
Abstract
Metallabenzenes are recognized as a unique class of aromatic compounds, not only of structural and theoretical interest but also as platforms to design powerful transformations. Here, we report the successful transmutation of a metallabenzene for pyridine synthesis. This ‘metal-to-nitrogen swapping’ process utilizes readily available ruthenabenzene phosphonium salts and commercially available 2-aminopyridines under mild conditions. The isolation of ruthena-azepines, containing a planar seven-membered aza-metallacycle, along with DFT calculations, supports the nitrogen insertion/metal deletion cascade driven by aromatization. Additionally, we investigate the tunable photophysical properties of the resulting pyridine phosphonium salts.
Similar content being viewed by others
Introduction
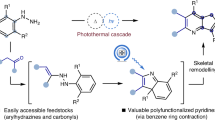
The emergence of single-atom skeletal editing has significantly spurred efficient and accurate synthetic approaches to ring system, which enables straightforward diversification of core ring structures of complex molecules (Fig. 1A)1,2,3,4,5,6,7,8. For example, replacing a carbon atom in benzene rings with a nitrogen, starting from aryl azides, has been developed to yield the corresponding pyridine derivatives9,10,11. As shown in Fig. 1B, 2-amino-azepines species, generated by the initial nitrogen insertion of the sequence, can undergo the formal “para”9, or “meta”-carbon deletion10 to produce aminopyridines and even “ipso”-carbon deletion11 to realize site-directable carbon-to-nitrogen transmutations. Given the multifaceted roles of pyridine core in biological and materials sciences12,13, aromatic nitrogen scanning offers a compelling strategy for directly modifying late-stage ring systems to access variously substituted pyridines. Generally, the functionalization of N-heterocyclic aromatics is relatively challenging, owing to the electron-deficient nature and the coordinative power of the nitrogen14. Traditional functionalization strategies of pyridines, such as direct C–H functionalization14,15, have mainly provided access to 2- or 4- substituted pyridines due to their electronically biased reactivity. Therefore, it’s not surprising that functionalization of pyridines at 3- or 5-possition is far less explored, although several elegant studies add to the success story very recently16,17. In recent late-stage functionalization (LSF) of pyridine area, formation of pyridine phosphonium salts and subsequent transformations are considered to be one of the most powerful methods to obtain a diverse array of valuable azine derivatives 18,19,20.

A Classification of single-atom skeletal editing. B Examples of nitrogen scans for the conversion of benzenes to pyridines. C Outline of metal-to-nitrogen transmutation allowing access to 3,5-bis(triphenylphosphonium) pyridines with tunable fluorescence.

A Synthesis of metalla-azepines 3a-3j by reactions of metallabenzene 1 with different 2-aminopyridines 2. B X-ray structures for the cations of compounds 3a, 3c, 3e, 3 f and 3i (thermal ellipsoids drawn with 50% probability level, the phenyl groups in PPh3 groups are shown as wireframe model for clarity) and the selected bond lengths (Å).
Metallabenzenes21,22,23,24,25, organometallic compounds consisting of one metal and five carbons in a six-membered aromatic ring, are an important chemical stepping stone for exploring the area of aromatic chemistry. The synthesis, structural characterization and theoretical studies in conjunction their aromaticity have been extensively documented26,27. Notably, recent examples of their use can be found in stoichiometric or catalytic processes, where metallabenzenes have served as transient intermediates28,29 or even as catalysts30 to generate polysubstituted carbocycles. Our group has developed a family of metallaaromatics with interesting aromatic and organometallic properties31,32,33,34,35,36,37,38. In terms of their structural characteristics, we envisage that metallabenzene can be utilized as starting heterocyclic aromatics of single-atom skeletal editing. In particular, if 3,5-disubstituted metallabenzenes can be exploited, this approach may enable heteroatom scanning to construct the 3,5-disubstituted pyridine motif. Herein, we describe this metal-to-nitrogen transmutation strategy to afford the corresponding 3,5-disubstituted pyridine phosphonium salts (Fig. 1C). During the course of this reaction, we isolate the stable metalla-azepine intermediate, leading to a comprehensive mechanistic investigation. The photophysical properties of pyridine phosphonium salts, tuned by the electronic perturbation with several substituents, are also investigated.
