
Abstract
Acute liver failure (ALF) is a life-threatening condition caused by rapid hepatocyte death and impaired liver regeneration. Here we show that extracellular vesicles engineered to express Signal Regulatory Protein Alpha (SIRP-EVs), produced via a scalable 3D bioreactor process with high yield and purity, exhibit significant therapeutic potential by targeting damaged cells and promoting tissue repair. SIRP-EVs block CD47, a crucial inhibitory signal on necroptotic cells, to enhance macrophage-mediated clearance of dying hepatocytes. They also deliver regenerative cargo from mesenchymal stem cells, reprogramming macrophages to support liver regeneration. In male animal models, SIRP-EVs significantly reduce liver injury markers and improve survival, demonstrating their dual-function therapeutic efficacy. By integrating the resolution of necroptosis with regenerative macrophage reprogramming, SIRP-EVs represent a promising platform for restoring liver function. These findings support the development of EV-based in vivo macrophage reprogramming therapies for ALF and other inflammation-driven diseases, paving the way for the clinical application of engineered EV therapeutics.
Similar content being viewed by others


Introduction
Acute inflammation, whether caused by infectious or non-infectious origins, often leads to severe outcomes, including organ failure1. Acute liver failure (ALF) exemplifies this issue, with a mortality rate exceeding 50%, largely due to limited therapeutic options2. ALF is characterized by the rapid death of hepatocytes, exacerbated by impaired clearance of these cells and insufficient regenerative responses, underscoring the urgent need for effective therapeutic strategies.
Recent studies highlight necroptosis, a form of regulated necrosis, as a significant contributor to ALF3,4,5. Triggered by inflammatory cytokines, necroptosis compromises cell membranes, releasing intracellular contents that further exacerbate inflammation6. While macrophages typically engulf dying cells via efferocytosis, necroptotic cells often evade this clearance by expressing high levels of CD47 a signal that prevents macrophage-mediated clearance7,8,9. Furthermore, inflammation triggered by uncleared necroptotic cells can polarize highly plastic macrophages towards a pro-inflammatory phenotype, perpetuating liver injury10,11. This barrier to resolution complicates inflammation control and hinders liver regeneration, underscoring the therapeutic challenges in managing ALF.
To address these unmet medical needs, we developed a dual-mode action therapeutic approach using extracellular vesicles expressing SIRPα, the natural ligand of CD47, derived from engineered human mesenchymal stem cells (SIRP-EVs). Initial analyses of ALF models revealed a direct correlation between necroptosis and CD47 expression, supported by comprehensive protein and transcriptomic analyses. We also established a rigorous SIRP-EV manufacturing process that ensures scalability, high purity, and yield, while retaining the inherent properties of MSCs. In addition, we explored the ability of SIRP-EVs to target both macrophages and necroptotic hepatocytes in ALF models. Our strategy effectively enhances macrophage functionality, specifically increasing phagocytic activity against necroptotic hepatocytes via CD47 blockade and promoting liver regeneration through the reprogramming of macrophages with MSC-derived cargo. This study demonstrates the therapeutic potential of SIRP-EVs in treating ALF, marking a significant advancement in addressing this critical healthcare challenge.
Results
Elevated CD47 expression in necroptotic hepatocytes hallmarks ALF
We developed various animal models of ALF to understand complexity, each model induced by different etiological factors. These models employ pharmacological agents such as Acetaminophen (APAP), Thioacetamide (TAA), Carbon tetrachloride (CCl4), and a combination of Lipopolysaccharide (LPS) with d-galactosamine (d-galN), each representing distinct pathways of hepatic injury. Specifically, APAP-induced hepatotoxicity results from its metabolic conversion into toxic metabolites. TAA and CCl4 induce liver damage through bioactivation to reactive intermediates causing hepatocellular damage. Meanwhile, the LPS/d-galN model mimics pathogen-induced liver failure12,13. These models provide valuable insights into the pathophysiological mechanisms of ALF and assist in identifying distinct therapeutic targets for various liver injury etiologies.
Our analysis showed significant increases in serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels across all ALF models, indicative of severe hepatic injury (Fig. 1A and Supplementary Fig. 1). Hematoxylin & eosin (H&E) staining and immunohistochemistry (IHC) on liver tissues revealed extensive cellular death and a marked increase in CD47 expression in all ALF models compared to normal liver tissue (Fig. 1A and Supplementary Fig. 1). Subsequent studies aimed to identify liver cell populations with elevated CD47 expression following ALF induction. Flow cytometry analysis revealed a significant upregulation of CD47 in hepatocytes from ALF conditions compared to normal (Fig. 1B and Supplementary Fig. 2A). This increase was further confirmed by western blot analysis of hepatocytes isolated from ALF-affected liver tissues, showing a notable enhancement in CD47 expression (Fig. 1C). Annexin V/7-AAD assays indicated a significant rise in the late apoptotic/necroptotic cell population in CD47-high hepatocytes, especially in the APAP-ALF model (Fig. 1D and Supplementary Fig. 2B). This was associated with overexpression of necroptotic markers, such as RIP3 and pMLKL, in ALF tissues (Fig. 1E and Supplementary Fig. 2C, D). Confocal microscopy analysis corroborated a direct correlation between CD47 expression and the pMLKL in ALF-affected liver tissues (Fig. 1F). These findings suggest a significant upregulation of CD47 expression in necroptotic hepatocytes across ALF models compared to normal liver.

A Biochemical evaluation (AST and ALT levels) of the ALF model after APAP induction (left). Representative images of H&E and CD47 staining of liver samples from an ALF model induced by APAP (right) (n = 4). B CD47 expression levels of cell populations within liver tissue (n = 4). C CD47 expression in hepatocytes from normal and APAP-ALF livers (n = 3). D Graphical representation of early apoptotic (Annexin V+/7-AAD−), late apoptotic/necroptotic (Annexin V+/7-AAD+), and necrotic (Annexin V−/7-AAD+) cell populations in liver hepatocytes from normal (n = 4) and APAP-ALF (n = 3) mice. E Expression of RIP3 and pMLKL in liver samples from normal and APAP-ALF groups. F Representative confocal images of liver sections from normal and APAP-ALF mice. Scale bar, 50 μm. G Scatter plot visualization of the Spearman correlation (R) between 121 necroptosis gene scoring and RNA levels of CD47 across samples. H STopover analysis to map the spatial overlap and interactions between cell types in liver tissue from normal and APAP-ALF mice. Highlights include 121 necroptosis gene scores (yellow) and macrophage RNA levels (blue), with overlapping areas shown in green. The plots below represent the three regions. I Violin plot visualizing SIRPα expression levels in different regions based on spatial correlation results. The numbers marked on the plot represent the median values of the SIRPα expression. Bar graph data are presented as mean ± SD. Statistical significance was determined by two-tailed unpaired Student’s t test (A), two-way ANOVA with Sidak’s post hoc test (B, D), and two-tailed test (G). Hepa hepatocytes, Immune immune cells, Endo endothelial cells. Source data are provided as a Source Data file.
Spatial transcriptomic (ST) analysis of liver tissue from the APAP-induced ALF model (APAP-ALF) demonstrated a distinct spatial association between necroptosis-related genes and CD47 expression, differentiating ALF liver tissues from normal tissues (Fig. 1G). In contrast, apoptosis-related genes showed little to no evident correlation with CD47 expression (Supplementary Fig. 3A). This analysis was conducted using a publicly available ST dataset (GEO accession number GSE223560), processed through the Seurat pipeline to focus on spatial features, including necroptosis and CD47 expression, with correlations assessed via Spearman analysis.
Using the CellDART algorithm14 cell types were mapped by integrating ST data with reference single-cell RNA-seq data, and STopover15 was employed to examine the colocalization of necroptosis- and macrophage-related gene expression spots. This analysis was expanded to identify spatial overlap between necroptosis and macrophage-related genes, which was more pronounced in ALF liver tissues than in healthy liver (Fig. 1H). However, there was no significant spatial overlap observed between apoptosis-related genes and macrophage-related genes in ALF liver compared to normal liver (Supplementary Fig. 3B). In addition, there was a significant increase in SIRPα expression at the colocalized sites of these genes (Fig. 1I). Previous studies have indicated that macrophages expressing high levels of SIRPα tend to be located around cells that overexpress CD479,16. Therefore, these results suggest that the CD47/SIRPα interaction, which inhibits efferocytosis in necroptotic hepatocytes, is upregulated in ALF and could serve as a therapeutic target for ALF.
3D bioreactor-based scalable purification process produces SIRP-EVs with high purity and preserves MSC characteristics
To address inflammatory diseases associated with upregulated CD47, we genetically engineered human bone marrow-derived MSCs to express SIRPα creating SIRPα-modified MSCs (SIRP-MSCs) through stable transduction with lentiviral vectors encoding the SIRPα. These cells retained the hallmark differentiation capabilities of naive MSCs, including differentiation into adipocytes, osteoblasts, and chondrocytes (Fig. 2A). Importantly, SIRP-MSCs maintained the expression of key MSC markers (CD73, CD90, CD105, and CD166) and did not express hematopoietic and endothelial markers (CD14, CD34, and CD45), as confirmed by flow cytometry (Fig. 2B). Multiplex ELISA analysis showed that the angiogenic cytokine secretion profiles of SIRP-MSCs were similar to those of non-modified MSCs (C-MSCs), indicating that the genetic modification did not alter the inherent properties of MSCs (Fig. 2C). Analysis of inter- and intra-donor variability showed consistent proliferation rates, EV yield, and the proportion of SIRPα-positive cells across biological replicates, demonstrating a high level of reproducibility in the modification process (Fig. 2D).

A Trilineage differentiation of engineered MSCs: adipogenic differentiation by lipid droplets (left), osteogenic differentiation by mineralized matrices (middle), and chondrogenic differentiation by cartilage matrix (right). B Surface marker expression on SIRP-MSCs post-transduction and control C-MSCs, confirming MSC phenotype with markers CD73, CD90, CD105, CD166, and absence of CD14, CD34, CD45. C Cytokine secretion profiles (FGF, HGF, IL-8, TIMP-1, TIMP-2, VEGF) of C-MSCs (n = 2 for bFGF; n = 3 for HGF, IL-8, TIMP-1, TIMP-2, and VEGF) and SIRP-MSCs (n = 2 for bFGF and HGF; n = 3 for IL-8, TIMP-1, TIMP-2, and VEGF). D Inter- and intra-donor variability in SIRP-MSCs shown by fold expansion 5 days post-thaw (left), nanoparticle concentration at final harvest (middle), and percentage of SIRPα-stained cells (right). Two independent vials from a single donor and one from a second donor were evaluated (n = 3). E Visualization of cells in 2D CellSTACKs and 3D bioreactor systems (Ambr250 and STR). F The time course measurements of particle concentration during two independent runs for each of the 2D CellSTACKs (2D), 3D Ambr250 (Ambr), and 3D STR (STR) systems. G Size distributions of particles using NTA from 2D CellSTACKs and 3D bioreactor systems (Ambr250 and STR). H SIRPα expression levels of MSCs before harvest from STR runs, with controls from Ambr and 2D systems (n = 2). I Schematic of the downstream processing flow for SIRP-EV isolation, encompassing clarification to sterile filtration. Created with BioRender.com (https://BioRender.com/r97a555). J–L In-process analytics results for SIRP-EV: J protein, (K) dsDNA, human serum albumin clearance, and (L) cumulative yield of SIRP-EV. M The purity of SIRP-EV per step is quantified as the particle count per mg protein. B–D, H, J–M Bar graph data are presented as mean ± SD. J, L, M Bars represent the mean of independent process replicate points using mean analytical test results. n = 1 for Harvest, Clar., Buffer Ex., and Sterile Filt.; n = 3 for Concen.; n = 2 for Guard Filt., and Chrom. K dsDNA and albumin tests included n = 3 technical test replicates. The numbers displayed on a bar graph represent the exact numerical values of the bar. Clar clarification, Concen concentration, Guard Filt guard filtration, Chrom chromatography, Buffer Ex buffer exchange, Sterile Filt sterile filtration. Source data are provided as a Source Data file.
