
Abstract
Earth-abundant TiO2 is a promising negative electrode for low-cost sodium-ion batteries (SIBs) owing to its high capacity, rapid (dis)charging capability, safe operation potential and nonflammability. Crystalline anatase TiO2 is not suitable for reversible Na+ (de)intercalation, but it displays pseudocapacitive response after repeated cycles. Herein, we find and demonstrate that ordered rocksalt (RS) NaTiO2 nanograins are in situ formed by electrochemically cycling with Na+ ions in anatase and amorphous TiO2. The in situ formed RS-NaTiO2 follows a solid-solution reaction with small volume changes of only 2.0%, that determines the pseudocapacitive “mirror-like” cyclic voltammetry curve with a couple of broad redox peaks at 0.75 V vs. Na+/Na, a high capacity of 253 mAh g−1, high-rate capability and thousands of stable cycles. The multistep crystalline-to-amorphous-to-RS transformations are able to be electrochemically activated during the aging process of assembled full cells. Our finding provides a direction for developing unconventional Ti-based high-performance active materials for SIBs with both high energy and power densities.
Similar content being viewed by others


Introduction
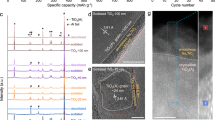
Electrochemical sodium-ion batteries (SIBs) arise extensive demands owing to the surge of large-scale energy storage markets and limited lithium resources1,2,3. The scalable applications of SIBs require abundant resources4,5, e.g. Si (28.2 wt.%), Fe (5.63 wt.%), Ti (0.56 wt.%), C (0.2 wt.%), P (0.105 wt.%), Mn (0.095 wt.%) and S (0.035 wt.%) on the Earth’s crust. Seeking safe and high-performance negative electrode materials for SIBs is of significant importance but is full of challenges. For example, the silicon and silicon oxides are electrochemically inactive for sodium-ion storage6. The conversion reaction of iron7 and manganese oxides8, and the alloying reaction of phosphorus9 and sulfur10 deliver large voltage hysteresis and poor cycling life, which is unsuitable for grid storage that requires high energy efficiency and long-term applications. Hard carbon negative electrodes display a high capacity of 300–400 mAh g−1 with a low potential plateau at ~ 0.1 V vs. Na+/Na, which is advantageous for high-energy SIBs11. Unfortunately, its sluggish sodiation kinetics and low potential plateau give rise to the growth of Na dendrites and even serious safety risk12. In addition to hard carbon, Ti-based materials are abundant and have various polymorphs and derivatives, including titanium dioxides13, titanates14, titanium phosphorous15, and Ti-MXenes16, for use in electrochemical sodium-ion storage (Fig. 1a and Supplementary Table 1)17,18. Among them, based on the full electrochemical activation of the Ti4+/Ti3+ redox couple (that is one Na+ per Ti), the titanium dioxides (TiO2) display a high capacity and appropriate operation potential19.

a Operation potential vs. capacity profiles of Ti-based negative electrode materials for SIBs. b CV curves of a-TiO2 and c-a’-RS-NaTiO2 at 0.2 mV s–1. The electrochemically formed c-a’-RS-NaTiO2 nanograins display a “mirror-like” CV shape with a couple of broad redox peaks at ~ 0.75 V vs. Na+/Na, which is quite different from the “box-like” shape a-TiO2 electrodes. c GCD curves of hard carbon, NaTi2(PO4)3 and c-a’-RS-NaTiO2 at 0.1 A g–1, indicating the promise of c-a’-RS-NaTiO2 for high-rate applications.
The intercalation of Na+ (or Li+) into active materials requires suitable diffusion channels and empty sites20,21. Anatase TiO2 (TiO2(A)) has empty octahedral vacancies for accommodating Li+ ions (a radius of 0.76 Å) with a reversible two-phase transformation, displaying a flat plateau at ~1.75 V vs. Li+/Li (that is − 1.29 V vs. standard hydrogen electrode (SHE))22. However, the initial intercalation of Na+ into nanosized TiO2(A) (~ 10 nm, Supplementary Fig. 1) displays an irreversible plateau at ~ 0.1 V vs. Na+/Na (− 2.61 V vs. SHE), indicating the infusing of Na+ in anatase lattices requires an additional electrochemical driving force (overcoming a large potential gap of ~ 1.32 V) compared to that of Li+ (Supplementary Fig. 2)19. Meanwhile, previous studies indicated that such intercalated Na+ ions compel its near-surface anatase phase (marked as c) into an electrochemically formed amorphous state (marked as a’), which was an irreversible c-to-a’ transformation19,23,24. Then, it was regarded that the near-surface a’-TiO2 reversibly reacts with Na+ in subsequent cycles. Nevertheless, pristine amorphous TiO2 (a-TiO2), composed of distorted Ti–O polyhedra in random networks, can also be chemically synthesized (Supplementary Fig. 1)25. The distorted open channels in a-TiO2 provide continuous intercalations of Na+ ions without phase transformations26,27, displaying “box-like” cyclic voltammetry (CV) curves (Fig. 1b) after 20 cycles27. Differently, the 20 times cycled TiO2(A) electrode displays a “mirror-like” CV curve with a couple of broad redox peaks (Fig. 1b). The different electrochemical behaviors indicate that something rather than the amorphous structure might determine the sodium-ion storage properties after long-term cycles, but it remains largely unclear.