Results and Discussion
Synthesis and characterization of metalla-azapines (3) by nitrogen insertion into metallabenzene (1)
Our design originated from the well-documented strategy that aryl azides could undergo a nitrene-internalization reaction to generate the seven-membered azepines with the aid of the introducing amine nucleophile (Fig. 1B)9,10,11. Although the archetypal metalla-cycloheptatrienes or aza-metalla-cycloheptatrienes have been identified as key intermediates in the transition-metal-catalyzed [2 + 2 + 2] cycloaddition process39,40,41,42,43 the related metalla-azepines are scarcely isolated in literature. We initially hypothesized that the aromatic nucleophilic substitution reaction (SNAr) of metallabenzene with amine nucleophile, followed by the nitrogen insertion, might be used to produce the desired metalla-azepines. Initial attempts to access metalla-azepines via reactions of metallabenzene 1 with alkylamine, such as diethylamine and triethylamine, or aniline proved unsuccessful, leading to decomposition of the metallacyclic framework and yielding complicated metal complexes along with a mixture of organic products. We anticipated that the use of pyridylamine instead could facilitate the intramolecular nucleophilic attack of amino group by coordination of pyridyl to the metal center, thus avoiding the decomposition of metallabenzenes.
We commenced our studies by using ruthenabenzene 1 and 2-aminopyridine 2a as the model substrates to establish the optimal reaction conditions. To our delight, the reaction of 1 with 2a in the presence of iodine and triethylamine led to the exclusive formation of the desired metalla-azapine 3a, which could be isolated as reddish-brown solid in 87% yield. As shown in Fig. 2A, under the above conditions, ruthenabenzene 1 readily reacted with a number of commercially available 2-aminopyridines to provide the corresponding substituted metalla-azapines in 73–87% yields. Electron-rich aminopyridines, such as alkylpyridines and alkoxypyridines gave satisfactory results (3b to 3 h), although the yield slightly dropped when the pyridine was substituted at the 3-position (3 d and 3e). The reaction also tolerates a phenyl group on the C4 carbon (3i), even with electron-withdrawing group, i.e., ester group (3j). In all cases, the yields are influenced by both electronic and steric factors. This discrepancy is likely due to the lower nucleophilicity of the amino group ascribed to the decreasing electron density and increasing steric hindrance of the substituted pyridines. 6-Substituted 2-aminopyridines or those bearing strong electron-withdrawing groups such as nitro, trifluoromethyl and cyano do not yield appreciable amounts of products, supporting this hypothesis.
The crystal structures of 3 were determined by X-ray crystallography. The OLEX drawings of 3a, 3c, 3e, 3 f and 3i are depicted in Fig. 2B including partial numbering scheme with 50% thermal ellipsoids and selected bond lengths provided in the figure caption. It is clear that the complex has a seven-membered metalla-azepine core with two substituted phosphonium groups. Each metal center was bound in a distorted octahedral coordination geometry, including two pyridyls, a PPh3 ligand and an iodine ligand. The planar geometric structures of metallacycles 3 were completely different from those of the nonplanar seven-membered aza-ruthenacycles described in the literature44,45,46. However, obviously different planarities of them deserve attention. The ruthena-azepine ring in 3a has a very small deviation from the least-squares plane of best fit (the maximum deviation is 0.035 Å shown by C4). In contrast, the decreased planarity was observed in other analogues derived from substituted 2-aminopyridines (mean deviation from the least-squares plane: 0.090 Å for 3c, 0.067 Å for 3 f, and 0.091 Å for 3i). This observation implies that the steric hinderance of the substituents in pyridyls has significant influence on the planarity of metallacycles. Additionally, the excellent planarity of 3e—the mean deviation from the least-squares plane is only 0.028 Å, is likely attributed to the intramolecular hydrogen bonding interaction between the hydrogen of α-carbon and the alkoxy oxygen. The tethered four-membered metallacycle causes elongation of the bond lengths, but the Ru1 − N3 bond length of metalla-azepine core remains within the range of single bonds (2.048 to 2.068 Å). The distances of the Ru−C bond and the C − C bonds within the seven-membered metallacycle are in a similar range to those of the ruthena-aromatics30,31,34. The bond lengths among these rings are notably averaged, indicating considerable electron delocalization. To the best of our knowledge, there is only one example of planar seven-membered conjugated aza-metallacycles in literature 47.
