
Abstract
Dihydroartemisinic acid (DHAA) converts into antimalarial drug artemisinin (ART) by auto-oxidation. High production of artemisinic acid (AA) has been achieved by fermentation of engineered Saccharomyces cerevisiae, and AA can be converted into ART through DHAA by chemical synthesis. However, there is no enzyme reported to catalyze the conversion of AA to DHAA. Here, we report a dihydroartemisinic acid dehydrogenase (AaDHAADH) from Artemisia annua L, which catalyzes the bidirectional conversion between AA and DHAA. An optimized mutant AaDHAADH (P26L) is obtained through site-directed mutagenesis and its activity toward AA is 2.82 times that of the original gene. De novo synthesis of DHAA is achieved in S. cerevisiae using the targeted optimized gene AaDHAADH (P26L). Furthermore, 3.97 g/L of DHAA is obtained by fermentation of engineered S. cerevisiae in 5 L bioreactor. The discovery of AaDHAADH provides a more convenient and efficient alternative route for ART biosynthesis.
Similar content being viewed by others

Introduction
Artemisinin (ART), a sesquiterpene endoperoxide, was isolated from the traditional Chinese medicine Artemisia annua L in the early 1970s by Chinese scientists1. ART has significant killing effects on Plasmodium falciparum and P. falciparum that are resistant to antimalarial drugs such as mefloquine1,2,3. So far, ART-based drugs are still the first-line antimalarial drugs for Artemisinin-based combination therapies (ACTs) recommended by the World Health Organization (WHO)2,3,4,5. Moreover, modern pharmacological research has revealed that artemisinins (ARTs) possess a growing number of biological activities, including anti-tumor, immunomodulation, anti-inflammatory, anti-viral, treatment of polycystic ovary syndrome and other pharmacological properties6,7,8,9,10,11. Therefore, the demand for ARTs is steadily increasing due to their broad therapeutic potential and effectiveness12. ART mainly exists in the glandular hairs of the above-ground parts of A. annua, with the highest content in the leaves13,14. At present, the source of ART still relies on direct extraction from plants, but its content in A. annua accounts for only 0.1–1.0% of the dry weight, which greatly limits its application in a high-demand market15,16,17. With the rapid development of synthetic biology, some natural active compounds including artemisinic acid (AA) that were originally synthesized only in specific species can now be produced in microorganisms such as Escherichia coli, yeast or heterologous plants18,19,20,21,22,23,24. Unfortunately, to date, no genetically engineered bacteria have been able to directly produce ART. Although heterologous plants can synthesize ART, the content is very low20,22. The bottleneck limiting biotechnological production of ART is that the terminal biosynthesis pathway, particularly the enzyme-catalyzed steps in ART remains unclear.
The biosynthetic pathway of ART is mainly divided into upstream and downstream pathways (Fig. 1). The upstream biosynthetic pathway involves the formation of farnesyl diphosphate (FPP), which is generated from isopentenyl pyrophosphate (IPP) and its isomer 2-methylallyl pyrophosphate (DMAPP), via the mevalonate (MVA) pathway or the 2-methylerythritol phosphate (MEP) pathway4,19,20,21. The enzymes involved in the upstream biosynthetic pathway of ART have been well characterized25,26,27. In the downstream biosynthetic pathway of ART, FPP is first converted into amorpha-4,11-diene (AD) by amorpha-4,11-diene synthase (ADS)28. Subsequently, AD is oxidized to artemisinol (AOH), artemisinic aldehyde (AO), and AA through the action of CYP71AV121. Cytochrome B5 (CYB5) can catalyze the conversion of AD into AOH, and AOH can further be oxidized to AO by alcohol dehydrogenase 1 (ADH1)19. Artemisinic aldehyde Δ11(13) double bond reductase (DBR2) catalyzes the reduction of AO to dihydroartemisinic aldehyde (DHAO)29. Additionally, aldehyde dehydrogenase (ALDH1) can catalyze the oxidation of AO or DHAO to produce AA and dihydroartemisinic acid (DHAA)30, respectively. DHAA can spontaneously oxidize to form ART3,19,21, but there is no enzyme reported to catalyze the conversion of AA to DHAA.

IPP: Iopentenyl pyrophosphate; DMAPP: Dimethylpropenyl pyrophosphate; FPP: Farnesyl pyrophosphate; AD: Amorpha-4,11-diene; AOH: Artemisinol; AO: Artemisinic aldehyde; AA: Artemisinic acid; ART-B: Artemisinin B; DHAO: Dihydroartemisinic aldehyde; DHAA: Dihydroartemisinic acid; ART: Artemisinin; IDI1: Isopentenyl isomerase; FPS: Farnesyl pyrophosphate synthase; ADS: Amorpha-4,11-diene synthase; CPR: Cytochrome P450 reductase; DBR2: Artemisinic aldehyde Δ11(13) double bond reductase; ALDH1: Aldehyde dehydrogenase. Black arrows indicated the routes that had been reported, while red arrows indicated the routes that were unclear.
The theoretical basis and practical application of semi-synthetic ART production methods are well-established. The process primarily consists of three steps: (1) Engineered strains with enhanced production of AA are constructed in microbial systems using synthetic biology techniques; (2) The Δ11(13) double bond of AA is chemically reduced to generate DHAA; and (3) DHAA serves as the starting material, which undergoes spontaneously oxidize to form ART19,21,31,32. The highest AA yield reported in engineered S. cerevisiae was 25 g/L19. However, when AA was chemically converted to DHAA, a considerable yield loss was caused by the formation of by-products19. In ART biosynthesis, the Km of DBR2 catalyzed AO to form DHAO is 19 μM while the Km of ALDH1 catalyzed AO to form AA is 2.58 μM29,30. As a result, the metabolic flows of ART biosynthetic pathway tend to generate AA rather than DHAA32.
Here, we identify an active enzyme dihydroartemisinic acid dehydrogenase (AaDHAADH) from A. annua that catalyzes the bidirectional conversion between AA and DHAA by employing a catalytic activity-guided protein purification approach combining proteomics and bioinformatics techniques. Through site-directed mutagenesis, we generate an optimized AaDHAADH variant that demonstrates 2.82-fold greater catalytic efficiency in DHAA biosynthesis compared to the wild-type enzyme. The production of DHAA in engineered S. cerevisiae reaches 3.97 g/L in 5 L bioreactor. The discovery and application of AaDHAADH provide a more convenient route to produce DHAA using synthetic biology methods, shorten the de novo DHAA synthesis pathway, and offer an alternative route for ART biosynthesis.
Results
Molecular cloning and functional characterization of AaDHAADH
There are many methods to elucidate the biosynthetic pathway of natural products, including candidate gene selection based on omics technology and bioinformatics analysis23,24,33,34,35, determination of gene function based on plant genetic transformation technology36,37, and pathway modularity based on the retrosynthetic analysis34,38, among others. In this study, we used a traditional enzymatic approach to screen and identify candidate genes24,29,39. The crude enzyme extracted from the leaves of A. annua was found to catalyze the conversion between AA and DHAA (Supplementary Fig. 1). This result indicated that the crude enzyme of A. annua likely contains one or two types of oxidoreductase, which could catalyze the bidirectional conversions of AA and DHAA29. AO is the intermediate metabolite in the ART biosynthetic pathway, sharing structural similarities with AA, and both contain α, β-unsaturated double bonds (Fig. 1 and Supplementary Fig. 2). Therefore, we speculated whether DBR2, a double bond reductase that catalyzes the conversion of AO to DHAO in ART biosynthesis (Supplementary Fig. 2)29, might also catalyze the bidirectional conversion between AA and DHAA. DBR2 was expressed and functionally verified in E. coli and N. benthamiana. It was found to be inactive towards both AA and DHAA in both systems (Supplementary Fig. 3). Based on the above results, we speculated that the crude enzyme of A. annua might contain other enzymes capable of catalyzing the bidirectional conversion between AA and DHAA (Supplementary Fig. 3F). Subsequently, the crude enzyme of A. annua was enriched by 80% ammonium sulfate (AS) precipitation, and further fractionated with dextran G50 gel column chromatography, dextran G25 gel column chromatography and DEAE chromatography under the guidance of catalytic activity (Fig. 2 and Supplementary Figs. 4–10). Finally, an active fraction, designated DEAE-2, was obtained (Supplementary Fig. 11).

A Purification flow chart of A. annua crude enzyme. B Bioconversion of AA to DHAA by purified active fraction with HPLC detection. C Bioconversion of DHAA to AA by purified active fraction with HPLC detection.
DEAE-2 active fractions were analyzed by mass spectrometry, identifying a total of 1261 proteins (Supplementary Data 1). Considering that the enzyme catalyzes bidirectional redox reactions, which likely belong to the oxidoreductase family29,40, we screened oxidoreductases from the proteomic data, identifying a total of 61 proteins (Supplementary Data 1). Evolutionary tree analysis was then conducted on these 61 oxidoreductases alongside key enzyme from the ART biosynthetic pathway, revealing that three proteins (A0A2U1QC71, A0A2U1KC90 (C90), and A0A2U1LV73 (V73)) clustered in the same branch as DBR2 (Supplementary Fig. 12). Amino acid sequence information of candidate enzymes was provided in Supplementary Table 1, A0A2U1QC71, a discovered active enzyme in this study, was named dihydroartemisinic acid dehydrogenase (AaDHAADH). The amino acid sequences of AaDHAADH, C90, and V73 were compared with DBR2 (Fig. 3A). Sequence alignment results showed that AaDHAADH and V73 both contained His, His and Tyr at position 180, 183 and 185, which were highly conserved sites in double-bond reductases29. Conserved amino acids, F69 and H239, in the substrate binding region of DBR2 were also found in AaDHAADH. However, the C-terminal amino acid residues of AaDHAADH were significantly different from those of DBR2, suggesting potential differences in catalytic activity and subcellular localization between the two enzymes29.

