
Abstract
Certain biological channels exhibit remarkable selectivity, effectively distinguishing between competing cations. If artificial membranes could achieve similar precision in differentiating competing ions from Li+, it could advance sustainable technologies in lithium extraction. In this study, we present a covalent organic framework (COF) membrane featuring a randomly oriented structure that enables selective separation of major competing ions from Li+. The random orientation results in narrow pores, which impart size-based selectivity among alkaline ions. Additionally, the COF incorporates sulfonic groups that preferentially bind to Na+ and K+, facilitating their transport while retaining Li+. These synergistic mechanisms endow the membrane with a selectivity beyond detection limit for K+ and Na+ over Li+. When driven by an electrical potential, the ion flux through the membrane is enhanced by over an order of magnitude. Notably, the membrane also permits the transport of Mg2+ and Ca2+ while still rejecting Li+, leveraging differences in their ion mobility. This work should advance the design and construction of biomimetic materials for the extraction of valuable species from seawater and other aqueous sources.
Similar content being viewed by others

Introduction
Biological ion channels transport target ions with high selectivity, which is crucial for the generation of action potential and many other psychological processes. Driven by both curiosity and potential practical applications in metal extraction, fit-for-purpose water treatment, and therapeutic agents, great efforts have been made to fabricate materials consisting of artificial ion channels in the last decades1,2,3,4,5. For example, to extract lithium from seawater6,7, a membrane that allows the separation of Li+ from other cations, particularly alkaline ions such as Na+, is highly demanded. For salt-lake lithium extraction, although Li+/Mg2+ separation remains important in current industrial processes8,9,10,11, Li+/Na+ and Li+/K+ separation is crucial for direct and greener lithium extraction in the future industry12,13. In principle, to sieve out Li+ from other alkaline ions, the membrane should either allow Li+ to pass but retain Na+ and K+, or allow Na+ and K+ to pass but retain Li+. However, both separation methods remain highly challenging. For the former method, the Li+/Na+ and Li+/K+ selectivity is demanded to be far larger than the Na+/Li+ and K+/Li+ ratio in seawater, respectively, i.e., 104 and 103, such that Li+ can be sieved out through a single-pass separation process. Otherwise, repeated separation processes are required14. For the latter method, the Na+/Li+ and K+/Li+ selectivity should be as high as possible, because unlike the former method, any Li+ leaked to the effluent cannot be recycled by multi-pass processes. Still, the latter method has an advantage. It may allow the separation of Li+ from all other competing ions (K+, Na+, Mg2+, Ca2+) with one membrane using electrodialysis since Li+ has the lowest electrical mobility.
In Nature, biological ion channels use a range of strategies to improve the ion selectivity. First, biological ion channels have small channel size (~0.3 nm) that can hinder the transport of larger hydrated ions. Second, their channels are lined with functional groups to promote the flux of target ions. For example, the biological potassium channels have a size of ~0.3 nm, and are lined with molecular recognition groups, showing almost perfect selectivity against other alkaline ions including Li+. To date, many artificial materials with small channel size and proper functional groups have been constructed for ion sieving5,15,16,17,18,19,20,21,22,23. Many of these materials can transport Li+ faster than Na+ and K+17,18,23,24,25,26. Yet, the selectivity was typically a few to a few tens, dramatically lower than the Na+/Li+ and K+/Li+ ratio in seawater. A few exceptions were demonstrated23, showing a selectivity up to 103, but were based on micro-sized materials and lacked the capability for scaled-up membrane separation. Alternatively, another series of membranes were constructed, which can impede the transport of Li+ but allow Na+ and/or K+ transport16,19,22,27,28. Still, the Na+/Li+ and K+/Li+ selectivity was also only up to a few hundreds, far from the required selectivity.
In bulk water, K+ and Na+ transport much faster than Li+ due to their smaller hydration sizes. Intuitively, realizing high Na+/Li+ and K+/Li+ selectivity might be easier than doing so for Li+/Na+ and Li+/K+. Indeed, we here demonstrate a membrane consisting of randomly oriented covalent organic frameworks (COFs) that exhibits high K+/Li+ and Na+/Li+ selectivity, on par with some biological ion channels. We chose COFs because they are widely used for molecular or ion separation11,29,30,31,32 due to their high porosity, tunable structure and chemistry, and the ease to form defect-free membranes. The high porosity is important for high flux. The tunable structure and chemistry allow versatile modulation of the ion selectivity. Yet, COFs suffer from the challenge of large channel/pore size in replicating the success of biological ion channels. COFs typically consist of channels or pores with a size > 0.5 nm, much larger than the biological ion channels and also much larger than alkaline ions30. In this work, we intentionally disrupted the orientation of the COFs, creating a membrane consists of randomly oriented microcrystalline COFs. At the crystalline boundaries, the channels are highly distorted and numerous necks with a size <0.32 nm are formed, forcing ions to at least partly dehydrate when entering the channel, and exerting high steric hinderance to Li+ which has a large hydration size. Furthermore, our COFs were functionalized with dense sulfonyl groups that can strongly attract target cations like biological ion channels do. Density functional theory calculation suggests that the sulfonyl groups have higher affinity to Na+ and K+ than to Li+18, consistent with previous results. In addition, these groups prefer the binding with dehydrated Na+, dehydrated K+, and hydrated Li+. This would strongly facilitate the transport of the already-dehydrated Na+ and K+, but would not do so for the dehydrated Li+. As a result, the membrane allows the transport of K+ and Na+ while completely blocking Li+. Interestingly, when applying an external electrical potential, Mg2+ and Ca2+ can also transport through the membrane due to their bivalency. This allows us to sieve out all major competing ions (K+, Na+, Mg2+, Ca2+) from Li+ with high selectivity using a moderate electrical potential.
