
Abstract
Wnt5a, a representative Wnt ligand that activates the β-catenin-independent pathway, has been shown to promote tumorigenesis. However, it is unclear where Wnt5a is produced and how it affects colon cancer aggressiveness. In this study, we demonstrate that Wnt5a is expressed in fibroblasts near the luminal side of the tumor, and its depletion suppresses mouse colon cancer formation. To characterize the specific fibroblast subtype, a meta-analysis of human and mouse colon fibroblast single-cell RNA-seq data is performed. The results show that Wnt5a is expressed in hypoxia-induced inflammatory fibroblast (InfFib), accompanied by the activation of HIF2. Moreover, Wnt5a maintains InfFib through the suppression of angiogenesis mediated by soluble VEGF receptor1 (Flt1) secretion from endothelial cells, thereby inducing further hypoxia. InfFib also produces epiregulin, which promotes colon cancer growth. Here, we show that Wnt5a acts on endothelial cells, inducing a hypoxic environment that maintains InfFib, thereby contributing to colon cancer progression through InfFib.
Similar content being viewed by others

Introduction
Colon cancer remains a significant global health concern, contributing to substantial morbidity and mortality worldwide1. Despite advances in the understanding of the molecular pathogenesis of this disease, much remains to be elucidated regarding the complex interplay of various cell types within the tumor microenvironment. Colon cancer typically arises from the accumulation of genetic and epigenetic alterations in colonic epithelial cells2. However, it has become clear that the tumor microenvironment exerts a profound influence on disease course and therapeutic outcomes3. Among cell types in the microenvironment, fibroblasts have emerged as key players in the development and progression of colon cancer4. Traditionally viewed as structural support cells, fibroblasts, often referred to as cancer-associated fibroblasts (CAFs), are now appreciated for their multifaceted roles in cancer biology.
Recent advancements in single-cell RNA sequencing (scRNA-seq) have revolutionized our ability to dissect the cellular diversity within tumors5,6. This technology allows us to discern previously obscured subpopulations of fibroblasts with varying expression patterns of genes associated with extracellular matrix deposition, cytokine signaling, and immune modulation. Such diversity within the fibroblast compartment suggests that different subtypes may contribute differently to tumor progression and treatment response4. Several papers show fibroblast diversity in colon cancer; however, there is no unified classification as in pancreatic cancer7, and different definitions are found among papers. Also, fibroblasts in colon cancer cannot be classified clearly by typical marker genes of fibroblast subtypes in pancreatic cancer, myCAF and iCAF8. Furthermore, it is not known whether a classification based on scRNA-seq from human specimens is homologous to that of fibroblasts in mouse colon cancer models. These situations have greatly hindered the development of therapies targeting CAFs that contribute to tumor malignancy.
Wnt5a, a member of the Wnt family proteins, has multifaceted roles in both physiological and pathological processes9,10,11. Unlike the canonical Wnt pathway, which primarily regulates cell fate and differentiation through β-catenin accumulation, Wnt5a predominantly activates non-canonical signaling cascades, driving diverse cellular responses independently of β-catenin accumulation. Wnt5a is expressed in tumor cells of various types of cancer, including gastric, prostate, and oral cancers, and malignant melanoma12,13,14,15, and can induce tumor cell proliferation and invasion, contributing to disease aggressiveness10,13,16. Meanwhile, Wnt5a expression has been shown to be associated with improved survival in some cancer types such as thyroid, breast, and Dukes B colon cancer17,18,19.
In addition to tumor epithelial cells, Wnt5a has been shown to be expressed in immune cells, such as mononuclear cells, dendric cells, and macrophages20,21 and induces the expression of the pro-inflammatory cytokines. Also, cytokine signaling can induce Wnt5a expression, suggesting that there is interplay between Wnt5a signaling and the inflammatory pathways22. Furthermore, Wnt5a is expressed in fibroblasts in mouse colon adenoma and inflammatory bowel disease models23,24, and colorectal CAFs25. However, it remains unclear how Wnt5a affects the properties of stromal and epithelial cells within the cell-cell communication centered on fibroblasts.
In this study, we first performed a large-scale, cross-disease meta-analysis of scRNA-seq data derived from human specimens, followed by cross-species analysis to characterize colon fibroblasts that are activated and transformed in pathological conditions. In light of the comprehensive fibroblast classification and the specific localization of Wnt5a, the induction mechanism of the Wnt5a-expressing fibroblasts and the role of Wnt5a in carcinogenesis were clarified.
Results
Wnt5a contributes to the development of colon cancer
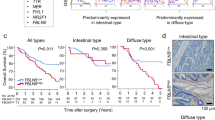
To determine the behavior of Wnt ligands in colon cancer, TCGA dataset of colon adenocarcinoma (COAD) and GTEx datasets were first used to identify the expression of 19 different Wnt ligands. Wnt5a was more highly expressed in the tumor than in the normal intestine, and its expression level was the highest among the Wnt ligands (Fig. 1a). Moreover, the gene expression profile of tumors from the azoxymethane (AOM)/dextran sodium sulfate (DSS) mouse model also revealed that Wnt5a was the most highly expressed Wnt ligand (Fig. 1b). Since the definitive effect of Wnt5a in colon cancer development has not been established9, we applied the AOM/DSS model to clarify the potential of Wnt5a in colon cancer. Wnt5aflox/flox (referred to as Wnt5afl/fl or control) mice were crossed to mice expressing a tamoxifen-inducible Cre to generate Wnt5afl/fl;CAG-Cre/ERT2Tg mice24 (referred to as Wnt5acKO or Wnt5a cKO). When tamoxifen was administrated 4 weeks before the AOM treatment, knockout (KO) of Wnt5a clearly suppressed tumor numbers at 20 weeks post-AOM administration (Fig. 1c, d).

a Expression profiles of 19 Wnt ligands in colon adenocarcinoma and normal colon from the TcgaTargetGtex dataset (adenocarcinoma, n = 288; normal, n = 308). Normalized count data are presented as a dot plot, and the color and size of dots represent the median level of each gene. b Expression profiles of 19 Wnt ligands obtained from bulk RNA-seq of mouse AOM/DSS tumors (n = 3 different tumors from 2 mice). Normalized count data are presented as a strip plot. c Experimental schedule of the AOM/DSS model. Tamoxifen was administered 4 weeks before the AOM treatment. Tm, tamoxifen. d (Left) Representative macroscopic images of tumors in colon from Wnt5afl/fl (control, n = 5) and Wnt5afl/fl;CAG-Cre/ERT2Tg (Wnt5a cKO, n = 7) mice induced by the experimental design shown in (c). (Right) The total number of tumors were counted, and the results are shown by a box plot. Scale bar, 10 mm. (Unpaired t-test, Two-tailed). e Experimental schedule of the AOM/DSS model. Tamoxifen was administered 4, 6, or 8 weeks after the AOM treatment. Tm, tamoxifen. f (Left) Representative macroscopic images of tumors in the colon from control and Wnt5a cKO mice treated with tamoxifen 6 weeks after the AOM treatment. (Right) The total number of tumors for each time period in which tamoxifen was administered (control, n = 27; 4wk, n = 13; 6wk, n = 13; 8wk, n = 5) were counted, and the results are shown by a box plot. Scale bars, 10 mm. (One-way ANOVA, Holm-Sidak’s multiple comparisons test). g The diameter of tumors from control (n = 28) and Wnt5a cKO (n = 18) mice treated with tamoxifen 6 weeks after the AOM treatment were measured, and the results are shown by a box plot. (Unpaired t-test, Two-tailed). h Tissue sections from control (n = 6 different tumors from 6 mice) or Wnt5a cKO (n = 5 different tumors from 3 mice)-derived tumors were stained with anti-Ki67 antibody and counterstained with hematoxylin, and the Ki67-positive cells were counted. The results are shown as the percentage of positively stained cells compared to the total number of cells and presented as a bar plot. Scale bars, 100 μm. (Unpaired t-test, Two-tailed). i Tissue sections from control and Wnt5a cKO-derived tumors or colonic mucosa from non-cancerous mice were stained with anti-Wnt5a antibody and counterstained with hematoxylin. Scale bars, 40 μm. j A scatter plot showing the correlation between signature scores of proliferation (x-axis) and WNT5A gene expression levels (y-axis) was obtained from TCGA-COAD patient dataset. A regression line was fitted using linear regression, and the shaded area represents the 95% confidence interval. The Spearman R- and P-values are shown on the top. k Tissue sections from human colon cancer specimens (n = 7) were stained with anti-Wnt5a antibody and counterstained with hematoxylin, and the proportion of Wnt5a-positive fibroblasts was calculated in the luminal surface and the core region. The core region was defined as the area located more than 1 mm away from the luminal surface toward the tumor center, while the area closer to the luminal surface than the core region was defined as the luminal surface region. The regions in the black solid squares in the upper panel are shown enlarged in the lower panels. The percentage of Wnt5a positive fibroblasts compared to the total fibroblasts is presented as a bar plot. Scale bars, (upper) 1 mm, (lower) 200 μm. (Unpaired t-test, Two-tailed). d, f, g In box and whiskers plots, the top and bottom horizontal lines represent the 75th and the 25th percentiles, respectively, and the middle horizontal line represents the median. The size of the box represents the interquartile range and the top and bottom whiskers represent the maximum and the minimum values, respectively. c, e Created in BioRender87.**, P < 0.01; *, P < 0.05; Source data are provided as a Source Data file.
Wnt5a is highly expressed in DSS-induced colitis and is known to contribute to the pathogenesis of colitis24. To exclude the possibility that suppression of DSS-induced inflammation by Wnt5a KO leads to inhibition of tumorigenesis, Wnt5a was knocked out after the inflammation caused by DSS was resolved and the intestinal mucosa had regenerated (Fig. 1e). Inflammatory cytokine expression was also found to have returned to normal levels at 6 weeks after the initiation of AOM/DSS treatment (Supplementary Fig. 1). Although tamoxifen was treated 2, 4, and 6 weeks after the completion of 7-days cycle of DSS, there was no difference in tumor suppressive effect for tumor numbers by Wnt5a cKO compared with the condition administrating tamoxifen 4 weeks before AOM treatment (Fig. 1f). KO of Wnt5a also resulted in reduced tumor size and decreased Ki67 staining in tumor areas (Fig. 1g, h). In the mouse tumor model, Wnt5a expression was limited in the normal intestinal tract, but clearly detected in the tumor lesion (Fig. 1i). The expression was higher in regions closer to the luminal side of the tumor than in the center of it, indicating the spatial identity of Wnt5a (Fig. 1i). These results suggest that Wnt5a contributes to tumorigenesis once DSS-induced inflammation settles down.
Next, we investigated the relationship between Wnt5a and colon cancer in human. Wnt5a expression and proliferation score showed a significant positive correlation in TCGA-COAD dataset (Fig. 1j). Furthermore, Wnt5a was localized to the stromal region adjacent to the luminal surface in human specimens (Fig. 1k and Supplementary Table 1). Therefore, these results indicate that Wnt5a is highly expressed in colon cancer tissues and prompt us to investigate the mechanism.
Wnt5a is expressed in fibroblasts in mouse and human colon cancer
It has been reported that Wnt5a is expressed in tumor stroma25,26, but its expression profile was not well investigated. To clarify the expression pattern of Wnt5a in mouse models, single-cell RNA-sequencing (scRNA-seq) analysis was performed on tumors at 20 weeks after AOM/DSS treatment by a droplet-based platform (Fig. 2a). To ensure generality, we also used a publicly available dataset of colon tumors from orthotopic transplantation model in which organoids derived from mice bearing tumors with mutations in Apc, Trp53, Kras, and Tgfbr2 genes were transplanted into the intestinal tract27 (Fig. 2a). After removing doublets, the data from both datasets were integrated, and cell type annotation was performed manually (Fig. 2b and Supplementary Fig. 2a, b). To investigate the presence of doublets between T cells and other immune cells, the expression of Cd3d, Cd3e, Cd4, and Cd8a was examined and compared with the expression patterns in a dataset related to immune cell infiltration in a mouse colon cancer model6 (Supplementary Fig. 2c, d). Doublets between T cells and other immune cells were not detected.

a Sample collection strategy for single-cell RNA-seq data analysis of whole cells in mouse colon cancer. Created in BioRender87. b UMAP embedding of 26,392 cells from mouse colon cancer models, colored by cell type. pDC plasmacytoid dendritic cell, cDC conventional dendritic cell, Sm.mus smooth muscle cell, Vas.endo vascular endothelial cell, Lym.endo lymphatic endothelial cell, NK natural killer cell, Tgd gamma delta T cell. c UMAP plot for Wnt5a. d Expression levels of Wnt5a across cell types are shown in a dot plot. Dot size indicates the proportion of expressing cells, colored by mean expression values in each cell type. e UMAP plots for Pdpn, Pdgfra, and Lum. f, g Tissue sections from AOM/DSS-derived tumors were stained with indicated antibodies and DAPI. f The region in the yellow solid square is shown enlarged in the bottom right panel (Scale bar for enlarged image, 20 μm). Scale bars, f 100 μm, g (upper) 100 μm, (lower) 50 μm.
Wnt5a was most strongly expressed in fibroblasts, where podoplanin (Pdpn) and PDGF receptor α (Pdgfra) were expressed close to the luminal surface (Fig. 2c–f). Since Pdpn was also expressed in lymphatic endothelial cells, it was confirmed that Lyve1, a lymphatic endothelial cell marker, does not colocalize with Wnt5a (Fig. 2g). A similar expression profile of Wnt5a, including the expression in squamous cells, was also observed in the scRNA-seq dataset of another mouse colon cancer model8 (Supplementary Fig. 2e–g). To determine the expression profile in human colon cancer, published dataset was re-analyzed28 (Supplementary Fig. 2h). As in mouse, Wnt5a were expressed in fibroblasts most strongly (Supplementary Fig. 2i), suggesting that an unknown Wnt5a action may occur via fibroblasts.
Human colon fibroblast atlas shows 6 distinct subtypes
The classification of fibroblasts or cancer-associated fibroblasts (CAFs) in colorectal cancer by the widespread use of scRNA-seq technology29 differed from study to study, and the similarity and commonality between reports or species was unknown. Therefore, it was necessary to establish a universal CAF classification for human colon cancer. The classification is different from that of previous studies in following three points: first, this is the largest meta-analysis of human colon cancer fibroblasts to our knowledge at present; secondary, we used healthy donor-derived samples as control, whereas previous studies have used adjacent normal samples28,30,31; thirdly, to evaluate whether classified subtypes were truly distinct subpopulations or were the same subpopulations in different activation status, datasets of inflammatory conditions where fibroblasts are activated as well as in tumors were included.
To characterize fibroblasts in-depth, we obtained 3 datasets of healthy donors and inflammatory bowel disease (IBD), and 4 datasets of colorectal cancer (CRC) (Fig. 3a, and Supplementary Tables 2–5). In addition to analyzing the expression profiles of DCN and LUM, which are marker genes for pan-fibroblasts, fibroblast signature scores were calculated for each cell based on the fibroblast marker gene set defined by Smillie et al. (Supplementary Fig. 3a). Then, in silico-selected fibroblasts were integrated, and meta-analysis of 47,328 fibroblasts from 157 specimens was performed (Supplementary Fig. 3b, and Supplementary Table 6). There were no significant differences in the results depending on the batch correction approaches used during the integration process (Supplementary Fig. 3c–e). Although many CAF subtypes have been identified as being induced specifically in the tumor environment previously, many of them were also induced in the inflammatory environment and only a limited group of cells were specifically observed in tumors (Fig. 3b, c).

