
Abstract
Whether and how regulatory events at the translation stage shape the cellular and metabolic features of thermogenic adipocytes is hardly understood. In this study, we report two hitherto unidentified cross-talk pathways between metabolic and translational regulation in beige adipocytes. By analysing temporal profiles of translation activity and protein level changes during precursor-to-beige differentiation, we found selective translational down-regulation of OXPHOS component-coding mRNAs. The down-regulation restricted to Complexes I, III, IV, and V, is coordinated with enhanced translation of TCA cycle genes, engendering distinct stoichiometry of OXPHOS and TCA cycle components and altering the related metabolic activities in mitochondria of thermogenic adipocytes. Our high-resolution description of ribosome positioning unveiled potentiated ribosome pausing at glutamate codons. The increased stalling is attributable to remodelled glutamate metabolism that decreases glutamates for tRNA charging during pan-adipocyte differentiation. The ribosome pauses decrease protein synthesis and mRNA stability of glutamate codon-rich genes, such as actin cytoskeleton-associated genes.
Similar content being viewed by others

Introduction
Thermogenic adipocytes have drawn great interest, given their specialised function of heat production and energy expenditure that are associated with their positive effects on ameliorating obesity-related metabolic disorders1. Thermogenic capacities of beige and brown adipocytes are primarily linked or attributed to their cellular morphologies and metabolic strategies. Unlike non-thermogenic white adipocytes, they harbour multilocular lipid droplets and densely packed mitochondria to presumably provide structural benefits for efficient energy burn and exploit uncoupled respiration from ATP synthesis to dissipate stored energy as heat2,3. The considerable similarities of brown and beige adipocytes, despite clear differences in their developmental lineages, imply that potent consensus programmes are involved in the acquisition of the thermogenic adipocyte-specific phenotypes4,5,6. In addition to the thermogenic characteristics, thermogenic adipocytes have been noted due to their phenotypic plasticities upon external stimuli, such as chronic cold exposure. Browning of white fat for adaptive thermogenesis involves de novo differentiation from adipocyte precursors or white-to-beige adipocyte conversion7,8. In either route, the cells differentiating into beige adipocytes undergo rapidly progressive and visually discernible changes in cellular morphology and metabolism, which unequivocally result from or in considerable changes in gene expression profiles9,10.
Delineating gene regulation governing thermogenic adipocyte differentiation may lead to an exquisite control over the thermogenic organs and a novel therapeutic intervention against obesity and metabolic disorders11. Numerous previous studies have attempted to explain transcriptional and epigenetic programmes underlying beige or brown adipocyte differentiation12,13,14,15. In addition to transcriptional and epigenetic controls, regulatory events after mRNA synthesis, such as translational controls, are critically involved in gene expression, which is supported by the previous studies showing that mRNA levels poorly correlate with protein levels in many biological contexts16,17. However, with regard to quantitative and genome-wide description of gene expression changes during thermogenic adipocyte differentiation, a very limited number of studies were performed to estimate alterations in translation activities or protein levels. Moreover, the available datasets were prepared by separate experiments at different times and in different laboratories, making them difficult to compare with each other and to find biologically significant changes. Simultaneous and parallel genome-wide evaluations of mRNA translation and protein expression along with mRNA expression will be useful to elucidate hidden gene regulation underlying the development and function of the thermogenic adipocytes.
Metabolism and gene expression reciprocally regulate each other, orchestrating cell proliferation, survival, and differentiation. Transcriptional and epigenetic programs for thermogenic adipocyte differentiation alter expressions of certain genes, such as Ucp1 and other mitochondrial protein-coding genes, which directly modulate cellular metabolism, including mitochondrial respiration, fatty acid oxidation, and tricarboxylic acid (TCA) cycle18,19,20,21. Changes in metabolites and metabolic pathways can directly or indirectly impact the transcriptional and epigenetic regulation of specific genes as well. Chromatin remodelling via DNA and histone modification that are mediated by metabolites of glycolysis, TCA cycle, and nucleotide synthesis is such an example22,23. Recent studies demonstrated that increase in α-ketoglutarate level via AMPK activation mediates DNA demethylation in the Prdm16 promoter, thereby enhancing PRDM16 expression for beige and brown adipocyte differentiation24,25. Given that thermogenic adipocytes require exquisite controls over various metabolic pathways to fulfil their specialised role of heat production by burning fats, such mutual interactions between metabolic and gene regulation possibly act as effective strategies to gain thermogenic adipocyte-specific phenotypes and functions. However, it is currently unclear whether and how metabolic activities and translation regulation influence each other during thermogenic adipocyte differentiation.
In this study, we elucidate two hitherto unknown cross-talk pathways between metabolic and translational controls underlying beige adipocyte differentiation: selective translational down-regulation of oxidative phosphorylation (OXPHOS) component-coding genes and potentiated ribosome stalling at glutamate (Glu) codons. From the simultaneous and parallel genome-wide evaluations of translation efficiencies and protein expressions during pre-to-beige differentiation, we found that translation activities of OXPHOS component-coding mRNAs remarkably declined. The down-regulation is restricted to Complexes I, III, IV, and V, which is attributable to the attenuated translation of mitochondrial ribosomes. The relatively higher expression of Complex II, a.k.a. succinate dehydrogenase (SDH), harmonises with translational up-regulation for other TCA cycle components, contributing to distinct stoichiometry and activities of OXPHOS and TCA cycle in mitochondria of thermogenic adipocytes. In addition, by virtue of high-resolution Ribo-seq data, we assessed ribosome positions across transcripts, which unveiled potentiation of ribosome stalling at glutamate codons. The elevated ribosome pauses at Glu codons is attributable to altered glutamate metabolism, such as a dramatic increase of glutamate-ammonia ligase (GLUL) which decreases available glutamates for charging of cognate tRNAs during pan-adipocyte differentiation. The potentiated stalling disrupts the downstream elongation of translating ribosomes, leading to decreases in protein production and mRNA stability of Glu-rich protein-coding genes, including actin cytoskeleton-associated genes. The rates of cellular differentiation and actin cytoskeleton formation can be controlled by manipulating glutamate consumption, highlighting the impact of the metabolic change-driven regulation on adipocyte differentiation.
Results
Genome-wide assessment of translation activities unveils attenuated translation of OXPHOS complex-coding mRNAs during beige adipocyte differentiation
In order to interrogate gene regulation at the RNA and protein levels for cellular differentiation of WAT browning, we quantitatively and genome-widely estimated the steady-state levels of translated RNAs and proteins, along with total RNAs by using Ribo-seq and tandem mass tag (TMT) mass spectrometry (MS), in parallel with RNA-seq (Fig. 1a and Supplementary Data 1). We prepared lysates of adipocyte lineage cells that were differentiating into beige adipocytes from WAT-derived precursors upon hormonal stimuli. To examine temporal dynamics of gene regulation during the differentiation, the lysates were taken at three different time points (0, 4, and 8 days post-differentiation induction, respectively) showing differential accumulation and expression of lipid droplets (Fig. 1a, left panels) and marker genes (Supplementary Fig. 1a, Plin1 and Ucp1), respectively. A lysate of one biological replicate at each time point was commonly shared in the construction of a single set of transcriptome (RNA-seq), translatome (Ribo-seq), and proteome (TMT MS) to avoid the effect of source inconsistency.

a Assessment of the steady-state levels and changes in mRNAs, translated mRNAs, and proteins of individual genes during the beige adipocyte differentiation to interrogate transcriptional and post-transcriptional controls. Oil Red O staining of differentiating cells was performed to track differentiation. Representative images among biological triplicates are presented. b Distribution of 5’ end of RPFs near the start and stop codons. Genes that have the most abundant RPFs in each dataset are shown. c Volcano plot of ribosome density fold change during differentiation (left) and cumulative distribution of normalised fold change of mRNA, translated mRNA, and protein levels for genes with decreased or increased ribosome density (ribosome density-differentially expressed genes; RD DEGs) (right). d Gene set enrichment analysis (GSEA) with ribosome density. Each circle represents a GO cellular component term. e Protein-protein interaction network and heat maps displaying ribosome density fold change of genes included in leading edge subsets from the three most significantly enriched terms by GSEA. f Ribosome density at each time point for each indicated MitoCarta pathway. g Normalised fold change of mRNA, translated mRNA, and protein levels for each indicated MitoCarta pathway. Statistical tests and tools: edgeR for FDR-adjusted p-values of (c); GSEApy for NES and FDR-adjusted p-values of (d); two-sided paired t-test for p-values of (f); and two-sided Kolmogorov-Smirnov test for p-values of (g). *p < 0.05, **p < 0.01, ***p < 0.001. Box plots of (f, g) include boxes that extend from first to third quartile with median line and whiskers with 1.5 x interquartile range.
Beige adipocyte differentiation accompanies massive and drastic accumulation of membranous organelles, including mitochondria and lipid droplets, which may disturb biochemical treatments to isolate ribosome-associated mRNAs that are subjected to Ribo-seq library construction. We hence optimised the Ribo-seq preparation protocol by examining various conditions for ribosome isolation and RNase digestion, which ensured high purities of the ribosome protected fragments (RPFs) with ~30 nt of lengths from the differentiating cells (Supplementary Fig. 1b). Our Ribo-seq libraries showed noticeably improved preservation of ribosome-mRNA interactions that were represented by coding sequence (CDS) enrichments and three nucleotide periodicities compared to the other Ribo-seq libraries prepared from adipocytes and adipose tissues (Fig. 1b and Supplementary Fig. 1c). The movements of translating ribosomes at a single nucleotide resolution were manifestly observable even across individual transcripts (Fig. 1b), which enabled in-depth analyses on the changes in ribosome positions along mRNAs that will be described later.
The profiles of transcriptomes, translatomes, and proteomes represent gene expression levels of three different states, total mRNAs, translated mRNAs, and proteins, respectively, in the differentiating cells. Principal component analyses of the three datasets revealed that the biological replicates of each time point were clustered together, and the clusters were segregated from each other, indicating that each group reproducibly describes characteristic gene expressions of total mRNAs, translated mRNAs, and proteins at individual time points of the differentiation (Supplementary Fig. 1d). Although certain dissimilarities were observed between sequencing- and mass spectrometry -based estimations, the profiles of translated mRNAs, rather than those of total mRNAs, exhibited higher correlations with those of cognate proteins (Supplementary Fig. 1e) in consistency with previous reports in different biological contexts26,27, suggesting that there are at least more than auxiliary roles of translation controls in determining protein abundances in the differentiating cells.
To evaluate whether translational regulation has a decisive role in gene regulation during beige adipocyte differentiation, we selected genes that exhibited significant increases or decreases in ribosome densities (i.e. translation efficiencies) during the differentiation (Fig. 1c, left panel, Supplementary Fig. 1f and Supplementary Data 2) and compared their changes in total mRNAs, translated mRNAs, and proteins27. Whereas the total RNA and protein level changes of these genes exhibited noticeable discrepancies (Fig. 1c, right panels, RNA and protein, and Supplementary Fig. 1g, upper panels), their translated mRNA and protein levels showed corresponding increases and decreases to each other (Fig. 1c, right panels, RPF and protein, and Supplementary Fig. 1g, lower panels). These results demonstrate that translational regulation critically affects protein expressions for certain genes in the cells undergoing beige adipocyte differentiation.
We performed gene set enrichment analyses (GSEAs) using ribosome densities of individual genes at 4 or 8 days post-induction (4 d or 8 d) and those of undifferentiated controls (0 d) to predict cellular and molecular functions that are modulated by translational regulation during beige adipogenesis (Fig. 1d, Supplementary Fig. 1h, i and Supplementary Data 3). Many gene sets that were significantly enriched (<0.1 FDR) in either pairwise comparison of 4 d versus 0 d or 8 d versus 0 d showed same direction of ribosome density changes in the other comparison, implying that translational up- or down-regulation for the gene sets consistently occur throughout the differentiation. We focused on three gene sets that exhibited profoundly significant enrichments (<10-3.5 FDR) in both comparisons: inner mitochondrial membrane protein complex (GO:0098800), respirasome (GO:0070469), and NADH dehydrogenase complex (GO:0030964). These gene sets with negative normalised enrichment scores are functionally related to each other and share many component genes, suggesting that a common set of genes involved in the gene set-representing functions are translationally down-regulated. Protein-protein interaction (PPI) network analysis28 of 97 genes leading the enrichments indicates that most of the leading genes (72 of 97 genes, ~74.2%) encode proteins that constitute OXPHOS complexes, highlighting attenuated translation for OXPHOS complex component genes during beige adipocyte differentiation (Fig. 1e).
Given that thermogenic adipocytes are characterised with dense mitochondria which require active synthesis of mitochondrial proteins during the differentiation, this finding was unexpected and surprising. Our RNA-seq datasets also showed potent transcriptional up-regulation of mitochondrial coding genes (Supplementary Fig. 1j), as with the previous transcriptome studies15,20. We then hypothesised that translational down-regulation for OXPHOS complex component genes possibly result in relatively lower rate of protein production than other mitochondrial protein-coding genes in beige adipocytes despite overall increases in mRNA levels by transcriptional activation throughout the differentiation. Indeed, compared to other mitochondrial protein-coding genes with higher ribosome densities, such as TCA cycle and fatty acid oxidation-related genes (Fig. 1f), protein productions of OXPHOS complex component genes were compromised (Fig. 1g). These observations support our hypothesis and indicate that differential translation activities of mitochondrial protein-coding genes can change stoichiometry of mitochondrial components and their related metabolic functions in the differentiating cells.