For the efficient isolation of SIRP-EVs from SIRP-MSCs, we employed a 3D bioreactor system utilizing microcarriers in the upstream process. The efficacy of this system was assessed by comparing the growth of SIRP-MSCs in both small-scale (250 ml) Ambr250 (Ambr) and large-scale (15 L) Biostat STR® (STR) bioreactors with traditional 2D CellSTACKs as the control. Daily inspections of the microcarrier suspensions in the bioreactor vessels (STR and Ambr) confirmed a consistent suspension of cells and microcarriers, ensuring an evenly mixed culture (Supplementary Fig. 4A). Critical process parameters such as temperature, dissolved oxygen, and agitation rates, were meticulously controlled. The metabolite profiles from the bioreactor vessels were closely aligned, indicating consistent process conditions (Supplementary Fig. 4B). Cells were monitored using a live stain, demonstrating clear attachment and expansion on microcarriers over a 10-day culture period (Fig. 2E). Daily measurements of particle count from the culture media, following the replacement of growth media with EV collection media, showed that 3D bioreactors produced significantly more EVs than 2D cultures (Fig. 2F, G and Supplementary Fig. 4C). In addition, the expression of SIRPα in MSCs remained stable throughout the collection period, up to the last day (Fig. 2H).
The scalability of our upstream process development was validated through experiments conducted in a 50 L STR bioreactor, with a 250 ml Ambr system serving as the scale-down. Comprehensive analyses including macroscopic and microscopic imaging, metabolite profiles, cell proliferation, cell viability, and EV counts per ml demonstrated consistency across scales, confirming a successful 200-fold scale-up in a 3D bioreactor (Supplementary Fig. 5). The downstream process comprised depth filter filtration for clarification, tangential flow filtration for concentration, chromatography for purification, followed by an additional tangential flow filtration for buffer exchange and further concentration, and concluding with sterile filtration prior to vialing (Fig. 2I). The final SIRP-EVs showed a significant cumulative reduction in double-stranded DNA (dsDNA) and albumin by 93% and 99%, respectively, with a 96% clearance of total protein content from the initial culture medium (Fig. 2J, K and Supplementary Fig. 6A–C). After the downstream process, the purity of the isolated SIRP-EVs was remarkably high, reaching 8 × 1011 particles per mg of proteins, with a cumulative yield, or particle recovery rate, of 50% (Fig. 2L, M and Supplementary Fig. 6D). These results demonstrate that our process development for the 3D-based EV isolation can efficiently separate SIRP-EVs with high purity and yield.
SIRP-EVs carry MSC-derived protein cargos and present a safe CD47 blockade
Naive MSC-derived EVs (C-EVs) are recognized for their effectiveness in treating inflammatory diseases through the transfer of miRNA and protein cargos17,18. To determine if SIRP-EVs replicate a similar MSC-derived cargo profile, we produced C-EVs using the same upstream and downstream processes, achieving high particle recovery rates and purity (Supplementary Fig. 7). Initial analysis of the RNA content using RNA electrophoresis revealed negligible RNA levels in SIRP-EVs (Supplementary Fig. 8A). To evaluate the potential impact of processing methods on RNA cargo and to perform more precise RNA quantification, we compared RNA quantities using the RiboGreen assay. This comparison was made between EVs isolated through research-grade processes—including serial centrifugation, tangential flow filtration, and ultracentrifugation—and those in C-EVs and SIRP-EVs isolated using our established 3D bioreactor-based process. The results indicated that EVs isolated through research-grade processes contained more RNA than those isolated using the 3D-based system (Supplementary Fig. 8B). These findings suggest that RNA content of EVs can vary significantly depending on the isolation and purification process used.
We utilized liquid chromatography with tandem mass spectrometry (LC-MS/MS) to analyze the protein profiles of SIRP-EVs, identifying 7391 proteins. This confirmed that the protein cargos of SIRP-EVs closely mirror those of C-EVs (Fig. 3A, B), while also demonstrating overexpression of SIRPα in SIRP-EVs (Fig. 3C). Further experiments were conducted to verify the EV nature of SIRP-EVs. Western blot analysis confirmed the presence of EV markers, including CD81, CD9, CD63, and TSG101, as well as the engineered expression of SIRPα, while confirming the absence of the non-EV marker, prohibitin19 (Fig. 3D and Supplementary Fig. 9). Cryo-transmission electron microscopy (cryo-TEM) revealed that SIRP-EVs, like C-EVs, are spherical nanoparticles characterized by a lipid bilayer (Fig. 3E and Supplementary Fig. 10). Dynamic light scattering (DLS) assays showed that SIRP-EVs, similar to C-EVs, exhibit a uniform particle size distribution with average diameters of approximately 200 nm, indicative of no particle aggregation and a high degree of monodispersity, as reflected by a polydispersity index close to 0.2 (SIRP-EV: 0.190, C-EV: 0.187) (Fig. 3F). Zeta potential (ZP) measurements recorded SIRP-EVs and C-EVs at −22.56 mV and −22.30 mV by nanoparticle tracking analysis (NTA), respectively, with both demonstrating over 95% lipidation, confirmed through MemGlow staining (Fig. 3G). Further analysis of the over-represented Gene Ontology Biological Processes (GOBPs) within the SIRP-EV protein cargo revealed enrichment of adhesion and binding proteins that promote directed and prolonged binding of C-EVs at sites of inflammation20 (Fig. 3H). Collectively, these findings demonstrate that SIRP-EVs maintain the fundamental characteristics of EVs, comparable to those of C-EVs.

A–C Protein profile comparisons and reproducibility between C-EVs and SIRP-EVs. A Scatter plot consistency and R2 values for protein identification across replicates; Venn diagram overlaps of EV type-specific proteins. B Correlation coefficient analysis of protein profile similarities between replicates. C Quantification of SIRPα overexpression in SIRP-EVs compared to C-EVs. D Protein expression of EVs to examine SIRPα, EV markers (CD81, CD9, CD63, and TSG101), and non-EV marker, prohibitin. E Representative cryo-TEM image of EVs. F Size distribution of EVs analyzed by DLS. G Zeta potential measurements of EVs analyzed using NTA, which denote the surface charge distribution (left, n = 3). MemGlow staining of EVs (right). H Network diagram displaying over-represented Gene Ontology Biological Processes (GOBPs) among the top 506 SIRP-EV proteins. Analysis shows the proportion of these proteins compared to the total, with significant GOBPs (P < 0.05) indicated by black and red lines for group and individual function significance, respectively. I Quantification of cell binding, following a 30-min co-incubation of Cy5.5-labeled C-EV and SIRP-EV with CD47 KO and CD47 WT cells (n = 3). Bar graph data are presented as mean ± SD. Statistical significance was determined by two-tailed unpaired Student’s t test (G), two-way ANOVA with Sidak’s post hoc test by comparing the groups treated with the same concentration of EVs (I). ****P < 0.0001. Source data are provided as a Source Data file.
To assess the targetability of SIRP-EVs toward CD47, we conducted in vitro binding assays using CD47 knockout (CD47 KO) and wild-type (WT CD47) cell lines. These assays showed that SIRP-EVs bind more selectively to WT CD47 cell line in a dose-dependent manner, compared to C-EVs, while no significant binding was observed in CD47 KO cell line (Fig. 3I). In addition, considering the known issues associated with CD47 antibodies—such as red blood cell (RBC) aggregation due to their Fc regions21—we carried out comparative in vitro hemagglutination assays between human CD47 antibodies and SIRP-EVs. Significantly, CD47 antibodies caused increased haziness indicating human RBC aggregation, while SIRP-EVs did not induce such adverse effects (Supplementary Fig. 11A–C). Further toxicity evaluations of SIRP-EVs, through intravenous administration (i.v.) in mice at doses exceeding therapeutic levels, showed no observable toxicity (Supplementary Fig. 11D). These results highlight SIRP-EVs as a promising alternative to traditional CD47 antibodies.
Intravenous delivery of SIRP-EV demonstrates preferential accumulation in CD47-overexpressing injured liver tissue
We investigated the biodistribution of SIRP-EV, focusing on their accumulation based on CD47 expression. Cy5.5 dye-labeled SIRP-EVs were administered intravenously to both normal mice and models with APAP-ALF. Initial verification using fast protein liquid chromatography confirmed that Cy5.5 dye was solely associated with the SIRP-EVs, with no free dye detected (Fig. 4A). In the APAP-ALF models, which overexpress CD47, these Cy5.5-labeled SIRP-EV accumulated more in the liver tissues of compared to those in normal mice (Fig. 4B, C).

A FPLC elution profiles of Cy5.5-labeled SIRP-EV, Cy5.5 dye, and SIRP-EV. Fluorescence was detected using a UV-Vis detector at 280 nm and 700 nm. B Ex vivo imaging of the organ distribution in mice 24 h after intravenous administration of 2.5 × 1010 Cy5.5-labeled SIRP-EVs. C, D Systemic delivery and uptake of EVs in mice liver tissues. C Dose-dependent accumulation of SIRP-EVs in liver tissues of APAP-ALF mice was observed within 24 h. The in vivo imaging system (IVIS) are shown (left), and quantitative fluorescence signals from ex vivo livers are presented (right). Representative images (n = 2) of independent repeated experiments. D A reduction in SIRP-EV accumulation in APAP-ALF liver tissues resulted from pre-blocking with the anti-CD47 antibody. Representative images (n = 2) of independent repeated experiments. Source data are provided as a Source Data file.
We further confirmed the CD47-dependent biodistribution of SIRP-EV by pre-treating with CD47 antibodies, which resulted in decreased accumulation of SIRP-EV in CD47-overexpressing liver tissues (Fig. 4D and Supplementary Fig. 12A). Intriguingly, SIRP-EVs displayed higher in vivo signals not only in the APAP-ALF model but also in normal mice 24 h post-injection, compared to C-EVs (Supplementary Fig. 12A). Furthermore, the use of an isotype control antibody (IgG) showed no reduction in liver accumulation of SIRP-EVs, further supporting the CD47-dependent nature of SIRP-EV accumulation in injured liver tissue (Supplementary Fig. 12B). These findings collectively underscore the selective and CD47-dependent accumulation of SIRP-EVs in APAP-induced liver injury.
SIRP-EV accumulates in CD47-overexpressing necroptotic hepatocytes and exhibits therapeutic efficacy in ALF
We assessed whether SIRP-EVs could accumulate in CD47-overexpressing necroptotic hepatocytes in ALF-induced liver tissue, promoting efferocytosis and liver regeneration. To do this, we conducted evaluations across various experimental schedules (Supplementary Fig. 13). Our results showed that SIRP-EV treatment significantly decreased CD47 expression in injured liver tissues (Fig. 5A). Furthermore, multiplex immunohistochemistry (IHC) provided deeper insights into CD47 expression dynamics within necrotic liver areas induced by APAP, which were primarily surrounded by macrophages (F4/80+) (Fig. 5B). In contrast, liver tissues treated with SIRP-EVs exhibited dispersed macrophages, reduced necrotic areas, and lower CD47 expression, accompanied by a significant decrease in the ratio of CD47+ necroptotic cells (Fig. 5C, D and Supplementary Fig. 14).

