
Abstract
Inorganic phosphate (Pi) has essential metabolic and structural roles in living organisms. The Pi exporter, XPR1/SLC53A1, is critical for cellular Pi homeostasis. When intercellular Pi is high, cells accumulate inositol pyrophosphate (1,5-InsP8), a signaling molecule required for XPR1 function. Inactivating XPR1 mutations lead to brain calcifications, causing neurological symptoms including movement disorders, psychosis, and dementia. Here, cryo-electron microscopy structures of dimeric XPR1 and functional characterization delineate the substrate translocation pathway and how InsP8 initiates Pi transport. Binding of InsP8 to XPR1, but not the related inositol polyphosphate InsP6, rigidifies the intracellular SPX domains, with InsP8 bridging the dimers and SPX and transmembrane domains. Locked in this state, the C-terminal tail is sequestered, revealing the entrance to the transport pathway, thus explaining the obligate roles of the SPX domain and InsP8. Together, these findings advance our understanding of XPR1 transport activity and expand opportunities for rationalizing disease mechanisms and therapeutic intervention.
Similar content being viewed by others

Introduction
Inorganic phosphate (Pi) is essential for all cells, serving critical roles in bioenergetics (ATP, GTP), metabolic regulation (i.e., glycolysis and oxidative phosphorylation), intracellular signaling pathways, cell proliferation (as part of DNA and RNA), and formation of structures such as bones and membranes1. Pi is also one of the most pervasive inhibitors of cellular enzymes, such as phosphatases, where it competes for binding to the active site. As one would expect for a metabolite of such central importance, the intracellular concentration and total body content of Pi are highly regulated2. Transporters that facilitate Pi translocation across cellular membranes play a crucial role in this regulation. Despite the importance of Pi homeostasis in human physiology, we have an incomplete understanding of the complex cellular mechanisms that govern Pi entry into and efflux from cells. In humans, there are two protein families of sodium (Na+)-dependent Pi importers, SLC20 and SLC343. A single Pi exporter has been identified – XPR1 (Xenotropic and Polytropic Retrovirus Receptor 1), also known as SLC53A1, as it belongs to the solute carrier (SLC) family of membrane transport proteins4.
In eukaryotes, including yeast, plants, and animals, control of Pi homeostasis is, in part, conferred through a family of SPX domain-containing proteins. These proteins sense and respond to increasing cellular Pi levels through the binding of inositol pyrophosphate (PP-InsP) signaling molecules to their SPX domain5,6. XPR1 is the only mammalian protein that contains an SPX domain, and consistent with this, its Pi transport function is a focal point for overall homeostatic maintenance of Pi balance in humans2,7,8,9. Recent findings have raised questions about whether XPR1 functions as a transport protein or if it instead plays a regulatory role10. A homolog in Drosophila, PXo, was also shown to play an important role in Pi homeostasis, where it marks multilamellar organelles termed PXo bodies, which are degraded following Pi starvation11. Several studies have demonstrated that in vivo XPR1 facilitated Pi export requires the PP-InsP, 1,5-bis-diphosphoinositol 2,3,4,6-tetrakisphosphate (1,5-InsP8 or more simply InsP8), and that other PP-InsPs, such as InsP6, 1-InsP7, and 5-InsP7 have no effect on its activity – clarifying that InsP8 is the physiologically-relevant ligand8,12,13. The concentration of InsP8 in mammalian cells is 100–500 nM, with cytosolic levels rising and falling in response to corresponding fluctuations in cytosolic Pi14,15,16. This correlation between cellular Pi and InsP8 levels has also been well documented in yeast and plants17,18. The mechanism through which the SPX domain of XPR1 discriminates between PP-InsP metabolites is not well understood. Isolated SPX domains have been shown to interact with negatively charged sulfate (SO42−) and PP-InsPs with minimal specificity and selectivity5,8,12. While the binding affinity of InsP8 towards the isolated SPX domain of XPR1 is higher than for other PP-InsPs8, given their cellular abundance, these differences in affinity are not sufficient to explain why only InsP8 supports Pi export through XPR1 in vivo19.
Highlighting critical roles in human physiology, aberrant XPR1 transport activity is associated with both brain disease and cancer. Loss-of-function mutations in XPR1 lead to primary familial brain calcification (PFBC), an inherited neurological disorder in which the formation of abnormal calcium phosphate deposits is causative of neuropsychiatric, cognitive, and movement disorders20,21,22,23. Clinical features include ataxia, Parkinsonism, psychosis, dementia, and cognitive dysfunction with diverse severity, even within the same family. Furthermore, these deleterious calcium phosphate deposits are also prevalent in ~ 20–30% of older adults with no genetic risk factor24. Also of importance for human health and disease, highly proliferative cells, such as cancer cells, have increased metabolism and high ATP and Pi needs. Overexpression of the Pi importer, SLC34A2, is a hallmark of ovarian and uterine cancers25,26. Preventing Pi export from these cancer cells via genetic or pharmacological XPR1 inhibition leads to Pi toxicity and cell death27,28. As such, inhibition of XPR1 is an attractive treatment option for these highly lethal cancers, especially as XPR1 antagonists as a targeted therapy have the potential to minimize side effects.
Through this work, we provide a molecular-level understanding of the mechanisms of Pi export through XPR1 and their regulation by elevated cellular InsP8 levels. We determine a series of single-particle cryo-electron microscopy (cryo-EM) structures of human XPR1, captured in distinct conformational states. This includes structures in complex with the substrate, Pi, and the physiologically relevant signaling molecule, InsP8. The structures and complementary functional studies reveal the mechanisms of activation by InsP8 orchestrated through its regulatory intracellular SPX domain. This work provides a framework for understanding XPR1 function, its transport mechanisms, its gating by InsP8, and how Pi export activity is modulated.
Results
Functional characterization of XPR1
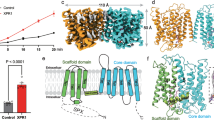
To allow for the measurement of Pi efflux through XPR1, we developed a cellular radioactive [32P] transport assay and measured a robust signal in hTERT RPE-1 cells (Fig. 1a). RPE-1 cells have several benefits. They have a normal karyotype, are non-tumorigenic, and have superior plate adherence characteristics, thereby decreasing nonspecific Pi release. To verify that the Pi efflux that we observed is dependent on endogenous XPR1, we generated XPR1 CRISPR/Cas9 knockout cells and used a previously characterized inhibitory peptide referred to as XRBD4. XPR1 is a cell-surface receptor for xenotropic and polytropic murine leukemia retroviruses (X- and P-MLV), and XRBD is a soluble ligand generated from the envelope-receptor-binding domain of X-MLV that is capable of inhibiting Pi export through XPR1. The export activity was minimal in XPR1 knockout cells, or when the cells were incubated with XRBD (Fig. 1a). Next, we stably reintroduced wild-type XPR1 to the knockout cells and measured the export of radiolabeled Pi. Pi export function is rescued in these cells (Fig. 1b). Though there is evidence that XPR1 can also localize to intracellular organelles9, these results confirm that functional XPR1 resides in the plasma membrane of these cells.

a Pi export through endogenous XPR1 in hTERT RPE-1 XPR1-knockout cells, and its inhibition by the viral peptide fragment XRBD. Data are represented as the mean ± SD (n = 3 samples). b Pi export capabilities were regained when wild-type XPR1 was reintroduced into the XPR1-knockout background. Data are represented as the mean ± SD (n = 3 samples). c, d Cartoon representation of the XPR1 structure with InsP8 bound and the SPX domain resolved, viewed parallel to the membrane plane (c) and perpendicular to the membrane from the extracellular space (d). The SPX domain is colored pink, the scaffold domain is colored teal, the linker helix is colored orange, and the transport domain is colored purple. Horizontal lines indicate the approximate boundaries of the plasma membrane. e–g Cryo-EM density maps of the XPR1 dimer, one monomer colored identically to (c, d) and the other monomer colored gray, in the absence of InsPs (e) and the presence of InsP6 (f) or InsP8 (g). Maps with a low filter threshold of 6 Å (colored white) are overlaid.
Architecture of XPR1
Recombinant full-length human XPR1 protein was purified in the detergent lauryl maltose neopentyl glycol (LMNG) and supplemented with glycol-diosgenin (GDN). The GDN was necessary to overcome the preferred orientation of the protein on the cryo-EM grids29. We determined single particle cryo-EM structures of XPR1 in distinct conformation states, and also when supplemented with substrate (Pi) and two PP-InsP signaling molecules (InsP6, which lacks biological activity, and InsP8 that we synthesized30, the physiological activator). The average resolutions of our structures range from 2.5 to 3.3 Å. The cryo-EM density maps are of sufficient quality to allow for the construction of atomic models of the protein and its corresponding substrates and ligands with good stereochemistry and correlation with the density (Supplementary Table S1 and Supplementary Fig. 6).