In this work, we reveal a multistep phase transformation from crystalline anatase to amorphous and then partially into rocksalt (RS) NaTiO2 nanograins (named c-a’-RS-NaTiO2) during repeated (de)sodiation of TiO2(A) nanograins, as demonstrated by synchrotron X-ray diffraction (XRD) and scanning transmission electron microscopy (STEM). This electrochemically-driven phase restructuration is beneficial for in situ creating suitable diffusion channels for reversible and rapid Na+ (de)intercalations. The formed RS-NaTiO2 undergoes a solid-solution reaction with a small lattice volume change of ~ 2.0% upon Na+ (de)intercalation, which determines the “mirror-like” CV curves and the sodium-ion storage capacities. The partial RS nanoregions are also observed in the cycled a-TiO2 electrode, but smaller RS regions and abundant amorphous regions lead to the “box-like” CV shape and lower capacity compared to those of c-a’-RS-NaTiO2. The operation potential of the c-a’-RS-NaTiO2 (Fig. 1c) is between those of hard carbon and NaTi2(PO4)3 negative electrodes, making it promising for both high-energy and high-power applications in grids28.
Results
Electrochemical c-a’-RS evolution of nanosized TiO2(A)
The TiO2(A) electrode shows a continued variation in the CV curves (Fig. 2a) and galvanostatic charge-discharge (GCD) profiles (Supplementary Fig. 3) with reduced voltage hysteresis during the initial 20 cycles. Ex situ characterizations were performed to explore the structure evolutions during long-term cycling. Ex situ XRD patterns (Fig. 2b) show the loss of crystalline diffraction peaks and the c-to-a’ transformation after 1 cycle, which is consistent with previously reported phenomena19,24. The destruction of long-range ordered structures after electrochemically driven Na+ intercalation was further demonstrated by ex situ synchrotron-radiation pair distribution function (SRPDF) (Fig. 2c), in which the correlation length decreased significantly from 50 to 16 Å after 1 cycle29. The Ti K-edge X-ray absorption near edge structure (XANES) spectra (Supplementary Fig. 4a) display changes in the split peaks for anatase30 into a single consolidated peak for the amorphous state23. Fourier transformed extended X-ray absorption fine structure (EXAFS) spectra reveal that the distances of first neighbor Ti–O pairs (1.54 Å) and Ti–Ti pairs (2.51 Å) expand to 1.59 Å and 2.76 Å after 1 cycle (Supplementary Fig. 4b), indicating the electrochemically driven distortion and expansion of the octahedral coordination structure for accommodating Na+ ions31.

a CV curves of the TiO2(A) electrode for initial 20 cycles at 0.2 mV s–1. b Ex situ XRD patterns of TiO2(A) for the initial 20 cycles. The disappearance and emergence of a series of peaks indicate the c-a’-RS phase transformation. c Ex situ SRPDF spectra of the TiO2(A) electrodes for pristine anatase, after 1 cycle and after 20 cycles, respectively. HRTEM images of (d) the pristine anatase, (e) after 1 cycle, and (f) after 20 cycles, respectively. Insets are the corresponding SAED patterns. HAADF-STEM images and inset FFT patterns of (g) the pristine anatase, (h) after 1 cycle, and (i) after 20 cycles, respectively. The evolutions of local atomic arrangements also indicate the c-a’-RS phase transformation.
Notably, the diffraction peaks at 36.8°, 43.3°, and 62.5° gradually emerge after 3, 10, and 20 cycles, indicating the formation of the RS phase (Fig. 2b). Moreover, the correlation length increases to 27 Å (Fig. 2c), demonstrating the partial restoration of order after 20 cycles. However, the active material retains relatively less ordering compared to that of the active material before cycling. The length of Ti–O and Ti–Ti pairs decrease to 1.55 and 2.54 Å, respectively (Supplementary Fig. 4b and Supplementary Table 2), confirming the reorganization of the atoms and the establishment of optimized structures through 20 cycles21,32.
Ex situ microscopy characterization was further applied to investigate the microstructural evolutions (Fig. 2d–i). The active material before cycling exhibits clear lattice fringes belonging to anatase phase in high-resolution transmission electron microscope (HRTEM) (Fig. 2d) and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images (Fig. 2g). After 1 cycle, it transforms into an amorphous structure, as confirmed by HRTEM (Fig. 2e), HAADF-STEM (Fig. 2h) and electron energy loss spectroscopy (EELS) (Supplementary Fig. 5), which is consistent with previously reported results19,23. After 20 cycles, partial crystalline nanograins surrounded by amorphous regions are observed in the HRTEM image (Fig. 2f). The selected area electron diffraction (SAED) pattern (inset of Fig. 2f) and HAADF-STEM image (Fig. 2i) clearly display the formation of an ordering RS phase32, confirming the multistep c-a’-RS transformation. The free energy and volume change for anatase- and RS-NaxTiO2 were calculated based on the density functional theory (DFT) method (Supplementary Fig. 6). The electrochemically-driven intercalation of Na+ ions into anatase causes the huge volume expansion and lattices distortions, leading to the c-to-a’ transformation process. Subsequently, the free energy of RS-Na4Ti4O8 is lower than that of anatase-Na4Ti4O8, indicating the energetically favorable of forming RS phase.
