
Abstract
Early Triple negative breast cancer (eTNBC) is the subtype with the worst outcome. Circulating tumor DNA (ctDNA) is shown to predict the prognosis of breast cancer, but its utility in eTNBC remains unclear. 130 stage II-III female eTNBC patients receiving neoadjuvant chemotherapy (NAC) have been enrolled prospectively and subjected to ctDNA analysis. ctDNA at post-NAC (pre-surgery) and post-surgery, but not at baseline, is associated with worse prognosis. A threshold of 1.1% maximum variant allele frequency at baseline stratifies patients with different relapse risk, which is validated internally and externally. A systemic tumor burden model integrating baseline and post-surgery ctDNA is independently prognostic (p = 0.022). Combining systemic tumor burden with pathologic response identifies a highly curable subgroup and a subgroup of high-risk eTNBC patients. ctDNA surveillance during follow-up identifies patients with high relapse risk. In conclusion, systemic ctDNA analysis demonstrates the utility of a systemic tumor burden model of ctDNA in risk stratification of eTNBC patients, which may guide future treatment escalation or de-escalation trials.
Similar content being viewed by others

Introduction
Among different subtypes of breast cancer, triple negative breast cancer (TNBC) is most aggressive with the worst prognosis1. The five-year overall survival (OS) of TNBC is only 66% and 13% for regional and distant disease respectively, much worse than that of other subtypes2. The median time from diagnosis to distant metastasis for early TNBC (eTNBC) was merely 24 months3 and the median OS of metastatic TNBC patients was only ~14 months4. The rapid relapse and short survival after metastasis present a great challenge in treating eTNBC patients. Although the majority (~70%) of eTNBC can be completely cured by current standard therapies, the fear of possible relapse and the ensuing poor outcome remains a terrifying psychological burden for many, if not most, eTNBC patients. However, it might not be worthwhile to try to further lower the relapse rate by escalating treatment in all eTNBC patients. It is of paramount importance to develop new methods that can more precisely identify individual relapse risk for eTNBC patients.
Neoadjuvant chemotherapy (NAC) with or without immunotherapy is so far the best way to measure the response to systemic treatment and to gauge the individual relapse risk for eTNBC patients. The patients who achieved pathological complete response (pCR) after NAC had significantly better survival than those without pCR5,6. In the meta-analysis of 805 eTNBC patients with pCR from five neoadjuvant GBG/AGO-B trials, the 4-year disease-free survival (DFS) rate was 90.2%. Nevertheless, for the eTNBC patients with lobular type or clinically positive lymph node before NAC, the 4-year DFS was 70.1% or 85.6% respectively, indicating that pCR is only an indicator of lower relapse, but not a good marker of no relapse7. Moreover, the eTNBC patients with non-pCR after NAC still have 5-year event-free survival (EFS)/DFS as high as 62–70% if pembrolizumab or adjuvant capecitabine was used8,9, suggesting that the majority of non-pCR eTNBC patients are relapse-free and there is no need of excessive panic. Taken together, pCR after NAC is not a satisfactory marker of the prognosis of eTNBC patients. How to identify eTNBC patients with high relapse risk remains a critical and challenging clinical problem. A most recent paper showed that 5-year distant recurrence-free survival (DRFS) of TNBC patients who did not receive any chemotherapy was as high as 73%10, highlighting the necessity of individualized risk assessment of eTNBC patients. Biomarkers to better stratify the risk of eTNBC patients are in great need to guide clinicians to tailor precise escalation or de-escalation treatment.
Recently, circulating tumor DNA (ctDNA) has been shown to be an effective method to evaluate tumor burden at the molecular level in many types of cancer11,12,13,14. ctDNA detection provides a non-invasive method to monitor treatment response and minimal residual disease (MRD) in cancer15,16,17. ctDNA presence after radical surgery was shown to associate with a higher relapse risk in different subtypes of breast cancer18,19,20,21. The lack of ctDNA clearance during and after NAC was an indicator of poor response to NAC and metastatic recurrence in eTNBC22,23. Given its high-shedding feature, eTNBC was reported to have positive ctDNA in 73–100% patients before treatment12,22,24,25,26, suggesting a ctDNA threshold at baseline may be needed to separate low-risk patients from high-risk ones. However, recently the first interventional study based on ctDNA guided therapy in eTNBC did not achieve a positive result due to the high metastatic rate in MRD positive patients27.
Although ctDNA is promising in detecting early dissemination of cancer cells into blood, it remains controversial regarding the optimal method/gene panel, timing, frequency, threshold, and value of guiding adjuvant treatment when using ctDNA in eTNBC patients.
In this study, we did a prospective, longitudinal, dynamic ctDNA monitoring in 130 eTNBC patients who received NAC to determine the optimal timing, frequency, and threshold of ctDNA in differentiating the risk of eTNBC patients. By analyzing the ctDNA at different timepoints including baseline (pre-NAC), post-NAC (pre-surgery), post-surgery, and during follow-up, we correlated them with prognosis to develop a method that better stratify the eTNBC patients with different relapse risk, which can help future interventional trials to accurately select patients for escalation or de-escalation study.
Results
Patient clinicopathologic characteristics and ctDNA features
130 female patients with stage II–III TNBC received NAC followed by surgery were prospectively enrolled in this study (Table 1, Table S1, Fig. S1A). The median age of the study cohort was 44 years (range 25–65 years), with the median follow-up at 24.2 months (range 6.0–45.3 months) as of November 1st, 2023. Most patients were stage II (80.8%) and clinically lymph node positive (56.2%). After NAC, 44 (33.9%) patients achieved total pCR. 15 patients experienced recurrence (12 distant metastases and 3 local relapses) and 2 patients died of disease during follow-up.
Overall, 130 tumor tissues (including 123 core needle biopsy samples and 7 postoperative tumor samples when biopsy samples were insufficient for sequencing) were successfully sequenced using a 1021 cancer-related gene panel previously reported (Supplementary Data 1)13. A total of 625 blood samples, including 122 baseline (pre-NAC) samples, 113 post-NAC (pre-surgery) samples, 118 post-surgery samples taken 1–8 weeks after surgery or radiation and 272 follow-up samples (every 3–6 months after surgery) were subjected to parallel ultra-deep sequencing (>10,000×) using the same 1021-gene panel followed by tumor-guided ctDNA analysis (Fig. 1A, Fig. S1B).