The overall architecture of XPR1 will first be described for our samples incubated with InsP8 prior to structure determination, as these maps are the most complete. The structure of XPR1 is comprised of an N-terminal SPX domain and ten transmembrane ɑ-helices (TM1 to TM10) – the majority of the C-terminal tail (residues 633–696) is disordered in all our cryo-EM maps (Fig. 1c, d). XPR1 assumes a dimeric conformation. The membrane-embedded domain of the protein can be divided into two regions: the scaffold domain formed by TM1-4, which permits dimerization, and the transport domain formed by TM5-10, where the substrate translocation pathway lies. The two domains are connected by a helix that runs parallel to the inner leaflet of the membrane designated the linker helix. We detected particularly striking densities for a cholesterol molecule and a phosphatidyl lipid at the interface between these two domains (Supplementary Fig. 8a). The cholesterol molecule that we observed is lodged between aromatic residues (Tyr558 and Phe609) of TM9 and TM10 (Supplementary Fig. 8b). The phosphatidyl lipid occupies an internal cavity formed by TM2, TM3, TM4, the linker helix, and TM5 (Supplementary Fig. 8c).
Visualizing XPR1 in distinct conformational states
We obtained three distinct conformational states of XPR1 that are correlated with the presence and identity of PP-InsPs. In our InsP8-bound structures, the intracellular SPX domain, which is thought to bind InsP8 and be essential for XPR1 function9, is well ordered (Fig. 1g). These maps stand in stark contrast to those generated from the apo and InsP6-supplemented protein where this intracellular sensing domain cannot be resolved (Fig. 1e, f). By low-pass filtering these maps, we can conclude that the SPX domain is present but that in the absence of the physiological activator, InsP8, this region of XPR1 is highly dynamic. The movements are likely imparted by the flexible linker (residues 223–229) that connects the SPX and transmembrane domains.
When we supplemented high Pi (50 mM – physiological concentrations are ~ 1-2 mM) in addition to InsP6, we resolved a third state, following extensive particle classification, in which the SPX domain is resolved (Supplementary Fig. 9). The map quality for the SPX domain is poor, indicating that there is a large degree of heterogeneity in this region. However, bound InsP6 is visible. This conformation is characterized by an asymmetric SPX dimer that is positioned beneath the transmembrane domain of one of the monomers. We are doubtful that this conformation exists in cells. Our results suggest that non-physiological concentrations of Pi can influence XPR1 protein behavior, perhaps acting in concert with PP-InsPs.
A comparison of our XPR1 maps suggests that InsP8 is altering the dynamics of the SPX domain. A key finding is that InsP6, though similar in chemical structure, is not capable of recapitulating the stabilizing effect of InsP8. Structures of a nonhomologous yeast Pi importer that contains a SPX domain have been determined using cryo-EM, and revealed that its SPX domain is highly dynamic31. Furthermore, NMR indicates that movements of the SPX2 domain of the yeast heterooligomeric vacuolar transport chaperone (VTC) complex are altered by PP-InsP binding32. This further suggests that the dynamic nature of SPX domains may be a conserved feature and may be key to their mechanisms of action, even in proteins with diverse functions.
Dimerization is essential for XPR1 function
Our structures reveal that XPR1 is a homodimer, with dimerization being facilitated by a GxxxG ɑ-helical packing motif located in TM1 (Supplementary Fig. 10a)33. To determine the importance of the residues in the GxxxG motif, we mutated the two glycine residues to leucine and employed our previously described cellular radioactive transport assay. Our results demonstrate that dimerization is essential for the proper transport function of XPR1 (Supplementary Fig. 10b).
Whereas this dimer population is the most prevalent, we did observe an alternate dimer assembly in which one of the monomers (TM2-10) appears to be rotated via a large rigid-body movement with respect to the other (Supplementary Fig. 10c). Overlay of these structures reveals that the rotation is caused by a large rearrangement of TM1 (Supplementary Fig. 10d). Cholesterol is known to bind, stabilize, and modulate the activity of several transporters34. The cholesterol molecule that we observed copurified with XPR1 is absent when TM1 assumes this alternate position, suggesting that cholesterol may play a role in stabilizing TM1 and permitting dimerization and XPR1 function (Supplementary Fig. 10e). In its place, we observed a density for a phospholipid, whose presence implies that this hydrophobic pocket must be filled. Though the implications of our findings require further investigation, they suggest that dimerization may serve as a regulatory mechanism of XPR1 function, one in which cholesterol binding may play a modulatory role.
Substrate transport pathway
In our XPR1 maps, we observed three discrete densities in the transport domain that are consistent with Pi substrate (Fig. 2a). To facilitate the confident assignment of these densities as Pi ions and the identification of additional binding sites, we determined structures in the presence of a concentration of Pi on the high end of the physiological range (5 mM). The same three densities were present, and no others were identified. The strength of these densities varies between maps; however, it is apparent that Pi at least partially occupies these sites even when it is omitted during protein purification. This suggests that the nominal Pi in our expression and purification buffers is sufficient to populate the Pi-binding sites and implies high-affinity binding.

a The position of three Pi ions (pink spheres) helps define the substrate translocation pathway. b Tunnels (gray; calculated by MOLE) leading from the cytosol to Pi sites 1 and 2 and from Pi site 3 to the extracellular space. There is no direct path between Pi sites 2 and 3 in this conformational state. Oxygen, red. c Interactions with the Pi substrate in the three bindings sites with densities for Pi and the coordinating residues (gray mesh, 5σ contour). Nitrogen, blue; oxygen, red. d Export of radioactive 32P by XPR1-knockout RPE-1 cells stably expressing wild-type or mutant XPR1-mCer. These are PFBC disease mutations. Data are represented as the normalized mean ± SD (n = 3 samples).
The position of the Pi-binding sites defines the substrate transport pathway through XPR1. The substrate to be exported from cells first encounters site 1 – the substrate-binding site closest to the inside of the cell (Fig. 2b). Site 1 is connected to site 2 via a short and narrow tunnel and lies approximately at the midway point of the membrane-embedded substrate translocation pathway. The final site, site 3, lies adjacent to the extracellular space. The lengthy tunnel connecting Pi sites 2 and 3 is blocked in this conformational state by Trp573. These constrictions suggest that multiple rearrangements would be required for substrate in sites 1 and 2 to reach site 3 and the outside of the cell. Direct binding of negatively charged Pi is commonly promoted through positive electrostatic potential. Indeed, all three Pi ions are coordinated by two or more positively charged residues (site 1: Arg459, Arg466, Arg611; site 2: Arg570, Arg603, Arg604, site 3: Arg270, Arg448) (Fig. 2c). Several of these Arg residues are positioned via cation-π interactions with Tyr and Phe residues.
Mutation of amino acid residues in these binding sites is known to cause the neurological disorder PFBC due to a lack of function of XPR1 (see below)35,36,37. To ascertain the functional relevance of the interactions observed, we stably introduced three disease-causing mutations (R459C at site 1, R570C at site 2, and R448W at site 3) into our XPR1 CRISPR knockout cell line and measured Pi export activity. While the R448W mutation was tolerated, R459C and R570C have reduced transport function – R459C is nonfunctional and R570C has limited activity (Fig. 2d). All three mutants had fluorescence-detection size-exclusion chromatography (FSEC) profiles that were nearly identical to wild-type XPR1 (Supplementary Fig. 12a), indicating that they did not markedly affect protein expression, folding, or stability despite the dramatic residue changes. However, we cannot exclude the possibility that changes in subcellular location are affecting the function of these mutants. This indicates that Pi binding to the sites adjacent to the intracellular environment, site 1 and, to a lesser extent, site 2, is essential for substrate translocation. It also suggests that substrate bind to the site proximal to the extracellular space, site 3, plays a minimal role in XPR1 function. Therefore, the ability of Pi to enter the substrate translocation pathway is of utmost importance for productive transport.
Structural mechanisms of InsP8-dependent regulation
To determine the structural changes and molecular mechanisms governing InsP8-mediated activation of XPR1, we further analyzed our structures resulting from protein prepared in the presence of InsP8 (Fig. 3a). As was previously discussed, in these maps, the SPX domains have been rigidified, and the resolution is sufficient in this region for reliable modeling of both protein and ligand. Our structures reveal that four InsP8 molecules bind the XPR1 dimer at two unique sites (Fig. 3b). In these sites, the highly negatively charged InsP8 is embedded within surface pockets of positive electrostatic potential.