Selective translational down-regulation of OXPHOS component-coding genes is attributable to attenuated translation within mitochondria
Whether and how translation of mitochondrial protein-coding mRNAs are specifically and differentially regulated still remains elusive. One recent study revealed that mitochondrial protein-coding mRNAs exhibit characteristic subcellular localisation depending on their interaction with translation machinery or RNA binding proteins by exploiting one leading-edge technique, APEX-seq29. We exploited the APEX-seq data to re-analyse proximities of mitochondrial protein-coding mRNAs to outer mitochondrial membrane (OMM) upon cycloheximide (CHX) or puromycin (PUR) treatment (Supplementary Fig. 2b, left panel). Given that CHX stalls elongating ribosomes onto mRNAs and PUR dissociates ribosomes from mRNAs (Supplementary Fig. 2a), exclusive responsivenesses of OMM enrichment to CHX or PUR treatment (Supplementary Fig. 2b, right panel) represent ribosome dependent and independent OMM localisation of individual mRNAs, respectively. Mitochondrial protein-coding genes were categorised into three groups by OMM enrichment scores (Fig. 2a). Intriguingly, a portion of genes whose mRNAs were enriched in OMM via ribosome-independent interactions exhibited potent translational down-regulation during beige adipocyte differentiation (Fig. 2a, group III). Pathway and function enrichment analysis for these genes overrepresented OXPHOS subunit and mitochondrial ribosome terms of MitoCarta pathways (Fig. 2b), suggesting that mRNAs encoding constituent proteins of these complexes are localised to OMM possibly via interactions with non-ribosomal RBPs. Indeed, most genes that belong to these MitoCarta pathway terms showed OMM localisation in ribosome-independent manner (Fig. 2c) and decreases in ribosome densities in the cells undergoing differentiation (Fig. 1c,f and Supplementary Fig. 2c).

a Relationship of ribosome dependent and independent OMM enrichment with ribosome density fold change of mitochondrial protein-coding genes. b Enrichment analysis of group III genes with MitoCarta pathways. c Ribosome dependent and independent OMM enrichment of OXPHOS subunit and mitochondrial ribosomal protein-coding genes. d Heat maps and distribution of the fold change in ribosome density of OXPHOS complex component genes that constitute each OXPHOS complex at 8 d relative to 0 d. e Distribution of protein level fold change of OXPHOS complex subunits at 8 d relative to 0 d. For (d, e) values of biological triplicates were pooled for statistical tests. f Percentage of OXPHOS complex II protein level over total OXPHOS complex protein level at each time point. Replicate N = 3. g Transcriptional (x-axis) and translational (y-axis) regulation of OXPHOS complex subunits. Right plot shows the ribosome density fold change of Complex II subunits, mtDNA- and nDNA-originated subunits of Complexes I, III, IV, and V. h Protein level fold changes of MitoCarta pathways related to mitochondrial translation and metabolism. i A diagram depicting the hypothesis to explain selective translational regulation of OXPHOS complex expression during beige adipocyte differentiation. j Western blot of OXPHOS complex subunits during beige adipocyte differentiation with or without inhibition of mitochondrial translation by chloramphenicol treatment. Right bar plot shows the band intensity of SDHB, a Complex II subunit, relative to the total intensity of all five subunits. Representative images among five replicates are shown. Statistical tests and tools: two-sided Kolmogorov-Smirnov test for p-values of (a) GSEApy for odds ratio and FDR-adjusted p-values of (b); two-sided Mann-Whitney U test for p-values of (d, e, h) two-sided paired t-test for p-values of (f); and two-sided unpaired t-test for p-values of (j). *p < 0.05, **p < 0.01, ***p < 0.001. Point plots of (d, e) are shown as median with 50% interval. Bar plots of (f, j) are shown as mean with or without SD, respectively. Box plots of (a, h) include boxes that extend from first to third quartile with median line and whiskers with 1.5 x interquartile range. Source data are provided as a Source Data file.
The electron transfer chain of OXPHOS is composed of five distinct complexes in the inner mitochondrial membrane. We compared magnitudes of translational down-regulation for the genes encoding component proteins of the five complexes by estimating fold changes in ribosome densities (Fig. 2d and Supplementary Fig. 2d). Notably, translational suppression of Complex II protein-coding genes was hardly detected while the other complex component-coding genes evidently exhibited attenuated translation in the differentiating cells. These results imply that protein synthesis for Complex II is relatively more active than the other OXPHOS complexes during beige adipocyte differentiation, which possibly leads to faster and higher accumulation of Complex II than the other complexes in the differentiating cells. In agreement with the prediction, our proteome datasets revealed that the protein level changes of Complex II genes were higher than those of genes of other complexes (Fig. 2e), and the proportion of Complex II proteins among all OXPHOS complex proteins increased from ~6.4 % on day 0 to ~12.8 % on day 8 of the differentiation (Fig. 2f).
One noteworthy feature of Complex II is that its component proteins are entirely encoded by nuclear DNA (nDNA), whereas the other complexes are composed of proteins encoded by mitochondrial DNA (mtDNA) as well as nDNA. We noted that the intensities of translational suppression for the component genes of Complex I, III, IV, and V were much stronger when they are encoded by mtDNA rather than nDNA (Fig. 2d, g). mRNAs transcribed from the mitochondrial genome are translated by mitochondrial ribosomes within the organelles, and it has been previously reported that mitochondrial translation is synchronised with cytosolic translation30,31 and suppression of mitochondrial translation reduces cytosolic translation via an Atf4-dependent cascade32. Given these previous findings and our data showing the more pronounced translational suppression of mtDNA-encoded genes (Fig. 2g) and translation inhibition of mitochondrial ribosome genes (Fig. 2a,c and Supplementary Fig. 2c), we hypothesised that attenuation of mitochondrial translation possibly leads to the selective translational suppression of OXPHOS Complex I, III, IV, and V component genes during beige adipocyte differentiation (Fig. 2i). The analyses of our transcriptome, translatome and proteome datasets indicated significantly lower expression of genes that are involved in mitochondrial translation activities, compared with other mitochondrial function-related genes, supporting this scenario (Fig. 2h and Supplementary Fig. 2e). To validate our hypothesis, we treated the differentiating cells with chloramphenicol, an inhibitor of mitochondrial translation, and evaluated the abundances of OXPHOS complex proteins via western blotting (Fig. 2j). Upon chloramphenicol treatment, MTCO1, one of the mitochondrial genome-encoded proteins, showed obvious decreases, indicating successful inhibition of mitochondrial ribosome activities. Surprisingly, protein levels of NDUFB8, UQCRC2, and ATP5A (Complex I, III, and V component proteins, respectively) noticeably decreased upon the treatment, though their cognate mRNAs are translated by cytosolic ribosomes that are not targeted by chloramphenicol. In contrast, SDHB, a component protein of Complex II, did not show a significant difference upon the treatment. Taken together, these results demonstrate that suppression of mitochondria translation can attenuate protein expressions of not only mitochondrial genes but also nuclear genes of Complexes I, III, IV, and V in coordinated manner, without affecting expression of Complex II component-coding genes devoid of mitochondrial origins.
Differential translation of OXPHOS- and TCA cycle-associated genes contribute to thermogenic adipocyte-specific mitochondrial activities
Given that thermogenic and non-thermogenic adipocytes have distinguishably different metabolic activities that are associated with mitochondria, we wondered whether the selective translational controls of OXPHOS complex genes manifest during differentiation towards the other types of adipocytes. We estimated ribosome density changes of OXPHOS component-coding genes from the translatome and transcriptome data of the cells differentiating into brown and white adipocytes33 (Supplementary Fig. 3a). Interestingly, brown adipocytes exhibited translational down-regulation for the genes encoding component proteins of Complexes I, III, IV, and V, as with beige adipocytes, whereas white adipocytes showed even up-regulation for these genes (Fig. 3a, upper panels). Translation efficiencies of Complex II protein-coding genes were slightly enhanced or not altered in brown adipocytes while moderately declined in white adipocytes (Fig. 3a, lower panels, SDH). These results imply that attenuated translation of the genes encoding component proteins of OXPHOS complexes with mitochondrial origins is likely to be characteristic gene regulation of thermogenic adipocytes.

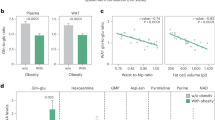
a Heat maps showing ribosome density fold change of mtDNA-originated OXPHOS complex subunits and TCA cycle components. Early and late indicate 4 d and 8 d compared to 0 d, respectively. b Fold changes of mRNA and translated mRNA levels of TCA cycle components and SDH relative to OXPHOS complex I, III, IV, and V during beige, brown, and white adipocyte differentiation. Values of biological replicates were pooled for statistical tests. c Western blot of OXPHOS complex subunits with the mitochondrial fraction of beige and white adipocytes. The right bar plot shows the band intensity of SDHB relative to the total intensity of all five subunits. Representative image among four replicates is shown. d Percentage of Complex II protein level over total OXPHOS complex protein level in beige, brown, and white adipocytes. Replicate N = 5. e Percentage of TCA cycle enzymes’ protein level over total MitoCarta protein level in beige, brown, and white adipocytes. Replicate N = 5. f, g Measurement of SDH activity (f) and Complex I activity g with the mitochondrial fraction of beige and white adipocytes. The activity of SDH and Complex I are depicted by a decrease in absorbance at 600 nm and an increase in absorbance at 450 nm, respectively. Replicate NBeige = 7, NWhite = 8 for (f) N = 9 for g. h Activity of SDH relative to activity of Complex I in beige and white adipocytes. i A diagram describing the hypothesis to explain differential translational regulation of TCA cycle and OXPHOS complex components in thermogenic adipocytes. Statistical tests: one-sided Mann-Whitney U test for p-values of b and two-sided unpaired t-test for p-values of c–g. *p < 0.05, **p < 0.01, ***p < 0.001. Point plots of b are shown as median with 50% interval. Bar plots of c–g and point plots of f, g are shown as mean (SD). Source data are provided as a Source Data file.
Complex II is a bifunctional enzyme that is involved in the succinate dehydrogenation of TCA cycle as well as electron transfer of OXPHOS. We noted that the genes encoding constituent proteins of the other TCA cycle enzymes exhibited enhanced translation activities in thermogenic adipocytes compared to white adipocytes (Fig. 3a, lower panels). Given that both OXPHOS and TCA cycle-associated genes are transcriptionally up-regulated in the cells differentiating into all types of adipocytes (Supplementary Fig. 3a, b), we hypothesised that the selective translational down-regulation of OXPHOS-associated genes and the overall translational up-regulation of TCA cycle-associated genes can alter the balances between the steady state levels of the proteins of OXPHOS and TCA cycle in thermogenic adipocytes, by adding another layer of differential gene expressions after the transcriptional changes. Indeed, when we normalised the RNA and RPF level changes of the TCA cycle and Complex II component-coding genes with those of the gene encoding constituent proteins of Complex I, III, IV, and V, the normalised RNA level changes of the TCA cycle-associated genes did not show noticeable differences among all types of adipocytes, but the normalised RPF level changes exhibited evident increases only in thermogenic adipocytes (Fig. 3b), supporting the scenario.
We, therefore, examined whether protein expressions of the OXPHOS and TCA cycle-associated genes follow the patterns of translational controls in thermogenic adipocytes and white adipocytes. To exclude the effect of overall transcriptional up-regulation of mitochondrial protein-coding genes, we isolated mitochondria from fully differentiated beige and white adipocytes and estimated protein levels of OXPHOS complexes from equal amounts of the mitochondrial lysates. Mitochondria of beige adipocytes showed a higher ratio of Complex II protein (SDHB) relative to other OXPHOS proteins, as compared with those of white adipocytes (Fig. 3c and Supplementary Fig. 3d), which is in line with our aforementioned results demonstrating that selective translational attenuation of OXPHOS component-coding genes can induce larger increases of Complex II proteins during thermogenic adipocyte differentiation (Fig. 2j). Additionally, we assessed the expression levels of other OXPHOS component proteins and TCA cycle enzyme proteins by analysing the proteomes of beige, brown, and white adipocytes34. The result showed higher percentages of Complex II proteins among all other OXPHOS proteins in beige and brown adipocytes compared to white adipocytes (Fig. 3d), augmenting our hypothesis. As for the comparison of TCA cycle proteins, given overall increases of mitochondrial proteins in thermogenic adipocytes (Supplementary Fig. 3e), we normalised the protein abundances of the TCA cycle-associated genes with those of whole mitochondrial protein-coding genes. The result indicated that beige and brown adipocytes had higher proportions of TCA cycle-related proteins among all mitochondrial proteins than white adipocytes (Fig. 3e and Supplementary Fig. 3f). Taken together, the relative protein levels of OXPHOS and TCA component genes between thermogenic and non-thermogenic adipocytes correspond to the differences between their translation controls of the genes.
We reasoned that the different stoichiometry of OXPHOS and TCA cycle components in mitochondria of thermogenic adipocytes can endow distinct activities of metabolic functions the components are involved in. To examine the possibility, we evaluated enzyme activities for NADH (1,4-Dihydronicotinamide adenine dinucleotide) oxidation and succinate dehydrogenation of Complex I and II, respectively, from beige and white adipocytes. As with the western blots, the enzyme activity assays were performed using equal amounts of mitochondrial lysates to exclude the effects of overall increases of mitochondrial proteins in thermogenic adipocytes. Mitochondria of beige adipocytes exhibited much higher activities of succinate dehydrogenation by Complex II, as compared with those of white adipocytes (Fig. 3f). Given that succinate dehydrogenation is one of key biochemical reactions of TCA cycles, this result supports that relatively higher expressions of TCA cycle proteins can lead to enhanced TCA cycle activities in individual mitochondria of thermogenic adipocytes. NADH oxidation activities of mitochondria from beige adipocytes were significantly lower than those from white adipocytes (Fig. 3g), which suggests that the activities of certain biochemical reactions of OXPHOS per mitochondrion can be attenuated by the relatively lower expressions of corresponding OXPHOS components. Complex II, a bifunctional enzyme acting as succinate hydrogenase as well, is presumably exempted from the translational down-regulation mediated attenuation of OXPHOS activities during beige adipocyte differentiation. This unequal regulation possibly works as an elaborate strategy to keep relatively higher activities of TCA cycles in mitochondria of thermogenic adipocytes versus those of non-thermogenic adipocytes (Fig. 3i). In agreement with this postulation, mitochondria isolated from beige adipocytes exhibited higher activities of Complex II/SDH relative to those of Complex I, as compared with white adipocytes (Fig. 3h). Moreover, it has previously been shown that extracellular succinates are rapidly metabolised by thermogenic adipocytes, and their oxidation by SDH is required for activation of thermogenesis19. The relatively larger increase in the amount and activity of Complex II/SDH per mitochondrion is likely a strategy to cope with that circumstance.