A Representative histological images of CD47 staining (left) and quantification of CD47-positive area (right) in liver tissue from normal and APAP-ALF mice. Box graph data are presented as mean ± SD. Box plot minima = 1.3, 5.2, 5.2; maxima = 6, 41.2, 39.1; center = 3.05, 22.8, 8.85. B Representative multiplex IHC images from APAP-ALF mouse liver after 4 × 109 SIRP-EVs treatment. The left panel shows a broader view, while the right panel focuses on a necrotic area, as delineated by the dashed line. C Representative multiplex IHC of APAP-ALF mouse liver tissue and D the quantification of CD47-positive necroptotic cells (CD47+pMLKL+) is shown on the right panel. Box graph data are presented as mean ± SD. Box plot minima = 8, 0; maxima = 52, 21; center = 22.5, 1. E Cell type-specific biodistribution of SIRP-EV in APAP-ALF livers (n = 4 per experimental group and n = 2 for the saline control group). F CD47 expression in liver tissue cell types after systemic treatment with SIRP-EVs compared to C-EVs (n = 4). G Representative confocal images of liver sections from mice 24 h post-induction with a 500 mg/kg APAP dose, treated with 4 × 109 SIRP-EVs. Cells were immunostained for F4/80 (green) and pMLKL (red), with white arrowheads indicating engulfed necroptotic cells. H Serum AST and ALT levels were determined 48 h after APAP injection in subjects receiving the indicated dose of SIRP-EV (AST: n = 7 for normal and NT, n = 6 for APAP-treated groups; ALT: n = 6 per group). I Representative multiplex IHC of APAP-ALF mouse liver treated with 4 × 109 SIRP-EVs. Quantification of Ki67-positive proliferative hepatocytes (Ki67+HNFα+) in APAP-ALF mouse liver is shown in the right panel. Box graph data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test (A, E, H), two-tailed unpaired Student’s t test (D, I) and two-way ANOVA with Sidak’s post hoc test (F). Hepa hepatocytes, Immune immune cells, Endo endothelial cells. Source data are provided as a Source Data file.
To confirm SIRP-EV accumulation in necroptotic hepatocytes, we administered fluorescently labeled EVs systemically in the APAP-ALF model and used flow cytometry to evaluate cellular uptake across cell types (Supplementary Fig. 15A). Myeloid cells, particularly Kupffer cells, are critical immune cells for the primary defense within liver tissue10,22. Kupffer cells are generally known to predominantly accumulate EVs within the liver23. Consequently, we identified that over 40% of Kupffer cells had EVs distributed within them. This result was consistent regardless of SIRPα expression on the EVs (Fig. 5E). Notably, SIRP-EVs appeared to reach hepatocytes more effectively compared to C-EVs (Fig. 5E). Furthermore, we observed that the accumulation of SIRP-EVs in hepatocytes was abolished when the cells were pre-treated with CD47-blocking antibodies (Supplementary Fig. 15B). These findings indicate that systemically administered SIRP-EVs achieve enhanced targeting of necroptotic hepatocytes that overexpress CD47, surpassing the targeting capabilities of C-EVs.
Interestingly, CD47, which was overexpressed in hepatocytes of the APAP-ALF model, showed a significant decrease following SIRP-EV treatment (Fig. 5F and Supplementary Fig. 16). To determine whether this CD47 reduction facilitated macrophage efferocytosis of necroptotic hepatocytes, we performed tissue staining and analyzed liver sections via confocal microscopy. Our findings revealed that SIRP-EV treatment significantly increased the presence of macrophages (F4/80+) and enhanced efferocytosis of necroptotic cells (pMLKL+) in liver tissues of APAP-ALF model. (Fig. 5G). Supporting this result, bone marrow-derived macrophages (BMDMs) exhibited enhanced phagocytosis of APAP-treated HepG2 cells and primary hepatocytes from APAP-ALF models incubated with SIRP-EVs compared to C-EVs (Supplementary Fig. 17).
Finally, we evaluated the therapeutic efficacy of SIRP-EVs in the APAP-ALF model. SIRP-EV administration led to a dose-dependent reduction in liver toxicity markers, such as AST and ALT (Fig. 5H). In addition, a single injection of SIRP-EV in the APAP-ALF model resulted in an increase in Ki67+ hepatocytes, indicative of liver regeneration (Fig. 5I). These findings suggest that SIRP-EVs not only bind to and suppress CD47 on necroptotic hepatocytes but also promote macrophage efferocytosis, resulting in enhanced therapeutic efficacy in a dose-dependent manner.
Single injection of SIRP-EV demonstrates therapeutic efficacy across multiple ALF models
To investigate the enhanced therapeutic efficacy mediated through SIRPα in our engineered EVs compared to C-EVs, we assessed their therapeutic potential in various ALF models (Supplementary Fig. 13). In the APAP-ALF model, histological analysis revealed that SIRP-EV treatment significantly reduced dead cell counts in damaged liver tissues (Fig. 6A, B). Treatment with SIRP-EVs also resulted in a higher ratio of monocyte-derived macrophages (MoMF) to the Ly6C low type and Kupffer cells, crucial for inflammation resolution, compared to C-EVs (Fig. 6C and Supplementary Fig. 18A–C). In addition, SIRP-EVs were associated with reductions in pro-inflammatory cytokine IL-6 levels and neutrophil infiltration, which are indicative of acute inflammation (Supplementary Fig. 18D, E). Moreover, SIRP-EV treatment led to statistically significant improvements in liver toxicity markers, AST and ALT, surpassing C-EVs in the APAP-ALF model (Fig. 6D).

A Representative histological images of H&E and TUNEL in liver tissue from normal and APAP-ALF mice after EVs treatment. B Quantification of TUNEL-positive cells. Box graph data are presented as mean ± SD. Box plot minima = 0, 0, 2, 1; maxima = 49, 332, 200, 58; center = 1, 28.5, 38.5, 6. C Flow cytometry determination of Ly6Chigh as a percentage of total liver CD11bhigh F4/80low MoMFs (n = 4 for Normal and APAP groups, and n = 5 for APAP + C-EV and APAP + SIRP-EV groups). D Serum AST (n = 7 for Normal and APAP + SIRP-EV groups, n = 8 for APAP group, and n = 9 for APAP + C-EV groups) and ALT (n = 8 for Normal, APAP + C-EV, and APAP + SIRP-EV groups, n = 9 for APAP group) levels were measured 48 h post-500 mg/kg APAP induction in subjects treated with 4 × 109 EVs. E Representative IHC images of α-SMA staining in liver tissues from mice that survived 120 h after induction with a 700 mg/kg dose of APAP. Quantification of α-SMA is shown in the right panel. Box represents the distribution of combined sample results across independent experiments. Box graph data are presented as mean ± SD. Box plot minima = 0.189, 0.884, 0.061; maxima = 9.321, 7.306, 4.731; center = 2,696, 2.406, 0.725. F–I Therapeutic effects of 9 × 108 EVs on an LPS/d-galN-induced ALF model. F Representative histological images of H&E and TUNEL staining in liver tissue from normal and LPS/d-galN-induced ALF mice after the indicated treatment. G Quantification of the TUNEL-positive area (n = 5 for PBS group, n = 10 for Normal, C-EV, and SIRP-EV groups). H Serum ALT levels were determined 8 h post-LPS/d-galN injection (n = 7 for PBS, C-EV, and SIRP-EV groups, n = 8 for the Normal group). I Kaplan–Meier survival curves for LPS/d-galN-induced ALF mice treated with EVs (n = 16 for LPS/d-galN group, n = 18 for LPS/d-galN+C-EV group, and n = 19 for LPS/d-galN+SIRP-EV group, across three independent experiments). J–L Therapeutic efficacy of 5 × 108 EVs in TAA-induced ALF models. J Representative histological images of H&E, TUNEL, and CD47 staining in liver tissue from both normal and TAA-induced ALF mice following EVs treatment. K Quantification of TUNEL-positive cells in liver tissue. Box graph data are presented as mean ± SD. Box plot minima = 0, 107, 150.8, 5; maxima = 11.6, 533.7, 429.3, 136; center = 0, 232.1, 284.3, 46.5. L Serum ALT (n = 5 per group) and total bilirubin (n = 6 per group) levels were determined 40 h post-TAA injection. Bar graph data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test (B–E, G, H, K, L). Source data are provided as a Source Data file.
Notably, SIRP-EV treatment resulted in higher survival rates in the high-dose APAP (700 mg/kg)-ALF model compared to C-EV and CD47 antibody treatments (Supplementary Fig. 19A, B). Given that ALF progression is often linked to liver fibrosis24 and non-efferocytosed damaged hepatocytes can induce fibrogenic activation25, we investigated whether SIRP-EVs could prevent fibrosis by facilitating the clearance of these damaged hepatocytes. Our findings revealed that SIRP-EV treatment notably mitigated liver fibrosis compared to C-EVs (Fig. 6E), underscoring the effectiveness of SIRP-EVs in targeting CD47+ necroptotic hepatocytes and broadening their potential applications in preventing ALF complications, including fibrosis.
In the LPS/DgalN-induced ALF prevention model, SIRP-EVs not only improved survival rates but also substantially reduced ALT levels and the ratio of dead cells in liver tissues more effectively than C-EVs (Fig. 6F–I and Supplementary Fig. 19C). Similarly, in the TAA-induced ALF model, SIRP-EVs exhibited superior therapeutic effects compared to C-EVs, as evidenced by lower dead cell ratios, reduced CD47 expression, and decreased serum ALT and bilirubin levels (Fig. 6J–L and Supplementary Fig. 19D). Overall, these results indicate that incorporating SIRPα into C-EVs significantly enhances their therapeutic efficacy.
SIRP-EV enhances therapeutic efficacy by transferring MSC protein cargos in ALF
We further verified that the therapeutic efficacy of SIRP-EV originates from their MSC-derived cargos by comparing them with SIRP-EVs isolated from HEK cells. We genetically modified HEK cells to express SIRPα, producing HEK cell-derived SIRP-EV(HEKSIRP-EV), and evaluated their efficacy in ALF models. SIRP-EVs were more effective than HEKSIRP-EVs, as demonstrated by a significant reduction in blood levels of AST and ALT (Fig. 7A). Furthermore, histological analysis demonstrated that SIRP-EVs significantly decreased dead cell counts and neutrophil infiltration compared to HEKSIRP-EV (Fig. 7B, C). This suggests that the enhanced therapeutic effects of SIRP-EV are not solely due to CD47 blocking but also involve other contributory factors.

A–F Therapeutic efficacy of 4 × 109 EVs in 500 mg/kg APAP-ALF models. A Serum AST (n = 8 for Normal, APAP + HEKSIRP-EV, and APAP + SIRP-EV groups, n = 10 for APAP group) and ALT (n = 8 for APAP + HEKSIRP-EV and APAP + SIRP-EV groups, n = 10 for Normal and APAP groups) levels were determined 48 h after APAP injection in subjects treated with EVs. B Representative histological images of TUNEL staining in liver tissue from normal and APAP-ALF models after the indicated treatment. Quantification of the TUNEL-positive area is shown in the right panel (n = 8 for the APAP group, n = 10 for the Normal group, and n = 13 for APAP + HEKSIRP-EV and APAP + SIRP-EV groups). C Flow cytometry determination of Ly6G-positive neutrophils as a percentage of the total liver CD45+ leukocytes (n = 7 for APAP + HEKSIRP-EV and APAP + SIRP-EV groups, and n = 8 for Normal and APAP groups). D The abundance of proteins associated with the prevention of cell death (PAPPA, SOD2), cell proliferation (FGF, HGF, WNT ligands), and angiogenesis (uPAR, uPA) in SIRP-EVs compared to HEKSIRP-EV (n = 3 per group). P values are annotated on the bar, ****P < 0.0001. E Principal component analysis (PCA) of RNA sequencing data from CD11b+ cells from liver tissues in APAP, APAP + HEKSIRP-EV, and APAP + SIRP-EV groups (n = 3 per group). F Heatmap of 404 representative genes from each gene cluster identified. G, H Therapeutic efficacy of 4 × 109 SIRP-EVs in APAP-ALF models with CD11b reduction. G Representative histological images showing CD47 staining in liver tissue from APAP-ALF models following the specified treatment. H Serum ALT levels were measured 48 h post-APAP injection in subjects receiving the indicated treatments (n = 5 for IgG+SIRP-EV and αCD11b + SIRP-EV groups and n = 6 for IgG and αCD11b groups). I Serum AST and ALT levels were measured 48 h after injection of 300 mg/kg APAP-ALF models in subjects receiving the indicated treatments (n = 3 for APAP, APAP + NAC, and APAP + SIRP-EV groups and n = 4 per Normal group). Bar graph data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test (A–C, H, I), two-tailed unpaired Student’s t test (D), Benjamini–Hochberg adjusted P values (F). Source data are provided as a Source Data file.
