
Abstract
Genetic risk prediction for non-European populations is hindered by limited Genome-Wide Association Study (GWAS) sample sizes and small tuning datasets. We propose JointPRS, a data-adaptive framework that leverages genetic correlations across multiple populations using GWAS summary statistics. It achieves accurate predictions without individual-level tuning data and remains effective in the presence of a small tuning set thanks to its data-adaptive approach. Through extensive simulations and real data applications to 22 quantitative and four binary traits in five continental populations evaluated using the UK Biobank (UKBB) and All of Us (AoU), JointPRS consistently outperforms six state-of-the-art methods across three data scenarios: no tuning data, same-cohort tuning and testing, and cross-cohort tuning and testing. Notably, in the Admixed American population, JointPRS improves lipid trait prediction in AoU by 6.46%–172.00% compared to the other existing methods.
Similar content being viewed by others

Introduction
Polygenic Risk Scores (PRS), the weighted sum of risk alleles across a collection of genetic variants, have seen active development for predicting complex traits in recent years. PRS have demonstrated their ability to identify individuals with high disease risk, which can be applied for disease monitoring, prevention, and treatment1,2,3,4. However, most PRS have been developed for European populations, due to the dominance of large European cohorts in Genome-Wide Associations Studies (GWAS)5, with non-European populations’ PRS exhibiting reduced accuracy in comparison6.
Several factors contribute to the reduced PRS prediction accuracy in non-European populations. First, both GWAS summary statistics for model training and individual-level datasets for parameter tuning are limited in these populations, hindering the development of population-specific PRS that rely solely on non-European datasets. In addition, distinctive genetic structures, such as varying numbers of single-nucleotide polymorphisms (SNPs) and different linkage disequilibrium (LD) patterns between European and non-European populations, constrain the transferability of European PRS prediction models to non-European populations. This gap in genetic risk prediction performance can exacerbate health disparities6,7, underscoring the urgent need to improve PRS prediction accuracy for non-European populations.
To address this need, there has been an increase in GWAS focused on non-European populations8,9,10,11,12,13,14,15,16, complemented by the development of various models tailored for multi-population PRS predictions17,18,19,20,21,22,23,24,25,26,27,28,29. These models can be categorized into two types: “auto methods” and “tune methods”. Auto methods, which utilize only GWAS summary statistics, can provide robust performance with or without individual-level tuning data20. However, neglecting individual-level tuning data when available may miss useful information that could further improve PRS prediction accuracy. But tune methods require an individual-level tuning dataset, limiting their applications in various data scenarios.
When individual-level tuning data are available, tune methods can enhance the prediction accuracy of PRS. However, their performance is significantly influenced by the quality and sample size of the tuning data, a challenge particularly pronounced in non-European populations. The quality of tuning datasets can be compromised by confounding factors and measurement errors of the phenotype being predicted and the chosen tuning dataset. In addition, various factors such as socio-economic status, age, and sex in the individual-level dataset can lead to varying PRS prediction accuracy across individuals30. Including individuals who are not effectively predicted by PRS due to these factors can adversely affect tuning weights and overall model performance. Small sample sizes further exacerbate the issue, potentially leading to unstable performance when PRS are applied to new datasets. Consequently, there remains a need for methods that not only provide accurate and robust predictions without tuning data but also can effectively utilize individual-level tuning data to achieve both efficiency and robustness.
Moreover, most published studies evaluate method performance by splitting a single individual-level data cohort into tuning and testing datasets17,19,20,21,22,23,24,29. However, in real-world PRS applications, such as predicting phenotypes for individuals in a medical center, tuning data from the same cohort are often unavailable. Instead, we may have no tuning data at all or only tuning data from other cohorts. Therefore, there is a need to benchmark PRS methods under diverse data scenarios, including no tuning data, tuning and testing data from the same cohort, and tuning and testing data from different cohorts. This comprehensive evaluation, which remains largely unexplored, is essential to ensure the robustness and applicability of multi-population PRS methods in practical settings.
In this study, we introduce JointPRS, a Bayesian framework that employs sparse distribution assumptions for genetic variant effect sizes, jointly models multiple populations, and incorporates automatically estimated chromosome-wise cross-population genetic correlations. JointPRS uses a continuous shrinkage (CS) prior to flexibly account for varying sparsity levels in the genetic variant effect sizes, which is also utilized in PRS-CSx19. However, JointPRS offers two distinct advantages: First, JointPRS incorporates cross-population genetic correlation structures, ensuring good performance even when only GWAS summary statistics are available for training. In contrast, PRS-CSx does not consider genetic correlations, limiting its prediction accuracy, particularly when tuning data are not available.
Moreover, the genetic correlation estimation method in JointPRS provides key advantages over existing methods. Unlike SDPRX20, which relies on externally derived genetic correlations from software like Popcorn31, JointPRS estimates genetic correlations within the model, preventing model mismatches and optimizing PRS performance. Compared to XPASS17, which employs the Method of Moments and may produce unconstrained genetic correlation estimates outside the valid range of [ − 1, 1], JointPRS utilizes a Bayesian estimation approach, ensuring valid and reliable correlation estimations. In addition, unlike MUSSEL and PROSPER22,23, which treat genetic correlations as tuning parameters and require additional tuning data, JointPRS relies solely on GWAS summary statistics, broadening its applicability across diverse data scenarios.
Second, when non-European individual-level tuning data are available, JointPRS adopts a data-adaptive approach that combines meta-analysis with tuning strategies. This approach addresses the challenge of small non-European tuning datasets, resulting in more robust performance. Conversely, the tuning method in PRS-CSx relies on the quality of individual-level tuning data, leading to unstable PRS performances. By leveraging these innovations, JointPRS enhances the robustness and accuracy of PRS across diverse populations.
The efficacy of JointPRS is evaluated through extensive simulations and applications to real data, including 22 quantitative traits and four binary traits across five populations (European (EUR); East Asian (EAS); African (AFR); South Asian (SAS); and Admixed American (AMR)) using the UK Biobank (UKBB)32 and All of Us (AoU) cohorts33. Evaluations are conducted under three data scenarios: [1] no tuning data, [2] tuning and testing data from the same cohort, and [3] tuning and testing data from different cohorts. JointPRS consistently outperforms other state-of-the-art methods17,19,20,22,23,24 across most traits in non-European populations and various data scenarios while maintaining model simplicity and achieving computational efficiency. Our results illustrate the contributions of appropriate sparse distribution, joint modeling of multiple populations, and genetic correlation structures to prediction accuracy. In addition, our findings highlight the importance of the data-adaptive approach for robust predictions when only a small tuning dataset is available. Moreover, our simulation studies underscore the impact of tuning sample sizes on PRS prediction accuracy. In conclusion, JointPRS offers a robust and accurate approach for genetic risk prediction in diverse populations, with or without tuning datasets. Our results also improve our understanding of the contributions of different types of information to impacting PRS prediction.
Results
Overview of JointPRS
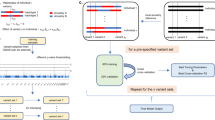
JointPRS is a Bayesian high-dimensional prediction framework designed for multiple populations with an additional data-adaptive approach to determine how to use tuning datasets when they are available. This framework operates in two key steps (Fig. 1): [1] model development for multiple populations, and [2] data-adaptive approach to select the optimal PRS between meta and tune versions when a tuning dataset is available. An independent testing dataset is used to evaluate prediction performance.

JointPRS operates under two data scenarios for PRS estimation: (1) No tuning data available: JointPRS-auto computes the auto version PRS using only GWAS summary statistics and LD reference panels from K populations. (2) Tuning data available: JointPRS leverages the tuning data in two distinct ways: (i) performs a meta-analysis for the meta version PRS; and (2) optimizes tuning parameters and performs a linear combination to generate the tune version PRS. The same tuning data are then used to select the optimal PRS between the meta and tune versions. This figure was created in BioRender. Xu, L. (2025) https://BioRender.com/4xidl7d.
Step1: JointPRS model development
In Step 1, various versions of the JointPRS model are trained using appropriate training inputs. First, the JointPRS model is introduced, followed by an explanation of each version and their application scenarios.
JointPRS Model
The JointPRS model has the following form:
where
Here, K is the number of populations, Nk denotes the sample size for population k, and S represents the number of SNPs that are available in at least one population. The vectors yk, Xk, βk, and ϵk correspond to the standardized phenotype vector, the column-standardized genotype matrix, the standardized effect size vector for SNPs, and residuals in population k, respectively. There may be missing elements in the genotype matrix and effect size vector, which will be discussed in detail later.
When SNP j is available for all K populations, the effect size βj for SNP j across K populations is modeled with a correlated Gaussian prior:
with
Here, Ψj represents the sample-size normalized effect size for SNP j shared across K populations. This shared effect, Ψj, is modeled using a continuous shrinkage prior, as implemented in the PRS-CSx framework19. Specifically, Ψj follows a gamma distribution Ψj ~ G(1, δj) with \({\delta }_{j} \sim G(\frac{1}{2},\phi )\). The global shrinkage parameter ϕ varies depending on the JointPRS version used, which will be detailed later. Additionally, we introduce the cross-population covariance matrix Σ in the model to capture shared patterns across populations. All parameters in the model are estimated automatically by the JointPRS algorithm using only GWAS summary statistics (“Methods”).
For SNPs available in only a subset of populations, the model truncates to include only those populations with the SNPs, using an index matrix (“Methods”).
JointPRS versions
The JointPRS modeling framework has three versions—auto, meta, and tune—each differing in terms of input data, model parameters, and modeling strategies (Table 1). Detailed illustrations and implementations of these different JointPRS versions are provided in the Methods section (“Methods”).
When tuning data are unavailable, JointPRS employs the auto version to construct the PRS, referred to as JointPRS-auto. When tuning data are available, JointPRS uses a data-adaptive approach to select between the meta version and the tune version, as detailed in Step2. This data-adaptive PRS approach is referred to as JointPRS. JointPRS-auto and JointPRS together cover data scenarios without and with tuning data, with the latter adaptively deciding how the available tuning data are used.
Step2: JointPRS data-adaptive approach
Step2 is performed only when the tuning data are available, aiming to select the optimal PRS between the meta version and the tune version. Directly evaluating the meta version using the tuning dataset can lead to overfitting, as the meta version already incorporates the tuning dataset into the original GWAS summary statistics during training. To mitigate this problem, we approximate the meta version’s performance by using the tuning data to evaluate the performance of the auto version, which employs the global shrinkage prior \({\phi }^{\frac{1}{2}} \sim {C}^{+}(0,1)\) with the original GWAS summary statistics. In addition, we evaluate the tune version by using \({\phi }^{\frac{1}{2}} \sim {C}^{+}(0,1)\), followed by a linear combination across populations, skipping the parameter selection step. The key difference in the evaluation of the two versions is whether a linear combination across populations is performed. This approach frames the choice between the meta version and the tune version as a model selection problem, using the tuning dataset to determine whether the linear combination across populations might be beneficial. This selection is achieved through the F-test for continuous traits and chi-squared test for binary traits. Detailed implementations of the data-adaptive approach are available in the Methods section (“Methods”).