a Sample collection strategy for single-cell RNA-seq data meta-analysis of fibroblasts from human colon. Created in BioRender87. b The density of cells per condition is plotted in UMAP embedding. c Abstracted graph of neighborhoods was superimposed on UMAP embedding using milopy package, where each node represents a neighborhood and is colored by their log2(fold change) between healthy state and disease (IBD and CRC) state. FC, fold change. d The optimal number of components for NMF was determined using an elbow plot. The maximum correlation (y-axis) refers to the highest correlation coefficient between the original data and the reconstructed data for a given number of components (x-axis). The optimal number of components, 8, is marked in orange. e For each program, the NMF feature weights, representing gene program activity scores, of each cell are plotted in UMAP embeddings. f The gene weights of the top 5 genes in each program are shown as a heatmap. g UMAP embedding of fibroblasts from human colon tissue, colored by cell type. Cell type annotation was defined based on the program activated in the cell type. h NMF feature weights of each cell are presented as a stacked violin plot. For visualization, the weights were min-max scaled across all cells for each program, with colors representing median weights. i (Upper) UMAP plot for WNT5A. (Lower) Expression levels of WNT5A across cell types are shown in a dot plot. Dot size indicates the proportion of expressing cells, colored by mean expression values in each cell type. j Expression levels of WNT5A in InfFib among the state of health are shown in a violin plot. k The proportion of fibroblast subtypes among the states of health is shown in a stacked bar plot. l The proportion of fibroblast subtypes in colon adenocarcinoma tissue and normal tissue adjacent to the tumor was inferred from bulk RNA-seq data of TCGA-COAD cohort using deconvolution method, and is visualized as a stacked bar plot. m The proportion of fibroblast subtypes in colon tissue from healthy donors and IBD patients was inferred from bulk RNA-seq data using deconvolution method, and is visualized as a stacked bar plot. n Spatial plots for InfFib cell density and WNT5A expression using two independent datasets of human colon cancer. The regions enclosed by the yellow dashed line indicate the areas where InfFib and expression of WNT5A are enriched near the luminal side.
Next, Nonnegative Matrix Factorization (NMF) profiling was performed to dissect gene programs independently from cell clusters, and eight gene programs were found to characterize the fibroblast expression data (Fig. 3d–f and Supplementary Table 7). CancerSpecific_Program was an active program in the tumor-specific cell populations identified in Fig. 3b, and the feature genes included COL10A1 and COL11A1 (Fig. 3e and Supplementary Fig. 4a). Bone morphogenic proteins (BMPs)_Program contained BMP5 and BMP4 as program-defining genes (Fig. 3e and Supplementary Fig. 4b). Chemotaxic_Program contained CCL8 and CCL11 as program specific genes (Fig. 3e and Supplementary Fig. 4c). It is known in the cornea that in response to inflammation, specific fibroblast populations that co-express the chemokine CCL11 and the adhesion molecule VCAM1 are induced32. The Chemotaxic_Program was found to reflect well the co-expression profile of CCL11 and VCAM1 (Supplementary Fig. 4d). Inflammatory_Program was enriched in subpopulations induced in inflammatory state (Fig. 3b, e). Myofibroblastic_Program was defined by typical genes defining myofibroblast33 (Fig. 3e and Supplementary Fig. 4e). Alarmin_Program included feature genes such as IL33, which is described as an alarmin (Fig. 3e and Supplementary Fig. 4f). IL33-producing cells are localized at the base of the intestinal crypts and are known to respond to bacterial infection, secrete alarmins, and induce type 2 immunity34. IBD_Program was specifically activated in IBD state, and included TNFAIP3, which is a target gene of NF-κB35, and captured the feature of NF-κB signaling (Fig. 3e and Supplementary Fig. 4g, h). FOS/JUN_Program was similar to the program defined as FOS/JUN factor (pS20) in the previous report28 (Fig. 3e and Supplementary Fig. 4i). Next, fibroblast categorization was performed by conventional clustering strategy. Fibroblasts were classified into six categories, and annotated based on the gene program activities as CancerSpecific fibroblast (CancerFib), BMPs fibroblast (BMPsFib), Chemotaxic fibroblast (ChemoFib), Inflammatory fibroblast (InfFib), Myofibroblast (MyoFib), and Alarmin fibroblast (AlarmFib) (Fig. 3e, g, h). The expression of NMF feature genes in each dataset, such as COL10A1 for the CancerSpecific_Program and IL11 for the Inflammatory_Program (Supplementary Fig. 4j, k), the gene program scores defining each subtype (Supplementary Fig. 5), and the proportions of each subtype (Supplementary Fig. 6a) did not show significant differences within the healthy, IBD, and CRC conditions across various datasets, indicating that the data integration was performed appropriately.
The InfFib population appears divided into upper and lower clusters in the UMAP plot (Fig. 3g). Inflammatory fibroblast or equivalent cell types identified in the original studies28,36 that provided the datasets for fibroblast atlas construction were also divided into upper and lower regions in the UMAP embedding (Supplementary Fig. 6b). Furthermore, the signature score for inflammatory fibroblast subtype c07-IL6, as defined in the report on cross-tissue fibroblast profiling37, showed separated high-score regions in the upper and lower areas of the UMAP embedding (Supplementary Fig. 6c, d). Meanwhile, when InfFib was defined separately in the upper and lower regions, InfFib_lower exhibited a higher IBD_Program (Supplementary Fig. 6e, f). In IBD, the proportions of InfFib_upper and InfFib_lower were nearly equal, whereas in CRC, most InfFib cells belonged to InfFib_upper (Supplementary Fig. 6g). Additionally, Wnt5a expression was higher in InfFib_upper, although both InfFib_upper and InfFib_lower showed high expression of Wnt5a within the fibroblast population (Supplementary Fig. 6h). Therefore, InfFib was defined as a single population with high Inflammatory_Program for the analysis in colon cancer.
According to previous reports, BMPsFib corresponds to WNT5B+ fibroblasts36, BMP producing fibroblasts28, or c15-SOX637, ChemoFib corresponds to WNT2B+ fibroblasts36, CCL8+ fibroblasts28, c02-ADAMDEC137, or immune-regulatory fibroblast38, MyoFib corresponds to c18-HHIP37, AlarmFib corresponds to c03-COL15A1 and c05-PI1637, which are collectively defined as progenitor-like fibroblasts, and CancerFib corresponds to c04-LRRC1537 (Supplementary Fig. 6c, i).
Conventional fibroblast classification often focuses on the difference between fibroblasts from physiological environments and those from the tumors, distinguishing them into normal fibroblasts and CAFs. While this binary approach is simple, it may not fully capture the biological nature of fibroblast subtypes. Therefore, we opted for a comprehensive analysis of fibroblasts under different colon conditions, including healthy, inflammatory, and cancerous settings, defining their underlying biological properties using NMF and subsequently classifying the cells.
Using the atlas of human colon fibroblasts that we established, we confirmed the expression of Wnt5a in BMPsFib as previously reported30,33, but found that Wnt5a is also strongly expressed in InfFib (Fig. 3i, j and Supplementary Fig. 6j). Integrated analysis of inflammatory disease and colon cancer revealed that InfFib is not disease-specific, but is induced across diseases (Fig. 3k). Deconvolution of bulk RNA-seq data from TCGA-COAD using scRNA-seq data as a reference revealed that the population of InfFib, as well as CancerFib, was increased in tumor lesions (Fig. 3l). Furthermore, cell proportion estimation based on bulk RNA-seq data from healthy donors and IBD patients39 revealed an increase of InfFib in IBD, consistent with the scRNA-seq results shown in Fig. 3k (Fig. 3m). Spatial transcriptome analysis of human colon cancer datasets40,41 revealed that Wnt5a was spatially colocalized with InfFib (Fig. 3n). From the above, cross-disease meta-analysis of colon fibroblasts with a comprehensive profiling based on NMF elucidated that Wnt5a was highly expressed in InfFib.
Mouse fibroblasts share common subsets with human
We then tested whether the classification in human fibroblasts could be adapted to mouse fibroblast data. For the establishment of mouse data, we performed scRNA-seq of fibroblasts isolated via FACS (fluorescence-activated cell sorting) from AOM/DSS model as an original dataset in this study. After quality control, 6796 cells were retained as fibroblasts. Also, we obtained publicly available scRNA-seq data from orthotopic transplantation model27 and 1754 cells were selected in silico as fibroblasts (Fig. 4a and Supplementary Fig. 7a). Fibroblasts from both models were integrated and used for downstream analysis (Supplementary Fig. 7b). Since it is difficult to adapt the classification across species on the basis of a limited number of marker genes, we projected the gene programs defined in human to mouse fibroblasts using NMFproj42 (Fig. 4b). We found that the cell-type specific gene programs CancerSpecific_Program, BMPs_Program, Inflammatory_Program, and Myofibroblastic_Program in human fibroblasts were enriched in specific subpopulations in mouse data (Fig. 4c and Supplementary Fig. 7c). On the other hand, Chemotaxic_Program and Alarmin_Program did not show an exclusive enrichment pattern, but showed a polarized activity pattern (Fig. 4c and Supplementary Fig. 7c). Although it is difficult to classify mouse fibroblasts solely on the basis of cell type-specific marker genes (e.g., Rspo3, Grem1, Wnt2, and Wnt2b) identified in human fibroblasts4,43 (Supplementary Fig. 7d), NMF-based gene programs can characterize clusters across species. Some program-specific genes exhibit unique expression patterns shared between humans and mice. For example, Il33 was expressed specifically in AlarmFib, and co-expression of Ccl11 and Vcam1 in ChemoFib were clearly observed as in human atlas (Supplementary Fig. 7e, f). Mouse fibroblasts were then classified using conventional clustering strategy and annotated based on the gene program activities (Fig. 4d). For validation of cell type classification of mouse fibroblasts, scRNA-seq data from healthy and DSS-colitis mice33 were projected in reference map (Fig. 4e). Fibroblasts from healthy mice were preferentially projected into all but CancerFib and InfFib, while fibroblasts from DSS-colitis mice were also projected into InfFib, suggesting the accuracy of the reference map (Fig. 4e). Furthermore, when human and mouse fibroblast data were analyzed integratively, the subtypes in each species were found to be highly homologous (Fig. 4f, g). Moreover, to perform profiling using a more comprehensive mouse colon fibroblast atlas, scRNA-seq dataset of fibroblasts from control, DSS-treated, and tumor model mice was integrated (Fig. 4h, Supplementary Fig. 7g and Supplementary Table 8). By applying NMF projection to this fibroblast atlas, it was able to define each fibroblast subtype more clearly through the enrichment of gene program activities (Fig. 4i–l).

a Sample collection strategy for single-cell RNA-seq (scRNA-seq) data meta-analysis of fibroblasts from mouse colon cancer models. For AOM/DSS model, fibroblasts were isolated by FACS to prepare samples for scRNA-seq. b Schematic representation of NMFproj. c UMAP plots for NMF-based programs. For each program, the weights of each cell are plotted in UMAP embeddings. The weights were calculated based on projected gene features from NMF of human colon fibroblast atlas dataset. d UMAP embedding of 8550 fibroblasts from mouse colon cancer model, colored by cell type. Cell type annotation was defined based on the program activated in the cell type. e UMAP embeddings for healthy mouse fibroblasts (left) and DSS-treated mouse fibroblasts (right) obtained from a previous report projected into the mouse tumor fibroblast reference map. f Human colon fibroblast atlas and mouse colon tumor fibroblasts data were projected in the same UMAP embedding. g Z-score normalized alignment scores between cell types related to (f) are represented in a correlation matrix plot. h Sample collection strategy for scRNA-seq data meta-analysis of fibroblasts from normal and inflamed mouse intestines, and intestinal mouse tumors. The two tumor-derived datasets are the same as those used in (a). i UMAP plots of mouse colon fibroblasts related to (h) for NMF-based programs. For each program, the weights of each cell are plotted in UMAP embedding. The weights were calculated based on projected gene features from NMF of human colon fibroblast atlas dataset. j UMAP embedding of 46,843 fibroblasts related to (h), colored by cell type. Cell type annotation was defined based on the program activated in the cell type. k UMAP embeddings of fibroblasts related to (j) for each disease states, colored by cell type. l The subtype proportion of fibroblasts in each condition related to (k) is plotted in a stacked bar plot. m Mean scaled expression levels of the top 5 cell type-specific genes in each fibroblast subtype are illustrated as a heatmap. For visualization, the expression levels were min-max scaled across all cells for each gene. a, h Created in BioRender87. Source data are provided as a Source Data file.
Cell type-specific genes of mouse fibroblast subtypes were defined based on the specificity of expression values for each gene among 6 subtypes which was quantified using the SPM (specificity measure) metric, an individualized scoring metric (Fig. 4m, Supplementary Fig. 7h and Supplementary Table 9). Another established method, scGeneFit, was used to select marker genes that retain the classification of cell types similar to that of the original embedding44. Many of the marker genes overlapped between the two independent methods, indicating the accuracy of selecting cell type-specific genes (Supplementary Fig. 7i and Supplementary Tables 9, 10).
Based on these findings, cross-species analysis revealed that human and mouse colon fibroblasts share the same subtypes, demonstrating the utility of mouse models for analyzing fibroblasts in human colon cancer.
Wnt5a expression is required for InfFib existence
As a result of scRNA-seq-based profiling of mouse colon cancer fibroblasts, Wnt5a was found to be highly expressed in BMPsFib and InfFib, similar to the results obtained in the human atlas (Fig. 5a). Immunostaining of the subtype marker genes showed that Wnt5a co-localizes with BMPsFib (Lama1) and especially InfFib (Mmp10) markers (Fig. 5b, c). On the contrary, MyoFib (Cnn1), AlarmFib (Ifi204), and CancerFib (Col10a1) markers didn’t co-localize with Wnt5a (Supplementary Fig. 8a). Localization analysis revealed that Wnt5a expression was confined to the stroma, which led us to consider the impact of Wnt5a on stromal remodeling. Therefore, we performed deconvolution on bulk RNA-seq data from colon tumors resected from AOM/DSS-treated Wnt5afl/fl and Wnt5acKO mice to estimate the composition change of cells constituting the tumor area. The proportion of InfFib was significantly attenuated among all the cell types in tumors from Wnt5acKO, while the percentage of total fibroblasts remained unchanged (Fig. 5d, e). Also, the mRNA expression of InfFib markers, Mmp10 and Stc1, decreased in tumors from Wnt5acKO mice (Fig. 5f). Consistent with the results, Mmp10- and Il11-, which is an InfFib marker in mouse tumors45, positive area facing the luminal side was considerably decreased when Wnt5a was depleted (Fig. 5g, h). Meanwhile, MyoFib, AlarmFib, and CancerFib were not affected by Wnt5a cKO (Supplementary Fig. 8b–d). Using the TCGA-COAD dataset, we examined the correlation between mRNA expression of Wnt5a and Inflammatory NMF program score and the percentage of InfFib estimated by the deconvolution method as shown in Fig. 3l, and found a significant positive correlation for both pairs (Fig. 5i). These results suggest that Wnt5a is required for the maintenance of InfFib in colon cancer.