A Amino acid sequence alignment of candidate enzymes and DBR2. B SDS-PAGE profile of candidate enzymes (This experiment was repeated independently three times with similar results). C Bioconversion of AA to DHAA by candidate enzymes with HPLC detection. D Bioconversion of DHAA to AA by candidate enzymes with HPLC detection. E Subcellular localization of AaDHAADH in A. thaliana protoplasts (This experiment was repeated independently three times with similar results). Source data are provided as a Source Data file.
The screened AaDHAADH, C90 and V73 were expressed and functionally verified in E. coli and N. benthamiana (Fig. 3B–D and Supplementary Fig. 13). Results showed that only AaDHAADH could catalyze the bidirectional conversion of AA and DHAA in both E. coli and N. benthamiana, while the other two candidate enzymes had no catalytic activity towards AA or DHAA. (Fig. 3C, D and Supplementary Fig. 13). Based on these results, we discovered an active enzyme, named AaDHAADH, in the ART biosynthetic pathway that catalyzes the bidirectional conversion between AA and DHAA.
Enzymatic characteristics of AaDHAADH
In order to increase the expression of AaDHAADH, we firstly investigated the optimal concentration of isopropyl-beta-D-thiogalactopyranoside (IPTG). We found that the optimal IPTG concentration was 0.6 mM (Supplementary Fig. 14). The optimal reaction pH and temperature of AaDHAADH was 7.5 and 30 °C respectively (Supplementary Fig. 15A–D). The optimal reaction time for AaDHAADH catalyzing AA to form DHAA was 4 h, while the optimal reaction time for catalyzing DHAA to form AA was 6 h (Supplementary Fig. 15E, F).
AaDHAADH was identified as a dehydrogenase involved in the ART biosynthesis. It not only catalyzed the reduction of the ∆11(13) double bond of AA to form DHAA, but also catalyzed the dehydrogenation of the ∆11(13) carbon atoms of DHAA to generate AA. Therefore, enzyme kinetic assays with AaDHAADH were performed using AA for the reduction reaction and DHAA for the oxidation reaction (Table 1 and Supplementary Fig. 15G, H). The optimized pH, temperature and reaction time for the AaDHAADH catalyzed reduction were 7.5, 30 °C and 4 h, respectively. Under the optimal conditions, the Km value of AaDHAADH for AA was 340 μM, with Kcat and Kcat/Km values of 0.004 min−1 and 1.1765 × 10−5 min−1. μM−1, respectively. The dehydrogenation reaction catalyzed by AaDHAADH, using DHAA as a substrate, was also analyzed. Maximum activity for the AaDHAADH-catalyzed oxidation of DHAA was detected at pH 7.5, 30 °C and 6 h. Under these conditions, the Km value of the AaDHAADH for DHAA was 532.35 μM, with Kcat and Kcat/Km values of 0.0014 min−1 and 2.6298 × 10−6 min−1. μM−1, respectively. These results indicated that AaDHAADH perferentially catalyzes the reduction of AA to DHAA over the oxidation of DHAA to AA. Therefore, AaDHAADH exhibited a stronger affinity for AA than for DHAA.
Amino acid sequence similarity between AaDHAADH and DBR2 is 94.32%, with the main difference located at the C-terminus. (Fig. 3A). Previous study has shown that DBR2 is primarily localized in the cytoplasm29. The subcellular localization of AaDHAADH was investigated using Arabidopsis protoplast transformation, revealing that it was primarily localized in the cytoplasm and nucleus (Fig. 3E). This indicated that although AaDHAADH and DBR2 shared high amino acid sequence similarity, there were subtle differences in their subcellular localization and functions. These differences may also explain their distinct substrate specificities.
Catalytic mechanism and directed modification of AaDHAADH
To clarify the catalytic mechanism of AaDHAADH, a computational docking program Auto Dock Tools-1.5.6 was used to investigate the structural basis of substrate binding in AaDHAADH. Based on the inferred conformations and binding affinities, the most favorable orientation of AA and DHAA in the AaDHAADH active site were determined (Fig. 4A, B). Molecular docking results revealed that AA interacted with P26, F69, Q101, W103, H180, Y185, and R232 in the active pocket of AaDHAADH while DHAA interacted only with F69, Q101, W103, H180, Y185, and R232, lacking interaction with P26. We found that P26 interacted with the carboxyl hydrogen of AA through intermolecular hydrogen bonds, which might explain why AaDHAADH exhibited stronger affinity for AA than DHAA. To verify our hypothesis, we conducted site-directed mutagenesis of AaDHAADH. Since proline is a non-polar aliphatic amino acid, we considered mutating it into a polar neutral amino acid to determine whether its catalytic activity would still remain. When P26 was mutated into S, M and T (Supplementary Fig. 16), the catalytic activity of the mutant strain towards AA was significantly reduced or completely lost (Supplementary Figs. 17 and 18). This result indicated that P26 of AaDHAADH was a key amino acid residue that determined catalytic activity. To optimize the catalytic activity of AaDHAADH and increase DHAA production, a total of 11 mutants (P26T, P26S, P26M, P26W, P26E, P26H, P26G, P26A, P26V, P26L, P26I) were generated through site-directed mutagenesis (Supplementary Fig. 16). We found that mutant AaDHAADH (P26L) exhibited the highest catalytic activity toward AA, which was 2.82 times that of the original strain, while its activity for catalyzing the conversion of DHAA to AA was only 0.03 times that of the wild type (Fig. 4C and Supplementary Figs. 17 and 18).

A The amino acid residues that bound to AA within 10 Å during the docking of AaDHAADH. B The amino acid residues that bound to DHAA within 10 Å during the docking of AaDHAADH. C The catalytic reactions of wild-type and mutant (P26L) of AaDHAADH.
The enzymatic properties and kinetic parameters of mutant AaDHAADH (P26L) towards substrate AA were further investigated (Supplementary Fig. 19). The optimal reaction conditions for AaDHAADH (P26L) to catalyze the conversion of AA to DHAA were pH 7.5, 30°C and 2 h. Under these conditions, the Km value of the AaDHAADH (P26L) for AA was 170.29 μM, with Kcat and Kcat /Km values of 0.0090 min−1 and 5.2850 × 10−5 min−1. μM−1, respectively (Table 1). The Molecular Mechanics Generalized Born Surface Area (MM-GBSA) method is related to the binding free energy of the docked complex, and the application of this method provides more accurate results than traditional molecular docking studies41. In this study, the MM-GBSA results were consistent with the molecular docking outputs from the complex of active enzyme AaDHAADH and its substrate AA or DHAA (Table 2). In terms of total binding energy, AaDHAADH exhibited stronger binding affinity for AA (ΔTOTAL = −23.10 ± 3.07 kJ mol−1) than for DHAA (ΔTOTAL = −25.27 ± 2.73 kJ mol−1). Additionally, the binding of mutant AaDHAADH (P26L) for AA (ΔTOTAL = −20.00 ± 2.42 kJ mol−1) also exceeded that of the wild-type AaDHAADH (ΔTOTAL = −23.10 ± 3.07 kJ mol−1). These computational results were consistent with the observed enzymatic kinetic parameters (Table 1).
Functional analysis of AaDHAADH in A. annua cells
Based on the detailed enzyme activity assays in E. coli and N. benthamiana systems, the functional characteristics of AaDHAADH were outlined. To further verify this function in A. annua, transgenic A. annua cells were generated through overexpression (OE) and RNA interference (RNAi)-mediated gene silencing33,37,42. After selection by hygromycin B (HB) selection, eight independent transgenic cell lines of AaDHAADH-OE, AaDHAADH (P26L)-OE and AaDHAADH-RNAi were obtained (Fig. 5 and Supplementary Figs. 20–26). Using qRT-PCR analysis, we selected three lines with elevated AaDHAADH expression form the AaDHAADH-OE or AaDHAADH(P26L)-OE transgenic cell lines and three lines with reduced AaDHAADH expression from the AaDHAADH-RNAi transgenic cell lines (Fig. 5A–C). Compared to the WT, significant changes in DHAA and ART production were observed in OE and RNAi transgenic A. annua cells, while AA content decreased in OE lines and slightly increased in RNAi lines (Fig. 5D–F and Supplementary Fig. 27). These findings provide direct evidence for the role of AaDHAADH in ART biosynthesis and confirm an alternative biosynthetic pathway for DHAA in A. annua.