Simulations
Simulation settings
Benchmarking methods
We compared the performance of JointPRS with six other state-of-the-art multi-population PRS methods: XPASS17, SDPRX20, PRS-CSx19, MUSSEL22, PROSPER23, and BridgePRS24(“Methods”). These methods can be grouped into two categories: auto methods and tune methods (Supplementary Fig. 1). Auto methods, including JointPRS-auto, XPASS, SDPRX, and PRS-CSx-auto, can be implemented using only GWAS summary statistics. When a tuning dataset is available, XPASS and SDPRX incorporate the tuning data with GWAS summary statistics to increase sample size. Tune methods, including JointPRS, PRS-CSx, MUSSEL, PROSPER, and BridgePRS, require a tuning dataset with corresponding tuning strategies for application. We benchmarked seven methods across various simulation settings, including different sample sizes for training, tuning, and testing datasets and various genetic architectures of a continuous trait.
Genetic dataset sample sizes settings
For the European population, we used the UKBB genotype data, which include 311,600 unrelated samples for training in the simulation. In addition, we used 10,000 simulated individuals for tuning and 10,000 for testing from a large simulation dataset21. Briefly, this simulated dataset comprises a total of 600,000 independent samples with reference LD based on the 1000 Genomes Project data34. There are 120,000 samples representing each of the five continental populations: EUR, EAS, AFR, SAS, and AMR.
For the non-European populations, we obtained all genotype datasets from the large simulated dataset21. We considered two training sample sizes: 80,000 and 15,000 samples. In each scenario, based on the availability and sample size of the tuning dataset, we examined the following situations:
-
1.
No tuning dataset available: We compared the performance of methods that only require GWAS summary statistics, including JointPRS-auto, XPASS, SDPRX, and PRS-CSx-auto.
-
2.
Tuning dataset available: We compared the performance of all methods: JointPRS, XPASS, SDPRX, PRS-CSx, MUSSEL, PROSPER, and BridgePRS, with varying tuning sample sizes (500, 2000, 5000, and 10,000).
In all scenarios, we evaluated the performance of all methods using 10,000 testing individuals. For all methods, we jointly modeled the maximum number of populations each method could utilize: five populations for JointPRS(-auto), PRS-CSx(-auto), MUSSEL, and PROSPER, and two populations for XPASS, SDPRX, and BridgePRS.
Simulation results
Simulation results (Fig. 2) demonstrate that JointPRS consistently performs well across various tuning dataset scenarios, non-European population training sample sizes, and genetic architecture settings. Compared to six alternative methods, JointPRS shows superior performance in accuracy and robustness, particularly when the training sample size is large (Ntrain = 80, 000).

a, b The total heritability was fixed at h2 = 0.4, with non-European training sample sizes set to Ntrain = 80, 000 and 15, 000. a No tuning dataset: Each dot represents the mean relative performance across five replicates, defined as \({R}_{{{\rm{other}}}\,{{\rm{methods}}}}^{2}/{R}_{{{\rm{JointPRS}}}\,{\mbox{-}}\,{{\rm{auto}}}}^{2}-1\), for one causal SNP proportion (p = 0.1, 0.01, 0.001, 5 × 10−4). b With tuning dataset: Each dot represents the mean relative performance across five replicates, defined as \({R}_{{{\rm{other}}}\,{{\rm{methods}}}}^{2}/{R}_{{{\rm{JointPRS}}}}^{2}-1\), for one causal SNP proportion and one tuning dataset size (Ntune = 500, 2, 000, 5, 000, 10, 000). Note that SDPRX cannot provide predictions for SAS and AMR as it does not provide the corresponding LD reference panels. Source data are provided as a Source Data file.
When a tuning dataset is not available, the results (Fig. 3 and Supplementary Table 1) show that JointPRS-auto outperforms other auto methods (XPASS, SDPRX, and PRS-CSx-auto), particularly when the non-European training sample size is large (Ntrain = 80, 000) and the traits are highly polygenic (p = 0.1, 0.01).

a, b The total heritability was fixed at h2 = 0.4. The proportion of causal SNPs was set to p = 0.1, 0.01, 0.001, and 5 × 10−4. The training sample sizes for non-European populations were set to aNtrain = 80, 000 and bNtrain = 15, 000. Each simulation scenario was repeated five times, and the mean values are presented in the bar plots. Only four auto methods were considered, with the best and second-best methods indicated by two stars and one star, respectively, in the corresponding bar plots. Note that SDPRX cannot provide predictions for SAS and AMR as it does not offer the corresponding LD reference panels. Source data are provided as a Source Data file.
Specifically, XPASS exhibits the worst performance in most situations, with JointPRS-auto showing improvements of 199.00% in EAS, 212.00% in AFR, 165.00% in SAS, and 253.00% in AMR. This indicates the importance of an appropriate sparse distribution assumption. Comparing JointPRS-auto with SDPRX involves a trade-off between modeling multiple populations and modeling exact genetic architecture. The significant improvement of JointPRS-auto over SDPRX (15.20% in EAS and 15.70% in AFR) with a large training sample size (Ntrain = 80, 000) and large causal SNP proportions (p = 0.1, 0.01) implies that modeling multiple populations may be more helpful as non-European training sample sizes increase in highly polygenic traits. Conversely, genetic architecture modeling becomes more critical with small training sample sizes (Ntrain = 15, 000) or large effect SNPs (p = 0.001, 5 × 10−4). Furthermore, SDPRX can only be applied to EAS and AFR populations because the LD reference data are only available for these two populations in SDPRX. Lastly, JointPRS-auto demonstrates an overall improvement of 30.40% in EAS, 35.20% in AFR, 32.40% in SAS, 31.30% in AMR over PRS-CSx-atuo. In addition, as the causal SNP proportion decreases (p = 0.1, 0.01, 0.001, 5 × 10−4) with fixed genetic correlation (ρ = 0.8), the improvement of JointPRS-auto over PRS-CSx-auto decreases, suggesting that the genetic correlation structure is more helpful for highly polygenic traits.
To evaluate the advantage of using automatically estimated chromosome-wise genetic correlation, we compared the performance of JointPRS-auto with JointPRS_popcorn, which relies on external genetic correlation estimates from Popcorn31. As shown in Supplementary Fig. 2, JointPRS-auto performs slightly worse but remains comparable to JointPRS_popcorn when the causal SNP proportion is large (p = 0.1). However, with a small causal SNP proportion (p = 5 × 10−4), JointPRS-auto significantly outperforms JointPRS_popcorn. These results suggest that utilizing automatically estimated chromosome-wise genetic correlation enhances the robustness of JointPRS.
We further investigated whether the improvement of JointPRS-auto over PRS-CSx-auto is related to the underlying cross-population genetic correlations. Five genetic correlation settings (ρ = 0, 0.2, 0.4, 0.6, and 0.8) were considered, with a large causal SNP proportion (p = 0.1) and a large non-European training sample size (Ntrain = 80, 000). Supplementary Fig. 3 demonstrates that the improvement of JointPRS-auto over PRS-CSx-auto increases as the genetic correlation increases.
When a tuning dataset is available, the results (Fig. 4 and Supplementary Figs. 4–6; Supplementary Tables 2–6) demonstrate that JointPRS outperforms six other methods (XPASS, SDPRX, PRS-CSx, MUSSEL, PROSPER, and BridgePRS). This advantage is particularly evident when the non-European population training sample size is large (Ntrain =80,000) and the trait is highly polygenic (p = 0.1,0.01), across various tuning sample sizes (Ntune = 500, 2000, 5000, and 10,000).

a–d, The total heritability was fixed at h2 = 0.4. The proportion of causal SNPs was set to p = 0.1 and 0.01. We evaluated four non-European populations, (a) EAS, (b) AFR (c) SAS, (d) AMR. The training sample sizes for non-European populations was set to Ntrain = 80, 000. The tuning sample sizes for the target population was set to Ntune = 500, 2000, 5000, and 10,000. Simulations in each scenario were repeated five times, and the mean values was presented in the bar plots. All seven methods were considered here, with the best and second-best methods denoted by two stars and one star, respectively, in the corresponding bar plots. Note that SDPRX cannot provide predictions for SAS and AMR as it does not provide the corresponding LD reference panels. Source data are provided as a Source Data file.
Specifically, JointPRS achieves the top performance for SAS and AMR across all simulation settings. For EAS and AFR, JointPRS is more advantageous when the causal SNP proportion is large (p = 0.1 and 0.01), being the best or second-best method in 15 out of 32 simulation settings in the EAS population and 13 out of 32 simulation settings in the AFR population, indicating JointPRS performed well for highly polygenic traits. When the causal SNP proportion is small (p = 0.001 and 5 × 10−4), JointPRS remains advantageous for the EAS population, being the best or second-best method in 11 out of 32 simulation settings, while SDPRX and MUSSEL are more preferred in the AFR population.
In addition, for different tuning dataset sizes (Ntune = 500, 2000, 5000, and 10,000), Supplementary Figs. 7–10 demonstrate that JointPRS, SDPRX, PRS-CSx, and BridgePRS have more stable performance than the other three methods (XPASS, MUSSEL, and PROSPER). One key reason for this is that MUSSEL and PROSPER use a super learning step, which involves tuning and training many more parameters. As a result, these methods require larger tuning sample sizes to perform well. When the tuning data are limited, the super learning step is more prone to overfitting, making the performance more sensitive to the size of the tuning dataset.
Moreover, we explored a scenario with a strong environmental component (h2 = 0.1). As shown in Supplementary Fig. 11, with a low heritability (h2 = 0.1), a high causal SNP proportion (p = 0.1), a large non-European training sample size (Ntrain = 80, 000), and a large non-European tuning sample size (Ntune = 10,000), PRS-CSx achieves the best performance. This suggests that advanced models such as JointPRS, SDPRX, and PROSPER may be more beneficial in moderate to high heritability scenarios.
Real data analysis
Data preparation
We collected GWAS summary statistics for 26 traits across five populations (EUR, EAS, AFR, SAS, and AMR) from various consortia. These traits were divided into four groups—GLGC traits, PAGE traits, BBJ traits, and binary traits—for reasons described in detail in the Methods section (Methods). Comprehensive information about these 26 traits is provided in the following section.