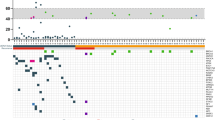
A Study schematic. Patients (n = 130) with stage II–III TNBC planned to receive NAC were enrolled. Peripheral blood samples were taken at the baseline (N = 122, 93.85%), post-NAC (pre-surgery) (N = 113, 86.92%), post-surgery (N = 118, 90.77%) timepoints and every 3–6 months during follow-up (N = 272). Created with BioRender.com. B Histogram showing the MVAF of each patient in the upper panel. Heatmap plot showing the baseline ctDNA profile (all detected variants) and the clinicopathological characteristics of each patient (n = 122) in the lower panel. C The association between baseline ctDNA detection and age (88% detected in age <40 versus 74% detected in age ≥40, p = 0.097), tumor size (76% detected in cT1/2 versus 92% detected in cT3/4, p = 0.100), lymph node (71% detected in cN negative versus 85% detected in cN positive, p = 0.064) status, Ki67 (64% detected in ki67 low versus 85% detected in ki67 high, p = 0.010). Chi square test was used for significance test (two-sided). D The association between baseline ctDNA concentration (MVAF) and age (median MVAF 0.7% in age <40 versus median MVAF 0.7% in age ≥40, p = 0.592), tumor size (median MVAF 0.5% in cT1/2 versus median MVAF 6.4% in cT3/4, p = 2.20E-07), lymph node status (median MVAF 0.2% in cN negative versus median MVAF 1.7% in cN positive, p = 0.0008), Ki67 (median MVAF 0.1% in ki67 low versus median MVAF 1.1% in ki67 high, p = 0.002). Wilcoxon test was used for significance test (two-sided). NAC neoadjuvant chemotherapy, cT clinical tumor category, cN clinical regional lymph nodes, pCR pathological complete remission. MVAF maximum variant allele frequency, Others other single-nucleotide variation, small insertions, and deletions. Multi-Hit multiple-hit. Source data are provided as a Source Data file.
In total, 1759 variants in 436 genes were detected in tumor tissue samples of 130 patients, with 2–76 variants (median 10 variants) per sample. The top 5 altered genes were TP53, MYC (CNV), PIK3CA, MCL1 (CNV), and MDM4 (CNV) (Fig. S2A).
Among 122 patients with baseline blood samples before any treatment, 96 (78.7%) of them had detectable ctDNAs. A total of 485 variants in 227 genes were detected, with a median of 3 variants per sample (range 1–39). TP53, PIK3CA, PTEN, LRP1B, and RB1 were the most frequently altered genes (Fig. 1B). Positive baseline ctDNA was significantly correlated with high Ki67 (≥50%) (p = 0.01), but not with age, tumor size, lymph node status (Fig. 1C). Nevertheless, larger tumors, positive lymph nodes and high Ki67 were associated with higher baseline ctDNA concentration (measured by maximum variant allele fraction, MVAF) (Fig. 1D).
Dynamic ctDNA response after NAC
It was reported that NAC could effectively clear ctDNA and thus decrease relapse28. In our cohort, the ctDNA positive rate dropped sharply from 79% to 23% (26/113) at the post-NAC (pre-surgery) timepoint (Fig. 2A). Patients with negative ctDNA at the post-NAC (pre-surgery) timepoints were more likely to achieve pCR or higher Miller–Payne grade than those with positive ctDNA at the post-NAC (pre-surgery) timepoint (Fig. 2B, Fig. S3A) indicating better local and systemic response to NAC in ctDNA negative patients. A significant drop of ctDNA concentration was also observed after NAC (p = 1.69E-14, Fig. 2C). The drop of ctDNA was significant in pCR patients (p = 1.86E-09 , Fig. S2B) as well as in non-pCR patients (p = 2.42E-12, Fig. S2C). We then further compared ctDNA concentration changes between pCR and non-pCR patients. There were similar ctDNA concentrations between pCR and non-pCR patients at the baseline (p = 0.76, Fig. 2D). However, non-pCR patients had significantly higher post-NAC (pre-surgery) ctDNA concentrations than pCR patients (Fig. 2D). Explorative analysis was performed to delineate ctDNA mutation profile and their change after NAC between patients with pCR and non-pCR. All TP53 mutations and PIK3CA mutations in blood were eradicated by NAC in pCR patients, while about 24% and 8% of non-pCR patients still had detectable post-NAC (pre-surgery) TP53 mutations and PIK3CA mutations respectively (Fig. 2E), suggesting that continued presence of TP53 or PIK3CA mutations indicates non-pCR status.

A Sankey plot depicting changes in the proportion of patients stratified by ctDNA status and pathologic response. B Association of post-NAC (pre-surgery) ctDNA status with pathologic response status. Fisher exact test was used for significance test (two-sided). C Comparison of ctDNA MVAF between baseline and post-NAC (pre-surgery) timepoints. Wilcoxon matched-pairs signed rank test was used for significance test (two-sided). D Comparison of ctDNA MVAF between pCR and non-pCR patients in baseline and post-NAC (pre-surgery) timepoints, respectively. Mann–Whitney tests were used for significance tests (two-sided). E Bar plots showing the prevalence of top 20 mutated cancer genes of ctDNA at baseline and post-NAC (pre-surgery) timepoints. Source data are provided as a Source Data file.
Furthermore, the post-surgery ctDNA positive rate further dropped to 16.9% (20/118), indicating the removal of primary tumor can further decrease ctDNA positivity in certain patients.
ctDNA status at different timepoints of treatment and the prognosis of TNBC
To determine the optimal timing of ctDNA sampling during the treatment of eTNBC, we analyzed the ctDNA positivity at different timepoints and correlated them with the EFS and DRFS.
The patients with positive baseline ctDNA showed a trend of worse EFS (Fig. 3A, p = 0.21, HR = 3.39, 95% CI: 0.44–26.3) and DRFS (Fig. 3B, p = 0.08, HR not calculable) than the ones with negative baseline ctDNA, but the differences were not statistically significant. Nevertheless, it is worthy of noting that none of the patients with negative baseline ctDNA had distant metastasis, indicating this subgroup of patients (21% of total, clinical stage: 13 IIA, 11 IIB and 2 IIIC) has very localized tumors and very low-risk of distant metastasis.