Aromaticity evaluation of metalla-azapines (3)
Ruthena-azepines 3a–3j display H5 and H3 signals of metallacycles in the range of 7.9–8.3 and 6.6–6.8 ppm, respectively. Compared to aromatic ruthenabenzene 131, these signals are substantially upfield shifted, indicating nonaromatic or weak aromatic character (vide infra). To gain further insight into the aromatic character of the ruthena-azepines 3, we performed nucleus-independent chemical shift (NICS)48,49 and anisotropy of the current (induced) density (ACID)50,51 analyses calculations at the B3LYP/def2-TZVP//B3LYP/6-31 G(d) (SDD for Ru, I, P, Cl) using model systems in which the phosphonium substituents of metallacycles were replaced with hydrogen atoms (see Fig. S4 in the Supplementary Information). Negative values of the out-of-plane NICS value (NICS(1)zz) and clockwise ring currents visualized by ACID plots have been demonstrated as the effective computational descriptors for diagnosing the aromaticity of metallacycles52,53,54. The parent ruthenabenzene 1 has a highly aromatic metallacycle core with a large negative NICS(1)zz value of −13.39 ppm, whereas the metallacycle ring of ruthena azepines 3a displays weak aromaticity with weakly negative NICS(1)zz value (−2.54 ppm). In addition, the ACID plot for ruthena-azepines 3a show random density vectors, making the ring current direction unrecognizable (See Fig. S4 in the Supplementary Information). The ACID plots are in agreement with the small negative NICS(1)zz values noted above, thus suggesting the nonaromatic (or weak aromatic) character of ruthena-azepines 3a. A ring-current analysis using the gauge-including magnetically induced current (GIMIC) method55,56,57,58,59 has also been performed (See Fig. S5 in the Supplementary Information), a method particularly effective for current patterns in complicated transition metal-containing π-conjugated structures60,61. A diatropic (paratropic) ring current strength is assigned to a positive (negative) value. Compared with distinctly aromatic ruthenabenzene 1, of which a net diatropic ring current of 7.09 nA/T is found, the diatropicity of 3a is obviously attenuated (2.31 nA/T).
Mechanism exploration experiments for the formation of metalla-azapines (3)
Generally, the seven-membered aza-metallacyclic intermediate is believed to be formed through the coordination and insertion of CN/CC multiple bonds into either a metallacyclopentadiene-like intermediate or an azametallacyclopentadiene-like intermediate in the metal-catalyzed [2 + 2 + 2] reactions39,40,41,42,43. However, the formation of ruthena-azepines 3 presumably proceeds through a ring expansion reaction via nitrogen insertion. To gain further insights into the reaction mechanism, we carried out control experiments. As shown in Fig. 3, green solutions of ruthenabenzene 1 were found to rapidly turn brownish to afford a triply iodide bridged dimer (4) in nearly quantitative yields in presence of excess I2. The isolation and characterization of 4 indicate that the reaction initially undergoes oxidation of metal center (see Fig. S2 in the Supplementary Information). Reaction of 4 with excess 2-aminopyridine afforded the complex 5a at room temperature as the sole product, which has been fully characterized, including X-ray diffraction analysis (see Fig. S3 in the Supplementary Information). Notably, complexes 5a proved to be competent in a nitrogen insertion with the aid of triethylamine, which delivered the desired ruthena-azepine product 3a. In the absence of triethylamine, 3a could not be detected, demonstrating the importance of base in enabling the reaction.

Control experiments and proposed mechanism for the formation of 3a.
Based on the mechanistic studies, a plausible mechanism for the formation of ruthena-azepines 3 is proposed as depicted in Fig. 3. The reaction begins by the oxidation of ruthenabenzene 1 by I2 yielding dimer 4, which opens some space of the metal center simultaneously. Subsequently, pyridyl coordination and nucleophilic addition of amino group produce Int A, which then undergoes elimination to form Int B along with the generation of H2. The released H2 gas is detected by residual gas analysis (see Fig. S3 in the Supplementary Information). The coordination and nucleophilic substitution reaction of the second 2-aminopyridine produces 5a. The dehydrogenation by triethylamine leads to the formation of nitrogen anion in Int C, facilitating the nitrogen insertion into the metallabenzene ring to form the corresponding metalla-benzazirine Int D. Finally, intramolecular rearrangement could generate metalla-azepine 3a. We hypothesize that the second nitrogen insertion cannot occur in parallel due to the destabilizing effect associated with the antiaromaticity of the corresponding eight-membered ruthenacycle. The DFT calculations support our prediction by demonstrating that the eight-membered ruthenacycle is anti-aromatic. This is indicated by positive NICS values and the presence of a counterclockwise current density flow in the ACID isosurface plot (see Fig. S4 in the Supplementary Information). A weak yet negative value of paratropic ring current indeed existed in the eight-membered ruthenacycle, as quantified via current-density studies using the GIMIC method (See Fig. S5 in the Supplementary Information). Therefore, the resulting antiaromaticity of the eight-membered ring would override the nucleophilicity of the nitrogen anion with the remaining of nonaromatic character of the seven-membered structure.