Quality control and data preparation
Quality control was conducted following the LDHub guidelines, using LDSC software to remove duplicate SNPs35,36. To ensure fair comparisons for different PRS models, we restricted SNP lists for each population to those available in reference panels used across all evaluated methods. The 1000 Genomes Project served as the reference panel for all methods. Detailed GWAS summary statistics, including trait names, sample sizes, and SNP numbers, are provided in Table 2 and Supplementary Table 7.
We calculated the pairwise cross-population genetic correlation using popcorn31 based on the GWAS summary statistics (Supplementary Table 8). Our analysis reveals that these 26 traits exhibit high cross-population genetic correlations, mostly ranging from 0.7 to 0.99. However, the genetic correlations between EUR and AFR, and EAS and AFR in breast cancer, were notably lower. This discrepancy is likely attributable to the small sample size of the breast cancer AFR GWAS summary statistics.
For tuning and benchmarking multi-population methods, we utilized individual-level genetic data from the UKBB and AoU Program. Detailed sample size information is available in Supplementary Tables 9 and 10, with data processing procedures including phenotype definition outlined in the Supplementary Notes (Individual-level Genetic Dataset Preparation section). Our primary goal is to assess prediction accuracy in non-European populations (EAS, AFR, SAS, and AMR). To ensure reliability, we have confirmed that there is no overlap between the training GWAS and the individual tuning and testing data from the UKBB or AoU for non-European populations. While we acknowledge the potential overlap between the EUR training GWAS and tuning and testing datasets, we do not consider this overlap to be a significant concern. Certain tune methods (MUSSEL, PROSPER, and BridgePRS) also require individual-level data for the EUR population; thus, we extracted data from 10,000 European individuals in the UKBB for these purposes.
Method comparison and evaluation scenarios
We compared the methods across all traits under three data scenarios:
-
1.
No tuning data: We used the UKBB data to evaluate the performance of four auto methods: JointPRS-auto, XPASS, SDPRX, and PRS-CSx-auto.
-
2.
Tuning and testing data from the same cohort: We performed 5-fold cross-validation in the UKBB data for seven methods: JointPRS, XPASS, SDPRX, PRS-CSx, MUSSEL, PROSPER, and BridgePRS.
-
3.
Tuning and testing data from different cohorts: We conducted tuning in the UKBB data and evaluation in the AoU data for the same seven methods.
The percentage of relative change of method B over A is defined as (metricB − metricA)/metricA*100%. This metric allowed us to assess the comparative performance of different PRS methods.
Benchmarking multi-population PRS methods when there are no tuning data
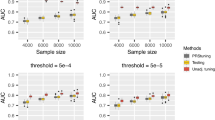
We first evaluated the performance of four auto methods (JointPRS-auto, XPASS, SDPRX, and PRS-CSx-auto) across 22 quantitative traits and four binary traits in the UKBB, focusing on four non-European populations (EAS, AFR, SAS, and AMR) in the absence of tuning data. The results (Fig. 5 and Supplementary Figs. 12–15 and Supplementary Tables 11 and 12) demonstrate that JointPRS-auto consistently outperforms the other three methods in terms of accuracy and robustness.

a–d, 26 traits from four categories were evaluated in UKBB when there is no tuning dataset: (a) GLGC traits, (b) PAGE traits (BBJ traits, (d) Binary traits. The R2 and AUC were used as evaluation metrics for quantitative traits and binary traits, and the values were presented in the bar plots. Only four auto methods were considered here, with the best and second-best methods denoted by two stars and one star, respectively, in the corresponding bar plots. Note that SDPRX cannot provide predictions for SAS and AMR as it does not provide the corresponding LD reference panels. Source data are provided as a Source Data file.
JointPRS-auto consistently outperforms XPASS in all four non-European populations across evaluated traits (Fig. 5a and Supplementary Figs. 12 and 13). The average improvement of JointPRS-auto over XPASS is 46.14% in the EAS population across 26 traits, 53.29% in the AFR population across 11 traits, 74.54% in the SAS population across four traits, and 88.48% in the AMR population across four traits. This implies that the sparse distribution in JointPRS-auto is more appropriate.
Moreover, JointPRS-auto and SDPRX are the top-performing methods in the EAS and AFR populations for many evaluated traits (Fig. 5 and Supplementary Figs. 12 and 14). However, JointPRS-auto shows more accurate and robust performance, with a 2.11% average improvement over SDPRX in the EAS population across 26 traits and a 17.86% average improvement in the AFR population across 11 traits. This indicates that JointPRS-auto achieves a balanced trade-off between modeling multiple populations and capturing complex genetic architectures, leading to more robustness across traits. Notably, SDPRX’s application is currently limited to the EAS and AFR populations due to the availability of LD reference data provided by SDPRX.
JointPRS-auto demonstrates superior performance compared to PRS-CSx-auto across all four non-European populations for the evaluated traits (Fig. 5 and Supplementary Figs. 12 and 15). Specifically, the average improvement of JointPRS-auto over PRS-CSx-auto is 17.82% in the EAS population across 26 traits, 27.85% in the AFR population across 11 traits, 22.70% in the SAS population across four traits, and 20.94% in the AMR population across four traits. This consistent improvement indicates that the automatically estimated chromosome-wise cross-population genetic correlation structure utilized by JointPRS-auto enhances PRS prediction accuracy in non-European populations, particularly in the absence of tuning data.
Furthermore, we analyzed the pairwise chromosome-wise genetic correlations estimated by JointPRS-auto across 22 continuous traits (Supplementary Fig. 16). We observed moderate genetic correlations between the European population and the other four populations (EAS, AFR, SAS, AMR). However, contrary to our simulation results, we did not find a positive relationship between the mean chromosome-wise genetic correlation and the improvement of JointPRS-auto over PRS-CSx-auto in real data (Supplementary Fig. 17). This discrepancy can be attributed to various factors influencing PRS prediction performance in real-world scenarios, such as GWAS sample sizes and the distinct genetic architectures of different traits. Nonetheless, our findings suggest that JointPRS-auto consistently outperforms PRS-CSx-auto.
Moreover, we compared the performance of JointPRS-auto and PRS-CSx-auto with another continuous shrinkage prior PRS model, Xwing-auto29, which accounts for local genetic correlations in the EAS population (“Methods”). This analysis utilized GWAS summary statistics from the EUR and EAS populations for 22 continuous traits. The results (Supplementary Fig. 18) demonstrate that despite Xwing-auto incorporating more localized genetic information, JointPRS-auto consistently outperformed Xwing-auto, showing an average improvement of 19.41% across the 22 traits in the EAS population. In contrast, Xwing-auto did not perform consistently better than PRS-CSx-auto across traits, with an average improvement of only 1.53%, compared to the 21.32% improvement of JointPRS-auto over PRS-CSx-auto. These findings suggest that while incorporating local genetic correlations can be beneficial for PRS modeling, the challenge of accurately estimating these correlations limits its effectiveness. The automatically estimated chromosome-wise genetic correlation approach proposed by JointPRS offers a more efficient and robust method for improving multi-population PRS prediction.
In conclusion, JointPRS-auto exhibits the most robust and accurate performance across 22 quantitative traits and four binary traits evaluated in the UKBB for scenarios without tuning data, confirming the importance of appropriate sparsity distributions, incorporating automatically estimated chromosome-wise cross-population genetic correlations, and simultaneously modeling multiple populations for accurate non-European PRS predictions.
Benchmarking multi-population PRS Methods When Tuning and Testing Data Come From the Same Cohort
We evaluated the performance of seven methods—JointPRS, XPASS, SDPRX, PRS-CSx, MUSSEL, PROSPER, and BridgePRS—across 22 quantitative traits and four binary traits in four non-European populations (EAS, AFR, SAS, and AMR). In this scenario, both the tuning and testing data originated from the same cohort. The UKBB dataset was split into five folds for each population, with each fold used for testing while the remaining folds for tuning, thus enabling a 5-fold cross-validation evaluation. The results (Fig. 6, Supplementary Figs. 19–25, Supplementary Tables 13 and 14) illustrate that JointPRS outperforms the other methods in terms of accuracy and robustness.

a–d 26 traits from four categories were analyzed in scenarios where tuning and testing data come from the same cohort (UKBB): (a) GLGC traits, (b) PAGE traits (c) BBJ traits, (d) Binary traits. A 5-fold cross-validation was conducted within UKBB. The relative performance of six existing methods compared to JointPRS was measured using \({R}^{2}/{R}_{{{\rm{JointPRS}}}}^{2}-1\) for quantitative traits and AUC/AUCJointPRS − 1 for binary traits across five folds. Results are displayed in violin plots, with the mean value across traits indicated by a black crossbar for each method within each trait category. Note that SDPRX cannot provide predictions for SAS and AMR as it does not provide the corresponding LD reference panels. Source data are provided as a Source Data file.
For GLGC traits, JointPRS and SDPRX are the top two performing methods in the EAS and AFR populations, whereas XPASS, PRS-CSx, MUSSEL, PROSPER, and BridgePRS had lower prediction accuracy. In the SAS population, JointPRS, PRS-CSx, MUSSEL, and PROSPER are the top-performing methods, while SDPRX cannot provide predictions. In contrast, XPASS and BridgePRS exhibit lower prediction accuracy. In the AMR population, JointPRS consistently outperforms XPASS, PRS-CSx, MUSSEL, PROSPER, and BridgePRS, with SDPRX again providing no predictions. In addition, a comparison between EUR and non-European populations across methods highlights a persistent performance disparity, as shown in Supplementary Fig. 26.
Regarding the PAGE traits, JointPRS, SDPRX, PRS-CSx, and PROSPER are the top-performing methods for the EAS population, whereas XPASS, MUSSEL, and BridgePRS show lower prediction accuracy. In the AFR population, JointPRS, PRS-CSx, and PROSPER are the top-performing methods, while XPASS, SDPRX, MUSSEL, and BridgePRS exhibit lower prediction accuracy.
For BBJ traits, JointPRS, SDPRX, PRS-CSx, and PROSPER are the top-performing methods, with XPASS, MUSSEL, and BridgePRS showing lower prediction accuracy.
In the case of binary traits, all methods perform similarly, though XPASS, SDPRX, MUSSEL, and BridgePRS show slightly worse performance in the EAS population.