Ribosome stalling at glutamate codons is potentiated during adipocyte differentiation
Our Ribo-seq datasets detected the precise information of ribosome-mRNA interactions at single nucleotide resolution even from individual transcripts (Fig. 1b), which enabled a thorough investigation into fine positions of ribosomes across mRNAs. A-site positions of ribosomes that are determined by the offset subtraction from individual RPFs can be exploited to identify ribosome stalling in the middle of translational elongation35,36,37. We evaluated ribosome pauses that were determined by aggregations of RPF tags in our datasets (Fig. 4a, left panel) and noted that ribosome stalling became intensified as the differentiation proceeded (Fig. 4a, right panel). Surprisingly, categorising the ribosome pauses according to amino acid codons at A-sites revealed a prominent elevation of ribosome stalling at glutamate (Glu) codons in the differentiating cells (Fig. 4b, d, Supplementary Fig. 4a and Supplementary Data 4): GAA and GAG codons encoding glutamate exhibited the most pronounced increases in stalling on day 4 and 8 of the differentiation, with the extent noticeably stronger at GAA codon than GAG codon (Fig. 4b, c and Supplementary Fig. 4b). We then exploited a statistical metric used in Stein et al.’s study37 to investigate specific codons exhibiting ribosome pauses during the differentiation. The number of codons with ‘differentiation-dependent stalling’ increased on day 4 and day 8, and Glu codons comprised the largest portions of them (Fig. 4e and Supplementary Data 5). These results demonstrate that the increases in ribosome pauses at Glu codon are predominant over those at other codons during beige adipocyte differentiation.

a Cumulative distribution of stalling score at each time point (left) and meta-gene analysis around sites with top 5% stalling score (upper right) or 47.5-52.5% stalling score (lower right). b Heat maps displaying A-site codon distribution of sites with top 5% stalling score, presented with counts (left) and their row z-scores (right). c Swarm plots showing A-site codon count of sites with top 5% stalling score. d Amino acid sequence preference of sites with top 5% stalling score at each time point. e Relative ribosome stalling of each codon site during beige adipocyte differentiation. The red dots in the left scatter plots indicate sites with differentiation-dependent ribosome stalling (odds ratio > 1, adjusted p-value < 0.01). Right pie charts depict the composition of A-site codon-encoded amino acids at differentiation-dependent stalling sites. p-values were calculated by two-sided Fisher’s exact test and adjusted by Benjamini-Hochberg method. f Amino acid sequence preference of sites where ribosome stalling was significantly increased at 8 d relative to 0 d. g Meta-gene analyses around each amino acid-coding codon group exhibit the differential ribosome occupancy at 8 d relative to 0 d in a nearby region of each codon. Small windows show the differential ribosome occupancy at upstream (left) or downstream (right) sites, as indicated by dashed boxes. h Coverage depth plots of RPF tags near exon 3 and 4 regions of Tef. In top and middle plots, the y-axis represents the average coverage depth of replicates at each indicated time point. In the bottom plot, the y-axis represents the average coverage depth normalised by the total depth of the Tef gene for each time point. i Swarm plots showing the count of A-site codon-encoded amino acids at sites with top 5% stalling score at each time point of beige adipocyte differentiation, in differentiated beige, brown, and white adipocytes, and in cold-treated inguinal white adipose tissue.
To examine whether and how the potentiated ribosome stalling at Glu codons affect translation elongation, we analysed movements of elongating ribosomes around Glu codons via meta-gene alignments of RPF tags (a.k.a. diricore analysis38) using our Ribo-seq datasets. Notably, ribosome occupancies exhibited dramatic surges at Glu codons in both comparisons of day 4 or 8 versus day 0 (Fig. 4g and Supplementary Fig. 4c, d), describing that elongating ribosomes get stuck when they confront Glu codons in the differentiating cells. Moreover, ribosome occupancies across the downstream region of Glu codons markedly declined in the comparisons, indicating compromised elongation of ribosomes passing through Glu codons. The signatures of impediment of ribosome elongation were most pronounced around Glu codons, though a weaker extent of enhanced stalling was observed around other amino acid codons, such as aspartate (Fig. 4f, g). The hindered movement of elongating ribosomes at Glu codons was observable even at individual transcript levels. For example, RPF tags aligned to Tef locus exhibited a substantial surge around the Glu codon, followed by noticeable decline in the downstream region on day 8 (Fig. 4h and Supplementary Fig. 4e). All these results collectively point to elevated stalling of ribosomes preferentially at Glu codons, hindering downstream movement of the ribosomes in the cells differentiating into beige adipocytes.
We next assessed whether the potentiated ribosome stalling at Glu codons was observed in other types of adipocytes by re-analyzing the Ribo-seq data in Martinez et al.’s study34. In spite of smaller numbers of the identified stalling sites due to the relatively lower depth and preservation of ribosome protected fragments (Fig. 1b), Glu codons exhibited the most pronounced ribosome stalling in brown and white adipocytes, which is similar to our observations in beige adipocytes (Fig. 4i, left and middle panels and Supplementary Fig. 4f). Notably, aspartate (Asp) codons showed comparable degrees of ribosome pauses to Glu codons in white adipocytes. To validate the preferential stalling at Glu codons in in vivo context, we examined ribosome stalling at amino acid codons in inguinal WATs (iWATs) that were exposed to cold temperature39. Despite a preference for Asp codons comparable to that for Glu codons, which is possibly attributed to white adipocytes comprising a proportion of the tissue13,40, Glu codons still exhibited most prominent ribosome pauses (Fig. 4i, right panel and Supplementary Fig. 4f). These results demonstrate that the potentiated ribosome stalling at Glu codons manifests in other types of adipocytes and adipose tissues, suggesting that the elevation of ribosome pauses is possibly attributed to a common mechanism that three different types of adipocytes share.
Potentiated ribosome stalling at Glu codon is attributable to remodelled glutamate metabolism, including potent increase of GLUL in adipocytes
Given that translation elongation involves incorporation of aminoacyl-tRNAs that are specified by codons of mRNA into ribosomes, we hypothesised that the elevated rates of ribosome stalling at Glu codons in the differentiated adipocytes are caused by diminished supply of glutamyl-tRNAs (Glu-tRNAs). The expression levels of glutamyl-prolyl-tRNA synthetase (Eprs1) did not show remarkable changes in our datasets (Supplementary Fig. 5a), indicating that the Glu codon-stalling is unlikely a direct outcome of altered expression of the aminoacyl-tRNA synthetase. Supply and steady-state level of aminoacyl-tRNA are affected by availability and cellular level of cognate amino acid. Moreover, it has been reported that intracellular depletion of certain amino acids resulted in ribosome pauses on the cognate codons in diverse biological contexts38,41,42,43. We hence assessed the expression changes of genes that are involved in glutamate metabolism during beige adipocyte differentiation (Fig. 5a and Supplementary Fig. 5a). The glutamate metabolism-associated genes exhibited considerable changes, indicating extensive remodelling of glutamate metabolism in the differentiating cells. We noted the most dramatic increase of Glul and moderate decrease of Gls, genes encoding glutamate-ammonia ligase (GLUL) and glutaminase (GLS) that converts glutamate (Glu) to glutamine (Gln) and Gln to Glu, respectively. Given that the other glutamate metabolism-associated genes can contribute to intracellular glutamate increase and decrease reversibly, the increase of GLUL and decrease of GLS had us speculate Glu to Gln conversion is accelerated in the differentiating cells, diminishing available glutamates for other uses, such as Glu-tRNA synthesis.

a Diagram showing how GLS, GLUL, GLUD1, GOT1, GOT2, GPT2, BCAT1, and BCAT2 mediate glutamate metabolism (top) and bar plots depicting their expression changes during beige adipocyte differentiation (bottom). Replicate N = 3 for each bar. b Glutamate to glutamine ratio during beige adipocyte differentiation with or without treatment of methionine sulfoximine (MSO), a glutamine synthetase (GLUL) inhibitor. Replicate Nnon-MSO = 3, NMSO = 2. c Changes in the ratios of charged versus uncharged tRNAs were measured in preadipocytes and beige adipocytes using a qPCR-based tRNA aminoacylation assay. Primers specific to tRNAGly(TCC), tRNAGln(CTG), and tRNAGlu(TTC) were used. Replicate N = 3. d Assessment of tRNAGlu(TTC) charging. Top panel shows northern blot of tRNAGlu(TTC) with deacylated sample (left) and lysates of 0 day (middle) or 8 days (right) of beige adipocyte differentiation. Bottom panel shows 5S and 5.8S rRNA stained with SYBR Gold. Representative images among three replicates are shown. e Schematic of the reporter assay for the doxycycline inducible Glu or Ala codon-rich EGFP expression (top) and flow cytometry results of 3T3-L1 cells showing changes in EGFP expression of the reporters whether doxycycline and BPTES treatment. (bottom). Replicate N = 3. f Bar plots displaying expression changes of genes involved in glutamate metabolism during white and brown adipocyte differentiation. Replicate N = 2. g A heat map showing single-nucleus expression of indicated genes in ASPCs or adipocytes. Each column of the heat map represents one nucleus, and columns are grouped into ASPCs (left) and adipocytes (right). h qRT-PCR results showing relative mRNA levels of adipogenic and thermogenic markers under differentiation with or without MSO treatment. Replicate NPparg2,Fabp4,Adipoq = 6, NUcp1,Cox8b = 2. Statistical tests: two-sided unpaired t-test for p-values of b, h; and two-sided paired t-test for p-values of c, e. *p < 0.05, **p < 0.01, ***p < 0.001, NS means ‘not significant’. Bar plots of a–c, e–g are shown as mean (SD). Source data are provided as a Source Data file.
To examine this scenario, we estimated the relative ratios of intracellular glutamate versus glutamine during the differentiation via a bioluminescence assay that detects glutamate/glutamine-mediated NADH production. The ratios of glutamate to glutamine exhibited noticeable decreases on day 4 and 8 (Fig. 5b), supporting enhanced Glu to Gln transition during the differentiation. The quantification of intracellular concentrations of Glu and Gln revealed decrease of Glu and increase of Gln in the differentiating cells, respectively (Supplementary Fig. 5c). The inhibition of GLUL/Glul using methionine sulfoximine (MSO), a chemical GLUL inhibitor, and siRNA targeting Glul mRNA (siGlul) increased the ratios of Glu relative to Gln (Fig. 5b and Supplementary Fig. 5c–f), underscoring the impact of GLUL in determining intracellular levels of glutamate and glutamine during adipocyte differentiation. Given that intracellular decrease of a specific amino acid can directly compromise efficient charging of the amino acid onto cognate tRNA, we performed tRNA charging assay38,44 and northern blot to evaluate the ratios between amino-acylated and free forms of glutamate tRNA (tRNAGlu) in the differentiating cells. Both results indicated reduction of glutamate-conjugated (amino-acylated) forms during the differentiation (Fig. 5c, d). These results collectively support our hypothesis that a decrease in net glutamate level caused by augmented Glu to Gln conversion leads to an inefficient supply of Glu-tRNAs during beige adipocyte differentiation.
We next examined whether hindering glutamate supply impedes protein synthesis of Glu codon-containing transcripts. To monitor protein production, we constructed reporter plasmids that harbour multiple Glu codons or Ala codons upstream of green fluorescence protein (GFP)-coding sequences (Fig. 5e, top panels). We transfected 3T3-L1 adipogenic precursor cell lines with the reporter plasmids, and then estimated GFP signals from individual cells upon treatment with BPTES, a GLS inhibitor, to decrease intracellular glutamate by blocking glutamate synthesis. GFP signals from the reporter genes with Glu codons not with Ala codons were attenuated (Fig. 5e, bottom panels) upon the GLS inhibition, supporting that translation of Glu codon-containing mRNAs can be preferentially hampered by intracellular glutamate decrease.
Given that the potentiated ribosome stalling at Glu codons was commonly observed in white and brown adipocytes as well as beige adipocytes, we wondered whether the alteration of glutamate metabolism including enhanced Glu to Gln transition, is preserved in the other types of adipocytes. We hence assessed expression changes of Glul, Gls, and other glutamate metabolism-associated genes during white and brown adipocyte differentiation33. The genes exhibited considerably similar patterns of up- or down-regulation in white and brown adipocytes (Fig. 5f and Supplementary Fig. 5b), demonstrating that the remodelling of glutamate metabolism commonly occurs during differentiation into all types of adipocytes. To investigate whether this finding is recapitulated in in vivo contexts, we exploited single-cell transcriptomic data of iWAT45. We examined expression levels of the glutamate metabolism-associated genes in adipocyte precursors and adipocytes within tissue microenvironments (Fig. 5g and Supplementary Fig. 5h, i). Glul and Gls expressions were considerably higher and moderately lower respectively in the adipocyte population compared to those in the precursor population, supporting a bias toward Glu to Gln conversion in mature adipocytes in the in vivo environment.
To evaluate the impact of the enhanced Glu to Gln conversion on adipocyte differentiation, we estimated expression levels of differentiation marker genes in the cells undergoing beige adipocyte differentiation when the GLUL-mediated conversion is dampened. Upon treatment with MSO (Fig. 5h) and siGlul (Supplementary Fig. 5g) to decrease intracellular GLUL activities, marker genes of common and thermogenic adipocyte markers, such as Pparg2, Fabp4, Adipoq, Ucp1, and Cox8b exhibited significantly compromised expressions during the differentiation. In addition, supplementation of glutamates for the differentiating cells not only recovered the decreased Glu to Gln ratio (Supplementary Fig. 5j) but also dampened the expression of the marker genes (Supplementary Fig. 5k). These results demonstrate that reversing the direct outcome of Glul up-regulation, intracellular glutamate reduction, can retard or attenuate the acquisition of adipocyte phenotypes, underlining the importance of Glu to Gln transition by GLUL for adipocyte differentiation.