Synthesis and characterization of pyridine phosphonium salts (7) by metal deletion of metalla-azapines (3) with AgOAc
The structural motif of ruthena-azepines 3 opens the possibility for subsequent metal deletion and thus the synthesis of pyridine phosphonium salts. To accomplish this, we used AgOAc to facilitate the desired reductive elimination to release the C − N coupled product. Indeed, when complexes 3 were treated with AgOAc in 1,2–dichloroethane (DCE) solvent at 60 °C followed by anion metathesis with excess NaBF4, reductive elimination occurred smoothly and provided the organic product 6 (Fig. 4A). Compounds 6 are soluble in common solvents and were unambiguously verified by spectroscopic and single-crystal X-ray analyses (see Fig. S7 in the Supplementary Information). As shown in Fig. 4B, the solid-state molecular structure of 6b features a six-membered ring in which two phosphonium substituents located at C2- and C4-possition. Notably, the ring carbon atom C5 have an oxygen atom attached, and the C5 − O1 bond distance (1.231(2) Å) is typical of carbonyl double bonds. An isotope labeling experiment was conducted using H218O to verify the oxygen of C5 was originated from trace water in the reaction system; and the isotope incorporation was confirmed by HRMS of 18O-6a (see Figs. S8 and S9 in the Supplementary Information). Thus, the carbonyl of ring is attributed to the over oxidation in the presence of silver(I) oxidant. Based on the isotope labeling experiment results, we think the oxidative dehydrogenation in the presence of AgOAc, is the driving force for this demetallization process (details see Fig. S6 in the Supplementary Information).

A Synthesis of compounds 7a-7m in two steps with isolation of the intermediate 6a-6j (dark blue) or one pot without the isolation of 6a-6j (black). B Selected X-ray structures for the cations of compounds 6 and 7 (thermal ellipsoils drawn with 50% probability level, the phenyl groups in PPh3 groups are shown as wireframe model for clarity) and the selected bond lengths (Å). C Isolation of intermediate 8a and 9a, and the conversion to 7a by stepwise heating up.
Compound 6 is only one electrophilic addition step away from the anticipated pyridine phosphonium derivative. Treatment with trimethyloxonium tetrafluoroborate (Me3OBF4) in DCE followed by workup gave compound 7 in moderate yield (see Fig. 4A). Alternatively, compound 7 can be prepared in one pot from ruthena-azepines 3 with sequential addition of AgOAc and Me3OBF4. The X-ray crystal structure analysis of compound 7 revealed the presence of a central planar six-membered heterocyclic system (Fig. 4B). A pyridyl group had migrated from ring nitrogen atom N1 to the substituted amino-pyridinyl nitrogen N2. The ring nitrogen-carbon bond lengths are quite different from those of the precursors 6. Compounds 7 all feature a delocalized six-electron π-system within the central pyridine moiety. The C − N bond length of 7 (1.309(4) − 1.354(4) Å) matches well with typical pyridine system. In line with the C − N bond length descending trend, the C − C bond length equalization increase from 6 to 7, which suggests increasing π-conjugation of the phosphonium substituted pyridine core from 6 to 7. It is noteworthy that the three N-heterocycles, i.e., methylated-pyridinium, phosphonium-substituted-pyridine skeleton, and linked N-heterocycle are conformationally twisted. The calculation (B3LYP/def2-TZVP//B3LYP/6-31 G(d) (SDD for P) gave negative NICS(1)zz values of −14.50 ppm(7a), consistent with its aromaticity. Alternative electrophile beyond Me3OBF4 was also investigated. When Et3OBF4 was added as electrophile, metalla-azepine 3a, and alkoxy or phenyl-containing substrates (3 g and 3i) displayed excellent efficiency, offering corresponding products in moderate yields (54–62%).
The successful isolation of 8a and 9a provides direct evidence that compounds 7 are formed through a intramolecular substitution reaction of methylated-pyridinium ring (see Fig. S11 in the Supplementary Information). As shown in Fig. 4C, compound 6a initially undergoes an electrophilic addition reaction with Me3OBF4 to give a methylation product 8a. Compound 8a could evolve to the pyridine derivative 9a via the intramolecular substitution of the pyridyl to the methylated pyridinium ring (Fig. S11). Further reaction of 9a with another molecule of Me3OBF4 would give the expected aromatization product 7a. Our computations reveal that the formation of 7 from 8 is exoergic by −48.5 kcal/mol (details see Fig. S11 and S12 in the Supplementary Information).