Results
Preparation of the COF membrane
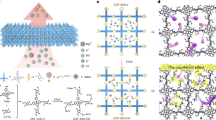
To synthesize the membrane, we first grew vertically aligned layered double hydroxide (LDH)33,34 nanosheets on an anodic aluminum oxide (AAO) substrate. Then the nanosheets were functionalized with –NH2 groups to guide the growth of COFs (Fig. 1a–c)35. The COF used in our work was in-situ synthesized from 1,3,5-triformylphloroglucinol (Tp) and 2,5-diaminobenzenesulfonic acid (Pa-SO3H) using single-phase hydrothermal method, having rich sulfonyl groups that can strongly attract target cations (Fig. 1d). The COF is termed as TpPa-SO3H, and the composite membrane consisting of randomly orientated COF is termed as r-TpPa-SO3H membrane (see Methods section for the membrane fabrication in detail). Figure 1e–h shows that after COF synthesis, the voids between LDH nanosheets (Fig. 1e, f) were filled, resulting in a smooth and defect-free surface (SEM images in Fig. 1g, h). Importantly, before and after COF synthesis, the change on the membrane thickness was insignificant (compare Fig. 1f and h), suggesting that the COF mostly grew between the LDH nanosheets. During the reaction, the structure of the LDH was kept stable (Figs. S1 and S2). In this work, the thickness of the COF was controlled to be around 6 µm unless otherwise specified, by controlling the height of the LDH nanosheet layer (see Methods section for detail). The mass ratio of COFs and LDH was calculated to be 3.94:1 (Table S1). Raman spectrum of the membrane exhibits the typical signals of the vibration of sp3-hybridized amorphous C atoms and the in-plane stretching of sp2-hybridized C atoms (Fig. S3). The Fourier transform infrared spectra also confirmed the successful synthesis of r-TpPa-SO3H COF (Fig. S4), while the X-ray diffraction (XRD) pattern shows the characteristic (100) peak of the COF36 (Fig. 1i).

a Schematic illustration of the fabrication process. Vertically aligned LDH nanosheets were grown on AAO substrate to disrupt the orientation of the COFs. b, c The COFs are randomly oriented and sandwiched between LDH nanosheets (b), forming narrow channels that allow ion sieving (c). d Molecular structure of the tested COF having sulfonyl groups, i.e., TpPa-SO3H. Our membrane is therefore termed r-TpPa-SO3H membrane. e, f Top- and cross-view of the LDH nanosheets grown on the AAO substrate. g, h Top-view (g) and cross-view (h) of the r-TpPa-SO3H membrane, showing a relatively smooth surface and dense structure. i XRD pattern of the composite membrane. j Cross-sectional high-resolution TEM image of the r-TpPa-SO3H, showing the randomly oriented COFs. k Magnified view of the L1-L4 regions in (j), showing that the COFs are crystalline in the few-nanometer scale but randomly oriented in the larger scale. l CO2 adsorption isotherm of the membrane showing a non-porous behavior (upper) and N2 adsorption isotherm showing almost no pores (lower).
To understand the orientation of the COFs, we took cross-sectional high-resolution transmission electron microscope (TEM) images of the r-TpPa-SO3H. Figure 1j shows that in the scale of tens of nanometer scale, the TpPa-SO3H is randomly oriented. Magnified view of the L1-L4 regions labeled in Fig. 1j further shows that the TpPa-SO3H is crystalline in the few-nanometer scale (Fig. 1k). The crystalline boundaries will highly distort the COF channels, which reduces the effective pore size of the membrane (sketch in Fig. 1c). From the TEM images, we cannot accurately find out the effective pore size. However, we note that to bridge different crystalline regions, the TpPa-SO3H structure must be deformed, creating pinholes with a size of at least one atom (see e.g., Fig. S5). It is reasonable to expect that the minimum effective pore size would be in the one-atom level, e.g., smaller than or close to 0.3 nm.
It is known that TpPa-SO3H typically have a laminar structure with horizontally aligned layers. To understand the importance of the vertical LDH for the growth of randomly oriented TpPa-SO3H, we also took TEM images of a TpPa-SO3H membrane grown on ‒NH2 functionalized alumina substrate using the same COF synthesis condition as for the synthesis of r-TpPa-SO3H between LDH (see Fig. S6 for characterizations). As shown in Fig. S6, most of the TpPa-SO3H layers are indeed horizontally aligned. We term this membrane h-TpPa-SO3H. These results confirm the importance of the LDH for the random orientation of TpPa-SO3H. For our material, the vertical nanosheet templates tend to guide the growth of vertically aligned COFs on the template surface35. Therefore, the competition between the pristine horizontal orientation and the guided orientation should facilitate the formation of random orientation.
We then characterized the porous structure of the r-TpPa-SO3H with the Brunauer-Emmett-Teller (BET) method using CO2 and N2 (Fig. 1l) as the probing gas molecules. Both results show a typical non-porous behavior. This essentially means that the effective pore size of the membrane is smaller than the sizes of N2 (0.32 nm) and CO2 (0.33 nm). This is in line with our estimation based on the TEM results. Such size is comparable to that of the biological potassium channels. We therefore can expect high alkaline ion selectivity.
As a control, we also performed BET experiments for the h-TpPa-SO3H membrane. The membrane clearly showed a porous structure, and the calculated pore size was 1.7 nm (Fig. S7), consistent with the reported value36, again highlighting the importance of the vertical LDH for the randomly oriented TpPa-SO3H and the consequent small pore size. We also conducted positron annihilation spectroscopy characterization37,38 of the membrane, which suggested a free volume radius of ~0.3 nm (Fig. S8), larger than that of the r-TpPa-SO3H membrane (Fig. S8) but much smaller than BET results, probably because the free volume radius represents half of the mean distance between structural defects rather than only the pore radius38.
High K+/Li+ selectivity
We then assembled the r-TpPa-SO3H membrane into a diffusion cell made of polytetrafluoroethylene (PTFE) to measure the ion selectivity (schematic illustration in Fig. S9). Later for potential practical application, we will assess the electrodialysis performance using the same device but with external voltage applied. The feed reservoir was filled with testing solutions and the permeate reservoir with deionized (DI) water. The ion concentration of interest was measured with inductively coupled plasma mass spectrometry (ICP-MS). By monitoring the changes of ion concentration with time, we can calculate the ion flux and thus the ion selectivity.
Before evaluating the selectivity of the membrane, we conducted several control experiments. First, we measured the K+ and Li+ flux through a blank AAO substrate (Fig. 2a). The K+/Li+ selectivity was negligible (see also Fig. S10 for various feed concentrations). Second, we measured the ion selectivity of AAO substrate after growing LDH nanosheets, which was also negligible, while the flux decreased significantly compared to that of the blank AAO substrate (Figs. 2b and S11 and S12). Third, we assessed whether the device or the r-TpPa-SO3H membrane was subject to any contamination since the ion concentration in the permeate could be down to µM level and is thus sensitive to contamination. To this end, we assembled the device with the r-TpPa-SO3H membrane and filled both reservoirs with DI water. Results in Fig. 2c shows that negligible amount of K+ and no Li+ ions were detected in the permeate in 60 min.

