
Abstract
Stable operation of the gas diffusion electrodes is key for industrial-scale electrochemical CO2 reduction (eCO2R). To enhance the electrolytic stability, we shield the Cu-coated gas diffusion electrode with a polycationic sheath via electrospinning and propose a Metal-Organic Double Layer (MODL) scheme to depict the triphasic interface. The as-fabricated electrode exhibits a high multi-carbon Faradaic efficiency of 91.2 ± 3.8%, along with operational stability for over 300 h at 300 mA cm−2 in an alkaline flow cell. In a membrane electrode assembly with pure water as the anolyte, it further achieves an ethylene Faradaic efficiency over 50% at 200 mA cm−2. Mechanistic investigations unveil that replacing hydrated cationic counter ions in the conventional double layer with hydrogen bond-woven polycationic groups in the MODL allows simultaneously tailoring the local electric field and interfacial water structure. This study introduces a molecular-level redesign of the electric double layer in eCO2R systems, achieving precisely tunable electrostatic characteristics and tailored chemical microenvironments while leveraging sustainable electrolysis systems to enable highly efficient and stable multi-carbon production.
Similar content being viewed by others

Introduction
Electrochemical CO2 reduction (eCO2R) powered by renewable energies offer a sustainable means to close the anthropogenic carbon cycle while generating value-added chemicals1,2,3. To maximize the mass and charge transport at the solid-liquid-gas interfaces, gas diffusion electrodes (GDEs) are required for carrying out eCO2R at industrially meaningful scale, either in the electrolyte-circulating flow cells or zero-gap membrane electrode assemblies (MEAs)4,5,6. The GDE is typically composed of a basal gas diffusion layer made of hydrophobic carbon paper or PTFE membrane and a microporous overlayer, on which Cu-based catalysts are often deposited to attain multi-carbon (C2+) products of higher economic values7,8,9,10. Despite the great progress witnessed in promoting the reaction kinetics of eCO2R, the stability of thus-composed GDEs remains formidable due to issues of corrosion11,12, flooding13,14, delamination15,16, and salt precipitation17,18. In a vicious spiral, salt precipitation occurs when the in situ generated hydroxides, even in neutral and acidic electrolytes, react with CO2 to form a substantial quantity of carbonates/bicarbonates8,19,20. These precipitants would then clog the gas permeation paths in GDE and restrain CO2 mass transport, exacerbating the side reaction of hydrogen evolution (HER) and the problem of water flooding.
Many studies have shown that applying organic modifiers to the Cu surface, or creating Cu/polymer interfaces, can effectively modulate local charge distribution and chemical environment towards enhanced eCO2R activity and stability8,21,22,23,24. On the one hand, polymers with the amino functional groups are often exploited to enrich and activate CO2 via Lewis base-acid interactions25, enhance local pH through their Brønsted basicity21,26, and augment the local electric field through charged moieties27. On the other hand, polymers consisting of N, O and F elements that are capable of donating hydrogen bond enable to regulate local hydrophilicity and stabilize reaction intermediates at the Cu surface through noncovalent interactions24,28,29. Furthermore, polymers with the electrostatically polarized backbone can gate ion passage through the Donnan effect, leading to the accumulation or depletion of certain charged species crucial for the activity, selectivity and stability of eCO2R30,31. Nonetheless, most of these polymer/Cu interfaces have been constructed through mixing of ionomers or polymeric blends, lacking a well-defined electric double layer (EDL), not to mention an explicit physical model to illustrate such a complex multiphasic interface. Thus, a vivid picture of the organics-modulated double layer structure is highly desirable, as is success in extending the operational stability.
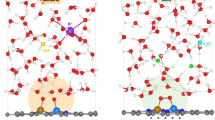
It is worth noting that in the sophisticate electrochemical systems mentioned above, the conventional Gouy-Chapman-Stern model becomes insufficient to depict EDL at the organics-modified electrode surface32,33. In this classic model, the distribution of counter ions near the electrode surface, as co-governed by the electrostatic interaction and thermal motion, is partitioned into an inner compact Stern layer and an exponentially decaying diffuse layer (Fig. 1a)32,34. The Stern layer can be further empirically divided into the inner and outer Helmholtz planes (IHP and OHP), with the former comprising mainly de-solvated species physically/chemically adsorbed onto the electrode surface34,35. In the presence of an organic overlayer, the OHP, however, could be drastically different from the conventional configuration and fine-tuned through varying the functional groups (Fig. 1b). For instance, the charge density inside OHP can be quantitatively modulated by cationic groups such as ammoniums and imidazoles introduced to the organic phase, and so that the local electric field in IHP can be regulated effectively27,36,37. In this context, an adaptive scheme is required to delineate such an EDL comprising an organic interphase, of which the molecular configuration can act on the reaction cascade in IHP.

a The classic Gouy–Chapman-Stern model. b The MODL scheme proposed in this study for eCO2R.
In the current work, we propose a metal-organic double layer (MODL) scheme to depict the polymer-modified Cu electrode used for eCO2R as shown in Fig. 1b. Therein, the functionalized organic phase spans the empirical OHP and diffuse layer, and thus the solvation and distribution of counter ions, as well as the solvent structure, are significantly different from the conventional EDL. The polymer-modified OHP further affects the molecular configuration and dynamics in IHP, ultimately steering the reaction cascade of eCO2R. In practice, we innovatively shield the GDEs with a thin film of quaternized polyvinyl alcohol via electrospinning, aiming to extend the electrocatalytic stability in both flow cell and MEA operated under industrially relevant conditions. Our design principle of the polymer structure is as follows. (1) The polymeric backbone, i.e. polyvinyl alcohol (PVA), should be readily gelated in the aqueous catholyte or in the membrane electrode to afford an intimate hydrated interface with tailored water structure; (2) the electrospun film should be mechanically robust to physically prevent catalyst delamination from GDE for extended durability; (3) Quaternary ammoniums are introduced as the tailing groups of side chains to regulate ion passage and modulate local electric field via creating a tunable MODL; (4) the aminated polymer should preserve abundant hydroxyls to maintain the hydrogen bonding network. As a result, the best GDE exhibits a high C2+ Faradaic efficiency (FE) of 91.2 ± 3.8%, along with operational stability for over 300 h at 300 mA cm−2 in an alkaline flow cell. In MEA using pure water as the anolyte, it further demonstrates a C2H4 FE of 53.0 ± 1.4% operated continuously at 200 mA cm−2 for over 20 h. In situ FT-IR, Raman and ab initio molecular dynamics (AIMD) simulations are carefully performed to interrogate the structure variables of the polymeric overlayer in modulating interfacial water structure and intermediates dynamics for steering the reaction pathways, highlighting the concerted role of ammoniums and hydroxyls in the MODL to boost both the selectivity and stability of multi-carbon synthesis. Consequently, by renovating the EDL with tunable electric properties and chemical functionalities, this study advances sustainable electrolysis technologies through innovative interfacial engineering and profound mechanistic understanding.
Results and discussion
Electrospun polycationic sheath to stabilize eCO2R in alkaline flow cell
The durability of physical-vapor-deposited copper films and the salt precipitation inside GDE constitute two of the major hurdles for robust eCO2R in flow cells (Supplementary Fig. 1). Once the GDE is clogged by carbonates/bicarbonates, hydrogen evolution dominates the electrode reactions due to restrained CO2 transport. To tackle these issues, we propose to apply a protective sheath on GDE to physically deter Cu delamination and block the passage of K+ cations involved in salt precipitation. We first simulated the potential-driven K+ diffusion near the electrode surface without and with a cation-blocking layer in 1 M KOH (Supplementary Fig. 2). In the case of unconstrained K+ transport, there was a notable rise of K+ concentration in the stern layer adjacent to the electrode surface, reaching up to 5 folds of that in the bulk electrolyte (Supplementary Fig. 2, left panel). By contrast, limiting the K+ transport in the flow channels resulted in a very low K+ concentration of 0.01 M near the electrode surface after applying the same negative bias for the same period (Supplementary Fig. 2, right panel). Thus, we envisage that the application of a cation-blocking overlayer might effectively reduce the K+ availability inside the GDE.
To materialize the simulated cation-blocking layer, we electrospun a series of quaternized polyvinyl alcohol films (denoted as PVAQAx, x represents the percentage of hydroxyls in PVA grafted with quaternary ammonium) in situ onto the electrodes of PTFE/Cu for assessing their potency in mediating eCO2R (Fig. 2a, upper panel). While the quaternary ammoniums (QAs) were incorporated to create a MODL and restrain the K+ passage, the hydroxyls on the side chains were deliberately annexed to afford an H-bonding network for interfacial gelation and intermediates stabilization. Moreover, the interwoven porous layer of quaternized PVA ensures the continuity of the MODL so that the charge and mass transfer therein are coherent. In such a continuum space, the electrostatic and microenvironmental properties within the EDL can effectively act on the CO2 reduction process at the Cu surface, allowing to adapt the classic EDL model under a global electrostatic and H-bonding framework (Fig. 2a, lower panel).