Overall, JointPRS consistently outperformed XPASS across all populations, with average improvements of 80.58% in EAS, 89.98% in AFR, 99.98% in SAS, and 166.84% in AMR. JointPRS demonstrated better accuracy and robustness over SDPRX in the majority of evaluated traits, with average improvements of 9.18% in EAS and 29.10% in AFR populations. Compared to PRS-CSx, JointPRS showed average improvements of 2.78% in EAS, 25.02% in AFR, 2.57% in SAS, and 27.31% in AMR populations. When compared to MUSSEL, JointPRS had average improvements of 20.15% in EAS, 47.83% in AFR, 5.98% in SAS, and 84.91% in AMR. JointPRS outperformed PROSPER with average improvements of 6.30% in EAS, 7.82% in AFR, and 9.73% in AMR, and PROSPER failed to converge for the lung cancer trait in some cross-validation folds. Finally, JointPRS demonstrated consistent superiority over BridgePRS, with average improvements of 605.46% in EAS, 291.07% in AFR, 31.06% in SAS, and 60.84% in AMR.
To summarize, JointPRS consistently demonstrates better performance compared to XPASS and BridgePRS. Methods such as SDPRX, PRS-CSx, MUSSEL, and PROSPER can exhibit top performance similar to JointPRS for certain traits and populations but perform considerably worse than JointPRS in others. Notably, tune methods tend to be less accurate in GLGC traits, while auto methods tend to be less accurate in PAGE traits.
We also summarize the selection results of JointPRS versions, the prediction accuracy of the meta and tune versions, and the relative improvement achieved by the data-adaptive approach over these two versions (Supplementary Figs. 27–31 and Supplementary Table 15). JointPRS selects different versions (tune or meta) depending on the trait, population, and tuning data, suggesting context-specific benefits of each version (Supplementary Table 15). Notably, our data-adaptive approach often identifies the more accurate version between the meta and tune versions, especially when there is a substantial difference in their prediction accuracies (Supplementary Figs. 28–31). In addition, the positive relative improvement of the selected version over the meta and tune versions underscores the robustness of the data-adaptive approach, as evidenced by the presence of extreme positive and the well-bounded negative dots in the plot (Supplementary Fig. 27). These findings are consistent with our previous observations that while existing tune and auto methods can perform well for specific traits, JointPRS demonstrates robust performance across all traits due to its data-adaptive approach.
In conclusion, JointPRS exhibits the most robust and accurate performance across 22 quantitative traits and four binary traits evaluated in the UKBB when the tuning and testing data come from the same cohort. This superior performance is attributable to its data-adaptive approach, which effectively integrates the strengths of both meta-analysis and tuning strategies.
Benchmarking Multi-population PRS Methods When Tuning and Testing Data Come From Different Cohorts
In this section, we study the performance of seven polygenic risk score (PRS) methods—JointPRS, XPASS, SDPRX, PRS-CSx, MUSSEL, PROSPER, and BridgePRS—across nine quantitative traits in two non-European populations (AFR and AMR). We consider the case where the tuning and testing data come from different cohorts: the UKBB dataset was used for tuning, while the AoU dataset was used for testing. The results (Fig. 7, Supplementary Figs. 32–38 and Supplementary Table 16) demonstrate that JointPRS outperforms the other methods in terms of both accuracy and robustness.

a, b Nine traits from two categories were analyzed in scenarios where tuning and testing data come from different cohorts (UKBB and AoU): (a) GLGC traits and (b) PAGE traits. UKBB data were used for tuning, while AoU data were used for testing. The relative performance of six existing methods compared to JointPRS was measured using \({R}^{2}/{R}_{{{\rm{JointPRS}}}}^{2}-1\) for quantitative traits. Results are displayed in violin plots, with the mean value across traits indicated by a black crossbar for each method within each trait category. Note that SDPRX cannot provide predictions for SAS and AMR, as it does not provide the corresponding LD reference panels. Source data are provided as a Source Data file.
For GLGC traits, JointPRS and SDPRX are the top two performing methods for the AFR population, whereas XPASS, PRS-CSx, MUSSEL, PROSPER, and BridgePRS have lower prediction accuracy. For the AMR population, JointPRS consistently outperforms XPASS, PRS-CSx, MUSSEL, PROSPER, and BridgePRS, with SDPRX failing to provide predictions. Regarding the PAGE traits, JointPRS and PRS-CSx are the top two performing methods, while XPASS, SDPRX, MUSSEL, PROSPER, and BridgePRS exhibit lower prediction accuracy.
Overall, JointPRS consistently outperformed XPASS across both AFR and AMR populations, with average improvements of 58.29% in AFR and 172.00% in AMR. JointPRS outperformed SDPRX with an average improvement of 29.14% in the AFR population. Against PRS-CSx, JointPRS showed average improvements of 7.03% in AFR and 10.55% in AMR. Compared to MUSSEL, JointPRS demonstrated average improvements of 16.85% in AFR and 71.72% in AMR. JointPRS also outperformed PROSPER in most traits, with average improvements of 8.71% in AFR and 6.46% in AMR. Finally, JointPRS consistently surpassed BridgePRS, with average improvements of 115.31% in AFR and 59.57% in AMR.
To summarize, XPASS and BridgePRS consistently perform worse than JointPRS. MUSSEL also performs worse than JointPRS in most traits. While SDPRX, PRS-CSx, and PROSPER exhibit top performances similar to JointPRS in some traits, JointPRS remains the only method with robust and accurate predictions across all traits and populations. Notably, tune methods tend to be less accurate for GLGC traits, whereas auto methods tend to be less accurate for PAGE traits, similar to the conclusions for the scenario where the tuning and testing data come from the same cohort.
It is also worth noting that prediction accuracy decreases when the tuning and testing data come from different cohorts. This scenario, often neglected in previous studies, should be emphasized to prevent overestimation of PRS prediction accuracy based on results where the tuning and testing data are from the same cohort. However, the relative performance of different methods remains similar between these two scenarios. Therefore, the optimal method selected when the tuning and testing data come from the same cohort is likely to be similar to the one selected when the tuning and testing data come from different cohorts.
In conclusion, JointPRS exhibits robust and accurate performance across various traits and populations in all three data scenarios, suggesting it as a promising method for multi-population PRS prediction.
Discussion
In this manuscript, we have introduced JointPRS, an efficient approach for accurate multi-population PRS construction, applicable even when only GWAS summary statistics are available. JointPRS integrates a continuous shrinkage model, incorporates a genetic correlation structure, and simultaneously models multiple populations. When a tuning dataset is available, it employs a data-adaptive approach, effectively combining the advantages of both meta-analysis and tuning strategies. Notably, despite its additional modeling of genetic correlation structures and data-adaptive approaches compared to PRS-CSx, the computational time of JointPRS is similar to PRS-CSx (Supplementary Table 17).
JointPRS was benchmarked against six state-of-the-art multi-population PRS methods: PRS-CSx19, PROSPER23, MUSSEL22, SDPRX20, XPASS17, and BridgePRS24. This benchmarking was conducted across three real data scenarios: no tuning dataset (UKBB), tuning and testing data from the same cohort (UKBB), and tuning and testing data from different cohorts (UKBB and AoU), covering 22 quantitative traits and four binary traits in four non-European populations (EAS, AFR, SAS, and AMR).
Consistent with previous studies21,22,23, our results show that no single method performs uniformly best across all traits and data scenarios. This highlights the critical importance of assessing the robust accuracy of each method.
In the absence of a tuning dataset, JointPRS-auto and SDPRX exhibited superior performance across various traits, with JointPRS-auto being more robust and accurate based on the average improvement over SDPRX. We further elucidated the advantages of the structural features within JointPRS-auto, including appropriate sparse distribution, cross-population genetic correlation, and multiple-population modeling. These benefits were validated through comparisons with XPASS, PRS-CSx-auto, and SDPRX. In addition, comparisons of JointPRS-auto, PRS-CSx-auto, and Xwing-auto29 show that the automatically estimated chromosome-wise genetic correlation structure in JointPRS-auto is advantageous to the no-correlation structure in PRS-CSx-auto and the local genetic correlation structure in Xwing-auto.
When a tuning dataset is available, JointPRS consistently demonstrates better or comparable performance across all traits, outperforming other methods that are less robust and accurate. This robust accuracy is attributed to the data-adaptive approach of JointPRS, which allows it to optimally select between meta-analysis and tuning strategies based on the available tuning data. In addition, we observed that when the tuning and testing data come from different cohorts, the prediction accuracy of all PRS methods decreases compared to when they come from the same cohort. This finding implies that for different PRS application scenarios, reliance solely on prediction accuracy from the same cohort may be overly optimistic.
We also evaluated all seven methods across extensive simulation settings, including varying training sample sizes, different tuning dataset scenarios (no tuning data and tuning data with varying sample sizes), varying causal SNP proportions, and varying cross-population genetic correlations. Across all simulation settings, JointPRS exhibited robust and accurate performance, particularly excelling with large training sample sizes, high causal SNP proportions, and large genetic correlations. In addition, JointPRS demonstrated robustness to varying tuning data sample sizes.
Despite its advantages, JointPRS has limitations. JointPRS is currently restricted to HapMap3 SNPs, consistent with methods like PRS-CSx, SDPRX, and BridgePRS19,20,24. However, studies such as CT-SLEB, MUSSEL, and PROSPER21,22,23 highlight that larger SNP lists could enhance prediction accuracy. The MEGA SNP array used in these studies expands variant coverage across multiple populations, offering benefits for non-European populations37. Integrating these variants into JointPRS could potentially improve its performance, especially for non-European populations. In addition, current PRS methods do not account for rare variants or large-effect variants. If we include these variants separately, as demonstrated in RICE and APOE modeling, prediction accuracy could be further improved38,39. Moreover, a disparity in prediction accuracy between European and non-European populations persists. This gap cannot be bridged solely by sophisticated statistical models and requires larger GWAS sample sizes for non-European populations6.
In addition, further exploration of additional techniques or information in PRS modeling will also be valuable, such as the transfer learning techniques proposed by TL-Multi and TL-PRS26,28, cross-trait information considered by XPXP18, fine-mapping information incorporated in PolyPred27, and GWAS subsampling to combine across PRS methods in PUMAS-SL40. However, these approaches may present additional challenges, and further investigations are required to evaluate their benefits.
In conclusion, we have proposed a robust and accurate method for incorporating GWAS summary statistics with possible tuning data. Our simulation and real data studies highlight crucial structures in PRS modeling and factors in real data that influence the accuracy and robustness of multi-population PRS methods. JointPRS stands out as a versatile and reliable method, suitable for diverse genetic prediction scenarios.
Methods
We confirm that our research complies with all relevant ethical regulations. All participants from the UK Biobank provided written informed consent (more information is available at https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/governance). The information of individuals from All of US included in our analyses has been collected according to All of Us Research Program Operational Protocol (https://allofus.nih.gov/article/all-us-research-program-protocol). The detailed consent process of All of Us is described on https://allofus.nih.gov/about/protocol/all-us-consent-process.
