
Abstract
Advancements in proton-exchange membrane water electrolyzer depend on developing oxygen evolution reaction electrocatalysts that synergize high activity with stability. Here, we introduce an approach aimed at elevating oxygen evolution reaction performance by enhancing the spatiotemporal coordination of oxygen radicals to promote efficient O-O coupling. A dense, single-atom configuration of oxygen radical donors within interconnected RuO2 nanocrystal framework is demonstrated. The stable oxygen radicals on gallium sites with adaptable Ga-O bonds are thermodynamically favorable to attract those from Ru sites, addressing dynamic adaptation challenges and boosting O-O coupling efficiency. The optimized catalyst achieves a low overpotential of 188 mV at 10 mA cm-2, operates robustly for 800 h at 100 mA cm-2 in acidic conditions, and shows a large current density of 3 A cm-2 at 1.788 V, with stable performance at 0.5 A cm-2 for 200 h, confirming its long-term viability in proton-exchange membrane water electrolyzer applications.
Similar content being viewed by others


Introduction
Proton-exchange membrane water electrolyzer (PEMWE) is pivotal for producing green hydrogen, essential for fostering a sustainable hydrogen economy1,2,3,4,5,6. However, the OER at the PEMWE anode, which impact on the commercial viability of PEMWE, influencing cost, energy efficiency, and operational lifespan, faces obstacles due to severe corrosive environments and sluggish reaction kinetics, challenging the development of active and long-lasting OER electrocatalysts7,8,9,10,11,12,13. Currently, noble metal nanomaterials, especially iridium and ruthenium, are the benchmarks in OER electrocatalysis, crucial for enhancing PEMWE’s commercial success by affecting cost, energy efficiency, and longevity14,15,16,17,18. Among these, ruthenium-based electrocatalysts offer a more economical solution, exhibiting superior intrinsic OER activity compared to more expensive iridium counterparts19,20,21. However, ruthenium’s propensity to form soluble RuO4 tetrahedral structures under acidic reaction conditions leads to rapid degradation22,23,24,25,26,27. Addressing this, there is a pressing need to engineer ruthenium-based electrocatalysts that maintain a delicate balance between activity and durability, thereby extending their practical application in industrial settings.
Effective regulating the OER reaction mechanism is essential to address these challenges, yet poses challenges in constructing electrocatalyst at atomic level. Two primary OER pathways have been established based on the premise that the OER cycle proceeds at a single active site: the adsorbate evolution mechanism (AEM) and the lattice oxygen oxidation mechanism (LOM)28,29,30,31,32,33. In AEM pathway, scaling relationships linking the binding energies of oxo-intermediates (e.g., *O, *OH, *OOH) result in a theoretical overpotential exceeding 370 mV34,35,36. Conversely, LOM typically exhibits a faster reaction rate due to the change in O-O coupling step by involvement of active lattice oxygen, which reduces the reaction energy barrier of rate determine step37,38,39. However, the release of lattice oxygen leads to oxygen vacancy creation, reducing the coordination number (CN) of metal cations (e.g., for active site of octahedral structure, its CN decreases from 6 to 4 upon O2 desorption) and promoting dissolution of metal species in the electrolyte, particularly in acidic environments. This results in rapid catalyst degradation40,41,42. Consequently, rational designing OER electrocatalysts that overcome the drawbacks of AEM and LOM is critical for enhancing acid OER performance.
It is crucial to emphasize that exploring another reaction pathways, such as the oxide path mechanism (OPM), could notablely enhance the catalytic activity and long-term stability of OER catalysts43,44,45. This mechanism facilitates the direct coupling of O-O radicals without a substantial decrease in coordination number (CN decrease greater than 2). Successfully achieving direct O-O radical coupling within the OPM requires fulfilling two stringent conditions. Firstly, from a spatial perspective, it is necessary for coexistence of adjacent active sites that display optimal oxygen radical adsorption energies, thereby thermodynamically favoring direct O-O coupling over the formation of -OOH intermediates. Secondly, from a temporal perspective, maintaining the synchronous presence of oxygen radicals at these adjacent active sites during the dynamic OER process is crucial, a factor that is often overlooked.
In the LOM, the adjacent lattice oxygen remains relatively stable, thereby enhancing the likelihood of O-O coupling following the deprotonation of -OH at metal active sites. In contrast, when oxygen radicals are generated by adjacent active sites, the dynamic evolution of these intermediates (*OH, *O, *OOH) crucially impacts the efficacy of O-O direct coupling. The synchronization of these kinetic processes is a significant hurdle to trigger the OPM pathway. We previously reported the Ru atom array featuring abundant homogeneous active site pairs and shortened the interatomic distances, which facilitate the simultaneous generation of adjacent oxygen radicals, thereby enabling O-O coupling46,47. However, the precise structural design requirements of the atomic arrays limit the choice of support materials and pose challenges in the broader application of OPM-like pathway. Therefore, the development of simpler and more versatile OPM-type electrocatalysts is of great significance.
Herein, we presented an advanced design principle for OPM based electrocatalyst, which employs a localized configuration of high-density single atom oxygen radical donors within interconnected RuO2 nanocrystal framework (iGaxRu1-xO2). The Ga single atoms, characterized by inherently low OER activity, predominantly generated oxygen radicals without progressing to -OOH or O2, which effectively alleviated the dynamic adaptation challenge of adjacent oxygen radical intermediates on temporal scale. Then, the oxygen radicals from Ga sites showed dynamically extensible Ga-O bonds, which thermodynamically more favorable to attract adjacent oxygen radicals on surrounding Ru sites, promoting O2 evolution through direct O-O coupling. Thus, the Ga-Ru heteroatom active site pairs effectively facilitate direct O-O radical coupling and enhances the OER process, realizing the transform of AEM pathway (observed in iRuO2) to OPM pathway (observed in iGa0.2Ru0.8O2 with high spatiotemporal coordination of oxygen radicals) (Fig. 1). The optimized catalyst demonstrated significant performance improvements, achieving low overpotentials of 188 mV and 219 mV at current densities of 10 mA cm−2 and 100 mA cm−2, respectively. Moreover, the optimized iGa0.2Ru0.8O2 catalyst maintained robust operation for 800 h at 100 mA cm−2 under acidic conditions, outperforming most reported OER catalysts. The optimized iGa0.2Ru0.8O2 based PEMWE achieved a high current density of 3 A cm−2 at 1.788 V, surpassing the U.S. Department of Energy’s 2025 target of 1.9 V at 3 A cm−2. Additionally, it exhibited long-term stability, maintaining performance at 0.5 A cm−2 for 200 h.

Schematic illustration AEM pathway of iRuO2 and OPM pathway of iGa0.2Ru0.8O2.