a Chemical structure of 1,5-InsP8. The characteristic pyrophosphate groups are highlighted in red. b XPR1 contains two positively charged pockets in which InsP8 (yellow sticks; oxygen, red) binds, forming a bridge between neighboring domains and locking XPR1 in a defined conformation with rigid SPX domains. The molecular surface is colored according to electrostatic potential: light gray regions are neutral; red, − 8 kTe−1; blue, + 8 kTe−1. c, d Close-up view of InsP8-binding site 1 (c) and InsP8-binding site 2 (d) with InsP8 densities (gray mesh, 5σ contour (c) and 3σ contour (d)). Nitrogen, blue; oxygen, red. e, f Structure-function analysis demonstrating the importance of the positively charged surface patches that bind InsP8 for transport function (binding site 1 (e) and binding site 2 (f)). Data are represented as the normalized mean ± SD (n = 3 samples). g Interactions between the Arg extension contributed by the transmembrane domain and the intracellular capture pocket. Dashed lines indicate electrostatic interactions. Nitrogen, blue; oxygen, red. h Structure-function analysis elucidating the import of Arg308, and its key role in stabilizing the SPX domain. Data are represented as the normalized mean ± SD (n = 3 samples).
The first InsP8 binding site is comprised of two binding interfaces, one contributed by monomer A and one contributed by monomer B (which we refer to as SPX + ve surface a and SPX + ve surface b, respectively) that form a pocket between the two neighboring SPX domains (Fig. 3c). SPX + ve surface a is comprised of Tyr22, Lys26, Tyr30, Arg157, Lys158, Lys161, Lys162, and Lys165 (Fig. 3c), and it is known to be functionally important for binding of negatively charged moieties to SPX domains, such as SO42− and InsP8, as well as InsP8-dependent Pi homeostasis5,9,38. SPX + ve surface b is formed by Arg211 and Arg219. The binding of InsP8 to this site stabilizes an SPX dimer by bridging this domain between monomers. This dimer configuration was not observed in the previous crystal structure of the SPX domain alone with InsP8 as the construct was truncated at residue 207 and, therefore, was missing SPX + ve surface b9. The importance of the 1- and 5-diphosphate groups of InsP8 for binding to site 1 is apparent (Fig. 3c): the diphosphates make critical polar contacts with XPR1, including for one pyrophosphate – Arg157 and Lys158 and for the other pyrophosphate – Lys26, Lys162, and Tyr22, as well as the backbone nitrogens of Lys2, Phe3, and Ala4.
The second InsP8 binding site is positioned both by an additional set of conserved residues within the intracellular SPX domain (which we designate as SPX + ve surface c) and a surface patch of positively charged residues contributed by the intramembrane scaffold and linker domains (referred to as the TM + ve surface) (Fig. 3d). Specifically, InsP8 at this site engages in electrostatic interactions with Lys129, Lys132, Lys213, Lys216, and Arg217 of SPX + ve surface c and Lys364 and Lys369 of the TM + ve surface. Residue Tyr368 likely positions Lys364 and Lys369 via cation-π interactions. The density for this second InsP8 molecule is weak in comparison to both the surrounding protein and the InsP8 molecule bound to site 1. We attribute this to the pocket being cavernous and able to accommodate numerous poses of InsP8. Of note, the phosphatidyl lipid that we found fills an internal cavity and has its headgroup positioned nearby to the InsP8-binding site (Supplementary Fig. 8c). Ultimately, binding of InsP8 to this site helps to bridge the SPX and transmembrane domains, further reducing the dynamics of the SPX domain and defining the relative positions of the SPX and transmembrane domains.
To determine whether the two binding sites that we have identified are responsible for InsP8-dependent activation, we analyzed Pi export for XPR1 proteins bearing single alanine substitutions at several of the identified InsP8-binding residues. The importance of the InsP8-binding site 1 interface on SPX + ve surface a has been previously appreciated5,38. To evaluate whether SPX + ve surface b is also critical, we mutated these arginine residues. Indeed, mutants R211A and R219A show diminished transport function, validating their critical role in the binding of InsP8 to the SPX domain of XPR1 and the importance, not just of a single positively charged surface, but of an entire positively charged pocket (Fig. 3e). Given the import of InsP8 binding to site 1 for proper transport function, we speculate that dimerization of the XPR1 transmembrane domain is essential because it brings the two interfaces that comprise the binding pocket into close proximity. For InsP8-binding site 2, single point mutations within the positively charged clusters on either surface had little effect on transport activity (Fig. 3f). This suggested to us that the major determinant of ligand recognition by this second site is the overall positive charge within these surfaces and the pockets they create.
To test this idea, we analyzed mutants lacking all positive charges within these pockets. For InsP8-binding site 1, this includes SPX + ve surface a (K26A, R157A, K158A, K161A, K162A, K165A) and SPX + ve surface b (R211A, R219A). For InsP8-binding site 2, this includes SPX + ve surface c (K129A, K132A, K213A, K216A, R217A) and TM + ve surface (K364A, K369A, K376A). We found that these XPR1 mutants can no longer or very minimally export Pi, nor can mutants in which all positive charges in the binding sites are converted to alanine (Fig. 3e, f). Together, these data indicate that binding of InsP8 to both pockets is necessary for XPR1 transport function and that all four of the electropositive surfaces that we have identified are important for InsP8 binding. Furthermore, our results demonstrate that InsP8 binding is not wholly intrinsic to the SPX domain of XPR1 but requires interactions with the neighboring transmembrane domain.
Further emphasizing the importance of the transmembrane domain of XPR1 in the InsP8 activation mechanism, our maps reveal that Arg308 extends from an intracellular loop of the transmembrane domain (the TM2-TM3 loop) down into the SPX domain where it is positioned in a pocket to enhance stability and help lock the SPX domain in this distinct state (Fig. 3g). This connection is strengthened by electrostatic interactions involving Glu137 and the backbone carbonyl groups of Ser136, Leu218, and Arg219. The hydrophobic residues Leu140 and Val220 also help define the pocket shape. Arg308 is critical for XPR1 transport activity (Fig. 3h). Three known PFBC disease mutations – S136N, L140P, and L218S are localized to the Arg capture pocket in the SPX domain and have a documented reduction in function (see below)23. Notably, in our structures where the SPX domain is disordered (apo or InsP6-supplemented), the Arg308 side chain is also not visible.
Communication between the two InsP8 sites and the Arg extension may be facilitated through the linker (residues 212–230) that connects the SPX domain to TM1. Specifically, Arg211 and Arg219 help form InsP8 site 1, whereas Lys213, Lys216, and Arg217 help form InsP8 site 2, and Leu218, Arg219, and Val220 help form the Arg extension and capture pocket.
The identification of how InsP8 binds provides additional support that it is the dynamics of the SPX domain that are crucial for the InsP8-dependent regulatory mechanisms of XPR1. Furthermore, it indicates how InsP8 locks the SPX domain into a single state. At physiological pH, InsP8 is highly negatively charged (−10 or −11)39, which allows it to interact with more than one positively charged protein site simultaneously. This allows for the bridging of surfaces that would otherwise repel each other. Our structures indicate that InsP8 binding at interfaces between the SPX monomers and SPX and transmembrane domains restricts movement of the intracellular SPX domain and stabilizes a defined functional state. Together, these observations illustrate how InsP8 co-factor binding significantly alters protein behavior.
InsP8-induced movements initiate Pi transport
Further classification of particles originating from the InsP8-supplemented samples yielded two different functional states. In both states, the SPX domains are engaged, and InsP8 is bound. The superposition of these states revealed that rearrangement of the C-terminal tail originating at His616 is a hallmark of the opening of an intracellular constriction in the Pi transport pathway (Fig. 4a). Highlighting the importance of this region, the C-terminal tail residues of XPR1 are highly conserved (Supplementary Fig. 7). We observed dimers in which one protomer is in an intracellular gate open (iO) state and the other in an intracellular gate closed (iC) state and in which both protomers adopt the iC state. As monomers have independent substrate translocation pathways and both XPR1 states can be visualized within the dimer, the protomers appear to have low cooperativity.

a Superimposition of the XPR1 structures in which the intracellular gate is open (iO) (yellow) and closed (iC) (purple). Pi ions are shown as pink spheres. b, c The C-plug of XPR1 blocks an otherwise open entrance to the substrate transport pathway. The intracellular gate is formed by Pro475, Met532, Cys620, and Glu622. The contour mesh of the tunnels (gray) calculated by MOLE. The radius at the narrowest constriction along the pathway is labeled. Oxygen, red; sulfur, green. d Tunnel radii plots of the entrance to the Pi translocation pathway in the iO and iC states. Tunnel radii generated by MOLE. The radius of a Pi ion is shown (dashed line). e, f Structural changes associated with XPR1 gating by InsP8. Transition from the iC (f) to iO (e) state is accompanied by a sequestration of the C-terminal tail by the SPX domain, thereby opening the gate. g Structure-function analysis demonstrating the importance of the intracellular domains on XPR1 transport activity. Data are represented as the normalized mean ± SD (n = 3 samples).
