
Abstract
Functionalization of carboranes, icosahedral boron−carbon molecular clusters, is of great interest as they have wide applications in medicinal and materials chemistry. Thus, site- and enantioselective synthesis of carboranes requires complete control of the reaction. Herein, we describe the asymmetric Rh(II)-catalyzed insertion reactions of carbenes into cage B–H bond of carboranes. This reaction thereby generates carboranes possessing a carbon-stereocenter adjacent to cage boron of the carborane, in excellent site- and enantioselectivity under mild reaction conditions. The fully computed transition structures of Rh(II)-catalyzed carbene insertion process through density functional theory are reported. These B–H insertion transition structures, in conjunction with topographical proximity surfaces analyses, visually reveal the region between the carborane and the phthalimide ligands responsible for the selectivities of this reaction.
Similar content being viewed by others
Introduction
Control of selectivity in reactions is of utmost importance in chemistry and the ultimate driving force for developing new reactions. Asymmetric catalytic reactions to control the stereoselectivity with chiral organic molecules, chiral auxiliaries, chiral reagents, and chiral metal complexes have been recognized as the most solid method in asymmetric synthesis1,2,3,4. Their syntheses involve breaking the point, axial, planar, and helical symmetry elements of symmetric molecules. Among these, transition metal-catalyzed enantioselective reactions have emerged as one of the most powerful approaches to get optically pure compounds2,4. To date, a huge myriad of chiral structures including central, axial, planar, and helical chirality have been achieved by catalytic asymmetric synthesis5,6. However, site-selective reactions to break the symmetry in hypersymmetric three-dimensional cluster compounds, such as the icosahedral carboranes, with exohedral stereocontrol is a formidable challenge due to various classes of chirality such as plane chirality, cage chirality, and carbon chirality adjacent to the cage carbon or boron.
Carboranes are icosahedral boron−carbon molecular clusters, attractive building blocks combining properties such as chemical and biological stability, lipophilicity and hydrophobicity solubility properties, hydridic B–H bonds, spherical geometry, and three dimensional σ-aromaticity7,8. Carboranes have been utilized in boron neutron capture therapy agents in medicine (Fig. 1a)9, as unique pharmacophores10, as ligands in transition metal catalysis to improve solubility, stability, and turnover numbers in catalysis11,12,13, and even confer unique and powerful structural and photooptical properties in supramolecular design and materials14,15,16,17. However, despite these distinctive functions that carboranes can confer, we are crippled in our ability to access the full potential of carboranes because we do not have means to site-selectively react on carboranes and build complexity rapidly.

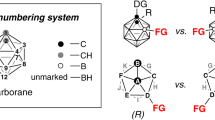
a Applications of carboranes in pharmaceutical chemistry. b Structure of o-carborane, relationship between boron and carbon, and possible regioisomers via B–H functionalization. c Timeline of chiral carboranes. d Direct approaches to setting carbon-stereocenter adjacent to cage boron of the carborane.
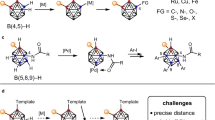
Accordingly, a variety of research and applications based on these facts have increased the interest in the site- and enantioselective functionalization on ten boron vertexes of carborane (Fig. 1b)18,19. However, significant advances in the synthesis of chiral molecules possessing carborane moieties have only recently been achieved despite of recent progress for the functionalization of carboranes20,21,22,23,24. For the first time, Kalinin and co-workers carried out the direct asymmetric synthesis (up to 32% e.e.) for o-carborane derivatives bearing chirality on C-proximity by Pd-catalyzed allylation reaction in the presence of chiral ligand (Fig. 1c)25. Recently, such o-carboranes were synthesized with high stereoselectivity via the Sharpless catalytic asymmetric dihydroxylation of C(1)-alkenyl o-carboranes26. If the substituents on the cage carbon atoms are different, substitution at B(3) position provides a pair of enantiomers possessing chiral center on the plane. In this regard, Krasnov and co-workers were the first to obtain planar-chiral 3-amino-1-methyl-1,2-dicarba-closo-dodecaborane in enantiomerically moderate form through a chiral resolution27. On the other hand, Xie, Qiu, and co-workers developed for the first time an enantioselective synthesis of chiral-at-cage o-carboranes through Pd-catalyzed intramolecular cross-coupling reactions with (R)-BI-DIME as a chiral ligand28. Furthermore, they reported Ir-catalyzed enantioselective B–H alkenylation with chiral phosphoramidite ligand, affording chiral-at-cage B(4)-alkenyl o-carboranes29. In these seminal reports, the functionalization was achieved either intramolecularly or made use of a directing group to control the site-selectivity. In contrast, there has been no success in achieving stereoselective introduction of an exohedral chiral center, which is a carbon-stereocenter adjacent to cage boron of the carborane, through direct B–H functionalization that is highly enantioselective and in the absence of any directing group. Rudimentary control of enantioselectivity using carboranes as the steric element or control of regioselectivity in B–H bond activation have been reported. However, true complexity building synthetic processes which can activate B–H bonds on carboranes in a regiocontrolled manner while simultaneously creating exohedral chiral centers are unknown at this time.