As the c-a’-RS transformation is complete, the electrochemically formed RS phase remains stable (Supplementary Fig. 7a), leading to well-overlapped CV curves during 20 to 100 cycles (Supplementary Fig. 7b). In addition, the transforming kinetics of the c-a’-RS transformation process is controllable by regulating the cycling rates (Supplementary Fig. 8) and temperatures (Supplementary Fig. 9). This c-a’-RS phase transformation is also observed when using carbonate electrolyte (Supplementary Fig. 10). When the nanosized TiO2(A) is cycled in 0.5–3.0 V vs. Na+/Na to avoid the c-to-a’ transformation, the crystalline anatase phase and related GCD profiles are unchanged after 200 cycles (Supplementary Fig. 11). This finding underscores the necessity of forming amorphous structures for further turning into the ordered RS phase after long-term cycling.
Sodium-ion storage mechanism of electrochemically formed RS-NaTiO2
The electrochemically formed RS-NaTiO2 (TiO2(A) after 20 cycles) at the fully sodiated state was further investigated. Synchrotron XRD Rietveld refinement (Fig. 3a) indicates that all diffraction peaks are indexed to the RS NaTiO2 (space group of Fm\(\bar{3}\)m, a = 4.20464 Å, Supplementary Table 3), which is similar to the electrochemically formed RS-Li3V2O5 (ref. 28), RS-Li3Nb2O5 (ref. 33) and RS-LiTiO2 (ref. 34) phase. The average size of the as-formed RS-NaTiO2 nanograins is ~3 nm, according to the Scherer formula19. The ex situ XRD patterns of c-a’-RS-NaTiO2 (Fig. 3b and Supplementary Fig. 12) show slight shifts in the diffraction peaks during desodiation and then sodiation, which is a solid-solution reaction mechanism. The changes in the diffraction peaks imply a very small volume change of ~ 2.0% (Supplementary Fig. 12), which is also confirmed by the unchanged ex situ SAED patterns (Supplementary Fig. 13). Ex situ Ti L-edge EELS spectra (Supplementary Fig. 14) and Ti 2p X-ray photoelectron spectroscopy (XPS) (Fig. 3c and Supplementary Table 4) support the reversible redox couple of Ti4+/Ti3+ during (de)sodiation. The sodiated sample has an average Ti valance of + 3.05, which is close to that of + 3 in RS-NaTiO2. After desodiation, the average Ti valance increases to + 3.75, indicating ~ 0.7 Na+ per Ti (corresponding to 234.5 mAh g−1), which is close to the reversible capacity of 253 mAh g−1 after 20 cycles (Supplementary Fig. 3a). The residual Na+ ions help to stabilize the RS phase. In addition, RS-NaTiO2 maintains diffraction peaks after 61 days of air exposure, indicating its air stability (Supplementary Fig. 15).

a Rietveld refinement synchrotron XRD pattern of c-a’-RS-NaTiO2 (TiO2(A) after 20 cycles) at the fully sodiated state. b Ex situ XRD patterns and (c) ex situ XPS spectra of c-a’-RS-NaTiO2 at different potentials, indicating a solid-solution (de)sodiation process. d STEM image, (e) schematic atomic configuration of the RS phase, (f) FFT pattern, and (g) Ti and Na element mapping of the fully sodiated c-a’-RS-NaTiO2. h Atomic position distribution spectra from (g). i Schematic of the sodium-ion storage mechanism: the electrochemically driven c-a’-RS transformation and the solid-solution reaction of in situ formed RS-NaTiO2 nanograins during cycling with Na+.
An ex situ HAADF-STEM image (Fig. 3d) illustrates the coexistence of the crystalline RS NaTiO2 nanograins and amorphous regions. The schematic atomic configuration (Fig. 3e) and fast Fourier transform (FFT) pattern (Fig. 3f) along the crystallographic zone axis of [\(\bar{1}\)10] clearly show the RS lattices. Atomic-resolution X-ray energy disperses spectroscopy (EDS) mapping (Fig. 3g) reveals no correlations between the Na and Ti signals in different directions (Fig. 3h and Supplementary Fig. 16), implying cation-disordering of the Na and Ti atoms at the octahedral (4a) sites. The octahedral sites are occupied by Na+ ions for sodiated RS-NaTiO2, while the vacancies remain after desodiation. The overall electrochemically driven structural evolutions of nanosized TiO2(A) during cycling with Na+ ions are schematically summarized in Fig. 3i. The initial intercalation of Na+ into anatase causes a c-to-a’ transformation, leading to a decrease in long-range order. Scrutinizing the continued changes in the CV curves (Fig. 2a), the anodic peak first emerged at the 2nd cycle during the extraction of Na+ from the sodiated materials. This phenomenon indicates the initial formation of RS-NaTiO2 when the Na+ ions occupy the octahedral sites. Upon repeated cycles with Na+ ions, the distorted Ti–O octahedra are continuously rearranged, leading to the continued formation of RS nanograins. The reversible Na+ extraction and intercalation between the RS-NaTiO2 and RS-Na1-xTiO2 follow a solid-solution reaction with the redox of Ti4+/Ti3+, finally leading to “mirror-like” CV curves with broad redox peaks.