Glutamate-rich protein-coding genes exhibit decreases in protein synthesis and mRNA stabilities during adipocyte differentiation
The diminished associations of ribosomes following their intensified pauses at Glu codons (Fig. 4g and Supplementary Fig. 4c, d) imply that the stalling leads to inefficient translation of mRNAs harbouring Glu codons. This interpretation is supported by attenuated protein expression of GFP reporter genes that contain multiple glutamate codons upon the blockage of glutamate synthesis (Fig. 5e). To investigate whether the scenario is valid for endogenous mRNAs, we first categorised genes according to glutamate codon frequencies in their CDS sequences (Glu enrichment rank). Gene sets harbouring higher Glu enrichment ranks showed more frequent ribosome stalling on day 4 and 8 of the differentiation (Fig. 6a and Supplementary Fig. 6a), demonstrating that Glu codons play a prominent role in provoking the ribosome pauses in the differentiating cells. We next examined whether the endogenous transcripts containing Glu codons exhibit compromised protein synthesis by assessing protein level changes of the corresponding proteins in the proteome datasets. To minimise the effects of RNA level changes, we separated the categorised genes into three groups with similar RNA level changes (average normalised RNA logFC: -0.85 ~ -0.25; -0.25 ~ 0.25; and 0.25 ~ 0.85). Within each group, negative correlations between Glu codon enrichments and protein level changes on both day 4 and day 8 versus day 0 were observed (Fig. 6b and Supplementary Fig. 6b), demonstrating that genes with more Glu codons showed more attenuated protein production in the differentiating cells. Given that the RNA level changes did not show noticeable positive or negative correlation with the Glu enrichment ranks within each group, the attenuated protein production for Glu-codon containing genes was unlikely attributed to RNA level changes. Taken together, these results indicate that Glu codon-rich genes have preferential increases in ribosome stalling and decreases in protein production during the differentiation, underpinning the stalling-mediated translational inhibition of Glu codon-rich mRNAs.

a Correlation between glutamate enrichment and ribosome stalling occurrence. Genes were ranked by glutamate proportion of protein they code and binned into 10 groups. Percentage of transcripts with at least one significantly increased stalling site was calculated for each group. b A scatter plot displaying protein expression change relative to RNA expression change for groups with different glutamate enrichment. Genes exhibiting RNA level changes in specific ranges (bottom: from −0.85 to −0.25, middle: from −0.25 to 0.25, top: from 0.25 to 0.85 of normalised RNA logFC) were grouped, and the average normalised logFC of RNA (grey) and protein (red) were calculated. c A scatter plot showing RNA and protein expression changes of groups with different glutamate enrichment. For each group, the average normalised logFC of RNA (grey) and protein (red) were calculated. d mRNA stability change of groups with different glutamate enrichment. e Gene list enrichment analysis of genes with significantly increased stalling sites at 8 d, included in the top 10% glutamate enrichment group. f Scatter plots showing stalled transcript percentage, average RNA log2FC, and average protein log2FC of cytoskeleton and actin-related gene sets. Distribution of other gene sets is displayed by contour plots. g, h Immunofluorescence images of F-actin (binary and red), PLIN1 (green), and nucleus (blue) in 3T3-L1 cells during beige adipocyte differentiation g or 2 days post-differentiation induction with or without BPTES h. i qRT-PCR results showing relative mRNA levels of adipogenic and thermogenic markers in beige and white adipocytes differentiated with or without cytochalasin D treatment. Replicate N = 3. Statistical tests and tools: two-sided linear least-squares regression for p-values of a–c; GSEApy for odds ratio and FDR-adjusted p-values of (e); and two-sided unpaired t-test for p-values of (i). *p < 0.05, **p < 0.01, ***p < 0.001. Shadings of a, b, c represent confidence intervals (CI). For d, the centre and radii of ellipses represent mean and standard error. Bar plots of (i) are shown as mean (SD). Source data are provided as a Source Data file.
Despite no significant correlation between RNA level change and Glu codon enrichment of the genes with narrow ranges of RNA level changes (Fig. 6b and Supplementary Fig. 6b), we observed weak but clear negative correlations (r ≈ -0.9) when the comparisons were extended to the entire range of RNA level changes (Fig. 6c and Supplementary Fig. 6c). These observations suggest that the ribosome stalling may possibly affect mRNA stabilities as well as translation activities of the Glu codon-containing transcripts in the differentiating cells. It has previously been reported that mRNA surveillance pathway (No-Go Decay) induces degradation of mRNAs harbouring stalled ribosomes46,47,48,49,50. To examine the enhanced decay of Glu codon-rich mRNAs, we took advantage of our RNA-seq libraries that were prepared by rRNA subtraction method, providing sequencing reads from pre- and mature- mRNAs. According to Alkallas et. al.’ s method51, we evaluated mRNA stability changes using discrepancies between exon- and intron-mapped reads, by separating stability- and transcription-driven RNA level changes during the differentiation (Supplementary Fig. 6 d and Supplementary Data 6). The estimated mRNA stability changes successfully detected elevated mRNA decay by miR-196a, a microRNA known to be involved in beige adipogenesis52, validating the efficacy of this estimation (Supplementary Fig. 6e). Notably, the gene sets with higher Glu enrichment ranks exhibited larger decreases in RNA stability on day 4 and day 8 (Fig. 6d), indicating that mRNA decay of Glu codon-rich genes was accelerated in the differentiating cells.
Given the differential decreases in protein production and mRNA stabilities of individual genes that are correlating with the enrichments of Glu codons, we tried to find cellular and molecular functions that sensitively respond to the potentiated stalling at Glu codons. To this end, we performed pathway and function enrichment analyses for the genes encoding Glu-rich proteins and exhibiting substantial increases in ribosome stalling in the differentiating cells. Interestingly, the result overrepresented multiple GO terms associated with cytoskeletal structures, especially actin cytoskeleton (Fig. 6e and Supplementary Data 7), suggesting that the expressions of genes that are involved in cytoskeletal structures and organisation, such as actin filament formation, are sensitively affected by the stalling at Glu codons. Indeed, cytoskeleton and actin-related genes exhibited decreases in mRNA stability and protein production during beige adipocyte differentiation (Fig. 6f). Actin-associated cytoskeletal structures are known to be disassembled during non-thermogenic adipocyte differentiation, and actin depolymerisation has been reported to enhance adipogenesis53,54,55, but the upstream mechanism underlying the disassembly process still remains elusive. We observed that actin proteins were densely polymerised in adipocyte precursors, and the polymerized structures were dismantled during beige adipocyte differentiation (Fig. 6g). Interestingly, blocking glutamate synthesis with BPTES treatment accelerated depolymerisation of the actin structures in the differentiating cells (Fig. 6h). Given that BPTES treatment diminished protein production of the Glu codon containing genes (Fig. 5e), the disassembly of actin cytoskeleton is possibly attributed to ribosome stalling at Glu codons of actin-associated protein-coding mRNAs. Lastly, we examined whether the depolymerisation of actin cytoskeleton enhances thermogenic adipocyte differentiation. Upon cytochalasin D treatment to disrupt actin polymerisation, adipogenic cells differentiating into beige adipocytes exhibited increases in their expressions of differentiation markers, as with the cells differentiating into white adipocytes (Fig. 6i). Taken together, these results suggest that manipulating glutamate metabolism can control pan-adipocyte differentiation possibly via disassembly of actin cytoskeletal structures by the stalling-mediated gene down-regulation.
Discussion
Although a number of previous studies have delineated transcriptional and epigenetic controls for thermogenic adipocyte differentiation12,13,14,15,56, post-transcriptional regulation underlying the cellular differentiation is less thoroughly understood. We provide the first single source-originated profiles of total RNA, translated RNA, and protein levels during beige adipocyte differentiation, which will be a valuable resource to study how gene regulations at the mRNA and protein levels shape gene expressions for the phenotype acquisition of thermogenic adipocytes, given the difficulties in comparing separately-constructed transcriptomes, translatomes, and proteomes of adipocytes and preadipocytes in the previous studies. Moreover, high-resolution profiles of well-preserved mRNA-ribosome interactions will enable further investigations into previously unknown gene regulation at translational elongation step and unidentified peptide coding regions that are authentically translated by ribosomes.
Given that thermogenic adipocytes have distinct cellular metabolisms such as uncoupled respiration, to perform their specialised roles of heat production by burning calories, it is not surprising that gene regulation and metabolic controls reciprocally influence each other for thermogenic adipocyte differentiation. Uncoupled respiration by UCP1 expression and demethylation of Prdm promoter by increase in α-ketoglutarate are such examples24,25. However, it still remains elusive whether and how translational regulation of specific genes modulates a certain metabolic pathway for the differentiation process and vice versa. In this study, we unveiled two previously-unreported cross-talk pathways between translational and metabolic controls during beige adipocyte differentiation: selective translational down-regulation of OXPHOS component-coding genes and increased ribosome pausing at glutamate codons (Fig. 7). The former one results in metabolic changes in thermogenic adipocytes, whereas the latter one results from altered metabolism during adipocyte differentiation.

During beige adipocyte differentiation, OXPHOS complexes and TCA cycle components are differently regulated at translation level, leading to distinct stoichiometry and altering the related metabolic activities in mitochondria. On the other hand, elevated glutamate consumption causes potentiated ribosome pausing at glutamate codons, decreasing protein synthesis and mRNA stability of glutamate codon-rich genes such as actin cytoskeleton-associated genes.
Thermogenic adipocytes are featured with an abundant population of mitochondria, which is presumably driven by potent and overall transcriptional up-regulation of mitochondrial protein-coding genes. By virtue of the elevated transcription, OXPHOS and TCA cycle component proteins exhibit increases as well in a view of an individual cell. Our study suggests that there is an additional layer of gene regulation at the translation stage that engenders differential expressions of the mitochondrial protein-coding genes. Translational down-regulation of OXPHOS complexes except Complex II results in smaller increases of these complexes while a larger increase of Complex II in thermogenic adipocytes (Fig. 7), as shown in the analyses of our translatome and proteome data. The selective attenuation of protein synthesis for Complexes I, III, IV, and V is attributable to weakened mitochondrial translation in the differentiating cells. It has been previously reported that a cross-talk between mitochondrial and cytosolic translation enables coordinated synthesis of subunits derived from different origins, serving as a quality control mechanism to ensure the proper assembly of OXPHOS complexes30,31. Our study reveals that the strategy can be exploited for the phenotype acquisition of thermogenic adipocytes.
The higher proportion of Complex II among other complexes underpins different stoichiometry among OXPHOS complexes in a view of an individual mitochondrion in thermogenic adipocytes. Given that Complex II/SDH acts as an essential component of TCA cycle, the relatively higher expression of Complex II genes may provide metabolic benefits for thermogenic adipocytes to perform their role in burning calories through the enhanced activity of TCA cycle. Acetyl-CoAs yielded by active fatty acid oxidation in thermogenic adipocytes need to be effectively consumed by TCA cycle to produce NADH and dihydroflavine-adenine dinucleotide (FADH2) for OXPHOS57. Our observation of translational up-regulation of TCA-associated genes during beige and brown adipocyte differentiation also supports this scenario. On the other hand, Complex II, succinate hydrogenase, may solely provide a benefit for thermogenic adipocyte metabolism. Previous studies reported that extracellular succinates significantly contribute to thermogenic respiration in brown adipose tissue via their oxidation by SDH19. The relatively larger increases of Complex II may be for priming the cells to cope with the situation.
While OXPHOS complexes play essential roles in ATP generation by running electron transfer chains, they produce reactive oxygen species (ROS), especially via electron leakage at Complexes I and III. Excessive accumulation of ROS impinges on mitochondrial function and homoeostasis, leading to damages on various cellular processes58. Given that thermogenic adipocytes harbour densely packed mitochondria for uncoupled respiration, the attenuated increase in OXPHOS complexes by translational down-regulation may possibly be a strategy for the cells to circumvent the mitochondrial stresses. Further investigation is needed to explain how mitochondria in thermogenic adipocytes monitor or cope with ROS stresses while they are actively running OXPHOS for heat production and energy expenditure.
Inadequate tRNA charging and ribosome stalling have been observed in several pathological contexts including leukaemia and kidney cancer, as well as in cellular stress conditions such as nutrient depletion and ageing37,38,42,44. In this study, we provided compelling evidence of ribosome stalling on glutamate codons upon a metabolic transition under physiological circumstances of a non-pathological context. Adipogenesis involves metabolic remodelling of preadipocytes, including increases in glucose uptake and net glutamine production59, which facilitate differentiation of the precursors towards non-thermogenic adipocytes60,61. We observed a higher ratio of glutamine to glutamate throughout beige adipocyte differentiation, which suggests that glutamate to glutamine transition is commonly critical for both thermogenic and non-thermogenic adipocyte differentiation. This notion is supported by the increases and decreases in catabolic and anabolic enzymes for glutamine, respectively, in the mature adipocytes regardless of the adipocyte lineages. Moreover, reversing glutamate to glutamine transition hampered differentiation into thermogenic adipocytes as well, underpinning the impact of the metabolic remodelling on adipocyte differentiation, along with the previous studies60,62. Our study reveals that the rewired glutamine-glutamate metabolism can lead to reduced intracellular levels of glutamate-charged tRNAs, thereby augmenting ribosome pausing at Glu codons. The elevated stalling at Glu codons can consequently decelerate protein synthesis and accelerate mRNA decay of Glu codon-rich genes, such as actin cytoskeleton-associated genes, during adipocyte differentiation, which is supported by decent correlations of protein production rates and mRNA stabilities with Glu codon enrichments of individual genes. Collectively, our findings elucidate a hitherto unidentified gene regulatory programme underlying adipocyte differentiation, which is implemented at the mRNA translation stage and is triggered by a specific change in the metabolic pathways during the cellular differentiation.