a–c Control experiments. a, b Concentration of K+ and Li+ in the permeate in response to permeation time, showing the low K+/Li+ selectivity of AAO substrate (a) and the LDH/AAO composite membrane (b). c To confirm the absence of contamination, we used DI water as the feed solution, and found trace amount of K+ contamination and no Li+ contamination. d Permeation of K+ through the r-TpPa-SO3H membrane when the feed solution contains 0.1 M K+. e Permeation of Li+ through the r-TpPa-SO3H membrane when the feed solution contains 0.1 M Li+, showing the completely blocked Li+ transport. f Comparison of the K+ and Li+ permeation rate, summarized from (d) and (e). g Permeation of K+ and Li+ through the r-TpPa-SO3H membrane when the feed solution contains 0.1 M K+ + 0.1 M Li+, showing high K+/Li+ selectivity. h Permeation rate of the K+ and Li+ when the feed solution contains different concentrations of equimolar K+ and Li+. i Permeation rate of the K+ and Li+ for membranes with different thickness of COFs when the feed solution contains 0.1 M K+ + 0.1 M Li+. h and i suggest the robustly high K+/Li+ selectivity. Error bars in all cases represent standard deviation of the data from at least three individual experiments.
Then we filled the feed reservoir with 0.1 M KCl or 0.1 M LiCl to evaluate the K+/Li+ selectivity of the r-TpPa-SO3H membrane. Figure 2d shows that the K+ permeate concentration increased steadily with time, which is a typical Fickian diffusion behavior. For other feed concentrations, similar increasing trend was observed (Fig. S13). The Li+ permeate concentration, however, was not detected in 60 min for all feed concentrations (Figs. 2e and S14), suggesting a very high K+/Li+ selectivity (summarized in Fig. 2f). To further confirm such selectivity, we mixed 0.1 M KCl and 0.1 M LiCl in the feed reservoir. For the binary solution, the K+ concentration showed a similar increasing trend while no Li+ flux was detected (Fig. 2g). For a more comprehensive study, we then varied the concentration of binary solution from 0.1 mM to 0.1 M for both K+ and Li+. For all these concentrations, no Li+ flux was detected while the K+ flux increased monotonically with the increasing concentration (Figs. 2h and S15). To assess the long-term stability of the selectivity, we conducted a continuous test for 120 h. Fig. S16 shows that during the test, no Li+ could be detected, and the K+ flux varied moderately but kept above 0.2 mol h−1 m−2. Moreover, we varied the thickness of the r-TpPa-SO3H (thickness of the AAO not included) by controlling the height of LDH (Fig. S17). For the thickness of 7 µm and 8 µm, high K+/Li+ selectivity was also observed (Fig. S18), while the K+ flux decreased with the increase of thickness (Fig. 2i). We also tried to tune the thickness below 6 µm by further reducing the LDH layer height. In this case, many LDH nanosheets merged (Fig. S19). In addition, the surface of the LDH layer became non-uniform, making the composite COF membrane defective (Fig. S19). These result in the loss of high selectivity (Fig. S18).
Importance of the random orientation
The blockage of Li+ transport could be achieved by rejecting Li+ at the entrance of the COF pores, or by completely adsorbing them inside the pores. The latter is unwanted for separation since the adsorption will saturate over time, resulting in the loss of selectivity. The fact that our membrane can stably function during long-time test should rule out the adsorption mechanism. Nevertheless, for a verification, we measured the amounts of lithium and potassium elements adsorbed by the membrane after 24 h of immersion in the binary solution. To do this, the membrane was microwave-digested in HNO3 solution, and the resultant solution was subjected to ICP-MS test. Results in Fig. 3a show that the amount of potassium is significantly higher than that of lithium. This clearly contradicts the adsorption mechanism since otherwise lithium should be more abundant than potassium. Note that although Li+ ions do not permeate through our membrane, some of them would still adsorb on the surface of the membrane, leading to the limited amount of lithium shown in Fig. 3a. Therefore, we conclude that the high K+/Li+ selectivity should result from the rejection of the relatively large and hydrated Li+ by the narrow COF pores. This is different from previous COFs9,39,40 used for Li+ separation for which the channels were engineered to allow fast Li+ transport instead of rejection.

a We measured the K+ and Li+ content inside the membrane after permeation experiments. Li+ content was much lower than the K+ content, confirming that the blocked Li+ transport is due to the rejection by narrow channels instead of Li+ adsorption inside the membrane. b, c To confirm the importance of narrow channels, we used horizontally aligned TpPa-SO3H membrane, termed h-TpPa-SO3H membrane for the K+/Li+ separation test (b). The membrane had nanometer-sized pores and showed low selectivity (c). d–f We also used vertically aligned TpPa-SO3H membrane, termed v-TpPa-SO3H membrane for the K+/Li+ separation test. d Schematic illustration of the membrane preparation and the test device. e Schematic illustration of the ion transport direction. f The v-TpPa-SO3H membrane is impermeable to any ion. g Permeability comparison of the tested COF materials, suggesting that the importance of the narrow channels induced by the random orientation for the high selectivity. Error bars in all cases represent standard deviation of the data from at least three individual experiments.
In our work, the narrow pores are created by the randomly oriented COFs. We further demonstrate that such orientation is crucial: horizontal orientation results in large channels and the absence of ion selectivity, while vertical orientation results in too-narrow channels and the absence of ion permeability. First, we tested the ion selectivity of h-TpPa-SO3H (Fig. 3b), which allows the ion transport through the pores with a size of 1.7 nm. Expectedly, both K+ and Li+ exhibited high flux but a low selectivity, as tested with the binary solution of 0.1 M KCl and 0.1 M LiCl (Fig. 3c) as well as other concentrations (Fig. S20).
In our previous work, we demonstrated that vertically-aligned COFs (termed v-TpPa-SO3H membrane) can completely block the transport of all cations and anions except protons41 since the interlayer spacings of the COFs are smaller than common ions. In this work, we used the h-TpPa-SO3H membrane to fabricate the v-TpPa-SO3H membrane (Fig. S21) using an established method42. The h-TpPa-SO3H membrane was first embedded in epoxy resin, followed by slicing into thin sheet to obtain the v-TpPa-SO3H membrane for ion transport tests (Fig. 3d, e). For a fair comparison, the thickness (11 μm) and width (1 cm) of the h-TpPa-SO3H was chosen in a way that the cross-sectional area of the v-TpPa-SO3H membrane (0.11 mm2) was in the same order of the test area of h-TpPa-SO3H membrane (0.25 mm2). For the v-TpPa-SO3H membrane, no K+ or Li+ permeation was detected (Fig. 3f). Overall, these experiments confirm that the narrow pores created by the random orientation are important for the high K+/Li+ selectivity (Fig. 3g).