a (Upper) UMAP plot of fibroblasts from mouse colon cancer models related to Fig. 4d for Wnt5a. (Lower) Expression levels of Wnt5a across cell types are shown in a violin plot. b, c Tissue sections from AOM/DSS-derived tumors were stained with indicated antibodies including anti-Lama1 (feature gene of BMPsFib) or anti-Mmp10 (feature gene of InfFib) antibody and DAPI. b The region in the yellow solid square is shown enlarged in the right panels. Scale bars, b (left) 100 μm, (right) 20 μm, c 50 μm. d The relative change in abundance of whole cell types between control and Wnt5a cKO-derived AOM/DSS-induced tumors (n = 4 per group) was inferred from bulk RNA-seq data of tumor tissues using the deconvolution method and is visualized as a bubble plot. The x-axis represents statistical significance, and the y-axis represents the cell type. The color of the bubble indicates the amount of change in the proportion of cells. FC fold change. (Multiple t-test, Two-tailed, Holm-Sidak’s multiple comparisons test). e The proportion of indicated cell types in control or Wnt5a cKO-derived AOM/DSS-induced tumors was inferred from bulk RNA-seq data of tumor tissues using the deconvolution method and is visualized as a stacked bar plot. f Expression levels of InfFib marker genes between control and Wnt5a cKO-derived AOM/DSS-induced tumors are shown in bar plots. mRNA expression values were obtained from bulk RNA-seq data used in (d). (n = 4; Unpaired t-test, Two-tailed). g, h Tissue sections from control (n = 6 lesions from 3 or 4 mice) or Wnt5a cKO (n = 6 lesions from 3 mice)-derived tumors were stained with indicated antibodies and DAPI. The results are shown as the percentage of (g) Mmp10- or (h) Il11-positive area compared to the total Pdpn-positive area proximity to the lumen (within 75 μm from the edge) and presented as a bar plot. Scale bars, 50 μm. (Unpaired t-test, Two-tailed). i Scatter plots showing the correlation between WNT5A gene expression levels (x-axis) and signature scores of the top 50 weighted genes from the Inflammatory_Program (y-axis; left) or InfFib proportion estimated in Fig. 3l (y-axis; right) were obtained from TCGA-COAD patient dataset. A regression line was fitted using linear regression, and the shaded area represents the 95% confidence interval. The Spearman R- and P-values are shown on the top. ***, P < 0.001; **, P < 0.01; *, P < 0.05; Source data are provided as a Source Data file.
InfFib is induced by hypoxia regulated by Wnt5a
To understand how Wnt5a regulates InfFib population, we tried to determine the mechanism by which InfFib increases in tumors. Trajectory inference was performed for the mouse scRNA-seq data using the partition-based graph abstraction (PAGA) method (Fig. 6a) and diffusion pseudotime (DPT) analysis (Fig. 6b), and found that there are two distinct pathways of differentiation toward InfFib, the BMPsFib pathway and the CancerFib pathway (Fig. 6a). As BMPsFib localized spatially close to InfFib facing the luminal surface (see Fig. 5b, c), we focused on the possibility of a lineage between the two subtypes. Inference of pseudotime and differentiation stage by DPT and CytoTRACE, respectively, along the trajectory revealed smooth transition between BMPsFib and InfFib (Fig. 6c, d). Next, pathway activity inference in each cell type identified that the hypoxia pathway was particularly activated in InfFib both in mouse and human data (Fig. 6e, f, and Supplementary Fig. 9a). Hypoxic regions were found near the luminal surface, where InfFib was enriched (Fig. 6g, h and Supplementary Fig. 9b). Furthermore, in TCGA-COAD dataset, the InfFib proportion correlated positively and BMPsFib proportion negatively with the signature score of the hypoxia gene set (Fig. 6i and Supplementary Table 11). Furthermore, correlation coefficients were calculated for all combinations of cell types and pathways. Among the fibroblast subsets, only the InfFib population showed a positive correlation with the hypoxia pathway (Supplementary Fig. 9c).

a (Left) Force-directed graph embedding for mouse colon cancer fibroblast single-cell RNA-seq (scRNA-seq) data related to Fig. 4a. Cell type labels were from Fig. 4d. (Right) PAGA plot on force-directed graph embedding. b Force-directed graph embedding depicting diffusion pseudotime. c Force-directed graph embedding colored by CytoTRACE scores. A lower score indicates more differentiated states. d Changes of marker gene expression, CytoTRACE score, and hypoxia activity along PAGA path during pseudotime progression are shown. Marker genes for InfFib, Il1rl1 and Mmp10; BMPsFib, Syt13 and Lama1. DPT, diffusion pseudotime. e, f Pathway activities in each fibroblast subtype from mouse (e) or human (f) scRNA-seq data related to Fig. 4d or 3g, respectively, were inferred and are represented in heatmaps. g, h FFPE (g) or fresh-frozen (h) tissue sections from AOM/DSS treated mice after injection of pimonidazole were stained with indicated antibodies and DAPI. Scale bars, g 100 μm, h 20 μm. i Scatter plots showing the correlation between signature scores of hypoxia (x-axis) and (left) InfFib or (right) BMPsFib proportion estimated in Fig. 3l (y-axis) were obtained from TCGA-COAD patient dataset. A regression line was fitted using linear regression, and the shaded area represents the 95% confidence interval. The Spearman R- and P-values are shown on the top. j WEHI-YH2 cells were treated with CoCl2 for 24 h and subjected to bulk RNA-seq. Signature scores for each subtype are represented as strip plots. (n = 3; Unpaired t-test, Two-tailed). k WEHI-YH2 cells were exposed to CoCl2 or 1% O2 for indicated duration. The mRNA levels of the indicated genes are represented in bar plots. (n = 3; One-way ANOVA, Holm-Sidak’s multiple comparisons test). l WEHI-YH2 cells were exposed to 20% or 1% O2 for 24 h, and Wnt5a was precipitated from conditioned media. The precipitates (CM) and total cell lysates (Lysate) were probed with different antibodies: the former was probed for anti-Wnt5a antibody, and the latter was probed for anti-Hsp90 antibody as a loading control. m Tissue sections from control or Wnt5a cKO-derived tumors (n = 3 different tumors) were stained with anti-hypoxyprobe antibody and DAPI. The results are shown as the percentage of hypoxyprobe-positive area compared to the total area proximity to the lumen (within 75 μm from the edge) and presented as a bar plot. Scale bars, 100 μm. (Unpaired t-test, Two-tailed). ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, P > = 0.05; Source data are provided as a Source Data file.
To prove the subtype switch due to hypoxia, mouse colon cancer fibroblasts WEHI-YH2 cells were exposed to chemically induced hypoxia (CoCl2 treatment), followed by the bulk RNA-seq to assess the overall changes in the expression profiles of marker genes for each fibroblast subtype. Hypoxia induced an increase in the gene expression profile of InfFib, accompanied by a decrease in the expression of BMPsFib markers (Fig. 6j). No significant changes in the expression of genes characteristic of CancerFib, ChemoFib, or AlarmFib were observed. In in vitro experiment, it is known that myofibroblastic feature is readily acquired, and this characteristic was attenuated following the switch to InfFib (Fig. 6j). Indeed, the analysis using selected specific genes revealed that hypoxia-induced InfFib feature genes, including Stc1 and Fbxo32, and Wnt5a were upregulated in WEHI-YH2 cells, and BMPsFib feature genes, including Lama1 and Sept4 were downregulated (Fig. 6k and Supplementary Fig. 9d, e). Hypoxia also induced the secretion of Wnt5a in the medium (Fig. 6l). Since hypoxia contributed to the induction of InfFib, Wnt5a was thought to be involved in the formation of the hypoxic environment. Supporting this idea, the hypoxic area facing the luminal surface was attenuated when Wnt5a was depleted (Fig. 6m). Moreover, using TCGA-COAD dataset, the expression of Wnt5a positively correlated with the signature score of the hypoxia gene set (Supplementary Fig. 9f).
Wnt5a and InfFib are induced by HIF2 and inflammatory signaling
Next, how hypoxia regulates InfFib was investigated. Hypoxia induces the expression of two types of transcription factors, HIF1 and HIF246. HIF1α, a subunit of HIF1, was localized to cancer cells in the AOM/DSS-induced tumors, while HIF2α, a subunit of HIF2, was present in the stromal cells in the luminal region of the tumor (Fig. 7a, b). Consistent with this, HIF2α was observed in the luminal region of human colon cancer, and it was specifically observed that HIF2α was activated in Wnt5a-expressing cells (Fig. 7c). This prompted us to investigate whether HIF2α was involved in the regulation of Wnt5a expression. Overexpression of HIF2α in X293T or WEHI-YH2 cells, but not HIF1α, induced Wnt5a mRNA expression, and knockdown of HIF2α suppressed hypoxia-induced Wnt5a expression (Fig. 7d, e). Publicly available data revealed that HIF2α localized near the transcription start site of Wnt5a, which was accessible chromatin region (Fig. 7f). HIF2α also localized to the regulatory region of Wnt5a in WEHI-YH2 cells under CoCl2 treatment confirmed by ChIP-qPCR analysis (Fig. 7g). Taken together, these results suggest that HIF2 functions as a transcription factor that upregulates Wnt5a expression.

a, b Fresh-frozen tissue sections from AOM/DSS-treated mouse tumors were stained with indicated antibodies and DAPI. Scale bars, 50 μm. c Tissue sections from human colon cancer specimens (n = 5) were stained with indicated antibodies. The tumor was divided into the luminal surface and the core region, and the proportion of fibroblasts with nuclear HIF2α was calculated for each. The results are shown as the percentage of fibroblasts with nuclear HIF2α compared to the total fibroblasts and presented as a bar plot. Scale bars, 100 μm. (Unpaired t-test, Two-tailed; P < 0.0001). d Either HIF1α or HIF2α were introduced into X293T (left) or WEHI-YH2 (right) cells. The mRNA levels of Wnt5a gene were measured by quantitative RT-PCR, and the results are represented in a bar plot. (n = 3; (left) One-way ANOVA, Holm-Sidak’s multiple comparisons test; (right) unpaired t-test, two-tailed; (left) P = 0.2657, P = 0.0378 from left to right; (right) P = 0.0133). e WEHI-YH2 cells were transfected with control or HIF2α siRNA for 36 h, and were exposed to 20% (normoxia) or 1% (hypoxia) O2 for 24 h. The mRNA levels of indicated genes were measured by quantitative RT-PCR, and the results are represented in bar plots. Scr, Scramble. (n = 3; One-way ANOVA, Holm-Sidak’s multiple comparisons test; (left) P < 0.0001, P < 0.0001, P = 0.0859, P < 0.0001, P < 0.0001 from left to right; (right) P = 0.0183, P = 0.0426, P = 0.0035, P = 0.0043, P = 0.0426 from left to right). f Cistrome Data Browser images show HIF2α ChIP-seq and ATAC-seq signals at Wnt5a gene loci. ChIP-seq and ATAC-seq data for mouse uterus were obtained from GSE234065. g Enrichment of HIF2α within the Wnt5a promoter region in WEHI-YH2 cells upon CoCl2 treatment for 24 h were measured by ChIP-qPCR. The results are shown as the percentage of input and presented as a bar plot. ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, P > = 0.05; Source data are provided as a Source Data file.
In addition to hypoxia, the inflammatory pathways, including the TNF-α/NF-κB pathway, were also activated and correlated positively with the proportion of InfFib (Fig. 6e, f, and Supplementary Fig. 9c). Indeed, NF-κB was activated in InfFib cells positive for Il11 of AOM/DSS tumors (Fig. 8a), and TNF-α increased the expression of InfFib marker gene signatures in WEHI-YH2 cells (Fig. 8b). Therefore, TNF-α/NF-κB signaling induces inflammatory fibroblastic characteristics. The TCGA-COAD dataset showed the positive correlation between inflammation score and InfFib proportion (Fig. 8c). Given the interplay between inflammation and hypoxia47, hypoxia would induce Wnt5a expression in fibroblasts under inflammatory background.

a Tissue sections from AOM/DSS-treated mouse tumors were stained with indicated antibodies and DAPI. Scale bars, 50 μm. b WEHI-YH2 cells were treated with TNFα (5 ng/mL) for 24 h and subjected to bulk RNA-seq. Signature scores for each subtype are represented as strip plots. (n = 3; **, P < 0.01; ns, P > = 0.05; Unpaired t-test, Two-tailed; CancerFib, P = 0.3638; BMPsFib, P = 0.0026; ChemoFib, P = 0.8453; InfFib, P = 0.0021; MyoFib, P = 0.0016; AlarmFib, P = 0.0901). c A scatter plot showing the correlation between signature scores of inflammation (x-axis) and InfFib proportion estimated in Fig. 3l (y-axis) was obtained from TCGA-COAD patient dataset. A regression line was fitted using linear regression, and the shaded area represents the 95% confidence interval. The Spearman R- and P-values are shown on the top. Source data are provided as a Source Data file.
Wnt5a secreted from fibroblasts impacts endothelial cells
Then, the mechanism by which Wnt5a promotes tumor growth was investigated. Many studies, reported that Wnt5a directly influences various tumor cells, contributing to cancer aggressiveness such as cell proliferation, migration, and invasion10. However, the effects of Wnt5a on colon cancer cells have not been fully elucidated. Conditioned medium (CM) containing Wnt5a (Wnt5a CM) and recombinant Wnt5a (rWnt5a) did not affect cell proliferation of colon cancer SW480 cells (Supplementary Fig. 10a, b). rWnt5a had no impact on migrative ability and expression of epithelial-mesenchymal transition (EMT)-related genes, neither (Supplementary Fig. 10c, d). Wnt5a CM did not affect the InfFib gene profiles in WEHI-YH2 cells (Supplementary Fig. 10e). Therefore, the indirect effects of Wnt5a on tumor formation via stromal cells were examined.
Based on the findings that Wnt5a contributes to the formation of hypoxic environment near the luminal side (Fig. 6m) and induces InfFib (Fig. 5d), the mechanism through which Wnt5a regulates hypoxia was investigated. We evaluated the distribution of blood vessels in the tumor area using anti-Cd31 antibody. Tumors derived from Wnt5acKO mouse showed enlarged vascular distribution in the region near the luminal surface compared to tumors derived from Wnt5afl/fl mouse (Fig. 9a) and that vessel formation was attenuated in the vicinity of Wnt5a (Fig. 9b). Indeed, Wnt5a CM suppressed ability of tube formation of HUVEC (Human Umbilical Vein Endothelial Cells) (Fig. 9c and Supplementary Fig. 11a). The similar results were obtained with rWnt5a (Fig. 9d and Supplementary Fig. 11b). Additionally, rWnt5a inhibited sprouting of HUVEC spheroid (Fig. 9e).