The tunable photophysical properties of the pyridine phosphonium salts (7)
Polycyclic aromatic compounds with twisted conjugation skeletons have been designed to alter the electronic density and regulate the extent of π-conjugation, leading to unique photophysical properties62,63. Consequently, the UV–vis absorption spectra and the fluorescence emission spectra of pyridine phosphonium salts 7 were studied (Fig. 5 and Fig. S13). For further comparisons, data for the selected examples of non-aromatic N-heterocycles 6 are also given in Table 1. The UV and fluorescence properties of 6a, 6 f, 6 g, 6i are almost identical, while the characteristic absorption bands of 7 were observed from 380 nm to 420 nm in DCM (see Fig. S13 in the Supplementary Information). The absorption maxima of selected compounds are noted in Table 1. In comparison to 6, 7 exhibited distinct fluorescent emissions, with the fluorescent intensities and wavelengths of twisted π-conjugate systems varying widely depending on the substituents on the pyridinium core. The compounds with electron-donating group (−OMe: 7 g, −OEt: 7 h) exhibited enhanced fluorescent emissions with comparison to 7a. The fluorescence of 7j, with electron-withdrawing carboxylate group, is greatly quenched. The tunable fluorescence behavior suggests that the three linked N-heterocycle core could serve as a fluorophore for further application in biosensing and bioimaging.

A Ambient and fluorescence images of selected N-heterocycles 6 (6a, 6 f, 6j, and 6i (upper) in DCM under natural light; (bottom) in DCM irradiated at 365 nm. B Ambient and fluorescence images of Pyridine Phosphonium Salts (7a, 7b, 7 d, 7 f, 7 g, 7 h, 7i; (upper) in DCM under natural light; (bottom) in DCM irradiated at 365 nm. C Normalized emission spectra of selected N-heterocycles 6 (6a, 6 f, 6j, and 6i). D Normalized emission spectra of 7a, 7b, 7 d, 7 f, 7 g, 7 h, 7i.
In summary, we developed an effective method for preparing a series of 3,5-disubstituted pyridine phosphonium salts and have investigated their structural characteristics and photophysical properties. Furthermore, we notably show an unusual nitrogen insertion to metallacycles resulting in very rare examples of planar metalla-azepine species. DFT calculations and the isolation of the key intermediates demonstrate that the aromatization is the driving force for the skeletal editing process. The transformation of aromatic metallabenzene phosphonium salts into the aromatic pyridine phosphonium salts achieves a metal to nitrogen swapping for the first time and lead to the peripherical editing of pyridine rings, resulting in tunable photophysical properties. Given the structural diversity and abundant properties of metalla-aromatics, we anticipate that our current results would open attractive opportunities for skeletal and peripherical dual editing and provide fresh eyes on tunable photophysical properties of twisted linked N-heterocyclic skeletons.
Methods
General information
Details of the synthesis and characterization of metallacycles (3a-3j, 4a, 5a) and N-heterocyclics compounds (6b, 6 d, 6j, 7a-7m, 8a and 9a) can be found in the Supplementary Information, pp. 5-41. For X-ray data of all the described compounds (3a, 3c, 3e, 3 f, 3i, 5a’, 6b, 6 d, 6j, 7a, 7 f, 7 g, 7i, 7j, 8a, 9a and [Ru]) see supplementary, pp. 104-128. For HRMS, 1H, 31P{1H} NMR, and 13C{1H} NMR spectra of compounds in this article, see Supplementary Information, pp 44-103 and supplementary Figs. 14–132.
General preparation procedure of compounds 3
A mixture of complex 1 (659 mg, 0.5 mmol), 2 (2.5 mmol), iodine (508 mg, 2.0 mmol), and Et3N (253 mg, 2.5 mmol) in dichloromethane (20 mL) was stirred at room temperature for 3 h to give a reddish brown solution. The volume of the mixture was reduced to ca. 2 mL under vacuum. The solution was purified by column chromatography on silica gel with dichloromethane/methanol = 50:1 as eluent to give the desired as solid.
General preparation procedure of compounds 6
To a Schlenk tube were added 3 (0.3 mmol), silver acetate (501 mg, 3 mmol) and 20 mL of 1,2-dichloroethane in nitrogen atmosphere. The reaction mixture was stirred for 12 h at 60 °C under nitrogen atmosphere. And then sodium tetrafluoroborate (329 mg, 3 mmol) was added to the reaction mixture for the counteranion exchange. The reaction system was collected by filtration and the volume of the filtrate was reduced to ca. 5 mL under vacuum. The filtrate was purified by column chromatography on silica gel with dichloromethane/methanol = 40:1 as eluent to give the desired as solid.