EFS (A) and DRFS (B) of 122 patients grouped by baseline ctDNA status. EFS (C) and DRFS (D) of 113 patients grouped by post-NAC (pre-surgery) ctDNA status. EFS (E) and DRFS (F) of 118 patients grouped by post-surgery ctDNA status. Survival analyses were performed using the Kaplan–Meier method. p values were calculated using the log-rank test. Hazard ratios (HR) and the corresponding confidence intervals (CI) were calculated using the Cox regression models. EFS event-free survival, DRFS distant recurrence-free survival. Source data are provided as a Source Data file.
The presence of positive ctDNA post-NAC (pre-surgery), as well as the ones post-surgery, were associated with significantly worse EFS (Fig. 3C, p = 0.002, HR = 4.34, 95% CI: 1.51–12.44; Fig. 3E, p = 0.002, HR = 4.63, 95% CI: 1.60–13.38) and DRFS (Fig. 3D, p < 0.001, HR = 6.18, 95% CI: 1.95–19.58; Fig. 3F, p < 0.001, HR = 6.25, 95% CI: 2.01–19.42), indicating their strong value in predicting the prognosis of eTNBC patients.
Post-surgery ctDNA represents the earliest MRD status and has been shown as a crucial marker to guide adjuvant chemotherapy in a randomized trial of early colon cancer29. However, it’s noteworthy that among the 12 eTNBC patients who had post-surgery ctDNA testing and later developed distant metastasis, only six of them had positive ctDNA at the post-surgery timepoint. Thus, the sensitivity of post-surgery ctDNA to predict metastasis was only 50%, indicating this single post-surgery timepoint is not good enough to select most high-risk eTNBC patients. This result is in line with the complexity of ctDNA dynamics throughout the management of eTNBC and underscores the need for a more comprehensive approach in using ctDNA to assess eTNBC prognosis.
Higher threshold of baseline ctDNA better stratifies the prognosis of eTNBC patients
Although the detection of baseline ctDNA in eTNBC patients could be as high as 73–100%12,21,22,24,25, yet only ~30% of such patients will develop relapse30. Furthermore, 21% of eTNBC patients had negative ctDNA in our study and none of them developed metastasis during follow-up, indicating there may be a threshold of baseline ctDNA that could distinguish high-risk from low-risk TNBC patients.
Using the data from our previous study25, an ROC model was constructed to explore the optimal cutoff value to distinguish high-risk and low-risk patients. The ROC model was statistically significant (AUC: 0.73, 95% CI: 0.5424–0.9176) with the optimal cutoff value being 1.1% of MVAF (Fig. 4A). Next, the cutoff value was applied into the present study. The patients with higher baseline ctDNA (defined as MVAF higher than 1.1%, 53/122, 43.4%) demonstrated significantly worse EFS (Fig. 4B, p = 0.014, HR = 4.49, 95% CI = 1.21–16.60) and DRFS (Fig. 4C, p = 0.002, HR = 13.02, 95% CI = 1.65–102.8), than the patients with lower baseline ctDNA (defined as MVAF lower than 1.1%, 69/122, 56.6%). The sensitivity and AUC of baseline ctDNA at 1.1% threshold were 90% and 0.75 respectively, better than any single timepoint without threshold (Table S2). For external validation, we employed the I-SPY2 cohort22 that enrolled eTNBC patients with similar clinical characteristics (Table S3). Per the calculation formula provided in the I-SPY2 study, the cutoff value of 1.1% MVAF was converted to 28.27 mean tumor molecules per milliliter (MTM/mL) using the median cfDNA extraction concentration (ng/mL) of the study. In this external validation, the patients with baseline ctDNA concentration higher than 28.27 MTM/mL had significantly worse DRFS (Fig. 4D, p = 0.002, HR = 2.55, 95% CI = 1.37–4.73) than the patients with ctDNA lower than 28.27 MTM/mL in the I-SPY2 trial. The specificity and AUC with threshold in I-SPY2 were better than the ones without threshold (Table S2), demonstrating the value of such a threshold of ctDNA.

A Receiver operating characteristic (ROC) curve for identifying optimal cutoff value of ctDNA MVAF at the baseline. Kaplan–Meier analysis of EFS (B) and DRFS (C) stratified by 1.1% MVAF in our cohort (n = 122). Kaplan–Meier analysis of DRFS (D) stratified by 28.27 MTM/mL in the I-SPY2 cohort (n = 129). Kaplan–Meier analysis of EFS (E) and DRFS (F) stratified by systemic tumor burden in our cohort (n = 119). Kaplan–Meier analysis of EFS (G) and DRFS (H) stratified by systemic tumor burden and pathologic response combined (n = 119). MTM mean tumor molecule. Stb systemic tumor burden, pCR pathologic complete response. Survival analyses were performed using the Kaplan–Meier method. p values were calculated using the log-rank test. Hazard ratios (HR) and the corresponding confidence intervals (CI) were calculated using the Cox regression models. Source data are provided as a Source Data file.
Systemic tumor burden with pathologic response to NAC identifies TNBCs with different relapse risk
It is known that effective NAC can eradicate not only local tumor, but also ctDNA in eTNBC patients. To reflect the initial pre-treatment circulating tumor burden as well as the efficacy of NAC and surgery, the results of ctDNA from baseline and post-surgery timepoints were integrated to build a systemic tumor burden model. The patients with high systemic tumor burden (63/119, 52.9%), defined as high baseline ctDNA (≥1.1% MVAF) or detectable ctDNA after surgery, had significantly inferior EFS (Fig. 4E, p = 0.029, HR = 3.76, 95% CI = 1.05–13.49) and DRFS (Fig. 4F, p = 0.004, HR = 11.05, 95% CI = 1.43–85.63) than other patients with low systemic tumor burden (baseline ctDNA <1.1% MVAF and non-detectable post-surgery ctDNA, 56/119, 47.1%). The systemic tumor burden model also exhibited a high sensitivity of 91.7% for DRFS (Table S3). Moreover, systemic tumor burden remained independently prognostic in a multivariate Cox model (p = 0.022, Table S4) after adjusted for clinical characteristics and pathologic response.