Although some reactions of carbenes with unsubstituted carboranes have been reported, the yield and site-selectivity were lackluster without stereoselectivity issue (see the Supplementary Information Fig. S1). For example, Jones reported the reaction of o-carborane with ethyl diazoacetate under irradiation, providing inseparable four regioisomers in combined 10% yield (12:7.2:4.8:1)30. Moreover, he found that methylene carbene could insert into B–H bonds of m-carborane under irradiation, producing inseparable regioisomeric mixture31. Sung and coworkers also demonstrated that the photolysis of o-carborane with diazomethane in a hexafluorobenzene leads to the formation of a mixture of four regioisomers through B–H insertion32. Reaction of p-carborane with methylene carbene under irradiation afforded B-alkylated product in low yield even giving single product due to identical ten B–H bonds. Therefore, the development of concise and efficient method that controls site- and enantioselectivity in functionalization of carborane is attractive and a significant challenge.
We describe herein the effective site- and enantioselective B–H functionalization, thereby generating o-, m-, and p-carboranes possessing an exohedral carbon-stereocenter, which is a carbon-stereocenter adjacent to cage boron of the carborane, in excellent enantioselectivity (99% e.e.), site-selectivity (>50:1 r.r.) and in excellent yields with broad substrate scope under mild reaction conditions (Fig. 1d). We are also pleased to report the fully quantum mechanically computed transition structures of this chiral dirhodium carbenoid insertion process into an icosahedral cage B–H bond of carboranes. Our density functional theory (DFT) results reproduce experimentally observed site- and enantioselectivity of the B–H functionalization. We employ a topographical tool to visualize and highlight the structurally subtle, but energetically critical, close contacts to elucidate the specific structural elements involved. This topographical proximity surfaces (TPS) analysis, coupled with NCI and EDA analysis, reveal the complex combination of interactions between the carborane and the phthalimide ligand responsible for the observed selectivities.
Results
Reaction optimization
To develop a site- and enantioselective carbene insertion reaction into B–H bond of o-carboranes, reaction conditions were extensively explored (see Supplementary Information Table S1 and S2 for details). Our investigation began with the reaction of 1,2-(DMPS)2-o-C2B10H10 (1a, DMPS dimethylphenylsilyl) with methyl 2-diazo-2-phenylacetate (2a) in dichloromethane (DCM) at 40 °C using various dirhodium tetracarboxylate catalysts. Upon detailed examination of these reactions, we found that two regioisomers [3aa-B(9) and 4aa-B(8)] were obtained as an inseparable mixture, while no other regioisomers were observed. The regioisomeric ratio (r.r.) of 3aa and 4aa and the enantiomeric excess (e.e.) of 3aa were determined through 1H NMR and chiral HPLC analysis, respectively. Among the achiral catalysts, Rh2(OAc)4 gave the mixture of regioisomers in 87% yield with 2.6:1 r.r. as racemate (entry 1). On the other hand, Rh2(oct)4 provided a relatively low yield (71%) but high site-selectivity (14:1) (entry 2). Furthermore, we investigated various chiral catalysts to achieve the enantioselective reaction (entries 3-11)33. As a result of examining chiral Rh(II) catalysts, it was revealed that Rh2(S-TCPTTL)4 showed quantitative yield, high site-selectivity (29:1), and enantioselectivity (25% e.e.) (entry 10). Rh2(R-BTPCP)4, the most sterically encumbering catalyst, gave trace amount of conversion of 1a, probably due to the steric effect of carborane cluster (entry 11)34. The enantioselectivity of the present reaction was affected by the solvents such as dichloroethane (DCE), cyclohexane, benzene, and trifluorotoluene (PhCF3). Especially, benzene and PhCF3 enhanced the enantioselectivity to 44% e.e. and 42% e.e., respectively, and then PhCF3 was chosen as an optimum solvent (entry 15)35. When the reaction temperature was lowered from 40 °C to 0 °C, the enantiomeric excess increased from 42% to 51% (entry 17). We were pleased to observe that quantitative yield was obtained even with 1.0 mol % catalyst loading (entry 19). When 2a (1.5 equiv) was used, the yield was reduced to 79% (entry 21).