a Schematics illustrating the fabrication of the PTFE/Cu/PVAQAx electrodes via thermal evaporation and electrospinning (top row), and the structure of MODL with OHP comprising a global electrostatic and H-bonding framework (bottom row). b FEs of C2H4 and C2+ products at various current densities demonstrated by PTFE/Cu/PVAQA54 in an alkaline flow cell. c Stability test on PTFE/Cu/PVAQA54 at 300 mA cm−2 in a flow cell. Error bars represent the standard deviation of three independent measurements, and 85% ohmic correction has been applied to the potential values.
Figure 2b, c showcases the eCO2R performance of one optimal example by adopting such an electrode configuration and material design. With a quaternization degree of 54%, the sandwich PTFE/Cu/PVAQA54 electrode exhibited a high Faradaic efficiency (FE) of 91.2 ± 3.8% for all C2+ products at a total current density of 300 mA cm−2 (U = −0.94 V vs. RHE), as well as a high C2H4 FE above 60% across a wide current range (Fig. 2b). Moreover, the composite electrode demonstrated an exceptional operational stability over 300 h at 300 mA cm−2 in the alkaline flow cell utilizing 1 M KOH as the electrolyte (Fig. 2c). With respect to the cathodic attributes of C2+ selectivity, partial current density, overpotential and operational stability (Supplementary Table 1)10,21,22,30,38, this performance ranks amongst states of the art reported in the literature and satisfies the techno-economic requirements for industrial eCO2R catalysts.
The electrostatic role of polycations in the MODL
To scrutinize the role of QAs introduced into the polymeric backbone, we began by optimizing the parameters of the PVAQAx films spun onto the PTFE/Cu electrodes (Supplementary Figs. 3–9). With a quaternization degree of 13%, PVAQA13 spun onto the PTFE/Cu electrode decreased the water contact angle from 132° to 26° (Supplementary Figs. 3, 4), evidencing a hydrophilic thin film favorable for water retention. All the PTFE/Cu/PVAQAx electrodes were thermally treated in air after the electrospinning process. It was found a modest annealing temperature of 140 °C could greatly enhance the physical adherence while maintaining structural integrity of the thin films on the Cu electrodes (Supplementary Figs. 5–7). The thickness of these PVAQAx films on the electrodes can be facilely modulated by varying the electrospinning time (Supplementary Fig. 8). Increasing the thickness of PVAQA13 from 14 to 86 μm witnessed a notable increase of C2+ products in eCO2R, especially at higher current densities (Supplementary Fig. 9). Further thickening the thin film to 132 μm did not gain any meaningful improvement in CO2 reduction efficiency, possibly due to the escalated interfacial resistance to augment the cathodic overpotential and HER, offsetting the benefits brought by the cationic polymer. Thus, in the following studies we controlled the thickness of all electrospun films roughly around 80 µm.
For a strict comparison, isopropanol-grafted PVA without the tailing ammoniums, namely PVAPA54 (Supplementary Fig. 10), was synthesized to qualitatively assess the function of QAs in PVAQA54 by excluding any possible interference from hydroxyls. Both electrospun films present similarly cross-linked fibrous morphology (Fig. 3a vs b) and thickness (Supplementary Fig. 11) but quite different IR signatures in the fingerprint regime (Supplementary Fig. 12). When tested for eCO2R, the PTFE/Cu/PVAPA54 cathode produced a maximum FE of 81.9 ± 3.4% for C2+ products at −0.87 V under a total current density of 200 mA cm−2 (Fig. 3c), which is inferior to the 91.2 ± 3.8% C2+ FE observed on PTFE/Cu/PVAQA54 at 300 mA cm−2 (Fig. 3d and Supplementary Fig. 13). At lower potentials a notable amount of CH4 and H2 was produced on PTFE/Cu/PVAPA54, whereas on PTFE/Cu/PVAQA54 ethylene and ethanol dominated the whole potential range. This indicates that the positively charged ammonium groups helped to boost C–C coupling at more negative bias. To assure that the produced liquid products such as ethanol and propanol were exclusively converted from CO2, rather than the decomposition of the PVAQA films, 1H-NMR spectra were taken, exhibiting doublets at 0.97 and 1.7 ppm corresponding to the 13C-labeled n-propanol and ethanol, respectively (Supplementary Fig. 14). More remarkably, at 100 mA cm−2 where the highest FEs of C2H4 were observed for both electrodes, PTFE/Cu/PVAPA54 can only sustain a stable electrolysis less than 30 h, while PTFE/Cu/PVAQA54 could last for at least 250 h without any notable decay of ethylene production (Supplementary Fig. 15).

a, b Top-view SEM images taken on the (a) PTFE/Cu/PVAPA54 and (b) PTFE/Cu/PVAQA54 electrodes. c, d FEs of different reduction products (left axis) under varying current densities for (c) PTFE/Cu/PVAPA54 and (d) PTFE/Cu/PVAQA54 with the corresponding potentials (right axis). Error bars represent the standard deviation of three independent measurements, and 85% ohmic correction has been applied to the potential values.
Cross-sectional SEM images were taken on the above PTFE/Cu/PVAPA54 and PTFE/Cu/PVAQA54 electrodes after prolonged electrolysis (Fig. 4a, b). EDX elemental mapping confirmed the sandwich architecture for both electrodes. For PTFE/Cu/PVAPA54 (Fig. 4a), the lack of O element in the PTFE/Cu substrate and the lack of N element in the PVAPA54 overlayer are expected from their chemical composition. The colocalization of F and K elements evidences the infiltration of K+ into the PTFE membrane during electrolysis. By contrast, there was negligible amount of K superimposed with F in the gas diffusion layer of the post-electrolytic PTFE/Cu/PVAQA54 electrode (Fig. 4b). Meanwhile, the prominent N distribution atop the Cu layer provides further evidence for the quaternized PVA film laid above. It is thus evident that the polycationic PVAQA54, when compared to the neutral PVAPA54, can effectively blockade K+ ions trafficking through the PTFE membrane via the Donnan exclusion. This argument was further consolidated by measuring the K+ conductivity in PVAQA54 and PVAPA54 through impedance measurements on symmetric stainless-steel cells utilizing 1.0 M KTFSI electrolyte (Fig. 4c). The ionic conductivity of K+ in PVAPA54 was calculated to be 14.36 mS cm−1, nearly 5 times of that in PVAQA54 (2.84 mS cm−1, Fig. 4d). Additionally, increasing the K+ concentration in the catholyte from 1 to 3 M while maintaining the same pH value had negligible impact on the eCO2R performance of PTFE/Cu/PVAQA54 except for a slightly lowered overpotential, further confirming the cation-shielding effect of PVAQA54 (Supplementary Fig. 16).

a, b Cross-sectional SEM and EDX elemental mapping images taken on the (a) PTFE/Cu/PVAPA54 and (b) PTFE/Cu/PVAQA54 electrodes after prolonged electrolysis for 20 h at 300 mA cm−2. c Electrochemical impedance spectra taken on symmetric stainless steel cells using PVAPA54 or PVAQA54 as the separating membrane and 1.0 M KTFSI as the electrolyte. d The ionic conductivity of K+ calculated for PVAPA54 and PVAQA54. The measurement of the ionic conductivity was conducted in a single instance. e The dynamics of K+ in simulated Cu/PEO/H2O, Cu/PVAPA/H2O and Cu/PVAQA/H2O interfaces, respectively. The statistics on Z coordinate are based on ~10 ps AIMD trajectories, which are utilized to analyze the average distribution profiles. f Calculated plannar-averaged Hartree potentials as a function of cell dimension normal to the Cu/PVAPA/H2O and Cu/PVAQA/H2O interfaces, respectively.
The impeded migration kinetics of K+ in PVAQA was further investigated using AIMD simulations (Fig. 4e). In these simulations, K+ cations were positioned at identical sites within the simplified MODL across the three modeled systems. Notably, only in PVAQA, the K+ cation was repelled out of the MODL within ~5 ps, enabling its dissolution into the bulk solvent (Supplementary Fig. 17). Conversely, in both PVAPA and PEO models, K+ cations descend within ~5 ps to the first water layer at IHP (Supplementary Figs. 18, 19). Figure 4f illustrates that within the polymer region in the MODL, PVAQA exhibits a significantly reduced work function compared to PVAPA, while the work function of other parts such as the metal slab and solvent are substantially similar. A smaller work function suggests a more pronounced tendency to lose electrons, and thus the positively charged PVAQA repels the K+ cations upwards. This further contributes to a reduction in the overall work function of the PVAQA system, aligning with the experimental observation of a smaller overpotential in driving eCO2R39.