Many studies have shown non-pCR status after NAC is strongly associated with worse EFS and overall survival in TNBC patients6,31,32. The patients who achieved pCR in our study also had markedly better EFS and DRFS (Fig. S3B, C). Thus, we tried to further refine prognosis stratification by combining systemic tumor burden with pathologic response. Not surprisingly, the patients with high systemic tumor burden and non-pCR status had the worst EFS and DRFS (Fig. 4G, H), suggesting this subgroup (10 distant metastasis out of 42 patients) is in urgent need of more effective adjuvant therapy after surgery. Among the 21 patients with high systemic tumor burden but reached pCR, only one patient had distant recurrence, indicating excellent response to NAC can significantly enhance the prognosis of TNBC patients with high systemic tumor burden. Among the 37 patients with low systemic tumor burden and non-pCR status, 3 patients had recurrence, including 2 local relapses in the ipsilateral breast after breast conserving surgery, and 1 distant metastasis. Moreover, the 19 patients with low systemic tumor burden and reached pCR had 100% EFS and DRFS (Fig. 4G, H), suggesting this subgroup is a highly curable population. Taken together, the combinational use of systemic tumor burden with pCR status after NAC provides an excellent tool to separate TNBC patients with high or low-risk of metastasis right after surgery, which may help to tailor personalized adjuvant treatment.
MRD surveillance after surgery precedes clinical imaging to detect relapse
Positive ctDNA after surgery represents un-eradicated tumor cells in the patients and the presence of MRD, which is associated with worse prognosis18,24. Next, we integrated longitudinal follow-up timepoints with the post-surgery timepoint. Patients remained ctDNA negative during follow-up (at least 2 timepoints) were defined as MRD negative, while patients with detectable MRD at any timepoints after surgery were defined as MRD positive. 82 of 130 patients had longitudinal MRD results, of which 44 were negative and 38 were positive. Patients with negative MRD had significantly better EFS (Fig. 5A, p = 0.002, HR = 7.70, 95% CI = 1.71–34.83) and 100% DRFS (Fig. 5B, p = 0.00011) than patients with positive MRD. The positive predictive value (PPV) and negative predictive value (NPV) of longitudinal MRD for DRFS were 29% and 100% respectively. MRD remained an independent prognostic factor in a multivariate Cox regression model after adjusting for clinicopathological characteristics (Table S5). Interestingly, two patients who had a local relapse in the ipsilateral breast after breast conserving surgery remained MRD negative during follow-up (Fig. 5C), suggesting that MRD surveillance is more effective in monitoring distant recurrence but not local relapse.

Kaplan–Meier analysis of EFS (A) and DRFS (B) stratified by longitudinal ctDNA MRD status in 82 patients. C Swimming plot showing the MRD test history of all 15 patients with recurrence. Survival analyses were performed using the Kaplan–Meier method. p values were calculated using the log-rank test. Hazard ratios (HR) and the corresponding confidence intervals (CI) were calculated using the Cox regression models. MRD micro residual disease. Source data are provided as a Source Data file.
Because MRD surveillance is quite expensive, it is valuable if we can further narrow down the subpopulation that needs close monitoring. Among the 38 MRD positive patients, 31 were high systemic tumor burden and 10 of them had distant metastasis (Table S6). The sensitivity, PPV, and NPV of systemic tumor burden are markedly higher than baseline ctDNA with the threshold, indicating that systemic tumor burden is a good marker in identifying MRD positive patients.
We then examined the temporal pattern of ctDNA MRD and the timing of recurrence during follow-up. 15 of 130 (11.5%) patients had regional or distant recurrence as of November 1st, 2023 (Fig. 5C), among which 14 patients had post-surgery ctDNA samples. 11 out of the 14 (78.6%) patients had positive MRD before clinical relapse. The median lead time between MRD positivity and clinical relapse was 3.4 months (range 0.3–12.4 months). MRD monitoring is a good tool for post-surgery surveillance of distant recurrence and may provide a window of opportunity to escalate the systemic treatment while micro-metastasis starts to appear.
Discussion
Several studies have investigated the prognostic value of ctDNA in breast cancer at various timepoints19. Yet, many questions still exist regarding the utility of ctDNA in the management of TNBC. To this end, we prospectively enrolled 130 eTNBC patients with a total of 625 blood samples before NAC, after NAC and after surgery. We demonstrated that a threshold for baseline ctDNA stratifies relapse risk more effectively in eTNBC patients, and we established a systemic tumor burden model utilizing both baseline and post-surgery ctDNA to independently predict relapse risk. By integrating systemic tumor burden with pCR status, we accurately identified patients with the highest risk requiring escalation therapy and those with very low relapse risk who could benefit from de-escalation. Furthermore, longitudinal ctDNA surveillance during follow-up showed that MRD was an independent prognostic marker in eTNBC patients. To our knowledge, it is so far the largest systemic ctDNA analysis throughout the course of management for eTNBC patients.
Our study demonstrated 79% ctDNA positivity in eTNBC patients before treatment, which was in line with previous studies demonstrating that ctDNA positivity was as high as 73–100% in eTNBC patients before treatment12,22,24,25. But many of them did not relapse later. Furthermore, none of the eTNBC patients with negative ctDNA before treatment in our study (21% of total) or I-SPY2 study (9% of total) developed distant recurrence, suggesting that a threshold of ctDNA at baseline may be useful in distinguishing high-risk from low-risk eTNBC patients. The DBCG 77 trials showed that about 50% of systemically untreated high-risk TNBC patients were free of recurrence at 10 years33. A recent study also showed that the 5-year DRFS of eTNBC patients who did not receive any chemotherapy was 73%10, suggesting that not all eTNBCs are systemic disease and need systemic treatments. Some stage IIIC TNBC patients in our study had negative baseline ctDNA and no relapse, further supporting that notion that ctDNA is superior to TNM staging in predicting the prognosis. The negative baseline ctDNA before treatment could be a good marker identifying the eTNBC patients that are mainly local disease with excellent prognosis, potentially avoiding excessive systemic treatment including NAC. Future interventional trials are needed to prove the feasibility of treatment de-escalation in such a group of eTNBC patients.
Although the positive baseline ctDNA did not significantly correlate with EFS and DRFS, the threshold of baseline ctDNA at 1.1% MVAF efficiently stratified high-risk and low-risk eTNBC patients, both in our study cohort and the I-SPY2 cohort. This study, for the first time, demonstrates the usefulness of a ctDNA threshold in stratifying risk for breast cancer. Coming larger cohorts with longer follow-up are warranted to further refine the value and corroborate the findings.