We next analyzed and compared the functional protein cargos known to promote regeneration20 in both types of EV, aiming to elucidate the differences in therapeutic efficacy between MSC-derived SIRP-EVs (SIRP-EVs) and HEK cell-derived SIRP-EVs (HEKSIRP-EVs). The analysis revealed that proteins associated with preventing cell death (PAPPA, SOD2), promoting cell proliferation (FGF, HGF, WNT ligands), and enhancing angiogenesis (uPAR, uPA) were more abundantly present in SIRP-EVs than in HEKSIRP-EVs (Fig. 7D).
Acute and severe liver injury drives substantial migration of myeloid cells into the liver, where they differentiate into macrophages that play essential roles in both exacerbating liver injury and promoting tissue repair alongside resident macrophages, such as Kupffer cells19,26. Given our biodistribution data demonstrating that SIRP-EVs were effectively distributed within myeloid cells (CD11b+ cells) and Kupffer cells (CD11b+F4/80high cells), we analyzed the regenerative efficacy of SIRP-EVs in these cells. Notably, transcriptomic analysis of liver CD11b+ cells in liver tissues from ALF models indicated that a single dose of SIRP-EV systemic treatment showed distinct genetic differences compared to both the HEKSIRP-EV and non-treated groups (Fig. 7E). Heatmap analysis of 404 statistically differentially expressed genes among the three groups demonstrated that SIRP-EV treatment significantly upregulated genes associated with cellular viability, detoxification/antioxidation, extracellular matrix (ECM) remodeling, immune modulation, and vascularization/perfusion, all of which are related to tissue regeneration (Fig. 7F).
To further elucidate whether the observed therapeutic effects of SIRP-EVs in ALF models are dependent on liver CD11b+ cells, we performed experiments using CD11b neutralizing antibodies (Supplementary Fig. 13). Consistent with previous studies26, ALF models showed a marked increase in CD11b+ cells in the liver and spleen compared to normal conditions (Supplementary Fig. 20). Treatment with CD11b neutralizing antibodies effectively reduced CD11b+ cell levels in the spleen to near-normal and significantly decreased liver CD11b+ cells relative to untreated APAP-ALF models, although levels remained higher than normal livers (Supplementary Fig. 20). Intriguingly, APAP-ALF models treated with the CD11b neutralizing antibody did not exhibit the reduction in serum ALT levels or the decreased hepatic CD47 expression observed in SIRP-EV-treated mice that received control IgG (Fig. 7G, H). These findings suggest that the therapeutic efficacy of SIRP-EVs in ALF may rely on macrophages, potentially not only by removing necroptotic hepatocytes through CD47 blockade but also by enhancing the regenerative capacity in liver tissue via MSC-derived cargo.
Currently, N-acetylcysteine (NAC) is the standard treatment for ALF caused by APAP overdosing in clinical settings, primarily preventing hepatocyte death27. However, its effectiveness depends on the timing of APAP exposure. Previous studies have shown that NAC loses its protective effects if administered more than 3 h after APAP exposure28. A comparative efficacy study between SIRP-EV and NAC was conducted by administering both therapeutics 8 h after APAP injection. The results demonstrated that SIRP-EV offered superior therapeutic benefits (Fig. 7I and Supplementary Fig. 21). This finding underscores the potential of SIRP-EV as a competitively advantageous alternative to current ALF treatments.
Discussion
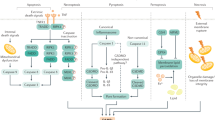
Acute inflammation impairs the resolution capabilities of immune cells and diminishes their inherent regenerative capacities, necessitating sophisticated therapeutic approaches29. This study introduces a dual-mode action therapeutic strategy utilizing SIRP-EVs derived from engineered MSCs. These SIRP-EVs not only enhance the phagocytic activity of liver macrophages to clear necroptotic hepatocytes via CD47 blockade but also promote liver regeneration by delivering MSC-derived cargo that reprograms macrophages to support tissue repair (Fig. 8).

SIRP-EVs mitigate necroptosis by targeting CD47 on necroptotic hepatocytes, modulate the function of macrophages, and enhance hepatocyte regeneration, ultimately promoting liver regeneration in ALF. Created with BioRender.com. (https://BioRender.com/w56l251).
As embryonically derived tissue-resident macrophages, Kupffer cells occupy niches that limit the differentiation of circulating CD11b+ monocytes into macrophages under homeostatic conditions30. However, under acute inflammation or severe injury, resident Kupffer cells become largely depleted, allowing CD11b+ monocytes from the bone marrow to migrate and repopulate the vacant niches30. These MoMF precursors possess an almost identical potential to develop into Kupffer cells and have been reported to play a critical role in both exacerbating and resolving inflammation during hepatic injury30,31. Unlike embryonic Kupffer cells, CD11b+ monocyte-derived Kupffer cells exhibit greater resistance to apoptosis and possess enhanced phagocytic and tissue repair capacities32.
In our ALF model, we observed a substantial depletion of Kupffer cells. However, SIRP-EV treatment increased the overall proportion of Kupffer cells and induced a shift of MoMFs toward a resolutive Ly6C low phenotype. Furthermore, SIRP-EVs promoted the clearance of CD47-overexpressing necroptotic hepatocytes by liver macrophages and significantly upregulated genes associated with tissue regeneration in liver CD11b+ cells. The observed reduction in therapeutic efficacy in a CD11b-diminished ALF model supports the hypothesis that CD11b+ cells are integral to SIRP-EV’s therapeutic mechanism. Our study does not definitively clarify whether the primary effectors in necroptotic cell clearance are embryonically derived or CD11b+ monocyte-derived macrophages, nor does it specify the roles of different CD11b+ cell types, such as monocytes, dendritic cells, and granulocytes. Future studies are needed to elucidate the specific cellular contributors to SIRP-EV’s therapeutic effects in liver regeneration and inflammation resolution.
Recent studies have identified necroptotic hepatocytes as a significant contributor to the exacerbation of ALF6. This study demonstrates that these necroptotic hepatocytes in ALF are characterized by CD47 overexpression, a previously unrecognized feature. Our findings indicate that blocking CD47 with SIRP-EVs reduces necroptotic hepatocytes and normalizes CD47 expression, suggesting its utility as a biomarker for ALF and potentially other diseases. Further support for this hypothesis is provided by spatial transcriptomic analysis, which reveals significant increases in SIRPα expression at sites of necroptosis and macrophage interaction. These results reinforce the potential of CD47 and SIRPα as biomarkers in severe acute inflammatory diseases.
The framework for EV production and purification has been established in previous studies33,34,35,36. This study demonstrates a robust method for producing high-purity, high-yield SIRP-EVs while preserving the intrinsic properties of MSCs. To accelerate the clinical translation of SIRP-EVs, we optimized a scalable manufacturing process to maximize EV yield using a 3D bioreactor system. Additionally, we developed a downstream process to concentrate EVs and remove non-EV impurities, such as nucleic acids released from cells and media proteins (e.g., albumin), to meet regulatory expectations37. This process achieved 93% and 99% reductions in nucleic acids and albumin, respectively, through filtration and chromatography, while maintaining high EV recovery. Using the particle-to-protein ratio (p/mg protein) as a purity metric38,39, we improved the ratio from 7.5E + 10 to 8.2E + 11 (~11-fold increase) and achieved a 50% recovery rate. This efficient, cost-effective approach offers a promising platform for developing advanced therapeutics using engineered EVs derived from MSCs.
Interestingly, the EVs produced by this method contained significantly low RNA content compared to those isolated through traditional research-grade EV production processes. While RNA is generally thought to be encapsulated within EVs, our findings suggest that RNA may be associated with the outside of EVs, warranting further investigation. Although numerous studies confirm the biological effects of miRNAs in MSC-derived EVs17, the RNA amount of ~1 ng in the SIRP-EVs used for our in vivo experiments challenges the notion that the therapeutic efficacy we observed can be explained by these RNAs. Therefore, our study shifts focus to the regenerative potential of MSC protein cargos but emphasizes that additional functional analyses of other MSC-derived cargos, such as DNA, RNA, and lipids are necessary.
EVs are increasingly recognized as promising drug delivery systems, noted for their high biocompatibility and ability to cross biological barriers40,41,42,43. However, they primarily accumulate in liver macrophages23. To address this issue, recent studies have enhanced EV targeting by forming a protein corona on their surface, specifically coating C-EVs with albumin to improve delivery to hepatocytes beyond macrophages23. In addition, surface engineering of EVs has been used to enhance targeted delivery to specific cells, thereby enhancing therapeutic efficacy44,45,46,47. Our findings prove that SIRPα expression on EVs not only mitigates necroptosis but also promotes effective distribution in CD47-overexpressing liver tissues and hepatocytes extending beyond macrophages in acute inflammatory conditions. This unique distribution of SIRP-EVs facilitates the delivery of MSC-derived regenerative factors and aids in blocking CD47 overexpression, which is observed in necroptotic cells. CD47 is not only overexpressed on pathological cells but also present on normal cells, posing challenges for therapies using CD47 antibodies48,49. These antibodies can inadvertently bind to normal cells, leading to unwanted clearance by immune cells that recognize the Fc region of antibodies. This limitation has narrowed the therapeutic window and contributed to clinical failures50,51. Importantly, SIRP-EVs, which lack Fc regions, do not present adverse side effects related to normal cell clearance.
Our research further indicates that even in normal mice, SIRP-EVs exhibit a prolonged in vivo presence compared to C-EVs. This finding implies that the presence of SIRPα on the EV surface may enhance their in vivo retention through interactions with CD47, which is expressed under normal conditions. In addition, previous studies have shown that SIRP-EVs bind to RBCs via CD47, and these RBC-bound SIRP-EVs can subsequently transfer to cancer cells that overexpress CD4721. This implies that RBCs may act as carriers, promoting circulation and enhancing targeted delivery to pathological sites. While these findings provide a strong basis, further validation through additional replicates of the biodistribution experiments will be important. More detailed and comprehensive pharmacokinetic studies will be necessary to demonstrate the prolonged circulation and targeted delivery of SIRP-EVs.
Studies of liver fibrosis in ALF have been limited due to the severity of clinical syndrome52,53. However, some research has demonstrated that delayed clearance of dead hepatocytes and subsequent regeneration after injury can amplify the fibrotic response25. We have shown that SIRP-EV not only has therapeutic efficacy in ALF but also provides evidence for a preventing effect on fibrosis progression after acute injury. While promising anti-fibrotic effects have been observed in experimental animal models, further mechanistic and fibrosis-related investigations are necessary to replicate these results in clinical trials.
In conclusion, this study introduces a dual-mode action approach utilizing SIRP-EVs for both inflammation resolution and regeneration promotion. Additional efforts are underway to improve our understanding, and further investigations are needed to definitively demonstrate how different cell types respond to SIRP-EV-mediated EV-cargo release. Although the primary focus has been on ALF, this strategy holds potential for application across a range of inflammatory conditions. In addition, the scalable and high-purity production process of these engineered MSC-derived EVs establishes a solid foundation for further research and development. This groundwork aims to facilitate clinical translation, potentially revolutionizing the management of acute inflammatory diseases.
Methods
Ethics statement
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Korea Institute of Science and Technology (KIST, KIST-IACUC-2021-154-5). Euthanasia was performed using gradually increasing concentrations of CO2 inhalation, in accordance with ethical guidelines.