In the iC state, the C-terminal tail residues Cys620 (via its backbone carbonyl) and Glu622 block Pi entry to the translocation pathway by introducing negative charge that repels the Pi ions and restricts its size (Fig. 4c, d and Supplementary Fig. 13b). A neighboring glycine residue, Gly621, also plays a critical role as it allows these residues to assume an optimal conformation. We define this region as the C-plug. This C-terminal tail position is stabilized through a cation-π interaction of Phe623, which projects into Pi-binding site 1, with Arg466 – one of the Pi coordinating residues (Fig. 4f). This conformation predicts that substrate binding to site 1 helps drive rearrangement from the iO to the iC state.
In contrast to the iC XPR1 structure, our iO structure shows an unplugging of the entrance of the Pi translocation pathway through a sequestering of the C-terminal tail by direct interactions with the SPX domain. This increases the radius of the entrance to the translocation pathway from ~ 1.5 Å to 2.9 Å, which is large enough to allow passage of a Pi ion (ionic radius 2.4 Å or ≥ 3 Å when hydrated40,41) (Fig. 4b, d). Furthermore, the entrance to the Pi permeation pathway is lined by residues Lys388 and Arg472 and thus is highly electropositive (Supplementary Fig. 13a). A critical salt bridge is formed between Arg627 of the C-terminal tail and Glu137 of the SPX domain, and a series of hydrophobic interactions also contribute to this interaction (Fig. 4e). The importance of this C-terminal tail region is further emphasized by the nearby location of several PFBC disease mutations, including N619D, R624H, and I629S (Fig. 5)35,42. Worth noting this key structural feature neighbors the interactions of the Arg308-containing intracellular loop that helps bridge the SPX and transmembrane domains.

Residues of which missense mutations are known to cause neurological disease phenotypes are represented as sticks. Nitrogen, blue; oxygen, red. The residues are clustered near protein regions critical for function, including (1) the substrate translocation pathway, (2) the Arg extension and capture pocket, and (3) the intracellular gate or C-plug.
Our structures predict that in the absence of an SPX domain (and InsP8) or a C-terminal tail, XPR1 will be unable to enter the iO state and will, therefore, be nonfunctional. Indeed, all our structures from samples without InsP8, whether the SPX domain is resolved or not, are in an iC state. Reports on whether the SPX domain is essential for function have been conflicting4,9,38,43,44. In contrast, the importance of the C-terminal tail is clear since deletion of this domain (∆612–696) renders XPR1 non-functional – although this may be attributed in part to protein misfolding and/or mislocalization35. To further assess the role of these intracellular domains on transport, we generated mutant XPR1 lacking the SPX domain (∆1–225) or the C-terminal tail (∆619–696). We expressed the mutants in XPR1-knockout cells and measured the export of radiolabeled Pi. XPR1 lacking the SPX domain was completely non-functional, indicating that this protein region is, indeed, essential for Pi transport (Fig. 4g). Consistent with previous studies, removing the C-terminal tail of XPR1 had a detrimental effect on protein behavior. The lack of an FSEC peak for this mutant suggests that it is either not expressed or that the expressed protein is not stable following detergent extraction (Supplementary Fig. 12e).
The mechanism of XPR1 activation is reminiscent of several other transporter families that conduct anions and for which their C-terminal tails are crucial for the transport cycle. For example, in the occluded state of the glutamate/gamma-aminobutyrate (GABA) antiporter GadC, the C-terminal tail is folded within the open binding cavity, thus blocking the substrate-binding site45. In SLC26A9, another transporter that mediates anion export activity, occupation of the intracellular pocket by its C-terminal tail leads to a change in electrostatics at the entrance of the translocation pathway and reduces accessibility from the cytosolic side, thereby altering substrate access46.
Given the detailed mechanisms of InsP8 activation and our structures of InsP6-supplemented protein, it is now apparent why InsP6 is unable to activate XPR1 transport function in vivo. At physiological concentrations of Pi, InsP6 is not capable of stabilizing the SPX domain, a necessity for conversion to the iO state and Pi export. It is plausible that InsP6 binds, though it is not possible to say with certainty due to the dynamics of the SPX domain and corresponding poor map quality. Regardless, it is apparent that InsP6, which has a lower negative charge than InsP8, cannot facilitate robust domain bridging. At extremely high concentrations of Pi (~20X cellular levels), we did obtain InsP6-bound structures where the SPX domain is resolved, albeit with poor resolution (Supplementary Fig. 9). The SPX dimer has an alternate placement relative to the transmembrane domains and adopts this asymmetric conformation because InsP6 and InsP8 binding are not analogous. We observed only two InsP6 molecules bound to the XPR1 dimer. One InsP6 binds only to SPX + ve surface a of monomer A. In the absence of a second interface, domain bridging is not possible. The second InsP6 binds to the SPX + ve surface a of monomer B and the TM + ve surface of monomer A, explaining why the position of the SPX domain relative to the transmembrane domain is different than what we observed for InsP8-bound XPR1. Though the dynamics of the SPX domain have been altered, the pose of this domain is not capable of releasing the C-plug and transitioning XPR1 to the iO state.
Our structures support a mechanism for the dependence of XPR1 function on InsP8, as depicted in Fig. 6. The intracellular regulatory gate (or C-plug) is formed by the C-terminal tail of XPR1. Upon InsP8 binding, the SPX domain becomes rigidified in a conformation that can sequester the C-terminal tail of XPR1, causing it to undergo a dramatic rearrangement away from the entrance of the translocation pathway. This leads to the opening of the intracellular gate. When open, Pi ions are free to enter the substrate translocation pathway and bind at site 1. We propose that the plugging of the entrance of the translocation pathway by the C-plug is a movement that is favored by the participation of Phe623 to substrate binding to site 1 and may help initiate the transition to the iC state. Even with InsP8-bound and the SPX domain engaged, the majority of our particles (~75%) reside in the iC state. This may be because in our structures, despite not introducing exogenous Pi, we find substrate binding site 1 is occupied. Lastly, our structures predict that a conformational change to a state open to the extracellular environment, substrate release, and completion of Pi export, requires relieving additional points of constriction along the substrate conduction pathway. This includes the constrictions caused by Trp573, which is located mid-way through the substrate tunnel, and several extracellular loops. While both the TM5-6 and TM9-10 loops occlude the release of Pi from substrate binding site 3, we hypothesize that the extracellular gate is formed by the TM9-10 loop, as the upper segments of TM5 and TM6 are stabilized by a disulfide bond between cysteines 415 and 440.

In the absence of InsP8, the entrance to the Pi translocation pathway is blocked by the C-plug (iC-eC). In this state, Phe623 is inserted into substrate-binding site 1. In the presence of InsP8, the SPX domain becomes rigidified, and the C-terminal tail of XPR1 can be sequestered by this domain, causing the intracellular gate to open (iO-eC). Structures from Yan et al. and Lu et al. (PDBs: 8X5E, 8YFD, and 8YF4) capture the opening of an extracellular gate (iC-eO), formed by Trp573 and the TM9-10 loop, to allow for Pi release44,50. InsP8 binding is not required to facilitate transitions to the iC-eO state. Only the SPX (pink) and transport (purple) domains of a monomer of XPR1 are drawn to improve clarity.
Discussion
XPR1 is the only mammalian SPX domain-containing protein. It is known to sense intracellular Pi concentrations by binding to InsP8, which allows for adjustments of its Pi export activity to meet the needs of the cell. Though InsP8 has emerged as a central cellular messenger, our understanding of the molecular mechanisms that govern protein regulation by this signaling molecule is limited47. A major advance was the discovery that PP-InsPs bind to and signal through SPX domains5. Our results demonstrate that when XPR1 binds InsP8, this signaling molecule aids in bridging domains, thereby restricting SPX domain heterogeneity. The ability of PP-InsPs to act as a bridging moiety between positively charged protein surfaces that would otherwise repel each other is well documented48,49. Here, we demonstrate that InsP8 can exploit this ability to act as a molecular glue. The binding pockets for the two InsP8 molecules are formed by four positively charged surfaces – only one of which had been previously identified through studies conducted with isolated SPX domain5,9. We have shown that InsP8 binds not only to the SPX domain but also to the transmembrane-embedded domain of XPR1. This is necessary to position the intracellular SPX dimer in the precise orientation relative to the transport domain, which is essential for opening the intracellular gate. This conformation is further stabilized by the Arg extension and capture pocket. Therefore, other protein domains, in addition to the SPX, are important for determining mechanisms of PP-InsP action. We propose that the signaling molecule first binds to InsP8-binding site 1, thereby forming the cavernous InsP8-binding site 2. Prior to this work, our molecular-level understanding of how PP-InsP binding stimulates protein function centered on the idea that binding relieves an inhibitory constraint imposed by the SPX domain. For example, in the yeast VTC, biophysical experiments support the notion that InsP8 disrupts an interaction between two SPX domains contributed by different proteins within the complex that block inorganic polyphosphate synthesis by the enzyme32. Our structures now suggest that modulation of SPX domain-containing proteins by PP-InsPs is mechanistically diverse – with XPR1 being the first example of a protein whose function is regulated by the ability of InsP8 to bridge domains and stabilize unique protein states, such as the iO conformation visualized here.