The post-surgery ctDNA status was vital as it reflected the earliest MRD status and was predictive of prognosis. However, the sensitivity of post-surgery ctDNA was only 50%, indicating that ctDNA turned negative temporarily after NAC and surgery in some high-risk patients. Using the post-surgery timepoint alone will miss a significant proportion of high-risk patients. To address this issue, we set out to explore how to identify high-risk patients from the beginning. Combining baseline ctDNA and post-surgery ctDNA status, we built a systemic tumor burden model that was independently prognostic in the Cox model. This systemic tumor burden model identifies high-risk patients from two angles: first, patients with high ctDNA burden at the baseline may have micro-metastasis in their body undetectable by imaging. Second, patients with positive ctDNA MRD after surgery indicated the tumor was refractory to NAC. We also found risk stratification can be further improved by combining systemic tumor burden and pathologic response. As expected, the patients with residual disease and a high systemic tumor burden had the highest relapse risks. Within 2 years of median follow-up, the DRFS dropped to less than 75%, suggesting more effective adjuvant treatments are needed for this subgroup. In contrast, non-pCR patients with a low systemic tumor burden had quite a favorable prognosis, indicating their residual disease is mainly local and surgery is sufficient to eradicate them. Some recent trials also tried to combine ctDNA status and pathological response to refine risk stratification12,22, but they didn’t include ctDNA MRD status after surgery, which is a crucial timepoint. We proved that not all non-pCR patients had high relapse risk and systemic tumor burden can be helpful to identify the curable subgroup after surgery. More importantly, our data showed that patients who had both low systemic tumor burden and reached pCR had an excellent prognosis just as the longitudinal MRD negative patients, matching the curable population.
Previous studies had reported positive MRD was associated with higher relapse risk across all breast cancer subtypes18,24, but only included a limited number of TNBC patients. Our study showed that MRD was an independent prognostic factor in a prospective cohort of eTNBC patients. Furthermore, we found that MRD negative patients had 100% DRFS. Avoiding over-treatment in this low-risk population may be of clinical significance. On the other hand, MRD positive TNBC patients had very high relapse risks, indicating better adjuvant treatments are needed to improve the outcome. The c-TRAK trial attempted to add immunotherapy into eTNBC based on ctDNA MRD status. However, the trial failed because 72% of patients had metastatic diseases upon ctDNA detection, with a median lead time at only 1.6 months27. The lead time between ctDNA detection and imaging was 3.4 months in our study, but ctDNA test results were not blinded from the patients and clinicians. Thus, CT or PET-CT scans were often performed right after positive ctDNA detection. Longitudinal MRD status is a useful surveillance tool with excellent prognostic value, but it may be too late for intervention because of the short lead time and the high metastatic rate for MRD positive patients. Our data suggest that, for high-risk patients, escalation therapy may be initiated immediately after surgery without waiting for MRD results. On the other hand, low-risk patients with negative baseline ctDNA may be suitable candidates for de-escalation therapy, potentially avoiding excessive chemotherapy and immunotherapy. Future clinical trials are needed in these settings.
This study has limitations, including relatively small sample size in training data, as well as the short follow-up period of the current study, which contributed to fewer recurrence cases than in the I-SPY2 trial22. It is also premature to analyze the overall survival data.
In conclusion, our study demonstrated the usefulness of baseline ctDNA threshold and the systemic tumor burden model to stratify the risk for eTNBC patients. It also confirmed the independent prognostic value of longitudinal ctDNA MRD in eTNBC patients. Moreover, combining systemic tumor burden and pCR status, this study highlighted how to precisely identify the patients with different relapse risk. These insights will inform the design of future trials exploring treatment escalation and de-escalation in eTNBC management. Future interventional studies are necessary to establish feasibility and further refine these strategies.
Methods
Study design and patient cohort
This prospective, observational, multicenter cohort study enrolled stage II–III TNBC patients treated with NAC at 9 participant hospitals from China between February 2020 and October 2022. This study was a follow-up study after a proof-of-concept study previously published25. The published study was an observational, prospective study, which enrolled patients from Sun Yat-sen Memorial Hospital between 2013 and 2015. The data of forty-four patients from the published study were used to define the threshold of baseline ctDNA in the present study. The present study was approved by the ethics committees of Sun Yat-sen Memorial Hospital and all participant hospitals (approval no. SYSEC-KY-KS-021[R1], Medical Ethics Committee of Sun Yat-sen Memorial Hospital, Sun Yat-sen University). Written informed consent was obtained from all participants. All data were deidentified before analysis.
136 patients registered for the study and a total of 130 patients were successfully enrolled (Fig. S1A).
Patients were eligible for enrollment if they were at least 18 years of age and had locally confirmed TNBC, newly diagnosed, previously untreated, stage II–III, scheduled to receive NAC. Patients with metastatic disease, a history of previous malignant tumor within 3 years, were excluded. All patients had image staging including but not limited to computed tomography (CT) scan, breast magnetic resonance (MR), ultrasound of abdomen and breast conducted by local centers to rule out distant metastasis before receiving NAC. TNBC was defined as estrogen receptor (ER) < 1%, progesterone receptor (PR) < 1%, HER2 IHC score 0–1 or HER2 IHC score 2 with a negative FISH result. The clinicopathological features of the study cohort were presented in Table 1. pCR was defined by the absence of invasive residual disease both in the breast and axillary lymph nodes following NAC. The NAC regimens and corresponding pathologic response were presented in Table S1. After completion of surgery, patients received follow-up every 3–6 months. The patients were followed for recurrence using routine follow-up visits every three months and imaging per standard of care.
Sample collection
Blood samples from each patient were collected at different timepoints, including baseline, post-NAC (pre-surgery), post-surgery, and every 3–6 months during follow-up (Fig. 1A, Fig. S1A). Baseline samples were collected after diagnosis and up to 1 day before the initiation of NAC. The post-NAC (pre-surgery) samples were collected 2 weeks after the last cycle of NAC and up to the day before surgery. The post-surgery samples were pre-defined as collected 1–8 weeks after surgery or radiotherapy. Also, the after-surgery samples of 2 patients were delayed up to two months after surgery, but not receiving RT in the meanwhile due to COVID-19 influences. The follow-up samples were defined as 3–6 months since the post-surgery timepoint.
