
Abstract
Electrocatalytic NO3− reduction (eNO3RR) is a sustainable method for purification of NO3− wastewater and NH3 recovery. Cu-based catalysts are promising for eNO3RR, but insufficient active hydrogen (*H) supply and *NO2 poison of active sites have hindered their performance, and the catalytic mechanism remains ambiguous. Here, we report oxide-derived copper nanosheet arrays (OD-Cu NSs) with residual lattice oxygen and lattice strains to enhance NH3 synthesis from eNO3RR. It is efficient for NH3 synthesis with high Faradaic efficiencies of 88.7-99.7% and maximum NH3 yield of 6.20 mmol·h−1·cm−2 at neutral solution, 10-140 mM NO3− and 50-1500 mA·cm−2. Experimental and theoretical results reveal that lattice oxygen regulates the electronic structure of OD-Cu NSs and promotes *NO2 conversion, while lattice strain enhances *H generation from water dissociation, resulting in the good performance for NH3 synthesis. The applicability of OD-Cu NSs is proved by the high recovery of ammonia compound from eNO3RR.
Similar content being viewed by others


Introduction
NH3 is an important chemical with extensive applications in agriculture, chemical industries, and the energy sectors1. However, NH3 synthesis currently relies on the conventional Haber-Bosch process2. It requires high temperature and pressure, which consumes about 1-2% of global energy and emits large amounts of CO23. Consequently, a green and low-energy method for NH3 synthesis is urgently needed. Electrocatalytic NO3− reduction (eNO3RR) has emerged as a promising alternative for NH3 synthesis owing to its mild operating conditions and low energy consumption (<0.5 kWh·\({{{{\rm{g}}}}}_{{{{{\rm{N}}}}}^{-1}}\))4. Moreover, eNO3RR can tackle NO3− pollution in water, one of the major environmental problems resulted from agricultural and industrial activities. It not only purifies water but also recoveries NH3 from NO3− rich wastewater.
NH3 synthesis from eNO3RR is highly dependent on electrocatalysts. Among the various catalysts reported, Cu-based catalysts are good candidates as the high electron density of Cu d-orbital is favorable for NO3− adsorption and the initiation of eNO3RR5. However, eNO3RR process involves complex steps, including proton-coupled electron transfer, the adsorption and hydrogenation of reactants and intermediates6. The eNO3RR on Cu-based catalysts is hindered by the inherent defects of *NO2 poison (intermediates for eNO3RR) and weak *H supply on Cu7,8. The Cu surface has abundant free electrons. It forms strong adsorption with *NO2 through the electron coupling effect9, poisoning the active sites10. Moreover, the hydrogenation of intermediates is also crucial for eNO3RR. Cu is known to be inefficient for *H generation from water dissociation due to its high energy barrier11. The slow *H supply limits the hydrogenation kinetics for eNO3RR. To enhance NH3 synthesis from eNO3RR, *NO2 conversion or *H supply has been regulated by the strategies such as metal/non-metal atom doping7,12,13, microstructure regulation14, design of atomically dispersed Cu15,16 and Cu/Cu2O heterojunction17,18. Despite the progresses have been made, simultaneous optimization of *NO2 conversion and *H supply to enhance NH3 electrosynthesis from eNO3RR has been rarely reported. Meanwhile, tailoring active sites that are both favorable for enhancing hydrogenation and modulating intermediate adsorption is difficult during catalyst synthesis.
Oxide-derived copper (OD-Cu) is a promising catalyst for electrocatalytic reduction19. Given the strong electron-withdrawing property of oxygen, residual lattice oxygen serves as a negatively charged center in the Cu lattice, which can limit the flow of free electrons, probably weakening *NO2 adsorption. However, previous studies have focused on the crystalline phases and facets of OD-Cu catalysts, overlooking the impact of residual lattice oxygen in eNO3RR. Moreover, OD-Cu has abundant lattice strains due to the oxygen atoms separating out from Cu oxide20. The lattice strains can modulate the activity and product selectivity of electrocatalysts by breaking the linear scaling relationship21. Li et al. observed lattice strains in OD-Cu, but their effect on catalysis was not investigated22. It was found later that lattice tensile strains on Cu could lead to hydrogen evolution23. However, the role of lattice tensile strain in *H supply from water splitting hasn’t been reported. Meanwhile, the catalytic mechanism of OD-Cu remains insufficiently understood. For example, in situ generated Cu/Cu2O interface, pure Cu or Cu2O phase have been reported as active sites for eNO3RR24,25,26,27. The precise active sites are still under debate.
In this work, oxide-derived copper nanosheet arrays (OD-Cu NSs) with residual lattice oxygen and lattice strains were designed to regulate *NO2 adsorption strength and *H supply during eNO3RR. The experimental and density functional theory (DFT) results revealed that residual lattice oxygen modulated the electronic state of the Cu surface, reducing *NO2 adsorption and facilitating its conversion. The lattice tensile strains enhanced *H supply from water dissociation. The resulted OD-Cu NSs achieved high Faradaic efficiencies (FEs) of 88.7–99.7% for NH3 production at neutral solution and 50–1500 mA·cm−2, surpassing the recently reported catalysts. Its potential application has been proved by the efficient recovery of NH3 and NH4Cl from NO3− reduction.
Results
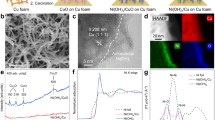
Synthesis and characterization of OD-Cu NSs
OD-Cu NSs were synthesized on copper foam via chemical oxidation and electrochemical reduction28 (Fig. 1a). The CuO nanosheets were grown on copper foam by oxidation in sodium hydroxide and ammonium persulfate solution. OD-Cu NSs were obtained by electrochemical reduction of CuO nanosheets, in which the reduction potential was determined from its cyclic voltammogram (Supplementary Fig. 1). As a comparison, copper nanosheet arrays (Cu NSs) were prepared by calcination of CuO nanosheets under H2. The calcination temperature was selected based on deep removal of lattice oxygen through H2-temperature programmed reduction (H2-TPR) test (Supplementary Fig. 2). Scanning electron microscopy (SEM) shows the ultrathin and smooth nanosheet morphology of OD-Cu NSs (Fig. 1b and Supplementary Fig. 3). However, the nanosheet surfaces of Cu NSs are rough due to the migration and agglomeration of surface atoms during thermal reduction29 (Fig. 1c and S3). High-resolution transmission electron microscopy (HRTEM) reveals both OD-Cu NSs and Cu NSs exhibit face-centered cubic (fcc) structures (Fig. 1d and e). Notably, the lattice spacing of the Cu (111) plane is 0.215 nm for OD-Cu NSs (Fig. 1f), which is larger than those of conventional Cu (0.208 nm) and Cu NSs (0.212 nm). The X-ray diffraction (XRD) patterns show the diffraction peaks of Cu NSs and OD-Cu NSs shift to lower angles in comparison to Cu foam (Fig. 1g), reflecting lattice tensile strains in Cu NSs and OD-Cu NSs as determined by the Bragg equation. The OD-Cu NSs have a lower diffraction angle than Cu NSs, which reveals the stronger tensile strains in OD-Cu NSs. The tensile strain value was calculated by the variation of crystal plane spacing. Compared to Cu foam, Cu NSs and OD-Cu NSs have tensile strains of 1.9% and 3.4%, respectively.