We revealed that manipulating glutamate consumption alters actin cytoskeleton formation and differentiation rates of pre-to-mature adipocytes, which underscores the impact of the glutamate and glutamine metabolism on adipocyte differentiation and proposes the potentials of controlling the regulatory pathways to cope with adipocyte-related metabolic disorders63. Moreover, metabolic controls of glutamate and glutamine are known to play critical roles in diverse pathological contexts, including cancer64 and neurodegenerative disease65. Given that ribosome stalling has been reported to occur at amino acid codons other than Glu codon upon cellular illnesses and stresses in the previous studies38,41,42,43,44,66, it will certainly be intriguing to examine whether ribosome pauses at Glu codons are implicated in the pathologies associated with glutamate and glutamine metabolisms.
Given that the differentially attenuated translation of OXPHOS is exclusive to thermogenic adipocyte lineages while potentiated ribosome stalling at Glu codons is common in pan-adipocyte lineages, we speculate the two regulatory events may independently take place in the differentiating cells. Indeed, the genes encoding Complex I, III, IV, and V did not exhibit strong Glu-codon enrichment and noticeably elevated stalling during the differentiation, supporting that weakened translation of the cognate mRNAs is not directly caused by ribosome pauses at Glu codons. However, we still cannot exclude that the two translational controls indirectly affect each other in a certain biological context. One recent study67 reported that inhibition of glutaminase (encoded by Gls) ameliorated obesity-associated metabolic abnormalities and triggered thermogenic gene programmes in iWATs, underscoring the impact of altered glutamate metabolism on thermogenic adipocyte differentiation. It will be intriguing to examine whether and how the ribosome pauses are involved in the acquisition of thermogenic adipocyte-specific phenotypes, including the different stoichiometry of OXPHOS per mitochondrion in the obesity-related condition.
Methods
Animal model
Four-week-old male C57BL/6 mice were purchased from Orient to isolate adipose precursors. Mice were maintained under temperature- (23 °C) and humidity-controlled (40–60%) conditions with free access to food (normal chow diet; LabDiet, #5053) and water on a 12-h light/12-h dark cycle. All mice are age and sex matched for individual experiments. All animal care and procedures were conducted according to the protocols and guidelines approved by the Soonchunhyang University Institutional Animal Care and Use Committee.
Isolation of adipocyte precursor cells and cell culture
Four-week-old male C57BL/6 mice were sacrificed with carbon dioxide (CO2) inhalation followed by cervical dislocation. Adipose precursor cells were isolated from inguinal white adipose tissue and then homogenised in 5 mL of digestion buffer containing collagenase 1 (Worthington, LS004196), bovine serum albumin, and Dulbecco’s modified Eagle’s medium (DMEM) at 37 °C for 30 min. After homogenisation, the mixtures were filtered through a 100 μm cell strainer and centrifuged at 400 g for 5 minutes. The isolated cells were subsequently resuspended in a culture medium consisting of 10% foetal bovine serum (FBS, Corning) and 1% penicillin/streptomycin (Thermo Scientific) in DMEM (Corning). For differentiation to beige adipocytes, cells were exposed to beige differentiation induction medium containing 2 μg/mL dexamethasone, 0.5 mmol/L 3-isobutyl-1-methylxanthine, 125 μmol/L indomethacin, 5 μg/mL insulin, 1 μmol/L 3,3′,5-triiodo-L-thyronine, and 0.5 μmol/L rosiglitazone (all from Sigma-Aldrich). After two days, the cells were maintained in media without 3-isobutyl-1-methylxanthine, indomethacin, and dexamethasone from beige differentiation induction medium68,69. For white adipocyte differentiation, cells were exposed to a white differentiation induction medium containing 2 μg/mL dexamethasone, 0.5 mmol/L 3-isobutyl-1-methylxanthine, 5 μg/mL insulin, and 0.5 μmol/L rosiglitazone for 2 days. Then, differentiated cells were maintained in a medium containing 5 μg/mL insulin. Beige and white maintenance media were replaced every two days. 3T3-L1 cells were cultured in a medium comprising DMEM with 10% calf serum (Hyclone) and 1% penicillin/streptomycin. These cells underwent the same differentiation induction mentioned earlier for beige-like differentiation. Beige-like differentiated cells were maintained in differentiation media without indomethacin and dexamethasone from the beige differentiation induction media. Media were changed every two days70,71. For chemical treatment, a chemical-containing medium was added to the cultured cells every two days until harvesting for analysis, unless otherwise indicated. To inhibit the mitochondrial translation, cells were treated with 200 μg/ml chloramphenicol for two days with daily changes of maintenance media. For measuring the glutamate/glutamine ratio and checking the adipogenic gene expression with qPCR, cells were treated with 1 mM methionine sulfoximine (Sigma-Aldrich, M5379) and 20 mM glutamate (Sigma-Aldrich, G1251) in maintenance media. To inhibit glutaminase, 3T3-L1 cells were treated with 20 μM BPTES (Sigma-Aldrich, SML0601). Adipocyte precursor cells were treated with white adipocyte differentiation media containing 0.02 μg/mL cytochalasin D (Sigma-Aldrich, C8273) to induce F-actin disassembly. For small interfering RNAs (siRNA)-mediated Glul knock-down (KD), siRNAs were purchased from BIONEER, and lipofectamine RNAiMAX (Thermo Scientific, 13778075) was used to transfect siRNA into the cells, according to the manufacturer’s protocol in a final siRNA concentration of 100 nM. To evaluate the effect of Glul siRNA treatment on intracellular glutamate/glutamine ratio, a mixture of 3 different Glul siRNAs was transfected into precursor adipocyte cells twice. To confirm the impact of Glul KD on adipogenesis, preadipocytes were transfected by Glul siRNA for a day, harvested, and seeded to achieve an appropriate confluency of cells for differentiation. Next day, differentiation into beige adipocytes was induced. During differentiation, cells were transfected with Glul siRNA on days 1 and 3, and harvested on day 4. siRNA sequences were provided in Supplementary Data 8.
Preparation of lysates for transcriptomes, translatomes, and proteomes
Cultured cells were washed two times with 1X Phosphate-base buffer (PBS) and then incubated for 30 min on ice with the lysis buffer (10 mM/mL Tris-HCl, pH 7.4 (Thermo Scientific, AM9850G, AM9855G); 5 mM/mL MgCl2 (Thermo Scientific, AM9530G); 100 mM/mL KCl (Thermo Scientific, AM9640G); 2 mM/mL dithiothreitol (DTT, Thermo Scientific, 707265 ML); 100 μg/mL cycloheximide (CHX, Sigma-Aldrich, C1988); 1% Triton X-100 (Sigma-Aldrich, T8787); 1X protease inhibitor (Sigma-Aldrich, 539134); 2 μL/mL SUPERase inhibitor (Thermo Scientific, AM2696); and 2 μL/mL RNase inhibitor (Thermo Scientific, AM2694). After centrifuging the lysates at 20000 g for 15 min at 4 °C, the layers of lipids formed upon centrifugation were removed, and the supernatant was separated into three portions for RNA-seq, Ribo-seq, and mass spectrometry.
Ribo-seq and RNA-seq
Preparation of Ribo-seq and RNA-seq libraries
For RNA-seq, the supernatant was treated with TRIzol LS (Thermo Scientific, 10296028) to isolate the RNA. The total RNAs were used for the generation of a transcriptome library (RNA-Seq) using the TruSeq Stranded Total RNA Library Prep Gold (Illumina, 20020599). For Ribo-seq, the supernatant was treated with 3 U/mL RNase I (Thermo Scientific, EN0601) for 30 min at 25 °C to digest mRNA regions not associated with ribosomes. The subsequent procedures followed the previous study72. The samples were passed through MicroSpin S400 HR Columns (Cytiva, 27-5140-01) and prewashed with a polysome buffer (20 mM HEPES-KOH, pH 7.4 (Sigma-Aldrich, H0887); 5 mM MgCl2; 100 mM KCl; 2 mM DTT; and 100 μg/mL CHX). The filtered samples of ribosome-protected mRNA-containing lysate excluding the digested mRNA fragments were treated with TRIzol LS to isolate the RNA. Ribosomal RNAs were removed using a Ribo-Zero Gold Kit of the TruSeq Stranded Total RNA Library Prep Gold protocol (Illumina, 20020599). The RNA samples were then precipitated in 100% ethanol (Thermo Scientific, A995-1) at -20 °C, labelled with radioisotope γ-P32 ATP (PerkinElmer, NEG-502H-1) using T4 Polynucleotide kinase (T4 PNK, New England Biolabs, M0201L), and separated on a 10% denaturing Urea-PAGE (Sigma-Aldrich, U5128) gel. The signals were visualised and analysed using an Amersham Typhoon Biomolecular Imager (GE Healthcare, v2.0.0.6) and MultiGauge (v3.0), respectively. Radioisotope signals around ~30 nucleotides region were excised and eluted in RNA elution buffer (0.3 M NaOAc, Thermo Scientific, AM9740; 2% sodium dodecyl sulphate (SDS), Biosesang, SR2003-050-00) followed by RNA precipitation. The isolated RNA samples were then dephosphorylated using Antarctic Phosphatase (New England Biolabs, M0289S) and relabeled with γ-P32 ATP using the T4 PNK reaction. The labelled samples were separated on a 10% denaturing Urea-PAGE gel to remove free ATPs, followed by gel excision and RNA extraction. Adaptor ligation and PCR amplification were performed using a RealSeq®-AC miRNA Library Kit (RealSeq Biosciences, 500-00048). The adaptor-ligated PCR products were purified with SPRIselect magnetic beads and then subjected to sequencing in the NovaSeq 6000 system.
Pre-processing and alignment for Ribo-seq and RNA-seq libraries
Pre-processing and alignment procedure is based on the previous study72 with few modifications. Adaptor sequences (Illumina TruSeq RA3) at the 3′ ends of sequenced Ribo-seq reads were trimmed, and reads between 20 nt and 40 nt in length were selected using cutadapt (v3.4) with the following options: ‘-u 1 --overlap 6 --minimum-length 20 --maximum-length 40 --trimmed-only’. Ribo-seq and RNA-Seq reads were mapped to common noncoding RNAs (ncRNAs), including ribosomal DNA complete repeating units (Rfam accession RF00001, RF00002, RF01960, and RF02543), signal recognition particle DNA (Rfam accession RF00017), and genomic tRNA (GtRNAdb release 19) using HISAT2 (v2.2.1) with the --very-sensitive option. Next, unmapped reads were aligned to the mouse genome (GENCODE GRCm39 Release M27) by HISAT2 (v2.2.1) with the following options: ‘--pen-noncansplice 0 --very-sensitive --no-softclip --rna-strandness F -k 32 --secondary’. Using intersectBed of BEDTools and FilterSamReads of Picard, reads mapped to the mouse repeat regions (according to the UCSC genome annotation database) were eliminated. To determine the representative isoform from several mRNA isoforms for each gene, the following priorities of the GENCODE annotation (release M27) column were used: ‘basic’, ‘CCDS’, and ‘APPRIS principal’.
Quantification of gene expression levels in Ribo-seq and RNA-Seq libraries
Using the featureCounts of Subread (v2.0.3), Ribo-seq and RNA-seq reads aligned to the exonic region were counted. Raw counts were normalised by the median of ratios method to minimise the bias by grouped regulation of mitochondrial protein-coding genes. Protein-coding genes that had over 20 normalised counts in all replicates of at least one condition were kept. As the final step, 1 pseudocount was added to prevent the exclusion of genes such as thermogenic marker Ucp1 from analyses. Ribosome density values were calculated by dividing normalised counts of Ribo-seq by normalised counts of RNA-seq.
Proteomics
Sample preparation for mass spectrometry
For the cell lysate aliquot for mass spectrometry (MS), the buffer exchange was performed with the MS-compatible solution (8 M Urea in 50 mM Tris-HCl, pH 8.0) through centrifugal filtration using 10 kDa molecular weight cutoff filter (Amicon Ultra-0.5 Centrifugal Filter Unit). Proteins were denatured with 5 mM DTT at 25 °C for 1 hr, and free thiol groups were alkylated with 12.5 mM Iodoacetamide at 25 °C for 1 hr in dark. Protein samples were treated with sequencing-grade trypsin (Promega, V5111) at 1:20 enzyme-to-protein ratio (w/w) overnight at 25 °C after dilution under 2 M Urea with 50 mM Tris-HCl, pH 8.0. The digestion was quenched by adding formic acid to reach 3% formic acid. Total peptide concentration of each sample was measured by BCA assay after desalting of the digest by solid phase extraction (SPE) cleanup. The same amount of peptide from each sample, including nine samples (triplicates of 0, 4, and 8 days post-differentiation induction) and two pooling samples made from nine samples, was then labelled with TMT-11plex reagents following the manufacturer’s protocol. After 1 hr incubation, 5% aqueous hydroxylamine solution was added to stop the labelling reaction. The 11 TMT-labelled samples were combined, dried, and reconstituted in water containing 0.1% formic acid. The combined TMT-11plex labelled sample was desalted with SPE, dried, and dissolved in water.