Results
Synthesis and characterization of iGaxRu1-xO2
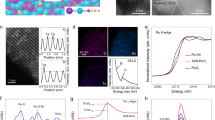
Gallium-doped interconnected ruthenium oxide nanocrystal (iGaxRu1-xO2, where x represents the molar ratios of Ga being 0.1, 0.2, and 0.3 for each sample) were synthesized using tannic acid and urea as the soft template (Supplementary Fig. 1). Scanning electron microscopy (SEM) images (Fig. 2a and Supplementary Fig. 2) revealed the nanosheet morphology of the iGaxRu1-xO2 and iRuO2 samples. The high-resolution transmission electron microscopy (TEM) (Fig. 2b and Supplementary Fig. 3) and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) images (Fig. 2c and Supplementary Fig. 4) displayed that iGa0.2Ru0.8O2 features interconnected nanocrystals (approximately 3 nm). Other iGaxRu1-xO2 and iRuO2 samples also exhibited two-dimensional (2D) nanosheet morphology with uniformly distributed elements (Supplementary Figs. 5–7). However, randomly dispersed isolated Ga0.2Ru0.8O2 nanoparticles were obtained without the addition of tannic acid, confirming the critical role of tannic acid in forming 2D interconnected nanostructures (Supplementary Fig. 8). The AC HAADF-STEM images (Fig. 2c, d) clearly showed lattice fringes corresponding to the crystallographic planes (110), (101), and (211). Energy dispersive spectroscopy (EDS) elemental mapping images demonstrated a homogeneous dispersion of Ga and Ru elements throughout the entire iGa0.2Ru0.8O2, with Ga exhibiting atomic-level dispersion (Fig. 2e). The elemental analysis based on EDS results revealed a Ga/Ru atom ratio of 23%, which is in agreement with the inputs ratio (1:4) and inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis (Supplementary Table 1). Atomic force microscopy (AFM) image showed that the iGa0.2Ru0.8O2 has a thickness of approximately 4 nm (Supplementary Fig. 9). The nitrogen adsorption-desorption isotherms for iGa0.2Ru0.8O2 exhibited a Type IV curve with H2-type hysteresis, indicative of a mesoporous structure with a pore size distribution about 3.5 nm (Supplementary Fig. 10).

a SEM, b TEM, c, d AC HAADF-STEM images of iGa0.2Ru0.8O2 catalyst. e EDS elemental mapping images and elemental analysis of Ga and Ru for iGa0.2Ru0.8O2 catalyst. f XRD patterns, and g high-resolution Ru 3d XPS of iRuO2, iGa0.2Ru0.8O2 and Com-RuO2 samples. FT-EXAFS spectra of h Ru K-edge for Ru foil, Com-RuO2, iRuO2 and iGa0.2Ru0.8O2, and i Ga K-edge for Ga foil, Ga2O3 and iGa0.2Ru0.8O2 samples. K, K shell. R, internal atomic radial distance. a.u. arbitrary units. Source data are provided as a Source Data file.
Powder X-ray diffraction (XRD) was utilized to elucidate the crystallographic structure of the synthesized samples. The iGa0.2Ru0.8O2 exhibited diffraction peaks congruent with those of both commercial RuO2 (Com-RuO2) and synthetic interconnected RuO2 (iRuO2), as depicted in Fig. 2f. Three prominent diffraction peaks corresponding to the (110), (101), and (211) planes of the rutile RuO2 phase (JCPDS no. 43-1027) were identified for iGaxRu1-xO2 (x = 0.1~0.3) (Supplementary Fig. 11)48,49,50. X-ray photoelectron spectroscopy (XPS) was employed to probe the surface chemical states of iGa0.2Ru0.8O2. In the high-resolution Ru 3d XPS spectrum of Com-RuO2, two characteristic peaks were identified at 281.65 eV and 285.82 eV, corresponding to the Ru 3d5/2 and Ru 3d3/2 of Ru4+, respectively (Fig. 2g and Supplementary Table 2). Additionally, there was a shift of ~0.12 eV toward lower binding energies for the Ru 3d5/2 peak in iGa0.2Ru0.8O2 compared to Com-RuO2, indicating a marginally reduced oxidation state of Ru in iGa0.2Ru0.8O2. For Ga 2p XPS spectrum of iGa0.2Ru0.8O2, two principal peaks were discerned at 1118.35 eV and 1145.20 eV, corresponding to the Ga 2p3/2 and Ga 2p1/2 of Ga3+, respectively (Supplementary Fig. 12)51,52,53. Moreover, the positive shift of 0.23 eV compared to those of Ga2O3 implies a strong electronic interaction between Ga dopants and surrounding Ru cations, which can be attributed to the difference in ion electronegativity between Ga3+ (1.579) and Ru4+ (1.848)54.
The X-ray absorption spectroscopy (XAS) was conducted on iGa0.2Ru0.8O2 and comparable samples to meticulously probe the electrocatalysts’ electronic structure and atomic coordination environments. The O K-edge soft XAS (sXAS) spectra of iGa0.2Ru0.8O2 provided insights into the excitation of oxygen core electrons from occupied O 1s states to unoccupied t2g and eg states of the Ru–O hybridized orbitals (Supplementary Fig. 13)55,56,57. Notably, the eg/t2g intensity ratio in iGa0.2Ru0.8O2 was elevated compared to iRuO2, indicating enhanced electron density within the eg orbitals relative to the t2g orbitals. Ru M-edge sXAS spectrum also showed a closed Ru4+ chemical valence for iGa0.2Ru0.8O2 (Supplementary Fig. 14). Furthermore, the Ru K-edge X-ray absorption near-edge structure (XANES) spectra of iGa0.2Ru0.8O2 and iRuO2 were positioned between those of Ru foil and Com-RuO2, suggesting an average valence state of Ru at approximately +3.9 (Supplementary Fig. 15). Additionally, the Ga K-edge XANES analysis revealed that the oxidation state of Ga in iGa0.2Ru0.8O2 (+3.2) is slightly higher than that of Ga3+ in Ga2O3, aligning with the XPS analyses (Supplementary Fig. 16). Figure 2h, i illustrated the Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra in R-space for the Ru and Ga K-edge of iGa0.2Ru0.8O2. The Ru K-edge FT-EXAFS spectrum of iGa0.2Ru0.8O2 is closely resemble those of Com-RuO2 and iRuO2. The FT-EXAFS fitting results suggest that the Ru-O bond length is maintained at 1.96 Å following Ga doping (Supplementary Fig. 17 and Table 3). The wavelet transformed EXAFS (WT-EXAFS) of iGa0.2Ru0.8O2 were observed at k ≈ 6 and 13 Å−1 exhibited intensity maximum, which can be attributed to the Ru-O and Ru-Ru (Ga) scattering paths, respectively (Supplementary Fig. 18). These feature peaks are weaker than those in Com-RuO2 samples, maybe due to the smaller nanoscale size of iGa0.2Ru0.8O2 and the mixed coordination shell of Ru-Ru/Ga45. In contrast, the FT-EXAFS spectrum at the Ga K-edge of iGa0.2Ru0.8O2 demonstrated coordination environment akin to that observed at the Ru K-edge of iGa0.2Ru0.8O2 (Fig. 2i and Supplementary Fig. 19). Furthermore, the Ga K-edge WT-EXAFS spectrum of iGa0.2Ru0.8O2 displayed a pronounced peak at approximately k ≈ 5 Å−1, indicative of Ga-O coordination, while no significant Ga-Ga interactions signatures (Supplementary Fig. 20). These findings emphatically confirm the predominance of a Ga-O monoatomic coordination in iGa0.2Ru0.8O210.
OER and PEMWE performance evaluation
The OER performance of various electrocatalysts was methodically evaluated within a conventional three-electrode configuration using 1 M HClO4 as the electrolyte. Geometrically normalized linear sweep voltammetry (LSV) curves revealed that iGa0.2Ru0.8O2 exhibited superior OER activity, evidenced by overpotentials of 188 mV and 219 mV at 10 mA cm−2 and 100 mA cm−2, respectively. These values surpassed those of iRuO2 (233 mV at 10 mA cm−2; 279 mV at 100 mA cm−2), iGa0.1Ru0.9O2 (199 mV at 10 mA cm−2; 237 mV at 100 mA cm−2), iGa0.3Ru0.7O2 (232 mV at 10 mA cm−2; 309 mV at 100 mA cm−2), Ga0.2Ru0.8O2 (299 mV at 10 mA cm−2; 429 mV at 100 mA cm−2), and Com-RuO2 (309 mV at 10 mA cm−2; 474 mV at 100 mA cm−2) (Fig. 3a and Supplementary Fig. 21). Notably, iGa0.2Ru0.8O2 demonstrated the lowest Tafel slope (33.9 mV dec-1) and the smallest charge transfer resistance (Rct = 4.3 Ω), indicating its enhanced charge transfer capabilities and improved OER kinetics compared to iRuO2 and Com-RuO2 (Fig. 3b, c).