In situ attenuated total reflection surface-enhanced infrared spectroscopy (ATR-SEIRAS, Supplementary Fig. 20) was employed to compare the dynamics of reaction intermediates evolved at the Cu/PVAQA54 and Cu/PVAPA54 interfaces (Fig. 5a–c). Apart from the common intermediates of *COOH, *CO, *COH and *OCCO/*OCCOH observed with Stark shift at ~1400, 2064, 1086 and 1285 cm−1, respectively, for eCO2R at the Cu surface40,41,42,43, the most prominent difference between the two samples lies in the H−OH bending of adsorbed water at 1646 cm−1 and the asymmetric stretching of *COO− at 1508 cm−1 (Fig. 5a vs b)44,45. For the former, the peak intensity is a collective manifestation of polarized water adsorption at IHP on the biased Cu surface, which intensifies with the decreasing potential (Fig. 5c) and agrees with the modern Bockris–Davanathan–Muller EDL model46. Apparently, the tailing ammoniums in PVAQA54 could effectively promote the adsorption of polarized water molecules, which are deemed more readily to be ionized31. As for the *COO− bands, while they are distinctive in the spectra of Cu/PVAPA54, decreasing with the increasing bias, on Cu/PVAQA54 they are barely discernable. Considering the following elemental steps in the early stage of eCO2R47,48,49,50:

a, b In situ ATR-SEIRAS spectra recorded while ramping down the potential from −0.4 to −2.0 V to monitor the evolution of intermediates binding on (a) Cu/PVAPA54 and (b) Cu/PVAQA54. c The trending of *COO− and H–OH peak intensities against the applied potential for Cu/PVAPA54 and Cu/PVAQA54. d, e In situ Raman spectra recorded from −0.2 to −1.2 V between 200 and 1200 cm−1 on (d) Cu/PVAPA54 and (e) Cu/PVAQA54. f The trending of surface pH against the applied potential for Cu/PVAPA54 and Cu/PVAQA54. g, h In situ Raman spectra recorded from OCP to −1.2 V between 2800 and 3900 cm−1 on (g) Cu/PVAPA54 and (h) Cu/PVAQA54. i The trending of various water peak intensities against the applied potential for Cu/PVAPA54 and Cu/PVAQA54. The potentials were recorded without iR correction for both in situ FT-IR and Raman studies.
the prominent *COO− bands observed on Cu/PVAPA54 strongly suggest the above chemical reaction step (Eq. (2)) is rate determining so that the species of *COO− are consumed slowly. By contrast, at the interface of Cu/PVAQA54 where water dissociates more facilely, the chemical step occurs quickly and therefore *COO− is difficult to be detected. This argument is further justified by the fact that the *COO− intensity on Cu/PVAPA54 decreased with the increasing bias, at which the ionization of water is expedited (Fig. 5c). The accelerated water dissociation and *COOH formation are conducive to the enrichment of OH− and *CO at the Cu/polymer interface, favoring the C–C coupling process.
Next, in situ surface-enhanced Raman spectroscopy (SERS) was performed on a flow-cell assembly (Supplementary Fig. 21) to monitor the chemical environment adjacent to the electrode surface. Both the Cu surfaces within Cu/PVAPA54 and Cu/PVAQA54 developed a superficial oxide layer upon air exposure, as evidenced by the two peaks at 517 and 620 cm−1 observed under open-circuit potential (OCP) (Fig. 5d, e)21,51. Under negative bias, two distinct peaks at 382 and 522 cm−1 emerged, respectively corresponding to the ν(Cu−CO) and ν(Cu−OH) stretching modes21,52. The observation of these intermediates well echoes the elementary steps mentioned above. Of particular note is the observation of a frustrated ρ(Cu−C−O) rotational mode at 272 cm−1 for Cu/PVAQA54 but barely notable for Cu/PVAPA54. This might be due to the QA-augmented local electric field, which enhanced the plasmon resonance effect at the Cu surface that led to an amplification of the Cu−CO vibration signal52. The adsorption of CO on the Cu surface was further confirmed by the stretching modes of C ≡ O at 2033–2049 cm−1 for both Cu/PVAPA54 and Cu/PVAQA54, with the former showing a faster intensity decay with the increasing bias (Supplementary Fig. 22). This might serve as additional evidence for the electric-field stabilized *CO intermediates in favor of C−C coupling.
The peaks at 1060 and 1011 cm−1 can be ascribed to species of CO32− and HCO3−, respectively9. Based on the following equilibrium:
the intensity ratio of CO32−/HCO3− could be used as a practical indicator for indexing local pH, which we pre-calibrated using a series of standard K2CO3/KHCO3 solutions (Supplementary Fig. 23). From quantitative peak analysis (Supplementary Fig. 24), the electrode of Cu/PVAQA54 showed a substantial increase in local pH from OCP to −1.2 V when compared to Cu/PVAPA54 (Fig. 5f). It is thus evident that the positive QA groups in the polymeric chain enable to enhance the local electric field for promoting water dissociation and electrostatically trap OH− anions, rendering a higher local pH in OHP. This matches the co-ion concentration profile illustrated in Fig. 1b and helps to sustain a basic environment for promoting C−C coupling. The effects of OH− trapping and H+ blocking were further confirmed by rotating disk electrode (RDE) measurements in Ar-saturated 0.25 M K2SO4/H2SO4 (pH=2) for Cu/PVAQA54 (Supplementary Fig. 25). The current plateaus between −0.8 V and −1.6 V in the linear scanning voltammetries correspond to HER limited by bulk proton (H3O+) transport53. Compared to Cu/PVAPA54, Cu/PVAQA54 shows a lower plateau current at each rotation rate, suggesting lower local H+ concentration presumably due to attenuated H3O+ mass transport to the Cu surface.
Delving into the water structure at molecular level, the O−H stretching of interfacial water in the Raman spectra between 2800–3900 cm−1 can be traced to three origins (Fig. 5g, h), including the 4-coordinated H-bonding water around 3260 cm−1 (4-HB·H2O), the 2-coordinated H-bonding water around 3450 cm−1 (2-HB·H2O), and the K+ ion-associated water at around 3610 cm−1 (K+·H2O)54,55. The intensity ratios of these water components are plotted in Fig. 5i against the applied potential. In general, the percentage of K+·H2O on Cu/PVAQA54 is much lower than that on Cu/PVAPA54, corroborating the cation-blocking effect of QAs. The percentage of 4-HB·H2O on Cu/PVAQA54 is notably higher than that on Cu/PVAPA54 across all potentials, implying a more matured H-bonding network that are better organized and less mobile. A similar phenomenon of QA-promoted 4-HB·H2O was also observed on Cu electrodes56. By contrast, 2-HB·H2O is more mobile and tends to gather on the Cu surface as the applied potential decreases (Fig. 5g, h)57. Notably, the ratio of 2-HB·H2O on Cu/PVAQA54 grew much faster with the decreasing potential, which is in line with the intensified H−OH bending observed in IR at lower potentials. Therefore, on Cu/PVAQA54 the MODL comprises mainly hydroxyl-interlinked 4-HB·H2O, concurrent with the faster emergence of 2-HB·H2O at higher bias. With the dissociation of water to produce H+ and OH− and the quick consumption of the former by eCO2R or HER, more OH− are yielded and accumulated within the MODL, which favorably reduces the energy barrier for the C−C coupling process58.
Quantitative studies on the quaternary ammoniums and hydroxyl contents
To further quantitatively interrogate the role of QA in modulating local electric field and thereby the eCO2R performance, we synthesized PVAQA13, PVAQA33 in addition to PVAQA54 by varying the QA content (Supplementary Figs. 26, 27). It should be noted that further increasing the QA content resulted in frequent nozzle clogging during electrospinning. At jtotal = 200 mA cm−2, the FEs of C2+ products were summed up to be 86.0 ± 1.5% for PTFE/Cu/PVAQA13 (U = −0.84 V), 86.9 ± 1.3% for PTFE/Cu/PVAQA33 (U = −0.82 V), and 86.8 ± 1.0% for PTFE/Cu/PVAQA54 (U = −0.80 V) (Supplementary Fig. 28). Despite of similar FEs, the linear decrease of overpotentials with the increasing QA content (Supplementary Fig. 29) corroborates the improved eCO2R activity and kinetics in the presence of QAs. However, when jtotal was raised to 400 mA cm−2, the partial current densities for total C2+ products were diverged to be 282.5, 325.3 and 340.5 mA cm−2 for PTFE/Cu/PVAQA13, PTFE/Cu/PVAQA33, and PTFE/Cu/PVAQA54, respectively (Supplementary Fig. 30). Consistent with the previous qualitative comparison between PVAPA54 and PVAQA54, here the quantitative analysis further supports the role of QAs in extending the high C−C coupling efficiency to lower potentials. Moreover, in situ ATR-SEIRAS revealed the gradual vanishment of the *COO− peak at 1508 cm−1 with the increasing QA content, concomitant with the notably intensified water peak at 1646 cm−1 (Supplementary Fig. 31). These results offer further quantitative evidence to support the polarized water adsorption and quick *COO− conversion on the Cu surface as regulated by QA. It is thus concluded that a higher QA content in the polymeric overlayer in collaboration with a more negative bias (corresponding to a more polarized MODL with a higher local electric field) could induce a more polarized and basic water structure that facilitates both CO2 hydrogenation and C−C coupling.