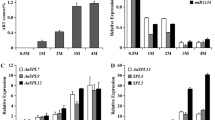
A Relative expression levels of AaDHAADH in overexpression of AaDHAADH (AaDHAADH-OE) transgenic A. annua cells compared with the wild-type (WT). B Relative expression levels of AaDHAADH in overexpression of AaDHAADH (P26L) (AaDHAADH (p26l)-OE) transgenic A. annua cells compared with WT. C Relative expression levels of AaDHAADH in RNAi of AaDHAADH (AaDHAADH-RNAi) transgenic A. annua cells compared with WT. D AA, DHAA and ART contents in AaDHAADH-OE transgenic A. annua cells compared with WT. 3, 5, and 7 correspond to AaDHAADH-OE-3, AaDHAADH-OE-5 and AaDHAADH-OE-7 in A), respectively. E AA, DHAA and ART contents in AaDHAADH (P26L)-OE transgenic A. annua cells compared with WT. 1, 4, and 7 correspond to AaDHAADH (P26L)-OE-1, AaDHAADH (P26L)-OE-4 and AaDHAADH (P26L)-OE-1-7 in B), respectively. F AA, DHAA and ART contents in AaDHAADH-RNAi transgenic A. annua cells compared with WT. 3, 5, and 6 correspond to AaDHAADH-RNAi-3, AaDHAADH-RNAi-5 and AaDHAADH-RNAi-6 in C), respectively. Three cell lines (in the red box) with higher or lower AaDHAADH expression levels were selected for downstream analysis. The bars denote means ± standard deviations (n = 3 biologically independent samples). *, ** and *** indicate significant differences from control at the levels of P < 0.05, P < 0.01 and P < 0.001, respectively, as determined by two-tailed student’s t-test. Source data are provided as a Source Data file.
De novo synthesis of DHAA in S. cerevisiae
In order to achieve large-scale production of DHAA, which is crucial for the application of semi-synthetic production of ART, we engineered DHAA-producing S. cerevisiae through three processes. To increase FPP production, we employed a genome integration approach to construct an expression module. This module enhanced the expression of key genes (tHMG1, ERG10, ERG19 and IDI1) in the endogenous MVA pathway of S. cerevisiae (Fig. 6A). Expression of ERG19 and IDI1 increased FOH production, with a 250 mL shake flask yield of 22.46 mg/L, representing an ~31.09-fold increase compared with initial strain BY4741 (Supplementary Figs. 28A and 29, strain FPP-1). tHMG1 and ERG10 were continued to be integrated based on FPP-1, the yield of FOH was 48.51 mg/L, a 48.51-fold increase compared to BY4741(Supplementary Figs. 28B and 29, strain FPP-2). To further enhance FPP yield, we optimized the strain by integrating the four genes into a multi-copy integration site (Supplementary Fig. 28C, strain FPP-3). The shake flask yield of strain FPP-3 was 75.15 mg/L, which was 107.35, 3.35 and 1.17 times higher than that of BY4741, FPP-1 and FPP-2, respectively (Supplementary Fig. 29). To construct a strain that produces AA from FPP, genes involved in AA biosynthesis (ERG20 from S. cerevisiae, ADS, CYP71AV1, CYB5, ADH1 and ALDH1 from A. annua and ATR1 from A. thaliana) were integrated into FPP-3, respectively (Supplementary Fig. 28D–F, strain AA-1, AA-2 and AA-3). The production of AA in strain AA-3 with a 250 mL shake flask was 1.65 g/L, increased by 79.35% and 9.27% compared with AA-1 (0.92 g/L) and AA-2 (1.51 g/L), respectively (Fig. 6B). Beginning with the AA-3 strain, we integrated AaDHAADH (P26L) into the genome and the shake flask yield of DHAA was 0.23 g/L. (Supplementary Fig. 28G, strain DHAA-1). To further increase DHAA production, the THI2 promoter (PTHI2) was used in addition to an additional copy of AaDHAADH (P26L) (Supplementary Fig. 28H, strain DHAA-2). The shake flask yield of strain DHAA-2 was 0.35 g/L, an increase of 52.17% compared with DHAA-1 (Fig. 6B).

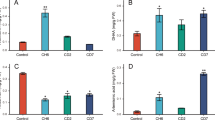
A Integration of key genes in the ART biosynthetic pathway. B Yields of AA or DHAA in shake flask fermentation (a tHMG1 + ERG10 + ERG19 + IDI1 + ERG20 + ADS + CYP71AV1. b CPR + ALDH1. c CYB5 + ADH1. d AaDHAADH (P26L). e PTHI2.). C Yields of DHAA and AA in strains DHAA-2 and AA-3. D Growth and metabolite production of strain DHAA-2 in a 5 L fermenter. The bars denote means ± standard deviations (n = 3 biologically independent samples). *, ** and *** indicate significant differences from control at the levels of P < 0.05, P < 0.01 and P < 0.001, respectively, as determined by two-tailed student’s t-test. P value for each comparison from top to bottom then left to right in (B): 0.0035 (**), 0.037 (*), 0.0021 (**). Source data are provided as a Source Data file.
To assess industrial viability, we performed fed-batch fermentation of strains AA-3 and DHAA-2 in a 5 L bioreactor. After 168 h, AA production reached 22.67 g/L, representing a 13.78-fold increase compared to the shake flask (Fig. 6C). Similarly, DHAA yield in the 5 L bioreactor reached 3.96 g/L at 204 h, a 10.31-fold increase over the shake flask output (Fig. 6C, D).
Reconstruction of ART biosynthesis in N. benthamiana
Since DHAA can be auto-oxidized to produce ART, we designed an ART production system in N. benthamiana using eight known functional proteins (tHMG1, ERG20, ADS, CYP71AV1, CPR, CYB5, ADH1, ALDH1) and the targeted optimized enzyme AaDHAADH (P26L) (Fig. 7A). Expressing ADS, CYP71AV1 and CPR in N. benthamiana resulted in AA production at 0.28 mg/g DW (Fig. 7B, D and Supplementary Fig. 30A). Introducing ADH1, ALDH1, and CYB5 increased AA production to 0.62 mg/g DW (Fig. 7B, D). Further optimization of the transient expression system by adding tHMG1 and ERG20 from the MVA pathway successfully increased AA yield to 1.19 mg/g DW (Fig. 7B, D). Adding the targeted optimized enzyme AaDHAADH (P26L) enable the DHAA production at 0.51 mg/g DW (Fig. 7C, D and Supplementary Fig. 30B). However, no ART was detected in the transiently expressed N. benthamiana (Supplementary Fig. 31). To accelerate DHAA auto-oxidation and ART production, we irradiated N. benthamiana leaves with UV, ultimately detecting ART at 0.041 mg/g DW (Fig. 7C, D).

A Biosynthetic route of ART in N. benthamiana. B Flow chart of the transient expression and products detection in N. benthamiana. C LC-MS for AA (EIC, m/z = 233.2) produced by transiently expressed enzymes in N. benthamiana. D LC-MS for DHAA (EIC, m/z = 235.2) and ART (EIC, m/z = 283.1) produced by transiently expressed enzymes in N. benthamiana. E AA, DHAA and ART contents of N. benthamiana leaves infiltrated with different combinations of genes. 1: ADS + CYP71AV1 + CPR. 2: ADH1 + ALDH1 + CYB5. 3: tHMG1 + ERG20. 4: AaDHAADH (P26L). 5: UV. The bars denote means ± standard deviations (n = 3 biologically independent samples). *, ** and *** indicate significant differences from control at the levels of P < 0.05, P < 0.01 and P < 0.001, respectively, as determined by two-tailed student’s t-test. P value for each comparison from top to bottom then left to right in D): 0.000679 (***), 0.001173 (**), 0.009464 (**), 0.03283 (*), 0.000775 (***). Source data are provided as a Source Data file.
Discussion
Chinese herbal medicine has a long history of utilizing natural active ingredients for medicinal purposes. However, elucidating complex biosynthetic pathways of natural products in plants remains highly challenging, as typically only one or a class of active enzymes can be identified at a time43. In contrast, fungi and bacteria have gene clusters that facilitate the elucidation of natural product biosynthetic pathways38,44. Similarly, research on the ART biosynthetic pathway is highly challenging, with the most recent discovery of active enzymes dating back to 200930. The regulation of key genes in ART metabolic pathways and metabolic flux constraints make the efficient production of DHAA in cell factories a significant challenge19,21,29,30,31,32.
Both traditional activity-directed purification and modern transcriptomics-based bioinformatics techniques have been successfully employed to identify elusive enzymes in plant natural product pathways23,24,34,38,39. However, transcriptomics-based methods typically require characterized enzymes as bait and gene predictions based on specialized metabolism for co-expression analysis39,45. This results in significant limitations in their application. In this study, we identified active enzymes catalyzing the conversion of AA to DHAA in A. annua by integrating activity-oriented purification, proteomics, and bioinformatics. Although activity-oriented purification is time-consuming, it enables efficient screening of low-abundance enzymes, reduces false positives, and adapts to a wide range of samples24,39. We identified one active fraction DEAE-2 from A. annua by activity-oriented purification (Supplementary Fig. 11). Through proteomic identification and bioinformatics analysis, we successfully determined the relevant candidate enzymes (Supplementary Fig. 12).