General preparation procedure of compounds 7
Procedure A (step by step): According to the corresponding preparation procedure of complexes 6, we first synthesized and isolated the complexes 6. And then we carried out next procedure to obtain complexes 7. To a Schlenk tube were added 6 (0.1 mmol), trimethyloxonium tetrafluoroborate (74 mg, 0.5 mmol) and 5 mL of 1,2–dichloroethane in nitrogen atmosphere. The reaction mixture was stirred for 12 h at 85 °C under nitrogen atmosphere. The reaction system was reduced to ca. 2 mL under vacuum, and then purified by column chromatography on silica gel with dichloromethane/ methanol = 20:1 as eluent to give the desired as solid. Procedure B (one pot): To a Schlenk tube were added 3 (0.1 mmol), silver acetate (167 mg, 1 mmol), sodium tetrafluoroborate (110 mg, 1 mmol) and 10 mL of 1,2-dichloroethane in nitrogen atmosphere. The reaction mixture was stirred for 12 h at 60 °C under nitrogen atmosphere. And then the reaction mixture was allowed to return to room temperature, without further purification. To the reaction mixture, trimethyloxonium tetrafluoroborate (148 mg, 1 mmol) was injected. The reaction mixture was stirred for 12 h at 85 °C under nitrogen atmosphere. The reaction system was collected by filtration and the volume of the filtrate was reduced to ca. 2 mL under vacuum and then purified by column chromatography on silica gel with dichloromethane/ methanol = 20:1 as eluent to give the desired as solid.
Data availability
All data relating to the full experimental procedures, spectral data for new complexes, crystallographic details, computational details, and Cartesian coordinates are provided in the Supplementary Information/ Supplementary Data file. The X-ray crystallographic coordinates data generated in this study have been deposited have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2370478 (3a), 2370486 (3c), 2370474 (3e), 2370485 (3 f), 2370483 (3i), 2370470 (5a’), 2370475 (6b), 2370479 (6 d), 2370473 (6j), 2370482 (7a), 2370476 (7 f), 2370477 (7 g), 2370481 (7i), 2370484 (7j), 2370472 (8a), 2370471 (9a) and 2370480 ([Ru]). The X-ray crystallographic data are available free of charge from The Cambridge Crystallographic Data Center via https://www.ccdc.cam.ac.uk/structures/. All data are available from the corresponding author upon request. Source data are provided in this paper.
References
Peplow, M. Almost magical’: chemists can now move single atoms in and out of a molecule’s core. Nature 618, 21–24 (2023).
Jurczyk, J. et al. Single-atom logic for heterocycle editing. Nat. Synth. 1, 352–364 (2022).
Liu, Z., Sivaguru, P., Ning, Y., Wu, Y. & Bi, X. Skeletal editing of (Hetero) arenes using carbenes. Chem. Eur. J. 29, e202301227 (2023).
Woo, J., Stein, C., Christian, A. H. & Levin, M. D. Carbon-to-nitrogen single-atom transmutation of azaarenes. Nature 623, 77–82 (2023).
Reisenbauer, J. C., Green, O., Franchino, A., Finkelstein, P. & Morandi, B. Diversification of indole skeletons through nitrogen atom insertion. Science 377, 1104–1109 (2022).
Liu, S. et al. Tunable molecular editing of indoles with fluoroalkyl carbenes. Nat. Chem. 16, 988–997 (2024).
Wu, F.-P. et al. Ring expansion of Indene by Photoredox-enabled functionalized carbon-atom insertion. Nat. Catal. 7, 242–251 (2024).
Lu, H. et al. Carbon–nitrogen transmutation in polycyclic arenol skeletons to access N-heteroarenes. Nat Commun 15, 3772 (2024).
Sundberg, R. J., Suter, S. R. & Brenner, M. Photolysis of ortho-substituted Aryl Azides in Diethylamine. Formation and Autoxidation of 2-Diethylamino-1H-azepine Intermediates. J. Am. Chem. Soc. 94, 513–520 (1972).
Patel, S. C. & Burns, N. Z. Conversion of aryl azides to aminopyridines. J. Am. Chem. Soc. 144, 17797–17802 (2022).
Pearson, T. J. et al. Aromatic nitrogen scanning by ipso-selective nitrene internalization. Science 381, 1474–1479 (2023).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the Structural Diversity, Substitution Patterns, and Fre-quency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Kallitsis, J. K., Geormezi, M. & Neophytides, S. G. Polymer electrolyte membranes for high-temperature fuel cells based on aromatic polyethers bearing pyridine units. Polym. Int. 58, 1226–1233 (2009).
Murakami, K., Yamada, S., Kaneda, T. & Itami, K. C−H Functionalization of Azines. Chem. Rev. 117, 9302–9332 (2017).
Maity, S., Bera, A., Bhattacharjya, A. & Maity, P. C−H Functionalization of Pyridines. Org. Biomol. Chem. 21, 5671–5690 (2023).
Cao, H., Cheng, Q. & Studer, A. Meta-selective C−H functionalization of pyridines. Angew. Chem. Int. Ed. 62, e202302941 (2023).
Chakraborty, S. & Biju, A. T. Directing group-free regiose-lective meta-C−H functionalization of pyridines. Angew. Chem. Int. Ed. 62, e202300049 (2023).