a Schematic illustration of Cu NSs and OD-Cu NSs synthesis. b–e SEM and HRTEM images of OD-Cu NSs (b, d) and Cu NSs (c, e), respectively. f Lattice spacings and of Cu NSs and OD-Cu NSs. g XRD patterns. h, i The strain field maps of OD-Cu NSs (h) and Cu NSs (i) at Exy, Exx, and Eyy based on HRTEM images. Source data for Fig. 1 are provided as a Source Data file.
The spatial distribution of tensile strains in the catalysts was analyzed by geometric phase analysis. OD-Cu NSs exhibit substantial lattice strains across shear strain (Exy), in-plane strain (Exx), and out-of-plane strain (Eyy). Their strain intensities are significantly higher than those in Cu NSs (Fig. 1h and i). This is consistent with the results of XRD and HRTEM. The tensile strains of Cu NSs may originate from the new bond formation, bond breaking, or bond elongation/deflection during the formation of nanosheets30. The more pronounced lattice strains in OD-Cu NSs are likely due to the escape of oxygen atoms that bound to tetrahedral interstitial sites, leading to further lattice expansion.
Time-resolved operando Raman spectroscopy was used to monitor the oxidation state of the catalyst surface during electro-reduction in the catalyst preparation process (Fig. 2a and Supplementary Fig. 4). The color of OD-Cu NSs surface gradually changes from its initial turquoise (CuO) to bright yellow (Cu) after reduction at −0.87 V (vs RHE) in Na2SO4 electrolyte. The Raman spectra at 0 min clearly show three peaks at 297, 343, and 614 cm−1, corresponding to the Ag, Bg1, and Bg2 modes of CuO31. As the reduction proceeds, the intensities of CuO peaks gradually decrease until the CuO phase completely disappears at 10 min. Notably, a new band at around 390 cm−1 is observed as the reduction continues, which corresponds to a multi-phonon process of lattice oxygen in Cu. It indicates the presence of residual lattice oxygen and the possible formation of a new Cu suboxide phase32. Moreover, the O 1 s XPS spectra (Fig. 2b) show that only surface-adsorbed oxygen is present in Cu foam and Cu NSs, whereas OD-Cu NSs exhibit a lattice metal-oxygen bond around 530.6 eV, confirming the presence of lattice oxygen in OD-Cu NSs33. Its lattice oxygen content is estimated to be 10.08 at%. The valence state of Cu can be determined from the positions of 1G and 3F peaks in the Cu Auger spectrum (L3M45M45 transition). The 1G peaks for Cu2+, Cu+, and Cu0 are usually located at 917.1, 915.8, and 918.0 eV, respectively. In addition, the 3F peak is prominent in the spectrum for Cu0 but not in the spectra for Cu2O and CuO34. As shown in Fig. 2c, the 1G peaks for Cu foam and Cu NSs are located at 918.0 eV with a distinct 3F peak, consistent with the characteristics of Cu0. Notably, the 1G peak for OD-Cu NSs shifts to approximately 916.5 eV, lower than those of Cu0 and Cu2+ but close to Cu+. Furthermore, the 3F peak related to Cu0 still appears in OD-Cu NSs. These results suggest that the valence state of OD-Cu NSs is Cuδ+ (0 <δ < 1).

a The operando Raman spectra (left panel) and time-resolved light microscopy images (right panel) of OD-Cu NSs surface at −0.87 V. b O 1 s XPS spectra and (c), Cu L3M45M45 Auger spectra of Cu foam, Cu NSs, and OD-Cu NSs. d The Cu K-edge XANES spectra. e the fitted valence states. f the corresponding k3-weighted FT-EXAFS spectra and (g), Morlet WT of the k3-weighted EXAFS data for Cu foil, Cu2O, CuO, Cu NSs, and OD-Cu NSs. Source data for Fig. 2 are provided as a Source Data file.
X-ray absorption near-edge structure (XANES) spectroscopy was used to investigate the electronic structure and coordination environment of catalysts. As displayed in Fig. 2d, Cu NSs and Cu foil exhibit insignificant differences in the absorption edge and transition intensity, indicating Cu NSs and Cu have similar coordination environments. However, OD-Cu NSs have higher near-edge absorption energy and lower 1 s → 4p transition intensity in comparison to Cu foil and Cu NSs, revealing an abnormal d-p hybridization in the Cu atoms of OD-Cu NSs35. It can be attributed to the reduced electron density of 4p orbitals caused by the strong electron-withdrawing property of residual lattice oxygen atoms36. In addition, the near-edge absorption energy of OD-Cu NSs is lower than that of Cu2O with a fitted valence state of 0.49, suggesting a mixed local environment for its Cu atoms (Fig. 2e). In the k3-weighted Fourier-transformed EXAFS (FT-EXAFS) spectra (Fig. 2f), a distinct Cu-O peak (1.5 Å) appears in OD-Cu NSs, confirming the presence of residual lattice oxygen. The peak intensity of the Cu-Cu bond (2.23 Å)37 in OD-Cu NSs is significantly lower than those of Cu foil and Cu NSs, indicating that the Cu-Cu coordination unsaturation of OD-Cu NSs is increased due to the presence of lattice oxygen atoms. Moreover, the Cu-Cu peaks in OD-Cu NSs and Cu NSs are slightly shifted to a larger radial distance, consistent with the existence of lattice tensile strains23. The Morlet wavelet transform (WT) plots show that both Cu NSs and OD-Cu NSs exhibit the highest peaks at ~7Å−1 (Fig. 2g), corresponding to the Cu-Cu shell. OD-Cu NSs show a distinct Cu-O shell at ~4.5 Å−1, further supporting the presence of residual lattice oxygen in OD-Cu NSs. The EXAFS fitting (Supplementary Fig. 5, 6 and Supplementary Table 1) indicates that the Cu-Cu coordination number (CN) in OD-Cu NSs is 8.7, significantly lower than those in Cu NSs (9.9) and Cu foil (12), which can be explained by that the residual lattice oxygen in OD-Cu NSs reduces the saturation of Cu-Cu coordination. In addition, the average coordination number of Cu-O shell in OD-Cu NSs is 1.0 ± 0.2, revealing the partial saturation of Cu atoms by O atoms in OD-Cu NSs.