a LSV curves for iRuO2, iGa0.1Ru0.9O2, iGa0.2Ru0.8O2, iGa0.3Ru0.7O2, Ga0.2Ru0.8O2 and Com-RuO2 catalysts (100% iR correction, where R was estimated to be 7.0 ± 0.7Ω). b Tafel slopes plots, c EIS plots for iRuO2, iGa0.2Ru0.8O2 and Com-RuO2 catalysts. d The chronopotentiometric curves without iR correction at 100 mA cm−2 for iGa0.2Ru0.8O2 catalysts [Insert: (I) photograph of the H-type electrochemical cell (50 mL volume in per compartment, in which the anode and cathode sides are separated by a Nafion 115 membrane, and the electrolyte was collected and replaced with fresh electrolyte every 50 h.) and (II) the chronopotentiometric curves at 100 mA cm−2 for Com-RuO2 catalysts]. e Polarization curves of the PEMWE using iGa0.2Ru0.8O2, iRuO2, Ga0.2Ru0.8O2 and Com-RuO2 as anode [Insert: photograph of the PEM electrolyzer device]. f Various overpotential contributions to the polarization curves of different catalysts at various current densities. g Chronopotentiometric curve of the PEMWE using optimized iGa0.2Ru0.8O2 catalyst operated at 0.5 A cm−2 [Insert: chronopotentiometric curve of Com-RuO2 at 0.5 A cm−2 in the PEMWE system]. Source data are provided as a Source Data file.
To comprehensively investigate the influence of Ga doping on OER performance, the double-layer capacitance (Cdl) and electrochemically active surface area (ECSA) were meticulously quantified to ascertain the intrinsic OER activity of each catalyst (Supplementary Figs. 22, 23). The specific activity at an applied potential of 1.46 V was found to follow the sequence: iGa0.2Ru0.8O2 (0.150 mA cm−2) > iRuO2 (0.008 mA cm−2) > Com-RuO2 (0.004 mA cm−2). The mass activity for iGa0.2Ru0.8O2 is 2302 mA mgRu−1, which is 38 times higher than iRuO2 (61 mA mgRu−1) and 256 times higher than Com-RuO2 (9 mA mgRu−1) (Supplementary Fig. 24). These results distinctly demonstrate the beneficial impact of Ga doping on enhancing the intrinsic catalytic activity for OER under acidic conditions.
The durability of iGa0.2Ru0.8O2 was investigated using the chronopotentiometry method under high currents of 100 mA cm−2 in an acid electrolyte of 1 M HClO4 (Fig. 3d). The Com-RuO2 suffered from a rapid activity decay within 20 h in the H-type electrolytic cell. Excitingly, the iGa0.2Ru0.8O2 electrocatalyst lasted for 800 h without significant potential variation. We quantified the ionic concentrations of Ru and Ga in the electrolyte using inductively coupled plasma-mass spectrometry (ICP-MS) during the OER stability tests (Supplementary Fig. 25). It can be observed that at high current densities of 100 mA cm−2, Ru and Ga dissolve rapidly in the initial period. However, the dissolution rate of iGa0.2Ru0.8O2 decreases over time, and even after 800 h, approximate 90% of the active Ru component is still retained, which demonstrates the stability of the catalyst. These results suggested that the atomic level Ga doping and the interconnected nanostructure can stabilize the rutile RuO2, which surpassing the performance of most previously reported Ru-based electrocatalysts (Supplementary Table 4).
A PEMWE was assembled using iGa0.2Ru0.8O2 as the anodic electrocatalyst. The optimized iGa0.2Ru0.8O2-based PEMWE achieved a current density of 3 A cm−2 at 1.788 V, surpassing both previously reported PEMWEs and the U.S. Department of Energy’s 2025 target of 3 A cm−2 at 1.9 V (Fig. 3e, Supplementary Fig. 26 and Table 5)25,58. Furthermore, the performance hierarchy of the PEMWE was observed as iGa0.2Ru0.8O2 > iRuO2 > Ga0.2Ru0.8O2 (isolated nanoparticle) > Com-RuO2, underscoring the enhancements conferred by the nanosheet structure and atomic Ga doping in improving PEMWE performance. To understand the enhanced performance of the iGa0.2Ru0.8O2-based PEMWE, an overvoltage subdivision was employed, distinguishing between kinetic (ηkin), ohmic (ηohm), and mass transport (ηmass) potential losses (Fig. 3f)59,60,61. For iGa0.2Ru0.8O2 and Ga0.2Ru0.8O2-based PEMWE, ηkin was found to play a dominant role in performance at lower current densities. Notably, the ηmass for Ga0.2Ru0.8O2-based PEMWE increased linearly with current density and exceed ηkin at a current density of 4.4 A cm−2, highlighting the significant impact of mass transfer limitations on PEMWE performance under conditions of high current density and substantial gas evolution. In contrast, the ηmass of iGa0.2Ru0.8O2-based PEMWE is always smaller than ηkin at the same current density. These findings underscore the advantages of the nanosheet morphology of iGa0.2Ru0.8O2 in enhancing mass transfer efficiency during demanding electrochemical conditions, thereby bolstering the performance of PEMWE. The stability of optimized iGa0.2Ru0.8O2-based PEMWE was further substantiated using the chronopotentiometry method at a current density of 0.5 A cm−2, with the system operating continuously for 200 h, thereby surpassing the durability metrics of most previously reported PEMWE (Fig. 3g and Supplementary Table 5). The chronopotentiometric stability test at 1 A cm−2 also affirmed the catalyst’s robust performance over a period of 100 h at higher current density (Supplementary Fig. 27). Additionally, the results of variable current stability tests within the current density range of 1–5 A cm−2 also showcased the nice stability of the PEM based on the optimized iGa0.2Ru0.8O2 electrocatalyst (Supplementary Fig. 28). These results firmly establish iGa0.2Ru0.8O2 as a highly effective acidic OER electrocatalyst, showcasing its potential for industrial-scale applications in PEMWE.