Electrochemical a-to-RS evolution of a-TiO2
To determine the origins of the differences in the electrochemical properties between cycled TiO2(A) and a-TiO2 (Fig. 1b), detailed ex situ XRD, SRPDF and STEM characterizations of a-TiO2 were also conducted, as shown in Fig. 4. The a-TiO2 electrode shows a “box-like” CV curve for the 1st cycle (Fig. 4a), which is different from the initial c-to-a’ of TiO2(A) (Fig. 2a). There are no significant redox peaks during the initial 10 cycles, which is consistent with the previously reported a-TiO2 arrays27 and spheres35. Even after 20 cycles, only small broad peaks emerge at ~ 0.75 V vs. Na+/Na (Fig. 4a). EXAFS spectra (Supplementary Fig. 17 and Supplementary Table 5) reveal that the distance of the first neighbor Ti–O and Ti–Ti pairs are 1.60 and 2.69 Å, respectively, which are larger than those of crystalline anatase (Supplementary Fig. 4). The large Ti–O polyhedra in a-TiO2 accommodate Na+ intercalation, leading to the “box-like” CV curve at the 1st cycle. After 20 cycles, the Ti–O and Ti–Ti pairs are shortened to 1.56 Å and 2.55 Å, respectively. Ex situ SRPDF spectra (Fig. 4b) reveal a continuous extension of the correlation length from 7 Å (before cycling) to 9 Å (after 1 cycle) and then to 12 Å (after 20 cycles), suggesting an increase in long-range order. However, laboratory X-ray did not detect diffraction patterns from the in situ formed RS-NaTiO2 nanograins for a-TiO2 after cycling (Fig. 4c). Ex situ TEM and STEM were further performed (Fig. 4d–i). The chemically synthesized active material before cycling is amorphous (Fig. 4d, g). After cycling with Na+, the RS nanograins and amorphous regions are observed (Fig. 4e, f). The amorphous to RS-NaTiO2 (a-to-RS) phase transformation is confirmed by diffraction rings in the SAED pattern (insets of Fig. 4e, f) and atom arrangements in the STEM image (Fig. 4h, i).

a Consecutive CV curves of a-TiO2 electrode for initial 20 cycles at 0.2 mV s–1. b Ex situ SRPDF spectra of the pristine a-TiO2, after 1 and 20 cycles. c Ex situ XRD patterns of a-TiO2 during 20 cycles. HRTEM images of (d) pristine a-TiO2, (e) after 1 cycle, and (f) after 20 cycles, respectively. The insets are the corresponding SAED patterns. Atomic-resolution HAADF-STEM images and the inset FFT patterns of (g) the pristine a-TiO2, (h) after 1 cycle, and (i) after 20 cycles. The a-TiO2 also partially transforms into RS nanograins after cycling with Na+.
The a-TiO2 undergoes a transformation to the RS phase after 1 cycle, which is attributed to the pristine amorphous nature of a-TiO2 with disordered Ti–O polyhedra26,33,36. However, the correlation length of 12 Å for cycled a-TiO2 is shorter than that of 27 Å for cycled TiO2(A) (Supplementary Fig. 18), indicating that the a-RS-NaTiO2 (cycled a-TiO2 with formed RS-NaTiO2) electrode has much smaller ordered domains than the c-a’-RS-NaTiO2 electrode37, which is consistent with the ex situ XRD results (Figs. 2b and 4c). In contrast to the a-to-RS transformation of chemically synthesized a-TiO2 with random Ti–O networks, the in situ multistep c-a’-RS transformation from the ordered Ti–O octahedra in TiO2(A) results in larger RS nanograins than those in cycled a-TiO2, which significantly affects their sodium-ion storage properties26.
Sodium-ion storage performance of electrochemically in situ formed RS-NaTiO2
The c-a’-RS-NaTiO2 displays a “mirror-like” CV curve with a couple of broad peaks (Fig. 5a), whereas a-RS-NaTiO2 displays a “box-like” shape (Fig. 5b). Using i = avb (ref. 19), the fitted b-values of the two electrodes are both equal to 1.0 (Supplementary Fig. 19a–d). Meanwhile, the simulated capacitive contribution13 of c-a’-RS-NaTiO2 and a-RS-NaTiO2 electrodes are 92.4% and 88.6% at 1.0 mV s–1 (Supplementary Fig. 19e, f), respectively. The kinetics analysis indicates pseudocapacitive behavior of the RS and amorphous regions. The main distinctions between the c-a’-RS-NaTiO2 and a-RS-NaTiO2 electrodes are their sizes of RS nanograins and residual amorphous regions. It has been reported that electrochemically formed RS-Li3Nb2O5 (ref. 33) and RS-LiTiO2 (ref. 34) displayed analogous “mirror-like” dQ/dV profiles according to their solid solution reactions with small voltage hysteresis, which is consistent with the electrochemical properties of RS-NaTiO2. The measured response currents are mainly derived from the RS nanograins and amorphous regions (Supplementary Fig. 20). The capacity contributions from RS nanograins and amorphous regions at different cycles are further analyzed (Supplementary Fig. 21a–f). The capacity contribution from RS nanograins gradually increases from 34.1% at the 3rd cycle (Supplementary Fig. 21a, b), to 56.4% at the 10th cycle (Supplementary Fig. 21c, d), and to 70% at the 20th cycle (Supplementary Fig. 21e, f), suggesting the growth of RS nanograins during cycling with Na+. After 20 cycles, the RS nanograins get stable. Owing to the smaller RS nanograins and more residual amorphous regions in the a-RS-NaTiO2 electrode (Fig. 4f and inset of Fig. 5b), it displays the “box-like” CV curves (Fig. 5b). However, the c-a’-RS-NaTiO2 electrode has large and abundant RS nanograins (Fig. 2f and inset of Fig. 5a), displaying broad redox peaks from the solid-solution reaction of the RS phase with small voltage hysteresis (Fig. 5a).