The catalytic function of AaDHAADH was both in vitro using E. coli and in vivo through systematic functional assays in N. benthamiana and A. annua cells. Combining transient expression in N. benthamiana with stable expression in A. annua provides a robust strategy for validating the function of plant-derived enzymes such as AaDHAADH. This approach ensures result reliability, enables detailed analysis of enzyme regulation and metabolic roles, and overcomes the limitations of traditional heterologous expression systems46. These methodologies are essential for advancing our understanding of plant enzyme functions and their biotechnological and natural product synthesis applications. In this study, we obtained transgenic A. annua cell lines with overexpression and RNAi. During initial screening, we observed that some A. annua cells grew slowly or even died (Supplementary Figs. 21–23). When the HB concentration was increased to 20 mg/L, newly transformed cells emerged after 10 days (Supplementary Fig. 24). When fresh cells were transferred to medium containing 30 mg/L HB, the transgenic cells grew as vigorously as normal cells (Supplementary Figs. 25 and 26). This phenomenon arises from selective pressure in the culture medium, where untransformed cells undergo browning and necrosis, while transformed cells maintain normal proliferation. Thus, the initial phase represents primary selection, while the subsequent stage involves the propagation of successfully transformed cells with normal growth. We also measured the levels of AA, DHAA, and ART in transgenic A. annua cells. Consistent with effective AaDHAADH overexpression or RNAi in each transgenic cell line, we observed significant changes in DHAA and ART production. In AaDHAADH-OE cells, DHAA levels increased to 1.23-fold of WT (Fig. 5D), while in AaDHAADH-RNAi cells, DHAA levels decreased to 0.26-fold of WT (Fig. 5F). In AaDHAADH (P26L)-OE cells, DHAA levels further increased to 1.56-fold of WT and 1.29-fold of AaDHAADH-OE (Fig. 5E). Additionally, ART levels in both OE and RNAi cells mirrored the changes in DHAA (Fig. 5D, E and F). AA levels decreased in AaDHAADH-OE and AaDHAADH (P26L)-OE cells (Fig. 5D, E), but increased in AaDHAADH-RNAi cells (Fig. 5F). These results confirm AaDHAADH as a key oxidoreductase in DHAA biosynthesis in A. annua. AaDHAADH catalyzes the conversion of AA to DHAA in A. annua cells. The accumulation of DHAA correlates with increased ART levels, confirming DHAA as a key intermediate in ART biosynthesis.
We have demonstrated that AaDHAADH functions as a sesquiterpene oxidoreductase and optimized its catalytic specificity for converting AA to DHAA in ART biosynthesis. Efficient DHAA production in heterologous systems is crucial for the semi-synthetic production of ART. Although DHAA production in S. cerevisiae has been achieved, researchers reduced or blocked AA production through enzyme modifications, diverting metabolic flux to DHAA and achieving a higher DHAA/AA ratio32. However, the primary metabolic flux in ART biosynthesis favors AA production over DHAA19,29,30. Despite a higher DHAA/AA ratio (9.84), the DHAA yield in a 5 L bioreactor reached only 1.7 g/L, which may not meet market demands. Our research aligned with the natural metabolic process of ART biosynthesis, producing DHAA from high-yield AA. This represents an efficient pathway for DHAA production. Although the DHAA/AA ratio was lower, we achieved a DHAA yield of 3.97 g/L in a 5 L fermenter. In comparison, our research offers significant advantages, and the discovery and application of AaDHAADH demonstrate strong industrial potential.
However, a major bottleneck in synthetic biology for ART production is the elucidation of its terminal biosynthetic pathway4,19. Since the mechanism underlying the formation of ART’s peroxide bridge remains unclear, direct ART production in heterologous systems is currently unfeasible. Therefore, further research is needed to elucidate the terminal biosynthetic pathway of ART. Natural product biosynthetic pathways are typically analyzed using transcriptomics and gene co-expression analysis4,34,43. Some researchers integrate metabolomics to identify compound accumulation sites, followed by transcriptomics analysis of specific tissues to expedite the discovery of relevant active enzymes34. However, metabolic processes are dynamic, and it is often unclear whether metabolites are synthesized at their detection sites or transported from other locations. To analyze the biosynthetic pathways of complex natural products like ART and ginkgolide, it is essential to first identify their biosynthetic active sites43. Existing studies have demonstrated that A. annua glandular secretory trichomes (AaGSTs) are the sites of ART biosynthesis and accumulation13,47,48. Comparative transcriptome sequencing of AaGSTs and other A. annua tissues, combined with bioinformatics methods like gene co-expression analysis, could help identify relevant active enzymes. This approach may help elucidate the terminal biosynthesis of ART.
Methods
Plant materials
A. annua seedlings (2-week-old) were procured from Youyang Wuling Mountain Germplasm Center (Chongqing, China) and acclimatized in controlled-environment growth chambers (25 °C, 16/8 h photoperiod) for 3 weeks prior to enzymatic extraction. N. benthamiana seeds (Guangzhou Danoan Biotech) were surface-sown on moistened potting mix (Pindstrup substrate), covered with transparent humidity domes, and germinated under 25 °C constant illumination (4-day germination threshold). Cotyledon-stage seedlings were transplanted to individual containers (10 cm diameter) containing fertilized soil matrix. Greenhouse-grown plants (25 °C, 60% RH, 16/8 h photoperiod) underwent 4-week maturation before experimental processing. A. thaliana seeds (Guangzhou Lige Biotech) were propagated using identical growth protocols as N. benthamiana.
Analysis of AA, DHAA and ART using HPLC, GC-MS and LC-MS system
Qualitative and quantitative analysis of AA, DHAA and ART were performed by comparing retention times and mass spectrum peak with standards using HPLC (Thermo UltiMate 3000), GC-MS (GC system: Agilent Technologies 7890B; MSD: Agilent Technologies 7977B) and LC-MS (Agilent Technologies 1260–6130). The HPLC analytical conditions were as follows: Samples were separated using a YMC-Pack C18 column (150 × 4.6 mm, 5.0 μm) and the column temperature was set at 35 °C. The flow rate was 0.3 mL/min. The mobile phases were 0.1% trifluoroacetic acid aqueous solution (A) and acetonitrile (B), and the gradient was as follows: 30% B for 2 min; 30–70% B for 8 min; 70% B for 15 min; 70–100% B for 10 min; 100% B for 10 min followed by a decrease to 30% B for 0.1 min and re-equilibration of the column for 4.9 min with 30% B. The injection volume was 20.0 μL and the sampler temperature was set at 25 °C. GC-MS analytical conditions were as follows: Samples were separated using a HP-5MS Ultra Inert column (30 m × 0.25 mm × 0.25 μm). The injection volume was 2 μL, splitless. Ion mode (SIM) was chosen, and eight ions characteristic to the products (121, 162,189, 204, 218, 220, 234 and 236) were used for detection. And the gradient was as follows: 60°C for 1 min; increased to 170 °C at a rate of 12 °C/min; 170°C for 12 min followed increased to 300 °C at a rate of 10 °C/min; post run time was set as 3 min. The LC-MS analytical conditions were as follows: Samples were separated using a YMC-Pack C18 column (150 × 4.6 mm, 5.0 μm) and the column temperature was set at 35 °C. The flow rate was 0.3 mL/min. The mobile phases were ultrapure water (A) and acetonitrile (B), and the gradient was as follows: 30% B for 2 min; 30–60% B for 8 min; 60% B for 25 min; 60-100% B for 10 min; 100% B for 10 min followed by a decrease to 30% B for 0.1 min and re-equilibration of the column for 4.9 min with 30% B. The injection volume was 5.0 μL. The negative ion mode was used for the detection of AA and DHAA, and the positive ion mode was used for the detection of ART.
Extraction of crude enzyme from A. annua
Appropriate amount of vigorously growing leaves of A. annua were taken and rinsed under tap water for 10 min. The leaves were put into a mortar and quickly ground into powder in liquid nitrogen. The ground powder was taken and placed in a sterile centrifuge tube, and protein extraction buffer (PBS stock solution which containing 10 mM DTT, 5 mM EDTA, 5 mM PMSF and 1 μg/mL of phosphoprotease inhibitor.) was added at a mass-to-volume ratio of 1:5. Extract was incubated for 5 h at 4 °C and 80 × g. The mixture was centrifuged at 12,000 × g at 4 °C for 15 min. Supernatant was passed through 0.22 μm microporous filter, and then dialyzed (1KD) in PBS solution at 4 °C for 24 h to completely remove small molecule compounds. The protein concentration was determined using ultra-trace nucleic acid protein detector. Extracted protein was immediately used for downstream experiments or stored at -20 °C for later use.
Activity-oriented protein purification and isolation
First, ammonium sulfate (AS) precipitation was used to fractionate the crude enzyme of A. annua, and the 80% AS fraction was collected. After adding AS powder, the mixture was shaken at 4 °C and 60 × g for 2 h, then centrifuged at 15,000 × g and 4 °C for 15 min. The supernatant was discard and pellet was resuspend in protein extraction buffer, and dialyzed (1KD) in PBS solution at 4 °C for 24 h to completely remove (NH4)2SO4.