Mechanism analysis for enhanced OER performance
To explore the mechanism behind the enhanced performance of iGa0.2Ru0.8O2, extensive characterizations were performed to analyze the structural and chemical state transformations of iGa0.2Ru0.8O2 after OER (iGa0.2Ru0.8O2-spent). The TEM images of iGa0.2Ru0.8O2-spent showed the preservation of its 2D interconnected nanocrystalline structure (Fig. 4a). The EDS elemental mapping confirmed the uniform Ga doping within the crystal lattice of iGa0.2Ru0.8O2 after OER (Fig. 4b). The unchanged characteristic peak positions in the XRD pattern of iGa0.2Ru0.8O2-spent affirmed its robust crystalline structure under acidic OER conditions (Fig. 4c). These findings elucidated the robustness and structural integrity of iGa0.2Ru0.8O2. XPS analysis of the Ga 2p core level in iGa0.2Ru0.8O2-spent showed a shift towards lower binding energies by 0.95 eV, indicative of increased electron density at the Ga sites (Fig. 4d). Similarly, Ga K-edge XANES of iGa0.2Ru0.8O2-spent exhibited a significant shift towards lower energy in the absorption edge, compared to the fresh sample, corroborating an increase in electron density at Ga sites. The quantitative analysis of the Ga K-edge XANES reveals that the oxidation state of Ga is reduced to +2.8 after OER (Supplementary Fig. 29). The alteration in valence state is indicative of the involvement of Ga in the OER process. In contrast, the Ru 3d XPS spectra between iGa0.2Ru0.8O2-spent and the fresh sample remained consistent (Fig. 4e). Ru K-edge XANES quantitative analysis of iGa0.2Ru0.8O2-spent demonstrate that Ru oxidation state remains at +3.9. Ru M-edge sXAS analysis of iGa0.2Ru0.8O2-spent were consistent with the XPS results (Supplementary Figs. 30 and 31). Conversely, a distinct positive shift of 0.20 eV in the Ru 3d XPS spectra and a 0.43 eV positive shift in Ru M-edge sXAS spectra for iRuO2 demonstrated progressive oxidation of Ru, emphasizing the distinct behavior of the Ru species in iRuO2 (Fig. 4f and Supplementary Fig. 32). These comprehensive findings highlight Ga’s crucial role in stabilizing the valence state of Ru within iGa0.2Ru0.8O2 during the OER process, contributing to the overall robustness and efficacy of the material in harsh electrochemical environments.

a TEM image, b HAADF-STEM and corresponding EDS mapping images of iGa0.2Ru0.8O2 after the OER test. c XRD patterns, d Ga 2p, e Ru 3d XPS spectra for iGa0.2Ru0.8O2 before and after the OER test. f High-resolution Ru 3d XPS spectra of iRuO2 before and after the OER test. a.u. arbitrary units. Source data are provided as a Source Data file.
The local coordination environment in iGa0.2Ru0.8O2 during the OER was investigated using operando Ru K-edge and Ga K-edge FT-EXAFS measurements (Supplementary Fig. 33). The analysis showed that the applied potential increased from the open circuit potential (OCP) to 1.6 V, the average Ru–O bond length in iGa0.2Ru0.8O2 remained constant at 1.96 Å, while the corresponding coordination number (CN) slight decreased from 5.7 to 5.4 (Fig. 5a, c, d and Supplementary Table 6). The slight decrease in CN suggests that the desorption of reaction intermediates (e.g., *O2) likely occurs at Ru sites during the OER. In contrast, the average Ga–O bond length in iGa0.2Ru0.8O2 lengthened from 1.96 to 2.02 Å, and the corresponding CN increased from 5.5 to 6.2 (Fig. 5b–d, and Supplementary Table 7). The observed increasement in CN and bond length for Ga–O in iGa0.2Ru0.8O2 indicated a probability of Ga site participation in the OER process. The Ga sites are likely involved in the evolution of reaction intermediates (e.g., *O), and the elongation in bond length between Ga and these intermediates may enhance synergistic catalytic actions with neighboring active sites.

Operando FT-EXAFS results at different applied potentials for a Ru K-edge, and b Ga K-edge of iGa0.2Ru0.8O2 catalysts. Summary of c the Ga-O, Ru-O coordination number and d bond length of iGa0.2Ru0.8O2 at different applied potentials according to the quantitative fitting results. e Operando SR-FTIR spectra of iRuO2 and iGa0.2Ru0.8O2 in HClO4-H216O for different applied potentials. Operando DEMS results in 0.5 M H2SO4-H218O and 0.5 M H2SO4-H216O for f iRuO2 and g iGa0.2Ru0.8O2 catalysts. a.u. arbitrary units. Source data are provided as a Source Data file.
To further elucidate the OER pathways, isotopically labeled operando synchrotron radiation Fourier transform infrared (SR-FTIR) spectroscopy was conducted. In HClO4-H216O electrolyte, a prominent peak at 1031 cm−1 was detected in the SR-FTIR spectra of iRuO2, which can be attributed to *OOH, a typical reaction intermediate in the AEM pathway (Fig. 5e, left)40,62. However, distinctive absorption peak at 1137 cm−1 and 1107 cm−1 were observed in the operando SR-FTIR spectra of iGa0.2Ru0.8O2 when the potential reached the OER region (≥1.4 V) (Fig. 5e, right). In the isotopically labeled HClO4-H218O electrolyte, the operando SR-FTIR spectra of iGa0.2Ru0.8O2 were shifted to the main peak position in the OER region to 1099 cm−1 (Supplementary Fig. 34). When the electrolyte was replaced with HClO4-D2O, the main peak position of the operando SR-FTIR spectra for iGa0.2Ru0.8O2 in the OER region remained at approximate 1140 cm−1. These results confirm the generation of linearly bonded superoxol species (M-O-O) rather than oxyhydroxide species (M-O-O-H) during the OER process47,63,64,65. Therefore, it can be surmised that iGa0.2Ru0.8O2 electrocatalysts follow a reaction pathway which involvement of the oxygen radical direct coupling step that differs from the conventional AEM. Operando differential electrochemical mass spectroscopy (DEMS) experiment with isotope labeling was employed to elucidate the OER mechanisms for iGa0.2Ru0.8O2. Initially, multiple LSV cycles ranging from 1.2 to 1.6 V versus RHE were executed using iGa0.2Ru0.8O2 and iRuO2 in an H218O electrolyte. According to established literature, the emergence of 32O2 gas products indicated the direct coupling of oxygen radicals during the OER in an H218O medium46,47. Contrarily, only mass signals for 34O2 and 36O2 were detected from iRuO2, suggesting adherence to a typical AEM pathway (Fig. 5f and Supplementary Fig. 35). In stark contrast, the detection of 32O2 signals from iGa0.2Ru0.8O2 suggested direct coupling of 16O-16O between two neighboring active sites (Fig. 5g and Supplementary Fig. 36).
To differentiate these mechanisms, further LSV cycles were performed in H216O for the 18O-labeled iGa0.2Ru0.8O2 and iRuO2. In typical AEM, 32O2 and 34O2 are observed for the 18O-labeled electrocatalysts in H216O (Fig. 5f and Supplementary Fig. 37). Conversely, the detection of 36O2 signals suggested direct coupling of surface adsorbed 18O during an OPM-type OER process (Fig. 5g and Supplementary Fig. 38). The 18O-labeled iGa0.2Ru0.8O2 generated signal ratio of 36O2:34O2:32O2 is about 1:46:953. The occurrence of 36O2 signals in the OER process for iGa0.2Ru0.8O2 thus shows that this catalyst predominantly follows the OPM pathway, characterized by direct oxygen radical coupling between two adjacent active sites.
To elucidate the intrinsic effects of Ga doping on the OER performance of iGa0.2Ru0.8O2, comprehensive Density Functional Theory (DFT) calculations were conducted. The model for single-atom Ga doped RuO2 (110) was constructed (Supplementary Fig. 39). Figure 6a, b illustrated the OPM at the Ga-Ru and Ru-Ru dual active sites of iGa0.2Ru0.8O2, detailing six elementary reaction steps. The associated Gibbs free energy changes for these steps are depicted in Fig. 6c.

Structures for key intermediates of OPM reaction pathways for a Ga- Ru site and b Ru-Ru site of iGa0.2Ru0.8O2. c The Gibbs free energy (ΔG) diagrams of OPM Ga-Ru and OPM Ru-Ru reaction pathways for iGa0.2Ru0.8O2. TS stands for the transition state. Colors in the figure: light green balls are Ga; light blue balls are Ru; red balls are O; light pink balls are H. Source data are provided as a Source Data file.