Deconvolution of the CV curves for (a) the c-a’-RS-NaTiO2 and (b) a-RS-NaTiO2 electrodes at 0.2 mV s–1. c Rate performance and (d) cycling performances of the c-a’-RS-NaTiO2 and a-RS-NaTiO2 electrodes. e GITT curves and corresponding logarithm DNa+ vs. NaxTiO2 plots of a-TiO2 and (f) TiO2(A) electrodes for the 1st, 2nd and 20th sodiation processes. The in situ-formed RS nanograins display fast Na+ diffusion abilities.
The rate and cycling performance of the a-RS-NaTiO2 and c-a’-RS-NaTiO2 electrodes are further discussed in Fig. 5c, d. The a-TiO2 and TiO2(A) electrodes deliver close initial sodiation capacities. However, during 2 − 20 cycles, a-TiO2 delivers a much lower capacity and quicker capacity fading, which is attributed to the irreversible reaction of the chemically synthesized amorphous materials with lots of defects26,27. When the RS nanograins get stable after 20 pre-cycles, the c-a’-RS-NaTiO2 electrode delivers 253, 224, and 200 mAh g–1 at 0.1, 1.0, and 4.0 A g–1, respectively, which are superior to those of the a-RS-NaTiO2 electrode (Fig. 5c). In addition, the c-a’-RS-NaTiO2 electrode delivers capacity retention of 72% after 5000 cycles at 2 A g–1 (Fig. 5d), which is better than that of 49% for the a-RS-NaTiO2 electrode.
The galvanostatic intermittent titration technique (GITT) was employed to investigate the Na+ diffusivity before and after cycling38. Relying on the open frameworks in a-TiO2, the diffusion coefficient is 10–10 ~ 10–11 cm2 s–1 and remains relatively unchanged during 20 cycles (Fig. 5e). In contrast, the initial intercalation of Na+ into TiO2(A) accompanied by the c-to-a’ transformation has a low Na+ diffusivity of 10–15 ~ 10–16cm 2–1. After partial transformation into the RS phase, the apparent Na+ diffusion coefficient increases dramatically by five orders of magnitude up to 10–10 cm2 s–1 (Fig. 5f), which is related to its high-rate capability. Electrochemical impedance spectroscopy (EIS) results of TiO2(A) during the initial 20 cycles show stable SEI resistance (RSEI) and decreased charge transfer resistance (Rct) for the subsequent a’-RS transformation (Supplementary Fig. 22 and Supplementary Table 6), confirming the enhanced kinetics by the in situ formation of RS phase. Previous works reported that TiO2(A) nanomaterials displayed surface-dependent specific capacities, which were determined by the ~ 5 nm thickness of near-surface activation regions39. Owing to the sluggish kinetics of the initial c-to-a’ transformation (Fig. 5f), the thickness of the near-surface amorphous regions is limited19,39. Ex situ TEM results (Supplementary Fig. 23) demonstrate the coexistence of anatase, amorphous, and electrochemically-formed RS phases in the TiO2(A)-25 nm nanoparticles after 20 cycles. Thus, TiO2(A) with larger grains delivers surface-dependent capacities and characteristic “mirror-like” CV curves after 20 cycles (Supplementary Fig. 24).
We directly assemble a TiO2(A)||Na3V2(PO4)3 full cell (N/P ratio of 0.95, Supplementary Fig. 25a) without any presodiation treatments. It displays continuously changing GCD profiles (Supplementary Fig. 25b, c), indicating that the electrothermal self-activation of the TiO2(A) negative electrode into the RS phase can be processed through the aging process in industry40. The assembled SIBs demonstrate fast-charging and high-power delivering properties (Supplementary Fig. 25d).
Discussion
In summary, we have demonstrated the in situ formation of nanosized RS-NaTiO2 from TiO2(A) through electrochemically driven multistep c-a’-RS transformations. The initial Na+ intercalation into crystalline anatase compels amorphization, and this sluggish kinetics limits the thickness of the near-surface amorphous regions that determine the surface-dependent capacities. More importantly, such amorphous regions transform into partially ordered RS-NaTiO2 nanograins after repeated cycles. The (de)intercalation of Na+ ions in RS-NaTiO2 follows a solid-solution reaction with the redox couple of Ti3+/Ti4+, resulting in pseudocapacitive “mirror-like” CV curves with a couple of broad redox peaks at 0.75 V vs. Na+/Na. After the in situ formation of the RS phase, the Na+ diffusivity increases dramatically, which is related to its high-rate capabilities. The volume changes of the RS lattices are only 2.0% during (de)sodiation, enabling excellent cycling stability. We prove that the electrochemical c-a’-RS transformation can be activated in full cells without any presodiation treatments, showing the potential for practical applications. Overall, the excellent sodium-ion storage properties of titanium dioxides are attributed primarily to the RS-NaTiO2 phase, which has the most promising high-rate and long-life sodium-ion storage performance. Our findings open a direction for developing Ti-based electrode materials for both high-energy and high-power SIBs.
Methods
Material synthesis
Anatase TiO2 (TiO2(A)) and amorphous TiO2 (a-TiO2) samples were synthesized through hydrolysis and annealing methods41. Tetrabutyl titanate (16 mL, purity: > 99%, Macklin) was injected into 0.1 M potassium chloride (> 99%, Aladdin) aqueous solution /ethanol mixture (400 mL at a volumetric ratio of 1:200) under magnetic stirring for 10 min. Then, the mixture was set aside for another 24 h to stratify the precipitate. The precipitates were centrifuged and washed with ethanol and deionized water and then dried in an oven at 80 °C for 24 h to obtain precursor. Subsequently, the precursor powders were annealed in air at 300 °C or 400 °C for 2 h to obtain nanosized a-TiO2 or TiO2(A), respectively.