Dextran G50 gel column was used to continue chromatography on the 80% AS precipitated fraction. Dextran G50 packing was load into the column (column volume is about 80 mL) in the chromatography cabinet (4 °C). The constant flow pump, nucleic acid protein detector and computer device were connected. The mobile phase was PBS solution and the flow rate was 0.50 mL/min. Sample was filtered with a 0.22 μm micropore filter, and the loading amount was 5 mg. From the chromatogram, it was seen that there were two peaks, which were named G50-1 and G50-2 in the order of peak time. The two components were collected separately, dialyzed (1KD) in ultrapure water at 4 °C for 24 h, and freeze-dried (-80 °C, 0.1 Pa) in vacuum freeze dryer. The lyophilized sample was remelted with an appropriate amount of protein extraction buffer and used for downstream experiments. Dextran G25 gel column chromatography and sample processing were the same as above.
Finally, the DEAE anion exchange column was used to continue purifying the active components. DEAE-Sepharose Fast Flow packing was loaded into the column (column volume ~200 mL) in chromatography cabinet (4 °C) with a flow rate of 2 mL/min. Sample was filtered with a 0.22 μm micropore filter, and the loading amount was 20 mg. A total of three gradients of 0.03 M Tris+0.30 M NaCl, 0.03 M Tris+1.00 M NaCl, and 0.03 M Tris+2.00 M NaCl were used for elution, which named DEAE-1, DEAE-2 and DEAE-3 respectively. The three components were collected separately, dialyzed (1KD) in ultrapure water at 4 °C for 24 h, and freeze-dried (−80 °C, 0.1 Pa) in vacuum freeze dryer. The lyophilized sample was remelted with an appropriate amount of protein extraction buffer and used for downstream experiments.
In vitro activity verification of active ingredients during protein purification
A 1000 μL reaction system containing protein extraction buffer (pH 7.4), 100 μg crude protein extract (or different chromatographic components) and 400 μM AA/DHAA were used respectively. Crude enzyme extract incubated in boiling water bath for 5 min was used as control group. Reaction system was incubated at 30 °C and 220 × g for a total of 4 h. At the end of the reaction, 20 μL hydrochloric acid solution with pH=2 and 800 μL ethyl acetate were added for extraction. Upper organic phase was collected after centrifugation at 12,000 × g and 25 °C for 5 min. The organic phases were combined and passed through 0.22 μm organic microporous filter. Organic phase was dried with a nitrogen blower, 100 μL of chromatographic methanol was added to remelt, and waited for HPLC detection.
Proteomic identification of active components
DEAE-2 active ingredients were processed for proteomic identification. Peptides were digested by trypsin and analyzed using LC-MS/MS (Thermo EASY nLC and Thermo Scientific Q Exactive™). The raw files obtained from mass spectrometry were processed and analyzed using Max Quant software (version 1.6.2.10) for database matching, yielding the final identification results. The mobile phase and mass spectrometric operational parameters employed in the LC-MS/MS analysis, along with the database search criteria, were systematically documented in Supplementary Tables 2–4. For the detailed procedures of mass spectrometry experiments, please see Supplementary Method 1.
Phylogenetic tree analysis and amino acid sequence alignment
The amino acid sequence of key genes in the downstream biosynthetic pathway of ART and the screened 61 redox family genes were obtained from NCBI, uniport and other public databases (Supplementary Data 2). Amino acid sequences were aligned and the Neighbor-Joining (NJ) trees were built using MEGA11 (Molecular Evolutionary Genetics Analysis version) with the following parameters: at least 1000 bootstrap replications, poisson model, uniform rates and complete deletion (Supplementary Tables 5 and 6; Supplementary Data 3 and 4). The detailed process of phylogenetic tree construction can be found in Supplementary Method 2.
Heterologous expression of candidate enzymes in E. coli and purification
RNA from A annua L. was extracted by plant total RNA extraction kit, and was reverse transcribed to obtain cDNA. The full length CDS of AaDHAADH (GeneBank ID: PWA95605.1), C90 (GeneBank ID: PWA95629.1), V73 (GeneBank ID: PWA52903.1) and DBR2 (GeneBank ID: KC505370.1) were amplified from A. annua L. cDNA using primers listed in Supplementary Data 5 was cloned into the pET−28b (+)-MBP plasmid. The recombinant plasmids were first transformed into E. coli DH5α which was grown on LB solid medium with 50 μg/mL kanamycin. After overnight incubation in the 37 °C incubator, single clones were selected for colony PCR verification. And 1–2 positive colonies were selected and placed in 5 mL LB liquid medium (50 μg/mL kanamycin) for overnight culture (37 °C and 220 × g). Cultures were extracted for plasmids and sequenced. Correctly sequenced recombinant plasmids were transferred into E. coli BL21 (DE3) which was grown on LB solid medium with 50 μg/mL kanamycin and 34 μg/mL chloramphenicol. After overnight incubation in the 37 °C incubator, single clone was selected and placed in 5 ml LB liquid medium (50 μg/mL kanamycin and 34 μg/mL chloramphenicol) for overnight culture (37 °C and 220 × g). And 1 mL cultures were transferred to 100 mL LB liquid medium containing antibiotics (50 μg/mL kanamycin and 34 μg/mL chloramphenicol) and isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM until the OD600 reached 0.4-0.6 (37 °C and 220 × g). The cultures were grown at 16 °C with shaking at 220 × g for about 16 h.
Cells were harvested by centrifugation at 8000 × g and 4 °C for 15 min, and 5 mL PBS (pH=7.4) was added to resuspend the cells. Resuspend cells were disrupted on ice for 10 min at 50% amplitude, 5 sec on and 5 sec off. After centrifugation at 8000 × g at 4 °C for 15 min, the supernatant was collected. Protein was purified through a nickel column.
Activity and enzymatic characteristics of AaDHAADH
In order to test the redox activity of catalyzing the interconversion of AA and DHAA, a 1000 μL reaction system containing protein extraction buffer (pH 7.4), 100 μg purified protein (AaDHAADH, C90, V73 and DBR2) and 400 μM AA or DHAA were used respectively. Purified protein incubated in boiling water bathing for 5 min was used as control group. Reaction system was incubated at 30 °C and 220 × g for 4 h. At the end of the reaction, 20 μL hydrochloric acid solution with pH=2 and 800 μL ethyl acetate were added respectively. Upper organic phase was collected by centrifugation at 12,000 × g and 25 °C for 5 min. Extraction was repeated with three times by adding 800 μL ethyl acetate, the organic phases were combined and passed through 0.22 μm organic microporous filter. The organic phase was dried with a nitrogen blower, and 100 μL chromatographic methanol was added to remelt, and waited for HPLC detection.
For determination of the effects of pH, starting solution (protein extraction buffer) was adjusted into different pH (4.5, 6, 7.5, 9 and 10.5) by adding 2 M HCl or 2 M NaOH, and all other procedures were performed using standard conditions. For reaction time analysis, reaction times were adjusted to 1, 2, 4, 6 and 8 h respectively. To determine the effect of temperature on this reaction, the reaction temperatures were set to 25, 30, 35 and 40 °C in different shakers. Enzyme kinetics of AaDHAADH to AA or DHAA were then analyzed at these optimal pH, temperature and reaction time conditions. Different concentrations of AA (0.2–2 mM) and DHAA (0.2–2 mM) were used for the analysis of enzyme kinetics. The enzymatic activity was calculated based on the amount of product produced.
Transient expression and functional characterization of candidate enzymes in N. benthamiana
The full length CDS of AaDHAADH, C90, V73 and DBR2 were amplified from A. annua. cDNA using primers were listed in Supplementary Data 5 and cloned into the pBI121-EGFP plasmid. Recombinant plasmids were transferred into GV3101 which was grown on YEB solid medium with 50 μg/mL kanamycin and 25 μg/mL rifampicin. After incubation in the 28 °C incubator about 60 h, single clone was selected for colony PCR verification. And 1–2 positive colonies were selected and placed in 5 ml YEB liquid medium (50 μg/mL kanamycin and 25 μg/mL rifampicin) for overnight culture (28 °C and 220 × g). The bacterial solution was centrifuged at 2000 × g and 4 °C for 20 min. Cells were resuspended by adding 5 mL of infection solution (contains 50 mM MES, 2 mM Na3PO4, 27.8 mM glucose, 10 mM MgCl2 and 0.1 mM acetosyringone in ddH2O). The resuspension was further centrifuged at 2000 × g and 4 °C for 20 min. Cells were resuspended to OD600 = 0.6 by adding infection solution and then activated for 2 h at 28 °C and 220 × g. The activated infection solution was injected into tobacco leaves and incubated at 25 °C in the dark for 24 h. The infected N. benthamiana was then continued for 24 h under conditions of 16 h of light and 8 h of darkness. Then, the substrate AA or DHAA was injected into the N. benthamiana leaves and cultured for 48 h. N. benthamiana leaves were collected and quickly ground into powder in liquid nitrogen. And 5 mL of ethyl acetate was added and placed on an ultrasonicator for 30 min to extract small molecule compounds. The organic phase was collected and concentrated to dryness, and then 100 μL of chromatographic methanol was added for GC-MS detection.