a Tissue sections from control (n = 3 different tumors from 2 mice) or Wnt5a cKO (n = 3 different tumors from 2 mice)-derived tumors were stained with anti-Cd31 antibody and DAPI. The results are shown as the percentage of Cd31 positively stained area compared to the total Pdpn-positive area proximity to the lumen (within 75 μm from the edge) and presented as a bar plot. Scale bars, 200 μm. (Unpaired t-test, Two-tailed). b Tissue sections from control or Wnt5a cKO-derived tumors were stained with indicated antibodies and DAPI. Scale bars, 50 μm. c HUVEC were cultured in control or Wnt5a CM and subjected to tube formation assay. After incubation for 6 h, number of segments and total length of tubes were quantified for each condition, and the results are presented as bar plots. Scale bars, 300 μm. (n = 5; Unpaired t-test, Two-tailed). d HUVEC were treated with rWnt5a and subjected to tube formation assay. After incubation for 20 h, number of segments and total length of tubes were quantified, and the results are presented as bar plots. Scale bars, 300 μm. (n = 5; One-way ANOVA, Holm-Sidak’s multiple comparisons test). e HUVEC spheroids were seeded on Matrigel layer for sprouting assay. After incubation for 7 h with or without rWnt5a (400 ng/mL) treatment, length of the sprouts was measured and the results are presented as mean in a bar plot. Scale bars, 100 μm. (n = 4; Unpaired t-test, Two-tailed). f HUVEC were cultured in control or Wnt5a CM for 12 h. The mRNA levels of the sFlt1 are represented in a bar plot. (n = 3; Unpaired t-test, Two-tailed). g HUVEC were treated with rWnt5a for 12 h. The mRNA levels of the sFlt1 are represented in a bar plot. (n = 3; One-way ANOVA, Holm-Sidak’s multiple comparisons test). h After HUVEC were cultured with or without rWnt5a (400 ng/mL) treatment for 12 h, CM was collected and the concentration of sFlt1 was measured by ELISA. The results are represented in a bar plot. (n = 3; Unpaired t-test, Two-tailed). i HUVEC were treated with rWnt5a (400 ng/mL), CsA (10 μM), or both for 12 h. The mRNA levels of sFlt1 are represented in a bar plot. CsA, Cyclosporin A. (n = 3; One-way ANOVA, Holm-Sidak’s multiple comparisons test). j Expression levels of ROR1, ROR2, and RYK across cell types in Human Colon Cancer Atlas related to Supplementary Fig. 2h are shown in a dot plot. Dot size indicates the proportion of expressing cells, colored by mean expression values in each cell type. k HUVEC were treated with rWnt5a (400 ng/mL) for 12 h following transfection with control or RYK siRNA for 48 h. The mRNA levels of the indicated genes are represented in bar plots. Scr, Scramble; Cont, Control. (n = 3; One-way ANOVA, Holm-Sidak’s multiple comparisons test). l Tissue sections from control mouse-derived tumors were stained with indicated antibodies and DAPI. Scale bars, 20 μm. Yellow arrowheads indicate Cd31 positive endothelial cells. m PLA with anti-Wnt5a and anti-Ryk antibodies was performed on tissue sections from control mouse-derived tumors, and they were counterstained with DAPI. Scale bars, 20 μm. n Spatial plots for endothelial cell (Endo), InfFib, macrophage (Macro), monocyte (Mono) cell density in human colon cancer related to Fig. 3n and Supplementary Fig. 11f. The region where WNT5A is enriched is outlined with a yellow dashed line. o Scatter plots showing the correlation between cell density of two cell types within the region outlined by the yellow dashed line in (n). A regression line was fitted using linear regression, and the shaded area represents the 95% confidence interval. The Pearson R- and P-values are shown on the top. ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, P > = 0.05; Source data are provided as a Source Data file.
How Wnt5a acts on vascular endothelial cells was investigated. It was reported that Wnt5a secreted by macrophages induces the secretion of soluble VEGF receptor 1 (sFlt1) from endothelial cells, a splicing variant of Flt1, which inhibits angiogenesis by binding to VEGF48,49. Wnt5a CM or rWnt5a induced the mRNA expression and the secretion of sFlt1 in HUVEC (Fig. 9f–h). Moreover, cyclosporin A, a calcineurin inhibitor, inhibited Wnt5a-dependent sFlt1 expression, suggesting that its expression is induced by non-canonical Wnt signaling (Fig. 9i). Ror1/2 and Ryk function as a Wnt5a receptor10,50,51. Since the mRNA levels of Ryk were much higher than those of Ror1/2 in endothelial cells (Fig. 9j), we focused on Ryk. Depletion of Ryk suppressed Wnt5a-induced sFlt1 expression (Fig. 9k). Furthermore, endothelial cells expressed Ryk and localized in close proximity to Wnt5a-expressing fibroblasts (Fig. 9l). For direct evaluation of the cell-cell communication, Wnt5a ligand transmission was visualized using the PLA method52 (Fig. 9m). The interaction between Wnt5a and Ryk was confirmed in endothelial cells. Therefore, sFlt1 secreted from endothelial cells, triggered by Wnt5a, would be one of the mechanisms by which Wnt5a regulates hypoxic environment.
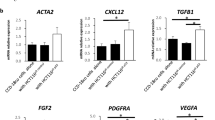
Moreover, in co-culture experiments with WEHI-YH2 fibroblasts and HUVECs, the expression of mRNAs for Wnt5a and InfFib marker genes, such as Mmp10 and Fbxo32 was increased (Supplementary Fig. 11c), suggesting that the interaction with endothelial cells shifts fibroblasts toward InfFib profile. Since the induction of InfFib suppresses angiogenesis through Wnt5a, endothelial cells may inhibit vessel formation through their interaction with fibroblasts.
Considering that Wnt5a and Wnt5b exhibited similar activities on cancer cells53,54, rWnt5b also showed inhibitory effect on angiogenesis (Supplementary Fig. 11d). However, BMPsFib, which express Wnt5b, located away from the region facing the luminal surface (Fig. 5b). Moreover, analysis of scRNA-seq data revealed that Wnt5b expression in InfFib is significantly lower than Wnt5a expression (Supplementary Fig. 11e). Taken together, these findings suggest that Wnt5b may have similar abilities to Wnt5a in the vessel formation but does not affect angiogenesis at the luminal surface of the tumor.
As shown in Supplementary Fig. 2i, Wnt5a is most strongly expressed in fibroblasts in human colon cancer, but its expression is also observed in macrophages and monocytes. To confirm the source of Wnt5a that interacts with endothelial cells, the localization of endothelial cells, InfFib, macrophages, and monocytes was analyzed using the spatial data presented in Fig. 3n (Fig. 9n and Supplementary Fig. 11f–h). Correlation analysis of cell density among these cell types revealed the strongest positive correlation between endothelial cells and InfFib (Fig. 9o and Supplementary Fig. 11i). This indicates that InfFib, rather than macrophages or monocytes, is the cell type closely localized to endothelial cells. Given that Wnt ligands, including Wnt5a, are difficult to freely diffuse in the extracellular space due to lipid modification55, these findings suggest that Wnt5a derived from InfFib interacts with endothelial cells. Thus, one mechanism by which Wnt5a suppresses angiogenesis could be by causing endothelial cells to produce sFlt1 in a localized region near the luminal surface, thereby exacerbating hypoxia.
Wnt5a-secreting fibroblasts impact tumor cells through epiregulin
Finally, we investigated why maintaining the proportion of InfFib by Wnt5a is necessary for tumor growth. Since the CM from InfFib cultured under the hypoxic condition stimulated the proliferation of CACO-2 cells (Fig. 10a), secretory factors specifically expressed in InfFib were examined. As betacellulin (BTC), heparin-binding EGF-like growth factor (HBEGF), neuregulin (NRG1), epiregulin (EREG), and amphiregulin (AREG) were feature genes of InfFib of either mouse or human, this led us to consider that InfFib was the source of epidermal growth factor (EGF) family56 (Fig. 10b–d). Among them, expression of Ereg, Hbegf, and Btc in AOM/DSS tumors was repressed in those derived from the Wnt5acKO mice (Fig. 10e and Supplementary Table 12). When WEHI-YH2 cells were treated with hypoxia or CoCl2, or overexpressed with HIF2α, Ereg mRNA increased (Fig. 10f). As HBEGF and BTC didn’t show high expression in human fibroblasts (Fig. 10c), the tumor-promoting effect of EREG was examined. EREG CM stimulated CACO-2 cell proliferation (Fig. 10g), and recombinant EREG (rEREG) promoted the growth of AOM/DSS-derived organoids (Fig. 10h), whose proliferation was otherwise inhibited by hypoxia (Fig. 10i). Taken together, EREG could be one of the mechanisms by which InfFib stimulates tumor growth. In addition, pathway analysis of DEGs obtained from bulk RNA-seq of AOM/DSS-induced tumors arising from Wnt5afl/fl and Wnt5acKO mice revealed that MAPK signaling was attenuated in the Wnt5acKO group (Fig. 10j). Also, in organoids stimulated with EREG, phosphorylation of ERK, a marker of MAPK activation, was observed, similar to EdU incorporation (Fig. 10k). Moreover, TCGA-COAD dataset revealed that the expression of Wnt5a positively correlated with the signature score of the MAPK gene set (Fig. 10l).

a CACO-2 cells were cultured in CM prepared from WEHI-YH2 cells under 20% or 1% O2 conditions. Proliferative abilities are presented as mean ± SD. (n = 4; Unpaired t-test, Two-tailed). b, c Expression levels of EGF ligand family across fibroblast subtypes in mouse (b) and human (c) single-cell RNA-seq data related to Fig. 4d or 3g, respectively, are shown in dot plots. Dot size indicates the proportion of expressing cells, colored by mean expression values in each cell type. d UMAP plot of cells from mouse colon cancer models related to Fig. 2b for Ereg. e Expression levels of Ereg between control and Wnt5a cKO-derived AOM/DSS-induced tumors are shown in a bar plot. mRNA expression values were obtained from bulk RNA-seq data used in Fig. 5d. (n = 4; Unpaired t-test, Two-tailed). f WEHI-YH2 cells were exposed to 1% O2 or CoCl2 for 48 h, or overexpressed with HIF2α. The mRNA levels of Ereg are represented in bar plots. (n = 3; Unpaired t-test, Two-tailed). g CACO-2 cells were cultured in CM prepared from WEHI-YH2 cells expressing EREG. Proliferative abilities are presented as mean ± SD. (n = 4; Unpaired t-test, Two-tailed). h Organoids established from AOM/DSS-derived tumors were treated with rEREG or rEGF and cultured for 6 days. Representative phase-contrast images are shown. Scale bars, 500 μm. i Organoids established from AOM/DSS-derived tumors treated with or without rEREG were exposed to 20% or 1% O2 for 48 h. The left panels are phase-contrast images. Mean area of organoids in each condition was measured for three experiments, and the results are represented in a bar plot. Scale bars, 500 μm. (n = 3; One-way ANOVA, Holm-Sidak’s multiple comparisons test). j Differentially expressed genes between control and Wnt5a cKO-derived AOM/DSS-induced tumors were subjected to pathway activity inference using decoupleR. mRNA expression values were obtained from bulk RNA-seq data used in Fig. 5d. k Organoids established from AOM/DSS-derived tumors were treated with 200 ng/mL rEREG for 2 days and EdU. Organoids were stained with indicated antibodies and DAPI, and representative images are shown. Scale bars, 20 μm. l Scatter plots showing the correlation between signature scores of MAPK (x-axis) and WNT5A gene expression levels (y-axis) were obtained from TCGA-COAD patient dataset. A regression line was fitted using linear regression, and the shaded area represents the 95% confidence interval. The Spearman R- and P-values are shown on the top. m Violin plots showing the mRNA expression levels of indicated genes among CMS subtypes in TCGA-COAD patient dataset. The width of each violin indicates the kernel density estimation of the data distribution. The central white dot represents the median, the thick bar in the center corresponds to the interquartile range (IQR), and the thin line extending from the bar denotes the data range within 1.5 times the IQR. (CMS1, n = 65; CMS2, n = 133; CMS3, n = 51; CMS4, n = 95; One-way ANOVA, Holm-Sidak’s multiple comparisons test). n Graphical abstract of this study. Created in BioRender87. ***, P < 0.001; **, P < 0.01; *, P < 0.05; Source data are provided as a Source Data file.
To demonstrate the clinical importance of EREG expression in colon cancer, we further analyzed the TCGA dataset. Although Wnt5a expression or InfFib abundance was not directly involved in aggressive phenotype that influence patient prognosis (Supplementary Fig. 12), Wnt5a may play a critical role in colon cancer progression (Fig. 1c–k). Meanwhile, CMS (consensus molecular subtype) classification is the best-known categorization of colon cancer for patient management2. Wnt5a expression was higher in CMS4 compared to other subtypes (Fig. 10m). While EREG was also significantly upregulated in CMS4, its highest expression was observed in CMS2, with only a slight difference from CMS4 (Fig. 10m). Additionally, CMS4 is characterized by a fibroblastic stroma, TGF-β signaling, and therapy resistance. Taken together, these results suggest that Wnt5a and InfFib contribute to the proliferative aspect of CMS4 tumors.
In conclusion, Wnt5a is suggested to suppress angiogenesis by regulating sFlt1 expression in endothelial cells, thereby maintaining hypoxia near the luminal surface. Hypoxia induces the expression of InfFib feature genes, and Wnt5a expression in InfFib is also enhanced through the activation of HIF2, which is induced under hypoxic conditions. This suggests that Wnt5a may enhance a positive feedback loop for InfFib maintenance. Additionally, it was revealed that not only hypoxia but also an inflammatory background induces the expression of feature genes defining InfFib, indicating that multiple factors regulate fibroblast subtypes. Furthermore, InfFib is likely to promote tumor growth in colon cancer under hypoxic conditions by secreting EREG. This study has elucidated part of the mechanism by which Wnt5a behaves within the tumor milieu and indirectly contributes to tumor malignancy.
Discussion
Wnt5a is a representative ligand, which activates the β-catenin-independent pathway of Wnt signaling10,57. Genetic alterations of the APC and CTNNB1(β-catenin) genes, which are essential components of the β-catenin-dependent pathway, are frequently observed in colon cancer cells and the mutations have been shown to activate cancer stem cells2,58. Wnt5a is also highly expressed in cancers with aberrant activation of β-catenin signaling, but its significance is not clearly understood. In this study we proposed that in colon cancer progression, Wnt5a-secreting InfFib increases through HIF2α due to hypoxia under the inflammatory conditions. Then secreted Wnt5a inhibits angiogenesis, leading to further hypoxia and a further increase in Wnt5a-secreting InfFib. The increased Wnt5a-secreting InfFib secretes EREG and promotes cancer cell proliferation (Fig. 10n).
One of the significant revelations from our study is the identification of the prominent role of InfFib as Wnt5a-secreting fibroblasts in colon cancer. To this end, we have characterized fibroblasts by integrating human scRNA-seq datasets. To comprehensively understand the nature of colon fibroblasts, we first performed a large-scale meta-analysis of fibroblasts derived from human specimens in this study. Importantly, data from healthy donors and IBD patients were added to the colon cancer samples. Since the pathologies of the two diseases, colon cancer and IBD, are very different, they are not usually analyzed in an integrated manner. However, from the biological perspective of fibroblast distribution and activation, they might have many parts in common. Given that inflammation is a strong stimulus for fibroblast activation, data from IBD patients was essential to comprehensively assess whether activated fibroblasts in tumors require tumor-specific stimuli or can also arise in non-tumor inflammatory contexts. Integrating these data made it possible to capture the whole picture of colon fibroblasts.
Another challenge in this study was to examine whether mouse models reflect the profiles of human colon fibroblasts. The NMF application enabled us to identify gene programs of fibroblasts. Furthermore, the projection of NMF gene features allows human and mouse comparison, which is an important evaluation method when applying scRNA-seq-based categorization to model organisms. While the proportions of subtypes were not consistent between human and mouse, major characteristics and localization were shared across subtypes. Therefore, mouse models are considered suitable as analytical models for studying human colon fibroblasts, except for the limitation that the proportions of each subtype cannot be compared in absolute term. Additionally, it can potentially facilitate the development of targeted therapies or predictive biomarkers for human colon cancer based on fibroblast subtypes.