In a parallel study to quantitatively assess the role of hydroxyls in the polymeric backbone, electrospun PVAx (x = degree of hydroxylation) films were fabricated and further compared to the hydroxyl-free polyethylene oxide (PEO). The morphology and chemical functionalities of the as-spun PVA and PEO films were confirmed by SEM (Supplementary Fig. 32) and IR spectra (Supplementary Fig. 33). Among the three PVA electrodes of different hydroxylation degree, PTFE/Cu/PVA98 presented the highest C2+ FE of 82.6 ± 1.2% at the total current density of 200 mA cm−2 (U = −0.87 V), followed sequentially by PTFE/Cu/PVA91 with the maximum C2+ FE of 78.9 ± 1.8% at 100 mA cm−2 (U = −0.73 V) and PTFE/Cu/PVA78 with the maximum C2+ FE of 74.5 ± 1.0% at 100 mA cm−2 (U = −0.72 V) (Supplementary Fig. 34). For the PEO-shielded Cu electrode, the main reduction product gradually shifted from C2H4 at lower current densities (e.g. 50 and 100 mA cm−2) to CH4 at higher current densities (e.g. 300 and 400 mA cm−2), with the maximum C2+ FE of 54.8 ± 2.8% observed at 100 mA cm−2 (Supplementary Fig. 35). Further comparing the i–V profiles of PTFE/Cu/PVA98, PTFE/Cu/PVA91 and PTFE/Cu/PVA78 to that of PTFE/Cu/PEO, all the hydroxyl-containing polymers exhibited a significantly lower voltage to afford the same current density (Supplementary Fig. 36). Clearly, the pendant hydroxyl groups in PVA are beneficial for improving both the eCO2R activity and C2+ production. Nonetheless, none of the PVAx films beats PVAQAx in terms of C2+ production, highlighting the synergic effect of hydroxyls and ammoniums to concertedly promote the C−C coupling process in eCO2R. Similar to the cases of PVAPA54 (Fig. 5a) and PVAQA13 (Supplementary Fig. 31a), in situ ATR-SEIRAS spectroscopy revealed the decreasing *COO− intensity at 1508 cm−1 in the presence of a modest water peak at 1646 cm−1 for all Cu/PVAx electrodes (Supplementary Fig. 37). This, again, signifies the correlation between polarized water adsorption and *COO− conversion (Supplementary Fig. 38). In situ Raman spectra displayed more prominent Cu−CO signals on Cu/PVA98 than on Cu/PEO across the whole potential range applied (Supplementary Fig. 39), suggesting that the hydroxyl groups from PVA helped to stabilize *CO, likely through the formation of direct O−H···O≡C and indirect H−O···H−O−H···O≡C hydrogen bonds48. This argument was further quantitatively supported by in situ ATR-SEIRAS in comparing the *COatop signals at 2064 cm−1 for samples of Cu/PEO and Cu/PVAx (Supplementary Fig. 40). In general, both the QA and hydroxyl groups are conducive to stabilizing *CO on the Cu surface.
AIMD calculations to unveil the concerted roles of quaternary ammoniums and hydroxyls
To help better understand the MODL with an intricate water structure at the Cu/polymer interface to tailor local chemical environment and intermediates of eCO2R, we constructed molecular models to represent the experimentally synthesized thin films of PEO, PVAPA and PVAQA covering the copper layer immersed in aqueous electrolyte (Supplementary Figs. 41–43). In our simulations, we introduced two CO molecules onto the Cu(111) surface and added 55 explicit water molecules to simulate the water structure and *CO configuration at the intimate Cu/polymer interfaces. The AIMD calculation was carried out based on the recently developed first-principles scheme CK2P dynamic constant potential module (CDCPM), utilizing a slow-growth scheme59 combined with the thermodynamic integration (TI) method60 to assess the reaction free energy (ΔG) and free energy barriers (ΔG‡) of C−C coupling at these multiphasic interfaces under working conditions. Further details of the modeling approach are outlined in the methodology section and Supplementary Data 1.
We first examined the H-bonding network at these interfaces by calculating the statistical distribution of hydrogen bonds perpendicular to the polymer-covered copper surfaces (Supplementary Figs. 44–47). At the Cu/PEO/H2O interface, we observed a gap zone indicating a depleted H-bonding network (marked by the shaded areas in Supplementary Fig. 47). In contrast, both Cu/PVAPA/H2O and Cu/PVAQA/H2O interfaces do not exhibit such depletion of the H-bonding network. The shaded area covers the MODL region occupied by the polymer down to the edge of IHP, suggesting that the hydroxyl groups in the latter two polymers enhance water connectivity and H-bonding network within the interfaces. This enhancement is crucial for the formation of C2+ products61, as a recent study shows that hydrogen bonds can promote C−C coupling by stabilizing the CO dimer62. This explains why the PTFE/Cu/PVAPA54 electrocatalyst, serving as a prototype for one of our computed triphasic interface models, shows improved eCO2R activity and higher C2+ production compared to another prototype, PTFE/Cu/PEO. The number of hydrogen bonds in the Cu/PVAQA/H2O interface within the shaded area is higher than Cu/PVAPA/H2O, confirming that the QA groups could further enrich the H-bonding network through orientated water arrangement63, which is in line with the Raman results in Fig. 5g, h.
We then explored the influence of the QA groups in promoting polarized adsorption of water molecules on the Cu surface, which is critical for water ionization. Our analysis of the water dipole orientations at the Cu/PVAPA/H2O and Cu/PVAQA/H2O interfaces revealed distinct patterns, as illustrated in Supplementary Fig. 48. At the Cu/PVAPA/H2O interface, the \(\overline{\cos }({{{\rm{\theta }}}})\) value of the water layer is positive at ~2.2 Å and turns negative from ~2.5 Å. This indicates water’s orientation changes, suggesting disorder of the water layer. In contrast, at the Cu/PVAQA/H2O interface the distribution of \(\overline{\cos }({{{\rm{\theta }}}})\) remains negative, indicating that the interfacial water molecules tend to be orderly aligned with the hydrogen atoms pointing downwards (lower right inset). We attribute the polarized arrangement of the water molecules to the larger capacitance of the Cu/PVAQA/H2O interface (in accordance with the more polarized MODL), as evidenced by the capacitance measurements shown in Supplementary Fig. 49. The positively charged QA groups augment the local electric field, and thus the PTFE/Cu/PVAQA54 electrode exhibited a higher double-layer capacitance compared to the PTFE/Cu/PVAPA54 electrode (0.601 vs 0.145 F cm−2). In addition to promoted ionization, such a polarized alignment of water molecules could foster their interactions with oxygenated intermediates via hydrogen bonds, aiding in C−C coupling57. For example, the AIMD trajectory analysis (Supplementary Fig. 50) demonstrates that at the Cu/PVAQA/H2O interface, *CO intermediates maintained optimal distances for coupling (less than 3.5 Å), influenced by the dense H-bonding network and polarized water adsorption64. Moreover, the charge disparity between two *CO intermediates is greater at the Cu/PVAQA/H2O interface, induced by the local electric field65, and this can substantially promote C−C coupling66.
We further compared the energetics of the C−C coupling reaction (*CO + *CO → *OCCO) at the Cu/PVAPA/H2O and Cu/PVAQA/H2O interfaces at a working potential of -0.9 V vs RHE and a pH value of 14 (Fig. 6a, b). With the synergistic effect of QA and hydroxyl groups, the C−C coupling reactions occurring at the Cu/PVAQA/H2O interface are further boosted. The ΔG‡ and ΔG for C−C coupling are 0.79 and 0.75 eV on Cu/PVAQA/H2O interface, significantly lower than the 1.07 and 1.01 eV on Cu/PVAPA/H2O. This reduction in both ΔG‡ and ΔG translates to a significant increase in activity, aligning with experimental observations.

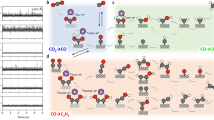
a, b Free energy change as a function of reaction coordinates for the reaction: *CO + *CO → *OCCO under −0.9 VRHE and pH 14 on (a) Cu/PVAPA/H2O and (b) Cu/PVAQA/H2O. Insets are the atomic structures of the initial (IS), transition (TS) and final states (FS) for the process. c Schematics to illustrate the synergic role of ammoniums and hydroxyls in the MODL for boosting multi-carbon synthesis. d Summary of the in situ FT-IR spectra between 1350 and 1700 cm−1 for Cu/PVAPA54 and Cu/PVAQAx. e Demonstration of pure-water MEA with CP/Cu/PVAQA54 at 200 mA cm−2. The potentials presented in Fig. 6d and the cell voltages shown in (e) were recorded without iR correction.