Focusing on the Ga-Ru dual active sites, the initial reaction step involves the endothermic transfer of a proton from free water to form an OH* intermediate at the Ga site, with a Gibbs free energy increase of 0.23 eV (Step A0 → A1, detailed in Supplementary Table 6). This is followed by a proton-coupled electron transfer that generates an adsorbed oxygen radical (*O), transitioning from A1 to A2. A similar sequence of adsorption and proton-coupled electron transfer occurs at the neighboring Ru site, leading to the formation of two adjacent O radicals (Steps A2 → A3 → A4). These radicals then couple to form a bridged O2* complex (M-O-O-M), overcoming a kinetic barrier of 0.81 eV (Step A4 through the transition state TS1 to A5). This coupling step is exothermic, releasing 1.46 eV. The final step involves the desorption of O2* to release molecular O2, accompanied by a free energy decrease of 0.91 eV (Step A5 to A6).
Comparatively, the kinetic overpotential for Ru-Ru dual active sites is measured at 1.03 eV, which is 0.22 eV higher than that of the Ga-Ru sites. Additionally, the energy profiles for the AEM pathway were quantified for iGa0.2Ru0.8O2 (Supplementary Fig. 40), revealing that the kinetic overpotential for AEM is substantially higher at 1.38 eV, making it 0.57 eV more energy-intensive than the OPM pathway for the Ga-Ru dual sites. These findings substantiate that iGa0.2Ru0.8O2 preferentially follows the OPM pathway, facilitated by the synergistic catalysis at Ga and Ru sites. Therefore, it can be inferred that the Ga sites play a pivotal role in directing the reaction pathway from the AEM to OPM. This regulatory function underscores the critical influence of Ga doping in modulating the energetic landscape and kinetic barriers, thereby optimizing the catalytic efficiency of OER processes.
Discussion
In this work, we presented a strategy for the rational design acidic OER electrocatalysts based on the OPM pathway, employing a localized configuration of single atom oxygen radical donors within 2D interconnected rutile RuO2 nanocrystal framework. The atomic Ga within RuO2 lattice frameworks addressed the dynamic adaptation of adjacent oxygen radical intermediates, facilitating direct O-O radical coupling by enhancing the spatiotemporal coordination of adjacent oxygen radicals. Consequently, the optimized catalyst delivered significant OER performance enhancements, achieving low overpotentials of 188 mV and 219 mV at current densities of 10 mA cm−2 and 100 mA cm−2, respectively. Moreover, it also maintained robust operation for 800 h at 100 mA cm−2 in acidic conditions, outperforming most reported advanced OER catalysts. In practical applications, the optimized catalyst based PEMWE achieved an impressive current density of 3 A cm−2 at 1.788 V, maintaining stable performance at 0.5 A cm−2 for 200 h, demonstrating its long-term viability. Therefore, this work provides a design principle for the rational design OPM based electrocatalyst to overcome the activity-stability trade-off for proton exchange membrane water electrolysis.
Methods
Chemical
Tannic acid was purchased Macklin Biochemical Co., Ltd. Ruthenium trichloride (RuCl3), Gallium nitrate hydrate (Ga(NO3)3·xH2O), Urea (CO(NH2)2) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Water-18O (H218O, RG, 98 atom % 18O) was purchased from Adamas. Perchloric acid (HClO4, 70–72%) was purchased from Greagent. Sulfuric acid (H2SO4, AR, 95.0–98.0%) was purchased from Sinopharm Chemical Reagent Co., Ltd. Nafion perfluorinated resin solution (5 wt%) and commercial ruthenium oxide (Com-RuO2) were purchased from Sigma-Aldrich. Carbon paper was purchased from Suzhou Sinero Technology Co., Ltd. The deionized (DI) water (18 MΩ cm−1) was prepared by passing water through an ultrapure purification system.
Materials synthesis
Synthesis of iRuO2, iGa0.1Ru0.9O2, iGa0.2Ru0.8O2, iGa0.3Ru0.7O2 and Ga0.2Ru0.8O2 samples. Firstly, 250 μL of 200 mg mL−1 of RuCl3 solution, 300 μL of 50 mg mL−1 of Ga(NO3)3-xH2O solution, 1 g of urea and 0.2 g of tannic acid were added to a quartz tube and stirred thoroughly. After continuous heating at 150 °C for 15 min, the resulting solid mixture was heated to 450 °C in a muffle furnace at a rate of 3 °C min−1 and kept at this temperature for 6 h to obtain the iGa0.2Ru0.8O2 sample. The iRuO2, iGa0.1Ru0.9O2 and iGa0.3Ru0.7O2 samples were prepared according to the same method, except that the Ga(NO3)3-xH2O input was changed to 0, 130 and 500 μL, respectively. Ga0.2Ru0.8O2 sample were synthesized by the same method as iGa0.2Ru0.8O2 except that no tannic acid was added.
Materials characterization
Scanning electron microscopy (SEM) images were obtained using field emission scanning electron microscope (Hitachi SU8010, Japan). Transmission electron microscopy (TEM) images, HAADF-STEM and EDS elemental mapping analysis were acquired using Talos F200S operated at 200 kV. AC HAADF-STEM characterization was performed on JEOL ARM200F and Themis ETEM installed with a CEOS probe corrector. Powder XRD was obtained using Bruker D8 Advance. The nanoporous structures were analyzed by nitrogen adsorption−desorption isotherms using the Autosorb IQ instrument (Quantachrome, USA). Before measurements, the catalysts were degassed in a vacuum at 180 °C for 10 h. Nitrogen adsorption-desorption isotherm data were then measured and collected at 77 K. The specific surface areas were calculated by Brunauer-EmmettTeller (BET) method and the pore size distribution was calculated by using the Barrett-Joyner-Halenda (BJH) model. The chemical state of samples was examined using X-ray photoelectron spectroscopy (XPS) with Thermo ESCALAB 250XI device. All the XPS spectra were calibrated to the C 1 s line at 284.8 eV and analyzed using the Gauss-Lorentz model in Advantage software. The metal content was detected with ICP-OES (Prodigy plus) and EDS elemental analysis (Talos F200S). Synchrotron sXAS spectra of the sample were collected on beamline BL02B02 at the Shanghai Synchrotron Radiation Facility (SSRF), which included Ru M-edge and O K-edge XAS information. Before collecting the XAS data, the chamber was evacuated for approximately 120 min.
XAFS measurements and data analysis
XAS measurements were conducted at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility, operating with a 3.5 GeV storage ring and injection currents of 220 mA. A Si (111) double-crystal monochromator was employed for the measurements. The Ru K-edge (22,117 eV) and Ga K-edge (10,367 eV) spectra of ex-situ samples were recorded in transmission mode, with the absorption edge positions calibrated using corresponding elemental foils. For operando XAS measurements, Ru K-edge and Ga K-edge spectra were acquired using catalyst-coated carbon paper within an operando XAS electrochemical cell. The spectra were collected in fluorescence mode with a Lytle detector, requiring over 40 min to obtain high-quality data at each applied potential. To investigate the evolution of Ru and Ga sites under oxygen evolution reaction (OER) operating conditions, XAFS spectra were recorded across a series of applied potentials. At each potential, a stabilization period of 10 min was applied prior to data collection. Multiple pre-scans were performed to confirm structural stabilization and spectral consistency before acquiring high-quality XAFS spectra for subsequent analysis. The collected XAFS data were processed following standard procedures using the ATHENA module of the IFEFFIT software package66. Quantitative curve fitting was conducted in R-space and Fourier-transform k-space ranges using the ARTEMIS module of IFEFFIT. During the fitting process, the overall amplitude reduction factor (S02) was fixed at the optimal value determined from foil data fitting. Structural parameters, including coordination number (CN), interatomic distance (R), Debye-Waller factor (σ2), and edge-energy shift (ΔE0), were allowed to adjust to achieve the best fit.