ALF animal models
Male C57BL/6NCrlOri mice (7-week-old) used in all experiments were purchased from Orient Bio (South Korea). The mice were maintained under 12 h light/12 h dark cycle at 22 ± 2 °C and 50 ± 5% humidity in pathogen-free animal facility of KIST. Mice were fed a chow diet (LabDiet, 5053). The mice received intraperitoneal (i.p.) injections of APAP (300 mg/kg, 500 mg/kg or 700 mg/kg, Sigma-Aldrich, A7085) after 16 h of fasting and then were returned to a normal diet. For LPS/d-galN, TAA, and Carbone tetrachloride (CCl4) inducing ALF models, male C57BL/6N mice (7-old-week) received i.p. injection of LPS (10 µg/kg, Sigma-Aldrich, L2630) and d-galN (700 mg/kg, Sigma-Aldrich, G0500) simultaneously, TAA (300 mg/kg, Sigma-Aldrich, 163678) and CCl4 (1 mL/kg, Sigma-Aldrich, 289116). To treat ALF models, the mice received intravenous (i.v.) injections of EVs (0.25 × 109–4 × 109 EV particles) via the tail vein, 8 h after APAP induction, 2 h before LPS/d-galN induction, and 16 h after TAA induction. N-acetylcysteine (300 mg/kg, Sigma-Aldrich, A7250, i.v.) and anti-CD47 antibodies (10 mg/kg, BioXCell, BE0007, i.p.) were administered 8 h following APAP induction. All mice were maintained in the pathogen-free animal research facility of the Korea Institute of Science and Technology (KIST).
Biochemistry, cytokine, hematology, and coagulation analysis
For biochemistry analysis (AST, ALT, ALP, Albumin, Total bilirubin), the whole blood was collected through retro-orbital bleeding with micro-Hematocrit capillary tubes into EDTA tubes (Gsmeditech, 123105) for plasma, and serum tubes (Gsmeditech, 111205) for serum. To remove cells and clots, the blood in collection tubes was centrifuged at 2000× g for 20 min, once for serum and twice for plasma. The biochemistry was analyzed using AU480 Chemistry Analyzer (Beckman Coulter). For cytokines analysis, serum was collected as described above and IL-6 was measured using the mouse IL-6 Quantikine ELISA kit (R&D System, M600B-1). For hematology and coagulation analysis, the whole blood was collected through cardiac puncture and blood samples were distributed into appropriate tubes [EDTA tube (BD microtainer, BD365974) for hematology (RBC, hematocrit, hemoglobin) analysis and trisodium citrate 0.109 M (V-TUBETM, 630187) for coagulation analysis (PT, aPTT)] and analyzed by Sysmex XN-9000 (Sysmex) for hematology and CA-1500 (Sysmex) for coagulation test.
Immunohistochemistry
The paraffin blocks of liver tissues fixed with 4% formalin were prepared, and sliced tissue samples were stained through Bond-X automatic slide stainer (Vision Biosystem) with primary antibody; CD47 (1:150, GeneTex GTX53912), RIP3 (1:200, Affinity Biosciences, AF7942), pMLKL (1:200, Affinity Biosciences, AF7420). The CD47-positive area and fibrosis (α-SMA) were quantified as a percentage of the stained area relative to the whole area of samples. TUNEL-stained samples were analyzed to quantify TUNEL-positive cell area relative to the total sample area.
Flow cytometry
Hepatic NPCs and primary hepatocytes were isolated from the mouse models following the liver tissue single-cell dissociation method. Isolated single NPCs were pre-stained with Zombie Aqua™ (1:500, BioLegend, 423102). Then, the cells were pre-incubated with anti-mouse CD16/CD32 (1:50, BD PharmingeTM, 553142) and stained with fluorescence-conjugated antibodies for liver immune cell analysis. Antibodies were purchased from BioLegend [PC7-anti-CD45.2 (1:80, 109830), APC-anti-CD11b (1:80, 101212), BV785-anti-CD11b (1:40, 101243), BV421-anti-CD31 (1:40, 102424), BV605-anti-Ly6G (1:80, 127639), FITC-anti-F4/80 (1:200, 123108) and PB-anti-Ly6C (1:333, 128014)]. To analyze the cell death phase, primary hepatocytes were stained with FITC-Annexin V and 7-AAD (1:20, BioLegend, 640922). The stained cells were analyzed by Beckman CytoFLEX flow cytometry and Kaluza Analysis 2.1.
Confocal microscopy
For confocal microscopy, liver tissues extracted from APAP-ALF models were immediately frozen in liquid nitrogen. The frozen tissues were then cryo-sectioned, which were fixed with 4% paraformaldehyde (Biosesang, P2031-050-00) and underwent a washing process four times for 5 min each, followed by blocking in 3% BSA/PBS solution for 1 h at room temperature. The primary antibodies [pMLKL (1:200, Abcam, ab196436), CD47 (1:40, R&D system, AF1866), and F4/80 (1:200, Bio-Rad, MCA497)] were applied to stain slides using a 1% BSA/PBS solution. After overnight incubation with the primary antibodies, samples underwent a washing process four times 5 min each. The secondary antibodies [Anti-rabbit Alexa Fluor 647-conjugated antibody (1:1000, Jackson ImmunoResearch, AB_2338072), Anti-goat Alexa Fluor 488-conjugated antibody (1:1000, Jackson ImmunoResearch, AB_2340439) and Anti-rat Alexa Fluor 488-conjugated antibody (1:1000, Jackson ImmunoResearch, AB_2338361)] were then applied using 1% BSA/PBS solution. After the washing process, the samples were mounted with DAPI Fluoromount-G solution (SouthernBiotech, 0100-20). The microscopic images were gained with confocal microscopy (Zeiss, LSM800).
Western blot
To analyze the expression of CD47, SIRPα, EV markers (CD9, CD63, CD81, and TSG101), and prohibitin, a western blot assay was employed. Protein concentrations of each sample were quantified using the BCA protein assay to ensure the loading of identical amounts of protein. Samples were lysed with RIPA buffer (Cell Signaling Technology) and mixed with Laemmli sample buffer (GenDEPOT). Initially, gels were run at 70 V for 10 min, followed by 100 V for 90 min to 2 h. Proteins were transferred to a PVDF membrane at 110 V for 90 min. After transferring proteins, membranes were blocked with a 5% skim milk solution or 5% BSA solution in 1× TBST for 1 h at room temperature. Then membranes were incubated with the primary antibody [CD47 (1:500, R&D System, AF1866), β-actin (1:10000, R&D System, MAB8929), SIRPα (1:4000, inhouse development), CD9 (1:1000, System Biosciences, EXOAB-CD9A-1), CD63 (1:500, Abcam, ab68418), CD81 (1:1000, Santa Cruz Biotechnology, sc-166029), TSG101 (1:2000, Abcam, ab125011), and prohibitin (1:500, Novus Biologicals, H00005245-M01)] in a 5% skim milk solution or 3% BSA overnight at 4 °C on a low-speed setting. Following primary antibody incubation, membranes were washed with 1× TBST, changing the wash buffer every 10 min, for 1 h at room temperature on a high-speed setting. Then membranes were incubated with the secondary antibody [horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Abcam, ab6728), anti-rabbit IgG (Abcam, ab6721) and anti-goat IgG (Thermo Fisher Scientific, 31402)] in a 5% skim milk solution for 1 h at room temperature on a low-speed setting. After incubation with secondary antibodies, the membrane was processed with enhanced chemiluminescence (ECL) western blotting substrate (Thermo Fisher Scientific, MA, USA), and the image was gained with the Amersham Image Quant™ 800 system. The uncropped scans of the blots have been provided in the Source Data file or the Supplementary Information files.
EV biodistribution
EVs were labeled with sulfo-Cyanine5.5-NHS ester or sulfo-Cyanine5.5 carboxylic acid (Lumiprobe, 27390) to assess the biodistribution of the EVs. Each EV was labeled with Cy5.5 dyes at a ratio of 5 × 1010 particles per 1 μg of Cy5.5 dye and incubated at 4 °C overnight. Zeba™ Spin Desalting Columns (ThermoFisher, A57762) were utilized to purify the labeled EVs and eliminate free Cy5.5 dyes. The fluorescence intensity of Cy5.5-NHS conjugated EVs was analyzed using a microplate. The efficiency of Cy5.5 labeling was determined using Fast Protein Liquid Chromatography (FPLC) (Cytiva, AKTApurifierTM) and Superdex 200 Increase 10/300 GL columns (Cytiva, GE28-9909-44). To assess the biodistribution of EVs, 7-week-old male C57BL/6NCrlOri mice fed an alfalfa-free diet (Teklad, 2918 C) were utilized for the APAP-ALF model analysis. 8 h post-APAP induction, Cy5.5-labeled samples at concentrations of 5 × 108, 2.5 × 109, or 2.5 × 1010 were administered intravenously. To assess the CD47-mediated targeting effect of SIRP-EVs, anti-CD47 antibodies (BioXCell, BE0007) and IgG2a isotype control antibodies (BioXCell, BE0090) (10 mg/kg, i.p.) were injected 30 min prior to the administration of 2.5 × 1010 Cy5.5-labeled EVs. After 24 h in the injection of EVs, organs were excised for ex vivo analysis to determine organ accumulation. The ex vivo biodistribution of EVs was visualized using the IVIS Spectrum (Caliper Life Sciences, IVIS® Lumina Series III). The diet of the mice was completely switched to alfalfa-free feed. For the evaluation of a single-cell-based EV distribution assay, liver tissues from the ALF model were dissociated using the gentleMACS™ Octo Dissociator with Heaters (Miltenyi Biotec, 130-096-427). Subsequently, individual cell populations, including hepatocytes, endothelial cells, myeloid cells, Kupffer cells, and MoMFs, were isolated in accordance with the liver tissue single-cell dissociation method. Further, the isolated single cells were labeled with an antibody panel of flow cytometry, and the Cy5.5 signals from each single cell were detected by CytoFLEX flow cytometry.
Spatial transcriptomics
Processing of ST data
This study used a publicly available spatial transcriptomic (ST) dataset (accession number GSE223560) of APAP-induced liver regeneration, focusing on liver samples from three healthy control mice and two collected 24 h post-APAP induction. All ST data were processed using the Seurat pipeline (version 4.0.2) in R (version 4.0.5). To integrate multiple datasets, the functions SelectIntegrationFeatures, FindIntegrationAnchors, and IntegrateData were employed. After normalization, spatial features were analyzed by examining associations such as the relationship between necroptosis and CD47 expression by calculating Spearman correlation coefficients.
Necroptosis and apoptosis signature score
The necroptosis or apoptosis signature scores were calculated to assess its correlation with CD47 expression in the ST data. Using the AddModuleScore function in Seurat, we computed necroptosis gene set scores for each spatial spot. Spearman correlation was then applied to evaluate the association between necroptosis or apoptosis signature scores with CD47 expression across spots. The list of genes was obtained from https://www.genome.jp/entry/pathway. The necroptosis-related gene list is provided in Supplementary Table 1, and the list of apoptosis-related genes is represented below: Aifm1, Apaf1, Bad, Bak1, Bax, Bbc3, Bcl2l11, Bid, Hrk, Pmaip1, Cycs, Casp3, Fas, Fasl, Diablo, Htra2, Endog, Trp53.
Cell type mapping on ST data
Spatial cell types were mapped by integrating spatial transcriptomics data with reference single-cell RNA-seq data specific to APAP-induced liver damage and regeneration, available at https://zenodo.org/records/603587354. For this integration, the CellDART algorithm was used14. In the mapping process, the number of marker genes for each cell type, as defined by the scRNA-seq data, was set to 30, while other parameters were kept at default to generate cell type maps on the ST data.