Taken together from the above experimental and computational results, it can be clearly seen that the incorporation of both hydroxyls and ammoniums into the polymer chain allows to synergistically promote the C2+ selectivity at high current densities (Supplementary Fig. 51). These groups lead to the enhanced local electric field, polarized water adsorption and enriched H-bonding within the MODL, all of which benefit efficient C−C coupling by following the cascade illustrated in Fig. 6c. Specifically, the remarkable eCO2R performance of PTFE/Cu/PVAQA54 with high C2+ production and long-term operational stability in alkaline electrolyte can be primarily attributed to the following aspects: (1) The thermally treated polymeric films with good adherence to the electrode prevent physical delamination of Cu, resulting in better mechanical and electrochemical stability; (2) the positively charged QAs effectively blockade K+ permeation into GDE, mitigating salt precipitation; (3) the grafted QAs induces a stronger local electric field under negative bias to afford a more polarized and basic water structure that facilitates intermediates hydrogenation (Fig. 6d) and C−C coupling; (4) The abundant hydroxyls furnish an extensive H-bonding network for stabilizing *CO through direct/indirect O−H···O interactions and lowering the energy barrier of C−C coupling.
eCO2R in Pure-water MEA
Lastly, inspired by the lack of K+ cations to participate eCO2R at the Cu/PVAQA54 interface while still achieving a high C2+ selectivity, we are intrigued by the feasibility to employ PVAQA54 as the cationic membrane to furnish a benign micro-environment in MEA utilizing pure water as the anolyte (Supplementary Fig. 52). We postulate that the Cu-coated and PVAQA54-shielded carbon paper electrode (CP/Cu/PVAQA54) should inherit all the aforementioned benefits in alkaline electrolyte, including the substitution of K+ ions by QAs for double-layer construction to maintain a high local electric field, the formation of an extensive H-bonding network for intermediates stabilization, the electrostatically trapping of OH− to afford a high local pH for suppressing HER and lowering the energy barrier of C−C coupling, and more importantly, the escalated water dissociation at the Cu surface to expedite the rate-determining step. At an operating current of 1 A (accounting for a current density of 200 mA cm−2), the electrolyzer was able to achieve a C2H4 FE of 53.0 ± 1.4% and last for more than 20 h (Fig. 6e), ranking among the best results reported in literature8,38,67,68. After the prolonged electrolysis, the CP/Cu/PVAQA54 cathode exhibited negligible structure and morphology changes (Supplementary Figs. 53, 54), despite that the total cell voltage changed to −5.24 V from the initial −4.03 V. Further considering the stable operation of PFTE/Cu/ PVAQA54 in the alkaline flow cells, we conjecture that the electrode stability should not be the cause for the MEA failure. Instead, the MEA degradation could be mainly attributed to the gradual dehydration and loss of ionic conductivity of the Sustainion membrane, which would eventually lose contact with the CP/Cu/PVAQA54 electrode and even rupture after prolonged electrolysis (Supplementary Fig. 55). Nevertheless, both the energy efficiency and CO2 utilization of the MEA setup are superior to those of the flow cell (Supplementary Figs. 56, 57).
In summary, in this study, cationic polymers were electrospun in situ onto the Cu electrode to simultaneously address the stability issues of GDE and create a MODL for electrostatic and microenvironmental tuning. Among the various polymers investigated, the one with the highest hydroxyl and ammonium contents demonstrated a high C2+ FE of 91.2 ± 3.8% and long-term stability over 300 h at an operational current density of 300 mA cm−2 in the alkaline flow cell. Comprehensive control studies and exquisite IR and Raman characterizations were carried out to illuminate the synergy from hydroxyls and ammoniums in the MODL for boosting multi-carbon synthesis. It was found that the positively charged QAs could not only blockade K+ permeation into the GDE to suppress salt precipitation, but also induce a stronger local electric field under negative bias to render a more polarized and basic water structure that facilitates intermediates hydrogenation and C−C coupling. The latter is further bolstered by the extensive H-bonding network for *CO stabilization, in virtue of the abundant hydroxyls appended to the polymeric backbone to afford a gelatinous interface. Owing to the substitution of K+ ions by QAs for double-layer construction, the CP/Cu/PVAQA54 electrode further demonstrated a marked performance of pure-water MEA, achieving a C2H4 FE of 53.0 ± 1.4% and lasting for more than 20 h under 200 mA cm−2.
It is worth noting that the sandwich electrodes fabricated here through physical vapor deposition and electrospinning are easily scalable and adaptable for industrial electrolyzers. Meanwhile, it serves as a universal method to modify the electrode surface with the desired functionality, as well as a paradigm to configure MODL for studying the organics-modulated EDL. While the polycationic ammoniums can be further extended to imidazoles, piperidines and pyridines, the hydroxyls can expand to amines, carboxylates and esters, etc. In short, by leveraging the selective ion permeability and tunable electrostatic field inside the MODL, this study offers a facile but potent strategy to tailor the local chemical environment on the Cu electrodes, paving the way for electrochemical CO2 reduction with industrially meaningful yield and stability. Further efforts should be paid to enhancing the current collecting efficiency of the sandwich electrodes by minimizing the interfacial and ohmic resistances, optimizing the device configuration and process variables of MEA, and narrowing down the MODL parameters, both theoretically and experimentally, for product-orientated multi-carbon synthesis.
Methods
Materials
PTFE membrane (PF-1F-F100100) was purchased from Cobeter Filtration Equipment Co., Ltd. Carbon paper (YLS-30T) was obtained from Suzhou Sinero Technology Co., Ltd. Cu nuggets (>99.99%) were purchased from Jiangxi Ketai Advanced Materials Co., Ltd. KOH (95%), polyvinyl alcohols (alcoholysis degree: 78%, 91% and 98%) and polyethylene oxide (average molecular weight ~600,000) were obtained from Shanghai Macklin Biochemical Co., Ltd. Propylene oxide (99.5%) and Glycidyl trimethylammonium chloride (95%) were purchased from Aladdin Reagent (Shanghai) Co., Ltd. Hydrochloric acid (HCl, 36.0%-38.0%) was purchased from Shanghai Lingfeng Chemical Reagent Co., Ltd. All materials were used as received without further purification. The distilled water (>18.0 MΩ) was purified with a Sartorius arium mini ultrapure water system.
Preparation of evaporated Cu on gas diffusion layers (GDL)
Cu was evaporated onto the PTFE (PF-1F-F100100) membrane or carbon paper (YLS-30T) in a UHV thermal evaporator (PD400, Wuhan Pudi Vacuum Technology Co., Ltd). 2.0 g of Cu nuggets were placed in a Tungsten boat inside the evaporation chamber. The deposition was performed under a base pressure of 10−7 Torr with the substrate for sample loading heated to 50 °C and kept rotating at 50 rpm during evaporation for uniform coating. The evaporation rate was 1 Å s−1, and the Cu loading was 0.2 mg cm−2.
Preparation of PTFE/Cu/PVAQAx and CP/Cu/PVAQAx
6.0 g of polyvinyl alcohol (PVA) was added to 60 mL of deionized water under stirring for 8 h at 70 °C. Then, 0.073 mol of glycidyl trimethylammonium chloride was added to the above solution and fully stirred for 12 h. Afterwards, the solution was fed into a 20 mL syringe equipped with a stainless-steel nozzle, connected to a high-voltage power supply set at 15 kV. The solution was then sprayed at a rate of 7 mL min−1 from an N-20 needle placed 10 cm away from the aluminum foil-coated cylinder receiver rotating at 500 rpm. Prior to that, the composite of PTFE/Cu or CP/Cu was mounted onto the aluminum foil. During electrospinning, the chamber temperature was kept at 40 °C, while the humidity was maintained about 20%. After peeling off from the aluminum foil, the electrospun PTFE/Cu/PVAQA54 and CP/Cu/PVAQA54 electrodes were obtained after heating to 140 °C for 4 h. For comparison, PVAQA13 and PVAQA33 were synthesized following the same procedure except 0.034 and 0.046 mol of glycidyl trimethylammonium chloride were used, respectively.
Preparation of PTFE/Cu/PVAPA54
6.0 g of polyvinyl alcohol (PVA) was dissolved into 60 mL of deionized water under stirring for 8 h at 70 °C. Then, 0.073 mol of propylene oxide and 1.2 mL of 1 M HCl were added to the above solution and stirred for 4 h at 70°C. Afterwards, the solution was used as the precursor for electrospinning following the same procedure as that of PTFE/Cu/PVAQA54.
Preparation of PTFE/Cu/PVA98
6 g of polyvinyl alcohol with a degree of hydroxylation of 98% (PVA98) was dissolved into 60 mL of deionized water under stirring for 8 h at 70 °C. Afterwards, the solution was used as the precursor for electrospinning following the same procedure as that of PTFE/Cu/PVAQA54. For comparison, PTFE/Cu/PVA91 and PTFE/Cu/PVA78 were synthesized following the same procedure except 6.3 g of PVA91 and 6.7 g of PVA78 were used, respectively. The polyvinyl alcohol with different degree of hydroxylation used in this paper were purchased from Shanghai Macklin Biochemical Co., Ltd.
Preparation of PTFE/Cu/PEO
6.0 g of polyethylene oxide (PEO) was dissolved into 60 mL of deionized water under stirring for 8 h at 70 °C. Afterwards, the solution was used as the precursor for electrospinning following the same procedure as that of PTFE/Cu/PVAQA54.
Structural characterizations
The surface morphology and micro-structure of all samples were observed using a dual-beam electron microscope (SEM, FEI Scios). The chemical functionality was examined by Fourier-transform infrared spectroscopy (FT-IR, Nicolet iS50). The contact angle was measured by precise automatic contact angle measurement instrument (JC2000DM) at room temperature.