The eNO3RR performance of OD-Cu NSs
The eNO3RR performance was evaluated in an H-type cell under ambient conditions. Considering the practical scenarios, a neutral Na2SO4 electrolyte was selected. The linear sweep voltammograms (LSV) of OD-Cu NSs (Supplementary Fig. 7) show a distinct current response for 10 mM NO3− at potential more negative than −0.1 V (vs RHE), indicating its good activity for eNO3RR. NO3− conversion to NH3 was further evaluated at −0.3 V to −0.8 V. The NH3 was quantitatively detected by colorimetric method and validated by 1H-NMR (Supplementary Fig. 8). Notably, the NH3 yields of OD-Cu NSs are higher than those of Cu NSs and Cu foam under all the applied potentials (Fig. 3a). At −0.7 V, OD-Cu NSs produce NH3 at the maximum yield of 0.38 mmol·h−1·cm−2, which is 1.4 times higher than that of Cu NSs, highlighting the significant role of lattice oxygen and tensile strain in enhancing eNO3RR. The \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) of OD-Cu NSs exhibit a volcano-type trend with the variation of potential (Fig. 3b). At −0.3 V, a high concentration of nitrite is accumulated as *NO2 can’t be efficiently converted, resulting in low \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) (Fig. 3c). As expected, its \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) is significantly increased, and nitrite is rapidly converted at more negative potentials. At −0.6 V, the \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) of OD-Cu NSs is 99.7%, significantly higher than those of Cu NSs (74.2%) and Cu foam (44.2%). The FE of NO2− is only 1.1% for OD-Cu NSs, much lower than 25.3% for Cu NSs and 28.7% for Cu foam (Fig. 3d and Supplementary Fig. 9). The NO2− concentration of OD-Cu NSs is also significantly lower than those of Cu NSs and Cu foam (Supplementary Fig. 10). It suggests that OD-Cu NSs can effectively promote nitrite conversion and alleviate *NO2 poison at the active sites. The generation of byproducts such as NH2OH, N2H4, and NOx is negligible for eNO3RR on OD-Cu NSs based on the results of online differential electrochemical mass spectrometry (DEMS) and colorimetric methods (Supplementary Fig. 11–14). Considering that OD-Cu NSs and Cu NSs have similar morphology and crystalline structure, the better performance of OD-Cu NSs can be contributed from its residual lattice oxygen and tensile strains, which increase the active sites and result in higher electrochemical active surface area (Supplementary Fig. 15, 16). The \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) of OD-Cu NSs decreases at more negative potentials due to the occurrence of hydrogen evolution. In addition, OD-Cu NSs are also efficient for NH3 production at lower electrolyte concentrations (0.05-0.1 M) with \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) of 91.2-96.7% (Supplementary Fig. 17). To assess the denitrification performance of OD-Cu NSs, a batch test of NO3− conversion was conducted at −0.6 V. As shown in Fig. 3e, almost all the nitrogen (10 mM NO3−) is converted to NH3 after 75 min, and the concentrations of NO3−-N (0.80 ppm) and NO2−-N (4.16 ppm) are well below the World Health Organization’s (WHO’s) drinking water standards38. The isotope labeling eNO3RR experiment was performed by using K15NO3 as a nitrogen source (Fig. 3f). The quantitative results for 15NH4+ were almost identical to those for 14NH4+ from 14NO3− reduction (Supplementary Fig. 18), which proves that NH3 is generated from NO3− reduction instead of other nitrogen sources.

a NH3 yields and (b), corresponding FEs for 10 mM NO3− reduction on Cu foam, Cu NSs, and OD-Cu NSs. c, d The product distribution for OD-Cu NSs (c) and Cu NSs (d). e Time-dependent concentrations of NO3−, NO2−, and NH3 during the reduction of 10 mM NO3− over OD-Cu NSs. f 1H NMR spectra of samples from OD-Cu NSs catalyzed 15NO3– or 14NO3– reduction. g LSV curves and (h), nitrate removal capacities and NH3 production rates of OD-Cu NSs at 10−140 mM NO3−. i Stability of OD-Cu NSs for 14 successive cycles of 10 mM NO3− reduction at −0.6 V (Na2SO4 electrolyte). Error bars represent the standard deviation from three independent measurements. All the potentials have not been iR-corrected. Source data for Fig. 3 are provided as a Source Data file.
Nitrate concentrations usually vary from ten to more than a hundred millimole per liter in industrial wastewaters39,40. It is crucial for the catalyst to exhibit excellent nitrate-to-ammonia conversion performance across a wide range of NO3− concentrations. Herein NH3 synthesis performance of OD-Cu NSs was further explored under 10–140 mM NO3−. The LSV curves show that the current density remarkably increases with the rise of NO3− concentration (Fig. 3g), indicating the good eNO3RR performance of OD-Cu NSs across a broad range of NO3− concentrations. Its NH3 synthesis from 10–140 mM NO3− was examined at 50 ~ 1500 mA·cm−2, considering that \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) normally declines under high current density41. As shown in Fig. 3h, OD-Cu NSs maintain high \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) across a wide range of NO3− concentrations, exceeding 88.7% even at a current density above 1000 mA·cm−2. Furthermore, both the NH3 yield and denitrification capability are improved with increasing NO3− concentration and current density. At 140 mM NO3− and 1500 mA·cm−2, the \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) is 88.9% with NH3 yield of 6.20 mmol·h−1·cm−2 (105.44 mg·h−1·cm−2) and NO3− removal rate of 91.87 mg·h−1·cm−2 NO3−-N. Notably, OD-Cu NSs outperform the recently reported electrocatalysts with higher NH3 yield while keeping an equal or even higher \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) under similar conditions (10–100 mM NO3−, Supplementary Table. 2). Meanwhile, OD-Cu NSs keep high FE for NH3 production during 14 h of NO3− reduction (10 mM, Fig. 3i and Supplementary Fig. 19), demonstrating the good durability of OD-Cu NSs for NH3 synthesis. After the stability test, OD-Cu NSs retain the initial nanosheet morphology and lattice strains (Supplementary Fig. 20a, b). Furthermore, XPS and Cu L3M45M45 Auger spectra prove the well-maintained lattice oxygen and Cu valence state after 14 cycles of eNO3RR, which reveals its good stability (Supplementary Fig. 20c, d). Meanwhile, in situ Raman spectra confirm the well-maintained lattice oxygen for OD-Cu NSs (Supplementary Fig. 21). The OD-Cu NSs are stable for eNO3RR without structure reconstruction.
The catalytic mechanism of OD-Cu NSs
The reduction of NO3− to NH3 involves a complex 8 electrons and 9 protons transfer process. For Cu-based catalysts, there are significant challenges in the conversion and hydrogenation of intermediates such as *NO2, leading to limited eNO3RR performance42. To investigate the ability of OD-Cu NSs for the conversion of intermediates, the rate constants for NO3− conversion to NO2− (k1, Supplementary Fig. 22, 23) and NO2− conversion to NH3 (k2, Supplementary Fig. 24, 25) were calculated to assess the reaction kinetics7. Meanwhile, the onset potentials for NO3− and NO2− reduction on OD-Cu NSs, Cu NSs, and Cu foam were compared (Supplementary Fig. 26). As can be seen from Fig. 4a, OD-Cu NSs have the highest k1 and k2 values among the three catalysts, which proves that OD-Cu NSs can realize rapid NO3− → NO2− and NO2− → NH3 conversion. Notably, OD-Cu NSs have the lowest onset potential for NO2− reduction, which further demonstrates its excellent NO2− conversion performance. This may be attributed to the presence of residual lattice oxygen, which limits free electrons on the Cu surface and facilitates *NO2 desorption and conversion (Fig. 4b, right panel). To illustrate the influence of lattice oxygen on *NO2 conversion, OD-Cu with lattice oxygen and tensile strain of 1.9% (OD-Cuoxide) was prepared (Supplementary Fig. 27a, b). The OD-Cuoxide has the same tensile strain value as Cu NSs, but it exhibits significantly higher \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) and lower nitrite accumulation than Cu NSs for eNO3RR under the same conditions (Supplementary Fig. 27c, d), demonstrating the crucial role of lattice oxygen in promoting *NO2 conversion and NH3 production. Although Cu NSs have a lower onset potential for NO3− reduction than OD-Cu NSs, its k1 and k2 values are lower than those of OD-Cu NSs. This may be caused by that the metallic property of Cu NSs favors NO3− reduction, but the generated NO2− poisons the active sites and reduces the reaction rate (Fig. 4b, left panel). For Cu foam, both NO3− and NO2− reduction require high overpotentials with the lowest k1 and k2 values. This is consistent with its worst eNO3RR performance.