Our structures indicate that there are at least two constrictions along the XPR1 ion conduction pathway that we refer to as the intracellular and extracellular gates. We demonstrate that in the absence of InsP8, the resting state of the intracellular gate, formed by the C-plug, is closed (iC-eC). The binding of InsP8 triggers a conformation transition that facilitates the opening of the intracellular gate (iO-eC). In addition to the iC-eC and iO-eC XPR1 states, a third state, in which the extracellular gate is open (iC-eO), has recently been observed (Fig. 6)44,50. Extracellular gate opening does not require InsP8 or stabilization of the SPX domain. In the iC-eO state, a large rearrangement of TM9 that simultaneously repositions Trp573, relieving a narrow constriction between Pi binding sites 2 and 3, provides an unobstructed path for Pi efflux from Pi-binding site 2 to the extracellular space. However, a constriction along the substrate translocation pathway between Pi binding sites 1 and 2 remains, and the intracellular gate is closed (i.e., the radius of the substrate entry tunnel is smaller than the ionic radius of Pi) (Supplementary Fig. 14).
There are several important outstanding questions regarding the mechanisms of Pi transport by XPR1. First, it will be beneficial to conduct additional unitary ion flux rate measurements. Current data on the transport rate of XPR1 varies greatly, perhaps suggesting that it is dependent on the cell type or physiological state4,44,50,51,52. Another critical unresolved question is whether the opening of the intracellular and extracellular gates is coupled. Questions also remain regarding the driving force of Pi export through XPR1, particularly since an increase in external Pi concentration has been shown to accelerate transport4. Finally, it will be interesting to determine whether protein-protein interactions, such as the reported interaction between XPR1 and Kinase D-interacting substrate of 220 kDa (KIDINS220)27, modulate the function of XPR1. Ultimately, in-depth biophysical characterization will be necessary to fully interpret the structural data and elucidate precisely how XPR1 effluxes Pi and its physiological consequences in different cell types and in normal and disease states.
Over the past decade, six causative genes for PFBC have been identified, two of which are Pi transporters (SLC20A2 and XPR1), establishing a direct link between the dysregulation of Pi homeostasis and disease20. Why these abnormal calcifications form is still yet to be discovered. Furthermore, no disease-modifying therapies are available for brain calcification, nor have definitive pharmacological targets been identified. Our structural elucidation of the Pi exporter XPR1 provides a detailed, mechanistic understanding of how PFBC disease mutations cause impaired Pi efflux and homeostasis, and neurological disease (Fig. 5). Both agonists and antagonists of XPR1 have potential therapeutic applications, with antagonists being explored as targeted ovarian and uterine cancer drug treatments27,28. To address biological questions and eventually illuminate novel therapies for the treatment of brain calcifications, cancer, and other disorders affected by Pi imbalance, such as kidney disease53, access to specific chemical probes that modulate XPR1 function is essential. However, aside from the physiological activator InsP8 and the inhibitory viral peptide XRBD, no such compounds currently exist. Our structures suggest that Pi mimics that could block the ion conduction pathway or prevent structural rearrangements during substrate transport are worth exploring. Our findings also indicate that modulators capable of disrupting the SPX domain stabilization that results when InsP8 binds might be effective antagonist drugs. Our findings provide a mechanistic rationale for understanding how Pi transverses the membrane through XPR1 and InsP8 binding permits Pi entry to the translocation pathway from the intracellular environment. This may facilitate the design of drugs that selectively modulate aberrant XPR1 activity under a variety of pathophysiological conditions.
Methods
Generation of CRISPR knockout constructs and cell lines
We generated an IL25 knockout hTERT RPE-1 cell line (ATCC, CRL-4000), as a positive control (referred to as wild-type in the text), and a XPR1 knockout RPE-1 cell line. sgRNAs (listed in the accompanying Supplementary Data file) were synthesized by IDT and cloned into pLentiCRISPRv2-mCherry (Addgene, 99154). The plasmid was simultaneously linearized by BsmBI (Fisher Scientific, FERFD0454) and dephosphorylated by Alkaline Phosphatase (Fisher Scientific, EFREF0651). The sgRNAs were phosphorylated using T4 Polynucleotide Kinase (NEB, M0202S) and annealed in preparation for ligation (NEB, M2200) into the vector. The constructs were validated by Sanger sequencing. The sgRNA expressing vectors, along with lentiviral packaging plasmid psPAX2 and envelop plasmid pMD2.G-VSVG, were transfected into LentiX 293 T (Takara, 632180) cells using Lipofectamine 3000 (Thermo Fisher Scientific, L3000015). Virus-containing supernatant was collected 48 h after transfection and passed through a 0.45-μm filter. RPE-1 cells in 6-well tissue culture plates were infected with the virus and 4 μg/ml Polybrene (Santa Cruz Biotechnology, sc-134220). After 24 h, fresh virus was added. After 48 h, the virus was removed, and the cells were allowed to recover overnight. Cell lines were selected using fluorescence-activated cell (FACs) sorting conducted by the MSK Flow Cytometry Core Facility. TIDE (Tracking of Indels by Decomposition) analysis (primers listed in the accompanying Supplementary Data file) was used to verify CRISPR-guided gene disruption54. The total efficiency was 91.9% for the IL25 knockout and 96.1% for the XPR1 knockout. All cell lines were grown in DMEM medium (MSK Media Facility) containing 2 mM Pen/Strep supplemented with 10% fetal bovine serum (MSK Media Facility) and were maintained at 37 °C and 5% CO2.
Generation of stable cell lines
Genes encoding wild-type or mutant XPR1 were cloned into the lentiviral expression vector pLenti-GIII-CMV (Applied Biological Materials). The constructs encode for the full-length protein followed by a C-terminal mCerulean tag (all primers listed in the accompanying Supplementary Data file). All constructs were validated by full plasmid sequencing. The cDNA expressing vectors along with lentiviral packaging plasmid psPAX2 and envelop plasmid pMD2.G-VSVG were transfected into LentiX 293 T cells using Lipofectamine 3000 transfection reagent (Thermo Scientific, L3000015). Virus-containing supernatant was collected 48 h after transfection and passed through a 0.45 μm filter. XPR1-knockout RPE-1 cells in 6-well tissue culture plates were infected with the virus and 4 μg/ml Polybrene (Santa Cruz Biotechnology, sc-134220). After 24 h, fresh virus was added. After 48 h, the virus was removed, and the cells were allowed to recover overnight. Cell lines were selected by 20 μg/ml puromycin (Sigma, P8833).
Cloning, expression, and purification of XRBD
The XRBD of X-MLV envelope protein (strain NZB, Genebank #K02730, 32-238) fused to a C-terminal mouse Fc tag was synthesized by Twist Biosciences and cloned into a pcDNA vector with a N-terminal signal peptide (MKHLWFFLLLVAAPRWVLS). XRBD-mFc protein was expressed in Expi293F cells (Thermo Scientific, A14527) using transient transfection. After 4 days, the supernatant was collected and dialyzed into TBS buffer. Protein was purified using Protein G resin (GenScript, L00209), concentrated to 2.5 mg/ml in TBS buffer, and stored at -80°C.
Radioactive Pi transport assays
Radioactive 32P substrate efflux experiments were carried out as follows. For FSEC analysis55, 5.0 × 106 RPE-1 cells were seeded per well in 10 ml DMEM medium in 10 cm dishes, and for the transport assay, 2.5 × 105 RPE-1 cells per well in 1 ml DMEM medium in 12-well plates. Both assays were performed 24 hours later. The cells designated for analysis by FSEC were resuspended in 20 mM Tris, pH 7.5, 200 mM NaCl, and 1:1,000 dilution of Protease Inhibitor Cocktail Set III (EDTA free, Calbiochem), lysed using sonication (8x10 s), then solubilized by supplementation with 10 mM of the detergent lauryl maltose neopentyl glycol (LMNG, Anatrace, NG310) and 2 mM cholesteryl hemisuccinate (CHS, Anatrace, CH210). Samples were rotated for 1 h at 4 °C and then centrifuged at 18,200 × g for 1 hr at 4 °C before FSEC analysis. The FSEC buffer contained 20 mM Tris, pH 7.5, 200 mM NaCl, 0.02 mM LMNG, and 0.004 mM CHS. In parallel, to load the radioactive substrate, the cells designated for the transport assay were incubated in DMEM containing 0.5 µCi/mL [32Pi] (Revity, NEX053001MC) for 20 min at 37 °C. Then, the cells were washed three times with DMEM medium. At t = 0 min, 400 µl of DMEM medium supplemented with 10 mM Pi, pH 7, was added. The reaction was carried out for 1 h at 37 °C. An aliquot of medium was removed. Cells were washed three times in cold PBS (MSK Media Facility) and subsequently lysed using 1 ml of 1% Triton X-100 (Sigma, T9284). Two samples per well (250 µl of extracellular media; 750 µl of lysed cells) were transferred to a scintillation vial with 5 ml of scintillation cocktail (Perkin Elmer, 6013327). Radioactivity was measured with a liquid scintillation analyzer (Perkin Elmer, Tri-Carb 2910-TR). Efflux is graphed as a percentage of the [32Pi] that was accumulated by each cell culture prior to the efflux assay using GraphPad Prism 10. All transport assay data are reported as means ± standard deviation from 3 replicates. For structure function analyzes, the data are reported as normalized means ± standard deviation (with error propagation from the data transformation). The efflux relative to wild-type for each cell line was normalized using the equation ((mutant-knockout)/(wild-type-knockout)). All mutants were tested in at least 3 independent experiments. During assay incubations, some intracellular [32Pi] is likely converted through oxidative phosphorylation and glycolysis into organic phosphates that are not transported by XPR1. Therefore, the efflux of [32Pi] is not expected to be 100%.