In situ ATR-SEIRAS measurements
The ATR-SEIRAS measurements were conducted in a two-compartment spectro-electrochemical cell comprising three electrodes, including a Si prism deposited with a layer of 150 nm Au film as the working electrode (The deposition of Au film was performed under a base pressure of 10−7 Torr with the substrate for sample loading heated to 300 °C and kept rotating at 50 rpm during evaporation. The evaporation rate was 0.5 Å s−1.), a platinum-wire as the counter electrode, and a standard Ag/AgCl (in saturated KCl solution) electrode as the reference. The catalyst ink containing 0.8 mg Cu nanoparticles (scraped off from the Cu-evaporated carbon paper) and 0.2 mg carbon black was casted onto the above-prepared silicon/Au prism covering a rounded area of 1.2 cm in diameter. Subsequently, a polymeric overlayer of desired functionality with the thickness of around 80 μm was deposited onto the prism surface via electrospinning. All electrochemical tests were carried out in CO2-satuated 0.1 M KHCO3 aqueous solution with a constant flow of CO2. Before the electrochemical measurements, the working electrode was initially activated by repeatedly running CVs from −0.1 to −1.1 VRHE at a scanning rate of 5 mV s−1 until the system stabilized. Then, the cathodic potential was swept from −0.4 to −2.0 (V vs. RHE) with an interval of 0.2 V. The cell potentials were recorded without iR correction. All the ATR-SEIRAS spectra were recorded by a Fourier Transform Infrared Spectrophotometer (FT-IR, Nicolet iS50, Supplementary Fig. 20) equipped with a mercury cadmium telluride detector.
In situ electrochemical Raman measurements
Raman spectra were collected using a commercially available Raman flow cell (Gaoss Union CO31-3) with the open-space microscope setup as illustrated in Supplementary Fig. 21. For in situ measurements, Raman spectra were acquired with a 633 nm excitation laser and 800 grooves/mm diffraction grating. The power of the laser was kept at 5 mW. Each Raman spectrum took four repetitions (30 s per acquisition) using a low numerical aperture (NA) objective (SOPTOP, 50×). This procedure ensured the reproducibility of Raman signals through the averaging of a large number of molecules in the excitation volume. During the measurement, a pump was utilized to circulate the electrolyte, maintained at 10 mL min−1, while the CO2 flow rate was controlled at 30 mL min−1 with a mass flow controller. The working electrodes were the aforementioned synthesized CP/Cu/polymer (Cu loading was 0.2 mg cm−2, and the thickness of the deposited polymer layer was 80 μm.), while graphite electrodes and Ag/AgCl were employed as the counter and reference electrode, respectively. 0.1 M KHCO3 was used as the catholyte in all tests. We perform at least three parallel tests at independent positions to ensure the uniformity of the spatial distribution and avoid unreliable data caused by local over-enhancement.
Electrochemical measurements in alkaline flow cell
In this study, electrochemical measurements in a flow cell (Supplementary Fig. 58) were conducted using a three-electrode configuration and an electrochemical workstation (CHI660E) via galvanostatic methods. The working electrode comprised a PTFE/Cu/polymer structure. Given the poor conductivity of PTFE, copper tapes were applied around the perimeter of the working area (1 × 1 cm2) prior to polymer deposition to enhance the current collection efficiency (Supplementary Fig. 4). The catholyte and anolyte chambers were separated by an anion exchange membrane (Fumasep FAB-PK-130, thickness: 130 μm, size: 2 × 2 cm2). The membrane needs to be activated in a 1 M KOH solution for more than 8 h before use. Gas and catholyte were separated by the gas diffusion electrode. A nickel foam of 0.8 mm in thickness was used as the anode. The Ag/AgCl reference was plugged into the center of the catholyte chamber through a top-drilled hole. Silicone gaskets with 1 × 1 cm2 window were placed to enable adequate sealing of each chamber. The high-purity CO2 was flowed into the gas chamber with a constant flow rate of 30 cm3 min−1 monitored by a digital mass flow controller (Horiba). The electrochemical measurements were carried out in 1 M KOH with a constant flow rate of 20 mL min−1 by using a dual-channel peristaltic pump. The pH value of the 40 mL of 1 M KOH electrolyte exhibited a negligible change from 14.0 to 13.8 as the current density increased from 100 to 500 mA cm−2 over an approximately 80-min testing period (Supplementary Fig. 59). For flow cell studies, the resistance between the working and reference electrodes was measured using the electrochemical workstation. Specifically, the procedure involved: (1) determining the voltage at various current densities through the galvanostatic method; (2) inputting these voltage values into the iR compensation program of the workstation to calculate the resistance. 85% ohmic correction on the flow cell data was based on the resistance between the working electrode and the reference electrode, which is typically in the range between 1 to 3 ohms depending on the distance between the two electrodes, the electrolyte concentration, and the salt precipitation. All the potentials were converted to RHE, according to the equation: E (vs RHE) = E (vs Ag/AgCl) + i × (0.85 × R) + 0.059 × pH + 0.198 (Ag/AgCl vs RHE). To enhance the measurement accuracy, we calibrated the reference electrode before each testing. Specifically, we employed the open-circuit potential method to measure the potential difference between the reference electrode and a standard reference electrode. This allowed us to determine the true potential of the reference electrode, which was subsequently incorporated into the potential calculations for correction. The gas products were quantitatively analyzed using gas chromatography (Agilent 7890B). The liquid products were collected and quantitatively analyzed by 1H NMR spectroscopy with H2O suppression. Specifically, 400 µL of the electrolyte was collected and mixed with 100 µL D2O and 50 µL dimethyl sulfoxide (20 mM), which was used as the internal standard.
MEA measurements using pure water as the anolyte
In the pure-water MEA tests, the CP/Cu/PVAQA54 cathode and IrO2/Ti anode were separated by an anion-exchange membrane (Sustainion X37-50 grade 60, thickness: 50 μm, size: 2.5 × 2.5 cm2). The membrane needs to be soaked in a 1 M KOH solution for at least 24 h for full activation. Before use, the membrane was repeatedly soaked in deionized water for three times, each lasting for 5 min. The gas flow channel was supplied with 35 cm3 min−1 humidified CO2 while the anode was circulated with pure water with a flow rate of 2 mL min−1. The cell voltages were recorded without iR correction. To prepare the IrO2/Ti anode, a Titanium mesh (2.5 × 2.5 cm2) was treated in oxalic acid (0.5 mol L−1) for 1 h to remove the surface oxide layer. After washing with water and ethanol, the titanium mesh was soaked in IrCl3/isopropanol solution (90 mg/27 mL) containing 10% HCl for 5 min, and then dried at 100 °C for 10 min and calcinated in air at 500 °C for 30 min. The soaking procedure was repeated until the loading of IrO2 on the titanium mesh reached 1 mg cm−2. Finally, the anode was soaked several times in the Sustainion solution (5 wt%) and dried before use.
Faradaic efficiency of gas products
FEa: Faradaic Efficiency of the product a
\(v\): CO2 gas flow rate (L s−1)
Ca: Volumefraction of the product a detected by GC
\(A\): Geometric area of the electrode (cm2)
Vm: Molar Volume (22.4 L mol−1)
Za: Electrons transferred for reduction to product a
\(F\): Faradaic Constant (C mol−1)
jtotal: Total current density during CO2 bulk electrolysis (A cm−2)
Faradaic efficiency of liquid products
in which Nx is the number of electrons transferred for liquid product x, \(F\) is the Faradaic constant, cx is the concentration of liquid product x determined by 1H NMR, Vx is the volume of the electrolyte and Qtotal is the total charge.
Partial current density
Ionic conductivity
in which \(L\) is the thickness of the polymers, \(S\) is the contact area, and \(R\) is the charge transfer resistance.
Double-layer capacitance
in which \({S}_{{\mbox{cv}}}\) is the area enclosed by the \({\mbox{CV}}\) curve, \(v\) is the scan rate, \(U\) is the voltage window, and \({S}_{{\mbox{e}}}\) is the geometrical area of electrode.
CO2 conversion
in which \({\mbox{CE}}\) is the carbon efficiency, \({f}_{x}\) is the molar rate of formation for product \(x\), \(A\) is the electrode’s geometric area, \({f}_{{{\mbox{CO}}}_{2}}\) is the CO2 molar flow rate, \({Q}_{{\mbox{total}}}\) is the total charge, \({{\mbox{FE}}}_{x}\) is the FE of product \(x\), \(F\) is the Faradaic constant, \({N}_{x}\) is the number of electrons transferred for product \(x\), and \(t\) is the electrolysis reaction time.
Full-cell energy efficiency
The full-cell energy efficiency was determined using the equation with OER as the anodic reaction (\({E}_{{\mbox{OER}}}^{{\theta }}\) = 1.23 V vs. RHE).
in which \({E}_{{\mbox{OER}}}^{{\theta }}\) and \({E}_{x}^{{\theta }}\) are the thermodynamic potentials (versus RHE) for OER and eCO2R to product x, respectively, \({{\mbox{FE}}}_{x}\) is the FE of product x, \({{\mbox{EE}}}_{{\mbox{full}}-{\mbox{cell}}}\) is the measured cell voltage.