Josephitis, C. M., Nguyen, H. M. & McNally, A. Late-stage C-H functionalization of azines. Chem. Rev. 123, 7655–7691 (2023).
Kelly, C. B. & Padilla-Salinas, R. Late stage C-H functionalization via chalcogen and pnictogen salts. Chem. Sci. 11, 10047–10060 (2020).
Dolewski, R. D., Hilton, M. C. & McNally, A. 4-Selective pyridine functionalization reactions via heterocyclic phosphonium salts. Synlett 29, 8–14 (2018).
Thorn, D. L. & Hoffmann, R. Delocalization in metallocycles. Nouv. J. Chim. 3, 39–45 (1979).
Bleeke, J. R. Metallabenzenes. Chem. Rev. 101, 1205–1227 (2001).
Landorf, C. W. & Haley, M. M. Recent advances in metallabenzene chemistry. Angew. Chem., Int. Ed. 45, 3914–3936 (2006).
Wright, L. J. Metallabenzenes and metallabenzenoids. Dalton Trans. 15, 1821–1827 (2006).
Chen, J. & Jia, G. Recent development in the chemistry of transition metal-containing metallabenzenes and metallabenzynes. Coord. Chem. Rev. 257, 2491–2521 (2013).
Fernandez, I., Frenking, G. & Merino, G. Aromaticity of metallabenzenes and related compounds. Chem. Soc. Rev. 44, 6452–6463 (2015).
Wright, L. J. Metallabenzenes An Expert View (Wiley, New York, 2017).
Schrock, R. R. et al. Formation of Cyclopentadienyl complexes from tungstenacyclobutadiene complexes and the X-ray crystal structure of an η3-Cyclopropenyl Complex, W[C(CMe3)C(Me)C(Me)](Me2NCH2-CH2NMe2)Cl3. Organometallics 3, 1574–1583 (1984).
Wei, W. et al. 2 + 2 + 1 + 1] Cycloaddition for de novo synthesis of densely functionalized phenols. Angew. Chem. Int. Ed. 62, e202307251 (2023).
Gupta, S. et al. Ruthenabenzene: a robust precatalyst. J. Am. Chem. Soc. 143, 7490–7500 (2021).
Zhang, H. et al. Synthesis and characterization of stable ruthenabenzenes. Angew. Chem. Int. Ed. 45, 2920–2923 (2006).
Luo, M. et al. Reactions of isocyanides with metal carbyne complexes: isolation and characterization of metallacyclopropenimine intermediates. J. Am. Chem. Soc. 139, 1822–1825 (2017).
Zhuo, Q. et al. Multiyne chains chelating osmium via three metalcarbon σ bonds. Nat. Commun. 8, 1912 (2017).
Zhuo, Q. et al. Constraint of a ruthenium-carbon triple bond to a five-membered ring. Sci. Adv. 4, eaat0336 (2018).
Zhou, X. et al. Successive modification of polydentate complexes gives access to planar carbon- and nitrogen-based ligands. Nat. Commun. 10, 1488 (2019).
Lu, Z. et al. Access to tetracyclic aromatics with bridgehead metals via metalla-click reactions. Sci. Adv. 6, eaay2535 (2020).
Cai, Y. et al. Electrophilic aromatic substitution reactions of compounds with craig-möbius aromaticity. Proc. Natl. Acad. Sci. USA. 118, e21023101 (2021).
Lin, Z., Cai, Y., Zhang, Y., Zhang, H. & Xia, H. Heterocyclic Suzuki–Miyaura coupling reaction of metalla-aromatics and mechanistic analysis of site selectivity. Chem. Sci. 14, 1227–1233 (2023).
Varela, J. A. & Saa, C. Construction of Pyridine Rings by Metal-Mediated [2 + 2 + 2] Cycloaddition. Chem. Rev. 103, 3787–3802 (2003).
Domínguez, G. & Pérez-Castells, J. Recent advances in [2 + 2 + 2] cycloaddition reactions. Chem. Soc. Rev. 40, 3430–3444 (2011).
Roglans, A., Pla-Quintana, A. & Sola, M. Mechanistic STUDIES OF TRANSITION-METAL-CAtalyzed [2 + 2 + 2] cycloaddition reactions. Chem. Rev. 121, 1894–1979 (2021).
Huh, D. N., Cheng, Y. K., Frye, C. W., Egger, D. T. & Tonks, I. A. Multicomponent syntheses of 5- and 6- Membered aromatic heterocycles using Group 4−8 transition metal catalysts. Chem. Sci. 12, 9574–9590 (2021).
Lv, Z.-J., Liu, W. & Zhang, W.-X. Progress of Azametallacyclopentadienes in the New Century. Chem. Eur. J. 29, e202204079 (2023).