JointPRS Model Development and Estimation
In the JointPRS model, we assume that the SNP effect size βj for SNP j across K populations is modeled with a correlated Gaussian prior when SNP j is available for all K populations:
with
Here, we can further express the covariance matrix Σ as the transformation of the correlation matrix
Here \({\rho }_{{k}_{1}{k}_{2}}\) denotes the cross-population genetic correlation between populations k1 and k2. Here, k1, k2 ∈ {EUR, EAS, AFR, SAS, AMR}, k1 ≠ k2. In addition, Ψj is the sample-size normalized effect size for SNP j shared across K populations. This shared effect Ψj is modeled by a continuous shrinkage prior following the PRScsx model and assumed to follow the gamma distribution \({\Psi }_{j} \sim G(1,{\delta }_{j}),\,{\delta }_{j} \sim G(\frac{1}{2},\phi )\)19. Here, the global shrinkage parameter ϕ is dependent on the JointPRS version we choose.
For SNP j available only in a subset of populations, this model is truncated to include only populations that contain SNP j.
Therefore, we can use the following index matrix T to encode the SNP missing patterns across the whole genome for different populations:
Each SNP j is assumed to follow a sum(Tj)-dimension multivariate correlated Gaussian model, with the truncated covariance matrix keeping only rows and columns corresponding to \({T}_{j}^{k}=1\) for k = 1, …, K.
The residual component ϵk is assumed to follow a Gaussian distribution, denoted by \({{{\boldsymbol{\epsilon }}}}^{k} \sim N(0,{\sigma }_{k}^{2}{I}_{{N}_{k}})\). The variance component \({\sigma }_{k}^{2}\) here is assumed to follow a non-informative Jeffreys prior with its density distribution expressed as \(f({\sigma }_{k}^{2})\propto {\sigma }_{k}^{-2}\). We note that the \({\sigma }_{k}^{2}\) are also the diagonal elements of the covariance matrix, thus avoiding the convergence issue. Since different populations do not share samples, we can assume the independence of ϵk across populations. Moreover, since the correlation matrix is symmetric, we only assume all upper triangle elements in the correlation matrix: ρ12, ρ13, ⋯ , ρK−1K to follow a uniform distribution Uniform(0, 1) and estimate each correlation pair automatically from the GWAS summary statistics.
The following sections provide a detailed illustration of the JointPRS versions, JointPRS data preparation, and JointPRS parameter estimation procedures.
JointPRS Versions
Auto Version
The auto version exclusively utilizes GWAS summary statistics and does not require any parameter tuning. Specifically, the input data consists of the original GWAS summary statistics, with the global shrinkage parameter ϕ assumed to follow a standard half-Cauchy prior, \({\phi }^{\frac{1}{2}} \sim {C}^{+}(0,1)\). This version streamlines the process by eliminating the need for additional tuning data or parameter adjustments.
Meta Version
The meta version leverages meta-analysis to integrate the original GWAS summary statistics with tuning datasets, utilizing the METAL software41. This results in updated GWAS summary statistics as input data. Similar to the auto version, the meta version assumes the global shrinkage parameter ϕ follows a standard half-Cauchy prior \({\phi }^{\frac{1}{2}} \sim {C}^{+}(0,1)\). The meta version does not require parameter tuning while integrating the tuning data through GWAS summary statistics updates.
Tune Version
In contrast to the meta version, the tune version uses the original GWAS summary statistics as input data and chooses the global shrinkage parameter ϕ from a broader range: \({\phi }^{\frac{1}{2}} \sim {C}^{+}(0,1)\) and the set ϕ ∈ {10−6, 10−4, 10−2, 1} using the tuning data. More specifically, this version performs a linear combination across populations for each candidate ϕ, selecting the optimal ϕ to derive the optimal weights. This approach ensures the model is finely tuned to achieve accurate performance for each population based on the tuning data.
Data preparation in JointPRS model
We first need to obtain the GWAS summary statistics, which are the marginal least squares effect size estimates for S SNPS in K populations \({\widehat{{{\boldsymbol{\beta }}}}}^{1},{\widehat{{{\boldsymbol{\beta }}}}}^{2},\cdots \,,{\widehat{{{\boldsymbol{\beta }}}}}^{K}\).
Here, yk, Xk, and Nk denote the standardized phenotype vector, the column-standardized genetic matrix in population k, and the GWAS summary statistic sample size, respectively. Note that some SNP data might be missing in each population, and this issue will be carefully addressed in the subsequent algorithm.
We also need the LD matrix \({D}^{k}=\frac{{{{{\boldsymbol{X}}}}^{k}}^{T}{{{\boldsymbol{X}}}}^{k}}{{N}_{k}}\) for each population k, utilizing either the data from the 1000 Genomes Project phase 334 or the UKBB32, as adopted from PRS-CSx19. Due to computational efficiency, the entire genome will be partitioned into Lk independent LD blocks based on the reference data for each population k. During each MCMC iteration, SNP effect sizes are updated sequentially for each population k within each LD block lk. We simplify the notation lk to l during the updates, representing the block variable in the current updating population k.
In the current updating population k, \({{{\boldsymbol{\beta }}}}_{(l)}^{k}={({\beta }_{({l}_{1})}^{k},{\beta }_{({l}_{2})}^{k},\cdots,{\beta }_{({l}_{{s}_{l}})}^{k})}^{T}\) represents the effect size vectors for SNPs in block l of population k, with sl indicating the number of SNPs in block l of population k. The marginal effect size estimates for SNPs in block l across K populations are denoted by \(({\widehat{{{\boldsymbol{\beta }}}}}_{(l)}^{1},{\widehat{{{\boldsymbol{\beta }}}}}_{(l)}^{2},\cdots \,,{\widehat{{{\boldsymbol{\beta }}}}}_{(l)}^{K})\). In addition, \({{{\boldsymbol{D}}}}_{(l)}^{k}\) denotes the LD matrix for SNPs in block l of population k, whereas \({{{\mathbf{\Psi }}}}_{(l)}=diag({\psi }_{({l}_{1})},{\psi }_{({l}_{2})},\cdots \,,{\psi }_{({l}_{{s}_{l}})})\) represents the shrinkage matrix for SNPs in block l, and the covariance matrix for SNPs in block l of population k are denoted by \(({{{\mathbf{\Sigma }}}}_{({l}_{1})},{{{\mathbf{\Sigma }}}}_{({l}_{2})},\cdots \,,{{{\mathbf{\Sigma }}}}_{({l}_{{s}_{l}})})=({\Psi }_{{l}_{1}}{{\boldsymbol{M}}}{{\mathbf{\Sigma }}}{{\boldsymbol{M}}},{\Psi }_{{l}_{2}}{{\boldsymbol{M}}}{{\mathbf{\Sigma }}}{{\boldsymbol{M}}},\cdots \,,{\Psi }_{{l}_{{s}_{l}}}{{\boldsymbol{M}}}{{\mathbf{\Sigma }}}{{\boldsymbol{M}}})\).
MCMC and MH Algorithm in JointPRS model
We use 1000 × K MCMC iterations with the first 500 × K steps as burn-in, as suggested by PRS-CSx19. For each iteration step, we update parameters by the following procedure:
-
1.
Update \({{{\boldsymbol{\beta }}}}^{1},{{{\boldsymbol{\beta }}}}^{2},\cdots \,,{{{\boldsymbol{\beta }}}}^{K}\,| \,\left({\widehat{{{\boldsymbol{\beta }}}}}^{1},{\widehat{{{\boldsymbol{\beta }}}}}^{2},\cdots \,,{\widehat{{{\boldsymbol{\beta }}}}}^{K}\right),\left({\sigma }_{1}^{2},{\sigma }_{2}^{2},\cdots \,,{\sigma }_{K}^{2}\right),{{\boldsymbol{D}}},{{\mathbf{\Psi }}},{{\mathbf{\Sigma }}},{{\boldsymbol{M}}}\):
For the current updating population k in block l, we update the posterior effect size by the following:
$$\begin{array}{rcl}&&{{{\boldsymbol{\beta }}}}_{(l)}^{k}\,| \,{{{\boldsymbol{D}}}}_{(l)}^{k},\widehat{{{{\boldsymbol{\beta }}}}_{(l)}^{k}},{\sigma }_{k}^{2} \sim MVN({{{\boldsymbol{\mu }}}}_{(l)}^{k},{{{\mathbf{\Sigma }}}}_{(l)}^{k}),\\ &&{{{\boldsymbol{\mu }}}}_{(l)}^{k}=\frac{{N}_{k}}{{\sigma }_{k}^{2}}\cdot {{{\mathbf{\Sigma }}}}_{(l)}^{k}\cdot \left(\widehat{{{{\boldsymbol{\beta }}}}_{(l)}^{k}}-\frac{{\sigma }_{k}^{2}}{\sqrt{{N}_{k}}}\cdot {{{\boldsymbol{A}}}}_{(l)}^{k}\cdot {{{\boldsymbol{N}}}}_{sqrt}^{k}\right),\\ &&{{{\mathbf{\Sigma }}}}_{(l)}^{k}=\frac{{\sigma }_{k}^{2}}{{N}_{k}}{\left({{{\boldsymbol{D}}}}_{(l)}+{\sigma }_{k}^{2}\cdot diag\left(\frac{{\widetilde{\Sigma }}_{kk}^{{t}_{j}}}{{\psi }_{({l}_{j})}}\right)\right)}^{-1}.\end{array}$$(4)Here,
$$\begin{array}{rcl}&&{{{\boldsymbol{A}}}}_{(l)}^{k}=\left(\begin{array}{cccccc}\frac{{\widetilde{\Sigma }}_{k1}^{{t}_{1}}}{{\psi }_{({l}_{1})}}{\beta }_{({l}_{1})}^{1}&\cdots \,&\frac{{\widetilde{\Sigma }}_{kk-1}^{{t}_{1}}}{{\psi }_{({l}_{1})}}{\beta }_{({l}_{1})}^{k-1}&\frac{{\widetilde{\Sigma }}_{kk+1}^{{t}_{1}}}{{\psi }_{({l}_{1})}}{\beta }_{({l}_{1})}^{k+1}&\cdots \,&\frac{{\widetilde{\Sigma }}_{kK}^{{t}_{1}}}{{\psi }_{({l}_{1})}}{\beta }_{({l}_{1})}^{K}\\ \vdots &\ddots &\vdots &\vdots &\ddots &\vdots \\ \frac{{\widetilde{\Sigma }}_{k1}^{{t}_{1}}}{{\psi }_{({l}_{{s}_{l}})}}{\beta }_{({l}_{{s}_{l}})}^{1}&\cdots \,&\frac{{\widetilde{\Sigma }}_{kk-1}^{{t}_{{s}_{l}}}}{{\psi }_{({l}_{{s}_{l}})}}{\beta }_{({l}_{{s}_{l}})}^{k-1}&\frac{{\widetilde{\Sigma }}_{kk+1}^{{t}_{{s}_{l}}}}{{\psi }_{({l}_{{s}_{l}})}}{\beta }_{({l}_{{s}_{l}})}^{k+1}&\cdots \,&\frac{{\widetilde{\Sigma }}_{kK}^{{t}_{{s}_{l}}}}{{\psi }_{({l}_{{s}_{l}})}}{\beta }_{({l}_{{s}_{l}})}^{K}\end{array}\right),\\ &&{{{\boldsymbol{N}}}}_{sqrt}^{k}={\left(\begin{array}{cccccc}\sqrt{{N}_{1}}&\cdots &\sqrt{{N}_{k-1}}&\sqrt{{N}_{k+1}}&\cdots &\sqrt{{N}_{K}}\end{array}\right)}^{T},\\ &&\widetilde{{{\mathbf{\Sigma }}}}=\left(\begin{array}{cccc}{\widetilde{\Sigma }}_{11}&{\widetilde{\Sigma }}_{12}&\cdots \,&{\widetilde{\Sigma }}_{1K}\\ {\widetilde{\Sigma }}_{21}&{\widetilde{\Sigma }}_{22}&\cdots \,&{\widetilde{\Sigma }}_{2K}\\ \vdots &\vdots &\ddots &\ddots \\ {\widetilde{\Sigma }}_{K1}&{\widetilde{\Sigma }}_{K2}&\cdots \,&{\widetilde{\Sigma }}_{KK}\end{array}\right)={{{\mathbf{\Sigma }}}}^{-1}={\left(\begin{array}{cccc}{\sigma }_{1}^{2}&{\sigma }_{12}&\cdots &{\sigma }_{1K}\\ {\sigma }_{21}&{\sigma }_{2}^{2}&\cdots &{\sigma }_{2K}\\ \vdots &\vdots &\ddots &\vdots \\ {\sigma }_{K1}&{\sigma }_{K2}&\cdots &{\sigma }_{K}^{2}\end{array}\right)}^{-1},\\ &&{\widetilde{{{\mathbf{\Sigma }}}}}^{{t}_{j}}={\left({{{\mathbf{\Sigma }}}}^{{t}_{j}}\right)}^{-1}\,{{\rm{the}}} {{\rm{inverse}}} {{\rm{of}}} {{\rm{the}}} {{\rm{covariance}}} {{\rm{matrix}}} {{\rm{for}}} {{\rm{non}}}-{{\rm{missing}}} {{\rm{populations}}} {{\rm{in}}} {\rm{SNP}}\,\,j.\end{array}$$ -
2.