AIMD calculations
The modeled systems consisting of 55 water molecules, periodic polymers and two CO molecules on Cu(111) (Supplementary Figs. 41–43) were used in the current work to simulate the electrochemical environment in aqueous solution. The slab of Cu (111) was constructed using periodic 4 × 6 or 4 × 8 unit cells with three atomic layers. The lattice parameter was optimized to be 3.57 Å for bulk Cu. All the water molecules, CO molecules, polymers and the top layers of Cu substrate were allowed to relax, whereas the bottom two layers were fixed during all simulations.
All AIMD calculations were performed within the hybrid Gaussian and plane waves (GPW) framework69 as implemented in the CP2K/Quickstep electronic structure and molecular dynamics software package. The expansion of the electron density in the auxiliary plane wave basis was truncated using an energy cutoff of 400 Ry. The core electrons were modeled by Goedecker–Teter–Hutter (GTH) pseudopotentials70,71 with 11, 4, 6, 5, and 1 valence electrons for Cu, C, O, N, and H, respectively. The revised formulation of the Perdew-Burke-Ernzerhof density functional approximation (RPBE)72 was applied in conjunction with DFT-D3 dispersion corrections73, which has been proven suitable for metal-water interface and adsorption simulations74,75,76. Traditional matrix diagonalization was used to solve the Kohn−Sham equations and the electronic structure convergence was further accelerated by applying Fermi−Dirac smearing using an electronic temperature of 1000 K as well as the efficient ELPA library of diagonalization routines77.
In our analysis of H bond counts, we have set specific criteria for bond length and bond directionality. The bond length standard is defined as the distance between the hydrogen atom and the electronegative atom (namely oxygen or nitrogen) being in the range of 1.5 to 2.5 Å. As for the bond directionality, the standard is established as the angle formed between the donor atom (the atom to which the hydrogen is covalently bonded), the hydrogen atom itself, and the acceptor atom (the electronegative atom with a lone pair of electrons) being greater than 120 degrees. we analyzed the water dipole orientations by calculating the average \(\overline{\cos }({{{\rm{\theta }}}})\) distribution profiles along the AIMD trajectories78, where θ represents the angle between the water dipole vector (\(\widetilde{d}\)) and the surface normal (\(\widetilde{z}\)), as illustrated in the inset of Supplementary Fig. 48. Both the H bond calculations and water dipole orientation analyses are based on 10 ps AIMD trajectories. The initial 3 ps are considered the equilibration process, and data from the last 7 ps are utilized to analyze the average distribution profiles.
The slow-growth approach59 within the constrained molecular dynamic framework, were performed with constrained reaction coordinate of the C–C bond length, i.e., collective variables (CV), using the SHAKE and RATTLE algorithms79. The increment of 0.0008 Å fs−1 for CV (dcoord) is set to drive the chemical reaction. The mean constraint force (Fc), required to constrain the atoms at each increment value, was evaluated from individual AIMD trajectory. To determine the free energy profile of the process, the potential of mean force (PMF) was calculated by integrating Fc over the reaction coordinate80:
To gain the constant potential results, our recently developed CK2P dynamic constant potential module (CDCPM) method was adopted. This model uses several layers of explicit water molecules in conjunction with implicit solution to solvate the reaction species. The electrons are coupled with a fictitious potentiostat81 so that the Fermi level of the system fluctuates around a constant and the number of electrons evolves following the grand-canonical distribution at the preset electrode potential. The net electronic charges are balanced by the ionic charges in the implicit solution, keeping the system charge neutral.
Data availability
All data supporting the findings of this study are available in the article and its Supplementary Information. Source data are provided with this paper.
References
Zhao, Y. et al. Conversion of CO2 to multicarbon products in strong acid by controlling the catalyst microenvironment. Nat. Synth. 2, 403–412 (2023).
Dinh, C.-T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
García de Arquer, F. P. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 367, 661–666 (2020).
Lees, E. W. et al. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat. Rev. Mater. 7, 55–64 (2021).
Wang, M. et al. Hydrophobized electrospun nanofibers of hierarchical porosity as the integral gas diffusion electrode for full-pH CO2 electroreduction in membrane electrode assemblies. Energy Environ. Sci. 16, 4423–4431 (2023).
Wakerley, D. et al. Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers. Nat. Energy 7, 130–143 (2022).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Li, W. et al. Bifunctional ionomers for efficient co-electrolysis of CO2 and pure water towards ethylene production at industrial-scale current densities. Nat. Energy 7, 835–843 (2022).
Wang, Z. et al. Localized alkaline environment via in Situ electrostatic confinement for enhanced CO2-to-ethylene conversion in neutral medium. J. Am. Chem. Soc. 145, 6339–6348 (2023).
Chen, Y. et al. Efficient multicarbon formation in acidic CO2 reduction via tandem electrocatalysis. Nat. Nanotechnol. 19, 311–318 (2024).
Iglesias van Montfort, H. P. et al. Non-invasive current collectors for improved current-density distribution during CO2 electrolysis on super-hydrophobic electrodes. Nat. Commun. 14, 6579 (2023).
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Moss, A. B. et al. In operando investigations of oscillatory water and carbonate effects in MEA-based CO2 electrolysis devices. Joule 7, 350–365 (2023).
Wheeler, D. G. et al. Quantification of water transport in a CO2 electrolyzer. Energy Environ. Sci. 13, 5126–5134 (2020).
Grosse, P. et al. Dynamic transformation of cubic copper catalysts during CO2 electroreduction and its impact on catalytic selectivity. Nat. Commun. 12, 6736 (2021).
Zhang, Z. et al. Two ships in a nottle” design for Zn-Ag-O catalyst enabling selective and long-lasting CO2 electroreduction. J. Am. Chem. Soc. 143, 6855–6864 (2021).
Sassenburg, M. et al. Zero-gap electrochemical CO2 reduction cells: challenges and operational strategies for prevention of salt precipitation. ACS Energy Lett. 8, 321–331 (2023).
Garg, S. et al. How alkali cations affect salt precipitation and CO2 electrolysis performance in membrane electrode assembly electrolyzers. Energy Environ. Sci. 16, 1631–1643 (2023).
Endrodi, B. et al. Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolyzers. Nat. Energy 6, 439–448 (2021).
Disch, J. et al. High-resolution neutron imaging of salt precipitation and water transport in zero-gap CO2 electrolysis. Nat. Commun. 13, 6099 (2022).
Chen, X. et al. Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes. Nat. Catal. 4, 20–27 (2020).
Li, F. et al. Molecular tuning of CO2-to-ethylene conversion. Nature 577, 509–513 (2020).
Song, D. et al. Electrochemical CO2 reduction catalyzed by organic/inorganic hybrids. eScience 3, 100097 (2023).
Coskun, H. et al. Biofunctionalized conductive polymers enable efficient CO2 electroreduction. Sci. Adv. 3, 1700686 (2017).
Wei, X. et al. Highly selective reduction of CO2 to C2+ hydrocarbons at copper/polyaniline interfaces. ACS Catal. 10, 4103–4111 (2020).
Cheng, J. et al. Proton shuttling by polyaniline of high Bronsted basicity for improved electrocatalytic ethylene production from CO2. Angew. Chem. Int. Ed. 62, e202312113 (2023).
Wang, J. et al. Selective CO2 electrochemical reduction enabled by a tricomponent copolymer modifier on a copper Surface. J. Am. Chem. Soc. 143, 2857–2865 (2021).
Deng, W. et al. Crucial role of surface hydroxyls on the activity and stability in electrochemical CO2 reduction. J. Am. Chem. Soc. 141, 2911–2915 (2019).
Xie, J. et al. Metal-free fluorine-doped carbon electrocatalyst for CO2 reduction outcompeting hydrogen evolution. Angew. Chem. Int. Ed. Engl. 57, 9640–9644 (2018).
Kim, C. et al. Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings. Nat. Energy 6, 1026–1034 (2021).
Zhao, Y. et al. Industrial-current-density CO2-to-C2+ electroreduction by anti-swelling anion-exchange ionomer-modified oxide-derived Cu nanosheets. J. Am. Chem. Soc. 144, 10446–10454 (2022).
Stern, O. Zur Theorie Der Elektrolytischen Doppelschicht. Elektrochem. Angew. Phys. Chem. 30, 508–516 (1924).
Helmholtz, H. Ueber einige Gesetze der Vertheilung elektrischer Ströme in körperlichen Leitern, mit Anwendung auf die thierisch‐elektrischen Versuche. Ann. Phys. 165, 211–233 (1853).
Wu, J. Understanding the electric double-layer structure, capacitance, and charging dynamics. Chem. Rev. 122, 10821–10859 (2022).
Grahame, D. C. The electrical double layer and the theory of electrocapillarity. Chem. Rev. 41, 441–501 (1947).
Feaster, J. T. et al. Understanding the Influence of [EMIM]Cl on the suppression of the hydrogen evolution reaction on transition metal electrodes. Langmuir 33, 9464–9471 (2017).