Electrochemical measurements
The OER performance was evaluated using a standard three-electrode system equipped with an electrochemistry workstation (CS310M) in an O2-saturated 1 M HClO4 electrolyte (pH = 0.08 ± 0.01), which was freshly prepared by diluting ~8.6 mL of HClO4 into 100 mL of DI water. A 3 mm glassy carbon electrode, an Ag/AgCl electrode and a Pt net serving as the work, reference and counter electrode, respectively. To prepare the electrocatalysts ink, 5.0 mg of the catalyst was dispersed in a mixture of 1 mL deionized water and ethanol (in a 1:3 volume ratio) containing 50 μL Nafion solution as a binder. After thorough ultrasonication for at least 30 min, the homogeneous ink was carefully dropped onto a glassy carbon electrode (0.07 cm2) allowed to dry completely at room temperature. The mass loadings of the electrocatalysts were optimized approximately 0.2 mg cm−2 on glassy carbon (approximately 4.0 mgRu cm−2 on carbon paper for stability test). LSV was conducted with a scanning rate of 10 mV s−1 within the potential range of 1.20–1.80 V versus the Reversible Hydrogen Electrode (RHE). Stability tests were performed in an H-type electrochemical cell (approximately 50 mL volume in per compartment, in which the anode and cathode sides are separated by a Nafion 115 membrane, Nafion 115 was sequentially treated with H2O2 and 1 M H2SO4 at 80 °C for 1 h). The electrolyte was collected and replaced with fresh electrolyte every 50 h. The Ag/AgCl reference electrode was calibrated by measuring the RHE potential under electrolytes are saturated with a high purity H2 atmosphere using a Pt wire as the working electrode. Polarization curve was measured with the scan rate of 1 mV s−1, and the potential at which the current becoming zero is taken to be the thermodynamic potential (vs. AgCl) for the hydrogen electrode reaction. The thermodynamic potential of hydrogen electrode reaction was finally obtained by averaging 2 sets of data (0.197 ± 0.001 V). Therefore, in 1 M HClO4 electrolyte, the relationship between the RHE potential (ERHE) and the Ag/AgCl potential electrode (EAg/AgCl) was established as ERHE = EAg/AgCl + 0.197 V (Supplementary Fig. 41). The overpotential (η) was determined as η = ERHE - 1.23 V (the measurements were performed once). Potential was precisely referenced using the RHE with iR compensation, where i represents the measured current and R signifies the uncompensated resistance (100% iR correction, where R was determined to be 7.0 ± 0.7 Ω). The non-iR corrected LSV curves were provided in Supplementary Fig. 42.
Calculation of the Tafel slopes
The Tafel slope is calculated by the Tafel equation:
where the slope, b, is further defined as
and the constant a is given by
In this study, η and a represents the overpotential and the Tafel constant, respectively. b corresponds to the Tafel slope, the variable i represents the current, T reprents the temperature in kelvin (K), R denotes the ideal gas constant, α denotes the transfer coefficient, and F represents the Faraday constant. Moreover, nb indicates the number of electrons transferred before the rate-determining step, υ is the number of rate-determining steps in the overall reaction, and β (typically taken as 0.5) is the symmetry factor47.
Electrochemically active surface area (ECSA)
The ECSA value was calculated from according to the following equation:
Here, the double-layer capacitance (Cdl) was determined from the scan-rate-dependent CVs measurements at various scan rates (20, 40, 60, 80 and 100 mV s−1) over the approximate potential window of 1.10–1.20 V versus RHE in 1 M HClO4. The specific capacitance (Cs), which represents the capacitance per unit area under identical electrolyte conditions, a typically reported value within the range of 0.015–0.110 mF cm−2 in acidic solution. In this work, a representative value of 0.035 mF cm−2 was chosen67.
Calculation of specific area activity
The specific area activity was determined by normalizing the ECSA for different catalysts. The specific current density per ECSA (js) was computed from:
Where i is the measured current.
Calculation of mass activity
The mass activity of electrocatalysts was evaluated by the following equation:
Where i is the measured current, mRu is the Ru mass.
Operando SR-FTIR measurement
Operando SR-FTIR measurement was conducted at beamline BL01B of the National Synchrotron Radiation Facility in Hefei, China, using a custom-designed top-plate cell from the Liu research group68,69,70. The as-prepared electrocatalyst ink was drop onto a carbon paper electrode and subsequently air-dried. We conducted isotopically labeled operando SR-FTIR spectroscopy using H218O, D2O, and H216O based electrolytes, respectively. To minimize loss of infrared signal, only a thin layer of aqueous electrolyte was thoughtfully coated onto the carbon paper working electrode. The SR-FTIR data acquisition was conducted in a reflection mode, ensuring the capture of high-quality spectra. With a spectral resolution of 4 cm−1, precise spectral analysis was achievable. Background spectra of the working electrode were recorded under open circuit potential conditions before collecting spectra at specified voltages for adsorbed species analysis. The recorded spectra were processed and analyzed by the OPUS software.
Operando DEMS measurement
The operando DEMS measurement consists of two main steps.
The first step is labeling 18O stage. The catalyst was drop-coated onto an Au film sputtered on a porous polytetrafluoroethylene film to prepare a working electrode. This electrode was integrated into a three-electrode system within a custom-built cell: a platinum wire served as the counter electrode, Ag/AgCl as the reference electrode, and N2 (or Ar) saturated 0.5 M H2SO4-H218O as the electrolyte. Multiple LSV tests were conducted at RHE 1.20–1.60 V, while mass spectrometry recorded stable signals corresponding to gas products 32O2 (16O16O), 34O2 (16O18O) and 36O2 (18O18O).
The second step is probing 18O stage. The catalyst was removed from the H218O environment and thoroughly rinsed with deionized water and dry to eliminate residual H218O solution on the catalysts surface. The 18O-labeled catalyst-loaded work electrode was reintegrated into the same three-electrode system for DEMS measurements. The setup included a platinum counter electrode, Ag/AgCl reference electrode, and N2 (or Ar) saturated 0.5 M H2SO4-H216O as the electrolyte. Then multiple LSV tests were performed in the RHE 1.20-1.60 V, while the stable signals of the gas products 32O2 (16O16O), 34O2 (16O18O) and 36O2 (18O18O) were collected by mass spectrometry.
PEMWE measurements
The membrane electrode assembly was prepared using Hyproof HPM-2080X membrane (Membrane thickness-80 ± 2.5 μm; Proton conductivity-81 mS cm−1; Reinforcement material-Expended Polytetrafluoroethylene) by the catalyst-coated membrane method with an effective geometric area of 1 cm × 1 cm (1 cm2). The commercial Pt/C (60 wt%) was used as the cathode for the hydrogen evolution reaction, and the optimized electrocatalysts was used as the anode for the oxygen evolution reaction. The membrane with optimized electrocatalysts coated were hot pressed to establish the MEA under 140 °C with a pressure of 1 tonne for 1 min. Pt coated Ti mesh were used as gas diffusion layer. During the test, a flow of water preheated to 80 °C was supplied to the anode side. LSV polarization curves were collected by electrochemical workstation (CS310M) with power amplifier at a scan rate of 5 mV s−1, typically between 1.2 and 2.2 V.