High pH reversed-phase liquid chromatography for peptide fractionation
The dissolved TMT-11plex labelled sample was fractionated using a XBridge BEH Shield RP18 Column (130 Å, 2.5 μm, 4.6 × 150 mm, Waters) on NexeraXR HPLC (Shimadzu) with a 70 min gradient from 5% to 95% mobile phases B at a flow rate of 1.0 mL/min. Mobile phase A consisted of 5 mM ammonium formate in 100% water, and mobile phase B consisted of 5 mM ammonium formate in 95% acetonitrile; both buffers were adjusted to pH 10.0 with ammonium hydroxide. A total of 40 fractions were collected using an FRC-10A fraction collector (Shimadzu) after the start of elution in an interval of 1 min for each fraction. The initially collected 42 fractions were concentrated into 11 fractions. The secondary fractions 1 to 10 were composed of the initial fractions with intervals of 10 fractions. For example, the initial fractions 1, 11, 21, and 31 were assigned to the secondary fraction 1; the initial fractions 2, 12, 22 and 32 fractions were assigned to the secondary fraction 2. The final secondary fraction 11 comprised the initial fraction 41 and 42. The concentrated fractions were dried and kept at -80 °C until LC-MS/MS analysis.
Liquid chromatography TMT mass spectrometry
Eleven fractions were analysed using an LC-MS/MS system consisting of an Easy nLC 1200 (Thermo Scientific) and an Orbitrap Fusion Lumos mass spectrometer with a nano-electrospray source (EASY-Spray Sources). Peptides were first trapped in a precolumn (C18, 75 μm × 2 cm, nanoViper, Acclaim PepMap100, Thermo Scientific) and then applied to an analytical column (C18, 75 μm × 50 cm PepMap RSLC, Thermo Scientific) at a flow rate of 250 nL/min. The mobile phases were composed of 100% water (A) and 80% acetonitrile and 20% water (B), each containing 0.1% formic acid. The LC gradient began with 5% B, ramped to 7% B over 5 min, followed by 25% B over 78 min and increased to 40% B for 13 min and to 95% B over 5 min, then was held constant for 8 min, and ended with 5% B over 1 min. After a gradient, the column was re-equilibrated with 5% B for 10 min before the next run. The voltage applied to produce an electrospray was 1900 V. The Orbitrap Fusion Lumos was operated in data-dependent mode, automatically switching between MS and MS/MS with a 3 sec cycle time. Full scan MS spectra (400-2000 m/z) were acquired with a maximal ion injection time of 100 ms at a resolution of 120,000 and an automatic gain control (AGC) target value of 4.0 × 105. MS/MS spectra were acquired from 110 m/z at a resolution of 60,000 with a high energy collision dissociation (HCD) of 37.5% normalised collision energy within 1.2 Da isolation window. AGC target value was 5.0 × 104 with maximal ion injection time of 105 ms. The exclusion time for previously fragmented ions was 30 s within 10 ppm.
Protein identification and quantitation
The Integrated Proteomics Pipeline using built-in search engines (IP2, v6.5.5, Integrated Proteomics) was utilised for data analysis with the UniProt mouse protein database (January, 2022, Reviewed 17,102 proteins). The reversed sequences of all proteins were appended into the database for calculation of false discovery rate (FDR). ProLuCID was used to identify the peptides with a precursor mass error of 5 ppm and a fragment ion mass error of 50 ppm. Trypsin was selected as the enzyme, with two potential missed cleavages. TMT modification (+229.1629) at the N-terminus and lysine residue by the labelling reagent and carbamidomethylation at cysteine were chosen as static modifications. Oxidation at methionine was chosen as variable modification. Reporter ions were extracted from small windows (±20 ppm) around their expected m/z in the HCD spectrum. The output data files were filtered and sorted to compose the protein list using the DTASelect (The Scripps Research Institute, USA) with two and more peptides assignments for protein identification and a false positive rate less than 0.01.
A quantitative analysis was conducted using Census in the IP2 pipeline (Integrated Proteomics, USA) with only the unique peptides. The intensity at a reporter ion channel for a protein was calculated as the sum of this reporter ion’s intensities from all constituent peptides from the identified protein73. Reversed and potential contaminant proteins were removed. For the protein quantification, intensities of proteins were normalised by the median of ratios method.
Oil Red O staining
Cells were fixed with 4% paraformaldehyde solution (SolMate, SM-P01) for 30 min and washed twice with PBS. Cells were then incubated with 60% isopropanol (Supelco, 109634) for 5 min and stained with a filtered working solution of 60% Oil Red O (Sigma-Aldrich, O0625) for 20 min. After washing with water for three times, images were acquired using a microscope (Olympus, CKX53).
Western blotting
Total cell lysates were prepared using the lysis buffer (50 mM Tris-HCl pH 7.4, 400 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS). The protein samples were separated by SDS-PAGE and transferred to nitrocellulose membranes (Cytiva, 10600002). The membrane was blocked in 5% BSA and 0.1% Tween 20 in PBS for 1 hr and incubated with primary antibodies overnight at 4 °C. After several washes, the membrane was incubated with HRP-conjugated secondary antibodies for 40 min at RT. Protein bands were detected using Clarity Western ECL Blotting Substrates (Bio-Rad, 170-5060) on a ChemiDoc Imaging System. The membrane was rinsed with TDW 3 times after antibody detection and stained with Ponceau S (Sigma-Aldrich, P7170). Ponceau S staining image was produced using the ChemiDoc Imaging System. The antibodies used for western blotting in this study were: HSP90 (Cell Signaling Technology, 4874; 1:4000), UCP1 (Abcam, ab10983; 1:2000), PLIN1 (Perilipin A, Sigma-Aldrich, p1998; 1:2000), OXPHOS Rodent Cocktail (Abcam, ab110413; 1:2000), GLUL (Cell Signaling Technology, 80636; 1:1000), Goat anti-Rabbit IgG (H + L) Secondary Antibody, HRP (Thermo Scientific, 31460; 1:4000), Goat anti-mouse IgG (H + L) secondary antibody, HRP (Thermo Scientific, 31430; 1:4000).
Mitochondria isolation
Beige and white differentiated adipocytes were homogenised using a Dounce homogeniser to isolate the mitochondria, following the protocol provided by the Mitochondria Isolation Kit for Cultured Cells (Thermo Scientific, 89874). Differential centrifugation was subsequently employed to isolate mitochondrial fractions, utilising reagents A and C provided by the kit.
Mitochondrial OXPHOS complex activity assay
Mitochondrial OXPHOS complex I and II activities were measured with Complex I Enzyme Activity Microplate Assay Kit (Colorimetric) (Abcam, ab109721) and Succinate Dehydrogenase Activity Assay Kit (Colorimetric) (Abcam, ab228560), respectively. Assays were performed according to the manufacturer’s instructions. The isolated mitochondria from beige and white adipocytes were diluted with PBS. Their protein concentrations were measured using BCA assay with small aliquots. After that, for the OXPHOS complex I, 5 μg mitochondrial lysates per well was incubated for 3 h in an antibody-coated plate for immunocapture. After incubation, the plate was washed twice, and NADH substrate and dye were added. OD450 nm was recorded for each well at every 1 min for 30 min using BioTek Epoch Microplate Reader, and Complex I activity was calculated as the increase in OD450 nm in each well between 10 min and 30 min time points. For the OXPHOS complex II, 50 μg of isolated mitochondria was loaded in a plate, and SDH substrate mix and electron probe were added in each well. OD600 nm was recorded for each well at every 1 min for 1 hr using BioTek Epoch Microplate Reader. Complex II activity was determined as the decrease in OD600 nm between 2.35 min and 12.53 min. With the DCIP standard curve, the amount of reduced DCIP during reaction time can be calculated with the Δ OD value. This value reflects SDH activity. OD values were estimated with BioTek Epoch Microplate Reader. The time windows that showed linear decrease or increase were selected for calculation of activity, and the slope was calculated with linear least squares regression. The relative activity values were calculated from dividing the activity values of both beige and white adipocytes by average activities of white adipocytes.
Glutamine/Glutamate measurement
Glutamine/Glutamate-Glo™ Assay kit (Promega, J8021) was used to measure the ratio of glutamine and glutamate during the differentiation of beige adipocytes. Intracellular glutamine/glutamate levels were determined from cell lysates in the 12-well plate scale, following a modified manufacturer’s instruction. Cells were washed twice with cold PBS and treated with 150 μl PBS and 75 μl inactivation solution I (0.3 N HCl) for 5 min. Next, 75 μl Tris solution I (450 mM Tris pH 8.0) was added and incubated for 1 min. Lysates were collected and diluted 20-fold with PBS. The sample was divided into two designated wells of a white luminometer 96-well plate to measure ‘total glutamine-plus-glutamate’ and ‘glutamate-only’. For the glutamine-plus-glutamate measurement, glutaminase in the glutaminase buffer was added. For the glutamate-only measurement, glutaminase buffer without glutaminase was added. Glutamate and glutamine at various concentrations are also measured to be used as standards for sample quantification. After incubation at RT for 40 min, Glutamate Detection Reagent was added to each well. The samples were incubated at RT for 1 h, and luminescence was measured using the GloMax Discover Microplate Reader (Promega, GM3000).
Quantitative real-time PCR
Total RNA was extracted from cells using TRIzol (Life Technologies, 15596018) and subsequently treated with DNase I (Takara Bio, 2270 A). cDNA was synthesised with 0.5 μg of DNase-treated RNA using ReverTra Ace qPCR RT Master Mix (Toyobo, FSQ-201). cDNA was amplified with SYBR Green real-time PCR Master Mix (Toyobo, QPK-201) and analysed by QuantStudio Real-Time PCR system. The mRNA level of the individual target gene was determined with normalisation to 36B4 level. Primer sequences were provided in Supplementary Data 8.
tRNA aminoacylation assay
Total RNA samples were prepared in 0.3 M acetate/10 mM EDTA buffer (pH 4.5) after isolation by TRIzol (Life Technologies, 15596018) according to the manufacturer’s protocol. RNA samples were treated with either 50 mM NaCl (control) or 50 mM NaIO4 (oxidised condition) in 100 mM acetate buffer (pH 4.5) for 20 min, subsequently quenched with 100 mM glucose for 20 min, purified with Microspin G-50 columns (Cytiva, 27-5330-01), and then precipitated with ethanol. The precipitated RNA was resuspended in 50 mM Tris-Cl (pH 9.0) and incubated for 50 min at 37 °C for deacylation. The deacylation reaction was quenched with acetate buffer. RNA was precipitated and ligated with a 5’ adenylated DNA adaptor, using truncated KQ mutant T4 RNA ligase 2 (New England Biolabs, M0373) for 3 h at 25 °C38. The reverse transcription was performed with the SuperScriptIV First-Strand Synthesis System (Thermo Scientific, 18091050) using the specific primer complementary to the DNA adaptor. Relative tRNA levels were analysed by qRT-PCR using tRNA isodecoder-specific forward primer and reverse primer complementary to the junction of the 3’ end of tRNA and the adaptor42,44. Yeast tRNAPhe (Sigma-Aldrich, R4018) was added to each sample and used for the normalisation. Primers were designed based on reference tRNA sequences retrieved from the GtRNA database74. Adaptor and primer sequences are provided in Supplementary Data 8.
tRNA northern blot
For tRNA northern blot, TRIzol-purified total RNA pellet was dissolved in ultrapure water and divided in half. One half was stored as final concentration of 10 mM NaOAc (pH 4.5)/1 mM EDTA buffer for acylated samples, and the other half was diluted in final concentration of 100 mM Tris-HCl (pH 9.0) for deacylation control. Deacylation of tRNA was done at 37 °C for 1 h and subsequently quenched to pH 5.3 by addition of pH 4.5 NaOAc. Deacylated control was re-precipitated with ethanol and suspended in 10 mM NaOAc (pH 4.5)/1 mM EDTA buffer. For acid urea polyacrylamide gel electrophoresis (acid Urea-PAGE), acylated RNA samples and deacylated control were mixed with 2X acidic loading buffer (0.1 M NaOAc pH 4.5, 8 M Urea, 0.05% Bromophenol blue, 0.05% Xylene Cyanol), denatured at 65 °C for 15 min, and quickly cooled down on ice. 17.2 cm long, 1 mm thick 10% acid Urea-PAGE gel (10% polyacrylamide [19:1 acrylamide/bisacrylamide], 8 M Urea, 0.1 M NaOAc pH 4.5) were casted for 2 h at room temperature after adding of ammonium persulfate (APS) and tetramethylethelenediamine (TEMED), and were pre-electrophoresed by 205 V (12 V/cm) constant for 1 hr at 4 °C prior to sample loading. Samples were loaded in the middle part of the wells, and the wells adjacent to the loaded wells were loaded with the same amount of blank RNA to prevent band curving. Electrophoresis was carried out for 6.5 - 8 h at 4 °C by 80 mA constant until the tRNA region was well separated. While the gel was running, excessive bubbles around the wells were removed every 1 hr, and the running buffer was replenished after 5 h of running. After gel running, RNA quality check prior to the transfer was done with SYBRTM Gold (Thermo Scientific, S11494) staining following the manufacturer’s protocol. tRNA region of the gel was excised and transferred to BrightStarTM-Plus Positively Charged Nylon Membrane (Thermo Scientific, AM10104) by semi-dry transfer using 0.5X Tris-Borate-EDTA (TBE) buffer at 2.5 mA/cm2 for 100 min. tRNAs were UV-crosslinked by irradiating 360 mJ/cm2 twice, and the membrane was baked at 80 °C for at least 30 min. Probes for tRNAGlu(TTC) were 5’-[32P] radiolabeled with [γ-P32]-ATP by T4 PNK (NEB, M0201L), purified using phenol, and finally dissolved in Tris-EDTA (TE) buffer. Membrane was pre-hybridised with sperm DNA at 37 °C for 30 min, and probe hybridisation was carried out at 37 °C for at least 2 h in PerfectHyb Plus Hybridisation Buffer (Sigma-Aldrich, H7033). After the hybridisation, the membrane was quickly washed three times with Sol I (2X SSC, 0.05% SDS), mildly rocked in Sol I for 30 min twice, and then mildly rocked in Sol II (0.1X SSC, 0.1% SDS) for 15 min three times. The membrane was exposed to the Fuji Imaging Plate, and the image was scanned by Amersham Typhoon.