Topological overlap analysis on ST data
To investigate spatial colocalization patterns between the necroptosis or apoptosis signatures with macrophages in liver tissue, topological overlap analysis was performed using the STopover method15. This approach identifies topologically defined regions within the tissue that show local activation or accumulation of specific markers and assesses the extent of overlap between these regions. The proportion of each region relative to the overall tissue was evaluated by counting ST spots in both overlapped and non-overlapped areas. In addition, SIRPα expression within these regions was analyzed to further characterize cellular interactions.
Cell culture
The 293FT cells (HEK293 cell derivatives) were purchased from Thermo Fisher Scientific (R70007) and maintained in Dulbecco’s modified Eagle’s medium with high glucose (DMEM, Cytiva, SH30243.01) supplemented with 10% fetal bovine serum (FBS, Gibco, 12483-020) and 1% antibiotic–antimycotic (A.A., Gibco, 15240-062). To generate a stable cell line, Plat-E cells were used to produce a retrovirus containing a DNA sequence of interest with a puromycin resistance gene. When the confluency of 293FT cells reached 80–90%, Plat-E-derived retrovirus transduction was performed. Plat-E cells and 293FT cells were grown at DMEM with 10% FBS and 1% antibiotic–antimycotic, and virus-transduced 293FT cells were grown at puromycin-added medium with the same medium of 293FT cell media. The hBM-MSCs (RoosterBio) were maintained with RoosterNourish™ (RoosterBio) in CellBIND® Polystyrene CellSTACK® (Corning) for 2D cell culture. The CD47 KO cell line (Abcam, ab266324) and CD47 WT cell line (Abcam, ab244449) were purchased from Abcam and maintained following the noted manufacturer’s handling procedure. To acquire the mouse BMDMs, mouse bone marrow-derived primary cells were differentiated and maintained in accordance with the previously reported method55,56.
2D cell culture-based EV production and purification
The SIRPα transduced 293FT cells and MSCs were utilized to produce EVs (HEKSIRP-EV, C-EV, and SIRP-EV). Once the cell confluency reached 90%, the medium was replaced with serum-free medium composed of DMEM, 1% GLUTAMAX™ (Gibco, 35050061), and 1% A.A or RoosterCollect™-EV (RoosterBio). The supernatant was collected after 48 h and subjected to sequential centrifugation at 300× g for 10 min, 2000× g for 10 min, and 10,000× g for 30 min. The supernatant was then filtered and purified using the KrosFlo® KR2i Tangential Flow Filtration System equipped with hollow fiber filters (TFF, Repligen, Spectrum Labs). The samples were subsequently concentrated and diluted with PBS. Post-TFF, the samples were sterilized by filtration and centrifuged at 150,000× g for 1.5 h to gain pelleted EVs. The EV pellets were resuspended in PBS containing a proteinase inhibitor cocktail (Sigma-Aldrich, 535140).
Engineered MSC production
Human bone marrow-derived MSC (hBM-MSC) master cell banks (Passage 0, RoosterBio) were plated in RoosterNourishTM (RoosterBio) in T-75 flasks (Corning) and incubated at 37 °C, 5% CO2. After 2 days, cells were harvested and counted for seeding to the T-75 flasks. To engineer EVs displaying SIRPα, we generated a SIRPα variant capable of binding both human and mouse CD47 with high affinity21,40,43,57, fused to a type 1 transmembrane domain to ensure effective surface expression on EVs58. Lentiviral transduction was performed in RoosterGEM™ (Genetic engineering medium, RoosterBio) with lentivirus (LV) encoding the SIRPα construct manufactured by Flash Therapeutics. LV volume for transduction was determined by titer and the multiplicity of infectivity (MOI). For LV transduction, the culture medium was replaced with a transduction medium that contained LV and RoosterGEM™, while control MSCs used RoosterGEM™ without LV. After incubation of cells with RoosterGEM™ for 24 h, the media was exchanged to RoosterNourish™. No rinses were performed before medium replenishment. The cells were maintained for 24 h, seeded into appropriate vessels, and expanded for 4–5 days until the cell confluency reached 80%. The cells were harvested, formulated in Cryostor 5 (Biolife Solutions), filled into 2 mL cryovials (Corning), and cryopreserved at passage 2 using a controlled rate freezer and stored in the vapor phase of liquid nitrogen. These vials are considered the SIRP-MSC from which all seed train expansions started. SIRP-MSCs were analyzed for expansion ability, MSC characteristics, MSC markers, and SIRPα expression. Mesenchymal stem/stromal cells (MSCs) used in this study were originally derived from human bone marrow aspirate obtained from healthy consenting adult donors from within the United States.
3D bioreactor-based upstream process for EV condition media generation
SIRP-MSCs were thawed and expanded in CellBIND® Polystyrene CellSTACK® Chamber (Corning) for two passages prior to bioreactor expansion. For generation of the seed train, each passage was considered for expansion of 4 or 5 days to reach a confluency of 80–95%. Optimization studies were previously conducted to establish the critical process parameters for scaling up the process59,60,61. SIRP-MSCs from the seed train and microcarriers were inoculated into the Ambr®250 vessels (Sartorius) or STR®50 bioreactor vessels (Sartorius). After inoculation of the Ambr®250 bioreactor, the temperature was maintained at 37 °C, dissolved oxygen (DO) at 100%, and airflow rate at 24 mL/min with 5% CO2 in the overlay. In the STR®50 bioreactor, the temperature was set to 37 °C, DO control to 50%, and air flow rate to 1.5 L/min with 5% CO2 in the headspace. RoosterNourish™ was supplemented with a RoosterReplenish™ medium feed (RoosterBio) on day 3 of expansion, per the manufacturer's instructions. The cells were expanded for 5 days. At the end of the expansion phase, the medium was removed, the cells/microcarriers were washed twice, and then a protein-free, chemically defined low particulate medium, RoosterCollect™-EV (RoosterBio) was added (250 ml final volume in Ambr, 15 L or 50 L in STR50) to the bioreactors to collect EVs at the end of the 5-day culture period. Cell counts and metabolites were measured daily during the expansion phase to monitor cell health. The particle count (particles/mL) was obtained regularly from the bioreactors. Macroscopic and microscopic images of the cells/microcarriers suspension were obtained throughout the process. At the end of the EV collection phase, the bioreactor impellers were stopped, and microcarriers were allowed to settle to the bottom of the vessel. After settling, the conditioned media containing EVs was collected through pipetting in the Ambr250 or pumped via the dip tube in the STR50 bioreactor single-use disposable bag. Particle count (particles/mL), particle size and distribution, and SIRPα content were measured. For 2D control flasks served as controls for cell expansion and EV collection, transduced MSCs were seeded into CellBIND® flasks. Cells were cultured for 5 days in an incubator at 37 °C and 5% CO2. At the end of the expansion, the media was aspirated, and the adherent cells were washed before being exchanged into RoosterCollect-EV media for EV collection. TrypLE™ (ThermoFisher) was used when needed for cell harvest from flasks or microcarriers.
Downstream purification of EV
SIRP-EVs were purified from conditioned medium collected from bioreactors utilizing technologies that are scalable, GMP compatible, and consistent with equipment found at traditional biopharma CDMOs. The conditioned media was treated with Agent V (RoosterBio) per the manufacturer’s instructions prior to clarification. Agent V-treated conditioned media was clarified to remove cell debris by pumping through a depth filter (Sartorius). Pre-filter pressure was continuously monitored and maintained per the manufacturer’s recommendations. The clarified medium was concentrated up to 10-fold by volume by filtering through tangential flow filtration (TFF) (Repligen). After concentration, buffer exchange was performed via continuous diafiltration to prepare the concentrated EV solution for chromatography. The concentrated solution was further purified with an AKTA Avant150 chromatography system (Cytiva) using multimodal chromatography resin (Cytiva). The system flow rate, pressures, conductivity, and column UV were controlled and monitored throughout the runs. SIRP-EVs were collected in the flow through fractions, which was consistent throughout scales. The resulting purified EVs were formulated in a sucrose-containing salt buffer via a second TFF diafiltration step (Repligen), then filter sterilized via 0.2 µm membrane (Sartorius) filtration. The resultant purified EV solution was filled into particle-free cryovials (NEST) and stored at −80 °C. To confirm the removal of impurities during the downstream process, intermediate products were analyzed, with total protein measured by Bradford assay (Thermo Fisher), double-stranded DNA (dsDNA) measured by PicoGreen dsDNA assay, and albumin measured using the human albumin ELISA kit (Abcam).
RNA analysis
The total RNAs in cells and EVs were isolated utilizing miRNeasy micro kit (Qiagen, 217084). To detect various ranges of RNA in samples, RNA electroporation was applied with two standard ladders (Thermo Fisher, SM1831, and SM1821), and the gels were detected by GelDoc Go Imaging System (Bio-Rad). The amount of RNA was quantified using the Quant-it™ RiboGreen RNA assay kit (Invitrogen, R11490), which was applied to RNase-free DNases pre-treated samples, and fluorescence was detected using VICTOR Nivo™ multimode microplate reader (PerkinElmer, HH35000500).
EV characterization
The size distribution of EVs was analyzed using Dynamic Light Scattering (DLS) with a Zetasizer Nano S90 (Malvern Panalytical). The PDI values were calculated using the Malvern Zetasizer software. The EV particle counts, and zeta potential (ZP) were measured with Nanoparticle Tracking Analysis (NTA) with ZetaView system (Particle Metrix). Data analysis was conducted using ZetaView software version 8.05.16 SP7. Prior to measuring the size distribution and ZP of the EV samples, auto-alignment was completed using a 100 nm polystyrene beads standard solution (WithInstrument, 700074). Before loading the samples, it was ensured that the number of detected particles was below 10, followed by a washing process to commence sample analysis. The background buffer for measurement of size distribution was sample buffer, and deionized water (DW) was used for ZP background buffer. To explore the structure of EVs, Transmission electron microscopy imaging was obtained using the cryo-TEM (FEI Tecnai F20 G2) equipment, and the image analysis was conducted. To quantify protein amount in EVs, BCA assay or Bradford assay was applied, and the results were converted into EV purity (particle/mg). To confirm the percentage of lipid-bound content, MemGlow™ dye was applied and optimized to a working concentration. The dyed samples were evaluated with NanoFCM, and stained events were used to calculate the ratio of bound lipid.
Proteomic profiling of EVs
For the transcriptomic analysis, C-EV, HEKSIRP-EV and SIRP-EV samples (n = 3), were employed followed by LC-MS/MS analysis. The identified proteins and their quantitative values in each batch were used to compare differences between batches. The number of proteins analyzed for each sample type was 7391 for both C-EV and SIRP-EV.
For sample lysis, EV samples were added chilled acetone with four volume of samples and incubated at −80 °C for 90 min. The reacted samples were centrifuged, removed supernatant, and dried. The dried samples were resuspended in 8 M urea in 100 mM ammonium bicarbonate and subjected to sonication for 5 min to lyse the precipitated samples. After centrifugation at 16,000 rpm for 5 min, the supernatant was transferred to a new tube, and the protein concentration was measured using a BCA assay kit to adjust the same amount of protein to be analyzed. For protein reduction and alkylation, dithiothreitol (DTT) was added to the EV lysates at a final concentration of 10 mM, and the mixture was incubated at 37 °C with shaking at 450 rpm for 30 min to reduce the proteins. After reduction, IAA (iodoacetamide) was added at a final concentration of 25 mM, and the mixture was incubated in the dark at room temperature for 30 min for alkylation. For protein digestion, the alkylated protein samples were diluted with 100 mM ABC (ammonium bicarbonate) to decrease the urea concentration to below 1 M. Trypsin digestion was performed at a 1:25 enzyme:protein ratio, and the reaction was allowed to proceed for 16 h at 37 °C. The trypsin reaction was quenched by adding TFA (trifluoroacetic acid) to a final concentration of 1% TFA. The prepared samples were Tandem Mass Tag (TMT) labeled, and TMT labeled 100 μg of samples were fractionated into 12 fractions to be analyzed by Nano LC-MS/MS system (Thermo Dionex Ultimate 3000 coupled with Thermo Orbitrap Exploris 480). DDA (Data Dependent Analysis) was performed using SAGE software to match mass spectrometry spectra with human protein sequence information provided by the UniProt database. Database search parameters were restricted to semi-tryptic peptides with length of 7–50 amino acids. Missed cleavage was allowed up to one and cysteine alkylation and methionine oxidation were included in peptide modifications. FDR (False Discovery Rate) was applied under 1% at the spectrum, peptide, and protein levels and precursor isolation purity were 0.7 or higher. Gene Ontology Biological Processes (GOBPs) among the top 506 SIRP-EV proteins was established through FunRich software v.3.1.3. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD060047.