For each sample, 20 mL of peripheral blood were collected into two 10 mL Streck tubes. Within 3 days of collection, samples were centrifuged at 1600 × g for 10 min at room temperature to separate plasma and peripheral blood lymphocytes (PBLs). The supernatant was then transferred to microcentrifuge tubes and subjected to another centrifugation at 16,000 × g for 10 min at 4 °C to remove cell debris. Finally, the plasma and PBLs samples were stored at −80 °C until analysis.
Matched tumor tissues were obtained from formalin-fixed paraffin-embedded (FFPE) tumor tissue specimens. All tumor tissue samples were examined under a microscope to ensure at least 20% of tumor cells present before downstream analysis (Supplementary Data 2). For 123 patients, paired tumor biopsy tissues taken from baseline were used. For 7 patients with insufficient biopsy tissues for sequencing, tumor tissues obtained from surgery were used.
Sequencing method
Sample preparation and processing
DNA Extraction
Genomic germline DNA was extracted from PBLs with QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Genomic tumor DNA was extracted from FFPE tumor tissue specimens with ReliaPrep FFPE gDNA Miniprep System (Promega, Madison, WI, USA). Cell free DNA (cfDNA) was further isolated from 8 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany). The size distribution and concentration of cfDNA were assessed via an Agilent 2100 BioAnalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA).
Library construction and sequencing
Briefly, genomic DNA from tumor tissues and PBLs was sheared to fragments with a peak of 200~250-bp with a Covaris S2 ultrasonicator (Covaris, Woburn, MA, USA) before library preparation. Then, indexed NGS libraries were constructed from 20–80 ng of cfDNA, 400 ng of paired germline DNA from PBLs, and 400 ng of matched tumor DNA using the Hieff NGS Ultima DNA Library Prep Kit for MGI (Yeasen Biotechnology, Shanghai, China). Unique identifiers (UMIs) were labeled on each double-stranded DNA of cfDNA.
The libraries were hybridized to a customized probe set (Integrated DNA Technologies, Inc) based on a 1.5 Mb region of 1021-gene panel (Supplementary Data 1). After hybridization, libraries were sequenced on a DNBSEQ-T7RS sequencer (MGI Tech, Shenzhen, China) or Gene+Seq-2000 sequencing system (Geneplus, Suzhou, China) with 100-bp paired-end reads. Averagely, 40 Gb data were generated for cfDNA samples and 5 Gb data for tissue DNA, and 1 Gb data for PBLs DNA. Experimental procedures were carried out per manufacturers’ protocols under restricted quality control and assessment.
Sequencing data analysis and variant calling
Preprocessing and alignment
After filtering the terminal adapter and removing low-quality sequence (defined as >50% N rate and >50% bases with Q < 5) from raw data, Burrows-Wheeler Aligner (BWA; version 0.7.12-r1039) was used to align the clean reads to the reference human genome (GRCh37.p13 (GCF_000001405.25)).
Tumor somatic variant calling
Patient-specific somatic variants were identified by analyzing the sequencing data of primary tumors and matched PBL samples. Tumor somatic single-nucleotide variants (SNVs) and small insertions and deletions (InDels) were called by realDcaller (version 1.8.1; Geneplus-Beijing, inhouse) and TNscope (v3.8.0; Sentieon Inc.) as previously reported15,34. CNVKit (v0.9.11, Geneplus-Beijing, inhouse) and NCsv (version 0.2.3; Geneplus-Beijing, inhouse) were applied to profiled copy number variations (CNVs) and structural variants (SVs), respectively15,35,36.
Tumor-informed ctDNA calling
For tumor-informed ctDNA variant calling, we generated customized tumor-variant-based bioinformatic pipelines (Fig. S1B). Matched tumor derived mutations of each patient were put into the SNV calling software. Then, cfDNA BAM files were thoroughly analyzed by realDcaller (version 1.8.1; Geneplus-Beijing, inhouse) and TNscope (v3.8.0; Sentieon Inc.) for precise variant positions and base change types. To maximize the sensitivity of residual mutation detection, a single read containing the tumor-matched variant was treated as a suspected mutation signal for subsequent filtration. The filtration strategies to ensure the specificity of the detection by eliminating the background noise and clonal hematopoiesis were as follows: (1) UMI eliminated PCR and sequencing errors. UMIs were ligated to individual DNA molecules before polymerase chain reaction (PCR) amplification. The duplicated UMI-supported reads distinguished true low-frequency mutations from background noise, thus reducing PCR and sequencing errors. Variants meeting the following criteria are considered eligible: (1) ≥1 duplex consensus sequence support and at least single-stranded consensus family has ≥2 members, or (2) ≥2 single-stranded consensus family support, and each single-stranded consensus family has ≥2 members. (2) Position-specific background error filter model: Our in-house background error filter model was constructed of cfDNA samples from 1000 healthy individuals (defined as no history of cancer or other notable diseases). These samples underwent the same testing and analytic process. The VAF of candidate variants statistically higher than the background mutation rate was considered as true variations. The analytical validation of this background error filter model has been previously described in earlier publications13,15. (3) Deep sequencing of PBLs removed clonal hematopoiesis. The majority of clonal hematopoiesis were non-recurrent. Hence, paired white blood cells of plasma samples were examined simultaneously to identify the individual clonal hematopoiesis mutations. Variants identified in tumor tissue and plasma samples, but not in PBLs, were considered as true tumor-specific variations.
A tissue-derived variant in plasma was considered a true mutation when it met all the three filtration steps.
CNVs detection
Parallel sequencing data from 20–30 PBL samples, processed using the same experimental protocols as matched tumor and plasma samples, were used as the baseline for the copy number analysis. Then, CNVs identification was performed using the CNVkit (v0.9.11, Geneplus-Beijing, in-house) software. The human genomic regions targeted by the 1021-gene panel were uniformly divided into 120-bp windows of PBL, tumor, and plasma samples. Each window’s depth ratio of cfDNA relative to the baseline of PBL samples was calculated separately. After that, the copy number was corrected by considering the relevant influence factors, including GC content and repeat sequence regions. Finally, the copy number alterations of each gene were integrated and calculated as previously reported (6). Copy number alterations were classified as amplification when copy number ≥2.6 and deletion when ≤1.4.