a The onset potentials and reaction constants for NO3− conversion to NO2− (k1) and NO2− conversion to NH3 (k2) on OD-Cu NSs, Cu NSs, and Cu foam in 0.5 M Na2SO4 + 10 mM NO3−/NO2− (Error bars represent standard deviation from three independent measurements). b Schematic diagram of lattice oxygen-mediated intermediate adsorption on Cu surface. c The operando ATR-FTIR spectra and 2D FTIR contour map. d the ANO2/ANO2 + ANH2OH and (e), in situ Raman spectra of different interfacial water during eNO3RR on OD-Cu NSs at different potentials. f Wavenumber shift of dangling O-H with potential changes. g NH3 yields and KIE values for eNO3RR at −0.6 V using H2O and D2O as solvents. Cyclic voltammograms of OD-Cu NSs in (h), Ar-saturated 0.5 M Na2SO4 under different starting potentials and (i), different NO3− concentrations. j EPR spectra of *H signal for OD-Cu NSs, Cu NSs, and Cu foam with and without the presence of nitrate (−0.6 V). All the potentials have not been iR-corrected. Source data for Fig. 4 are provided as a Source Data file.
The in situ ATR-FTIR spectroscopy was employed to probe the dynamic evolution of intermediates generated from eNO3RR on OD-Cu NSs (Supplementary Fig. 28). As shown in Fig. 4c, the characteristic peak at 1650 cm−1 corresponds to the bending vibration of *H2O in the thin electrolyte layer43. Concurrently, the strong absorption peak at 3390 cm−1 is attributed to the *O-H stretching. It implies that OD-Cu NSs are favorable for the adsorption and decomposition of water to produce *H. The band at 3250 cm−1 can be assigned to *NH2 bending44. The intermediate at ~ 1430 cm−1 is attributed to *NH2OH, a key intermediate for NH3 formation43. Meanwhile, the downward band at 1240 cm−1 belongs to *NO2. As the potential is shifted from −0.3 V to −0.7 V, the peak intensity of *NO2 increases gradually and suddenly flattens out at −0.7 V, indicating that *NO2 undergoes a rapid conversion. It is noteworthy that the ratio of *NO2 peak area to the total area of *NO2 and *NH2OH decreases sharply with the potential negatively shifted (Fig. 4d). Meanwhile, the peak intensity of *NH3 (1100 cm−1) exhibits a progressive enhancement at more negative potentials. However, both Cu NSs and Cu foam exhibit distinct *NO2 signals, and the peak intensity of *NO2 almost keeps unchanged with the negative shift of potential (Supplementary Fig. 29). These results indicate that OD-Cu NSs are more efficient for converting *NO2 to NH3.
The *H supply from water dissociation is a key step for NH3 production in neutral solution45. In situ Raman spectroscopy was used to analyze the water peak at 2800 ~ 4000 cm−1. By Gaussian fitting of the interfacial water peak, tetrahedrally coordinated H2O (tetra-H2O, 3230 cm−1), trihedrally coordinated H2O (tri-H2O, 3425 cm−1) and dangling O-H bond (3600 cm−1) of H2O46 can be found. The dangling O-H is a weak hydrogen bond configuration, which tends to adsorb on the catalyst surface and dissociate to generate *H. Its position is red-shifted as the potential decreases (Fig. 4e), indicating preferential dissociation of dangling O-H on OD-Cu NSs47. A weaker shift of the dangling O-H peak under more negative potential is observed on Cu NSs, and a negligible shift is observed on Cu foam compared to OD-Cu NSs (Supplementary Fig. 30, 31). It is noteworthy that the slopes of dangling O-H peak displacement show a trend of OD-Cu NSs (−110 cm−1·V−1) > Cu NSs (−85 cm−1·V−1) > Cu foam (−3 cm−1·V−1) (Fig. 4f), suggesting that OD-Cu NSs are more conducive to *H generation from water dissociation. OD-Cu NSs and Cu NSs display significantly enhanced ability for water dissociation to *H relative to Cu foam. Both Cu NSs and Cu foam don’t contain lattice oxygen, but Cu NSs have tensile strains compared to Cu foam. It is reasonable to speculate that tensile strains play a key role in enhancing *H supply from water dissociation.
The kinetic isotope effect (KIE) of H/D (H2O/D2O) for eNO3RR was further examined to validate the role of lattice tensile strains in accelerating H2O dissociation. The KIE value is a good indicator of proton transfer kinetics, providing insights into the rate-determining step (RDS). KIE > 1 indicates that proton transfer is involved in the RDS, while KIE ≈ 1 suggests proton transfer is not a limiting factor48. As shown in Fig. 4g, the NH3 yield of Cu foam is significantly decreased after the replacement of H2O by D2O and its KIE value is 2.47, indicating the limited proton supply for eNO3RR. Cu NSs show a smaller change in NH3 yield and current density after the replacement of H2O by D2O (Supplementary Fig. 32) and a KIE value of 1.69, suggesting that the proton supply capacity of Cu NSs is enhanced by the introduction of tensile strains. Interestingly, the NH3 yield and current density (Supplementary Fig. 33) of OD-Cu NSs almost keep unchanged after the replacement of H2O by D2O, and its KIE value is only 1.05. This phenomenon may be due to the fact that OD-Cu NSs have stronger tensile strains, which promotes water dissociation and provide sufficient *H for hydrogenation. Moreover, the linear relationship between tensile strain and KIE value further confirms that tensile strain accelerates *H generation (Supplementary Fig. 34). The *H generation ability of catalysts was investigated by measuring *Hads peak intensity via cyclic voltammogram at different initial potentials49. OD-Cu NSs exhibit stronger *Hads peak intensity and larger intensity enhancement than Cu NSs as the applied potential decreases, whereas negligible *Hads peak appears for Cu foam (Fig. 4h and Supplementary Fig. 35, 36). It further confirms the vital role of tensile strain in promoting *H generation. By increasing the NO3− concentration for cyclic voltammogram test, the *Hads intensity on OD-Cu NSs is reduced obviously (Fig. 4i), indicating that *H plays a crucial role in promoting NO3− reduction. Its good *H generation and utilization ability is also proved by EPR spectra (Fig. 4j). OD-Cu NSs exhibit stronger *H signal intensity than Cu NSs and Cu foam (−0.6 V). Its *H signal almost disappears after the addition of nitrate. In contrast, the *H consumption is slower for Cu NSs and Cu foam. It confirms the accelerated *H supply and utilization of OD-Cu NSs for nitrate reduction.