Cell binding assay
For SIRPα in vitro cell binding analysis, each set of EVs was labeled with sulfo-Cyanine5.5-NHS ester (Lumiprobe, 27320) at a ratio of 5 × 1010 particles per 25 ng of Cy5.5 dyes and incubated at 4 °C overnight. The Zeba™ Spin Desalting Columns (ThermoFisher, A57762) were employed to purify the labeled EVs and eliminate free Cy5.5 dyes. The fluorescence intensity of Cy5.5-NHS ester conjugated EVs was analyzed using a microplate reader (PerkinElmer, VICTOR Nivo™) to compare the fluorescence intensity among samples. In total, 2 × 105 HEK CD47 WT (wild type, Abcam, ab24449) or KO (knockout, Abcam, ab266324) cells were mixed with 200 μL of 3% BSA and incubated at 4 °C for 15 min. The samples were then centrifuged at 450× g for 3 min at 4 °C, and the supernatants were discarded. The pellets were resuspended in 200 μL of the desired concentrations of C-EV or SIRP-EV (5 × 108, 1 × 109, or 2.5 × 109) and incubated at 4 °C for 15 min. Following another centrifugation at 450 g for 3 min at 4 °C, the supernatants were discarded. The pellets were washed with 200 μL of 1% BSA in PBS and centrifuged at 450× g for 3 min. The final pellets were resuspended in 200 μL of PBS and analyzed by flow cytometry.
In vitro phagocytosis assay
The fully differentiated BMDMs were labeled with 1 µM CellTracker Green CMFDA (Invitrogen, C7025) for 30 min at room temperature in the dark and the excess dye was removed by following centrifugation. The stained BMDMs were subsequently seeded 24 h before the assay. HepG2 cells (Korean Cell Line Bank, 72650) treated with 40 µM APAP or primary hepatocytes isolated from 300 mg/kg APAP-ALF model mice were labeled with 0.5 µM CellTracker Deep Red (Invitrogen, C34565). BMDMs and either HepG2 cells or primary hepatocytes were co-cultured in the presence of C-EVs or SIRP-EVs at a cell ratio of 1:2 at 37 °C. After the incubation, all cells were collected, washed, and analyzed using flow cytometry. Phagocytosis levels were quantified by calculating the percentage of HepG2 cells (Deep Red+ cells) within the total BMDM population (Green CMFDA+ cells). For analysis of phagocytic activity, the MFI of Deep Red in primary hepatocytes internalized by BMDMs was quantified.
Hemagglutination assay
For confirming RBC aggregation by EVs or anti-CD47 antibody (Santa Cruz Biotechnology, sc-21786), washed human RBCs (Innovative Research, IWB3ALS40ML) were incubated with Alsever’s solution (Sigma-Aldrich, A3551) and centrifuged at 600× g for 5 min at 4 °C, twice. The pellets were then resuspended in PBS and centrifuged again at 600× g for 5 min at 4 °C. Subsequently, the pellets were prepared in a 5% RBC/PBS solution. The samples were incubated with the indicated quantities of samples for 4 h at 37 °C in U-shaped bottom 96-well microplate. The aggregations were analyzed by flow cytometry or microscopy (Nikon, ECLIPSE Ts2).
Liver tissue single-cell dissociation
To analyze CD47 expression, necroptotic hepatocyte, cell type-specific biodistribution of EVs, and CD11b+ cell depletion, primary hepatocytes and hepatic non-parenchymal cells (NPCs) were isolated using the gentleMACS™ Octo Dissociator with Heaters (Miltenyi Biotec, 130-096-427) with gentleMACS™ Perfusers (Miltenyi Biotec, 130-128-151), gentleMACS™ Perfusion Sleeves (Miltenyi Biotec, 130-128-752), and Liver Perfusion Kit (Miltenyi Biotec, 130-128-030). For the liver immune cell flow cytometry analysis, NPCs were isolated from HBSS-perfused livers. The livers were transferred to gentleMACS™ C Tubes (Miltenyi Biotec, 130-096-334) and dissociated into single-cell suspension with DMEM containing DNase (Roche, 10104159001) and collagenase (Sigma, C5138) at 37 °C utilizing gentleMACS™ Octo Dissociator with Heaters (Miltenyi Biotec, 130-096-427). Dissociated liver tissues were filtered through 70-μm cell strainers (SPL, 93070). Cell suspensions were centrifuged at 300× g for 5 min at 4 °C. NPC pellet was resuspended in 5 mL RBC lysis buffer (BioLegend, 420301) and incubated for 5 min at 4 °C. DPBS (Welgene, LB001-02) was added, and suspensions were centrifuged at 300× g for 5 min in 4 °C. The prepared NPC pellets were resuspended with DPBS for analysis.
Bulk RNA sequencing analysis
To compare the RNA expression, the APAP (500 mg/kg) induced mice were treated with HEKSIRP-EV or SIRP-EV, and the livers were harvested 24 h after APAP induction. The harvested livers were dissociated into cells following liver tissue single-cell dissociation protocol, and CD11b-positive cells were isolated using CD11b MicroBeads UltraPure (Miltenyi Biotec, 130-126-725). Total RNA from CD11b-positive cells was prepared using Trizol reagent (Invitrogen, 15596026). The quality of RNA (RNA integrity) was assessed using an Agilent Technologies 2100 Bioanalyzer. The size of the RNA library was determined using an Agilent Technologies 2100 Bioanalyzer with a DNA 1000 chip, and the RNA library was quantified following the Illumina qPCR Quantification Protocol Guide. To prepare aligned reads, the trimmed RNA reads were mapped to the reference genome using the HISAT2 program, and then the aligned reads were processed for transcript assembly with the StringTie program. Gene expression profiles were calculated by the read count, normalized FPKM (Fragments Per Kilobase of transcript per Million mapped reads)/RPKM (Reads Per Kilobase of transcript per Million mapped reads), and TPM (Transcripts Per Kilobase Million). To compare different gene expressions among the groups, the significant genes were identified by applying adjusted P value with the Benjamini–Hochberg procedure and categorized into the five gene clusters (Cellular viability, Detoxification/Antioxidation, ECM Remodeling, Immune Modulation, and Vascularization and Perfusion). The list of 404 genes (Source Data Fig. 7F) was applied for DEG (Differentially Expressed Genes) analysis. For the DESeq2 analysis, the negative binomial Wald Test (nbinomWaldTest) was conducted with RLE (Relative Log Expression) normalized counts.
CD11b+ myeloid cell reduction in the APAP-induced ALF model
An antibody depletion model of CD11b+ myeloid cell under the APAP induction was used to assess the therapeutic efficacy of CD11b+ myeloid cells. 5 mg/kg of CD11b+ myeloid cell depletion antibody (BioXCell, BE0007) and IgG antibody (BioXCell, BE0090) were intraperitoneally injected 16 h before 500 mg/kg dose of APAP induction. In all, 4 × 109 EVs were intravenously injected 8 h after APAP induction. To test its depletion for pre-test, liver, and spleen tissues were harvested 24 h after APAP induction and dissociated into the single-cell populations utilizing the gentleMACS™ Octo Dissociator with Heaters. The isolated single cells were further stained with a flow cytometric antibody panel. To determine the therapeutic efficacy in the reduction of CD11b+ myeloid cells, serum and liver tissues were harvested at 48 h post-APAP induction and analyzed biochemistry (ALT), tissue staining (CD47 IHC) following the same protocol in “Methods”.
Multiplex immunohistochemistry
The paraffin blocks of liver tissues fixed with 4% formalin were prepared to 4-μm thick slides. The tissue slides underwent sequential steps for staining, heat-drying, dewaxing, and antigen retrieval. After blocking with antibody diluent/block, the slides were stained with primary antibody [F4/80 (1:150, Cell Signaling Technology, 70076), HNF-4α (1:200, R&D system, PP-H1415-0C), CD47 (1:150, GeneTex, GTX53912), Ki67 (1:1000, Abcam, ab16667), pMLKL (1:100-400, Affinity Biosciences, AF7420)]. Then, tyramide signal amplification was applied to visualize the antigen. The samples were treated with Bone Epitope Retrieval 1 to remove bound antibodies before staining the next antigen. The process from the blocking to retrieval was repeated to stain each primary antibody. Nuclei were visualized with DAPI. The CD47+ necroptotic cells were quantified as a percentage relative to the total cells, and Ki67+ cells were quantified relative to the total area (cell/mm2).
Statistical analysis
GraphPad Prism 10 was utilized for statistical analysis. Two-tailed unpaired Student’s t tests were used to compare the two groups. One-way analysis of variance (ANOVA) with Tukey’s post hoc test was employed for comparisons between multiple groups, and two-way ANOVA with Sidak’s post hoc test was used for analyzing multiple parameter samples. The survival rate was analyzed using Kaplan–Meier log-rank analysis. The proteomic analysis data was evaluated by Pearson’s correlation coefficient. Spearman correlation was applied to spatial transcriptomic analysis. Heatmap analysis was evaluated by Benjamini–Hochberg adjusted P values. The values were expressed as the mean ± SD. for samples. *P < 0.05, **P < 0.01, ***P < 0.001. Additional information on materials and methods is included in the supplementary information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All relevant data generated in this study are available within the article and its Supplementary Information files, Raw data files. The source data is provided with this paper. The publicly available spatial transcriptomic (ST) dataset has been deposited to the National Center for Biotechnology Information Gene Expression Omnibus (GEO) with the accession number GSE223560. The reference single-cell RNA-seq data specific to APAP-induced liver damage and regeneration can be accessed at Zenodo via the dataset identifier [https://zenodo.org/records/6035873]. The proteomics data are included in the source data files and have also been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD060047. The bulk RNA sequencing data are included in the source data and under accession number GSE281951 or SRA. Source data are provided with this paper.
References
Chen, L. et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9, 7204–7218 (2018).
Stravitz, R. T. & Kramer, D. J. Management of acute liver failure. Nat. Rev. Gastroenterol. Hepatol. 6, 542–553 (2009).
Deutsch, M. et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 6, e1759 (2015).
Jaeschke, H., Ramachandran, A., Chao, X. & Ding, W.-X. Emerging and established modes of cell death during acetaminophen-induced liver injury. Arch. Toxicol. 93, 3491–3502 (2019).
Zhao, H. et al. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 6, e1975 (2015).
Schwabe, R. F. & Luedde, T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 15, 738–752 (2018).
Gerlach, B. D. et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. 27, 525–539 (2020).
Gerlach, B. D. et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab. 33, 2445–2463.e8 (2021).
Shi, H. et al. CD47-SIRPα axis blockade in NASH promotes necroptotic hepatocyte clearance by liver macrophages and decreases hepatic fibrosis. Sci. Transl. Med. 14, eabp8309 (2022).
Weston, C. J., Zimmermann, H. W. & Adams, D. H. The role of myeloid-derived cells in the progression of liver disease. Front. Immunol. 10, 893 (2019).
Wen, Y., Lambrecht, J., Ju, C. & Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol. 18, 45–56 (2021).
Hefler, J. et al. Preclinical models of acute liver failure: a comprehensive review. PeerJ 9, e12579 (2021).
Bélanger, M. & Butterworth, R. F. Acute liver failure: a critical appraisal of available animal models. Metab. Brain Dis. 20, 409–423 (2005).