Importance of the functional groups
Although the size exclusion results in high K+/Li+ selectivity, it should also result in low K+ permeability. Therefore, the size exclusion alone cannot account for our experimental results. The molecular interaction between the COF channel and the ions may also play a vital role. To understand such interaction, we conducted density functional theory calculations of the ion-COF binding energy. Hydration water molecules were taken into consideration. For a more comprehensive exploration, we studied three alkali ions, including K+, Na+, and Li+. Obviously, the region with the lowest surface electrostatic potential in the TpPa-SO3H structure is around the sulfonic acid group (Fig. 4a), which is the most likely region for binding cations. We found that the binding energy of the Li+ ion to the COF unit (−0.56 eV) is significantly smaller than that of the Na+ and K+ ions (−0.81 and −0.77 eV, respectively) (Fig. 4b and Table S2). The changes in the atomic charge before and after the interaction between the hydrated ion and the sulfonic acid group could also reflect the strength of the interaction between the two. Only the atomic charge of Li+ ion experienced nearly no change after binding (Fig. 4c–f and Table S3), suggesting that the Li+ ions had almost no direct charge transfer with the TpPa-SO3H. The same conclusion could also be visually verified in the electron density difference maps (Fig. S22), which showed that the region where the electron density altered was mainly the hydrated shell around the Li+ ion rather than the ion itself. Furthermore, the maps of intermolecular weak interactions clearly revealed that the Li+ ion maintained its hydration structure (Fig. 4g). On the contrary, Na+ and K+ ions interacted directly with the ‒SO3H groups with their hydration shell partly removed (Fig. 4h, i). The stronger binding of Na+ and K+ ions resulted from interaction between oxygen atoms on the sulfonic group and aldehyde group, which is confirmed by the bond order analysis (Fig. S23 and Table S4). In addition, the ab-initio molecular dynamics simulations showed that Li+ ions move slower than both K+ and Na+ ions in the vicinity of the sulfonic acid groups due to the carrying of the large hydration shell (Fig. S24b). The radial distribution function curve indicated the tightness of the hydrated shell, also consistent with the order of Li+ > Na+ > K+ (Fig. S24c).

a Electrostatic potential map of TpPa-SO3H. b Binding energy between the cations and the TpPa-SO3H structural unit, showing that Li+ has the weakest affinity to TpPa-SO3H. c Variations of Hirshfeld-I atomic charge of cations after binding with TpPa-SO3H, confirming that Li+ has the weakest affinity. d–f Hirshfeld-I atomic charge maps of the three cation-COF complexes. The cations were marked by arrows. K+ and Na+ interact directly with the TpPa-SO3H while Li+ interacts with TpPa-SO3H through hydration water molecules. g–i Non-covalent interaction regions (green isosurfaces, isovalue = 0.01) between TpPa-SO3H and Li+, Na+, K+, respectively, also suggesting that K+ and Na+ interact directly with the TpPa-SO3H while Li+ interacts with TpPa-SO3H through its hydration shell.
We recall that the transition state theory can be invoked to estimate the K+/Li+ and Na+/Li+ selectivity (Sel),
where ΔE is the energy barrier difference between two competing ions (K+ and Li+, or Na+ and Li+). kB and T have their usual meaning. ΔE is the sum of the difference in dehydration energy barrier (ΔEdehy) and ion-COF binding energy (ΔEbind). Clearly, due to the size difference, the dehydration energy barrier is the highest for Li+ and lowest for K+. Since the COFs are randomly stacked, it is notoriously difficult to quantify the dehydration energy barrier. However, considering that the difference of hydration energy between these ions is in the order of 100 eV and these ions may be partially dehydrated, we can roughly estimate that the dehydration energy difference would be in the order of 10−1 to 100 eV43,44. The ΔEbind, on the other hand, can be directly derived from Fig. 4b. For K+ and Li+, ΔEbind = −0.21 eV.
Even when the ΔEdehy is down to −0.1 eV, the selectivity would be 174844. If we use a moderate ΔEdehy of −0.5 eV, the selectivity would be 2 × 1010, thus explaining the experimental selectivity which is beyond detection limit.
For Na+ and Li+, a similar conclusion can be drawn, considering that ΔEbind for Na+ and Li+ is −0.25 eV, even larger than that for K+ and Li+. This means that the Na+/Li+ selectivity should also be beyond detection limit.
Obviously, the large binding energy between K+ or Na+ and the TpPa-SO3H should facilitate the high flux K+ and Na+ transport. In addition, we should realize that the preferential interaction between the TpPa-SO3H and dehydrated Na+ and K+ ions is also highly favored for the Na+ and K+ transport, considering that hydration shell of the alkali ions is indeed expected to be partly removed when entering the small COF pores. On the contrary, the preferential interaction between the TpPa-SO3H and hydrated Li+ ions is disfavored for the transport of dehydrated Li+ ions. This would further enhance the K+/Li+ and Na+/Li+ selectivity.
To further validate the importance of the ‒SO3H groups, we prepared a membrane by replacing the monomer Pa-SO3H with Pa (p-phenylenediamine) while keeping other synthesis conditions the same. The membrane was not permeable to any ion (Fig. S25). This is consistent with our mechanism that the ions experience high energy barrier when entering the pores and the ‒SO3H preferably facilitates the transport of K+ and Na+. In summary, we can conclude that the ion selectivity is attributed to both the size exclusion effect and the ion-specific interaction with the ‒SO3H groups. The former strongly hinders the transport of Li+ but less so against Na+ and K+, and the latter promotes the transport of K+ and Na+ but not Li+.
Highly selective Na+/Li+ sieving
We further notice that Na+ also binds with the TpPa-SO3H strongly, as K+ does. This suggests the possibility to sieve out Na+ from Li+. We then tested the Na+/Li+ separation performance of our membrane. First, we conducted several control experiments, including the separation with AAO membrane (Fig. 5a), LDH/AAO composite membrane (Fig. 5b), and a blank diffusion experiment with DI water (Fig. 5c), to rule out the effect of AAO substrates, LDH, and contamination, respectively. Then we tested the diffusion of Na+ through the r-TpPa-SO3H membrane. Results in Fig. S26 and Fig. 5d–f show for a wide range of feed concentrations, the Li+ permeation was not detected while the permeation of Na+ increased steadily with the feed concentration (summarized result in Fig. 5g), showing the high selectivity. Moreover, we used a more complex mixture containing various electrolytes, i.e., KCl, NaCl, LiCl, MgCl2, and CaCl2, each with a concentration of 0.1 M, as the feed solution, and assessed the ion selectivity (Fig. 5h). High K+ or Na+ selectivity against Li+, Mg2+, and Ca2+ was observed (Fig. 5i). This is expected since hydrated divalent ions are even larger than hydrated Li+ (Table S5). We also tested seawater (composition shown in Table S6). Results in Fig. S27 still suggested the high selectivity against Li+. Overall, these results suggest that the high K+/Li+ and Na+/Li+ selectivity can be robustly kept in complex environments.