To evaluate the effect of external Pi concentration on XPR1 transport activity, DMEM medium with no Pi (Gibco cat#: 11971025) and no FBS, supplemented with 0, 1, 5, 10, or 20 mM Pi, pH 7, was substituted. We found no appreciable difference in Pi export by XPR1 (Supplementary Fig. 11a, b). This warrants further investigation, considering that Giovannini et al. measured that Pi export was increased ~ 2-fold when the extracellular Pi concentration was increased from 0 to 10 mM4. A time course experiment (30 min, 1 h, 1 h 30 min, and 2 h) was also carried out (Supplementary Fig. 11d). The 1 h time point gives a robust signal but does not reflect initial XPR1 efflux. Additionally, the high external Pi concentration did not significantly affect the intracellular Pi concentration (Supplementary Fig. 11c). To measure intracellular Pi, 7.5 × 105 RPE-1 cells were seeded per well in 2 ml DMEM medium in 6-well plates. 24 h later, cells were incubated in DMEM medium (with no Pi or FBS) supplemented with 10 mM Pi for 1 h at 37 °C. Subsequently, cells were washed three times in TBS and lysed using 250 μl of TBS with 0.5% Tween20 (Sigma, P9416) in the presence of cOmplete Protease Inhibitor Cocktail (Roche, 11697498001). Cellular debris was removed by centrifugation (18,200 x g for 1 h at 4 °C). The Pi levels in the lysate were measured using the Malachite Green Phosphate Assay Kit (Sigma, MAK307). Total protein concentration was determined using the BCA Protein Assay (Thermo Scientific, 23228). Intracellular Pi levels are graphed as the interpolated Pi concentration normalized by the total protein concentration per sample. Data is reported as means ± standard deviation from 3 replicates.
Cloning, expression, and purification of human XPR1
Human XPR1 was selected as a candidate for protein purification and structure determination using FSEC screening55. The full-length human XPR1 cDNA (synthesized by Twist Biosciences) was cloned into a modified pEG BacMam expression vector (addgene, 160686) containing a C-terminal mCerulean and Strep-tag (WSHPNFEK) proceeded by a Precision Protease cleavage site (LEVLFQ/GP) to facilitate its removal.
XPR1 was expressed in Expi293F cells using transient transfection. Briefly, 100 µg of plasmid DNA was incubated with 300 µg of polyethylenimine (PEI 25,000) (Polysciences, 23966-1) for 30 min at room temperature, then added to 100 ml of cells at a density of 2.0 × 106 cells/ml. Cells were supplemented with 10 mM sodium butyrate after 20 h, harvested 44 h post-transfection, and stored at − 80 °C.
The cell pellet was re-suspended in buffer containing 25 mM Tris, pH 7.5, 200 mM NaCl, and a cOmplete Protease Inhibitor Cocktail Tablet. 10 mM LMNG (Anatrace, NG310) was added to the cell lysate, and the mixture was rotated at 4 °C for 1 h 30 min to extract XPR1 from membranes. The sample was centrifuged at 50,000 × g for 50 min at 4 °C, and the supernatant was filtered through a 0.22 μm polystyrene membrane (Millipore Sigma). The sample was incubated with Strep-Tactin XT 4Flow high-capacity resin (IBA Lifesciences, 2-5030-002) at 4 °C for 1 h with rotation. At room temperature, beads were collected on a column, washed with buffer containing 25 mM Tris, pH 7.5, 200 mM NaCl, 0.02 mM LMNG, and 0.017 mM glycol-diosgenin (GDN, Anatrace, GDN101), and the protein as eluted with an identical buffer supplemented with 10 mM Biotin (IBA Lifesciences, 2-1016-002). The C-terminal mCerulean/Strep tag was removed by rotating the sample overnight at 4°C with PreScission Protease. XPR1 was further purified using a Superose 6 Increase size-exclusion column (Cytiva) in 25 mM Tris, pH 7.5, 200 mM NaCl, 0.02 mM LMNG, and 0.017 mM GDN. For the InsP6-supplemented sample, 1 mM InsP6 (Sigma, P8810) was added to all purification buffers. For the high Pi- and InsP6-supplemented sample, all purification buffers contained 50 mM Na Phosphate, pH 7.4, and 150 mM NaCl (instead of 25 mM Tris, pH 7.5, and 200 mM NaCl) and 1 mM InsP6. For the InsP8-supplemented sample, 100 µM InsP8 (synthesized by the Jessen lab30) was added to the protein prior to gel filtration. For the Pi- and InsP8-supplemented sample, 5 mM Na Phosphate, pH 7.4, was added to all purification buffers and 100 µM InsP8 was added to the protein prior to gel filtration. Peak fractions were pooled and concentrated to 8–12 mg/ml using a 100-kDa concentrator (Amicon Ultra, Millipore Sigma).
Cryo-EM sample preparation and data acquisition
Cryo-EM grids were prepared using a FEI Vitrobot Mark IV (Thermo Fisher Scientific) by applying 3 μl of purified XPR1 to a glow-discharged holy carbon gold QUANTIFOIL 1.2/1.3 grid (400 mesh, Electron Microscopy Sciences) and blotting for 4 to 5 s at 4 °C and 100% humidity, then plunge freezing in liquid ethane. Cryo-EM datasets were collected on FEI Titan Krios microscopes (Thermo Fisher Scientific) operated at 300 kV housed in the MSK Richard Rifkind Center for Cryo-EM. Images were recorded in an automated fashion on a K3 Summit Direct Electron Detector (Gatan) in super-resolution counting mode with a super-resolution pixel size of 0.413 Å (physical pixel size of 0.826 Å) at a dose rate of 15 e-/pixel/s using SerialEM or a Falcon 4i Direct Electron Detector (Thermo Fisher) with a Selectris X Energy Filter (Thermo Scientific) in counting mode with a physical pixel size of 0.725 Å at a dose rate of 11.6 e-/pixel/s using EPU. The defocus range was − 0.7 to − 1.7 µm. Data collection statistics are shown in Supplementary Table S1.
Electron microscopy data processing
Image processing was performed using cryoSPARC (v4.5.0 or v4.5.1)56. The videos were gain-corrected, Fourier-cropped by two (0.826 Å) (when using the K3 camera only), and aligned using whole-frame and local-motion-correction algorithms within cryoSPARC. The cyroSPARC validation tool was used for FSC calculations and reliable resolution estimation.
Apo sample
Supplementary Fig. 1 shows the cryo-EM workflow for this dataset. A total of 5500 super-resolution movies of apo XPR1 were collected using a K3 camera. Blob-based auto-picking was implemented to select initial particles. Several rounds of two-dimensional (2D) classification were carried out, and the best 2D classes (those with easy-to-identify transmembrane helices) were manually selected to generate two initial three-dimensional (3D) models – the dimer and rotated dimer. False-positive selections and contaminants were excluded through iterative rounds of heterogeneous classification using the models generated from the ab initio algorithm as well as several decoy classes representing noise. Particles contributing to these dimer and rotated dimer maps were combined and used for Topaz training and picking using neural networks, resulting in 4,266,672 particles57. Heterogeneous classification of these particles, followed by 3D classification using a focus mask that excludes detergent micelle density, was used to separate well-aligning particles into dimer and rotated dimer classes. Stacks of 431,220 particles for the dimer and 124,969 particles for the rotated dimer were refined using non-uniform refinement and local refinement, yielding reconstructions of 2.52 Å and 3.06 Å, respectively.