Bae, S. et al. CellDART: cell type inference by domain adaptation of single-cell and spatial transcriptomic data. Nucleic Acids Res. 50, e57–e57 (2022).
Bae, S. et al. STopover captures spatial colocalization and interaction in the tumor microenvironment using topological analysis in spatial transcriptomics data. Preprint at biorxiv https://doi.org/10.1101/2022.11.16.516708 (2022).
Giatromanolaki, A. et al. Expression of CD47 and SIRPα macrophage immune-checkpoint pathway in non-small-cell lung cancer. Cancers 14, 1801 (2022).
Roefs, M. T., Sluijter, J. P. G. & Vader, P. Extracellular vesicle-associated proteins in tissue repair. Trends Cell Biol. 30, 990–1013 (2020).
Lee, I. et al. Small extracellular vesicles as a new class of medicines. Pharmaceutics 15, 325 (2023).
Welsh, J. A. et al. Minimal information for studies of extracellular vesicles (MISEV2023): from basic to advanced approaches. J. Extracell. Vesicles 13, e12404 (2024).
Van Balkom, B. W. M., Gremmels, H., Giebel, B. & Lim, S. K. Proteomic signature of mesenchymal stromal cell-derived small extracellular vesicles. Proteomics 19, e1800163 (2019).
Kim, Y. K. et al. Advantage of extracellular vesicles in hindering the CD47 signal for cancer immunotherapy. J. Control Release 351, 727–738 (2022).
Bilzer, M., Roggel, F. & Gerbes, A. L. Role of Kupffer cells in host defense and liver disease. Liver Int. 26, 1175–1186 (2006).
Liam-Or, R. et al. Cellular uptake and in vivo distribution of mesenchymal-stem-cell-derived extracellular vesicles are protein corona dependent. Nat. Nanotechnol. 19, 846–855 (2024).
Dechêne, A. et al. Acute liver failure is associated with elevated liver stiffness and hepatic stellate cell activation. Hepatology 52, 1008–1016 (2010).
An, P. et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 11, 2362 (2020).
Feng, D. et al. Monocyte-derived macrophages orchestrate multiple cell-type interactions to repair necrotic liver lesions in disease models. J. Clin. Investig. 133, e166954 (2023).
Prescott, L. F. et al. Intravenous N-acetylcystine: the treatment of choice for paracetamol poisoning. Br. Med. J. 2, 1097–1100 (1979).
Arnold, K. et al. Design of anti-inflammatory heparan sulfate to protect against acetaminophen-induced acute liver failure. Sci. Transl. Med. 12, 1–12 (2020).
Starkey Lewis, P. et al. Alternatively activated macrophages promote resolution of necrosis following acute liver injury. J. Hepatol. 73, 349–360 (2020).
Guilliams, M. & Scott, C. L. Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol. 17, 451–460 (2017).
Flores Molina, M. et al. Distinct spatial distribution and roles of Kupffer cells and monocyte-derived macrophages in mouse acute liver injury. Front. Immunol. 13, 994480 (2022).
Li, W., Yang, Y., Yang, L., Chang, N. & Li, L. Monocyte-derived Kupffer cells dominate in the Kupffer cell pool during liver injury. Cell Rep. 42, 113164 (2023).
Silva, R. M. et al. Anion exchange chromatography-based platform for the scalable purification of extracellular vesicles derived from human mesenchymal stromal cells. Sep Purif. Technol. 310, 123238 (2023).
Watson, D. C. et al. Scalable, cGMP‐compatible purification of extracellular vesicles carrying bioactive human heterodimeric IL‐15/lactadherin complexes. J. Extracell. Vesicles 7, 1442088 (2018).
Busatto, S. et al. Tangential flow filtration for highly efficient concentration of extracellular vesicles from large volumes of fluid. Cells 7, 273 (2018).
Estes, S., Konstantinov, K. & Young, J. D. Manufactured extracellular vesicles as human therapeutics: challenges, advances, and opportunities. Curr. Opin. Biotechnol. 77, 102776 (2022).
Yang, H. Establishing acceptable limits of residual DNA. PDA J. Pharm. Sci. Technol. 67, 155–163 (2013).
Brennan, K. et al. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 10, 1039 (2020).
Webber, J. & Clayton, A. How pure are your vesicles? J. Extracell. Vesicles 2, 19861 (2013).
Koh, E. et al. Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis. Biomaterials 121, 121–129 (2017).
Kim, G. B. et al. Harnessing oncolytic extracellular vesicles for tumor cell-preferential cytoplasmic delivery of misfolded proteins for cancer immunotherapy. Small 19, e2300527 (2023).
Yang, Y. et al. Virus-mimetic fusogenic exosomes for direct delivery of integral membrane proteins to target cell membranes. Adv. Mater. 29, https://doi.org/10.1002/adma.201605604 (2017).
Cho, E. et al. Comparison of exosomes and ferritin protein nanocages for the delivery of membrane protein therapeutics. J. Controlled Release 279, 326–335 (2018).
Nam, G.-H. et al. Emerging prospects of exosomes for cancer treatment: from conventional therapy to immunotherapy. Adv. Mater. 32, e2002440 (2020).
Herrmann, I. K., Wood, M. J. A. & Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 16, 748–759 (2021).
Kim, G. B. et al. Xenogenization of tumor cells by fusogenic exosomes in tumor microenvironment ignites and propagates antitumor immunity. Sci. Adv. 6, eaaz2083 (2020).
Kim, Y. K., Choi, Y., Nam, G.-H. & Kim, I.-S. Functionalized exosome harboring bioactive molecules for cancer therapy. Cancer Lett. 489, 155–162 (2020).
Willingham, S. B. et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 109, 6662–6667 (2012).
Cui, L. et al. Activation of JUN in fibroblasts promotes pro-fibrotic programme and modulates protective immunity. Nat. Commun. 11, 2795 (2020).
Jiang, Z., Sun, H., Yu, J., Tian, W. & Song, Y. Targeting CD47 for cancer immunotherapy. J. Hematol. Oncol. 14, 180 (2021).
Maute, R., Xu, J. & Weissman, I. L. CD47-SIRPα-targeted therapeutics: status and prospects. Immuno-Oncol. Technol. 13, 100070 (2022).
He, Y. et al. Mechanisms of fibrosis in acute liver failure. Liver Int. 35, 1877–1885 (2015).
Nissim, O. et al. Liver regeneration signature in hepatitis B virus (HBV)-associated acute liver failure identified by gene expression profiling. PLoS ONE 7, e49611 (2012).
Ben-Moshe, S. et al. The spatiotemporal program of zonal liver regeneration following acute injury. Cell Stem Cell 29, 973–989.e10 (2022).
Nam, G.-H. et al. An optimized protocol to determine the engulfment of cancer cells by phagocytes using flow cytometry and fluorescence microscopy. J. Immunol. Methods 470, 27–32 (2019).
Nam, G.-H. et al. Combined Rho-kinase inhibition and immunogenic cell death triggers and propagates immunity against cancer. Nat. Commun. 9, 2165 (2018).
Weiskopf, K. et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91 (2013).
Kim, I. S., Park, S., Nam, G. H. & Lee, I. Surface-engineered extracellular vesicles and therapeutic uses thereof. US Patent 20230381226A1, United States Patent and Trademark Office (2023).
Lenzini, S. et al. Exosomes/EVs: developing a microcarrier stirred tank process for large-scale hMSC-EV production. Cytotherapy 24, S91 (2022).
Wagey, R. et al. Mesenchymal stem/stromal cells: suspension culture of human mesenchymal stromal cells on dissolvable microcarriers in an animal component-free culture system. Cytotherapy 24, S71–S72 (2022).
Kirian, R. D. et al. Scaling a xeno-free fed-batch microcarrier suspension bioreactor system from development to production scale for manufacturing XF hMSCs. Cytotherapy 21, S71–S72 (2019).
Acknowledgements
This research was supported by SHIFTBIO INC., Korean Fund for Regenerative Medicine funded by Ministry of Science and ICT, and Ministry of Health and Welfare (Grant Number: 23C0111L1, Republic of Korea, Participant: S.K. (Seohyun Kim), Y.K.K., S.K.(Seonghyun Kim), Y.-S.C., I.L., J.K., Y.C., and G.B.K.), a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant Number: RS-2023-KH136648, Participant: S.K. (Seohyun Kim), Y.K.K., S.K.(Seonghyun Kim), Y.-S.C., I.L., J.K., Y.C., G.B.K., M.K.J, M.K., S.P., and G-H.N.; RS-2023-KH140007, Participant: M.K.J, M.K., S.P., and G-H.N.), a grant of the BIG3 Project, funded by the Ministry of SMEs and Startups, Republic of Korea (Grant Number: RS-2022-TI022422, Participant: S.K. (Seohyun Kim), Y.K.K., S.K.(Seonghyun Kim), I.L., J.K., and G.B.K.), and National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (Grant Number: RS-2017-NR022964, Participant: I.-S.K). Production of LV was outsourced to Flash Therapeutics (Toulouse, France). Quantifying total RNA amount was outsourced to Macrogen (Seoul, Republic of Korea). Multiplex immunohistochemistry (multiplex IHC) was conducted by outsourcing to PrismCDX Co., Ltd. (Gyeonggi-do, Republic of Korea). The biochemistry, hematology, and coagulation analysis were outsourced to DKKorea (Seoul, Republic of Korea). Cryo-TEM (FEI Tecnai F20 G2) imaging was conducted at Korea Institute of Science and Technology Advanced Analysis Center (Seoul, Republic of Korea). Proteomic profiling was analyzed by outsourcing to Bertis (Gyeonggi-do, Republic of Korea).
Author information
Authors and Affiliations
Contributions
S.K. (Seohyun Kim), G.-H.N., G.B.K., and I.-S.K. conceived the idea. S.K. (Seohyun Kim), Y.K.K., G.-H.N., and G.B.K. designed the research. S.-Y.P. designed the genetic constructs and performed the cloning. I.K. T.D.S., J.J., E.Z., and S.L. developed the manufacturing process. S.K. (Seohyun Kim), Y.K.K., J.K., M.K., S.P., M.K.J., and Y.C. performed the in vitro experiments. S.K. (Seohyun Kim), Y.K.K., S.K. (Seonghyun Kim), Y.-S.C., H.J., Y.C., and G.B.K. the in vivo experiments. J.W. supported in vivo experiments. S.K. (Seohyun Kim), Y.K.K., S.K. (Seonghyun Kim), I.L., J.K., T.D.S., J.J., E.Z., S.L., H.C., J.P. and G.B.K. analyzed the results. S.K. (Seohyun Kim), Y.K.K., I.K., G.B.K., and G.-H.N. wrote the manuscript with input from all authors. G.B.K., G.-H.N., and I.-S.K. co-led the study.
Corresponding authors
Ethics declarations
Competing interests
I.-S.K. and G.-H.N. are the co-founders and have stock interest in SHIFTBIO INC. S.K. (Seohyun Kim), Y.K.K., S.K. (Seonghyun Kim), Y.-S.C., I.L., H.J., J.K., Y.C., and G.B.K. are employees of SHIFTBIO INC. T.D.S., J.J., E.Z., and S.L. are employees of RoosterBio, Inc. H.C. are the co-founder and have stock interest in Portrai, Inc. J.P. is an employee of Portrai, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Takashi Matozaki, Yasuyuki Saito and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, S., Kim, Y.K., Kim, S. et al. Dual-mode action of scalable, high-quality engineered stem cell-derived SIRPα-extracellular vesicles for treating acute liver failure. Nat Commun 16, 1903 (2025). https://doi.org/10.1038/s41467-025-57133-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57133-w
This article is cited by
-
Dual-ligand engineered exosome regulates WNT signaling activation to promote liver repair and regeneration
Nature Communications (2025)
-
Identification of key biomarkers associated with necroptosis and immune infiltration in hepatitis B virus-related acute-on-chronic liver failure
Scientific Reports (2025)