a–c Control experiments. a, b Concentration of Na+ and Li+ in the permeate in response to permeation time, showing the low Na+/Li+ selectivity of AAO substrate (a) and the LDH/AAO composite membrane (b). c To confirm the absence of contamination, we also used DI water as the feed solution, and found no Na+ or Li+ contamination. d–f Permeation of Na+ and Li+ through the membrane when the feed solution contains Na+ and Li+ with concentrations of 0.001 M (d), 0.01 M (e), and 0.1 M (f) for each ion. g Summarized permeation rate for the tested binary mixtures of various concentrations, consistently showing the high Na+/Li+ selectivity. h Permeation of various cations through the membrane when the feed solution contains all five cations, each having a concentration of 0.1 M. i Permeation rate summarized from the data in (h), showing the robust selectivity in the solution with complex composition. Error bars in all cases represent standard deviation of the data from at least three individual experiments.
Electrodialysis performance
In practical applications, diffusion dialysis is seldom used due to the relatively low flux. The facts that our membrane retains Li+ and that Li+ has the lowest electrical mobility compared to competing cations suggest that our membrane may be suitable for electrodialysis. Compared to nanofiltration, electrodialysis often shows higher energy efficiency because the external force is only used to drive the transport of ions instead of both ions and water45,46,47. We then used a pair of Ag/AgCl electrodes to apply bias voltage across the reservoirs, with the working electrode on the permeate side. Such electrodes can store and release Cl– ions reversibly, maintaining the electroneutrality in both reservoirs during electrodialysis48. We first tested the K+/Li+ binary system (Figs. 6a, b and S28), with each cation having a concentration of 0.1 M. We found that the K+ flux increased gradually with the increase of voltage (Fig. 6b), up to 10.01 mol h−1 m−2 at the voltage of 3 V, 30 times of that in diffusion dialysis. The Li+ flux was kept at zero. For the Na+/Li+ binary (Figs. 6c, d and S29) and K+/Na+/Li+ ternary system (Fig. S30), similar results were found.

a, b Sieving out K+ from Li+ by electrodialysis. a Permeation of K+ and Li+ under a bias voltage of 3 V. b Permeation rate of K+ and Li+ under different bias voltages. Feed solution: 0.1 M K+ + 0.1 M Li+. The results for 0 V are the results of diffusion dialysis. c, d Sieving out Na+ from Li+ by electrodialysis. c Permeation of Na+ and Li+ under a bias voltage of 3 V. d Permeation rate of Na+ and Li+ under different bias voltages. Feed solution: 0.1 M Na+ + 0.1 M Li+. a–d shows the high K+/Li+ and Na+/Li+ selectivity under electrodialysis. e, f Sieving out Mg2+ and Ca2+ from Li+ by electrodialysis. e Permeation of Mg2+, Ca2+ and Li+ under a bias voltage of 4.5 V when the feed solution contains 0.1 M Mg2+ + 0.1 M Ca2+ + 0.1 M Li+, showing the high Mg2+/Li+ and Ca2+/Li+ selectivity. f Permeation rate of Mg2+, Ca2+ and Li+ under different bias voltages. g–i Sieving out competing cations from Li+ in salt lake brine. g Permeation of cations under diffusion dialysis. h Permeation of cations under a bias voltage of 4.5 V. i Summarized permeation rate for (g) and (h), showing the potential of our membrane for Li+ extraction from salt lake brine using electrodialysis. Error bars in all cases represent standard deviation of the data from at least three individual experiments.
In the diffusion dialysis experiments, Mg2+ and Ca2+ could not transport through the membrane. However, leaving Li+ contaminated with divalent ions is unwanted in lithium extraction. We note that Mg2+ and Ca2+ experience an electrical force twice that of Li+. In bulk water, Mg2+ and Ca2+ also transport faster than Li+ under an electric field. Therefore, it is possible that we can selectively transport Mg2+ and Ca2+ while retaining Li+ using a sufficiently large bias voltage. Indeed, using a ternary Mg2+/Ca2+/Li+ system (0.1 M for each cation) and a bias voltage of 4.5 V, we found that Mg2+ and Ca2+ can indeed permeate through the membrane while no Li+ can (Fig. 6e). When we decreased the voltage to 3 V or lower, Mg2+ and Ca2+ were also blocked (Fig. 6f). At a higher voltage of 6 V, the flux of Mg2+ and Ca2+ increased. However, Li+ also transported through the membrane.
To put our results into the context of practical application, we tested the separation of Li+ from a salt-lake (Qinghai Lake, composition shown in Table S7) brine. Using diffusion dialysis, K+ and Na+ could be separated out while Mg2+, Ca2+, and Li+ were retained (Fig. 6g). Using electrolysis with a voltage of 4.5 V, however, all competing ions could transport through the membrane, leaving only Li+ retained, showing high Li+ selectivity (Fig. 6h). Compared to diffusion dialysis, the electrodialysis shows much higher flux (Fig. 6i).
Discussion
High selectivity is characteristic of biological ion channels and is crucial for lithium extraction applications yet remains challenging for previous artificial materials. Our work demonstrates a method to replicate the success of biological ion channels, by using randomly oriented COFs to create small pore size and using sulfonic groups to facilitate the transport of target ions, e.g., Na+ and K+. Through electrodialysis, the permeability of Na+ and K+ can be further enhanced by tens of times. In all cases, the Li+ can be completely blocked, allowing us to sieve out Na+ and K+ from Li+. Moreover, electrodialysis even allows us to mobilize divalent ions and maintain the Li+ retainment performance at a moderate voltage. Such selectivity is exclusive in previous works. It is noteworthy that our COF membrane was grown on the AAO substrate which should be replaced with other substrates, e.g., porous alumina ceramics, in scalable applications. Our work offers a possibility to realize membrane separation of Li+ from various brines by filtering out all concentrated competing cations.