Subcellular localization of AaDHAADH
AaDHAADH was cloned into pCAMBIA2300-EGFP using the primers listed in Supplementary Data 5 to create a C-terminal EGFP fusion protein. Protoplasts were extracted from 4-week-old and normally growing leaves of A. thaliana49. Then 10 ng of the recombinant plasmid containing AaDHAADH, 100 μL of A. thaliana protoplasts and 100 μL of 40% PEG4000 were mixed together and incubated in a water bath at 25 °C for 20 min. The reaction was quenched by adding 1 mL of W5 solution (Supplementary Table 7), followed by centrifugation at 4 °C and 400 × g for 5 min. Finally, 1 mL of W5 solution was added to gently resuspend the pellet and incubated in the dark at 25 °C for 12 h. After the incubation, the protoplasts were centrifuged at 4 °C and 400 × g for 5 min, and then photographed using a laser confocal system. The detection parameters were as follows: GFP excitation wavelength was 488 nm, and chloroplast excitation wavelength was 514 nm.
Molecular docking
AaDHAADH was modeled by SwissModel66 using DBR2 as template. The ligands AA and DHAA were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov/), and then energy minimized with Molecular Mechanics (MM2) until a minimum root-mean-square (RMS) gradient of 0.100 was achieved (https://www.chemdraw.com.cn/). AutoDock 4.2 was used for docking experiments. Molecular graphics were rendered with PyMOL (version 2.3.2).
Mutagenesis and expression of AaDHAADH in E. coli
Overlapping gene fragments were separately amplified using AaDHAADH as a template with primers containing the desired single and combined amino acid substitutions (Supplementary Data 5). The fragments were simultaneously cloned into pET-28b (+)-MBP vector for expression as described above.
Free energy analysis by molecular mechanics generalized born surface area (MM-GBSA) calculation
The free energy of the ligand-protein complexes of AaDHAADH and its mutant AaDHAADH (P26L) with substrates AA or DHAA were determined using the MM-GBSA method via the SiBioLead web server. The GROMACS utility GMX-MMPBSA v1.5.6 was used for the estimation of the binding free energy50. The following formula 1 was used to determine the total energy component:
where ΔTOTAL is the total free energy component achieved by summing ΔGSOLV, the total solvation free energy, and ΔGGAS, the total gas phase free energy.
Overexpression and RNAi of AaDHAADH in A. annua cells
A. annua cell line was established in our laboratory from leaf-derived callus induction37,42. Subsequent cultivation was performed on fresh MS solid medium containing 1 mg/L 6-BA and 1 mg/L NAA, with subculturing at two-week intervals under dark condition at 25 °C (Supplementary Fig. 26)37,42. The full-length coding regions of AaDHAADH and AaDHAADH(P26L) were cloned and inserted into pCAMBIA2300-EGFP driven by the CaMV35S promoter to generate overexpression recombinant plasmids containing the target genes. The RNAi fragment, corresponding to the 128 bp-427 bp of AaDHAADH (Supplementary Fig. 20), was cloned into pCAMBIA1300 and driven by the CaMV35S promoter to construct an RNAi recombinant plasmid. These vectors were introduced into A. tumefaciens GV3101, and positive transformants were cultured in YEB medium supplemented with rifampicin (25 mg/L) and kanamycin (50 mg/L) at 28 °C until OD600 = 0.6. The bacterial suspension was transferred to centrifuge tubes and centrifuged at 4000 × g, 4 °C for 20 min. Cells were collected and resuspended in infiltration buffer (200 μM acetosyringone, 10 mM anhydrous magnesium chloride, 0.02% silwet L-77 and 10 mM morpholineethanesulfonic acid monohydrate) to OD600 = 0.6 and activated at 28 °C, 220 × g for 2 h. A. annua cells (20 g, fresh weight) were transferred to a 250 mL conical flask containing 50 mL of the infiltration solution for 5 min and co-cultured on MS medium (containing 1 mg/L 6-B, A and 1 mg/L NAA) for 3 d. After 3 days, the recombinant cells were washed 3 times with sterile water and then washed with 600 mg/L timentin (Tim) to remove A. tumefaciens. The washed recombinant cells were cultured in MS medium (containing 1 mg/L 6-BA, 1 mg/L NAA, and 300 mg/L Tim) at 25 °C in the dark for 15 days. During the culture period, the cells were photographed every 5 days to monitor their recovery. Subsequently, the recombinant A. annua cells were screened by culturing them with HB for 15 days per round. Three rounds of screening were performed using HB concentrations of 10 mg/L, 20 mg/L, and 30 mg/L in sequential order. Quantitative real-time PCR was used to detect the overexpression and silencing of AaDHAADH in transgenic cell lines, and LC-MS was used to analyze the changes in the contents of AA, DHAA and ART in transgenic cell lines. Primers used in this study were summarized in Supplementary Data 5.
Quantitative real-time PCR
Total RNA was extracted from transgenic A. annua cells using the Plant Total RNA Isolation Kit (Vazyme RC411-01). cDNA was synthesized using the Hiscript III 1st Strand cDNA Synthesis Kit (+gDNA wiper) (Vazyme R312-01). Gene expression was determined by quantitative real-time PCR using All-In-One 5× RT MasterMix (ABM G592) on a QuantStudio7 Flex Real-Time PCR System (Thermo Fisher Scientific). Data were then presented according to the 2-ΔΔCT method and normalized using the gene encoding β-actin.
Reconstruction of DHAA biosynthesis in S. cerevisiae
ERG10 (GeneBank ID: 856230), ERG19 (GeneBank ID: 855889), ERG20 (GeneBank ID: AF112881), IDI1 (GeneBank ID: 851557) and tHMG1 (GeneBank ID: AF142473), the key genes in the MVA pathway, were cloned through the gDNA of S. cerevisiae BY4741 as a template. ERG10 + tHMG1 and ERG19 + IDI1 were constructed into the expression vectors pESC-TRP and pESC-URA respectively, and forming the recombinant plasmids pESC-TRP-ERG10 + tHMG1 and pESC-URA-ERG19 + IDI1. The expression fragments TRP-tHMG1-ERG10 and URA-IDI1-ERG19 were cloned using two expression plasmids as templates respectively. Then they were inserted into the integration vectors pCDF-Trp1 and pCDF-Ty4 between the restriction enzyme sites BamH1 and SacI respectively, and form the integrating vector pCDF-Trp1-tHMG1-ERG10 and pCDF-Ty4-URA-IDI1-ERG19. Then, the integrate fragments TRP1-tHMG1-ERG10 and TY4-URA-IDI1-ERG19 were cloned using two integrate plasmids as templates respectively. The integrated fragment TY4-URA-IDI1-ERG19 was integrated into the genome of S. cerevisiae BY4741, and was spread on SD-URA YNB plates and cultured in a 30 °C constant-temperature incubator for about 72 h. Single colony was picked for yeast colony PCR verification, and the positive colony was named FPP-1 and stored at -80 °C for later use. The integrated fragment TRP1- tHMG1-ERG10 was integrated into the genome of FPP-1, and was spread on SD-URA + TRP YNB plates and cultured in a 30 °C constant-temperature incubator for about 72 h. Single colony was picked for yeast colony PCR verification, and the positive colony was named FPP-2 and stored at -80 °C for later use. The engineered strains FPP-1, FPP-2 and initial yeast BY4741 were inoculated into 5 mL YNB liquid medium, which contained corresponding amino acid deficiencies, and cultured overnight at 30 °C and 220 × g until the OD600 = 1.0. The seed liquid (200 μL) was transferred from the test tube to a shake flask containing 30 mL of YPD liquid medium, and 3 mL n-dodecane were added to perform a 96 h biphasic fermentation. After the fermentation completed, the fermentation broth was transferred to a 50 mL centrifuge tube and centrifuged at 4 °C and 8000 × g for 15 mins. The upper organic phase was taken out and awaited for GC-MS detection. In order to further improve the production of FPP, we next tried to integrate several key genes in the MVA pathway into the same copy site. The single restriction site BglII was designed to be added between the integration homology arm and terminator of the integration vector pCDF-TY4-URA-IDI1-ERG19. Then through homologous recombination, the expression module tHMG1-ERG10 was inserted into the BglII site of pCDF-TY4-URA-IDI1-ERG19 to form a integration vector pCDF-TY4-URA-IDI1-ERG19-ERG10-tHMG1. The integrated fragment pCDF-TY4-URA-IDI1-ERG19-ERG10-tHMG1 was cloned and integrated into the genome of S. cerevisiae BY4741, and strain FPP-3 was obtained by screening on the YNB plate with the corresponding SD-URA.