Density functional theory (DFT) calculation was employed to further elucidate the mechanism of residual lattice oxygen and lattice strain for regulating the eNO3RR activity of OD-Cu NSs. Based on the good matching between FT-EXAFS data of OD-Cu NSs and the simulated EXAFS result of atypical suboxide copper in previous works50, we constructed the OD-Cu model (Cu4O) (Supplementary data 1). To investigate the impact of residual lattice oxygen on eNO3RR, the tensile strain was not introduced into the computational model. The impact of residual lattice oxygen on the electronic state of the Cu surface was investigated (Supplementary Fig. 37). As shown in the electron localization function (ELF) map (Fig. 5a), the surface electrons on OD-Cu that contain residual lattice oxygen are more localized compared to those on Cu surface51. It implies that the valence electrons in OD-Cu are less free to move across the surface, thereby weakening the electronic coupling between the catalyst surface and *NO2. This is conducive to reduce adsorption strength of *NO2 intermediate. The applied potential has insignificant influence on the electronic structures of OD-Cu and Cu under the investigated conditions (Supplementary Fig. 38). The adsorption energy of *NO2 on OD-Cu and Cu surface were further calculated (Supplementary Fig. 39). As depicted in Fig. 5b, the adsorption of *NO2 on OD-Cu surface (−1.28 eV) is weaker than that on Cu surface (−1.62 eV), which is attributed to the strong electron-withdrawing effect of subsurface lattice oxygen on OD-Cu. It diminishes the electron transfer between its surface and the *NO2 (Fig. 5c), thus alleviating the poison of active sites.

a ELF maps and (b) adsorption energies of NO2− on Cu and OD-Cu surfaces. c The adsorption model and charge density difference of Cu and OD-Cu (accumulation and deletion of electrons are shown in blue and yellow). d Gibbs free energy for H2O dissociation to *H on Cu-0%, Cu-1.9% and Cu-3.4% surfaces. e Schematic diagram of eNO3RR promoted by lattice oxygen-mediated electron regulation and lattice strain-mediated water dissociation. f Gibbs free energy diagrams for eNO3RR on the surfaces of Cu and OD-Cu. Source data for Fig. 5 are provided as a Source Data file.
To explore the influence of tensile strain on *H generation from water dissociation, DFT calculation was conducted on the models of Cu (111) with 0%, 1.9%, and 3.4% lattice tensile strains (denoted as Cu-0%, Cu-1.9%, and Cu-3.4%) based on the tensile strains observed in OD-Cu NSs and Cu NSs (Supplementary Fig. 40–43 and Supplementary data 1). The water adsorption energy is −0.37 eV for Cu-0%, which decreases to −0.48 eV for Cu-1.9% and −0.82 eV for Cu-3.4%. Water adsorption is facilitated on the Cu surface with the rise of tensile strains. It may be due to the altered electronic structure by the increased tensile strains, leading to an upward shift of its d-band center (Supplementary Fig. 44). The upshifted d-band center strengthens the interaction between the catalyst surface and water, improving the adsorption of water molecules. The facilitated water adsorption favors subsequent water dissociation to produce *H. As shown in Fig. 5d, Cu-3.4% exhibits a low energy barrier (ΔG*H2O = 0.11 eV) for H2O dissociation to *H, while ΔG*H2O increases to 0.22 eV for Cu-1.9% and 0.32 eV for Cu-0%, indicating that larger tensile strains are more favorable for *H generation. In addition, the protonation of the surface oxygen atoms of OD-Cu is thermodynamically unfavorable in comparison with eNO3RR (Supplementary Fig. 45), which is unlikely to influence *NO2 conversion and water decomposition. Both theoretical and experimental results confirm that the electronic modulation mediated by residual lattice oxygen effectively promotes *NO2 conversion and mitigates active site poison. Concurrently, tensile strains improve the water dissociation capability of Cu, providing sufficient *H for hydrogenation. The synergistic effects of residual lattice oxygen and tensile strains enhance NH3 synthesis on OD-Cu NSs (Fig. 5e).
To explore the reaction pathway for eNO3RR, the reaction intermediates were identified by in situ Raman and DEMS. In the Raman spectra (Supplementary Fig. 46), *NO3 stretching peak is observed at 1050 cm−1 under open circuit potential (OCP). When negative potential is applied, *NO3 peak gradually disappears, while two new Raman bands appear at 1326 cm−1 and 1591 cm−1, corresponding to the antisymmetric stretching of *NO2 and *NH3, respectively. The signal intensity of *NO2 first increases and then disappears with the negative shift of potential. It reveals the generation of *NO2 and its transformation to ammonia during eNO3RR. The online DEMS shows a strong signal at m/z = 17, which is attributed to NH3 (Supplementary Fig. 11). The other signals at m/z = 16, 33, 44, 15, and 30 can be assigned to NH2, NH2OH, N2O, NH and NO, respectively. The signal intensities of these intermediates are 1–4 orders of magnitude lower than that of NH3, which may be related to the rapid conversion of these intermediates. These results are consistent with in situ ATR-FTIR data in Fig. 4c. Based on the intermediates detected, the eNO3RR pathway and the reaction-free energies on OD-Cu and Cu surfaces were explored by DFT calculation. As shown in Fig. 5f, the reaction begins with the adsorption of NO3− onto the catalyst surface, and the formed *NO3 is spontaneously converted to *NO2 via N-O bond breaking. The *NO2 hydrogenation to *NO2H is an uphill process with the free energy of 0.11 eV on the OD-Cu surface, significantly lower than that on the Cu surface (0.60 eV), highlighting the role of residual lattice oxygen in promoting *NO2 conversion and suppressing nitrite accumulation. Subsequently, the *NO2H is converted to *NO, followed by *NO reduction to *NOH. The formed *NOH is further reduced to *NH3 via the pathway of *NOH → *NHOH → *NH2OH → *NH2 → *NH3 or *NOH → *N → *NH → *NH2 (downhill process). NH3 is generated after *NH3 desorption from the catalyst surface. Notably, the rate-determining step (RDS) on OD-Cu is *NO → *NOH. Its energy barrier is 0.37 eV, which is considerably lower than 0.60 eV for the RDS of Cu (*NO2 → *NO2H). Meanwhile, the effect of tensile strains on reducing the energy barrier of RDS is insignificant for OD-Cu (Supplementary Fig. 47). These results demonstrate that the lattice oxygen of OD-Cu significantly reduces the energy barrier for eNO3RR and promotes NH3 production.