Characterization
Powder X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Advance X-ray diffractometer (Cu Kα radiation, λ = 1.54056 Å). Raman measurements were performed using a laser wavelength of 532 nm to acquire data in the high-resolution Raman spectrometer (HORIBA HR Evolution). The Brunauer‒Emmett‒Teller (BET) specific surface area was measured via nitrogen sorption isotherms (Micromeritics Tristar 3020) at 77 K after degassing under vacuum at 150 °C for 6 h. Transmission electron microscopy (TEM) images were recorded via a Titan G2 60‒300. Scanning transmission electron microscopy (STEM) images and electron energy loss spectroscopy (EELS) spectra were obtained through double spherical aberration-corrected transmission electron microscopy (Titan Cubed Themis G2-300).
Electrode preparation
The TiO2, NaTi2(PO4)3 (Guangdong Qianjin Chemical Reagent Co.), and hard carbon (type 2, Kuraray) electrodes were prepared by mixing 85 wt.% active materials, 7 wt.% Ketjen black (EC-600JD, Nouryon), 4 wt.% carboxymethyl cellulose (CMC, > 99.5%, Canrd New Energy Technology Co.), and 4 wt.% styrene butadiene rubber (SBR, Canrd New Energy Technology Co.) in deionized water using an automatic mixer to form a slurry at room temperature in air, and then cast on Al foil (16 μm in thickness) using a doctor blade. For lithium-ion storage measurements, the slurry was cast on Cu foil (25 μm in thickness). The mass loading of active materials was controlled at 2 mg cm–2. The electrodes were dried at 60 °C in the atmosphere for 6 h and then at 100 °C in a vacuum oven for another 12 h. These electrodes were cut into 12 mm diameter discs using a punching machine. The cut disk electrodes were dried at 100 °C under vacuum overnight and then transferred to the Ar-filled grove box.
The Na3V2(PO4)3 (NVP, > 99.9%, Hubei Ennaiji Energy Technology Co.) electrode was made by mixing 90 wt.% NVP, 5 wt.% Ketjen black, and 5 wt.% polyvinylidene difluoride (PVDF, Arkema) in N-methylpyrrolidone (NMP, > 99%, Acmec) to form a slurry at room temperature in air and then cast on Al foil. The mass loading of NVP was ~ 4.5 mg cm–2. The NVP electrodes were dried at 120 °C in a vacuum oven for 12 h. The pressing, cutting, and drying processes of NVP electrodes are similar to the preparation of TiO2 electrodes.
Electrochemical measurements
Coin cells (CR2032) were assembled in an Ar-filled glove box with O2 < 0.1 ppm and H2O < 0.1 ppm. For measurements of sodium-ion storage, a fresh Na metal (> 99.9%, 0.6 mm in thickness, 15.8 mm in diameter, Sinopharm Chemical Reagent Co.) disk was used as the counter and reference electrode. 1 M NaPF6 in diglyme (80 μL, > 99.7%, DuoDuo Chemical Technology Co.) was used as the electrolyte to assemble each coin cell. Celgard 2325 (25 μm in thickness, 19 mm in diameter, Celgard) was used as the separator. As for lithium-ion storage measurement, a fresh Li metal disk (> 99.9%, 0.6 μm in thickness, 15.8 mm in diameter, Canrd New Energy Technology Co.) was used as the counter and reference electrode. 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC)/ethyl methyl carbonate (EMC) with a volume ratio of 1:1:1 (80 μL, > 99.7%, DuoDuo Chemical Technology Co.) was used as the electrolyte. For the electrochemical measurements of the half cells, the potential window was in 0.01–3 V vs. Na+/Na for TiO2(A), 1.2–3 V vs. Na+/Na for NaTi2(PO4)3, and 0.01–2 V vs. Na+/Na for hard carbon electrodes. The TiO2(A) | | NVP full cells were assembled by matching the capacity of the positive and negative electrodes, and 1 M NaPF6 in diglyme (80 μL) was used as the electrolyte and Celgard 2325 as the separator. The operating voltage window of full cells was set in 0.4–3.4 V. Cyclic voltammetry (CV) measurements were carried out by a potentiostat (VSP, Bio-Logic). Galvanostatic charge-discharge (GCD) tests were performed via the NEWARE Battery Test System (CT-4008T, China). For the cycling test, the coin cells were precycled at 0.1 A g–1 for 20 cycles and then cycled at 2 A g–1 to 5000 cycles. Thus, the capacity retention (%) was calculated based on the sodiation capacities at the 21st and 5000th cycles. The galvanostatic intermittent titration technique (GITT) was performed with a constant current discharge at 0.03 A g–1 for 10 min followed by relaxation for 1 h on the NEWARE Battery Test System. Electrochemical impedance spectroscopy (EIS) measurements were conducted on the Bio-Logic VSP potentiostat by a potentiostatic method with an applied amplitude of 10 mV in the range of 200 kHz to 10 mHz (10 points per decade of frequency). The cells were sodiated to 0.05 V vs. Na+/Na and reset 20 min before carrying out the EIS measurement. Unless otherwise specified, all the coin cells were placed in an incubator at a stationary temperature of 25 °C for electrochemical measurements. Two or three cells were tested for a single electrochemical experiment.