Reporter assay
Glu or Ala codon-rich EGFP reporter was constructed by incorporating the Glu or Ala codon-rich oligo and the EGFP fragment into the pLVX TetOne Puro vector (Clontech, 631849). EGFP sequence was obtained from a pLenti CMV GFP Hygro (Addgene, #17446) through PCR amplification. The Glu or Ala codon-rich oligo, coupled with a downstream flexible linker (GGGGSGGGGS) was synthesised by GENEWIZ, Inc, and IDT, respectively. The sequence of the Glu or Ala codon-rich oligo is detailed in Supplementary Data 8. For the reporter assay in 3T3-L1 cells, lentivirus expressing the reporter was generated by transfecting a mixture of Glu or Ala codon-rich EGFP, psPAX2 (Addgene, #12260), and pMD2.G (Addgene, #12259) plasmids into HEK293T cells using lipofectamine 3000 reagent (Thermo Scientific, L3000015). psPAX2 and pMD2.G were gifts from Didier Trono. At 72 h post-transfection, the supernatant was collected, centrifuged at 500 g for 10 min to remove cell debris, and concentrated by Lenti-X concentrator (Takara Bio, PT4421-2). The concentrated viral mix was centrifuged at 1500 g for 45 min at 4 °C, and the supernatant was removed. The viral pellet was resuspended with the appropriate media. 3T3-L1 cells in 6 well plates were infected with virus in the presence of 10 μg/mL of polybrene (Sigma-Aldrich, TR-1003-G). On the following day, the viral mix from the plates was replaced with the appropriate media. After 24 h, 3T3-L1 cells were harvested, centrifuged at 200 g for 5 min, and seeded in 12 well plates. One day later, cells were treated with 20 μM of BPTES (Sigma-Aldrich, SML0601) for 24 h. Subsequently, the expression of Glu or Ala codon-rich EGFP was induced by 1 μg/ml of doxycycline with 20 μM of BPTES for an additional 24 h. For flow cytometry, cells expressing Glu or Ala codon-rich EGFP were harvested and centrifuged at 200 g for 5 min. The cell pellet was resuspended in FluoroBriteTM Buffer (Thermo Scientific, A1896701) and put on ice. EGFP signal was measured using a FACSCanto II (Becton Dickinson And Company) with 488/530 nm (ex/em). Data was analysed using FlowJo (v10.9.0).
Immunocytochemistry
3T3-L1 cells were seeded on coverslips (Marienfeld Superior, 0101050) coated with 0.1% gelatin solution (Welgene, LS 023-01) in 6-well plates. One day later, differentiation was induced for 48 h using an induction media. The cells were then maintained in culture with the appropriate media for up to 8 days. Cells on the coverslips were fixed for 15 min with 4% formaldehyde (Thermo Scientific, 28908) in phosphate-buffered saline (PBS, Sigma-Aldrich, P4417) and rinsed twice with PBS. Permeabilization was performed by treating the cells with 0.5% Triton X-100 (Sigma-Aldrich, 99443) in PBS for 10 min, followed by three washes with PBS. Cells were blocked for 30 min using a solution containing 1% bovine serum albumin (BSA, BioShop Life Science, ALB001.100) and 0.05% Tween 20 (Sigma-Aldrich, P9416) in PBS, and washed three times with PBS. Cells were incubated with anti-PLIN1 antibodies (Sigma-Aldrich, P1998; 1:200) in the blocking solution for 1 hr at room temperature (RT), followed by incubation with anti-rabbit secondary antibodies (Thermo Scientific, A32731; 1:400), DAPI (Sigma-Aldrich, MBD0015; 1:2000), and rhodamine conjugated phalloidin (Thermo scientific, R415; 1:400) in the blocking solution for 1 hr. After washing, coverslips were mounted to microscope slides (Thermo Scientific, 1255015) with SlowFade Antifade Mountants (Thermo Scientific, MAN0018916). Imaging was performed using an LSM 710 confocal microscope (Carl Zeiss) and analysed using Zeiss Zen 2012 (blue edition, v1.1.1.0.) software. Binary images were produced using the image to binary art converter function provided by OnlineTools.
Bioinformatics
Reproducibility check of transcriptomes, translatomes, and proteomes by the principal component analysis
StandardScaler and PCA of scikit-learn (v1.2.2)75 were used to scale the normalised expression tables of transcriptomes, translatomes and proteomes to perform the principal component analysis.
Gene set enrichment analysis with ribosome density
To estimate the change of ribosome density during beige adipocyte differentiation, signal to noise ratio (μA-μB/σA + σB) was calculated by comparing 4 d vs 0 d or 8 d vs 0 d. Using the signal to noise ratio as the ranked metric, GSEAs were carried out by prerank of GSEApy (v1.0.6)76 with the following options: ‘min_size = 15, max_size = 500, permutation_num = 3000, seed = 6’. Gene set collections from MSigDB77 were applied for analyses.
Protein-protein interaction analysis
As a downstream analysis of the GSEA, STRING analysis was performed with leading edge subsets from the three terms most significantly and commonly enriched in both GSEA results. Genes included in subsets at least once were used for analysis. Analysis was performed by STRING v12.0 (string-db.org)28 with the following options: ‘network type = physical subnetwork, meaning of network edges = confidence, active interaction sources = (text mining, experiments, databases), minimum required interaction score = 0.4’. Then, information of the network and functional annotations were exported for visualisation.
Gene list enrichment analysis
Gene list enrichment analyses in this study were performed using the enrichr of GSEApy (v1.0.6)76. To prevent redundant enrichment of pathways or terms, MitoCarta pathways without any child pathways were selected from MitoCarta3.078 for analysis in Fig. 2b. With the same purpose, gene ontology terms with levels between 3 and 5 were used for analysis in Fig. 6e.
Determination of P-site and A-site codons of ribosome-protected fragments (RPFs)
Meta-gene analysis was performed for each length of RPFs. Based on the robust peaks of start codon and stop codon, offsets between 5’ end of RPFs and P- and A-site codons were determined. For RPFs of 30-33 nucleotides in length, P-site offsets of 12 and 13, and A-site offsets of 15 and 16 were used for the following analyses.
Calculation of subsequence (ribosomal A-site) abundance shift
The analysis followed the process described in Loayza-Puch et al.’s study38. A-site codons of RPFs were counted for each transcript, and transcripts that have over 200 RPFs in every sample were selected. Percentages of codon abundance at A-sites and their changes during differentiation were calculated at the transcript level. Average percentage changes of transcripts were applied for visualisation.
Calculation of stalling score
The calculation method of stalling score was based on the Stein et al.’s method to calculate pause score37 with some modifications. In the following procedure, count refers to the number of RPFs that have an A-site on the indicated codon position. Transcripts with CDS length of more than 100 nucleotides were initially selected. After exclusion of the first 10 and last 10 sense codons, transcripts were additionally filtered out with the following criteria: ‘over 90% of codons have no counts’ or ‘average total counts of replicates <100 in at least one condition’ or ‘average read depth of replicates <0.5 in at least one condition’. Stalling score of each codon position was calculated by dividing the counts of the codon position by the average read counts of codons in the same transcript. Only sites with stalling score > 0 for every condition were exploited for further analyses. Stalling scores of Reid et al.’s and Martinez et al.’s Ribo-seq libraries33,34 were calculated with the same procedure.
Identification of ribosome stalling sites dependent upon beige adipocyte differentiation
The method to identify differentiation-dependent ribosome stalling sites was based on the Stein et al.’s method37 with some modifications. After the same filtering steps in the stalling score calculation, read counts at each codon position were averaged across the replicates and rounded to the closest integer. For each codon position, a contingency table of counts of the codon and total counts of transcript except the codon in each condition (0 d and either 4 d or 8 d) was generated. Odds ratio was calculated by the following formula (n and N refers to counts of the codon and total counts of transcript except the codon, respectively. ‘day’ is 4 d or 8 d): Odds ratio = (nday / n0d) / (Nday / N0d). p-values were calculated by the two-sided Fisher’s exact tests with the contingency tables and corrected by Benjamini-Hochberg method.
Evaluation of ribosome occupancy around each codon
Differential ribosome occupancy was evaluated by the diricore analysis38. Transcripts that have at least one average count mapped to CDS per codon were used to calculate relative ribosome occupancy. Counts of each transcript were internally normalised by the total counts of the transcript. Then, for every codon position of each transcript, normalised counts from 150 nt upstream to 150 nt downstream were acquired. Average normalised counts of all acquired windows represent the relative ribosome occupancy. Differential ribosome occupancy was calculated by subtracting relative ribosome occupancies between different conditions.
Estimation of mRNA stability changes
Using the exonic and intronic counts, relative mRNA stability was calculated by REMBRANDTS51 with the linear biasMode and 0.6 stringency. Exonic and intronic reads were counted by the Counting Reads for Intronic and Exonic Segments (CRIES) pipeline, which is accessible through the github page of REMBRANDTS.
Reanalysis of publicly available APEX-seq dataset
From Fazal et al.’s study29, we utilised the data of mRNA enrichment near outer mitochondrial membrane (OMM) upon treatment of cycloheximide (CHX) or puromycin (PUR), which are identical with Cycloheximide_L2FC and Puromycin_L2FC from Supplementary Data 4 of their study, respectively. In Fig. 2 and Supplementary Fig. 2, ‘Ribosome dependent OMM enrichment’ score was calculated by subtracting ‘OMM enrichment upon PUR’ from the ‘OMM enrichment upon CHX’. ‘Ribosome independent OMM enrichment’ score is identical with ‘OMM enrichment upon PUR’. After calculation of enrichment scores, the table was merged with a table containing ribosome density values by Ensembl gene IDs for further analyses.
Re-analysis of publicly available Ribo-seq and RNA-seq libraries constructed using adipocyte lineage cells and adipose tissues
Ribo-seq and RNA-seq raw result files of Reid et al.’s and Martinez et al.’s studies33,34 were downloaded from Gene Expression Omnibus (GSE89108 and GSE198109, respectively). Except the adaptor sequence (Illumina TruSeq Read 1) for trimming, pre-processing, alignment, and gene expression quantification procedure was applied in the same way as applied to the libraries in this study. The only exception is that ‘over 5 normalised counts’ criterion is applied for Ribo-seq libraries of Reid et al.’s study due to relatively low sequencing depth.
Re-analysis of publicly available mass spectrometry libraries constructed using adipocyte lineage cells
Raw mass spectrometry files of Martinez et al.’s studies34 were downloaded from MassIVE (MSV000089022). To process the raw files, the DIA-NN software platform (v1.8.1) was used79. To construct the spectral library, 24 DDA raw data were searched using DIA-NN with fasta file (uniprot_all_Mus_musculus_20200810_reviewed_All_Novo_BAT_WAT_beige_smORFs_noBLASTp_final_PRTC) which Martinez et al. provided. Regarding the general parameters, digestion mode was set to specific with Trypsin/P protease, allowing maximum of one missed cleavage. Carbamidomethylation of cysteine was set as fixed modification. Oxidation of methionine and acetylation of the N-terminus were set as variable modifications. The parameters for peptide length range, precursor charge range, precursor m/z range, and fragment ion m/z range as well as other software parameters were used with their default values. 1% precursor false discovery rate (FDR) cutoff was applied, and MS1 and MS2 mass tolerances were automatically determined by DIA-NN.
To process the 15 DIA raw files, the ‘report-lib.tsv’ spectral library file constructed from 24 DDA raw data was used as a database for searching with DIA-NN. All other parameters were set the same as the DDA search method.
6495 and 4803 proteins were totally identified and commonly quantified respectively from 15 raw data (‘report-first-pass.pg_matrix.tsv’ generated by DIA-NN). Intensities of proteins were normalised by the median of ratios method.
For Supplementary Fig. 3f, to calculate the relative protein levels among mitochondrial proteins, mitochondrial proteins reported by MitoCarta3.078 were selected, and their intensities were normalised by the median of ratios method.
Re-analysis of mouse white adipose tissue single-nucleus transcriptome
A publicly available single-nucleus RNA-Seq dataset of mouse white adipose tissue45 was downloaded from Gene Expression Omnibus (GSE176171). Data processing was carried out using Seurat (v4.3.0.1)80 in R (v4.2.0). Cells with unique feature counts <200 or >5000, and >2% mitochondrial counts were removed. Then, data was normalised using the NormalizeData with the following options: ‘normalisation.method = “LogNormalize”, scale.factor = 10000’. 4000 most variable genes were identified by FindVariableFeatures with the ‘vst’ selection method. After scaling the data by applying ScaleData using all genes that passed the filtering, principal component analysis was performed with the variable genes. Neighbours and clusters were found by FindNeighbors and by FindClusters with 0.5 resolution, respectively. Uniform Manifold Approximation and Projection (UMAP) was performed with 30 neighbours and minimum distance 0.3.
Statistics
For the gene set enrichment analysis and gene list enrichment analysis, statistics calculated by ‘prerank’ and ‘enrichr’ of GSEApy (v1.0.6)76 were utilised, respectively. For the identification of genes with decreased or increased ribosome density, glmQLFit, glmQLFTest, and other functions of edgeR (v4.0.2)81 were used. Remaining statistical tests performed for each analysis were indicated in the figure legends. Paired t-test, unpaired t-test, Mann-Whitney U test, Fisher’s exact test, and linear least squares regression were carried out by ttest_rel, ttest_ind, mannwhitneyu, fisher_exact, and linregress of SciPy (v1.9.3)82, respectively. Locally weighted scatterplot smoothing (LOWESS) was performed by lowess of statsmodels (v0.13.5). NS is an abbreviation of ‘not significant’ and represents p-value > 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw RNA-seq and Ribo-seq data files have been deposited in Gene Expression Omnibus under the accession code GSE247715, and all raw mass spectrometry data files have been deposited at MassIVE under the accession code MSV000093396 (PXD046925). Source Data are provided with this paper. Publicly available datasets used in this study were downloaded from Gene Expression Omnibus and MassIVE under the accession code GSE8910833, GSE19810934, GSE17617145, and MSV00008902234. Source data are provided with this paper.