Based on this fibroblast subtype classification, we found that Wnt5a is expressed in BMPsFib and InfFib in IBD and colon cancer patients. Localization of each fibroblast subtype revealed that InfFib, similar to Wnt5a, has a specific localization near the luminal surface. Further analysis, including trajectory analysis, revealed the shift in fibroblast populations from BMPsFib to InfFib, partly due to hypoxia in the disease state. Thus, hypoxia was suggested to regulate the spatial identity of Wnt5a and InfFib. Consistently, it was reported that HIF2 induces Wnt5a expression in duodenal organoids after radiation by binding to a hypoxia response element in the Wnt5a promoter59. We demonstrated that HIF2 but not HIF1 directly upregulates Wnt5a expression in fibroblasts. Additionally, it is generally accepted that Wnt5a expression is induced under inflammatory conditions10,24. For instance, inflammation induces Wnt5a expression through signaling pathways involving TGF-β, IL-1β, TNFα, IFNγ, and IL-660. There is significant crosstalk between hypoxia and inflammation, where hypoxia induces inflammation and inflammation induces hypoxic states47. Hypoxic conditions outside the cancer setting, such as those in DSS-induced inflammation and IBD cases, would induce Wnt5a expression in fibroblasts. Indeed, the present study showed that the NF-κB and hypoxia pathways are activated in InfFib where Wnt5a is expressed. While it is possible that both signals influence each other, our data shows that hypoxia can directly induce Wnt5a expression in fibroblasts through HIF2.
Moreover, Wnt5a knockout mice analysis showed that Wnt5a contributes to fibroblast switching via regulation of angiogenesis during disease progression. These are consistent with the results that Wnt5a inhibits vascular endothelial growth factor-induced angiogenesis in lung cancer61. Wnt5a has been shown to have both angiogenesis-suppressing and -promoting effects62. For instance, sFRP2 induces nuclear translocation of NFATc3, which may be involved in Wnt5a-dependent Ca2+ signaling, and promotes angiogenesis63, while macrophage-derived Wnt5a inhibits wound angiogenesis49. These differences may be due to variations in the receptor selectivity of Wnt5a. Ror1 and Ror2 (Ror1/2) are well-known and major Wnt5a receptors10 and it was reported that hypoxia induces a more invasive Wnt5a-Ror2-positive phenotype in melanoma64. However, since the mRNA levels of Ryk were much higher than those of Ror1/2 in endothelial cells in colon cancer, we proposed the importance of the Wnt5a-Ryk pathway. This study revealed that the Wnt5a-Ryk pathway contributes to the suppression of angiogenesis. Thus, Wnt5a may have context-dependent effects on angiogenesis, influenced by the specific receptor profile of the cell type.
One might question why Wnt5a’s effects do not extend throughout the tumor, creating a uniformly hypoxic environment. This can be attributed to the biochemical properties of Wnt ligands. Wnt ligands are lipid-modified glycoproteins with limited diffusibility in the extracellular space. Consequently, in most tissues, Wnt proteins are thought to function primarily through interactions with neighboring cells55. This characteristic likely explains the formation of a localized hypoxic region near the luminal surface due to the restricted action of Wnt5a. Since InfFib is closely localized to endothelial cells rather than macrophages and monocytes, Wnt5a derived from InfFib could interact with endothelial cells due to its lipid modification even though macrophages and monocytes express Wnt5a.
There are conflicting reports indicating that Wnt5a shows a tumor-promoting or -suppressing role65. Taken together with the results that Wnt5a is necessary for the maintenance of InfFib, our study supports the former possibility. Namely, Wnt5a-secreting InfFib, but not Wnt5a itself, promoted the growth of colon cancer cells by secreting EREG, albeit indirectly. Although InfFib has been known to play a crucial role in tumor malignancy and therapy resistance by inducing the expression of pro-inflammatory mediators, extracellular matrix remodeling enzymes, and cytokines66, this study revealed that InfFib is a major source of EGF ligand family in fibroblasts, which are essential for tumor progression. EREG has been reported to enhance angiogenesis and vascular remodeling67 as well as to increase tubular formation in HUVEC, suggesting that EREG promotes angiogenesis by upregulating VEGFA expression in endothelial cells68. This may appear contradictory to the increase in vascularization in Wnt5a cKO tumors. However, we found that Wnt5a induces the expression of soluble Flt1 (sFlt1) in HUVEC. Since sFlt1 binds to VEGFA and inhibits VEGFA-dependent signaling, Wnt5a may suppress EREG-VEGFA-mediated angiogenesis near the luminal surface. On the other hand, EREG directly stimulates tumor cells, promoting colon cancer growth. Therefore, it is plausible that in Wnt5a cKO tumors where EREG expression is reduced, angiogenesis in regions adjacent to the luminal surface may be enhanced, whereas tumor growth is suppressed.
Hypoxia is generally considered tumor-suppressive due to reduced oxygen and nutrient supply, supporting the rationale for anti-angiogenic therapies. However, such therapies often show limited clinical efficacy69, suggesting the existence of another aspect of hypoxia. This study proposes that luminal hypoxia, maintained by Wnt5a, promotes tumor progression, as discussed above. Moreover, hypoxia induced by vascular depletion can enhance malignancy through mechanisms such as pro-angiogenic factor secretion (bFGF, PDGF), epithelial-mesenchymal transition (EMT), and an immunosuppressive microenvironment70. These findings suggest that hypoxia can play a tumor-promoting role under certain conditions.
These observations concerning the interplay between Wnt5a-secreting InfFib, hypoxia, and inflammation underscore the importance of understanding the diverse cellular components within the tumor microenvironment and their unique contributions to colon cancer progression (Fig. 10n). Our study uncovers roles of Wnt5a in colon cancer, emphasizing its multifaceted contributions to tumor progression through modulation of the tumor microenvironment and intercellular signaling pathways. In addition to colon cancer, Wnt5a expression in CAFs was observed in bladder-urothelial, ovarian, and pancreatic cancers71. These findings not only enhance our understanding of cancer biology but also provide a foundation for developing targeted therapeutic strategies aimed at disrupting Wnt5a-mediated pathways and Wnt5a-expressing fibroblasts.
Methods
Ethical approval
The research complies with all relevant ethical regulations. The protocol for the utilization of human specimens was approved by the Ethical Committee of Osaka University Graduate School of Medicine (The University of Osaka, Osaka, Japan; No. 13455). The protocols used for all animal experiments in this study were approved by the Animal Research Committee of Osaka University (The University of Osaka, Osaka, Japan; No. 21-048-1).
Patients and cancer tissues
Human colon cancer tissue specimens were used for immunohistochemical analysis. In this study, tissue samples were obtained from seven colon adenocarcinoma patients (two female and five male; 55 to 84 years) with stage IIa to IIIc who underwent surgical resection between April 2024 and May 2024 at Osaka University Hospital. Tumor tissues were fixed in 10% (v/v) formalin, embedded in paraffin, and sectioned at a thickness of 5 µm. The protocol for the utilization of human specimens was approved by the Ethical Committee of Osaka University Graduate School of Medicine (The University of Osaka, Osaka, Japan; No. 13455). The study was performed in accordance with committee guidelines and regulations. Written informed consent was obtained from all participants prior to sample collection. No compensation was provided to the participants. Details of the patient information are provided in Supplementary Table 1. The research using human samples was well-designed to evaluate the expression of a specific protein; however, since it was not primarily designed to investigate sex-based differences in its expression, a sex-based analysis was not performed.
Animals
The species used in this study was Mus musculus. Details of Wnt5afl/fl and Wnt5afl/fl;CAG-Cre/ERT2Tg mouse line generation were described previously24. The background strain of these mice is a C57BL/6 and 129 mixed strain. A stock solution of tamoxifen (4-hydroxitamoxifen; Sigma-Aldrich, St. Louis, MO, USA; Cat. T5648) in ethanol (20 mg/mL) was diluted in corn oil to 2 mg/mL, and 15 mg/kg of tamoxifen was administered to Wnt5afl/fl;CAG-Cre/ERT2Tg mice and control Wnt5afl/fl mice every two days for a total of three doses to induce the activation of the Cre-ERT2 recombinase and to remove the floxed Wnt5a coding region. Seven to eight-week-old male mice were used for all the experiments. Sex-based analysis was not performed in this study. Only male mice were used for AOM/DSS-induced colon cancer model, because it is known that tumor incidence rates are significantly higher in male mice than in female mice72. Mice were housed under SPF (specific pathogen-free) conditions, fed a chow diet, and provided with ad libitum access to water. They were maintained under a 12 h light/dark cycle, with the housing room temperature between 21.5 °C and 24.5 °C and humidity at 30–60%. The protocols used for all animal experiments in this study were approved by the Animal Research Committee of Osaka University (The University of Osaka, Osaka, Japan; No. 21-048-1). All animal experiments were carried out according to the guidelines for the care and use of experimental animals of the University of Osaka. For the AOM/DSS tumor model, the protocol approved by the committee defined humane endpoints as unexpected feeding disorders, a body weight loss exceeding 20% within a few days, and respiratory or circulatory distress associated with diarrhea or hematochezia. Prior to tumor sampling, it was confirmed that none of these conditions indicating pain or distress were present.
Colitis-associated cancer model in mice
Induction of colitis-associated carcinogenesis was performed as described previously73. Briefly, seven to eight-week-old mice were given a single intraperitoneal administration of 10 mg/kg azoxymethane (AOM; FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan; Cat. 011-20171). After 1 week since the administration of AOM, they were given 3% (w/v) dextran sulfate sodium (DSS; FUJIFILM Wako Pure Chemical Corporation; Cat. 199-08361) drinking water for 1 week, and 15 mg/kg tamoxifen solution was administered at the indicated periods. The tumor evaluation was conducted 20 weeks after AOM administration.
Cell culture
WEHI-YH2 cells were kindly provided by Dr. Antony Burgess (The University of Melbourne) in March 2021, and were grown in Dulbecco’s modified Eagle’s medium (DMEM; Nacalai Tesque, Kyoto, Japan; Cat. 08456-65) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA; Cat. 10270-106). HUVEC were purchased from PromoCell (Heidelberg, Germany; Cat. C-12208) in December 2022, and were maintained in Endothelial Cell Growth Medium 2 supplemented with supplement mix (PromoCell; Cat. D12018). HUVEC from passages 2 to 5 were used for experiments. SW480 cells were kindly provided by Dr. Wataru Yasui (Hiroshima University) in September 1997, and were grown in RPMI 1640 (FUJIFILM Wako Pure Chemical Corporation; Cat. 189-02025) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific; Cat. 10270-106). CACO-2 cells were purchased from RIKEN BioResource Research Center (BRC) Cell Bank (Ibaraki, Japan) in April 2013, and were grown in DMEM (Nacalai Tesque; Cat. 08456-65) supplemented with 20% fetal bovine serum (Thermo Fisher Scientific; Cat. 10270-106) and 1% non-essential amino acids (Thermo Fisher Scientific; Cat. 11140-050). Lenti-X 293 T (X293T) cells were purchased from Takara Bio (Shiga, Japan; Cat. 632180) in October 2011, and were grown in DMEM (Nacalai Tesque; Cat. 08456-65) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific; Cat. 10270-106). All cell lines were cultured in a 37 °C humidified incubator supplying 5% CO2 except for hypoxic condition, in which cells were incubated in a multi-gas incubator with 1% oxygen. For induction of chemical hypoxia, cells were treated with 200 μM CoCl2 (Nacalai Tesque; Cat. 09206-92). Mycoplasma testing was conducted for WEHI-YH2 and Lenti-X 293 T cells, and for the prevention of contamination by mycoplasma, cells were cultured with Normocin (Invivogen, San Diego, CA, USA; Cat. ant-nr-2). HUVEC, SW480 cells and CACO-2 cells were not tested for mycoplasma contamination. Authentication of HUVEC was provided by PromoCell. Lenti-X 293 T cells were authenticated using short tandem repeat profiling by BEX (Tokyo, Japan). WEHI-YH2, SW480, and CACO-2 cells were not authenticated.
Meta-analysis of human fibroblasts from public datasets
For meta-analysis of human colon fibroblasts related to Fig. 3a, scRNA-seq data of colon tissue generated by 10x platform were collected as described in Supplementary Tables 2–5. Data from tumor adjacent normal tissue were excluded, because the definition of adjacent normal tissue was not consistent across datasets. A breakdown of cell counts for each dataset is provided in Supplementary Table 6 and illustrated in Supplementary Fig. 3b. The datasets used for the meta-analysis of fibroblasts included some without specified sex information, making it impossible to conduct an accurate sex-based analysis. If the count matrix was available, quantified expression values were used. Otherwise, the expressions were quantified using Cell Ranger with pre-built reference refdata-gex-GRCh38-2020-A. Quantified expressions were pre-processed for each dataset and visualized using Scanpy (v1.8.1) and Python (v3.8.13). Doublets were detected using Scrublet (v0.2.1) and were removed. The filtered data from Uhlitz F. et al. were used as is without further doublet removal31. Cells with high level of mitochondrial gene expression, and with detected genes less than 200 were filtered out (sc.pp.filter_cells). Genes that were expressed in at least 3 cells were kept (sc.pp.filter_genes). Gene expression was normalized (sc.pp.normalize_total) and log-transformed (sc.pp.log1p). Highly variable genes were extracted (sc.pp.highly_variable_genes). Effects of the total counts of UMI and percentages of mitochondrial genes were regressed out (sc.pp.regress_out), and gene expression was scaled (sc.pp.scale). Then principal components and neighbors were calculated (sc.pp.pca, sc.pp.neighbors). The batch effect of samples was removed by BBKNN (sc.external.pp.bbknn), which was only used for the batch correction of different samples within a single dataset. Otherwise, principal components were calculated (sc.pp.pca), the batch effect of samples was removed by Harmony (sce.pp.harmony_integrate), and neighbors were computed (sc.pp.neighbors). Cells were embedded in a two-dimensional space with UMAP (sc.tl.umap) and clustered using Leiden algorithm (sc.tl.leiden). Cell clusters were annotated based on well-known marker genes: epithelial cells (EPCAM, KRT19); immune cells (PTPRC, CD52); endothelial cells (PECAM1); pericytes (RGS5, PDGFRB); and glial cells (S100B). To further refine cell type identification, we utilized sc.tl.score_genes from the Scanpy package to calculate signature scores based on marker gene sets. Marker gene sets were obtained from the top 100 genes reported by Smillie CS et al.36 for endothelial cells, pericytes, and fibroblasts. Similarly, top 20 cell type marker genes defined by Kinchen J. et al.33 were used for endothelial cells, pericytes, and glial cells. In addition, fibroblast clusters were extracted by confirming the ubiquitous expression of fibroblast markers such as DCN, LUM, and COL1A174. The data obtained from Korsunsky I et al.75 had already been filtered to include only fibroblasts, and it was used directly for the downstream analysis. These are illustrated in Supplementary Fig. 3a. Next, extracted fibroblasts were integrated across datasets. Pre-filtering and pre-process were performed as mentioned above, and hemoglobin and immunoglobulin genes were removed since they are common contaminants from ambient RNA as previously described76. Effects of the percentages of mitochondrial genes and ribosomal genes were regressed out (sc.pp.regress_out), and gene expression was scaled (sc.pp.scale). Then principal components were calculated (sc.pp.pca), the batch effect of samples was removed by Harmony (sce.pp.harmony_integrate), and neighbors were computed (sc.pp.neighbors). Additionally, to assess the impact of different batch correction methods, BBKNN was used instead of Harmony, as mentioned above. Alternatively, batch correction was performed using scVI, following the tutorial77. Ten datasets described in Supplementary Fig. 3c were set as batch covariate. Cells were embedded in a two-dimensional space with UMAP (sc.tl.umap) and clustered using Leiden algorithm (sc.tl.leiden). Fibroblasts were classified into six categories according to the degree of activation of the NMF programs. Details for NMF are described below.