For plasma samples, ctDNA status was defined as positive when at least one or more true tumor-specific variants were identified. The limit of detection by this assay is 0.1%, with a sensitivity of 97.85% and specificity of 98.3%.
In total, blood samples and tumor tissue samples from 130 patients were sequenced to a median depth with duplication of 15918× and 1834×, respectively, and a median depth without duplication of 5252× and 1431×, respectively.
Baseline maximum variant allele frequency cutoff value
First, data from our previous proof-of-concept study25 were used to calculate the MVAF cutoff value at baseline for the DRFS risk stratification. The training cohort from our previous published article was comprised of 44 early breast cancer patients who had received neoadjuvant chemotherapy and curative surgery in the breast cancer center of Sun Yat-sen memorial hospital between 2013 and 2015. The sequencing data from our previously published cohort and current cohort in the manuscript were generated using the same capture-based targeted next-generation sequencing (NGS) method that covers 1021 genes. Furthermore, the variant allele frequency (VAF) from these two cohorts was calculated using the same algorithm. The Youden Index (J) method was used to calculate the optimal MVAF cutoff value37,38.
The Youden Index is defined as
The \({Se}\left(c\right)\) represents sensitivity and at \(Sp\left(c\right)\) represents specificity at a given cutoff value. The cutoff value determined by Youden Index method is the cutoff value that achieves the maximum of \(J\), in other words, the optimal cutoff value is calculated by maximizing Se and Sp at different cutoff points. In our study, we aimed to precisely select the patients with high risk for future escalation therapy while trying to avoid low-risk patients being falsely classified into the high-risk group for over-treatment. Thus, we chose the Youden Index method that weights sensitivity and specificity equally.
To determine whether the cutoff value from our cohorts can be validated in an independent dataset, the eTNBC patients from the I-SPY2 cohort were used. However, I-SPY2 cohort did not provide VAF data, but provided the data of mutant tumor molecules (MTM) per milliliter in plasma that was calculated using the formula28:
Thus, we translated the MVAF cutoff value in our cohort to an MTM cutoff value with the formula:
The MTM cutoff value was 28.27 mutant tumor molecules per milliliter.
Statistical analysis
The correlations analyses of categorical variables were performed using Pearson chi square test. The differences between two groups of MVAF, which were continuous variables, were compared by the Mann–Whitney tests. The pROC package in R software was employed to construct the ROC models and calculate the cutoff point.
EFS was calculated from the day of diagnosis to the first local, regional, or distant recurrence or cancer specific death, whichever happened first. DRFS was calculated from the day of diagnosis to the first distant recurrence or cancer specific death. Survival analyses were performed using the Kaplan–Meier method and the log-rank test. Cox regressions were performed using the Fit Proportional Hazards Regression Model. Statistical analyses and data visualization were performed on SPSS 26.0 (IBM Corp.), GraphPad Prism (10.2.0) and R software (v. 4.3.2) with the following package: “survminer”, “ggplot2”, “pheatmap”, “pROC”. All p values are two-sided and p < 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All DNA sequencing data have been deposited in the Genome Sequence Archive (GSA-Human: HRA008824) that are available under controlled access [https://bigd.big.ac.cn/gsa-human/browse/HRA008824]. Access can be requested from Dr Qiang Liu (liuq77@mail.sysu.edu.cn). Access can be obtained for academic use only under a data transfer agreement and upon Ethics Committee approval. The timescale for this process is approximately 6 months and the data will be available for 1 year. To protect the privacy and confidentiality of our patients, identifiable clinical data are not made publicly available in a repository or in the supplementary material of the article. The individual deidentified participant data can be made available upon request to Dr Qiang Liu (liuq77@mail.sysu.edu.cn). The remaining data supporting the findings of this study are available within the article, Supplementary Information and Supplementary Data. Source data are provided with this paper.
Code availability
The source code for analyses can be accessed via: https://github.com/sue8149/NC2024.
References
Nelson, D. R., Brown, J., Morikawa, A. & Method, M. Breast cancer-specific mortality in early breast cancer as defined by high-risk clinical and pathologic characteristics. PLoS ONE 17, e0264637 (2022).
SEER*Explorer: An interactive website for SEER cancer statistics [Internet]. Surveillance Research Program, National Cancer Institute. Data source(s): SEER Incidence Data, November 2022 Submission (1975-2020), SEER 2022 registries. (2023 Apr 19. [updated: 2023 Nov 16].).
Zhang, Y. et al. Genomic features of rapid versus late relapse in triple negative breast cancer. BMC Cancer 21, 568 (2021).
Grinda, T. et al. Evolution of overall survival and receipt of new therapies by subtype among 20 446 metastatic breast cancer patients in the 2008-2017 ESME cohort. ESMO Open 6, 100114 (2021).
Liedtke, C. et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 41, 1809–1815 (2023).
Cortazar, P. et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164–172 (2014).
Huober, J. et al. Identifying breast cancer patients at risk of relapse despite pathological complete response after neoadjuvant therapy. NPJ Breast Cancer 9, 23 (2023).
Masuda, N. et al. Adjuvant capecitabine for breast cancer after preoperative chemotherapy. N. Engl. J. Med. 376, 2147–2159 (2017).
Schmid, P. et al. LBA18 Pembrolizumab or placebo plus chemotherapy followed by pembrolizumab or placebo for early-stage TNBC: updated EFS results from the phase III KEYNOTE-522 study. Ann. Oncol. 34, S1257 (2023).
Leon-Ferre, R. A. et al. Tumor-infiltrating lymphocytes in triple-negative breast cancer. JAMA 331, 1135–1144 (2024).
Reinert, T. et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 65, 625–634 (2016).
Parsons, H. A. et al. Circulating tumor DNA association with residual cancer burden after neoadjuvant chemotherapy in triple-negative breast cancer in TBCRC 030. Ann. Oncol. 34, 899–906 (2023).
Pan, Y. et al. Dynamic circulating tumor DNA during chemoradiotherapy predicts clinical outcomes for locally advanced non-small cell lung cancer patients. Cancer Cell 41, 1763–1773.e1764 (2023).
Tabernero, J. et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 16, 937–948 (2015).
Zhang, J. T. et al. Longitudinal undetectable molecular residual disease defines potentially cured population in localized non-small cell lung cancer. Cancer Discov. 12, 1690–1701 (2022).