Substrate scope
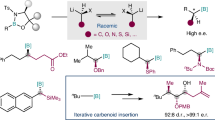
Based on these results, the substrate scope of o-carboranes was next investigated (Fig. 2). When unsubstituted, silyl- or benzyl-disubstituted o-carboranes (1a-1d) were treated with methyl 2-diazo-2-phenylacetate (2a), the yields of the desired products (3aa-3da) were all quantitative, but the selectivity was affected by the substituents of o-carborane. We found that 1,2-(TMS)2-o-C2B10H10 (1b; TMS trimethylsilyl) gave rise to 3ba in high site- and enantioselectivity (22:1 r.r. and 55% e.e.). After close examination of diazo substrate scope in the reaction with 1b, it is disclosed that substituents on aryl and ester group play an important role in enantioselectivity (3bb-3bo). As a result, 2,2,2-trichloroethyl 2-(4-bromophenyl)-2-diazoacetate (2d) underwent the B–H insertion reaction to produce the desired product 3bd in 96% yield with high site- and enantioselectivity (25:1 r.r. and 99% e.e.). This result indicates that trichloroethyl (TCE) group is very effective to the B–H insertion36. When there was no substituent on the aryl group, the desired 3be was obtained in 98% yield with 25:1 r.r. and 89% e.e., suggesting that para-substituents on aryl group are essential for excellent enantioselectivity. Then, we evaluated various electron-withdrawing groups on para-position of the aryl ring. TCE aryl diazoacetates possessing chloro, iodo, trifluoromethyl, ketone, and ester groups provided the corresponding B–H insertion products (3bf-3bj) in high yields ranging from 89% to 99% with excellent site- and enantioselectivity (up to >50:1 r.r. and 99% e.e.). p-Nitro-substituted diazoacetate (2k) was reacted with 1b at 40 °C, resulting in the formation of the desired product (3bk) in 69% yield with >50:1 r.r. and 93% e.e. In addition, a variety of electron-donating groups such as methyl, tert-butyl, phenyl, and methoxy group on para-position were tolerable, affording the desired carboranes (3bl-3bo) in high yields with site- and enantioselectivities. Diazo compounds possessing bromo, methyl, and methoxy groups on meta-position (2p-2r) and fluoro, bromo, and methyl groups on ortho-position (2s-2u) gave the corresponding products (3bp-3bu) in good to excellent site-selectivities, enantioselectivities (up to 97% e.e.), and yields (up to 99%). TCE aryl diazoacetates that possess 3,4-dichlorophenyl, 3,5-dimethylphenyl, and 2-naphthyl groups were also compatible with the present reaction conditions (3bv-3bx). TCE heteroaryl diazoacetates including thiophene and pyridine (2y and 2z) were successfully applied to the present reaction. The enantiomeric excesses of 3bk, 3bq, 3bt, and 3by were determined after desilylation because of the difficulty in separation of enantiomers.

a Optimum condition A: After 1 (0.20 mmol, 1.0 equiv) and Rh2(S-TCPTTL)4 (1.0 mol %) were dissolved in PhCF3 (1.5 mL), a solution of 2 (2.0 equiv) in PhCF3 (1.5 mL) was added over a period of 3 min at 0 °C under a N2 atmosphere. The reaction mixture was stirred for additional 10 min. b The e.e. of the product was determined after desilylation. c A solution of 2 in PhCF3 was added at 40 °C. d Crystal structure was obtained after transformation of ester to carboxylic acid. e 2d (3.0 equiv) was used. f Optimum condition B: After 1 (0.20 mmol, 1.0 equiv) and Rh2(S-TPPTTL)4 (2.0 mol %) were dissolved in PhCF3 (1.5 mL), a solution of 2 (2.0 equiv) in PhCF3 (1.5 mL) was added over a period of 3 min at 60 °C under a N2 atmosphere. The reaction mixture was stirred for additional 10 min. g When dirhodium-catalyzed reaction was conducted on a larger scale under optimum condition A, desilylation reaction was performed in one-pot.