Ex situ Synchrotron XRD, SRPDF and XAS characterizations
All synchrotron-based characterizations were carried out at the Advanced Photon Source (APS) at the Argonne National Laboratory. Synchrotron XRD and synchrotron-radiation pair distribution function (SRPDF) measurements were collected at beamline 11-ID-C (λ = 0.1173 Å). Synchrotron X-ray absorption spectroscopy (XAS) measurements were collected at beamline 12-BM-B with radiation monochromatized by a Si(111) double-crystal monochromator. In preparation for the ex situ synchrotron radiation tests, the coin cells at different cycled states were disassembled in an Ar-filled glove box, and the electrodes were soaked in diglyme for 3 h. Then, the active materials were separated from the current collector and carefully sealed in Capton tape for further testing.
Ex situ TEM, STEM and XPS characterizations
For ex situ TEM and STEM characterization, the coin cells were disassembled in an Ar-filled glove box, and the electrodes were washed with diglyme. Then, the active materials were carefully scratched from the Al foil, sonicated in diglyme, and dropped onto grids for TEM and STEM observations. Ex situ high-resolution (HR)TEM images, selected area electron diffraction (SAED) patterns, HAADF-STEM images, and EELS spectra were obtained via a double spherical aberration-corrected transmission electron microscope (Tian Cubed Themis G2-300). X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Kratos Axis Supra using a monochromatized Al Kα X-ray source (1486.7 eV). The XPS chamber was directly connected to the Ar-filled glove box to avoid air exposure during sample preparation and transfer.
First-principles calculations
All calculations on anatase TiO2 and rocksalt NaTiO2 were executed by the projector augmented wave (PAW)41 method within density functional theory (DFT), conducted in the Vienna ab initio Simulation Package (VASP)42,43. The generalized gradient approximation (GGA) in the form of the Perdew-Burke-Ernzerhof (PBE)44 was used to treat the exchange-correlation energy. Geometry optimizations were performed on rocksalt and anatase TiO2 structures using a 2a × 2b × 1c supercell, selected as the host framework for Na+ storage. A kinetic energy cutoff of 500 eV was used for wave functions expanded in the plane wave basis. Besides, spin polarization was considered. The energy (converged to 1.0 × 10−6 eV atom−1) and force (converged to 0.05 eV Å−1) were set as the convergence criterion for geometry optimization. For the Brillouin-zone sampling, a 4 × 4 × 4 k-points mesh was adopted to ensure convergence of the total energy.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files. All raw data generated during the current study are available from the corresponding authors upon request. Source data are also provided with this paper.
References
Zhao, C. L. et al. Rational design of layered oxide materials for sodium-ion batteries. Science 370, 708–711 (2020).
Rudola, A., Sayers, R., Wright, C. J. & Barker, J. Opportunities for moderate-range electric vehicles using sustainable sodium-ion batteries. Nat. Energy 8, 215–218 (2023).
Duffner, F. et al. Post-lithium-ion battery cell production and its compatibility with lithium-ion cell production infrastructure. Nat. Energy 6, 123–134 (2021).
Zhu, Z. X. et al. Rechargeable batteries for grid scale energy storage. Chem. Rev. 122, 16610–16751 (2022).
Cai, Z. J. et al. In situ formed partially disordered phases as earth-abundant Mn-rich cathode materials. Nat. Energy 9, 27–36 (2024).
Li, Y. et al. Multilevel gradient-ordered silicon anode with unprecedented sodium storage. Adv. Mater. 36, 2310270 (2023).
Ni, J. F., Sun, M. L. & Li, L. Highly efficient sodium storage in iron oxide nanotube arrays enabled by built-in electric field. Adv. Mater. 31, 1902603 (2019).
Yuan, Y. F., He, K. & Lu, J. Structure-property interplay within microporous manganese dioxide tunnels for sustainable energy storage. Angew. Chem. Int. Edit. 63, e202316055 (2023).
Tian, H. J. et al. Renaissance of elemental phosphorus materials: properties, synthesis, and applications in sustainable energy and environment. Chem. Soc. Rev. 52, 5388–5484 (2023).
Lao, M. M. et al. Alloy-based anode materials toward advanced sodium-ion batteries. Adv. Mater. 29, 1700622 (2017).
Li, Y. Q. et al. Origin of fast charging in hard carbon anodes. Nat. Energy 9, 134–142 (2024).
Feng, X. et al. Unlocking the local structure of hard carbon to grasp sodium-ion diffusion behavior for advanced sodium-ion batteries. Energ. Environ. Sci. 17, 1387–1396 (2024).
Wang, C. T. et al. Highly conductive hierarchical TiO2 micro-sheet enables thick electrodes in sodium storage. Adv. Funct. Mater. 34, 2301996 (2024).
Dong, S. Y. et al. Titanates for sodium-ion storage. Nanotoday 42, 101349 (2022).
Xu, C. et al. Carbon-coated hierarchical NaTi2(PO4)3 mesoporous microflowers with superior sodium storage performance. Nano Energy 28, 224–231 (2016).
Wang, X. et al. Pseudocapacitance of MXene nanosheets for high-power sodium-ion hybrid capacitors. Nat. Commun. 6, 6544 (2015).
Lou, S. F. et al. Ti-based oxide anode materials for advanced electrochemical energy storage: lithium/sodium ion batteries and hybrid pseudocapacitors. Small 15, 1904740 (2019).