Update \({\sigma }_{k}^{2}| {{\mathbf{\Psi }}},{{{\boldsymbol{\beta }}}}^{k},{\widehat{{{\boldsymbol{\beta }}}}}^{k},{{{\boldsymbol{D}}}}^{k}:\)
For the current updating population k, we update the variance by the following:
$${\sigma }_{k}^{2}| {{\mathbf{\Psi }}},{{{\boldsymbol{\beta }}}}^{k},{\widehat{{{\boldsymbol{\beta }}}}}^{k},{{{\boldsymbol{D}}}}^{k} \sim iG\left(\frac{{N}_{k}+{S}_{k}}{2},\frac{{N}_{k}}{2}\left\{1-2{\sum }_{l=1}^{L}{{{{\boldsymbol{\beta }}}}_{(l)}^{k}}^{T}{\widehat{{{\boldsymbol{\beta }}}}}_{(l)}^{k}\right.\right. \\ \left.\left.+{\sum }_{l=1}^{L}{{{{\boldsymbol{\beta }}}}_{(l)}^{k}}^{T}\left({{{\boldsymbol{D}}}}_{(l)}+{{{\mathbf{\Psi }}}}_{(l)}^{-1}\right){{{\boldsymbol{\beta }}}}_{(l)}^{k}\right\}\right).$$(5)Here, iG(α, β) is the inverse-gamma distribution with the probability density function
$$f(x;\alpha,\beta )=\frac{{\beta }^{\alpha }}{\Gamma (\alpha )}{\left(\frac{1}{x}\right)}^{\alpha+1}\exp \left(-\frac{\beta }{x}\right).$$ -
3.
Update each pair of correlation \({\rho }_{{k}_{1}{k}_{2}}\) from the upper triangle under different constraints. Based on the joint model we propose, we can estimate the cross-population correlation \({\rho }_{{k}_{1}{k}_{2}}\) for two populations k1 and k2 (k1 ∈ {EUR, EAS, AFR, SAS, AMR}, k2 ∈ {EUR, EAS, AFR, SAS, AMR}) by assuming:
$$\left(\begin{array}{c}{\beta }_{j}^{{k}_{1}}\\ {\beta }_{j}^{{k}_{2}}\end{array}\right) \sim N\left(0,{\Psi }_{j}{{\boldsymbol{M}}}\left(\begin{array}{cc}\sqrt{{\sigma }_{{k}_{1}}^{2}}&0\\ 0&\sqrt{{\sigma }_{{k}_{2}}^{2}}\end{array}\right)\left(\begin{array}{cc}1&{\rho }_{{k}_{1}{k}_{2}}\\ {\rho }_{{k}_{1}{k}_{2}}&1\end{array}\right)\left(\begin{array}{cc}\sqrt{{\sigma }_{{k}_{1}}^{2}}&0\\ 0&\sqrt{{\sigma }_{{k}_{2}}^{2}}\end{array}\right){{\boldsymbol{M}}}\right).$$We note that when we update \({\rho }_{{k}_{1}{k}_{2}}\), we only use SNPs shared by populations k1 and k2. And if we denote the posterior distribution of \({\rho }_{{k}_{1}{k}_{2}}\) as \({h}_{r}({\rho }_{{k}_{1}{k}_{2}})\), we have
$$\begin{array}{l}{h}_{r}({\rho }_{{k}_{1}{k}_{2}})\equiv f({{\boldsymbol{\beta }}}| {\rho }_{{k}_{1}{k}_{2}})\\ \propto {\left({\sigma }_{{k}_{1}}^{2}{\sigma }_{{k}_{2}}^{2}(1-{\rho }_{{k}_{1}{k}_{2}}^{2})\right)}^{-\frac{{S}_{{k}_{1}{k}_{2}}}{2}}\exp \left\{-\frac{1}{2}\cdot \frac{{N}_{{k}_{1}}{N}_{k2}}{{\sigma }_{{k}_{1}}^{2}{\sigma }_{{k}_{2}}^{2}(1-{\rho }_{{k}_{1}{k}_{2}}^{2})}\right.\\ \left.\cdot {\sum }_{j=1}^{{S}_{{k}_{1}{k}_{2}}}\frac{1}{{\Psi }_{j}}\left(\frac{{\sigma }_{{k}_{2}}^{2}}{{N}_{{k}_{2}}}{\beta }_{{k}_{1}}^{2}-2\frac{\sqrt{{\sigma }_{{k}_{1}}^{2}{\sigma }_{{k}_{2}}^{2}}\cdot {\rho }_{{k}_{1}{k}_{2}}}{\sqrt{{N}_{{k}_{1}}{N}_{{k}_{2}}}}{\beta }_{j}^{{k}_{1}}{\beta }_{j}^{{k}_{2}}+\frac{{\sigma }_{{k}_{1}}^{2}}{{N}_{{k}_{1}}}{\beta }_{{k}_{2}}^{2}\right)\right\}\end{array}.$$Here \({S}_{{k}_{1}{k}_{2}}=\frac{K\cdot (K-1)}{2}\) is a constant representing the total pairs of cross-population genetic correlation.
Since there is no closed-form distribution to update the correlation \({\rho }_{{k}_{1}{k}_{2}}\), we use the following MH algorithm:
Algorithm 1
MH algorithm for JointPRS
Ensure: δr = 0.05
1: while itr ≤ n_iter do
2: while 1 ≤ k1 ≤ K − 1 do
3: while k1 < k2 ≤ K do
4: \({\rho }_{{k}_{1}{k}_{2}}^{*}=Uniform({\rho }_{{k}_{1}{k}_{2}}-{\delta }_{r},{\rho }_{{k}_{1}{k}_{2}}+{\delta }_{r})\)
5: if \({\rho }_{{k}_{1}{k}_{2}}^{*}\in [0,cons]\) then
6: log_ratio = \(log({h}_{r}({\rho }_{{k}_{1}{k}_{2}}^{*}))-log({h}_{r}({\rho }_{{k}_{1}{k}_{2}}))\)
7: if \(\exp (\log\_{\mbox{ratio}})\ge random.Uniform(0,1)\) then
8: \({\rho }_{{k}_{1}{k}_{2}}={\rho }_{{k}_{1}{k}_{2}}^{*}\)
9: end if
10: end if
11: end while
12: end while
13: end while
Note: Here we choose cons = 0 when ϕ = 10−6 which is equivalent to the PRS-CSx method, to avoid convergence issue and choose cons = 0.99 for all other situations to consider the possible positive correlations between population k1 and k2.
Then we can also update the corresponding covariance pair \({\sigma }_{{k}_{1}{k}_{2}}=\sqrt{{\sigma }_{{k}_{1}}^{2}{\sigma }_{{k}_{2}}^{2}}\cdot {\rho }_{{k}_{1}{k}_{2}}\).
-
4.
Update Ψj∣βj, σ2, δj:
For each SNP j, we update the corresponding shrinkage parameter by the following:
$$ {\Psi }_{j}\,| \,{{{\boldsymbol{\beta }}}}_{j},{{\boldsymbol{M}}}{{\mathbf{\Sigma }}}{{\boldsymbol{M}}},{\delta }_{j} \sim giG\left(a-\frac{K}{2},2{\delta }_{j},{{{{\boldsymbol{\beta }}}}_{j}}^{T}{\left({{\boldsymbol{M}}}{{\mathbf{\Sigma }}}{{\boldsymbol{M}}}\right)}^{-1}{{{\boldsymbol{\beta }}}}_{j}\right)\\ \equiv giG\left(a-\frac{K}{2},2{\delta }_{j},{{{{\boldsymbol{\beta }}}}_{j}}^{T}\widetilde{{{\mathbf{\Sigma }}}}{{{\boldsymbol{\beta }}}}_{j}\right).$$(6)Here, giG(λ, ρ, χ) is the three-parameter generalized inverse Gaussian distribution with the probability density function
$$f(x;\lambda,\rho,\chi )=\frac{{\left(\rho /\chi \right)}^{\lambda /2}}{2{K}_{\lambda }\sqrt{\rho \chi }}{x}^{\lambda -1}{e}^{-(\rho x+\chi /x)/2},\quad x > 0,\quad \rho > 0,\quad \chi > 0,$$where Kλ is the modified Bessel function of the second kind.