Theoretical calculation details
The Vienna ab initio Simulation Program (VASP) was used for the density functional theory (DFT) calculations71,72. In these calculations, the generalized gradient approximation (GGA) in the Perdew-Burke-Ernzerhof (PBE) form was employed, with a planewave basis set cutoff energy of 500 eV to ensure accurate results73. A 3 × 3 × 1 Monkhorst-Pack grid was utilized for Brillouin zone sampling during structure optimization74. The ion-electron interactions were described via the projector augmented wave (PAW) method75. The convergence criteria for structure optimization were set to ensure that the maximum force on any atom remain below 0.02 eV/Ǻ and that the total energy change be less than 1 × 10−5 eV. The DFT-D3 semiempirical correction was described via Grimme’s scheme method76. Given that the reaction occurs in an aqueous environment, the implicit solvent model (VASPsol) was adopted77. The Gibbs free energy change (ΔG) for each elemental step is defined by the equation78,79:
in this study, ΔE represents the adsorption energy as calculated by density functional theory. Moreover, other parameters such as ΔGpH represents the free energy correction of the pH, U represents the applied electrode potential, T represents the temperature, and ΔS represents the entropy change. Additionally, ΔZPE denotes the zero-point energy correction80.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information files. Source data are provided with this paper.
References
Li, A. et al. Atomically dispersed hexavalent iridium oxide from MnO2 reduction for oxygen evolution catalysis. Science. 384, 666–670 (2024).
Turner, J. A. Sustainable hydrogen production. Science. 305, 972–974 (2004).
Chong, L. et al. La- and mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science. 380, 609–616 (2023).
Hao, S. et al. Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotechnol. 16, 1371–1377 (2021).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2022).
Xue, Y. et al. Stabilizing ruthenium dioxide with cation-anchored sulfate for durable oxygen evolution in proton-exchange membrane water electrolyzers. Nat. Commun. 14, 8093 (2023).
Shi, Q., Zhu, C., Du, D. & Lin, Y. Robust noble metal-based electrocatalysts for oxygen evolution reaction. Chem. Soc. Rev. 48, 3181–3192 (2019).
Li, L., Wang, P., Shao, Q. & Huang, X. Recent progress in advanced electrocatalyst design for acidic oxygen evolution reaction. Adv. Mater. 33, 2004243 (2021).
An, L. et al. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 33, 2006328 (2021).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule. 7, 558–573 (2023).
Xu, L. et al. Amorphous mnruox containing microcrystalline for enhanced acidic oxygen-evolution activity and stability. Angew. Chem., Int. Ed. 63, e202405641 (2024).
Chen, D. et al. Bicontinuous RuO2 nanoreactors for acidic water oxidation. Nat. Commun. 15, 3928 (2024).
Huo, L. et al. Amorphous MnO2 lamellae encapsulated covalent triazine polymer-derived multi-heteroatoms-doped carbon for orr/oer bifunctional electrocatalysis. Adv. Mater. 36, 2312868 (2024).
Oh, A. et al. Topotactic transformations in an icosahedral nanocrystal to form efficient water-splitting catalysts. Adv. Mater. 31, 1805546 (2019).
Zhu, W. et al. Stable and oxidative charged Ru enhance the acidic oxygen evolution reaction activity in two-dimensional ruthenium-iridium oxide. Nat. Commun. 14, 5365 (2023).
Wen, Y. et al. Stabilizing highly active ru sites by suppressing lattice oxygen participation in acidic water oxidation. J. Am. Chem. Soc. 143, 6482–6490 (2021).
Yao, L. et al. Sub-2 nm irrunimoco high-entropy alloy with iridium-rich medium-entropy oxide shell to boost acidic oxygen evolution. Adv. Mater. 36, 2314049 (2024).
Chen, S. et al. Reconstructed ir‒o‒mo species with strong brønsted acidity for acidic water oxidation. Nat. Commun. 14, 4127 (2023).
Lu, S.-Y. et al. Synthetic tuning stabilizes a high-valence Ru single site for efficient electrolysis. Nat. Synth 3, 576–585 (2024).
Zhao, G. et al. Metallic Ru─Ru interaction in ruthenium oxide enabling durable proton exchange membrane water electrolysis. Adv. Mater. 36, 2404213 (2024).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304–313 (2019).
Klyukin, K., Zagalskaya, A. & Alexandrov, V. Role of dissolution intermediates in promoting oxygen evolution reaction at RuO2(110) surface. J. Phys. Chem. C. 123, 22151–22157 (2019).
Cherevko, S. et al. Dissolution of noble metals during oxygen evolution in acidic media. Chemcatchem. 6, 2219–2223 (2014).
Hodnik, N. et al. New insights into corrosion of ruthenium and ruthenium oxide nanoparticles in acidic media. J. Phys. Chem. C. 119, 10140–10147 (2015).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance pem electrolyzers. Nat. Commun. 14, 843 (2023).
Spöri, C., Kwan, J. T. H., Bonakdarpour, A., Wilkinson, D. P. & Strasser, P. The stability challenges of oxygen evolving catalysts: Towards a common fundamental understanding and mitigation of catalyst degradation. Angew. Chem. Int. Ed. 56, 5994–6021 (2017).
Hao, S. et al. Dopants fixation of ruthenium for boosting acidic oxygen evolution stability and activity. Nat. Commun. 11, 5368 (2020).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: From mechanistic understanding to reactor design. Joule. 5, 1704–1731 (2021).
Zhong, H. et al. Fundamental understanding of structural reconstruction behaviors in oxygen evolution reaction electrocatalysts. Adv. Energy Mater. 13, 2301391 (2023).
Zheng, X. et al. Deciphering cationic and anionic overoxidation: Key insights into the intrinsic structural degradation of catalysts. Adv. Energy Mater. 14, 2401227 (2024).
Wang, Q. et al. Long-term stability challenges and opportunities in acidic oxygen evolution electrocatalysis. Angew. Chem., Int. Ed. 62, e202216645 (2023).
Zhang, Y., Zhu, X., Zhang, G., Shi, P. & Wang, A.-L. Rational catalyst design for oxygen evolution under acidic conditions: Strategies toward enhanced electrocatalytic performance. J. Mater. Chem. A. 9, 5890–5914 (2021).
Lin, Y., Dong, Y., Wang, X. & Chen, L. Electrocatalysts for the oxygen evolution reaction in acidic media. Adv. Mater. 35, 2210565 (2023).
Huang, Z.-F. et al. Strategies to break the scaling relation toward enhanced oxygen electrocatalysis. Matter. 1, 1494–1518 (2019).
Rossmeisl, J., Qu, Z. W., Zhu, H., Kroes, G. J. & Nørskov, J. K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83–89 (2007).
Gao, G. et al. Recent advances in Ru/Ir-based electrocatalysts for acidic oxygen evolution reaction. Appl. Catal. B Environ. 343, 123584 (2024).
Yao, N. et al. Atomically dispersed ru oxide catalyst with lattice oxygen participation for efficient acidic water oxidation. Chem. 9, 1882–1896 (2023).
Huang, R. et al. Constructing the oxygen diffusion paths for promoting the stability of acidic water oxidation catalysts. Chem Catal. 3, 100667 (2023).
Wang, Y. et al. Unraveling oxygen vacancy site mechanism of rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 14, 1412 (2023).