To obtain an AA-producing S. cerevisiae chassis strain, ERG20, ADS (GeneBank ID: Q9AR04) and CYP71AV1 (GeneBank ID: Q1PS23) were cloned from A. annua cDNA. Then, ERG20-ADS-CYP71AV1 was inserted into the expression vector pESC-LEU to form the expression plasmid pESC-LEU-ERG20-ADS-CYP71AV1. Using the expression plasmid as a template, the expression fragment LEU-ERG20-ADS-CYP71AV1 was cloned and integrated into the BamHI and SacI sites of pCDF-Ty2 to form a integrated vector pCDF-Ty2-LEU-ERG20-ADS-CYP71AV1. The integrated fragment Ty2-LEU-ERG20-ADS-CYP71AV1 was cloned and integrated into the genome of strain FPP-3 to obtain strain AA-1. In order to further improve AA production, ALDH1 (GeneBank ID: JQ609276.1) and ART1 (GeneBank ID: 820589) were cloned from A. annua cDNA and A. thaliana cDNA, respectively. ALDH1-ART1 fragment was inserted into the expression vector pESC-His to form the expression plasmid pESC-His-ALDH1-ART1. The expression fragment His-ALDH1-ART1 was cloned and integrated into the vector pCDF-Trp1 to form an integrated vector pCDF-Trp1-His-ALDH1-ART1. The integrated fragment Trp1-His-ALDH1-ART1 was cloned and integrated into the genome of strain AA-1, and was spread on the YNB plate with the corresponding SD-URA-LEU-His and cultured in a 30 °C constant-temperature incubator for about 72 h. Single colony was picked for yeast colony PCR verification, and the positive colony was named AA-2 and stored at -80 °C for later use. ADH1 (GeneBank ID: JF910157.1) and CYB5 (GeneBank ID: JQ582841.1) were cloned from A. annua cDNA, respectively. The ADH1-CYB5 fragment was inserted into the expression vector pESC-Trp to form the expression plasmid pESC-Trp-ADH1-CYB5. The expression fragment Trp-ADH1-CYB5 was cloned and integrated into the vector pCDF-Ty1 to form an integrated vector pCDF-Ty1-Trp-ADH1-CYB5. The integrated fragment Ty1-Trp-ADH1-CYB5 was cloned and integrated into the genome of strain AA-2, and was spread on the YNB plate with the corresponding SD-URA-LEU-His- Trp and cultured in a 30 °C constant-temperature incubator for about 72 h. Single colony was picked for yeast colony PCR verification, and the positive colony was named strain AA-3.
On the basis of the strain AA-3, the targeted optimized gene AaDHAADH (P26L) was integrated to obtain a DHAA-producing S. cerevisiae chassis strain which named strain DHAA-1(Supplementary Fig. 28G). To further increase DHAA production, the promoter PTHI2 was introduced and the copy number of the gene AaDHAADH (P26L) was increased (Supplementary Fig. 28H). Gene fragment PTHI2 + 2*AaDHAADH (P26L) was inserted into the expression vector pESC-Trp-ADH1-CYB5, it was then integrated into strain AA-2, and was spread on the YNB plate with the corresponding SD-URA-LEU-His-Trp and cultured in a 30 °C constant-temperature incubator for about 72 h. Single colony was picked for yeast colony PCR verification, and the positive colony was named strain DHAA-2. Primers used in this study were summarized in Supplementary Data 5. The plasmid constructs employed in this study were detailed in Supplementary Data 6 and 7.
Production of S. cerevisiae strains incorporating key genes in a 5 L fermenter
First, 10 μL of the engineered S. cerevisiae strain was added to 5 mL of YNB liquid medium with corresponding amino acid deficiencies, and cultured at 30 °C and 220 × g until OD600 = 1 to obtain a first-level seed liquid. And 2 mL first-level seed liquid was added to 100 mL of YNB liquid medium containing the corresponding defects, and continued to be cultured at 30 °C and 220 × g until OD600 = 1 to obtain grade 2 seed liquid. 3 L of YPD fermentation medium was prepared and added to the fermenter, and sterilized at 121 °C for 20 mins. After the fermentation liquid cools to room temperature, the second-level seed liquid was added. The dissolved oxygen in the fermentation broth was controlled at 35% through rotation speed and ventilation. The pH of the fermentation broth was maintained at around 6.0 by adding acid or alkali. The fermentation temperature was controlled at 30 °C, followed by fed-batch fermentation for 10 d. An appropriate amount of fermentation broth was taken out every 12 h to monitor the OD600 value of the bacterial broth and the content of the corresponding products.
Reconstruction of ART biosynthesis in N. benthamiana
For transient transformation in N. benthamiana, tHMG1 (GeneBank ID: AF142473), ERG20 (GeneBank ID: AF112881), ADS, CYP71AV1, CPR (GeneBank ID: JF910157.1), ADH1, ALDH1, CYB5, and AaDHAADH(P26L) were cloned into the destination vector pCAMBIA2300-EGFP using homologous recombination and transformed into A. tumefaciens GV3101. Positive transformants were cultured in YEB medium supplemented with rifampicin (25 mg/L) and kanamycin (50 mg/L) at 28 °C until OD600 = 0.6. The bacterial suspension was transferred to centrifuge tubes and centrifuged at 4000 × g, 4 °C for 20 min. Cells were collected and resuspended in infiltration buffer (200 μM acetosyringone, 10 mM anhydrous magnesium chloride, 50 mM morpholineethanesulfonic acid monohydrate, 20 mM glucose and 2 mM trisodium phosphate anhydrous) to OD600=0.6 and activated for 2 h at 28 °C and 220 × g. A. tumefaciens strains carrying the vector of the target gene were infiltrated into N. benthamiana leaves using a 1 mL syringe. After 96 h, leaves were collected for LC-MS analysis. N. benthamiana infected with A. tumefaciens were exposed to UV light (1.5 mW cm-2) for 1 h/d at 20 cm below the inverted ultraviolet transilluminator (302 nm; Analytik-Jena). Primers used in this study were summarized in Supplementary Data 5. The plasmid constructs employed in this study were detailed in Supplementary Data 6 and 7.
Statistical analysis
Unless specifically described, three biological replicates were performed for each quantification, and the numerical values were depicted as means ± standard. Statistical differences between the control and experimental groups were evaluated by the two-tailed student’s t-test. In all cases, P-values < 0.05 were considered significant. *, ** and *** indicate significant differences from control at the levels of P < 0.05, P < 0.01 and P < 0.001, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All genes used in this study are publicly available: ERG10 [https://www.ncbi.nlm.nih.gov/gene/?term=856230], ERG19 [https://www.ncbi.nlm.nih.gov/gene/?term=855889], ERG20 [https://www.ncbi.nlm.nih.gov/nuccore/AF112881.1/], IDI1 [https://www.ncbi.nlm.nih.gov/gene/?term=851557], tHMG1 [https://www.ncbi.nlm.nih.gov/nuccore/AF142473.1], ADS [https://www.ncbi.nlm.nih.gov/protein/Q9AR04], CYP71AV1 [https://www.ncbi.nlm.nih.gov/protein/Q1PS23], ALDH1 [https://www.ncbi.nlm.nih.gov/nuccore/JQ609276.1], ADH1 [https://www.ncbi.nlm.nih.gov/nuccore/JF910157.1], CYB5 [https://www.ncbi.nlm.nih.gov/nuccore/JQ582841.1], CPR [https://www.ncbi.nlm.nih.gov/nuccore/JF910157.1], AaDHAADH [https://www.ncbi.nlm.nih.gov/protein/PWA95605.1], C90 [https://www.ncbi.nlm.nih.gov/protein/PWA34386.1], V73 [https://www.ncbi.nlm.nih.gov/protein/PWA52903.1] and DBR2 [https://www.ncbi.nlm.nih.gov/nuccore/KC505370.1]. The LC-MS/MS data of the DEAE-2 active fraction used in this study have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the accession code PXD062563. Source data are provided with this paper.
References
Tu, Y. Artemisinin-a gift from traditional Chinese medicine to the world (Nobel lecture). Angew. Chem. -Int. Ed. 55, 10210–10226 (2016).
Wang, J. et al. Artemisinin, the magic drug discovered from traditional Chinese medicine. ENGINEERING 5, 32–39 (2019).
Ma, N., Zhang, Z., Liao, F., Jiang, T. & Tu, Y. The birth of artemisinin. Pharmacol. Ther. 216, 107658 (2020).
Xu, J. et al. 50th anniversary of artemisinin: from the discovery to allele-aware genome assembly of Artemisia annua. Mol. PLANT 15, 1243–1246 (2022).
Shen, Q. et al. The genome of Artemisia annua provides insight into the evolution of Asteraceae family and artemisinin biosynthesis. Mol. PLANT 11, 776–788 (2018).
Qiu, F. et al. Immunoregulation by Artemisinin and its derivatives: a new role for old antimalarial drugs. Front. Immunol. 12, 751772 (2021).
Lu, X. & Efferth, T. Repurposing of artemisinin-type drugs for the treatment of acute leukemia. Semin. CANCER Biol. 68, 291–312 (2021).
Kiss, E. et al. Artemisinin-treatment in pre-symptomatic APP-PS1 mice increases gephyrin phosphorylation at Ser270: a modification regulating postsynaptic GABAAR density. Biol. Chem. 403, 73–87 (2022).
Lai, H., Sasaki, T. & Singh, N. P. Targeted treatment of cancer with artemisinin and artemisinin-tagged iron-carrying compounds. EXPERT Opin. Ther. TARGETS 9, 995–1007 (2005).
Kong, Z., Liu, R. & Cheng, Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 109, 2043–2053 (2019).
Liu, Y. et al. Artemisinins ameliorate polycystic ovarian syndrome by mediating LONP1-CYP11A1 interaction. SCIENCE 384, eadk5382 (2024).
Firsov, A. et al. Heterologous biosynthesis of Artemisinin in Chrysanthemum morifolium ramat. SEPARATIONS 8, 75 (2021).
Olofsson, L., Lundgren, A. & Brodelius, P. E. Trichome isolation with and without fixation using laser microdissection and pressure catapulting followed by RNA amplification: Expression of genes of terpene metabolism in apical and sub-apical trichome cells of Artemisia annua L. PLANT Sci. 183, 9–13 (2012).