Gene expression decomposition using NMF
The log-transformed and normalized gene expression matrix of highly variable genes (HVG) were used for NMF analysis. The detailed method is described in the previous report42. Briefly, we applied NMF implemented in scikit-learn (v0.24.2) to normalize gene expression of HVGs and got a decomposed cell feature matrix and gene feature matrix. To determine the number of factors, we utilized the measurements, explained variance and the proportion of overlapped highly variable genes (POH) to select conditions with less duplication while maintaining as much information as possible.
Sample preparation for scRNA-seq of mouse tissue
The large intestines within 2 cm of the anus were opened and the fecal content was removed. Tumors were incubated in HBSS containing 5 mM EDTA for 20 min at 37 °C with shaking at 110 rpm. Tumor tissues were then cut into small pieces and incubated with RPMI 1640 (FUJIFILM Wako Pure Chemical Corporation; Cat. 189-02025) containing 4% fetal bovine serum (Thermo Fisher Scientific; Cat. 10270-106), 1 mg/mL Collagenase D (Roche Diagnostics, Basel, Switzerland; Cat. 11088866001), 0.5 mg/mL Dispase II (Roche Diagnostics; Cat. 04942078001), and 40 μg/mL DNase I (Roche Diagnostics; Cat. 11284932001) for 45 min at 37 °C with shaking at 110 rpm. The digested tissues were washed with DMEM (Nacalai Tesque; Cat. 08456-65) containing 10% fetal bovine serum (Thermo Fisher Scientific; Cat. 10270-106) and filtered through a 70 μm cell strainer, followed by a 40 μm cell strainer. Cell suspension was incubated with anti-CD45, anti-CD326, anti-CD31, and anti-Pdpn antibodies, and FACS (fluorescence-activated cell sorting)-sorted for live CD45-CD326-CD31-Pdpn+ cells (Supplementary Fig. 13), followed by scRNA-seq. Samples sorted for live cells by FACS, without selection based on cell-specific markers, were also subjected to scRNA-seq.
Single-cell RNA-sequencing
For the construction of single-cell RNA-seq library, Chromium Next GEM Single Cell 3’ Library and Gel Bead Kit v3.1 (10x Genomics, Pleasanton, CA, USA) was used according to the manufacturer’s instructions. Libraries were sequenced on a NovaSeq 6000 (Illumina, San Diego, CA, USA), and the resulting raw data were processed by Cell Ranger (10x Genomics).
scRNA-seq data analysis of total cells in the mouse tumor
The scRNA-seq data presented in Fig. 2a were obtained from two different sources. One dataset is original to this study and derived from tumor of AOM/DSS model mice. The other one, derived from an orthotopic transplantation mouse model, was obtained from publicly available data27. Quantified expressions were pre-processed and visualized using Scanpy (v1.8.1) and Python (v3.8.13). Doublets were detected using Scrublet (v0.2.1) and were removed (Supplementary Fig. 2a). Cells with high levels of mitochondrial gene expression and with detected genes less than 200 were filtered out (sc.pp.filter_cells). Genes that were expressed in at least 3 cells were kept (sc.pp.filter_genes). Gene expression was normalized (sc.pp.normalize_total) and log-transformed (sc.pp.log1p). Highly variable genes were extracted (sc.pp.highly_variable_genes), and gene expression was scaled (sc.pp.scale). Then principal components were calculated (sc.pp.pca), the batch effect of samples was removed by Harmony (sce.pp.harmony_integrate), and neighbors were computed (sc.pp.neighbors). Cells were embedded in a two-dimensional space with UMAP (sc.tl.umap) and clustered using Leiden algorithm (sc.tl.leiden). Cell clusters were annotated based on well-known marker genes shown in Supplementary Fig. 2b. To further refine cell type identification, CellTypist78 was used to transfer cell type labels from the model, “Adult_Mouse_Gut,” to mouse colon cancer total cell data. “Best match” mode was used with default parameters. Furthermore, using the dataset of immune profile from MC38 tumor-bearing mice6, the expression of T cell markers was evaluated across different cell types. Additionally, to independently evaluate Wnt5a expression across cell types, scRNA-seq data derived from healthy mouse and AOM/DSS and Lrig1CreERT2/+;Apc2lox14/+ mouse model was used8. The analysis followed the same workflow as described above. While the classification was performed in the same manner as the original data, modifications were made based on the expression of marker genes shown in Supplementary Fig. 2b and the results of label transfer using CellTypist. Specifically, “macrophage” was further subdivided into macrophage, cDC, and pDC, “T cell” into Cd4T, Cd8T, and Tgd, and “CAF” into fibroblast and pericyte.
scRNA-seq data analysis of mouse tumor fibroblasts
The scRNA-seq data presented in Fig. 4a were obtained from two different sources. One dataset is original to this study and includes fibroblasts isolated from AOM/DSS model mice. Fibroblasts were isolated via FACS as described in Supplementary Table 8 after tumor harvest, followed by scRNA-seq. The second dataset was derived from an orthotopic transplantation mouse model as described in the “scRNA-seq data analysis of total cells in the mouse tumor” section. Quantified expressions were pre-processed and visualized using Scanpy (v1.8.1) and Python (v3.8.13). Doublets were detected using Scrublet (v0.2.1) and were removed. Cells with high levels of mitochondrial gene expression, and with detected genes less than 200 were filtered out (sc.pp.filter_cells). Genes that were expressed in at least 3 cells were kept (sc.pp.filter_genes). Gene expression was normalized (sc.pp.normalize_total) and log-transformed (sc.pp.log1p). Highly variable genes were extracted (sc.pp.highly_variable_genes). Effects of the percentages of mitochondrial genes and ribosomal genes were regressed out (sc.pp.regress_out), and gene expression was scaled (sc.pp.scale). Then principal components were calculated (sc.pp.pca), the batch effect of samples was removed by Harmony (sce.pp.harmony_integrate) for data obtained in this study, and neighbors were computed (sc.pp.neighbors). Cells were embedded in a two-dimensional space with UMAP (sc.tl.umap) and clustered using Leiden algorithm (sc.tl.leiden). The method for fibroblast selection is described in detail in the “Meta-analysis of mouse fibroblasts from public datasets” section. Next, extracted fibroblasts were integrated across datasets. Pre-filtering was performed as mentioned above. Effects of the total counts were regressed out (sc.pp.regress_out), and gene expression was scaled (sc.pp.scale). Then, principal components were calculated (sc.pp.pca), the batch effect of samples was removed by Harmony (sce.pp.harmony_integrate), and neighbors were computed (sc.pp.neighbors). Cells were embedded in a two-dimensional space with UMAP (sc.tl.umap) and clustered using Leiden algorithm (sc.tl.leiden). For gene program quantification of mouse fibroblasts, NMFproj was used according to the tutorial42. Mouse fibroblasts were factorized with 8 NMF programs defined in the human colon fibroblasts, and cell type labels were assigned based on the program activity.
Meta-analysis of mouse fibroblasts from public datasets
For meta-analysis of mouse colon fibroblasts related to Fig. 4h, scRNA-seq data of mouse colon tissue was collected as described in Supplementary Table 8 including those originally obtained in this study. If the count matrix was available, quantified expression values were used. Otherwise, the expressions were quantified using Cell Ranger with pre-built reference refdata-gex-mm10-2020-A. Quantified expressions were pre-processed for each dataset and visualized using Scanpy (v1.8.1) and Python (v3.8.13). Doublets were detected using Scrublet (v0.2.1) and were removed except for Ho YT. et al. where cells were sorted into the plate using single-cell purity mode of FACS instrument79. Cells with high level of mitochondrial gene expression, and with detected genes less than 200 were filtered out (sc.pp.filter_cells). Genes that were expressed in at least 3 cells were kept (sc.pp.filter_genes). Gene expression was normalized (sc.pp.normalize_total) and log-transformed (sc.pp.log1p). Highly variable genes were extracted (sc.pp.highly_variable_genes), and gene expression was scaled (sc.pp.scale). Then, principal components and neighbors were calculated (sc.pp.pca, sc.pp.neighbors). The batch effect of samples was removed by BBKNN (sc.external.pp.bbknn), which was only used for the batch correction of different samples within a single dataset. Otherwise, principal components were calculated (sc.pp.pca), the batch effect of samples was removed by Harmony (sce.pp.harmony_integrate), and neighbors were computed (sc.pp.neighbors). Cells were embedded in a two-dimensional space with UMAP (sc.tl.umap) and clustered using Leiden algorithm (sc.tl.leiden). Cell clusters were annotated based on well-known marker genes: epithelial cells (Epcam, Krt19); immune cells (Ptprc, Cd52); endothelial cells (Pecam1); pericytes (Rgs5, Pdgfrb); glial cells (S100b); and Cajal cells (Ano1).
To further refine cell type identification, we utilized sc.tl.score_genes from the Scanpy package to calculate signature scores based on marker gene sets. Marker gene sets were obtained from the top 20 genes reported by Kinchen J. et al.33 for epithelial cells, immune cells, vascular endothelial cells, lymphatic endothelial cells, pericytes, glial cells, and smooth muscle cells / Cajal cells. In addition, fibroblast clusters were extracted by confirming the ubiquitous expression of fibroblast markers such as Pdgfra and Lum74. Next, extracted fibroblasts were integrated across datasets. Pre-processing was performed as mentioned above. Then, principal components were calculated (sc.pp.pca), the batch effect of samples was removed by Harmony (sce.pp.harmony_integrate), and neighbors were computed (sc.pp.neighbors). Cells were embedded in a two-dimensional space with UMAP (sc.tl.umap) and clustered using Leiden algorithm (sc.tl.leiden). For gene program quantification of mouse fibroblasts, NMFproj was used according to the tutorial42. Mouse fibroblasts were factorized with 8 NMF programs defined in the human colon fibroblasts, and cell type labels were assigned based on the program activity in the same manner as described in the “scRNA-seq data analysis of mouse tumor fibroblasts” section.
Cross-species analysis
SAMap was performed for integrative analysis of human and mouse fibroblast scRNA-seq data following the tutorial80. Human genome GRCh38.p13 and mouse genome GRCm39 were used to run tblastx analysis. Raw counts from scRNA-seq were loaded in SAMap object and pre-processed using sam.preprocess_data. Then, the SAMap algorithm was performed with the default parameter using sam.run to determine cross-species neighbors. Integrated data were visualized in the UMAP space. The alignment scores between cell types are used to show the homology between human and mouse in a matrix plot.
Trajectory analysis
Trajectory analysis for mouse colon cancer fibroblasts data was conducted using the Partition-based Graph Abstraction (PAGA)81 function in Scanpy. Pre-processing steps, including normalization, highly variable gene selection, and scaling, were performed as described in the Methods “scRNA-seq data analysis of mouse tumor fibroblasts” section. To integrate data across different batches, Harmony (sce.pp.harmony_integrate) was applied. After integration, a neighborhood graph was constructed with sc.pp.neighbors, and cluster connectivity was calculated using sc.tl.paga. PAGA builds a graph representation of cellular clusters, where each node represents a cluster, and edges reflect the connectivity strength between clusters based on transcriptomic similarity. Also, diffusion pseudotime (DPT) analysis82 was performed using sc.tl.dpt with the default parameters. DPT calculates a diffusion-based distance metric, referred to as DPT distance, with respect to the root cell. BMPsFib was set to the root cell in this study. Additionally, CytoTRACE was calculated to assess cellular differentiation potential based on the number of expressed genes per cell83. The results from PAGA, DPT, and CytoTRACE were integrated and projected onto a force-directed graph using sc.pl.draw_graph.
Cell-type deconvolution of clinical RNA-seq data
Gene expression data of tumor lesions and adjacent normal sites from colon adenocarcinoma patients were obtained from UCSC Xena Hub (cohort: TCGA Pan-Cancer (PANCAN),”Batch effects normalized mRNA data”), and expression profiling of colon tissue from healthy donors and Crohn’s disease patients were obtained from GSE 16487139. Normalized transcript expression values in natural scale were used as input data for prediction. To prepare the training data for the analysis of TCGA cohort, “Human Colon Cancer Atlas” was used with modification28. Cells annotated as “Fibro” in ClusterMidway were removed from the atlas, and the expression profile from “human colon fibroblast atlas” which was established in this study, was integrated. Data from tumor-adjacent normal tissue were excluded from the reference scRNA-seq dataset because the definition of adjacent normal tissue was not consistent across datasets. To prepare the training data for the analysis of IBD dataset, single-cell profiles of colon tissue established previously was used with modification36. The cell population defined as fibroblasts in the original study was replaced with the expression profile from “human colon fibroblast atlas.” Cell-type deconvolution was performed using Scaden (v1.1.2) with default parameters.
Cell deconvolution of Visium samples
For investigating the spatial localization of specific cell subtypes, Cell2location was applied using previously published Visium datasets according to the tutorial84. For the preparation of scRNA-seq reference data, “Human Colon Cancer Atlas” was used with modification28. Cells annotated as “Fibro” in ClusterMidway were removed from the atlas, and the expression profile from “human colon fibroblast atlas” which was established in this study was integrated.
Preparation of bulk RNA-seq sample and RNA-sequencing
The intestinal tract within 2 cm of the anus was sampled and only the tumor mass was harvested from tumor-bearing mice 20 weeks after AOM administration. Tumor tissues were homogenized using BioMasher II (Nippi, Tokyo, Japan; Cat. 320 103) and total RNAs were isolated using NucleoSpin RNA (MACHEREY-NAGEL, Dueren, Germany; Cat. 740955.250). For experiments investigating changes in fibroblast subtypes in vitro, WEHI-YH2 cells were treated with 5 ng/mL TNFα (FUJIFILM Wako Pure Chemical Corporation; Cat. 203-15263) or 200 μM CoCl2 (Nacalai Tesque; Cat. 09206-92) for 24 h, and total RNA was isolated using Maxwell RSC instrument with Maxwell RSC simplyRNA cells kit (Promega, Madison, WI, USA; Cat. AS1390). Library preparation was performed using a TruSeq stranded mRNA Library Prep kit (Illumina) according to the manufacturer’s instructions. Sequencing was performed on a DNBSEQ-G400 (MGI Tech, Shenzhen, China) in a paired-end mode (2×101 nt).
Bulk RNA-seq data analysis
Sequenced reads were preprocessed by Trim Galore! (v0.6.7) and quantified by Salmon (v1.4.0) with the flags gcBias and validateMappings. GENCODE vM26 (GRCm39) annotation was used as the transcript reference. All procedures were implemented using the RNA-seq pipeline ikra (v2.0.1)85 with default parameters. The quantified transcript-level scaled TPM was summarized into a gene-level scaled TPM using tximport (v1.22.0). DESeq2 (v1.34.0) was used to generate normalized counts. Signature score for mouse fibroblast subtype was defined as the mean z-score of the list of marker genes for each subtype (Supplementary Table 9). Genes with a DESeq2 normalized count of less than 10 in more than half of the samples were excluded from the list of genes.