Methods
Chemicals
1,3,5-Triformylphloroglucinol (Tp, 98%), 3-aminopropyltriethoxysilane (APTES, 98%), Al(NO3)3·9H2O (99%), ammonium fluoride (NH4F, 96%), 1,4-dioxane (99%), and mesitylene (99%) were purchased from Aladdin Co., Ltd. p-Phenylenediamine (PDA, 99%), Co(NO3)2·6H2O (98.5%), urea (99%), acetone (99%), ethanol (99%), KCl (99%), LiCl (97%), NaCl (99%), CaCl2 (99%), and MgCl2·6H2O (98%) were supplied by Sinopharm Chemical Reagent Co., Ltd. 2,5-Diaminobenzenesulfonic acid (Pa-SO3H, 98%), acetic acid (HOAc, 36%), dichloromethane (DCM, 99%), dimethylformamide (DMF, 98%) and toluene (99%) were purchased from Meryer Co., Ltd. AAO supporting substrates (12 mm of diameter, 1 mm thick, 80–120 nm of pore size) were purchased from Hefei Puyuan-nano Technology Co., Ltd. All chemicals were bought with high purity and used as received without further purification.
Fabrication of the LDH template
Typically, Co (NO3)2·6H2O (244.4 mg), Al (NO3)3·9H2O (105.0 mg), urea (168.2 mg) and NH4F (148.2 mg) were dissolved in 35 mL DI water to prepare the precursor solution of CoAl-LDH in a Teflon-lined stainless-steel autoclave. The porous AAO supporting substrate was then cleaned by sequentially rinsing with acetone, ethanol, and DI water. After that, the air dried AAO substrate was immersed vertically into the precursor solution. The autoclave was then sealed and heated at 120 °C for 24 h in an oven. After naturally cooled to room temperature, the AAO substrate was taken out, washed with DI water, and then dried at room temperature. Through the above hydrothermal reaction, the LDH template was obtained. The thickness of the LDH layer was typically 6 μm and can be adjusted by controlling the concentration of precursors. The concentration was increased by 50% to obtain a thickness of 7 μm, or 100% to obtain a thickness of 8 μm. To assess the stability of the LDH during COF synthesis, the LDH template was mixed with the synthesis solution containing no COF monomers (i.e., 1.6 mL of 3 M HOAc, 8 mL of dioxane, and 8 mL of mesitylene) at 120 °C for 3 days, followed by characterizations.
Fabrication of the r-TpPa-SO3H COF membrane
The LDH template was firstly treated by an oxygen plasma apparatus (Harrick, PDC-002-HP) for 3 min under a power of 45 W to generate ‒OH terminal groups on the surfaces of CoAl-LDH nanosheets. Then the treated LDH template was placed in 35 mL toluene with APTES (17.5 μL) for 24 h at room temperature, to obtain the –NH2 monolayer for guiding the COF growth. Afterwards, the –NH2 functionalized LDH template was mounted in a Teflon-lined stainless-steel autoclave containing the COF precursor solution, which was obtained typically by adding diamine monomers 2,5-diaminobenzenesulfonic acid (Pa-SO3H, 28.2 mg), aldehyde monomers 1,3,5-triformylphloroglucinol (Tp, 21.0 mg), acetic acid (HOAc, 3 M, 1.6 mL) in 16 mL mixed solvent of dioxane (8 mL) and mesitylene (8 mL), and then reacted at 120 °C for 72 h. After naturally cooling down to room temperature and thoroughly washing with dioxane and ethanol, the TpPa-SO3H COF membrane (termed as r-TpPa-SO3H) was finally obtained, with COF densely synthesized inside the LDH nanosheet layer. The COF membrane thickness can be adjusted by controlling the height of the LDH layer, that is, by increasing or decreasing the concentration of the LDH precursor solution.
Fabrication of the h-TpPa-SO3H and v-TpPa-SO3H COF membranes
To prepare the h-TpPa-SO3H membrane, an aluminum substrate was treated with oxygen plasma (Harrick, PDC-002-HP) for 3 min under a power of 45 W to generate ‒OH groups. Then the treated substrate was functionalized with ‒NH2 groups by placing in a solution containing 35 mL toluene and 17.5 μL APTES at room temperature for 24 h. The substrate was then installed in a Teflon stainless steel autoclaved kettle containing the COF precursor solution identical to the solution used to synthesize the r-TpPa-SO3H COF membrane. The subsequent reaction procedure is the same as for the synthesis of r-TpPa-SO3H. The h-TpPa-SO3H membrane can be peeled off from the aluminum substrate by soaking in ethanol.
The v-TpPa-SO3H membrane was prepared by using a h-TpPa-SO3H membrane. In detail, the as-prepared h-TpPa-SO3H membrane was embedded in epoxy resin, followed by slicing into thin sheet with diamond saw42.
Fabrication of the r-TpPa COF membrane
The r-TpPa membrane was fabricated by using the same template-guided procedure as that of the r-TpPa-SO3H COF membrane, except that the precursor solution of COF was prepared by adding diamine monomers p-phenylenediamine (Pa, 16.2 mg), aldehyde monomers Tp (21.0 mg), HOAc (3 M, 3.2 mL) in 16 mL of dioxane.
Material characterization
SEM characterizations were carried out with a field-emission scanning electron microscopy (Hitachi S-4800, Japan), while the element mapping was proceeded using energy-dispersive X-ray spectroscopy (EDS) integrated with the SEM. XRD patterns were measured using XRD diffractometer (Bruker D8 Advance, Germany). Raman spectrum was recorded by Raman spectrometer (Thermo Scientific DXR3). The porous structures of the membrane were investigated by Auto Fast Specific Surface Area Analyzer (APAP 2460, America). The free volume radius of the membranes was measured by Positron Annihilation Lifetime Spectroscopy (DPLS 300). To observe the orientation, the COF membranes were sampled by Focused Ion Beam Scanning Electron Microscope (FEI Scios 2 HiVac) and was observed by Transmission Electron Microscopy (FEI Talos F200X G2).
Ion selectivity measurement
The COF membrane was mounted between two PTFE reservoirs to construct the self-made permeation device. During the ion-selective permeation experiments, one reservoir was filled with 10 mL feed solution containing chloride salt(s) of single or multiple cation components including KCl, LiCl, NaCl, CaCl2, and MgCl2 (equimolar for each cation, typically 0.1 M unless otherwise specified) while 10 mL DI water was added in the permeate reservoir. The concentration of cation(s) in the permeate solution was measured by ICP-MS (Agilent 7850) over the permeation time.
