Mycobacterium tuberculosis H37Ra. J. Bacteriol. 190, 1329–1334 (2008).
Scheres, S. H. W. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290 (2017).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Pettersen, E. F. et al. UCSF chimera - a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 66, 213–221 (2010).
Rigsby, R. E. & Parker, A. B. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem Mol. Biol. Educ. 44, 433–437 (2016).
Acknowledgements
This work was funded by the National Key R&D Program of China (2023YFC2308200), the National Natural Science Foundation of China (32270037, 82311530689, 32270192, 82072240, 32000025), the Jiangsu Qinglan Project to J.S. and the Natural Science Foundation of Jiangsu Province (BK20211302, SBK2023030145), the landmark talent training project of Nanjing University of Chinese Medicine (RC202404), and the special fund project of Nanjing Drum Tower hospital for the transformation of scientific and technological achievements (202404).
We appreciate Shenghai Chang at the Center of Cryo-Electron Microscopy in Zhejiang University School of Medicine, Guangyi Li, Liangliang Kong, Jialin Duan, and Yun Song of the Electron Microscopy System at the National Facility for Protein Science in Shanghai (NFPS), Shanghai Advanced Research Institute, Chinese Academy of Sciences, China for providing technical support and assistance in data collection. We thank the Experiment Center for Science and Technology, Nanjing University of Chinese Medicine for experimental assistance. We thank the Core Facilities, Zhejiang University School of Medicine for technical support.
Author information
Authors and Affiliations
Contributions
W.L., J.S., and F.Y. designed the experiments. Q.S., Z.Z.F., T.Y.L., Z.H.Y., S.M.X., F.G., and L.X.X. performed molecular cloning, protein expression and purification, enzymatic determination and cryo-EM sample assembly. A.J.W., J.S., Z.Z.F., and L.Q.X performed cryo-EM sample preparations and data acquisition. W. L. and F. Y. analyzed the cryo-EM data and determined the structures. J. S., W.L., J.L., J.P.Z., and K.D. performed formal analysis. J.S., W.L., and G.P.Z. wrote the paper with help from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Dibyendu Sarkar and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shi, J., Feng, Z., Song, Q. et al. Structural insights into transcription regulation of the global OmpR/PhoB family regulator PhoP from Mycobacterium tuberculosis. Nat Commun 16, 1573 (2025). https://doi.org/10.1038/s41467-025-56697-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56697-x
This article is cited by
-
Vitamin B6 form produced by Lactobacillus induces metabolic disorder and suppresses multi-pathogenic bacteria
Communications Biology (2025)