Pathway activity inference
Pathway activity inference with scRNA-seq or bulkRNA-seq data was performed using decoupleR (v1.2.0) and PROGENy as a curated collection of pathways and their target genes. Top 50 responsive genes ranked by p- value were used for pathway enrichment score calculation in the multivariate linear model (mlm) method.
Analysis of gene expression profiles from open-source clinical RNA-seq data
Transcript expression data of colon adenocarcinoma patients were obtained from UCSC Xena Hub. In comparing tumor and normal expression, cohort: TCGA TARGET GTEx (RSEM norm_count) was used. Otherwise, cohort: TCGA Pan-Cancer (PANCAN) (Batch effects normalized mRNA data) was used for normalized expression data. To evaluate NMF program and signaling pathway activities in bulk RNA-seq cohorts, signature scores were calculated. For Inflammatory_Program scores, top 50 weighted genes in the Inflammatory_Program were used. For cell proliferation, hypoxia, and MAPK signaling score, gene sets obtained from MSigDB under dataset ID M16210, M5891, and M10792, respectively, were used. The signature score was defined as the mean z-score of the z-score transformed data.
Published ChIP-seq and ATAC-seq data analysis
HIF2α ChIP-seq data and ATAC-seq data obtained from mouse uteri were collected from the Gene Expression Omnibus (GEO) with accession number GSE234065. Processed BigWig files were loaded into Integrative Genomics Viewer (IGV) using GRCm38 (mm10) as a reference genome. WashU Epigenome Browser (https://epigenomegateway.wustl.edu/) was used for visualization.
Plasmid construction and generation of stable cell lines
To generate WEHI-YH2 cells stably expressing mouse Wnt5a (mWnt5a), human HIF2α (hHIF2α), and mouse Ereg (mEreg), WEHI-YH2 cells were transfected with pPB-CAG-mWnt5a-IRES2-BSD, pPB-CAG-hHIF2α-IRES2-BSD, or pPB-CAG-mEreg-IRES2-BSD, and pCMV-hyPBase using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific; Cat. 11668019). Then cells were selected with 10 μg/mL Blastcidin (Invivogen; Cat. ant-bl-05). To construct PiggyBac transposon vector expressing mWnt5a, hHIF2α, and mEreg standard recombinant DNA techniques including In-Fusion cloning method (Takara Bio; Cat. 639649) were used. mEreg gene was synthesized by Eurofins Genomics (Tokyo, Japan). pcDNA3-HA-HIF1α (#18949) and pcDNA3-HA-HIF2α (#18950) were obtained from Addgene (Watertown, MA, USA) for transient gene expression and gene cloning.
Immunohistochemical analysis
The intestinal tract within 2 cm of the anus was sampled, opened, stretched using insect pins, fixed for 24 h at 4 °C in PBS containing 4% (w/v) paraformaldehyde, and embedded in paraffin. Paraffin-embedded samples were sectioned at 5-μm thickness. Heat-induced epitope retrieval was performed using Decloaking Chamber NxGen (Biocare Medical, Walnut Creek, CA, USA). Target Retrieval Solution, Citrate pH6 (Dako, Carpentaria, CA, USA; Cat S1699) or HistoVT One (Nacalai Tesque; Cat. 06380-76) was used for antigen retrieval solution. In DAB staining, tissue peroxidase activity was blocked with Dako REAL Peroxidase Blocking Solution (Dako; Cat. S2023) for more than 1 h at 4 °C. To block non-specific antibody binding sites, the sections were then incubated with Blocking One Histo (Nacalai Tesque; Cat. 06349-64) or Protein Block, Serum-Free (Dako; Cat. X0909) for more than 4 h at 4 °C. Next, tissue sections were incubated with antibody solution diluted with Antibody Diluent, Background Reducing solution (Dako; Cat. S3022). In DAB staining, HRP-conjugated antibody was used as secondary antibody and diaminobenzidine (DAB; Dako; Cat. K3468) was used as a chromogen. The tissue sections were then counterstained with Tissue-Tek Hematoxylin 3 G (Sakura Finetek Japan, Tokyo, Japan; Cat. 9131-4 P). In immunofluorescence staining, fluorescent dye (Alexa Fluor 488, 546, 647; Thermo Fisher Scientific)-conjugated antibodies were used as secondary antibodies, and a coverslip was mounted on a slide using Vibrance Antifade Mounting Medium with DAPI (Vector Laboratories, Newark, CA, USA; Cat. H-1800) for PFA fixed samples or VECTASHIELD Mounting Medium with DAPI (Vector Laboratories; Cat. H-1200) for MeOH fixed samples. Primary antibodies and their final concentration for IHC studies were described in Supplementary Table 13. Fluorescence images were viewed and analyzed under an LSM880 laser scanning microscope (Carl Zeiss, Jana, Germany) equipped with objective Plan-Apochromat 10x/0.45 M27 (Carl Zeiss), objective Plan-Apochromat 20x/0.8 M27 (Carl Zeiss), and objective Plan-Apochromat 40x/1.4 Oil DIC M27 (Carl Zeiss), ZEN black edition software (Carl Zeiss), and ZEN blue edition software (Carl Zeiss). Bright-field images were viewed and analyzed under an OLYMPUS BX53 microscope (Evident, Tokyo, Japan) equipped with objective UPlanSApo 4x/0.16 (Evident) and objective UPlanSApo 10x/0.40 (Evident), DP73 microscope digital camera (Evident), cellSens Standard (Evident), and NanoZoomer-SQ (Hamamatsu Photonics K.K., Shizuoka, Japan). For fluorescence images, contrast and brightness were uniformly adjusted across the entire image for all comparative images using either ZEN Blue Edition or PowerPoint. The positive areas for Mmp10, Il11, Cnn1, Ifi204, Col10a1, Cd31, Pdpn, HIF2α, and hypoxyprobe were quantified using the “area quantification” module of HALO (Indica Labs; Albuquerque, NM, USA). Ki67 stain positivity was quantified using “cytonuclear” module of HALO (Indica Labs).
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation (ChIP) was performed using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, Beverly, MA, USA; Cat. 9003) according to the manufacturer’s instructions, except for using SureBeads Protein G Magnetic Beads (Bio-Rad, Hercules CA, USA; Cat. 161-4023) (100 μL) for immunoprecipitation. The immunoprecipitated chromatin was purified using DNA Purification Buffers and Spin Columns (Cell Signaling Technology; Cat. 14209) and analyzed by quantitative RT-PCR. The primers used are described in Supplementary Table 14.
Knockdown of protein expression by siRNA
The siRNA sequences used in this study are shown in Supplementary Table 15. WEHI-YH2 cells were transfected with siRNA (20 nM) against the genes of interest using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific; Cat. 13778150).
Quantitative RT-PCR
Total RNAs from cultured fibroblasts were extracted using the Maxwell RSC instrument with Maxwell RSC simplyRNA cells kit (Promega; Cat. AS1390). Total RNA (1 μg) was reverse transcribed using ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan; Cat. FSQ-201). Quantitative RT-PCR was performed with Luna Universal qPCR Master Mix (New England Biolabs, Ipswich, MA, USA; Cat. M3003). The primers used for quantitative RT-PCR are listed in Supplementary Table 16.
Immunofluorescence and proximity ligation assay (PLA)
For sample preparation, unfixed tumor tissues were frozen in an O.C.T. Compound (Sakura Finetek; Cat. 4583). Freshly frozen tissues were sectioned at 10 μm and fixed for 10 min at -20 °C in MeOH. To block non-specific antibody binding sites, the sections were then incubated with Blocking One Histo (Nacalai Tesque; Cat. 06349-64) or Protein Block, Serum-Free (Dako; Cat. X0909) for 1.5 h at 4 °C. Next, tissue sections were incubated with antibody solution (anti-Wnt5a and anti-Ryk antibodies) diluted with Antibody Diluent, Background Reducing solution (Dako; Cat. S3022) for 10 h at 4 °C, followed by incubation with goat anti-rat antibody (Abcam, Cambridge, UK; Cat. Ab7092) for 10 h at 4 °C. Then, sections were incubated with the Duolink In Situ PLA probes anti-rabbit PLUS and anti-goat MINUS (Sigma-Aldrich; Cat. DUO92002 and DUO92006) for 1 h at 37 °C. Ligation and amplification steps were performed using Duolink In Situ Detection Reagents Red (Sigma-Aldrich; Cat. DUO92008) according to the manufacturer’s instructions. For counterstaining, a coverslip was mounted on a slide using VECTASHIELD Mounting Medium with DAPI (Vector Laboratories; Cat. H-1200).
Detection of hypoxia in tissue
Hypoxyprobe (pimonidazole hydrochloride; Hypoxyprobe, Burlington, MA, USA; Cat. HP1-100KIT) solution were intraperitoneally injected to AOM/DSS-induced tumor-bearing mice at 60 mg/kg. One h and 45 min after injection, tumor tissues were harvested. The method for preparation of tissue sections was described in the Methods “Immunohistochemical analysis” section. Anti-hypoxyprobe (Hypoxyprobe; Cat.PAB27) was used for detection of hypoxyprobe adducts.
ELISA
Conditioned media were collected from HUVEC exposed to recombinant Wnt5a for 12 h. Next, the conditioned media were centrifuged at 1200 x g at 4 °C for 3 min to remove cell debris, and the supernatant was subjected to ELISA. ELISA for soluble Flt1 was performed using Human VEGFR1/Flt-1 ELISA Kit (R&D Systems, Minneapolis, MN, USA; Cat. DVR100C) according to the manufacturer’s instructions. Absorbance was measured at the specified wavelength using Synergy HTX Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA). Quantification was performed based on the standard curve generated from serial dilutions of the reference standard using GainData, a tool hosted on the web platform (https://www.arigobio.com/elisa-calculator), with the default parameter.
Evaluation of secreted Wnt5a protein
Conditioned media were collected from WEHI-YH2 cells exposed to 1% or 20% O2 for 48 h. Next, the conditioned media were centrifuged at 500 x g for 2 min at 4 °C to remove cell debris, and were incubated with 50 μL of Blue Sepharose 6 Fast Flow (Cytiva, Marlborough, MA, USA; Cat. 17094801) (50% slurry), rotating for 1 h at 4 °C. After incubation, proteins were eluted from the beads with Laemmli’s sample buffer for 5 min at 98 °C. Denatured proteins were separated by SDS-PAGE and transferred onto Amersham Protran 0.45 μm NC (Cytiva; Cat. 10600002) membranes. Membranes were blocked with 5% skim milk in TBST at room temperature for 1 h. Anti-Wnt5a and anti-Hsp90 antibodies were incubated for 12 h at 4 °C, followed by incubation with HRP-conjugated secondary antibodies for 1 h at room temperature. Chemiluminescent signals were detected using ECL Western Blotting Detection Reagents (Cytiva; Cat. RPN2106) or ECL Select Western Blotting Detection Reagents (Cytiva; Cat. RPN2235), and images were captured using the ImageQuant LAS 4000 mini (Cytiva). All images were processed uniformly, with contrast and brightness adjustments applied equally across the entire blot using a LAS 4000 mini built-in analysis tool.
Tube formation assay
HUVEC (1.5e04 cells/well) were plated onto Matrigel (Corning, One Riverfront Plaza, NY, USA; Cat. 354230) and cultured in Endothelial Cell Basal Medium supplemented with 0.5 % fetal calf serum, 1.25 ng/mL recombinant human EGF, 2.5 ng/mL recombinant human basic FGF, 5 ng/mL IGF, 0.125 ng/mL VEGF-165, 0.25 μg/mL ascorbic acid, 5.625 μg/mL heparin, 0.05 μg/mL hydrocortisone (PromoCell; Cat. D12018). The following recombinant proteins were administered where indicated; Wnt5a (R&D Systems; Cat. 645-WN-010) and Wnt5b (purified as previously described54). Cells were incubated for 6 or 20 h to allow tube formation, and phase-contrast images were captured using an OLYMPUS CKX53 microscope (Evident) equipped with objective UPlanFL N 4x/0.13 iPC (Evident) and objective Plan N 2x/0.06 (Evident), and STD3 microscope digital camera (Visualix, Hyogo, Japan). To minimize the meniscus effect near the well edges, regions at a fixed distance from the center were cropped for ensuring consistent quantification. Images were quantified using “Angiogenesis Analyzer” for ImageJ86. Number of segments and the total length of tubes in the analyzed area were quantified. Junctions corresponds to nodes (pixels with 3 neighbors) or group of fusing nodes, and segments corresponds to elements delimited by two junctions.
Sprouting assay
HUVEC spheroids were generated by culturing HUVEC in Endothelial Cell Growth Medium 2 supplemented with supplement mix (PromoCell; Cat. D12018) in low-attachment U-shaped-bottom 96 well plate (Thermo Fisher Scientific; Cat. 174925) for 18 h. Collected spheroids were then seeded onto Matrigel (Corning; Cat. 354230) layer. After culturing for 7 h in the same medium as described above, phase-contrast images were captured and the length of sprouts were measured using ImageJ.
Cell proliferation and wound healing assay
For quantification of proliferative ability, CyQUANT NF Cell Proliferation Assay (Thermo Fisher Scientific; Cat. C35006) was used according to the manufacturer’s instructions. Fluorescence was measured using Synergy HTX Multi-Mode Microplate Reader (BioTek). For evaluating migrative ability, wound healing assay was performed using Culture-Insert 2 well (ibidi GmbH, Bayern, Germany; Cat. 80209) according to the manufacturer’s instructions. Quantification of wound healing assay was performed using WimScratch, a tool hosted on the web platform Wimasis (www.wimasis.com). This platform provides automated, objective assessment of wound healing. Images were uploaded to Wimasis.com, where the WimScratch tool analyzed wound edges and calculated wound closure percentage.
Preparation and cultivation of organoids from AOM/DSS model mice
Tumor mass were isolated and homogenized using a gentleMACS dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). After passing the cells through a 40-μm sterile filter, cells were suspended in 30 μL Matrigel solution (Corning; Cat. 354230) and Matrigel domes were formed in a 24-well culture plate. Complete growth medium consisted of Advanced DMEM/F12 (Thermo Fisher Scientific; Cat. 12634010), 1 x N-2 Supplement (Thermo Fisher Scientific; Cat. 17502048), 1 x B-27 Supplements (Thermo Fisher Scientific; Cat. 17504044), 100 ng/mL recombinant mouse Noggin (R&D Systems; Cat. 1967-NG-025), 1mM N-Acetylcysteine (Sigma-Aldrich; Cat. A7250) and 10 μM Y27632 (FUJIFILM Wako Pure Chemical Corporation; Cat. 036-24023). For the evaluation of EGF ligand family in organoid growth, 100 ng/mL recombinant human EGF (Thermo Fisher Scientific; Cat. PHG0311) or 200 ng/mL recombinant human EREG (BioLegend, San Diego, CA, USA; Cat. 550204) were used. Phase-contrast images were captured, and the organoid area was quantified using BZ-X800 Analyzer (Keyence, Osaka, Japan). The average area for each experiment was calculated. For EdU staining, organoids were treated with 10 μM EdU for 2 h. Organoids in Matrigel were resuspended in iPGell (Genostaff, Tokyo, Japan; Cat. PG20-1), fixed for 24 h at 4 °C in PBS containing 4% (w/v) paraformaldehyde, and embedded in paraffin. Paraffin-embedded samples were sectioned at 5-μm thickness. Labeled DNA was detected using Click-iT EdU Cell Proliferation Kit (Thermo Fisher Scientific; Cat. C10338) according to the manufacturer’s instructions.