Liang, S., Wang, X., Cheng, Y. J., Xia, Y. Q. & Peter, M. B. Anatase titanium dioxide as rechargeable ion battery electrode - a chronological review. Energy Storage Mater. 45, 201–264 (2022).
Wei, Q. L. et al. Surface-redox sodium-ion storage in anatase titanium oxide. Nat. Commun. 14, 7 (2023).
Viswanathan, V. et al. The challenges and opportunities of battery-powered flight. Nature 601, 519–525 (2022).
Zhu, X. H. et al. LiMnO2 cathode stabilized by interfacial orbital ordering for sustainable lithium-ion batteries. Nat. Sustain. 4, 392–401 (2021).
Lee, D. H. et al. Engineering titanium dioxide nanostructures for enhanced lithium-ion storage. J. Am. Chem. Soc. 140, 16676–16684 (2018).
Xu, Z. L. et al. Engineering solid electrolyte interphase for pseudocapacitive anatase TiO2 anodes in sodium-ion batteries. Adv. Funct. Mater. 28, 1802099 (2018).
Li, K. K. et al. Evolution of the electrochemical interface in sodium ion batteries with ether electrolytes. Nat. Commun. 10, 1248 (2019).
Wang, X. T. et al. Two-dimensional amorphous TiO2 nanosheets enabling high-efficiency photoinduced charge transfer for excellent SERS activity. J. Am. Chem. Soc. 141, 5856–5862 (2019).
Kang, J. X. et al. Recent progress of amorphous nanomaterials. Chem. Rev. 123, 8859–8941 (2023).
Zhou, M. et al. Amorphous TiO2 inverse opal anode for high-rate sodium ion batteries. Nano Energy 31, 514–524 (2017).
Liu, H. D. et al. A disordered rock salt anode for fast-charging lithium-ion batteries. Nature 585, 63–67 (2020).
Stratford, J. M. et al. Correlating local structure and sodium storage in hard carbon anodes: insights from pair distribution function analysis and solid-state NMR. J. Am. Chem. Soc. 143, 14274–14286 (2021).
Li, J. T. et al. Revolutionizing lithium storage capabilities in TiO2 by expanding the redox range. ACS Nano 17, 21604–21613 (2023).
Hua, X. et al. Revisiting metal fluorides as lithium-ion battery cathodes. Nat. Mater. 20, 841–850 (2021).
Wang, Q. D. et al. Chemical short-range disorder in lithium oxide cathodes. Nature 629, 341–347 (2024).
Barnes, P. et al. Electrochemically induced amorphous-to-rock-salt phase transformation in niobium oxide electrode for Li-ion batteries. Nat. Mater. 21, 795–803 (2022).
Baudrin, E. et al. Structural evolution during the reaction of Li with nano-sized rutile type TiO2 at room temperature. Electrochem. Commun. 9, 337–342 (2007).
Deng, C. J. et al. Amorphous and crystalline TiO2 nanoparticle negative electrodes for sodium-ion batteries. Electrochim. Acta. 321, 134723 (2019).
Gao, Q. et al. Direct evidence of lithium-induced atomic ordering in amorphous TiO2 nanotubes. Chem. Mater. 26, 1660–1669 (2014).
Zheng, T., Hu, P. F., Wang, Z. C. & Guo, T. Q. 2D amorphous iron selenide sulfide nanosheets for stable and rapid sodium-ion storage. Adv. Mater. 35, 2306577 (2023).
Chien, Y. C. et al. Rapid determination of solid-state diffusion coefficients in Li-based batteries via intermittent current interruption method. Nat. Commun. 14, 2289 (2023).
Song, G. R. et al. Physical interpretations of diffusion-controlled intercalation and surface-redox charge storage behaviors. Energy Storage Mater. 61, 102859 (2023).
Wei, Q. L. et al. An ultrahigh-power mesocarbon microbeads|Na+-diglyme|Na3V2(PO4)3 sodium-ion battery. Adv. Mater. 34, 2108304 (2022).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phy. Rev. B 59, 1758–1775 (1999).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phy. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Perdew, J., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22479123, 22179113, and 52431009) and the Fundamental Research Funds for the Central Universities (Grant No. 20720230028). This research used resources from the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, which operates for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Q.L.W. is also thankful for the support of the Xiaomi Young Talents Program/Xiaomi Foundation. We sincerely acknowledge the guidance and support of Dr. Khalil Amine (Argonne National Laboratory) in this work.
Author information
Authors and Affiliations
Contributions
Q.L.W., D.F.T., and D.-L.P. proposed the idea. D.F.T. and Q.L.W. wrote the manuscript. D.F.T., Z.R.Y., and S.C.F. performed the material syntheses, structural characterizations, electrochemical testing, and data processing. R.H.Y. performed the STEM and EELS. Y.L.J. performed the DFT calculations. J.T.L., S.L., and T.Y.L. performed the synchrotron measurements. J.T.L., X.J.H., Q.S.X., L.Q.M., and D.-L.P. reviewed and edited the article. L.Q.M., D.-L.P., and Q.L.W. supervised the project. All the authors contributed to the discussion of the results and writing of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yao Xiao, and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, D., Yu, R., Jiang, Y. et al. Electrochemically in situ formed rocksalt phase in titanium dioxide determines pseudocapacitive sodium-ion storage. Nat Commun 16, 2015 (2025). https://doi.org/10.1038/s41467-025-57310-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57310-x