-
5.
Update δj∣Ψj:
For each SNP j, we update the distribution parameter by the following:
$${\delta }_{j}\,| \,{\Psi }_{j} \sim G(a+b,{\Psi }_{j}+\phi ).$$(7)Here, G(c, d) is the Gamma distribution with the probability density function
$$f(x;c,d)=\frac{{(d)}^{c}}{\Gamma (c)}{(x)}^{c-1}{e}^{-d\cdot x}.$$
The detailed proof for the above updating procedure can be found in the Supplementary Notes (MCMC and MH algorithm in the JointPRS Section).
JointPRS Data-Adaptive Approach Implementation
When a tuning dataset is available, JointPRS employs a data-adaptive approach to select the PRS between two versions: the meta version and the tune version.
Meta version
For the meta version in JointPRS, we utilized the SNP effect size estimates, denoted as \({\hat{{{\boldsymbol{\beta }}}}}_{{{\rm{meta}}},t}\), for the target population t. This estimate was applied to the tuning dataset to obtain the PRS estimation result:
To address overfitting issues noted in Section "Overview of JointPRS", we use \({{\mbox{PRS}}}_{{{\rm{auto}}},t}={{\boldsymbol{X}}}{\hat{{{\boldsymbol{\beta }}}}}_{{{\rm{auto}}},t}\) to approximate the performance of PRSmeta,t. Here \({\hat{{{\boldsymbol{\beta }}}}}_{{{\rm{auto}}},t}\) is the SNP effect estimate from the auto version for the target population t.
Tune Version
For the tune version, SNP effect size estimates, \({\hat{{{\boldsymbol{\beta }}}}}_{{{\rm{tune}}},k}(\phi )\), were obtained for each of the K populations (k = 1, 2, ⋯ K). These estimates were applied to the tuning dataset to generate K PRS estimation results for each ϕ ∈ {10−6, 10−4, 10−2, 1, auto} as follows:
For convenience in model comparison, we set ϕ = auto and use PRStune,k(auto) ≡ PRSauto,k to approximate the tune version PRS for each population k.
Model comparison
After establishing the two models and their approximation to avoid overfitting, we compare the performance of the two approximated versions on the tuning data using the F-test. Specifically, the model comparison between the tune version and the meta version approximately tests whether the full model is statistically significant compared to the submodel, as follows:
We use residual sum of squares (RSS) from standard linear regression of the above two models (RSSauto,t and RSSauto,1:K, respectively) to evaluate the reduction in RSS:
The right-tail p-value of this F-test statistic is assessed with F(K − 1, #{tuning data samples} − K), using 0.05 as the p-value threshold. The full model is accepted only when the p-value is less than 0.05.
Maximum likelihood estimates (MLE) are obtained for the above two logistic regression models. We then calculate the χ2-test statistic:
The right-tail p-value of this χ2-test statistic is assessed with χ2(K − 1), using 0.05 as the p-value threshold. The full model is accepted only when the p-value is less than 0.05. Here, \(\log {{\mathcal{L}}}\,{\mbox{(submodel)}}\,\) represents the log-likelihood of the submodel, and \(\log {{\mathcal{L}}}\,{\mbox{(full model)}}\,\) represents the log-likelihood of the full model.
The algorithm to implement the JointPRS data-adaptive approach can be found in the Supplementary Notes (Data-Adaptive Approach in JointPRS Section).
Overview of Existing Methods
XPASS
The XPASS method17 jointly integrates GWAS summary statistics and LD structures from two populations through a bivariate Gaussian distribution. The genetic correlation structure is incorporated into the model to facilitate the information transference from the auxiliary to the target population. The following model illustrates its idea for the prior on the effect size of each SNP j for population 1 and population 2.
This method is further augmented by considering population-specific effects using selected SNPs based on the P+T procedure, treating them as fixed effects during the estimation. Although this method does not need tuning parameters, when a tuning dataset is available, a linear combination of scores from both populations will be performed to derive the final score for the target population in our analysis.
SDPRX
The SDPRX method20 establishes a hierarchical Bayesian framework, jointly modeling GWAS summary statistics and LD structures from two populations. This framework comprises four components to characterize the genetic architecture, identifying SNPs as no effect, being population-specific, or being shared across two populations. The essential component of this method is the shared component, which uses a mixture of bivariate Gaussian distributions, coupled with a precalculated genetic correlation to approximate the true shared structure. The following equation represents the prior on the effect sizes for each SNP j for population 1 and population 2.
Although this method does not need tuning parameters, in the presence of a tuning dataset, a linear combination of scores from both populations will be performed to obtain the final score for the target population in our analysis.
PRS-CSx(-auto)
The PRS-CSx model19, an extended Bayesian model of the PRS-CS framework42, integrates GWAS summary statistics and LD structures from multiple populations by utilizing a shared continuous shrinkage prior for each SNP j in population k as the following equation.
In scenarios when a tuning dataset is unavailable, the model leverages a full Bayesian approach to estimate the global shrinkage parameter ϕ0.5 ~ C+(0, 1). However, when a tuning dataset is available, PRS-CSx evaluates a predefined set of global shrinkage parameters ϕ ∈ {10−6, 10−4, 10−2, 1, auto}. For each parameter in the set, the obtained scores for each population will be linearly combined to obtain the final score for each population. This integration relies on the tuning dataset, and the shrinkage parameter that leads to the best-performing combined scores will then be selected based on the prediction accuracy of the tuning dataset.
MUSSEL
The MUSSEL method22 jointly models GWAS summary statistics and LD structures from multiple populations, utilizing the following multivariate spike-and-slab prior with an incorporated genetic correlation structure for the effect size of each SNP j in each block (J).
This method requires a tuning dataset due to the presence of two sets of tuning parameters within the model: the causal SNP proportion and heritability in each population \({h}_{k}^{2},{p}_{k}\,(k=1,\cdots \,,K)\) and the between-population correlation ρij (i ≠ j, i = 1, ⋯ , K, j = 1, ⋯ , K). In addition, a super learning step, which selects linear methods from Lasso, Ridge, Elastic net, and Linear regression, is introduced to further integrate the scores from various populations and tuning parameters.
PROSPER
The PROSPER method23 integrates GWAS summary statistics and LD structures from multiple populations, leveraging a linear regression with a combination of Lasso and ridge penalties for estimation in order to consider the spraity of genetic effect sizes and the similarity across populations. The objective function to optimize the effect size vectors of SNP i in K populations can be represented by the following equation
This method requires a tuning dataset to determine the tuning parameters associated with these penalties \(({\delta }_{i},{\lambda }_{i},{c}_{{i}_{1}{i}_{2}},i,{i}_{1},{i}_{2}=1,c\ldots M)\). A further ensemble step (also called super learning, which selects linear methods from Lasso, Ridge, Elastic net, and Linear regression) is implemented to combine PRS scores generated across different penalty parameters and populations.
BridgePRS
The BridgePRS method24 integrates GWAS summary statistics from two populations to consider shared and population-specific SNP effects for the target population. In the first stage, it models the SNP effect sizes for each population under the following zero-centered Gaussian prior
In the second stage, it utilizes the auxiliary population (population 1) to determine the prior of the target population under the following Gaussian model
And they finally linearly combine the PRS from the target population with the above joint PRS based on the ridge regression fit.
XWing(-auto)
XWing method29 integrates GWAS summary statistics and LD information from multiple populations, incorporating portable genetic effects identified through quantifying local genetic correlation between populations with continuous shrinkage prior to construct cross-population PRS. The algorithm consists of three main steps: Firstly, Xwing detects local genetic correlations between a target population and an auxiliary population using a scan statistic approach. Secondly, Xwing incorporates the identified correlated regions as an annotation into for the SNP effect sizes modeling:
Here, λf(j),k is the annotation-dependent shrinkage parameter that allows differential regularization based on whether the SNP is annotated or not.
The global shrinkage parameter ϕ was set to ϕ ∈ {10−6, 10−4, 10−2, 1, auto} Population-specific PRS models are generated for each target-auxiliary population pairing. Finally, X-Wing combines all generated PRS models via an innovative repeated learning strategy to estimate optimal linear combination weights.
Statistics & reproducibility
Simulation settings
We simulated the true effect sizes using a spike and slab model for five populations: EUR, EAS, AFR, SAS, and AMR:
We considered four different sparsity levels for the proportion of causal SNPs: p = 0.1, 0.01, 0.001, and 5 × 10−4. The total heritability of common SNPs was set to h2 = 0.4, and the cross-population genetic correlation was set to ρ = 0.8 in our primary analysis. We also conducted two secondary analyses. The first evaluated a low-heritability scenario with h2 = 0.1, while keeping the causal SNPs proportion fixed at p = 0.1 and the genetic correlation at ρ = 0.8. The second analysis explored four genetic correlation settings ρ = 0, 0.2, 0.4, 0.6, and 0.8, with fixed causal SNPs proportion p = 0.1 and heritability h2 = 0.4. The total number of SNPs was S = 1, 203, 063, based on the HapMap3 SNP list43. We generated five replicates of the effect sizes for each setting.
Using these parameters and effect sizes, we utilized GCTA-sim44 to generate phenotypes for analysis in the training, tuning, and testing datasets. PLINK245 was used to derive GWAS summary statistics in the training dataset. For the GWAS summary statistics provided for each method, we extracted SNPs common to all methods’ LD reference panels across all populations, resulting in 717, 985 SNPs. This intersection ensures that each method used the same number of SNPs in the GWAS data for fair comparisons.
Real data analysis
We collected GWAS summary statistics for 26 traits across five populations (EUR, EAS, AFR, SAS, and AMR). Each trait was initially categorized as either continuous or binary and further subdivided based on the number of populations available, yielding four distinct groups:
-
1.
GLGC traits (4 continuous): HDL-cholesterol (HDL), LDL-cholesterol (LDL), total cholesterol (TC), and triglycerides (logTG), each with GWAS data from five populations (EUR, EAS, AFR, SAS, and AMR).
-
2.
PAGE traits (5 continuous): Height, body mass index (BMI), systolic blood pressure (SBP), diastolic blood pressure (DBP), and platelet count (PLT), each with GWAS data from three populations (EUR, EAS, and AFR).
-
3.
BBJ traits (13 continuous): White blood cell (WBC), neutrophil (ENU), lymphocyte (LYM), monocyte (MON), eosinophil (EOS), red blood cell (RBC), hematocrit (HCT), mean corpuscular hemoglobin (MCH), mean corpuscular volume (MCV), hemoglobin (HB), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and γ-glutamyl transpeptidase (GGT), each with GWAS data from two populations (EUR and EAS).
-
4.