Tan, X., Hou, X., Rogge, T. & Ackermann, L. Ruthenaelectro-Catalyzed Domino three-component alkyne annulation for expedient isoquinoline assembly. Angew. Chem., Int. Ed. 60, 4619–4624 (2021).
Zhang, B., Duan, Y., Zhang, X. & Guo, S. Uncommon Carbene-to-Azole ligand rearrangement of N-heterocyclic carbenes in a ruthenium system. Chem. Commun. 57, 6879–6882 (2021).
Li, B. et al. Ruthenium-catalyzed oxidative coupling/cyclization of isoquinolones with alkynes through C-H/N-H activation: mechanism study and synthesis of Dibenzo [a, g] quinolizin-8-one derivatives. Chem. Eur. J. 18, 12873–12879 (2012).
Bailey, B. C., Fan, H., Huffman, J. C., Baik, M.-H. & Mindiola, D. Room temperature ring-opening metathesis of pyridines by a transient Ti≡C Linkage. J. Am. Chem. Soc. 128, 6798–6799 (2006).
Schleyer, Pv. R. C. et al. Nucleus-independent chemical shifts: a simple and efficient aromaticity probe. J. Am. Chem. Soc. 118, 6317–6318 (1996).
Chen, Z. et al. Nucleus-Independent Chemical Shifts (NICS) as an aromaticity criterion. Chem. Rev. 105, 3842–3888 (2005).
Herges, R. & Geuenich, D. Delocalization of electrons in molecules. J. Phys. Chem. A 105, 3214–3220 (2001).
Geuenich, D., Hess, K., Köhler, F. & Herges, R. Anisotropy of the Induced Current Density (ACID), a general method to quantify and visualize electronic delocalization. Chem. Rev. 105, 3758–3772 (2005).
Frogley, B. J. & Wright, L. J. Recent advances in metallaaromatic chemistry. Chem. Eur. J. 24, 2025–2038 (2018).
Chen, D., Hua, Y. & Xia, H. Metallaaromatic chemistry: history and development. Chem. Rev. 120, 12994–13086 (2020).
Zhang, Y. et al. Metalla-aromatics: planar, nonplanar, and spiro. Acc. Chem. Res. 54, 2323–2333 (2021).
Jusélius, J., Sundholm, D. & Gauss, J. Calculation of current densities using gauge-including atomic orbitals. J. Chem. Phys. 121, 3952–3963 (2004).
Fliegl, H., Taubert, S., Lehtonen, O. & Sundholm, D. The gauge including magnetically induced current method. Phys. Chem. Chem. Phys. 13, 20500–20518 (2011).
Sundholm, D., Fliegl, H. & Berger, R. J. F. Calculations of magnetically induced current densities: theory and applications. WIREs Comput. Mol. Sci. 6, 639–678 (2016).
Sundholm, D., Dimitrova, M. & Berger, R. J. F. Current density and molecular magnetic properties. Chem. Commun. 57, 12362–12378 (2021).
GIMIC, version 2.2.1 The program can be freely downloaded from https://zenodo.org/records/8183038, 2023.
Rabe, A., Wang, Q. & Sundholm, D. Unraveling the enigma of Craig-type Möbius-aromatic osmium compounds. Dalton Trans. 53, 10938–10946 (2024).
Blasco, D. & Sundholm, D. The aromatic nature of auracycles and diauracycles based on calculated ring-current strengths. Dalton Trans. 53, 10150–10158 (2024).
Jin, L. et al. Chiral dinitrogen ligand enabled asymmetric Pd/Norbornene cooperative catalysis toward the assembly of C–N axially chiral scaffolds. Nat. Commun. 15, 4908 (2024).
Wang, Z. et al. Construction of Cationic Azahelicenes: regioselective three-component annulation using in situ activation strategy. Angew. Chem. Int. Ed. 59, 23532–23536 (2020).
Acknowledgements
H.Z. acknowledged the funding from the National Natural Science Foundation of China (Grant No. 22171234, 22471225). We gratefully thank Prof. Haiping Xia (Southern University of Science and Technology, China) and Prof. Shunhua Li (Xiamen University, China) for their critical and insightful discussion.
Author information
Authors and Affiliations
Contributions
H.Z. designed and conceived the project. Y.Z. and F.H. performed the experiments. Y.Z., Z.Y., and H.Z. analyzed and interpreted the experimental data. Y.C. and X.Z. performed the theoretical calculations. All authors have given approval to the final version of the manuscript. ⊥Y.Z. and F.H. contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Erhong Hao, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Han, F., Yin, Z. et al. Fluorescent pyridine phosphonium salts via transmutation of metallabenzenes. Nat Commun 16, 3632 (2025). https://doi.org/10.1038/s41467-025-58855-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58855-7