Encouraged by these results, a wide range of carboranes were investigated in the reaction with 2,2,2-trichloroethyl 2-(4-bromophenyl)-2-diazoacetate (2d) to verify if the excellent site- and enantioselectivity would be maintained. When o-C2B10H12, 1,2-dibenzyl- and 1,2-dimethyl-o-C2B10H10 (1c-1e) were treated with 2d, the desired products (3cd-3ed) were obtained in high yield with 99% e.e. However, these substrates exhibited inferior site-selectivity (4.9:1 ~ 6.3:1 r.r.), suggesting that silyl groups on the cage carbon of the carborane play a critical role. To demonstrate the versatility of these Rh-catalyzed cage B–H insertion reactions, we examined whether the substrates possessing substituent on the cage boron could be employed. It is noteworthy that both 1f and 1g were smoothly converted to the desired products (3fd and 3gd) in 89% and 97% yields, respectively, without any regioisomers. Although 3,6-diphenyl-o-C2B10H10 (1f) showed 35% enantioselectivity, 1-methyl-7,11-diphenyl-o-C2B10H9 (1g) exhibited excellent enantioselectivity (99% e.e.). The structures of (R)-3bd and (R)-3gd were confirmed by X-ray crystallography (see Supplementary Information Table S5 and S6). Crystal structure of (R)-3gd was obtained after transformation of ester to carboxylic acid because of difficulty in crystal formation.
Next, we applied the present method to m- and p-carboranes (Fig. 2). Gratifyingly, the carbenes on phthalimido Rh catalyst smoothly underwent B–H insertion reactions with m- and p-carboranes. When m-C2B10H12 (1h) was treated with 2d under optimum reaction conditions, the corresponding product 3hd was obtained in 76% yield with excellent enantioselectivity (99% e.e.). 1,7-(TMS)2-m-C2B10H10 (1i) was transformed to the desired product (3id) in 92% yield with 95% e.e. with 3.0 equivalents of 2d. p-Carborane (1j) having equivalent ten B–H bonds can react with two or more carbenes to give multialkylated products. To suppress repetitive B–H insertion reaction, steric influence of the rhodium catalyst was enhanced, and it was revealed that Rh2(S-TPPTTL)4 is suitable for mono-selective B–H insertion reactions of p-carboranes, affording 3jb-3jd in good yield with high enantioselectivity (up to 97% e.e.). The structures of (R)-3hd and (R)-3jd were confirmed by X-ray crystallography (see Supplementary Information Table S7 and S8).
To prove the practicability of the present catalytic procedure, the B–H insertion reaction of o-carboranes was examined on a large scale using 1.01 g (3.50 mmol) of 1b. After completion of Rh-catalyzed B–H insertion reaction, the one-pot desilylation reaction was successfully carried out, leading to the desilylated products 5 and 6 in high yields (85% and 91%, respectively) with excellent enantioselectivity (99% e.e.).
CD spectra, reactivity comparison, and synthetic applications
Circular dichroism (CD) spectra of (R)-3bd and (S)-3bd obtained with Rh2(S-TCPTTL)4 and Rh2(R-TCPTTL)4 catalyst exhibited unambiguously mirror images to each other, indicating a pair of enantiomers (Fig. 3a). Furthermore, the absolute configuration of (R)-3bd and (S)-3bd was confirmed by X-ray crystallography (see Supplementary Information Table S5 and S9).

a CD spectra and X-ray crystal structures of (R)-3bd and (S)-3bd. b Comparison of relative reactivity with carbenophiles under the optimum condition A. c Transformation of B–H insertion products. Reaction conditions: (i) 5 (0.2 mmol), DIBAL-H (2.2 equiv) in DCM (6.0 mL) at -78 °C to 25 °C for 2 h. (ii) 5 (0.2 mmol), Zn (10.0 equiv) in AcOH (4.0 mL) at 25 °C for 48 h. (iii) 6 (0.2 mmol), phenyl acetylene (1.5 equiv), Pd2dba3 (5.0 mol %), XPhos (10.0 mol %), CuI (10.0 mol %) in Et3N (1.0 mL) at 80 °C for 12 h. (iv) 6 (0.2 mmol), PhNH2 (1.5 equiv), Pd2dba3 (5.0 mol %), XPhos (10.0 mol %), NaOt-Bu (1.5 equiv), 4 Å molecular sieve (100.0 mg) in toluene (2.0 mL) at 50 °C for 3 h.
To examine the reactivity of carboranes with rhodium carbenoids, competition experiments were conducted with various carbenophiles using 1.0 equivalent of 2d under the optimum reaction conditions (Fig. 3b). First, we initiated competition experiment with 1,4-cyclohexadiene (1,4-CHD) that rapidly undergoes allylic C–H insertion reactions with rhodium carbenoids37. As a result, 7 was obtained in 70% yield without the formation of 3bd, suggesting that reactivity of 1,4-CHD is strong compared to 1b. Next, competition reaction of 1b with dioxolane furnished 3bd and 8 in 21% and 47% yields, respectively. This result implies that reactivity of dioxolane has slightly better than that of 1b. Finally, since 3bd was only produced from competition experiment of 1b and tetrahydrofuran (THF), relative reactivity order of these carbenophiles could be listed as follows 1,4-CHD >> dioxolane > TMS-carborane (1b) >> THF.