Statistics and reproducibility
No statistical method was used to predetermine sample size. Data were excluded where the investigators noted a technical error that may have affected results. Sample selection for immunohistochemical staining was performed randomly. In vitro experiments using cell lines were not randomized since the studies were controlled by the same cell line receiving different treatments. The investigators were not blinded to allocation during experiments and outcome assessment. Biological replicates are replicates on independent biological samples versus technical replicates using the same starting samples. All experiments in this study were repeated using biological replicates. A minimum of three biological replicates were used for quantitative analysis, and the results are presented as the mean. The numbers of biological replicates are stated in each figure legend. The microscopy observation data in Figs. 1i, 2f, g, 5b, c, 6g, h, 7a, b, 8a, 9b, l, m, and 10k, and Supplementary Figs. 8a and 9b represent typical results from two independent experiments using three different tissue sections. The Student’s t-test or Mann-Whitney test was used to determine if there was a significant difference between the means of two groups. One-way analysis of variance (ANOVA) with Holm-Sidak’s multiple comparisons test was used to compare three or more group means. Statistical analysis was performed using GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA); P < 0.05 was considered statistically significant, and the specific P-values are provided in the figure legends or a Source Data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data generated in this study have been deposited in the DDBJ Sequence Read Archive (DRA) database under accession code DRR538779, DRR538780, DRR538781, DRR538782, DRR538783, DRR538784, DRR538785, DRR538786, DRR538787, DRR538788, DRR538789, DRR538790, and DRR538791. The scRNA-seq data used in this study are available in the Gene Expression Omnibus (GEO) database under accession code GSE114374, GSE132465, GSE134255, GSE148794, GSE154863, GSE166555, GSE172261, and GSE178341; in the Single Cell Portal (SCP) database under accession code SCP259 and SCP738; in the European Nucleotide Archive (ENA) database under accession code PRJEB34105; in the following link [https://lambrechtslab.sites.vib.be/en/pan-cancer-blueprint-tumour-microenvironment-0]. Spatial transcriptomic data used in this study are available in the GEO database under accession code GSE225857; in the Genomic Expression Archive (GEA) database under accession code E-GEAD-619. Transcript expression data of colon adenocarcinoma patients were obtained from UCSC Xena Hub [https://xena.ucsc.edu/]. In comparing tumor and normal expression, cohort: TCGA TARGET GTEx (RSEM norm_count) was used. Otherwise, cohort: TCGA Pan-Cancer (PANCAN) (Batch effects normalized mRNA data) was used for normalized expression data. Transcript expression data of colon tissues from healthy donors and Crohn’s disease patients are available in the GEO database under accession code GSE164871. The ChIP-seq and ATAC-seq data used in this study are available in the GEO database under accession code GSE234065. Source data are provided with this paper.
References
Haggar, F. A. & Boushey, R. P. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 22, 191–197 (2009).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 (2013).
Brugger, M. D. & Basler, K. The diverse nature of intestinal fibroblasts in development, homeostasis, and disease. Trends Cell Biol 33, 834–849 (2023).
Joanito, I. et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat. Genet. 54, 963–975 (2022).
Zhang, L. et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459. e429 (2020).
Ohlund, D. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596 (2017).
Vega, P. N. et al. Cancer-associated fibroblasts and squamous epithelial cells constitute a unique microenvironment in a mouse model of inflammation-induced colon cancer. Front Oncol 12, 878920 (2022).
Sun, G. et al. WNT5a in colorectal cancer: Research progress and challenges. Cancer Manag. Res. 13, 2483–2498 (2021).
Kikuchi, A., Yamamoto, H., Sato, A. & Matsumoto, S. Wnt5a: its signalling, functions and implication in diseases. Acta Physiol. (Oxf) 204, 17–33 (2012).
Kikuchi, A. & Yamamoto, H. Tumor formation due to abnormalities in the β-catenin-independent pathway of Wnt signaling. Cancer Sci 99, 202–208 (2008).
Kurayoshi, M. et al. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res 66, 10439–10448 (2006).
Yamamoto, H. et al. Laminin γ2 mediates Wnt5a-induced invasion of gastric cancer cells. Gastroenterology 137, 242–252 (2009).
Yamamoto, H. et al. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene 29, 2036–2046 (2010).
Weeraratna, A. T. et al. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 1, 279–288 (2002).
Matsumoto, S., Fumoto, K., Okamoto, T., Kaibuchi, K. & Kikuchi, A. Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J 29, 1192–1204 (2010).
Kremenevskaja, N. et al. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 24, 2144–2154 (2005).
Leris, A. C., Roberts, T. R., Jiang, W. G., Newbold, R. F. & Mokbel, K. WNT5A expression in human breast cancer. Anticancer Res 25, 731–734 (2005).
Dejmek, J., Dejmek, A., Safholm, A., Sjolander, A. & Andersson, T. Wnt-5a protein expression in primary dukes B colon cancers identifies a subgroup of patients with good prognosis. Cancer research 65, 9142–9146 (2005).
Blumenthal, A. et al. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 108, 965–973 (2006).
Valencia, J. et al. Wnt5a signaling increases IL-12 secretion by human dendritic cells and enhances IFN-γ production by CD4+ T cells. Immunol. Lett. 162, 188–199 (2014).
Pashirzad, M. et al. Role of Wnt5a in the pathogenesis of inflammatory diseases. J. Cell Physiol. 232, 1611–1616 (2017).
Van der Flier, L. G. et al. The Intestinal Wnt/TCF signature. Gastroenterology 132, 628–632 (2007).
Sato, A. et al. The Wnt5a-Ror2 axis promotes the signaling circuit between interleukin-12 and interferon-γ in colitis. Sci. Rep. 5, 10536 (2015).
Hirashima, T. et al. Wnt5a in cancer-associated fibroblasts promotes colorectal cancer progression. Biochem. Biophys. Res. Commun. 568, 37–42 (2021).
Liu, Q. et al. Wnt5a/CaMKII/ERK/CCL2 axis is required for tumor-associated macrophages to promote colorectal cancer progression. Int. J. Biol. Sci. 16, 1023–1034 (2020).
Kasashima, H. et al. Stromal SOX2 upregulation promotes tumorigenesis through the generation of a SFRP1/2-expressing cancer-associated fibroblast population. Dev. Cell 56, 95–110.e110 (2021).
Pelka, K. et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752.e4720 (2021).
Li, H. et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 49, 708–718 (2017).
Lee, H. O. et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 52, 594–603 (2020).
Uhlitz, F. et al. Mitogen-activated protein kinase activity drives cell trajectories in colorectal cancer. EMBO Mol. Med. 13, e14123 (2021).
Fukuda, K. et al. Alarmins from corneal epithelial cells upregulate CCL11 and VCAM-1 in corneal fibroblasts. Invest. Ophthalmol. Vis. Sci. 54, 5817–5823 (2013).
Kinchen, J. et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell 175, 372–386. e317 (2018).
Mahapatro, M. et al. Programming of intestinal epithelial differentiation by IL-33 derived from pericryptal fibroblasts in response to systemic infection. Cell Rep 15, 1743–1756 (2016).
Hymowitz, S. G. & Wertz, I. E. A20: from ubiquitin editing to tumour suppression. Nat. Rev. Cancer 10, 332–341 (2010).
Smillie, C. S. et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730.e722 (2019).
Gao, Y. et al. Cross-tissue human fibroblast atlas reveals myofibroblast subtypes with distinct roles in immune modulation. Cancer Cell 42, 1764–1783.e1710 (2024).
Lavie, D., Ben-Shmuel, A., Erez, N. & Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nature Cancer 3, 793–807 (2022).
Li, D., Liang, Y., Lu, J. & Tan, Y. An alternative splicing signature in human Crohn’s disease. BMC Gastroenterol 21, 420 (2021).
Wang, F. et al. Single-cell and spatial transcriptome analysis reveals the cellular heterogeneity of liver metastatic colorectal cancer. Sci. Adv. 9, eadf5464 (2023).
Hashimoto, M. et al. Spatial and single-cell colocalisation analysis reveals MDK-mediated immunosuppressive environment with regulatory T cells in colorectal carcinogenesis. EBioMedicine 103, 105102 (2024).
Yasumizu, Y. et al. Single-cell transcriptome landscape of circulating CD4(+) T cell populations in autoimmune diseases. Cell Genom 4, 100473 (2024).
Jasso, G. J. et al. Colon stroma mediates an inflammation-driven fibroblastic response controlling matrix remodeling and healing. PLoS Biol 20, e3001532 (2022).
Dumitrascu, B., Villar, S., Mixon, D. G. & Engelhardt, B. E. Optimal marker gene selection for cell type discrimination in single cell analyses. Nat. Commun. 12, 1186 (2021).
Nishina, T. et al. Interleukin-11-expressing fibroblasts have a unique gene signature correlated with poor prognosis of colorectal cancer. Nat. Commun. 12, 2281 (2021).
Pouyssegur, J., Dayan, F. & Mazure, N. M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443 (2006).
Pham, K., Parikh, K. & Heinrich, E. C. Hypoxia and Inflammation: Insights From High-Altitude Physiology. Front. Physiol. 12, 676782 (2021).
Stefater, J. A. et al. Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature 474, 511–515 (2011).
Stefater, J. A. et al. Macrophage Wnt-Calcineurin-Flt1 signaling regulates mouse wound angiogenesis and repair. Blood 121, 2574–2578 (2013).
Keeble, T. R. et al. The Wnt receptor Ryk is required for Wnt5a-mediated axon guidance on the contralateral side of the corpus callosum. J. Neurosci. 26, 5840–5848 (2006).
Miyashita, T. et al. Wnt-Ryk signaling mediates axon growth inhibition and limits functional recovery after spinal cord injury. J. Neurotrauma 26, 955–964 (2009).
Eubelen, M. et al. A molecular mechanism for Wnt ligand-specific signaling. Science 361, eaat1178 (2018).
Kurayoshi, M., Yamamoto, H., Izumi, S. & Kikuchi, A. Post-translational palmitoylation and glycosylation of Wnt-5a are necessary for its signalling. Biochem. J. 402, 515–523 (2007).
Harada, T. et al. Wnt5b-associated exosomes promote cancer cell migration and proliferation. Cancer Sci 108, 42–52 (2017).
Nusse, R. & Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Abud, H. E., Chan, W. H. & Jarde, T. Source and impact of the EGF family of ligands on intestinal stem cells. Front Cell Dev. Biol. 9, 685665 (2021).
Kikuchi, A., Yamamoto, H., Sato, A. & Matsumoto, S. New insights into the mechanism of Wnt signaling pathway activation. Int. Rev. Cell Mol. Biol. 291, 21–71 (2011).
Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Garcia Garcia, C. J. et al. HIF2 regulates intestinal Wnt5a expression. Front Oncol 11, 769385 (2021).
Lopez-Bergami, P. & Barbero, G. The emerging role of Wnt5a in the promotion of a pro-inflammatory and immunosuppressive tumor microenvironment. Cancer Metastasis Rev 39, 933–952 (2020).
Rapp, J. et al. Increased Wnt5a in squamous cell lung carcinoma inhibits endothelial cell motility. BMC Cancer 16, 915 (2016).
Shi, Y. N. et al. Wnt5a and its signaling pathway in angiogenesis. Clin. Chim. Acta. 471, 263–269 (2017).
Courtwright, A. et al. Secreted frizzle-related protein 2 stimulates angiogenesis via a calcineurin/NFAT signaling pathway. Cancer Res 69, 4621–4628 (2009).
O’Connell, M. P. et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov 3, 1378–1393 (2013).
McDonald, S. L. & Silver, A. The opposing roles of Wnt-5a in cancer. Br. J. Cancer 101, 209–214 (2009).
Davidson, S. et al. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol. 21, 704–717 (2021).
Riese, D. J. 2nd & Cullum, R. L. Epiregulin: roles in normal physiology and cancer. Semin. Cell Dev. Biol. 28, 49–56 (2014).
Yang, W. W. et al. Epiregulin promotes lung metastasis of salivary adenoid cystic carcinoma. Theranostics 7, 3700–3714 (2017).
Lugano, R., Ramachandran, M. & Dimberg, A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cellular and molecular life sciences: CMLS 77, 1745–1770 (2020).
Li, A. Q. & Fang, J. H. Anti‐angiogenic therapy enhances cancer immunotherapy: Mechanism and clinical application. Interdiscip. Med. 2, e20230025 (2024).
Luo, H. et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 13, 6619 (2022).
Lee, S. M. et al. The effect of sex on the azoxymethane/dextran sulfate sodium-treated mice model of colon cancer. J. Cancer Prev. 21, 271–278 (2016).
Tanaka, T. et al. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci 94, 965–973 (2003).
Sebastian, A. et al. Single-cell transcriptomic analysis of tumor-derived fibroblasts and normal tissue-resident fibroblasts reveals fibroblast heterogeneity in breast cancer. Cancers (Basel) 12, 1307 (2020).
Korsunsky, I. et al. Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Med 3, 481–518.e414 (2022).
Qian, J. et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res 30, 745–762 (2020).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Dominguez Conde, C. et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science 376, eabl5197 (2022).
Ho, Y. T. et al. Longitudinal single-cell transcriptomics reveals a role for Serpina3n-mediated resolution of inflammation in a mouse colitis model. Cell Mol Gastroenterol Hepatol 12, 547–566 (2021).
Tarashansky, A. J. et al. Mapping single-cell atlases throughout Metazoa unravels cell type evolution. Elife 10, https://doi.org/10.7554/eLife.66747 (2021).
Wolf, F. A. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol 20, 59 (2019).
Haghverdi, L., Buttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411 (2020).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 (2022).
Hiraoka, Y. et al. ikra: RNAseq pipeline centered on Salmon. (v1.2). Zenodo (2019).
Carpentier, G. et al. Angiogenesis Analyzer for ImageJ - A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay". Sci. Rep. 10, 11568 (2020).
Harada, A. https://BioRender.com/y41o288 (2025).
Acknowledgements
We thank Drs. D. Motooka and D. Okuzaki for analyzing the single-cell RNA-seq and bulk RNA-seq data. This work was funded by Grants-in-Aid for Scientific Research 2021-2022, No. 21K19407; 2023-2024, No. 23K18241 (to AK) and Grants-in-Aid for Scientific Research 2022-2023, No. 22K20791; 2023-2024, No. 23K14566 (to A.H.). This work was also supported by the Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), The University of Osaka to A.H., Y.Y., R.S., S.M., K.T., and A.K., Osaka Cancer Society to A.H., and in part by Grants-in-Aid for Scientific Research 2022-2024 No. 22K06932, and research grants from the Takeda Science Foundation, the Daiichi Sankyo Foundation, and the Mochida Foundation to T.N.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.H., A.K. Methodology: A.H., Y.Y., T.H., K.F., A.S., N.M., T.N. Investigation: A.H., Y.Y., T.H., K.F., A.S., N.M., R.S., S.M., K.T., E.M., H.K., A.K. Supervision: A.H., A.K. Writing—original draft: A.H., A.K. Writing—review & editing: A.H., Y.Y., T.H., K.F., A.S., N.M., R.S., S.M., K.T., H.K., A.K. Project administration: A.H., A.K. Funding acquisition: A.H., A.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fernando Calvo, Heng Xu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Harada, A., Yasumizu, Y., Harada, T. et al. Hypoxia-induced Wnt5a-secreting fibroblasts promote colon cancer progression. Nat Commun 16, 3653 (2025). https://doi.org/10.1038/s41467-025-58748-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58748-9