Faulkner, L. G., Howells, L. M., Pepper, C., Shaw, J. A. & Thomas, A. L. The utility of ctDNA in detecting minimal residual disease following curative surgery in colorectal cancer: a systematic review and meta-analysis. Br. J. Cancer 128, 297–309 (2023).
Rothe, F. et al. Circulating tumor DNA in HER2-amplified breast cancer: a translational research substudy of the NeoALTTO phase III trial. Clin. Cancer Res. 25, 3581–3588 (2019).
Garcia-Murillas, I. et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 7, 302ra133 (2015).
Olsson, E. et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 7, 1034–1047 (2015).
Oshiro, C. et al. PIK3CA mutations in serum DNA are predictive of recurrence in primary breast cancer patients. Breast Cancer Res. Treat. 150, 299–307 (2015).
McDonald, B. R. et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med. 11, eaax7392 (2019).
Magbanua, M. J. M. et al. Clinical significance and biology of circulating tumor DNA in high-risk early-stage HER2-negative breast cancer receiving neoadjuvant chemotherapy. Cancer Cell 41, 1091–1102.e1094 (2023).
Magbanua, M. J. M. et al. Circulating tumor DNA and magnetic resonance imaging to predict neoadjuvant chemotherapy response and recurrence risk. NPJ Breast Cancer 7, 32 (2021).
Garcia-Murillas, I. et al. Assessment of molecular relapse detection in early-stage breast cancer. JAMA Oncology 5, 1473–1478 (2019).
Li, S. et al. Circulating tumor DNA predicts the response and prognosis in patients with early breast cancer receiving neoadjuvant chemotherapy. JCO Precis. Oncol. 4, PO.19.00292 (2020).
Cavallone, L. et al. Prognostic and predictive value of circulating tumor DNA during neoadjuvant chemotherapy for triple negative breast cancer. Sci. Rep. 10, 14704 (2020).
Turner, N. C. et al. Results of the c-TRAK TN trial: a clinical trial utilising ctDNA mutation tracking to detect molecular residual disease and trigger intervention in patients with moderate- and high-risk early-stage triple-negative breast cancer. Ann. Oncol. 34, 200–211 (2023).
Magbanua, M. J. M. et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann. Oncol. 32, 229–239 (2021).
Tie, J. et al. Circulating tumor DNA analysis guiding adjuvant therapy in stage II colon cancer. N. Engl. J. Med. 386, 2261–2272 (2022).
Dent, R. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 13, 4429–4434 (2007).
von Minckwitz, G. et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 30, 1796–1804 (2012).
Consortium, I. S. T. et al. Association of event-free and distant recurrence-free survival with individual-level pathologic complete response in neoadjuvant treatment of stages 2 and 3 breast cancer: three-year follow-up analysis for the I-SPY2 adaptively randomized clinical trial. JAMA Oncol. 6, 1355–1362 (2020).
Jensen, M. B. et al. Mortality and recurrence rates among systemically untreated high risk breast cancer patients included in the DBCG 77 trials. Acta Oncol. 57, 135–140 (2018).
Yi, Z. et al. Molecular landscape and efficacy of HER2-targeted therapy in patients with HER2-mutated metastatic breast cancer. NPJ Breast Cancer 6, 59 (2020).
Li, J. et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics 28, 1307–1313 (2012).
Ai, X. et al. Clonal architecture of EGFR mutation predicts the efficacy of EGFR-tyrosine kinase inhibitors in advanced NSCLC: a prospective multicenter study (NCT03059641). Clin. Cancer Res. 27, 704–712 (2021).
Hajian-Tilaki, K. The choice of methods in determining the optimal cut-off value for quantitative diagnostic test evaluation. Stat. Methods Med. Res. 27, 2374–2383 (2018).
Hassanzad, M. & Hajian-Tilaki, K. Methods of determining optimal cut-point of diagnostic biomarkers with application of clinical data in ROC analysis: an update review. BMC Med. Res. Methodol. 24, 84 (2024).
Acknowledgements
We are deeply grateful to our patients who generously volunteered to participate in our study and the families behind them. We thank all our investigators, research nurses, and managing clinicians for their diligent work on behalf of our patients at the following centers, Peking University Shenzhen Hospital, First Affiliated Hospital of AFMU (Air Force Medical University), Renmin Hospital of Wuhan University, Cancer Hospital of Xinjiang Medical University, Daping Hospital, Tongji Hospital, Shenzhen Qianhai Shekou Free Trade Zone Hospital and Guangxi Medical University Cancer Hospital. This work was supported by the National Key R&D Program of China (2022YFC2505101, Q. Liu), National Natural Science Foundation of China (82061148016, Q. Liu; 82230057, Q. Liu; 82272859, Q. Liu; 82203087, Q. Liu; 82102729, S.Y.L.), Science and Technology Program of Guangzhou (202201020486, Q. Liu; SL2022A03J01155, L.J.), and Beijing Medical Award Foundation (YXJL-2020-0941-0760, Q. Liu).
Author information
Authors and Affiliations
Contributions
Q. Liu contributed to the design of the research. S.Y.L., Y.D.L., W. Wei, T.W., G.L., F.Y., J-H.O., Y.X., W. Wu, L.J., N.Y.R., Y.N., F.Y.Y., W.J.J., X.R.L., J.Z., H-W.Y., and C.G. contributed to data acquisition. Q. Li., F.L., Y.H.G., and X.Y. contributed to the experiments. Y.P.Y., M.Z.W., and S.Y.L. contributed to data analysis. S.Y.L., Y.D.L., and C.G. contributed to data interpretation. S.Y.L., Q. Li, F.L., and Q. Liu wrote the manuscript; and all authors critically reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Li, Y., Wei, W. et al. Dynamic ctDNA tracking stratifies relapse risk for triple negative breast cancer patients receiving neoadjuvant chemotherapy. Nat Commun 16, 2786 (2025). https://doi.org/10.1038/s41467-025-57988-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57988-z
This article is cited by
-
Prognostic and monitoring value of circulating tumor DNA at multiple clinical time points in breast cancer
Breast Cancer (2026)
-
Longitudinal deep learning models for tracking disease progression in ovarian cancer using PET/CT imaging and clinical reports
Physical and Engineering Sciences in Medicine (2025)
-
Circulating tumor DNA refines risk stratification of neoadjuvant therapy-resistant breast tumors
Nature Communications (2025)