Binary traits (4 binary): Type 2 diabetes (T2D), breast cancer (BrC), coronary artery disease (CAD), and lung cancer (LuC), each with GWAS data from two or three populations (EUR, EAS, and possibly AFR).
We analyzed 22 quantitative traits by examining how much variance in the phenotype was explained by each derived PRS. Specifically, we calculated
where SS0 and SS1 are the residual sums of squares for the null and full models, respectively. The null model included age, sex, and the top 20 genetic principal components (PCs), while the full model added the PRS as an additional covariate. For the four binary traits, we employed a logistic model and evaluated prediction accuracy using the area under the receiver operating characteristic curve (AUC).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The BBJ GWAS summary-level statistics data are available in the NBDC Human Database under accession codes hum0014-v36 [https://humandbs.dbcls.jp/en/hum0014-v36] and hum0197-v23 [https://humandbs.dbcls.jp/en/hum0197-v23]. The BCAC GWAS summary-level statistics data are available in the GWAS Catalog under accession code 29059683. The BCX GWAS summary-level statistics data are available in the GWAS Catalog under accession code 32888493. The CARDIoGRAM GWAS summary-level statistics data are available in the GWAS Catalog under accession code 21378990. The TRICL-ILCCO and LC3 GWAS summary-level statistics data are available in the GWAS Catalog under accession code 28604730. The DIAGRAM GWAS summary-level statistics data are available in the GWAS Catalog under accession code 28566273. The GIANT GWAS summary-level statistics data are available from the GIANT consortium [https://portals.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files]. The GLGC GWAS summary-level statistics data are available from the Global Lipids Genetics Consortium [https://csg.sph.umich.edu/willer/public/glgc-lipids2021/]. The ICBP GWAS summary-level statistics data are available in the GWAS Catalog under accession code 30224653. The PAGE GWAS summary-level statistics data are available in the GWAS Catalog under accession code 31217584. The UKBB Liver Enzymes GWAS summary-level statistics data are available in the GWAS Catalog under accession code 33972514. Supplementary Information files are provided with this paper. Source data are provided in this paper.
Code availability
All codes for simulation studies, real-data analysis, and the main figure plot are available on GitHub at https://github.com/LeqiXu/JointPRS_analysis46. Software implementing JointPRS is available at GitHub https://github.com/LeqiXu/JointPRS47. Other methods and their corresponding repositories include: XPASS at https://github.com/YangLabHKUST/XPASS, SDPRX at https://github.com/eldronzhou/SDPRX, PRS-CSx at https://github.com/getian107/PRScsx, MUSSEL at https://github.com/Jin93/MUSSEL, PROSPER at https://github.com/Jingning-Zhang/PROSPER, and BridgePRS at https://github.com/clivehoggart/BridgePRS.
References
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Seibert, T. M. et al. Polygenic hazard score to guide screening for aggressive prostate cancer: development and validation in large scale cohorts. BMJ 360, https://doi.org/10.1136/bmj.j5757 (2018).
Abraham, G., Rutten-Jacobs, L. & Inouye, M. Risk prediction using polygenic risk scores for prevention of stroke and other cardiovascular diseases. Stroke 52, 2983–2991 (2021).
Dehestani, M., Liu, H. & Gasser, T. Polygenic risk scores contribute to personalized medicine of parkinson’s disease. J. Pers. Med. 11, 1030 (2021).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019).
Martin, A. R. et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591 (2019).
Lewis, C. M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 1–11 (2020).
Graham, S. E. et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 600, 675–679 (2021).
Akiyama, M. et al. Characterizing rare and low-frequency height-associated variants in the japanese population. Nat. Commun. 10, 4393 (2019).
Wojcik, G. L. et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518 (2019).
Akiyama, M. et al. Genome-wide association study identifies 112 new loci for body mass index in the japanese population. Nat. Genet. 49, 1458–1467 (2017).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Chen, M.-H. et al. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell 182, 1198–1213 (2020).
Suzuki, K. et al. Identification of 28 new susceptibility loci for type 2 diabetes in the japanese population. Nat. Genet. 51, 379–386 (2019).
Ishigaki, K. et al. Large-scale genome-wide association study in a japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 52, 669–679 (2020).
Jia, G. et al. Genome-wide association analyses of breast cancer in women of african ancestry identify new susceptibility loci and improve risk prediction. Nat. Genet. 56, 819–826 (2024).
Cai, M. et al. A unified framework for cross-population trait prediction by leveraging the genetic correlation of polygenic traits. Am. J. Hum. Genet. 108, 632–655 (2021).
Xiao, J. et al. Xpxp: improving polygenic prediction by cross-population and cross-phenotype analysis. Bioinformatics 38, 1947–1955 (2022).
Ruan, Y. et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580 (2022).
Zhou, G., Chen, T. & Zhao, H. Sdprx: A statistical method for cross-population prediction of complex traits. Am. J. Hum. Genet. 110, 13–22 (2023).
Zhang, H. et al. A new method for multiancestry polygenic prediction improves performance across diverse populations. Nat. Genet. 55, 1757–1768 (2023).
Jin, J. et al. Mussel: Enhanced bayesian polygenic risk prediction leveraging information across multiple ancestry groups. Cell Genom. 4, https://doi.org/10.1016/j.xgen.2024.100539 (2024).
Zhang, J. et al. An ensemble penalized regression method for multi-ancestry polygenic risk prediction. Nat. Commun. 15, 3238 (2024).
Hoggart, C. J. et al. Bridgeprs leverages shared genetic effects across ancestries to increase polygenic risk score portability. Nat. Genet. 56, 180–186 (2024).
Amariuta, T. et al. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat. Genet. 52, 1346–1354 (2020).
Tian, P. et al. Multiethnic polygenic risk prediction in diverse populations through transfer learning. Front. Genet. 13, 906965 (2022).
Weissbrod, O. et al. Leveraging fine-mapping and multipopulation training data to improve cross-population polygenic risk scores. Nat. Genet. 54, 450–458 (2022).
Zhao, Z., Fritsche, L. G., Smith, J. A., Mukherjee, B. & Lee, S. The construction of cross-population polygenic risk scores using transfer learning. Am. J. Hum. Genet. 109, 1998–2008 (2022).
Miao, J. et al. Quantifying portable genetic effects and improving cross-ancestry genetic prediction with gwas summary statistics. Nat. Commun. 14, 832 (2023).
Mostafavi, H. et al. Variable prediction accuracy of polygenic scores within an ancestry group. Elife 9, e48376 (2020).
Brown, B. C., Ye, C. J., Price, A. L. & Zaitlen, N. Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet. 99, 76–88 (2016).
Sudlow, C. et al. Uk biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
of Us Research Program Investigators, A. The “all of us” research program. N. Engl. J. Med. 381, 668–676 (2019).
Consortium, G. P. et al. A global reference for human genetic variation. Nature 526, 68 (2015).
Zheng, J. et al. Ld hub: a centralized database and web interface to perform ld score regression that maximizes the potential of summary level gwas data for snp heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017).
Bulik-Sullivan, B. K. et al. Ld score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Bien, S. A. et al. Strategies for enriching variant coverage in candidate disease loci on a multiethnic genotyping array. PloS ONE 11, e0167758 (2016).
Williams, J. et al. Integrating common and rare variants improves polygenic risk prediction across diverse populations. Preprint at https://doi.org/10.1101/2024.11.05.24316779 (2024).
Leonenko, G. et al. Identifying individuals with high risk of alzheimer’s disease using polygenic risk scores. Nat. Commun. 12, 4506 (2021).
Zhao, Z. et al. One score to rule them all: regularized ensemble polygenic risk prediction with gwas summary statistics. Preprint at https://doi.org/10.1101/2024.11.27.625748 (2024).
Willer, C. J., Li, Y. & Abecasis, G. R. Metal: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Ge, T., Chen, C.-Y., Ni, Y., Feng, Y.-C. A. & Smoller, J. W. Polygenic prediction via bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Consortium, I. H. et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52 (2010).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. Gcta: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Chang, C. C. et al. Second-generation plink: rising to the challenge of larger and richer datasets. Gigascience 4, s13742–015 (2015).
Xu, L. JointPRS: A data-adaptive framework for multi-population genetic risk prediction incorporating genetic correlation. JointPRS data analysis codes. Zenodo https://doi.org/10.5281/zenodo.15060561 (2025).
Xu, L. JointPRS: A data-adaptive framework for multi-population genetic risk prediction incorporating genetic correlation. JointPRS software. Zenodo https://doi.org/10.5281/zenodo.15060557 (2025).
Wood, A. R. et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186 (2014).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 50, 1412–1425 (2018).
Vuckovic, D. et al. The polygenic and monogenic basis of blood traits and diseases. Cell 182, 1214–1231 (2020).
Pazoki, R. et al. Genetic analysis in european ancestry individuals identifies 517 loci associated with liver enzymes. Nat. Commun. 12, 2579 (2021).
Scott, R. A. et al. An expanded genome-wide association study of type 2 diabetes in europeans. Diabetes 66, 2888–2902 (2017).
Zhang, H. et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat. Genet. 52, 572–581 (2020).
Michailidou, K. et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017).
Schunkert, H. et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 43, 333–338 (2011).
McKay, J. D. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 49, 1126–1132 (2017).
Acknowledgements
This research was supported in part by NIH grant R01 HG012735 and NSF grant DMS 2310836. We thank Jiaqi Hu, Dr. Chi Zhang, and Dr. Yingxin Lin for their invaluable discussions. We are also grateful to Dr. Tian Ge, Dr. Jingning Zhang, Dr. Jin Jin, Jiacheng Miao, and Dr. Hanmin Guo for sharing their codes, LD reference panels, and for their suggestions on implementing various methods. Our research utilized the UK Biobank resource under approved data request (refs: 29900) and the All of Us resource. We gratefully acknowledge the UK Biobank and All of Us participants for their contributions, without whom this research would not have been possible. We also thank the National Institutes of Health’s All of Us Research Program for making available the participant data examined in this study.
Author information
Authors and Affiliations
Contributions
H. Zhao, L.G., and L.X. conceived and designed the study. L.G. and L.X. developed the statistical framework and algorithm. L.X. conducted all simulations and real-data analyses under the supervision of H. Zhao; H. Zhang and G.Z. assisted with simulations; G.Z., Y.D., and H. Zhang assisted with real-data analyses; L.X. developed the JointPRS software. L.X., G.Z., W.J., H. Zhang, L.G., and H. Zhao drafted the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, L., Zhou, G., Jiang, W. et al. JointPRS: A data-adaptive framework for multi-population genetic risk prediction incorporating genetic correlation. Nat Commun 16, 3841 (2025). https://doi.org/10.1038/s41467-025-59243-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59243-x