Su, H. et al. Tensile straining of iridium sites in manganese oxides for proton-exchange membrane water electrolysers. Nat. Commun. 15, 95 (2024).
Du, H. et al. A review of activating lattice oxygen of metal oxides for catalytic reactions: Reaction mechanisms, modulation strategies of activity and their practical applications. Appl. Catal. A Gen. 664, 119348 (2023).
Shi, Z. et al. Confined ir single sites with triggered lattice oxygen redox: Toward boosted and sustained water oxidation catalysis. Joule. 5, 2164–2176 (2021).
Zhu, W. et al. Direct dioxygen radical coupling driven by octahedral ruthenium–oxygen–cobalt collaborative coordination for acidic oxygen evolution reaction. J. Am. Chem. Soc. 145, 17995–18006 (2023).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659–23669 (2023).
Zhang, D. et al. Construction of Zn-doped RuO2 nanowires for efficient and stable water oxidation in acidic media. Nat. Commun. 14, 2517 (2023).
Chang, J. et al. Oxygen radical coupling on short-range ordered Ru atom arrays enables exceptional activity and stability for acidic water oxidation. J. Am. Chem. Soc. 146, 12958–12968 (2024).
Lin, C. et al. In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012–1023 (2021).
Su, J. et al. Assembling ultrasmall copper-doped ruthenium oxide nanocrystals into hollow porous polyhedra: Highly robust electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 30, 1801351 (2018).
Jin, H. et al. Dynamic rhenium dopant boosts ruthenium oxide for durable oxygen evolution. Nat. Commun. 14, 354 (2023).
Ping, X. et al. Locking the lattice oxygen in RuO2 to stabilize highly active ru sites in acidic water oxidation. Nat. Commun. 15, 2501 (2024).
He, J., Ye, Y., Zhang, D., Yao, K. & Zhou, M. Visualized gallium/lyticase-integrated antifungal strategy for fungal keratitis treatment. Adv. Mater. 34, 2206437 (2022).
Sun, G. et al. Metastable gallium hydride mediates propane dehydrogenation on H2 co-feeding. Nat. Chem. 16, 575–583 (2024).
Zhang, H. et al. An integrated self-healing anode assembled via dynamic encapsulation of liquid metal with a 3d ti3c2tx network for enhanced lithium storage. Energy Environ. Sci. 15, 5240–5250 (2022).
Li, K. & Xue, D. Estimation of electronegativity values of elements in different valence states. J. Phys. Chem. A. 110, 11332–11337 (2006).
Frati, F., Hunault, M. O. J. Y. & de Groot, F. M. F. Oxygen k-edge x-ray absorption spectra. Chem. Rev. 120, 4056–4110 (2020).
Miao, X. et al. Quadruple perovskite ruthenate as a highly efficient catalyst for acidic water oxidation. Nat. Commun. 10, 3809 (2019).
Liu, Y. et al. Electrosynthesis of chlorine from seawater-like solution through single-atom catalysts. Nat. Commun. 14, 2475 (2023).
Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the Ir/Nb2O5−x catalyst/support interfaces. Angew. Chem., Int. Ed. 61, e202212341 (2022).
Hyun Oh, J. et al. Self-supported electrodes to enhance mass transfer for high-performance anion exchange membrane water electrolyzer. Chem. Eng. J. 460, 141727 (2023).
Tao, L. et al. Mass-efficient catalyst layer of hierarchical sub-nanosheets on nanowire for practical proton exchange membrane electrolyzer. Joule. 8, 450–460 (2024).
Wan, L., Xu, Z., Xu, Q., Wang, P. & Wang, B. Overall design of novel 3d-ordered mea with drastically enhanced mass transport for alkaline electrolyzers. Energy Environ. Sci. 15, 1882–1892 (2022).
Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
Wang, B. et al. In-situ structural evolution of multi-site alloy electrocatalyst to manipulate intermediate for enhanced water oxidation reaction. Energy Environ. Sci. 13, 2200–2208 (2020).
Lu, X., Faguy, P. W. & Liu, M. In situ potential-dependent ftir emission spectroscopy: A novel probe for high temperature fuel cell interfaces. J. Electrochem. Soc. 149, A1293 (2002).
Vivek, J. P., Berry, N. G., Zou, J., Nichols, R. J. & Hardwick, L. J. In situ surface-enhanced infrared spectroscopy to identify oxygen reduction products in nonaqueous metal–oxygen batteries. J. Phys. Chem. C. 121, 19657–19667 (2017).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541 (2005).
Shen, B. et al. Facile fabrication of sensing electrode based on cofe-mofs/mxene for ultrasensitive detection of picomolar chloramphenicol. Talanta. 286, 127552 (2025).
Cheng, W. et al. Lattice-strained metal–organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy. 4, 115–122 (2019).
Su, H. et al. Dynamic evolution of solid–liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 142, 12306–12313 (2020).
Zhang, X. et al. Quick evolution of edge-shared metal-oxygen octahedrons for boosting acidic water oxidation. Nano Energy. 102, 107680 (2022).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 16, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 18, 3865–3868 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B. 13, 5188–5192 (1976).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B. 50, 17953–17979 (1994).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J.Chem. Phys. 132, 154104 (2010).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Rossmeisl, J., Logadottir, A. & Nørskov, J. K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 319, 178–184 (2005).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Yao, S., Lin, J., Yi, K., Liu, W. & Wang, M. Cu-modified invo4 photocatalysts for enhanced N2 fixation using chemical reagents and electroplating sludge as the cu source. Chem. Commun. 60, 1790–1793 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52225204, 52173233, 52272289 and 52402231), the Natural Science Foundation of Shanghai (23ZR1479200), the Innovation Program of Shanghai Municipal Education Commission (2021-01-07-00-03-E00109), the Natural Science Foundation of Shanghai (23ZR1479200), the Shanghai Scientific and Technological Innovation Project (24520712800), “Shuguang Program” supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (20SG33), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, the Fundamental Research Funds for the Central Universities (2232024Y-01) and the DHU Distinguished Young Professor Program (LZA2022001). We thank beamline BL01B of the National Synchrotron Radiation Facility in Hefei and Professor Qinghua Liu from the University of Science and Technology of China for his helpful suggestions with SR-FTIR measurement. The Synchrotron XAFS and sXAS experiments in this work were supported by BL14W, BL02B02, and User Experiment Assist System of Shanghai Synchrotron Radiation Facility (SSRF).
Author information
Authors and Affiliations
Contributions
H.W. and C.L. contributed equally to this work. H.W., X.L., and W.L. conceived the project design. H.W. prepared samples, analyzed the experimental data, and wrote the draft. L.T., J.S., X.W., B.M., X.P., H.Z., Y.Z. and C.L. assisted with PEMWE, SR-FTIR, AC HAADF-STEM, EXAFS, DEMS measurements, analysis and DFT calculation analysis. Y.S., H.U., Q.X and W.J. participated in the scientific discussion and revised the manuscript. H.W. and C.L. wrote and revised the manuscript with help from all authors. X.L. and W.L. supervised the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Haeseong Jang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, H., Lin, C., Tan, L. et al. Atomic Ga triggers spatiotemporal coordination of oxygen radicals for efficient water oxidation on crystalline RuO2. Nat Commun 16, 3976 (2025). https://doi.org/10.1038/s41467-025-58346-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58346-9
This article is cited by
-
Pursuing spatiotemporal coordination in electrocatalysis
Nature Reviews Chemistry (2025)
-
In situ infrared spectroscopy and sum frequency generation spectroscopy in energy catalysis: fundamentals, challenges, and perspectives
Discover Catalysis (2025)