Code availability
All custom codes used for the analyses of RNA-seq, Ribo-seq, and mass spectrometry datasets and for the visualization of experimental results in this study have been deposited in GitHub and Zenodo: https://github.com/DaehwaYoun/Beige_Ad and https://doi.org/10.5281/zenodo.1495827983.
References
Harms, M. & Seale, P. Brown and beige fat: development, function and therapeutic potential. Nat. Med 19, 1252–1263 (2013).
Bartelt, A. & Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 10, 24–36 (2014).
Sidossis, L. & Kajimura, S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 125, 478–486 (2015).
Wu, J. et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150, 366–376 (2012).
Kajimura, S., Spiegelman, B. M. & Seale, P. Brown and beige fat: Physiological roles beyond heat generation. Cell Metab. 22, 546–559 (2015).
Ikeda, K., Maretich, P. & Kajimura, S. The common and distinct features of brown and beige adipocytes. Trends Endocrin Met 29, 191–200 (2018).
Lee, Y. H., Petkova, A. P., Mottillo, E. P. & Granneman, J. G. In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab. 15, 480–491 (2012).
Rosenwald, M., Perdikari, A., Rülicke, T. & Wolfrum, C. Bi-directional interconversion of brite and white adipocytes. Nat. Cell Biol. 15, 659–667 (2013).
Nguyen, T. T. V., Vu, V. V. & Pham, P. V. Transcriptional factors of thermogenic adipocyte development and generation of brown and beige adipocytes from stem cells. Stem Cell Rev. Rep. 16, 876–892 (2020).
Inagaki, T., Sakai, J. & Kajimura, S. Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nat. Rev. Mol. Cell Biol. 17, 480–495 (2016).
Yoneshiro, T. et al. Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Investig. 123, 3404–3408 (2013).
So, J. et al. Chronic cAMP activation induces adipocyte browning through discordant biphasic remodeling of transcriptome and chromatin accessibility. Mol. Metab. 66, 101619 (2022).
Lee, S. et al. Remodeling of gene regulatory networks underlying thermogenic stimuli-induced adipose beiging. Commun. Biol. 5, 584 (2022).
Rajbhandari, P. et al. IL-10 Signaling remodels adipose chromatin architecture to limit thermogenesis and energy expenditure. Cell 172, 218–233.e217 (2018).
Brunmeir, R. et al. Comparative transcriptomic and epigenomic analyses reveal new regulators of murine brown adipogenesis. Plos Genet 12, e1006474 (2016).
Schwanhäusser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Vogel, C. & Marcotte, E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13, 227–232 (2012).
Shapira, S. N. & Seale, P. Transcriptional control of brown and beige fat development and function. Obes. (Silver Spring) 27, 13–21 (2019).
Mills, E. L. et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560, 102–106 (2018).
Timmons, J. A. et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. P Natl Acad. Sci. USA 104, 4401–4406 (2007).
Panic, V. et al. Mitochondrial pyruvate carrier is required for optimal brown fat thermogenesis. Elife 9, e52558 (2020).
Li, X., Egervari, G., Wang, Y., Berger, S. L. & Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 19, 563–578 (2018).
Martinez-Reyes, I. & Chandel, N. S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102 (2020).
Yang, Q. et al. AMPK/alpha-ketoglutarate axis dynamically mediates dna demethylation in the prdm16 promoter and brown adipogenesis. Cell Metab. 24, 542–554 (2016).
Tian, Q. Y. et al. Dietary alpha-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice. Aging Cell 19, e13059 (2020).
van Heesch, S. et al. The translational landscape of the human heart. Cell 178, 242–260 (2019).
Harnett, D. et al. A critical period of translational control during brain development at codon resolution. Nat. Struct. Mol. Biol. 29, 1277–1290 (2022).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Fazal, F. M. et al. Atlas of Subcellular RNA localization revealed by APEX-Seq. Cell 178, 473–490.e426 (2019).
Couvillion, M. T., Soto, I. C., Shipkovenska, G. & Churchman, L. S. Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499–503 (2016).
Richter-Dennerlein, R. et al. Mitochondrial protein synthesis adapts to influx of nuclear-encoded protein. Cell 167, 471–483.e410 (2016).
Molenaars, M. et al. A conserved mito-cytosolic translational balance links two longevity pathways. Cell Metab. 31, 549–563.e547 (2020).
Reid, D. W., Xu, D., Chen, P., Yang, H. & Sun, L. Integrative analyses of translatome and transcriptome reveal important translational controls in brown and white adipose regulated by microRNAs. Sci. Rep.-Uk 7, 5681 (2017).
Martinez, T. F. et al. Profiling mouse brown and white adipocytes to identify metabolically relevant small ORFs and functional microproteins. Cell Metab. 35, 166–183.e111 (2023).
Ingolia, N. T., Lareau, L. F. & Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 (2011).
Bartok, O. et al. Anti-tumour immunity induces aberrant peptide presentation in melanoma. Nature 590, 332–337 (2021).
Stein, K. C., Morales-Polanco, F., van der Lienden, J., Rainbolt, T. K. & Frydman, J. Ageing exacerbates ribosome pausing to disrupt cotranslational proteostasis. Nature 601, 637–642 (2022).
Loayza-Puch, F. et al. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530, 490–494 (2016).
Xie, R. et al. Activation of METTL3 promotes white adipose tissue beiging and combats obesity. Diabetes 72, 1083–1094 (2023).
Chi, J. Y. et al. Three-dimensional adipose tissue imaging reveals regional variation in beige fat biogenesis and PRDM16-dependent sympathetic neurite density. Cell Metab. 27, 226–236.e223 (2018).
Loayza-Puch, F. et al. TGFbeta1-induced leucine limitation uncovered by differential ribosome codon reading. EMBO Rep. 18, 549–557 (2017).
Thandapani, P. et al. Valine tRNA levels and availability regulate complex I assembly in leukaemia. Nature 601, 428–433 (2022).
Darnell, A. M., Subramaniam, A. R. & O’Shea, E. K. Translational control through differential ribosome pausing during amino acid limitation in mammalian cells. Mol. Cell 71, 229–243.e211 (2018).
Pavlova, N. N. et al. Translation in amino-acid-poor environments is limited by tRNA charging. Elife 9, e62307 (2020).
Emont, M. P. et al. A single-cell atlas of human and mouse white adipose tissue. Nature 603, 926–933 (2022).
Navickas, A. et al. No-Go Decay mRNA cleavage in the ribosome exit tunnel produces 5′-OH ends phosphorylated by Trl1. Nat. Commun. 11, 122 (2020).
D’Orazio, K. N. et al. The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during No Go Decay. Elife 8, e49117 (2019).
Doma, M. K. & Parker, R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440, 561–564 (2006).
Powers, K. T., Szeto, J. Y. A. & Schaffitzel, C. New insights into no-go, non-stop and nonsense-mediated mRNA decay complexes. Curr. Opin. Struc Biol. 65, 110–118 (2020).
Mishima, Y., Han, P. X., Ishibashi, K., Kimura, S. & Iwasaki, S. Ribosome slowdown triggers codon-mediated mRNA decay independently of ribosome quality control. Embo J. 41, e109256 (2022).
Alkallas, R., Fish, L., Goodarzi, H. & Najafabadi, H. S. Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer’s disease. Nat. Commun. 8, 909 (2017).
Mori, M., Nakagami, H., Rodriguez-Araujo, G., Nimura, K. & Kaneda, Y. Essential Role for miR-196a in brown adipogenesis of white fat progenitor cells. Plos Biol. 10, e1001314 (2012).
Nobusue, H. et al. Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat. Commun. 5, 3368 (2014).
Hansson, B. et al. Adipose cell size changes are associated with a drastic actin remodeling. Sci. Rep.-Uk 9, 12941 (2019).
Chen, L., Hu, H. M., Qiu, W. M., Shi, K. K. & Kassem, M. Actin depolymerization enhances adipogenic differentiation in human stromal stem cells. Stem Cell Res 29, 76–83 (2018).
Rodó, J. et al. Integrated gene expression profiles reveal a transcriptomic network underlying the thermogenic response in adipose tissue. Sci. Rep.-Uk 13, 7266 (2023).
Townsend, K. L. & Tseng, Y. H. Brown fat fuel utilization and thermogenesis. Trends Endocrin Met 25, 168–177 (2014).
Chen, Q., Vazquez, E. J., Moghaddas, S., Hoppel, C. L. & Lesnefsky, E. J. Production of reactive oxygen species by mitochondria - Central role of complex III. J. Biol. Chem. 278, 36027–36031 (2003).
Oates, E. H. & Antoniewicz, M. R. Coordinated reprogramming of metabolism and cell function in adipocytes from proliferation to differentiation. Metab. Eng. 69, 221–230 (2022).
Velickovic, K. et al. Targeting glutamine synthesis inhibits stem cell adipogenesis in vitro. Cell Physiol. Biochem 54, 917–927 (2020).
Hauner, H., Rohrig, K., Spelleken, M., Liu, L. S. & Eckel, J. Development of insulin-responsive glucose uptake and GLUT4 expression in differentiating human adipocyte precursor cells. Int J. Obes. Relat. Metab. Disord. 22, 448–453 (1998).
Okuro, K. et al. Glutamine deficiency induces lipolysis in adipocytes. Biochem Biophys. Res Commun. 585, 155–161 (2021).
Petrus, P. et al. Glutamine links obesity to inflammation in human white adipose tissue. Cell Metab. 31, 375–390.e311 (2020).
Altman, B. J., Stine, Z. E. & Dang, C. V. From krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634 (2016).
Andersen, J. V. et al. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 196, 108719 (2021).
Rubio, A., Ghosh, S., Mülleder, M., Ralser, M. & Mata, J. Ribosome profiling reveals ribosome stalling on tryptophan codons and ribosome queuing upon oxidative stress in fission yeast. Nucleic Acids Res. 49, 383–399 (2021).
Lecoutre, S. et al. Reduced adipocyte glutaminase activity promotes energy expenditure and metabolic health. Nat. Metab. 6, 1329–1346 (2024).
Lee, J. M. et al. Reduction in endoplasmic reticulum stress activates beige adipocytes differentiation and alleviates high fat diet-induced metabolic phenotypes. Biochim Biophys. Acta Mol. Basis Dis. 1867, 166099 (2021).
Aune, U. L., Ruiz, L. & Kajimura, S. Isolation and Differentiation of Stromal Vascular Cells to Beige/Brite Cells. Jove-J Vis Exp, 50191 (2013).
Karamanlidis, G., Karamitri, A., Docherty, K., Hazlerigg, D. G. & Lomax, M. A. C/EBPβ reprograms white 3T3-L1 preadipocytes to a brown adipocyte pattern of gene expression. J. Biol. Chem. 282, 24660–24669 (2007).
Xue, H. L. et al. Molecular signatures and functional analysis of beige adipocytes induced from in vivo intra-abdominal adipocytes. Sci. Adv. 4, eaar5319 (2018).
Kim, J. et al. SARS-CoV-2 infection engenders heterogeneous ribonucleoprotein interactions to impede translation elongation in the lungs. Exp. Mol. Med. 55, 2451–2552 (2023).
Raso, C. et al. Characterization of breast cancer interstitial fluids by TmT Labeling, LTQ-Orbitrap velos mass spectrometry, and pathway analysis. J. Proteome Res 11, 3199–3210 (2012).
Chan, P. P. & Lowe, T. M. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189 (2016).
Pedregosa, F. et al. Scikit-learn: machine learning in python. JMLR 12, 2825–2830 (2011).
Fang, Z., Liu, X. & Peltz, G. GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39, btac757 (2023).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Rath, S. et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Youn, D. et al. Cross-talks between metabolic and translational controls during beige adipocyte differentiation. Zenodo https://doi.org/10.5281/zenodo.14958279 (2025).
Acknowledgements
We thank the members of our laboratories for their discussions and assistance throughout this project. We are grateful to Dr. Kae Won Cho and Dr. Jong-Seok Moon (Soonchunhyang University) for discussion and technical advice. We also thank Kevin Dong Jun Min (Gwangju Institute of Science and Technology) for reading the manuscript and helpful comments. This work was supported through the Basic Science Research Program by National Research Foundation of Korea (NRF) (2020R1C1C1003667 and RS-2023-00209176 to J.C; 2019M3E5D3073092, RS-2023-00219563, and RS-2024-00345327 to M.L); and the ‘2022 Joint Research Project of Institutes of Science and Technology’ (to J.C.); This work was also supported by a GIST-CNUH research Collaboration grant funded by the GIST in 2024 (to J.C.) and a grant from the Korea Basic Science Institute (KBSI) (Research Grant No. A423100 to J.Y.L.). Imaging and flow cytometry analysis were performed using the Zeiss LSM 710 confocal microscope and the FACS Canto available at the Soonchunhyang Biomedical Science Core-facility of Korea Basic Science Institute (KBSI).
Author information
Authors and Affiliations
Contributions
M.L. and J.C. conceptualised the study; M.L. and J.C. provided the methodology; D.Y., J.Y.L., and J.C. performed the formal analysis; D.Y., B.K., D.J., J.Y.L., S.K., D.S., R.P.G., M.L., and J.C. participated in the investigation; J.Y.L., S.K., D.S., R.P.G., M.-W.L., J.H.S., Y.S.P., Y.K., C.-M.O., M.L., and J.C. provided the resources; D.Y., B.K., D.J., M.L., and J.C. wrote the original draft; M.L. and J.C. edited the manuscript; D.Y., B.K., and D.J. visualised the data; M.L. and J.C. administered the study; M.L. and J.C. supervised.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Youn, D., Kim, B., Jeong, D. et al. Cross-talks between Metabolic and Translational Controls during Beige Adipocyte Differentiation. Nat Commun 16, 3373 (2025). https://doi.org/10.1038/s41467-025-58665-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58665-x