Application for ammonia recovery
Given the excellent eNO3RR performance of OD-Cu NSs, the conversion of NO3− into high-purity, ammonia-based chemicals was explored to assess its potential for practical application. NH3 in the electrolyte after eNO3RR (100 mM NO3−, −1.1 A cm−2) was extracted via argon stripping at 50 °C (Fig. 6a). Approximately 95.0% of NH3 vapor is successfully removed from the electrolyte (Fig. 6b), indicating the system is effective for NO3− removal and NH3 recovery. 92.2% of the stripped NH3 vapor is captured through an acidic solution (HCl), and 82.9% of high-purity NH4Cl powder is collected after solvent evaporation (Fig. 6c and Supplementary Fig. 48). This demonstrates the feasibility of OD-Cu NSs-based eNO3RR system for NH3 recovery. For eNO3RR under industrial-scale current density, slow mass transfer and incontiguous NH3 production in static H-cell hinder its large-scale application. Flow cells can enhance mass transfer and provide a stable reaction microenvironment (e.g., local concentration and pH) for continuous NH3 production compared to H-cell. Therefore, we assembled the OD-Cu NSs catalyst into a membrane electrode assembly (MEA) to investigate its performance and long-term stability for NO3− reduction to NH3 under industrial-scale current. The setup of MEA is illustrated in Supplementary Fig. 49, 50. The total current for long-term operation was set at 1 A. As shown in Fig. 6d, OD-Cu NSs still achieve a \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) of 98.6% and NH3 yield of 1.15 mmol·h−1·cm−2 in the MEA system (30 mM NO3−). The \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) and NH3 yield remain ~ 90% of initial values with negligible potential decay after 100 h of operation, demonstrating the good stability of OD-Cu NSs for NH3 synthesis. Techno-economic analysis shows that NH3 electrosynthesis from nitrate wastewater is economically and technically feasible on OD-Cu NSs-based MEA system (Supplementary Fig. 51 and Supplementary Note 1). The effluent from the MEA was subjected to NH3 separation and conversion. The mass flow and product distribution of eNO3RR are shown in Supplementary Fig. 52. After eNO3RR, the nitrate removal rate reaches 98%, and 84% of nitrate is converted to NH4Cl, illustrating its applicability for scaled applications in nitrate wastewater treatment and resource utilization.

a Schematic illustration of the process for converting nitrate to high-purity NH4Cl. b The conversion efficiency for NH3 synthesis from the reduction of 100 mM NO3− at −1.1 A cm−2 (Error bars represent standard deviation from three independent measurements). c XRD pattern of the synthesized NH4Cl product. d Long-term stability for eNO3RR over OD-Cu NSs using a MEA system (30 mM NO3−, 1.0 A, 4.0 cm2, Na2SO4 electrolyte). Source data for Fig. 6 are provided as a Source Data file.
Discussion
OD-Cu NSs with residual lattice oxygen and lattice tensile strains were prepared by a simple method. The OD-Cu NSs demonstrated remarkable eNO3RR activity and stability with nearly 100% \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) at 10 mM NO3− and neutral solution. It was also efficient for NH3 electrosynthesis with \({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) of 88.7–99.7% and maximum NH3 yield of 6.20 mmol·h−1·cm−2 across a wide range of NO3− concentrations (10–140 mM) at 50–1500 mA·cm−2. It converted over 80% of 100 mM NO3− into high-purity NH4Cl. The superior performance of OD-Cu NSs was attributed to electron regulation mediated by residual lattice oxygen and water dissociation mediated by lattice strains, which accelerated *NO2 conversion and *H supply for NH3 synthesis from eNO3RR. This work broadens the mechanistic understanding and will open up new possibilities for enhancing electrocatalytic performance in other fields.
Methods
Chemicals
Cu foam (thickness: 2.0 mm) was purchased from Suzhou Keshenghe Metal Material Co., Ltd. Ammonium persulphate ((NH4)2S2O8, AR 98%) and Sodium hydroxide (NaOH, AR 99.9%) were obtained from Aladdin. All the chemicals were received and used without further purification. Deionized water (18.25 mΩ·cm−1) was used in all experiments.
Synthesis of OD-Cu NSs, Cu NSs and OD-Cuoxide
In a typical procedure, Cu foam was ultrasonically treated with 1 M hydrochloric acid and anhydrous ethanol for 15 min each to remove surface impurities. The Cu foam was then rinsed thoroughly with ultrapure water, dried under vacuum, and stored for later use. To prepare the oxidation solution, 16 g of sodium hydroxide was dissolved in ultrapure water (70 mL). Subsequently, 3.2 g of ammonium persulfate was added to the sodium hydroxide solution and dissolved using ultrasound treatment. The Cu foam was immersed in the prepared solution and oxidized at 60 °C for 20 min. After oxidation, the Cu foam was rinsed with ultrapure water, and CuO nanosheets were obtained. The OD-Cu NSs were prepared by electrochemical reduction of CuO nanosheets for 30 min in 0.5 M Na2SO4 electrolyte at −0.87 V vs RHE (All the potentials have not been iR-corrected in this work). As comparison, Cu NSs were prepared by annealing CuO nanosheets at 350 °C for 2 h under 10% H2/Ar atmosphere. OD-Cuoxide was prepared through direct pyrolysis of copper foam (450 °C for 2 h under Ar), followed by electrochemical reduction under the same conditions as OD-Cu NSs preparation.
Characterization
The morphology and structure of catalysts were examined using a scanning electron microscope (Hitachi S4800) and a transmission electron microscope (FEI Talos F200S). X-ray photoelectron spectroscopy (XPS) data were acquired with an ESCALAB 250Xi spectrometer. All spectra were calibrated by setting the C 1 s peak to 284.8 eV. Raman spectra were recorded on a Renishaw inVia Qontor confocal Raman spectrometer using a 633 nm laser. X-ray diffraction (XRD) patterns were collected on a Rigaku Ultima IV diffractometer. 1H NMR spectra were obtained using a BRUKER AVANCE NEO 600 M spectrometer. Ultraviolet-visible (UV-Vis) absorbance spectra were measured with a SP-756P spectrophotometer. The TPR-H2 experiment was carried out on an Autochem II 2920-MKS CirrusTM 3.
In situ, Raman measurements were performed using a customized Raman reaction cell and a CHI 760E electrochemical workstation. The conditions for the Raman test were the same as those of electrochemical tests, except that the electrolytes (50 mL) for the cathode and anode chambers were circulated outside the cell using a peristaltic pump.
In situ attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy was conducted using a Thermo Scientific iS50 FT-IR instrument equipped with a mercury cadmium telluride (MCT) detector. The experiments employed a custom-made infrared cell connected to a CHI 760E electrochemical workstation. The electrolyte solution consisted of 0.5 M Na2SO4 and 0.1 M KNO3. After subtracting the infrared background, spectra were recorded under applied potentials with Ag/AgCl as the reference electrode and platinum (Pt) as the counter electrode. The applied potential was from 0.2 V to −1.2 V.
The H2-TPR experiment was conducted on an Autochem II instrument. OD-Cu NSs were loaded into a quartz tube and pretreated by sweeping with Ar at 120 °C for 30 min to remove any adsorbed species. After cooling to 25 °C, the temperature was increased to 600 °C at a rate of 10 °C min−1 under a gas mixture of 10% H2/Ar. The H2 consumption during the process was continuously monitored and quantified using a thermal conductivity detector (TCD).
Electrochemical experiments
The eNO3RR was conducted in a customized H-type cell using a CHI1140C electrochemical workstation. The anodic and cathodic compartments were separated by a Nafion 117 membrane (DuPont). The Nafion 117 membrane was soaked in 5 wt% H2O2 solution at 80 °C for 1 h, followed by immersing in deionized water for 30 min. It was then treated with 0.5 M H2SO4 at 80 °C for 1 h and rinsed with deionized water for another 30 min before use. The as-synthesized electrocatalyst served as the working electrode. An Ag/AgCl electrode was used as the reference electrode, and a platinum plate was used as the counter electrode. 0.5 M Na2SO4 solution (pH = 6.8 ± 0.3) was used as the electrolyte for both the cathode and anode chambers, and 10–140 mM NO3− was added to the cathode chamber. The solution in the cathode chamber was stirred during the process of eNO3RR. All potentials were measured against the Ag/AgCl reference electrode and converted to the reversible hydrogen electrode (RHE) scale using the equation: E(vs. RHE) = E(vs. Ag/AgCl) + 0.1976 V + 0.0591 × pH.