InsP6-supplemented sample
Supplementary fig. 2 shows the cryo-EM workflow for this dataset. A total of 4116 super-resolution movies of InsP6-supplemented XPR1 were collected using a K3 camera. Blob-based auto-picking was implemented to select initial particles. False-positive selections were excluded through iterative rounds of heterogeneous classification using the dimer and rotated dimer apo models, as well as several decoy classes representing noise. Selected particles were used for Topaz training and picking using neural networks, resulting in 5,084,405 particles57. Heterogeneous classification of these particles, followed by 3D classification using a focus mask that excludes detergent micelle density, was used to separate well-aligning particles. A stack of 487,706 particles was refined using non-uniform refinement, yielding a reconstruction of 2.97 Å.
High Pi- and InsP6-supplemented sample
Supplementary Fig. 3 shows the cryo-EM workflow for this dataset. A total of 3981 super-resolution movies of high Pi- and InsP6-supplemented XPR1 were collected using a K3 camera. Blob-based auto-picking was implemented to select initial particles. Several rounds of 2D classification were carried out, and the best 2D classes (those with easy-to-identify SPX domains and transmembrane helices) were manually selected to generate initial 3D models. False-positive selections were excluded through iterative rounds of heterogeneous classification using the initial models as well as several decoy classes representing noise. Selected particles were used for Topaz training and picking using neural networks, resulting in 5,425,183 particles57. 2D classification and heterogeneous classification of these particles, followed by 3D classification using a focus mask that excludes detergent micelle density, was used to separate well-aligning particles. 3D variability analysis (3DVA) was used to assess heterogeneity within the particles. A stack of 100,408 particles was refined using non-uniform refinement, yielding a reconstruction of 3.30 Å. This subset of particles gave a reconstruction in which the SPX domain was resolved, thought the density is low resolution, and its orientation with respect to the transmembrane domain is likely to be non-physiological.
InsP8-supplemented sample
Supplementary Fig. 4 shows the cryo-EM workflow for this dataset. A total of 12,900 movies of InsP8-supplemented XPR1 were collected using a Falcon 4i camera. Blob-based auto-picking was implemented to select initial particles. Several rounds of 2D classification were carried out, and the best 2D classes (those with easy-to-identify SPX domains and transmembrane helices) were manually selected to generate an initial 3D model. False-positive selections were excluded through iterative rounds of heterogeneous classification using the initial models as well as several decoy classes representing noise. Selected particles were used for Topaz training and picking using neural networks, resulting in 5,312,652 particles57. Heterogeneous classification of these particles, followed by 3D classification using a focus mask that excludes detergent micelle density, was used to separate well-aligning particles. 3D classification and 3DVA, both with and without symmetry expansion, using a focus mask that excludes detergent micelle density, allowed for the separation of two unique C-terminal tail conformations – corresponding to the states iO and iC. Polished particle stacks were used for non-uniform refinement with global contrast transfer function (CTF) estimation, as well as higher-order tetrafoil, anisotropic magnification, and aberration corrections, local refinement, and reference-based motion correction. The final reconstructions of the iO/iC mixed dimer, 117,620 particles, and the iC dimer, 295,448 particles, were 2.92 Å and 2.75 Å, respectively.
Pi- and InsP8-supplemented sample
Supplementary Fig. 5 shows the cryo-EM workflow for this dataset. A total of 13,132 movies of Pi- and InsP8-supplemented XPR1 were collected using a Falcon 4i camera. Data processing was analogous to the InsP8-supplemented sample. The final reconstructions of the iO/iC mixed dimer, 116,612 particles, and the iC dimer, 338,543 particles, were 3.10 Å and 2.81 Å, respectively.
Model building, refinement, and validation
ModelAngelo was used to automatically build initial atomic models into the cryo-EM density maps58. Subsequently, the models were manually rebuilt using Coot to improve the fit within the density59. Atomic coordinates were refined against their corresponding density-modified maps using PHENIX real space refinement with geometric and Ramachandran restraints maintained throughout60. Model validation was performed using MolProbity61. Molecular graphics figures were prepared using UCSF Chimera62 or PyMOL. For electrostatic calculations, the APBS plugin in PyMOL was used63. To analyze and visualize tunnels in the XPR1 structures leading from Pi-binding sites to the surrounding solvent, MOLEonline was used64.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding author upon request. Cryo-EM density maps have been deposited to the Electron Microscopy Data Bank (EMDB) under the accession numbers EMD-47208 (apo XPR1), EMD-47209 (apo XPR1; rotated dimer); EMD-47210 (InsP6-supplemented XPR1), EMD-47207 (high Pi- and InsP6-supplemented XPR1); EMD-47211 (InsP8-supplemented XPR1; iO/iC state), EMD-47212 (InsP8-supplemented XPR1; iC state), EMD-47213 (Pi- and InsP8-supplemented XPR1; iO/iC state), EMD-47214 (Pi- and InsP8-supplemented XPR1; iC state). Atomic coordinates have been deposited in the Protein Data Bank (PDB) under IDs 9DVJ (apo XPR1), 9DVK (apo XPR1; rotated dimer); 9DVL (InsP6-supplemented XPR1), 9DVM (InsP8-supplemented XPR1; iO/iC state), 9DVN (InsP8-supplemented XPR1; iC state), 9DVO (Pi- and InsP8-supplemented XPR1; iO/iC state), 9DVP (Pi- and InsP8-supplemented XPR1; iC state). The following previously published PDB codes are referenced 8X5E, 8YFD, and 8YF4. All DNA constructs and cell lines described in this study are available upon request. The source data underlying Figs. 1a, b, 2d, 3e, f, h, 4g, and Supplementary Figs. 10b, 11a–d are provided as a Source Data file. Source data are provided in this paper.
References
Wagner, C. A. The basics of phosphate metabolism. Nephrol. Dial. Transplant. 39, 190–201 (2023).
Jennings, M. L. Role of transporters in regulating mammalian intracellular inorganic phosphate. Front. Pharmacol. 14, 1163442 (2023).
Hernando, N., Gagnon, K. & Lederer, E. Phosphate transport in epithelial and nonepithelial tissue. Physiol. Rev. 101, 1–35 (2021).
Giovannini, D., Touhami, J., Charnet, P., Sitbon, M. & Battini, J.-L. Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep. 3, 1866–1873 (2013).
Wild, R. et al. Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Science 352, 986–990 (2016).
Gerasimaite, R. et al. Inositol pyrophosphate specificity of the SPX-dependent polyphosphate polymerase VTC. ACS Chem. Biol. 12, 648–653 (2017).
Cheng, X. et al. (2024). Astrocytes modulate brain phosphate homeostasis via polarized distribution of phosphate uptake transporter PiT2 and exporter XPR1. Neuron 112, 3126–3142 (2024).
Li, X. et al. Control of XPR1-dependent cellular phosphate efflux by InsP8 is an exemplar for functionally-exclusive inositol pyrophosphate signaling. Proc. Natl. Acad. Sci. USA 117, 3568–3574 (2020).
Li, X. et al. Homeostatic coordination of cellular phosphate uptake and efflux requires an organelle-based receptor for the inositol pyrophosphate IP8. Cell Rep. 43, 114316 (2024).
Burns, D., Berlinguer-Palmini, R., & Werner, A. (2024). XPR1: a regulator of cellular phosphate homeostasis rather than a Pi exporter. Pflügers Arch. 476, 861–869 (2024).
Xu, C. et al. A phosphate-sensing organelle regulates phosphate and tissue homeostasis. Nature 617, 798–806 (2023).
Wilson, M. S., Jessen, H. J. & Saiardi, A. The inositol hexakisphosphate kinases IP6K1 and −2 regulate human cellular phosphate homeostasis, including XPR1-mediated phosphate export. J. Biol. Chem. 294, 11597–11608 (2019).
Moritoh, Y. et al. The enzymatic activity of inositol hexakisphosphate kinase controls circulating phosphate in mammals. Nat. Commun. 12, 4847 (2021).
Qiu, D. et al. Analysis of inositol phosphate metabolism by capillary electrophoresis electrospray ionization mass spectrometry. Nat. Commun. 11, 6035 (2020).
Qiu, D. et al. Capillary electrophoresis mass spectrometry identifies new isomers of inositol pyrophosphates in mammalian tissues. Chem. Sci. 14, 658–667 (2022).
Gu, C. et al. The significance of the bifunctional kinase/phosphatase activities of diphosphoinositol pentakisphosphate kinases (PPIP5Ks) for coupling inositol pyrophosphate cell signaling to cellular phosphate homeostasis*. J. Biol. Chem. 292, 4544–4555 (2017).
Chabert, V. et al. Inositol pyrophosphate dynamics reveals control of the yeast phosphate starvation program through 1,5-IP8 and the SPX domain of Pho81. ELife 12, RP87956 (2023).