DFT calculations
All density functional theory (DFT) calculations were performed on ORCA 6.0.0 program packages49. The optimized structures were conducted at B3LYP level of theory with def2-TZVP basis set50. All calculations have contained the Grimme’s D3 dispersion correction (BJ-damping) and taken the solvent effect into account51. The binding energy (Eb) between hydrated cations and COF structural unit was defined as:
where Etotal, ECOF, and Eion represent the energies of the complex, COF unit and hydrated ions, respectively.
The molecular electrostatic potentials52, Hirshfeld-I atomic charges53, non-covalent interaction (independent gradient model based on Hirshfeld partition) analysis54, electron density difference maps, and Mayer bond order analysis55 were achieved by Multiwfn 3.8(dev) program56, of which graphical presentations were generated with the help of VMD 1.9.3 software57.
AIMD calculations
The ab-initio molecular dynamics (AIMD) simulations were performed to study the mobility and structure of hydrated cations within COF. The COF single unit cell (lattice parameters a = 22.56 Å, b = 22.56 Å, c = 7.4 Å and α = β = 90.00°, γ = 120°) was optimized first and the Monkhorst-Pack method was used to generate the Brillouin zone integration from a Gamma centered 1 × 1 × 2 k-point mesh. All calculations employed the Perdew-Burke-Ernzerhof functional with Grimme’s D3 dispersion corrections58. The core electrons were applied by the Goedecker-Tetter-Hutter pseudo-potentials and the valence electrons were expanded using the Gaussian and Plane-Wave combined basis sets (DZVP-MOLOPT along with a plane wave basis set with 400 Ry cut-off energy)59,60. All calculations were performed with the time step of 1 fs on CP2K 2024.1 package61. The temperature was fixed at 298.15 K using the Nosé-Hoover thermostat with a damping constant of 100 fs. Periodic boundary conditions were applied in all directions.
The mean square displacement (MSD) was calculated by
where \(N\) is the total number of observed species in the unit cell, and \({r}_{i}\left(t\right)\) is the position of the \(i\)-th species at time \(t\).
Data availability
All data generated in this study are provided in the main manuscript and the Supplementary Information, and are available from the corresponding author upon request. Source data of the coordinates of the optimized structures are present. Source data are provided with this paper.
References
Fyles, T. M. Synthetic ion channels in bilayer membranes. Chem. Soc. Rev. 36, 335–347 (2007).
Hou, X., Guo, W. & Jiang, L. Biomimetic smart nanopores and nanochannels. Chem. Soc. Rev. 40, 2385–2401 (2011).
Matile, S., Vargas Jentzsch, A., Montenegro, J. & Fin, A. Recent synthetic transport systems. Chem. Soc. Rev. 40, 2453–2474 (2011).
Epsztein, R., DuChanois, R. M., Ritt, C. L., Noy, A. & Elimelech, M. Towards single-species selectivity of membranes with subnanometre pores. Nat. Nanotechnol. 15, 426–436 (2020).
Shen, J., Liu, G., Han, Y. & Jin, W. Artificial channels for confined mass transport at the sub-nanometre scale. Nat. Rev. Mater. 6, 294–312 (2021).
Li, Z. et al. Continuous electrical pumping membrane process for seawater lithium mining. Energy Environ. Sci. 14, 3152–3159 (2021).
Yang, S., Zhang, F., Ding, H., He, P. & Zhou, H. Lithium metal extraction from seawater. Joule 2, 1648–1651 (2018).
Peng, Q. et al. Extreme Li-Mg selectivity via precise ion size differentiation of polyamide membrane. Nat. Commun. 15, 2505 (2024).
Meng, Q.-W. et al. Enhancing ion selectivity by tuning solvation abilities of covalent-organic-framework membranes. Proc. Natl. Acad. Sci. USA 121, e2316716121 (2024).
Zhang, Y. et al. Congener-welded crystalline carbon nitride membrane for robust and highly selective Li/Mg separation. Sci. Adv. 10, eadm9620 (2024).
Sheng, F. et al. Efficient ion sieving in covalent organic framework membranes with Sub-2-nanometer channels. Adv. Mater. 33, 2104404 (2021).
Farahbakhsh, J. et al. Direct lithium extraction: a new paradigm for lithium production and resource utilization. Desalination 575, 117249 (2024).
Vera, M. L., Torres, W. R., Galli, C. I., Chagnes, A. & Flexer, V. Environmental impact of direct lithium extraction from brines. Nat. Rev. Earth Environ. 4, 149–165 (2023).
Wang, R., Alghanayem, R. & Lin, S. Multipass nanofiltration for lithium separation with high selectivity and recovery. Environ. Sci. Technol. 57, 14464–14471 (2023).
Guo, L. et al. Covalently functionalized nanopores for highly selective separation of monovalent ions. Adv. Mater. 36, 2307242 (2024).
Xin, W. et al. Biomimetic KcsA channels with ultra-selective K+ transport for monovalent ion sieving. Nat. Commun. 13, 1701 (2022).
Xin, W. et al. Nacre-like mechanically robust heterojunction for lithium-ion extraction. Matter 4, 737–754 (2021).
Wang, H., Lu, Z., Wu, Y., Ding, L. & Wei, Y. A lamellar MXene (Ti3C2Tx)/PSS composite membrane for fast and selective lithium-ion separation. Angew. Chem. Int. Ed. 60, 22472–22478 (2021).
Acar, E. T., Buchsbaum, S. F., Combs, C., Fornasiero, F. & Siwy, Z. S. Biomimetic potassium-selective nanopores. Sci. Adv. 5, eaav2568 (2019).
Zhang, H. et al. Ultrafast selective transport of alkali metal ions in metal organic frameworks with subnanometer pores. Sci. Adv. 4, eaaq0066 (2018).
Lao, J. et al. Spontaneous and selective potassium transport through a suspended tailor-cut Ti3C2Tx MXene film. ACS Nano 16, 9142–9149 (2022).
Ye, T. et al. Artificial sodium-selective ionic device based on crown-ether crystals with subnanometer pores. Nat. Commun. 12, 5231 (2021).
Ye, T. et al. Highly selective lithium transport through crown ether pillared angstrom channels. Angew. Chem. Int. Ed. 63, e202316161 (2024).
Warnock, S. J. et al. Engineering Li/Na selectivity in 12-Crown-4–functionalized polymer membranes. Proc. Natl. Acad. Sci. USA 118, e2022197118 (2021).