Salehi, M., Karimzadeh, G., Naghavi, M. R., Badi, H. N. & Monfared, S. R. Expression of artemisinin biosynthesis and trichome formation genes in five Artemisia species. Ind. CROPS Prod. 112, 130–140 (2018).
Banyai, W., Kirdmanee, C., Mii, M. & Supaibulwatana, K. Overexpression of farnesyl pyrophosphate synthase (FPS) gene affected artemisinin content and growth of Artemisia annua L. PLANT CELL TISSUE ORGAN Cult. 103, 255–265 (2010).
Chen, D. H., Ye, H. C. & Li, G. F. Expression of a chimeric farnesyl diphosphate synthase gene in Artemisia annua L. transgenic plants via Agrobacterium tumefaciens-mediated transformation. PLANT Sci. 155, 179–185 (2000).
He, Q. et al. Glandular trichome-specific expression of alcohol dehydrogenase 1 (ADH1) using a promoter-GUS fusion in Artemisia annua L. PLANT CELL TISSUE ORGAN Cult. 130, 61–72 (2017).
McCarty, N. S. & Ledesma-Amaro, R. Synthetic biology tools to engineer microbial communities for biotechnology. TRENDS Biotechnol. 37, 181–197 (2019).
Paddon, C. J. et al. High-level semi-synthetic production of the potent antimalarial artemisinin. NATURE 496, 528 (2013).
Ikram, N. K. B. K. et al. Stable production of the antimalarial drug artemisinin in the moss Physcomitrella patens. Front. Bioeng. Biotechnol. 5, 47 (2017).
Ro, D. K. et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. NATURE 440, 940–943 (2006).
Malhotra, K. et al. Compartmentalized metabolic engineering for artemisinin biosynthesis and effective malaria treatment by oral delivery of plant cells. Mol. PLANT 9, 1464–1477 (2016).
Wang, Y.-J. et al. Discovery and engineering of the cocaine biosynthetic pathway. J. Am. Chem. Soc. 144, 22000–22007 (2022).
Fu, R. et al. Versatility in acyltransferase activity completes chicoric acid biosynthesis in purple coneflower. Nat. Commun. 12, 1563 (2021).
Liu, H. et al. AaMYB108 is the core factor integrating light and jasmonic acid signaling to regulate artemisinin biosynthesis in Artemisia annua. NEW Phytol. 237, 2224–2237 (2023).
Cao, J. et al. Graphene enhances artemisinin production in the traditional medicinal plant Artemisia annua via dynamic physiological processes and miRNA regulation. PLANT Commun. 5, 100742 (2024).
Zheng, H. et al. Transcriptional regulatory network of high-value active ingredients in medicinal plants. TRENDS PLANT Sci. 28, 429–446 (2023).
Wallaart, T. E., Bouwmeester, H. J., Hille, J., Poppinga, L. & Maijers, N. C. A. Amorpha-4,11-diene synthase: cloning and functional expression of a key enzyme in the biosynthetic pathway of the novel antimalarial drug artemisinin. Planta 212, 460–465 (2001).
Zhang, Y. et al. The molecular cloning of artemisinic aldehyde Δ11(13) reductase and its role in glandular trichome-dependent biosynthesis of artemisinin in Artemisia annua. J. Biol. Chem. 283, 21501–21508 (2008).
Teoh, K. H., Polichuk, D. R., Reed, D. W. & Covello, P. S. Molecular cloning of an aldehyde dehydrogenase implicated in artemisinin biosynthesis in Artemisia annua. BOTANY 87, 635–642 (2009).
Paddon, C. J. & Keasling, J. D. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 12, 355–367 (2014).
Zeng, B.-X., Yao, M.-D., Wang, Y., Xiao, W.-H. & Yuan, Y.-J. Metabolic engineering of Saccharomyces cerevisiae for enhanced dihydroartemisinic acid production. Front. Bioeng. Biotechnol. 8, 152 (2020).
Qiu, F. et al. Biochemical and metabolic insights into hyoscyamine dehydrogenase. ACS Catal. 11, 2912–2924 (2021).
Hong, B. et al. Biosynthesis of strychnine. NATURE 607, 617 (2022).
Jiang, B. et al. Characterization and heterologous reconstitution of Taxus biosynthetic enzymes leading to baccatin III. SCIENCE 383, 622–629 (2024).
Ibdah, M. et al. Identification and characterization of three nearly identical linalool/nerolidol synthase from Acorus calamus. PHYTOCHEMISTRY 202, 113318 (2022).
Zhu, J. et al. Effects of dihydro-epi-deoxyarteannuin B on artemisinin biosynthesis, transcriptional profile and associated gene expression in suspension-cultured cells of Artemisia annua. Biochem. Eng. J. 160, 107633 (2020).
Yan, D. et al. Flavin-dependent monooxygenase-mediated 1,2-oxazine construction via Meisenheimer rearrangement in the biosynthesis of paeciloxazine. J. Am. Chem. Soc. 144, 4269–4276 (2022).
Gao, L. et al. FAD-dependent enzyme-catalysed intermolecular [4+2] cycloaddition in natural product biosynthesis. Nat. Chem. 12, 620 (2020).
Bortolotti, M., Polito, L., Battelli, M. G. & Bolognesi, A. Xanthine oxidoreductase: one enzyme for multiple physiological tasks. REDOX Biol. 41, 101882 (2021).
Kushwaha, P. P. et al. Phytochemicals present in Indian ginseng possess potential to inhibit SARS-CoV-2 virulence: a molecular docking and MD simulation study. Microb. Pathog. 157, 104954 (2021).
Zhu, J. et al. Inducing effect of dihydroartemisinic acid in the biosynthesis of artemisinins with cultured cells of Artemisia annua by enhancing the expression of genes. ScientificWorldJournal 2014, 293190 (2014).
Mehta, N., Meng, Y., Zare, R., Kamenetsky-Goldstein, R. & Sattely, E. A developmental gradient reveals biosynthetic pathways to eukaryotic toxins in monocot geophytes. Cell 187, 5620 (2024).
Wang, Z. et al. Cytochrome P450 mediated cyclization in Eunicellane derived diterpenoid biosynthesis*. Angew. Chem. 135, e202312490 (2023).
Medema, M. H. & Osbourn, A. Computational genomic identification and functional reconstitution of plant natural product biosynthetic pathways. Nat. Prod. Rep. 33, 951–962 (2016).
Xu, B. et al. Total biosynthesis of the medicinal triterpenoid saponin astragalosides. Nat. Plants 10, 1826–1837(2024).
Chen, M. et al. GLANDULAR TRICHOME-SPECIFIC WRKY 1 promotes artemisinin biosynthesis in Artemisia annua. NEW Phytol. 214, 304–316 (2017).
Zhao, Q., Li, M., Zhang, M. & Tan, H. Glandular trichomes: the factory of artemisinin biosynthesis. Med. Plant Biol. 3, 0–0 (2024).
Ryu, K. H., Huang, L., Kang, H. M. & Schiefelbein, J. Single-cell RNA sequencing resolves molecular relationships among individual plant cells. PLANT Physiol. 179, 1444–1456 (2019).
Rababi, D. & Nag, A. A top-down approach for studying the in-silico effect of the novel phytocompound tribulusamide B on the inhibition of Nipah virus transmission through targeting fusion glycoprotein and matrix protein. Comput. Biol. Chem. 112, 108135 (2024).
Acknowledgements
This work was supported by Key R&D Program of Shandong Province (No. 2022SFGC0105 to J.Z.), Guangzhou Science and Technology Plan Project (No. 2023A03J0613 to J.Z.), Natural Science Foundation of Guangdong province (No. 2024A1515010301 to J.Z.), National Natural Science Foundation of China (No. 32470418 to J.Z.).
Author information
Authors and Affiliations
Contributions
J.Z. guided, managed and supervised the smooth progress of this work. J.Z. revised the manuscript. R.Y. directed the project and revised the manuscript. Z.G. performed the screening of active enzymes, functional verification and investigation of enzymatic properties. Z.G. and Y.H. performed the catalytic mechanism and directional optimization of AaDHAADH. Z.G. established the AaDHAADH-OE or RNAi transgenic A. annua cell lines and detected the expression of AaDHAADH and the content of ARTs. Y. Zhou performed the construction of S. cerevisiae chassis strains FPP-1, FPP-2 and FPP-3. J.L. performed the construction of S. cerevisiae chassis strains AA-1 and AA-2. Z.G. performed the construction of S. cerevisiae chassis strains AA-3, DHAA-1 and DHAA-2. Z.G. performed shake flask fermentation and yield calculation for all S. cerevisiae chassis strains in the article. Z.G. performed fermentation and product yield tests on AA-3 and DHAA-2 S. cerevisiae chassis strains in a 5 L fermenter. Z.G. wrote the manuscript and generated figures. D.L. made appropriate revisions and improvements to the manuscript. Y. Zhang assisted with the fermentation experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors claim no competing interests.
Peer review
Peer review information
Nature Communications thanks Henrik Simonsen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, Z., Zhou, Y., Li, J. et al. Dihydroartemisinic acid dehydrogenase-mediated alternative route for artemisinin biosynthesis. Nat Commun 16, 3888 (2025). https://doi.org/10.1038/s41467-025-59312-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59312-1