Fan, J. et al. Immobilized tetraalkylammonium cations enable metal-free CO2 electroreduction in acid and pure water. Angew. Chem. Int. Ed. Engl. 63, e202317828 (2024).
She, X. et al. Pure-water-fed, electrocatalytic CO2 reduction to ethylene beyond 1,000 h stability at 10 A. Nat. Energy 9, 81–91 (2024).
Kahn, A. Fermi level, work function and vacuum level. Mater. Horiz. 3, 7–10 (2016).
Zhu, S. et al. Direct observation on reaction intermediates and the role of bicarbonate anions in CO2 electrochemical reduction reaction on Cu surfaces. J. Am. Chem. Soc. 139, 15664–15667 (2017).
Zheng, M. et al. Electrocatalytic CO2-to-C2+ with ampere-level current on heteroatom-engineered copper via tuning *CO intermediate coverage. J. Am. Chem. Soc. 144, 14936–14944 (2022).
Wei, D. et al. Decrypting the controlled product selectivity over Ag-Cu bimetallic surface alloys for electrochemical CO2 reduction. Angew. Chem. Int. Ed. Engl. 62, e202217369 (2023).
Perez-Gallent, E. et al. Spectroscopic observation of a hydrogenated CO dimer intermediate during CO reduction on Cu(100) electrodes. Angew. Chem. Int. Ed. 56, 3621–3624 (2017).
Firet, N. J. & Smith, W. A. Probing the reaction mechanism of CO2 electroreduction over Ag films via operando infrared spectroscopy. ACS Catal. 7, 606–612 (2016).
Wang, P. et al. Boosting electrocatalytic CO2-to-ethanol production via asymmetric C-C coupling. Nat. Commun. 13, 3754 (2022).
Li, G., Du, X., Qin, L. & Liu, J. Highly efficient removal of fluorine from groundwater by membrane capacitive deionization. Environ. Eng. Sci. 39, 352–363 (2022).
Hossain, M. D. et al. Reaction mechanism and kinetics for CO2 reduction on nickel single atom catalysts from quantum mechanics. Nat. Commun. 11, 2256 (2020).
Yan, H.-M. et al. Fast transformation of CO2 into CO via a hydrogen bond network on the Cu electrocatalysts. J. Phys. Chem. C 126, 7841–7848 (2022).
Liu, S. et al. Elucidating the electrocatalytic CO2 reduction reaction over a model single-atom nickel catalyst. Angew. Chem. Int. Ed. Engl. 59, 798–803 (2020).
Cheng, T. et al. Reaction mechanisms for the electrochemical reduction of CO2 to CO and formate on the Cu(100) surface at 298 K from quantum mechanics free energy calculations with explicit water. J. Am. Chem. Soc. 138, 13802–13805 (2016).
Yao, K. et al. Mechanistic insights into OC-COH coupling in CO2 electroreduction on fragmented copper. J. Am. Chem. Soc. 144, 14005–14011 (2022).
Shao, F. et al. In situ spectroelectrochemical probing of CO redox landscape on copper single-crystal surfaces. Proc. Natl Acad. Sci. USA 119, 2118166119 (2022).
Ooka, H. et al. Competition between hydrogen evolution and carbon dioxide reduction on copper electrodes in mildly acidic media. Langmuir 33, 9307–9313 (2017).
Liu, Z. et al. Interfacial water tuning by intermolecular spacing for stable CO2 electroreduction to C2+ products. Angew. Chem. Int. Ed. Engl. 62, e202309319 (2023).
Zhang, H. et al. Promoting Cu-catalysed CO2 electroreduction to multicarbon products by tuning the activity of H2O. Nat. Catal. 6, 807–817 (2023).
Gao, Z. et al. Promoting electrocatalytic CO2 reduction via anchoring quaternary piperidinium cations onto copper electrode. Electrochim. Acta 458, 142509 (2023).
Wang, Y. et al. Strong hydrogen-bonded interfacial water inhibiting hydrogen evolution kinetics to promote electrochemical CO2 reduction to C2+. ACS Catal. 14, 3457–3465 (2024).
Fan, M. et al. Cationic-group-functionalized electrocatalysts enable stable acidic CO2 electrolysis. Nat. Catal. 6, 763–772 (2023).
Martyna, G. J. et al. Nosé–Hoover chains: the canonical ensemble via continuous dynamics. J. Chem. Phys. 97, 2635–2643 (1992).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985).
Bagger, A. et al. Electrochemical CO reduction: a property of the electrochemical interface. J. Am. Chem. Soc. 141, 1506–1514 (2019).
Li, J. et al. Hydrogen bonding steers the product selectivity of electrocatalytic CO reduction. Proc. Natl Acad. Sci. USA 116, 9220–9229 (2019).
Rastogi, A. et al. in Thermodynamics-Physical Chemistry of Aqueous Systems (ed. Moreno-Piraján, J. C.) 351–364 (IntechOpen, 2011).
Chen, L. et al. Spatial-confinement induced electroreduction of CO and CO2 to diols on densely-arrayed Cu nanopyramids. Chem. Sci. 12, 8079–8087 (2021).
Kronberg, R. & Laasonen, K. Reconciling the experimental and computational hydrogen evolution activities of Pt(111) through DFT-based constrained MD simulations. ACS Catal. 11, 8062–8078 (2021).
Chen, L. et al. Carbene ligands enabled C–N coupling for methylamine electrosynthesis: a computational study. Energy Fuels 36, 7213–7218 (2022).
Zhuansun, M. et al. Promoting CO2 electroreduction to multi-carbon products by hydrophobicity-induced electro-kinetic retardation. Angew. Chem. Int. Ed. 62, e202309875 (2023).
Xue, L. et al. Dual-role of polyelectrolyte-tethered benzimidazolium cation in promoting CO2/pure water co-electrolysis to ethylene. Angew. Chem. Int. Ed. 62, e202309519 (2023).
Lippert, B. et al. A hybrid Gaussian and plane wave density functional scheme. Mol. Physi. 92, 477–488 (1997).
Goedecker, S., Teter, M. & Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 54, 1703 (1996).
Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 114, 145–152 (2005).
Hammer, B. et al. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413 (1999).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. chem. Phys. 132, 154104 (2010).
Sakong, S. & Groß, A. The electric double layer at metal-water interfaces revisited based on a charge polarization scheme. J. Chem. Phys. 149, 084705 (2018).
Kronberg, R. & Laasonen, K. Coupling surface coverage and electrostatic effects on the interfacial adlayer–water structure of hydrogenated single-crystal platinum electrodes. J. Phys. Chem. C 124, 13706–13714 (2020).
Sakong, S. et al. The structure of water at a Pt (111) electrode and the potential of zero charge studied from first principles. J. Chem. Phys. 144, 194701 (2016).
Marek, A. et al. The ELPA library: scalable parallel eigenvalue solutions for electronic structure theory and computational science. J. Phys.: Condens. Matter 26, 213201 (2014).
Li, P. et al. Revealing the role of double-layer microenvironments in pH-dependent oxygen reduction activity over metal-nitrogen-carbon catalysts. Nat. Commun. 14, 6936 (2023).
Ryckaert, J. P. et al. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Carter, E. A. et al. Constrained reaction coordinate dynamics for the simulation of rare events. Chem. Phys. Lett. 156, 472–477 (1989).
Bonnet, N. et al. First-principles molecular dynamics at a constant electrode potential. Phys. Rev. Lett. 109, 266101 (2012).
Acknowledgements
This work is supported by National Natural Science Foundation of China (Nos. 22072101 (Y.P.), 22075193 (Z.D.) and 22109099 (L.X.)), Natural Science Foundation of Jiangsu Province (No. BK20211306 (Y.P.) and BK20240807 (J.C.)), and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions. This research is also supported by the Australian Research Council through FT190100636 (Y.J.). Molecular modeling for this research were undertaken with the assistance of supercomputing resources provided by the Phoenix HPC service at the University of Adelaide and the National Computational Infrastructure (NCI), which is supported by the Australian Government.
Author information
Authors and Affiliations
Contributions
Y.P. conceived the idea and led the project. J.C. designed and performed the majority of the experiments and obtained most of the results, including material synthesis, characterization, electrochemical, and in situ FT-IR tests. L.C., L.J., and Y.J. carried out the DFT calculations. Y.Z. performed the polymers synthesis and electrospinning experiments. M.W. carried out the MEA tests. Z.Z. and Y.L. helped draw the mechanism diagram. Z.W. took the SEM images. M.M. performed the in situ Raman tests. Z.D. and Y.P. helped to polish the paper. Z.D., M.M., L.X., W.H. (Wei Hua), D.S., W.H. (Wenxuan Huo), W.Y., F.L., and Y.J. devoted to the data analysis. J.C., L.C., Z.D., Y.J., and Y.P. wrote the paper. All the authors have discussed the results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, J., Chen, L., Zhang, Y. et al. Metal-organic double layer to stabilize selective multi-carbon electrosynthesis. Nat Commun 16, 3743 (2025). https://doi.org/10.1038/s41467-025-59025-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59025-5