Analytical methods
The concentrations of NH4+, NO3−, and NO2− in the electrolyte (diluted to the appropriate concentration) before and after eNO3RR were determined via a colorimetric method.
NH4+ detection: NH4+ was determined by the Nessler’s reagent method. 100 µL NaKC4H4O6 solution (500 g/L) and 100 µL Nessler’s reagent were added into a 5 mL sample and mixed thoroughly. The absorbance was determined at 420 nm after 20 min.
NO2− detection: 100 µl of color developer (0.04 g of C12H14N2·2HCl, 0.8 g of sulfonamide, and 2 mL of 85% H3PO4 dissolved in 10 mL water) was added to 5 mL of diluted sample, and the reaction was carried out for 10 min at room temperature (20–25 °C). The absorbance was measured at 540 nm.
NO3− detection: 1.1 mL of color developer (1 M HCl and 0.8 wt% NH2SO3H) was added to 5 mL of diluted sample. The absorbance was measured at 220 nm and 275 nm after 5 min. The final absorbance value was obtained by the equation of A = A220nm-2A275nm.
N2H4 detection: The color reagent is consisted of 5.99 g of 4-dimethylaminobenzaldehyde (C9H11NO), 30 mL of concentrated HCl, and 300 mL of ethanol. The color reagent was added into sample solution (v:v = 1:1), and the mixture was stirred for 10 min at room temperature (20–25 °C). The absorbance was measured at 457 nm. The calibration curve was obtained from N2H4 solutions (in 0.1 mol L−1 HCl) with known concentrations.
NH2OH detection: 100 μL of acetate buffer (1.0 M sodium acetate + 1.0 M acetic acid), 100 μL, 4 mM ammonium ferric sulfate solution, and 100 μL, 10 mM 1, 10-phenanthroline ethanolic solution were sequentially added into 3 mL of sample, and the mixture was stood for 20 min at room temperature (20–25 °C). The absorbance was measured at 510 nm.
Isotope labeling experiments
The K15NO3 was used as an isotope-labeled N source for eNO3RR. For 1H NMR (BRUKER AVANCE NEO 600 M) quantification, the pH value of the sample (25 mL) after eNO3RR was adjusted to 2 by adding H2SO4. The C4H4O4 (0.01 g) was added as an internal standard. 50 μL of DMSO-d6 was added into 0.5 mL of the above solution before 1H NMR test. The calibration curve was obtained using the peak area ratio of 15NH4+-15N to C4H4O4 with 50, 100, 150, 200 mg L-1 of 15NH4+ solutions.
Ammonia yield and Faradaic efficiency
\({{{{\rm{FE}}}}}_{{{{{\rm{NH}}}}}_{3}}\) was obtained by Eq. (1), and the NH3 yield was determined via Eq. (2), FE of NO2− was calculated by Eq. (3).
Where F is the Faraday constant (96485 C mol−1), CNH3 is NH3 concentration (mol L−1), V is the volume of catholyte (L), Q is the quantity of charge (C), M is the relative molecular mass of NH3, t is the reaction time (s), I is the current (A), CNO2− is the NO2− concentration (mol L−1).
Density functional theory (DFT) calculation
All the calculations were performed with the projector-augmented plane-wave method implemented in the Vienna ab initio simulation package. The generalized gradient approximation proposed by Perdew-Burke-Ernzerhof (PBE) was selected for the exchange-correlation potential. The cut-off energy for the plane wave was set to 480 eV. The energy criterion was set to 10−4 eV in the iterative solution of the Kohn-Sham equation. All the structures were relaxed until the residual forces on the atoms have declined to less than 0.02 eV/Å. To avoid interlaminar interactions, a vacuum spacing of 20 Å is applied perpendicular to the slab. VASPsol code with an implicit solvation model was used to simulate the electrolyte solution to deal with the solvation effect. Here, differences in Gibbs free energy (ΔG) (Eq. 4) for intermediates in eNO3RR defined as:
where ΔG is the total energy difference between the slab and respective terminations computed by DFT-PBE. ΔEZPE and TΔS denote differences in zero-point energy and entropy between adsorbed states of reaction intermediates and gap phase, respectively. T is the room temperature (20–25 °C). ΔGU = −eU, in which U is the electrode potential.
Data availability
The data supporting the conclusions of this study are presented in the paper and the supplementary information. Source data are provided in this paper.
References
Inta, H. R. et al. Recent advances in electrocatalytic NOx reduction into ammonia. EES Catal. 1, 645–664 (2023).
Yin, H. et al. Alloying effect-induced electron polarization drives nitrate electroreduction to ammonia. Chem. Catal. 1, 1088–1103 (2021).
Cheng, X. F. et al. Coordination symmetry breaking of single-atom catalysts for robust and efficient nitrate electroreduction to ammonia. Adv. Mater. 34, 2205767 (2022).
Fan, Y. et al. Highly efficient metal-free nitrate reduction enabled by electrified membrane filtration. Nat. Water 2, 684–696 (2024).
Hu, T. et al. Theoretical insights into superior nitrate reduction to ammonia performance of copper catalysts. ACS Catal. 11, 14417–14427 (2021).
Zhao, R. et al. A Bi-Co corridor construction effectively improving the selectivity of electrocatalytic nitrate reduction toward ammonia by nearly 100%. Adv. Mater. 35, 2306633 (2023).
Wu, L. et al. Boosting electrocatalytic nitrate‐to‐ammonia via tuning of N-intermediate adsorption on a Zn-Cu catalyst. Angew. Chem. Int. Ed. 62, e202307952 (2023).
Yu, J. et al. Boosting electrochemical nitrate-ammonia conversion via organic ligands-tuned proton transfer. Nano Energy 103, 107705 (2022).
Wei, C. et al. Lattice oxygen-mediated electron tuning promotes electrochemical hydrogenation of acetonitrile on copper catalysts. Nat. Commun. 14, 3847 (2023).
Li, P. et al. Pulsed nitrate-to-ammonia electroreduction facilitated by tandem catalysis of nitrite intermediates. J. Am. Chem. Soc. 145, 6471–6479 (2023).
Luo, M. et al. Hydroxide promotes carbon dioxide electroreduction to ethanol on copper via tuning of adsorbed hydrogen. Nat. Commun. 10, 5814 (2019).
Fan, J. L. et al. Electrochemical reduction of nitrate to ammonia on ultra-stable amorphous Co-P electrocatalyst. J. Mater. Chem. A 12, 20077–20087 (2024).
Zhang, W. et al. Fluorine modification promoted water dissociation into atomic hydrogen on a copper electrode for efficient neutral nitrate reduction and ammonia recovery. Environ. Sci. Technol. 58, 7208–7216 (2024).
Wu, Q. et al. Efficient and selective electroreduction of nitrate to ammonia via interfacial engineering of B-doped Cu nanoneedles. Appl. Catal. B Environ. Energy 361, 124597 (2025).
Yin, H. et al. Electrocatalytic reduction of nitrate to ammonia via a Au/Cu single atom alloy catalyst. Environ. Sci. Technol. 57, 3134–3144 (2023).