Riemer, E. et al. ITPK1 is an InsP6/ADP phosphotransferase that controls phosphate signaling in Arabidopsis. Mol. Plant 14, 1864–1880 (2021).
Gu, C., Wilson, M. S. C., Jessen, H. J., Saiardi, A. & Shears, S. B. Inositol pyrophosphate profiling of two HCT116 cell lines uncovers variation in InsP8 levels. PLoS ONE 11, e0165286 (2016).
Xu, X. et al. The pathology of primary familial brain calcification: Implications for treatment. Neurosci. Bull. 39, 659–674 (2023).
Chen, S.-Y. et al. The genetics of primary familial brain calcification: A literature review. Int. J. Mol. Sci. 24, 10886 (2023).
Monfrini, E., Arienti, F., Rinchetti, P., Lotti, F. & Riboldi, G. M. Brain calcifications: Genetic, molecular, and clinical aspects. Int. J. Mol. Sci. 24, 8995 (2023).
Legati, A. et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet. 47, 579–581 (2015).
Yamada, M. et al. High frequency of calcification in basal ganglia on brain computed tomography images in Japanese older adults. Geriatrics Gerontol. Int. 13, 706–710 (2012).
Yin, B. W. T. et al. Monoclonal antibody MX35 detects the membrane transporter NaPi2b (SLC34A2) in human carcinomas. Cancer Immun. 8, 3 (2008).
Levan, K. et al. Immunohistochemical evaluation of epithelial ovarian carcinomas identifies three different expression patterns of the MX35 antigen, NaPi2b. Bmc Cancer 17, 303 (2017).
Bondeson, D. P. et al. Phosphate dysregulation via the XPR1–KIDINS220 protein complex is a therapeutic vulnerability in ovarian cancer. Nat. Cancer 3, 681–695 (2022).
Akasu‐Nagayoshi, Y. et al. PHOSPHATE exporter XPR1/SLC53A1 is required for the tumorigenicity of epithelial ovarian cancer. Cancer Sci. 113, 2034–2043 (2022).
Li, B., Zhu, D., Shi, H. & Zhang, X. Effect of charge on protein preferred orientation at the air–water interface in cryo-electron microscopy. J. Struct. Biol. 213, 107783 (2021).
Capolicchio, S., Wang, H., Thakor, D. T., Shears, S. B. & Jessen, H. J. Synthesis of densely phosphorylated Bis‐1,5‐diphospho‐myo‐inositol tetrakisphosphate and its enantiomer by bidirectional P‐anhydride formation. Angew. Chem. Int. Ed. 53, 9508–9511 (2014).
Schneider, S., Kühlbrandt, W. & Yildiz, Ö. Complementary structures of the yeast phosphate transporter Pho90 provide insights into its transport mechanism. Structure 32, 979–988 (2024).
Pipercevic, J. et al. Inositol pyrophosphates activate the vacuolar transport chaperone complex in yeast by disrupting a homotypic SPX domain interaction. Nat. Commun. 14, 2645 (2023).
Lemmon, M. A., Treutlein, H. R., Adams, P. D., Brünger, A. T. & Engelman, D. M. A dimerization motif for transmembrane alpha-helices. Nat. Struct. Biol. 1, 157–163 (1994).
Drew, D. & Boudker, O. Ion and lipid orchestration of secondary active transport. Nature 626, 963–974 (2024).
López-Sánchez, U. et al. Characterization of XPR1/SLC53A1 variants located outside of the SPX domain in patients with primary familial brain calcification. Sci. Rep. 9, 6776 (2019).
Tang, L.-O. et al. Biallelic XPR1 mutation associated with primary familial brain calcification presenting as paroxysmal kinesigenic dyskinesia with infantile convulsions. Brain Dev. 43, 331–336 (2021).
Guo, X. X. et al. Spectrum of SLC20A2, PDGFRB, PDGFB, and XPR1 mutations in a large cohort of patients with primary familial brain calcification. Hum. Mutat. 40, 392–403 (2019).
López-Sánchez, U. et al. Interplay between primary familial brain calcification-associated SLC20A2 and XPR1 phosphate transporters requires inositol polyphosphates for control of cellular phosphate homeostasis. J. Biol. Chem. 295, 9366–9378 (2020).
Kurz, L., Schmieder, P., Veiga, N. & Fiedler, D. One scaffold, two conformations: The ring-flip of the messenger insP8 occurs under cytosolic conditions. Biomolecules 13, 645 (2023).
Fraústo da Silva, J. J. R., and Williams, R. J. P. The Biological Chemistry of Elements: The Inorganic Chemistry of Life (1st Edition) (Oxford University Press, 1997).
Marcus, Y. Ionic radii in aqueous solutions. J. Solut. Chem. 12, 271–275 (1983).
Yoon, S., Chung, S. J. & Kim, Y. J. Primary familial brain calcification with XPR1 mutation presenting with cognitive dysfunction. J. Clin. Neurol. 20, 229–231 (2024).
Wege, S. & Poirier, Y. Expression of the mammalian Xenotropic Polytropic Virus Receptor 1 (XPR1) in tobacco leaves leads to phosphate export. FEBS Lett. 588, 482–489 (2014).
Yan, R. et al. (2024). Human XPR1 structures reveal phosphate export mechanism. Nature 633, 960–967 (2024).
Ma, D. et al. Structure and mechanism of a glutamate–GABA antiporter. Nature 483, 632–636 (2012).
Chi, X. et al. Structural insights into the gating mechanism of human SLC26A9 mediated by its C-terminal sequence. Cell Discov. 6, 55 (2020).
Gu, C., Li, X., Zong, G., Wang, H. & Shears, S. B. IP8: A quantitatively minor inositol pyrophosphate signaling molecule that punches above its weight. Adv. Biol. Regul. 91, 101002 (2024).
Trung, M. N., Furkert, D. & Fiedler, D. Versatile signaling mechanisms of inositol pyrophosphates. Curr. Opin. Chem. Biol. 70, 102177 (2022).
Sahu, S. et al. Nucleolar architecture is modulated by a small molecule, the inositol pyrophosphate 5-InsP7. Biomolecules 13, 153 (2023).
Lu, Y. et al. Structural basis for inositol pyrophosphate gating of the phosphate channel XPR1. Science 386, eadp3252 (2024).
Zhang, W. et al. Structural insights into the mechanism of phosphate recognition and transport by XPR1. Nat. Commun. 16, 18 (2025).
Yao, X.-P. et al. Analysis of gene expression and functional characterization of XPR1: a pathogenic gene for primary familial brain calcification. Cell Tissue Res. 370, 267–273 (2017).
Haykir, B., et al. The Ip6k1 and Ip6k2 kinases are critical for normal renal tubular function. J. Am. Soc. Nephrol. 35, 441–455 (2024).
Brinkman, E. K., Chen, T., Amendola, M. & van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168–e168 (2014).
Kawate, T. & Gouaux, E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681 (2006).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. methods 14, 290–296 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps. Nature 628, 450–457 (2024).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Real‐space refinement in PHENIX for cryo‐EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 98, 10037–10041 (2001).
Pravda, L. et al. MOLEonline: a web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 46, W368–W373 (2018).
Acknowledgements
We thank M. Alkareh, L. Cohen-Abeles, and J. Luo for their contributions to establishing the radioactive transport assay; R. Hite, D. Julius, C.D. Lima, S. Shuman, and members of the Hite and Diver laboratories for discussions; and M. J. de la Cruz of the Structural Biology core Facility at the Memorial Sloan Kettering Cancer Center (MSKCC) for help with data acquisition. M.M.D. is supported by the National Institutes of Health (NIH) National Cancer Institute (NCI) Cancer Center Support Grant P30-CA008748 and is a Josie Robertson Investigator. H.J.J. is supported by the Deutsche Forschungsgemeinschaft (DFG) under Germany’s excellence strategy (CIBSS, EXC-2189, Project ID 390939984). M.F.Y. receives funds from an NIH/National Institute of General Medical Sciences (NIGMS) T32 through the Weill Cornell Initiative for Maximizing Student Development (IMSD).
Author information
Authors and Affiliations
Contributions
Q.Z., M.F.Y., and M.M.D. designed and executed experiments, including protein expression and purification, cryo-EM data acquisition and image processing, atomic model building and refinement of XPR1 structures, as well as radioactive transport assays. N.J. and H.J.J. synthesized the InsP8. Q.Z., M.F.Y., M.M.D., and H.J.J. contributed to the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Daohua Jiang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Q., Yaggi, M.F., Jork, N. et al. Transport and InsP8 gating mechanisms of the human inorganic phosphate exporter XPR1. Nat Commun 16, 2770 (2025). https://doi.org/10.1038/s41467-025-58076-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58076-y
This article is cited by
-
The identification of XPR1 as a voltage- and phosphate-activated phosphate-permeable ion channel
Nature Communications (2025)