Guo, Y., Ying, Y., Mao, Y., Peng, X. & Chen, B. Polystyrene sulfonate threaded through a metal–organic framework membrane for fast and selective lithium-ion separation. Angew. Chem. Int. Ed. 55, 15120–15124 (2016).
Huang, L., Wu, H., Ding, L., Caro, J. & Wang, H. Shearing liquid-crystalline MXene into lamellar membranes with super-aligned nanochannels for ion sieving. Angew. Chem. Int. Ed. 63, e202314638 (2024).
Lu, J. et al. An artificial sodium-selective subnanochannel. Sci. Adv. 9, eabq1369 (2023).
Zhu, Q. et al. An isoporous ion exchange membrane for selective Na+ transport. J. Membr. Sci. 659, 120805 (2022).
Li, Y. et al. Laminated self-standing covalent organic framework membrane with uniformly distributed subnanopores for ionic and molecular sieving. Nat. Commun. 11, 599 (2020).
Wang, H. et al. Covalent organic framework membranes for efficient separation of monovalent cations. Nat. Commun. 13, 7123 (2022).
Wang, Z. F., Zhang, S. N., Chen, Y., Zhang, Z. J. & Ma, S. Q. Covalent organic frameworks for separation applications. Chem. Soc. Rev. 49, 708–735 (2020).
Yuan, S. S. et al. Covalent organic frameworks for membrane separation. Chem. Soc. Rev. 48, 2665–2681 (2019).
Liu, Y., Wang, N., Diestel, L., Steinbach, F. & Caro, J. MOF membrane synthesis in the confined space of a vertically aligned LDH network. Chem. Commun. 50, 4225–4227 (2014).
Liu, Y., Wang, N., Pan, J. H., Steinbach, F. & Caro, J. In situ synthesis of MOF membranes on ZnAl-CO3 LDH buffer layer-modified substrates. J. Am. Chem. Soc. 136, 14353–14356 (2014).
Fan, H. et al. High-Flux vertically aligned 2D covalent organic framework membrane with enhanced hydrogen separation. J. Am. Chem. Soc. 142, 6872–6877 (2020).
Peng, Y. et al. Mechanoassisted synthesis of sulfonated covalent organic frameworks with high intrinsic proton conductivity. ACS Appl. Mater. Interfaces 8, 18505–18512 (2016).
Zuo, P. et al. Near-frictionless ion transport within triazine framework membranes. Nature 617, 299–305 (2023).
Selim, F. A. Positron annihilation spectroscopy of defects in nuclear and irradiated materials- a review. Mater. Charact. 174, 110952 (2021).
Bing, S. et al. Bio-inspired construction of ion conductive pathway in covalent organic framework membranes for efficient lithium extraction. Matter 4, 2027–2038 (2021).
Meng, W. et al. Three-dimensional cationic covalent organic framework membranes for rapid and selective lithium extraction from saline water. Nat. Water 3, 191–200 (2025).
Li, Q. et al. Covalent organic framework interlayer spacings as perfectly selective artificial proton channels. Angew. Chem. Int. Ed. 63, e202402094 (2024).
Abraham, J. et al. Tunable sieving of ions using graphene oxide membranes. Nat. Nanotechnol. 12, 546–550 (2017).
Randles, J. E. B. The real hydration energies of ions. Trans. Faraday Soc. 52, 1573–1581 (1956).
Babu, C. S. & Lim, C. Theory of ionic hydration: insights from molecular dynamics simulations and experiment. J. Phys. Chem. 103, 7958–7968 (1999).
Gmar, S. & Chagnes, A. Recent advances on electrodialysis for the recovery of lithium from primary and secondary resources. Hydrometallurgy 189, 105124 (2019).
Yang, S., Wang, Y., Pan, H., He, P. & Zhou, H. Lithium extraction from low-quality brines. Nature 636, 309–321 (2024).
DuChanois, R. M. et al. Prospects of metal recovery from wastewater and brine. Nat. Water 1, 37–46 (2023).
Zhang, G. et al. Spontaneous lithium extraction and enrichment from brine with net energy output driven by counter-ion gradients. Nat. Water 2, 1091–1101 (2024).
Neese, F. Software update: the ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Zhang, J. & Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 36, 20323–20328 (2021).
Bultinck, P., Van Alsenoy, C., Ayers, P. W. & Carbo-Dorca, R. Critical analysis and extension of the Hirshfeld atoms in molecules. J. Chem. Phys. 126, 129 (2007).
Lu, T. & Chen, Q. Independent gradient model based on Hirshfeld partition: a new method for visual study of interactions in chemical systems. J. Comput. Chem. 43, 539–555 (2022).
Mayer, I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 97, 270–274 (1983).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Goedecker, S., Teter, M. & Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. 54, 1703–1710 (1995).
Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 114, 145–152 (2005).
Kühne, T. D., Iannuzzi, M., Ben, M. D., Rybkin, V. V. & Hutter, J. CP2K: an electronic structure and molecular dynamics software package I. quickstep: efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFB3805900, J.G.), the Natural Science Foundation of Shandong Province (ZR2023MB037, X.L.), National Natural Science Foundation of China (22272194, J.G.; 52273036, K.S.), Key R&D Project of Shandong Province (2022CXGC010302, J.G.), Natural Science Foundation of Shandong Province (ZR2021YQ12, J.G.), and QIBEBT/SEI/QNESL (S202303, J.G.). The authors acknowledge the use of DeepSeek to correct language errors.
Author information
Authors and Affiliations
Contributions
J.G. and X.L. conceived the idea. J.G., X.L., and K.S. supervised the project. S.B. and Z.M. conducted the experiments and simulations with help from L.Y., Q.L., J.X., S.S., and Y.Z. J.G. and X.L. wrote the manuscript with input from all other authors. S.B. and Z.M. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Dinesh Shetty, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bao, S., Ma, Z., Yu, L. et al. Randomly oriented covalent organic framework membrane for selective Li+ sieving from other ions. Nat Commun 16, 3896 (2025). https://doi.org/10.1038/s41467-025-59188-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59188-1
This article is cited by
-
Thiazole-conjugated covalent organic framework membranes enable ultraselective molecular desalination under strongly acidic conditions
Nature Communications (2026)
-
Energy-autonomous KCl extraction from brine via biomimetic covalent-organic-framework membranes and redox-driven ion transport
Nature Communications (2025)
-
Single acid extraction via covalent organic framework membranes featuring vertically asymmetric charge distribution
Science China Chemistry (2025)