Xie, M. et al. Periodic adjacent Pd-Fe pair sites for enhanced nitrate electroreduction to ammonia via accelerating proton relay. Adv. Energy Mater. 14, 2401717 (2024).
Fu, W. et al. Tuning mobility of intermediate and electron transfer to enhance electrochemical reduction of nitrate to ammonia on Cu2O/Cu interface. Chem. Eng. J. 433, 133680 (2022).
Zhou, J. et al. Achieving efficient and stable electrochemical nitrate removal by in-situ reconstruction of Cu2O/Cu electroactive nanocatalysts on Cu foam. Appl. Catal. B Environ. Energy 317, 121811 (2022).
Cheng, D. et al. The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts. Nat. Commun. 12, 395 (2021).
Wei, Z. et al. Enhancing selective electrochemical CO2 reduction by in situ constructing tensile-strained Cu catalysts. ACS Catal. 13, 4711–4718 (2023).
Jansonius, R. P. et al. Strain engineering electrocatalysts for selective CO2 reduction. ACS Energy Lett. 4, 980–986 (2019).
Li, C. W. et al. Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper. Nature 508, 504–507 (2014).
Kang, W. J. et al. Strain-activated copper catalyst for pH-universal hydrogen evolution reaction. Adv. Funct. Mater. 32, 2112367 (2022).
Wang, Y. et al. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Xiao, L. et al. Coupling electrocatalytic cathodic nitrate reduction with anodic formaldehyde oxidation at ultra-low potential over Cu2O. Energy Environ. Sci. 16, 2696–2704 (2023).
Li, Y. et al. Enhancement of nitrate-to-ammonia on amorphous CeOx-modified Cu via tuning of active hydrogen supply. Adv. Energy Mater. 14, 2303863 (2024).
Shi, Y. et al. In-situ reconstructed Cu/Cu2O heterogeneous nanorods with oxygen vacancies for enhanced electrocatalytic nitrate reduction to ammonia. Chem. Eng. J. 479, 147574 (2024).
Wei, X. et al. Formic acid electro-synthesis by concurrent cathodic CO2 reduction and anodic CH3OH oxidation. Angew. Chem. Int. Ed. 60, 3148–3155 (2021).
Matte, L. P. et al. Reduction-driven 3D to 2D transformation of Cu nanoparticles. Small 18, 2106583 (2022).
Dou, Y. et al. Graphene-like holey Co3O4 nanosheets as a highly efficient catalyst for oxygen evolution reaction. Nano Energy 30, 267–275 (2016).
Niveditha, C. V. et al. Comprehensive interfacial study of potentio-dynamically synthesized copper oxide thin films for photoelectrochemical applications. J. Electrochem. Soc. 163, H426 (2016).
Singhal, A. et al. Copper(I) oxide nanocrystals - one step synthesis, characterization, formation mechanism, and photocatalytic properties. Eur. J. Inorg. Chem. 2013, 2640–2651 (2013).
Eilert, A. et al. Subsurface oxygen in oxide-derived copper electrocatalysts for carbon dioxide reduction. J. Phys. Chem. Lett. 8, 285–290 (2017).
Tahir, D. et al. Electronic and optical properties of Cu, CuO and Cu2O studied by electron spectroscopy. J. Phys: Condens. Matter 24, 175002 (2012).
Sun, W. et al. An efficiently tuned d-orbital occupation of IrO2 by doping with Cu for enhancing the oxygen evolution reaction activity. Chem. Sci. 6, 4993–4999 (2015).
Kappen, P. et al. The state of Cu promoter atoms in high-temperature shift catalysts—an in situ fluorescence XAFS study. J. Catal. 198, 56–65 (2001).
Huang, K. et al. Boosting nitrate to ammonia via the optimization of key intermediate processes by low-coordinated Cu–Cu sites. Adv. Funct. Mater. 34, 2315324 (2024).
Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Su, L. et al. Electrochemical nitrate reduction by using a novel Co3O4/Ti cathode. Water Res. 120, 1–11 (2017).
Zheng, W. et al. Self-activated Ni cathode for electrocatalytic nitrate reduction to ammonia: From fundamentals to scale-up for treatment of industrial wastewater. Environ. Sci. Technol. 55, 13231–13243 (2021).
Chauhan, R. et al. Electrochemical denitrification of highly contaminated actual nitrate wastewater by Ti/RuO2 anode and iron cathode. Chem. Eng. J. 386, 122065 (2020).
Fang, J. Y. et al. Copper-based electrocatalysts for nitrate reduction to ammonia. Materials 16 (2023).
Zhang, R. et al. Electrochemical nitrate reduction in acid enables high-efficiency ammonia synthesis and high-voltage pollutes-based fuel cells. Nat. Commun. 14, 8036 (2023).
Xu, Y. et al. Sulphur-boosted active hydrogen on copper for enhanced electrocatalytic nitrate-to-ammonia selectivity. Angew. Chem. Int. Ed. 63, e202400289 (2024).
Fan, K. et al. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 13, 7958 (2022).
Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Li, C. Y. et al. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 18, 697–701 (2019).
Sawata, S. et al. Kinetic evidence based on solvent isotope effects for the nonexistence of a proton-transfer process in reactions catalyzed by a hammerhead ribozyme: Implication to the double-metal-ion mechanism of catalysis. J. Am. Chem. Soc. 117, 2357–2358 (1995).
Wang, K. et al. Intentional corrosion-induced reconstruction of defective NiFe layered double hydroxide boosts electrocatalytic nitrate reduction to ammonia. Nat. Water 1, 1068–1078 (2023).
Zhang, W. et al. Atypical oxygen-bearing copper boosts ethylene selectivity toward electrocatalytic CO2 reduction. J. Am. Chem. Soc. 142, 11417–11427 (2020).
Zhang, Y. et al. Low-coordinated copper facilitates the *CH2CO affinity at enhanced rectifying interface of Cu/Cu2O for efficient CO2-to-multicarbon alcohols conversion. Nat. Commun. 15, 5172 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22222601 and No. 22076019, Y.L.), and the Fundamental Research Funds for the Central Universities (DUT23LAB611, Y.L.).
Author information
Authors and Affiliations
Contributions
Q.W. and Y.L. conceived the idea. Q.W., X.F., and Y.L. designed the experiments. Q.W. performed the experiments. L.Q. helped with material characterization. Q.W., X.F., B.S., and X.Q. analyzed the data and discussed the results. Q.W. and Y.L. co-wrote the manuscript. All authors commented on the manuscript. Y.L. supervised the whole project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chun-Chih Chang, Zhihui Ai, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Q., Fan, X., Shan, B. et al. Insights into lattice oxygen and strains of oxide-derived copper for ammonia electrosynthesis from nitrate. Nat Commun 16, 3479 (2025). https://doi.org/10.1038/s41467-025-58811-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58811-5
This article is cited by
-
Spatiotemporally ordered topological transformation in layered double hydroxides enables synergistic mineralization of AsIII/Cd2+
Nature Communications (2026)
-
Highly dispersed Cu/WO3 heterojunctions featured by promoting hydrogen radical-mediated pathway for efficient nitrate reduction to ammonia
Science China Materials (2025)