To explore the application of these reactions, further functionalization of 5 and 6 was attempted. When 5 was treated with DIBAL-H, the corresponding alcohol 10 was obtained in 75% yield without erosion of the stereochemical fidelity. Trichloroethyl ester was successfully transformed to carboxylic acid 11 in 89% yield using zinc and acetic acid also with no erosion of enantiomeric excess. Enantiomeric excess of 6 was slightly deteriorated under coupling reaction conditions. As a result of Sonogashira and Buchwald-Hartwig cross-coupling reactions with 6, desired internal alkyne 12 and diaryl amine 13 were produced in 86% (89% e.e.) and 68% yields (86% e.e.), respectively.
Mechanistic studies
A deuterium labeling experiment revealed that B–H insertion reaction occurs through concerted mechanism because the deuterium atom substituted at B(9)-position of o-carborane 1b-[Dn] was transferred to the α-carbon adjacent to cage boron of the product 3bd-[Dn] without a change in the H/D ratio. When 1b-[Dn] or 1b was treated with H2O or D2O under the optimum reaction conditions, deuterium scrambling was not observed at all (Fig. 4a).

a Mechanistic experiments with deuterated starting material, H2O, and D2O. b A proposed catalytic cycle and quantum mechanically computed energies for the formation of (R)-3bd with respect to the dirhodium carbenoid I complex. c The topographic proximity surface (TPS) of the TMS groups against the isodensity surface of the dirhodium catalytic pocket ([Rh]- = Rh2(S-TCPTTL)4) reveals close contact ranging from 1.0-2.5 Å.
In addition to the experimental data, computations were also conducted to understand the site- and enantioselectivity of this dirhodium-catalyzed carbenoid B–H insertion reaction into icosahedral cage o-carborane using density functional theory (DFT). The applicability of DFT for studying dirhodium-catalyzed reactions and C–H bond insertions have been explored by others38,39,40,41,42,43,44,45,46,47,48. It is noteworthy that despite significant efforts by multiple research groups, these pioneering efforts reveal the enormous challenges and complexities involved with computing transition structures of large and conformationally flexible systems. Most computational studies of dirhodium carbenoid insertion processes have been rationalizations from ground state structures. To date, there are only two computed transition state studies involving the full dirhodium-catalyzed carbenoid insertion for C–H bonds, and none for B–H bond insertions using carboranes. Houk, Davies, and co-workers reported an enantioselective functionalization of a non-activated primary C–H bond using an alkyl substrate, but this study involved a relatively conformationally rigid catalyst and a judicious choice of QM/MM methodology to deal with the cost of computing such large structures42. Tantillo and co-workers reported ab initio molecular dynamics simulations to rationalize the origins of selectivity in a C–H functionalization involving an intramolecular 1,4-shift48. Herein, we are pleased to report the fully quantum mechanically computed transition structures of a chiral dirhodium-catalyzed carbenoid B–H insertion reaction of carboranes involving the complete experimentally used ligands and substrates with no structural simplifications. Our DFT results reproduce experimentally observed site- and enantioselectivity. In addition, we reveal a tool to visualize and highlight the structurally subtle, but energetically critical, steric close contacts in a topographical view to elucidate the specific functional groups and moieties. All computations and structures presented in this paper were performed at the PBE-D3BJ level of theory in conjunction with the LANL2DZ(Rh, Br, Cl) & 6-31 G* (for all other atoms) basis sets as implemented in Gaussian 16. CPCM(C6H6) solvation corrections were also used at 0 °C. Single point energy refinements were performed at the PBE-D3BJ level of theory with the Ahlrich def2-TZVP basis set (see Supplementary Information Computational Section).
The proposed catalytic cycle for the synthesis of product (R)-3bd begins with the decomposition of the diazo compound 2d by the dirhodium catalyst Rh2(S-TCPTTL)4 to afford the dirhodium carbenoid intermediate I (∆G = 0.0 kcal/mol) with the release of molecular nitrogen gas (Fig. 4b). The highly reactive dirhodium carbenoid I undergoes B–H insertion with the incoming o-carborane 1b, forming the major three-member transition state (TS) (II-TS(R)-B(9), ∆G‡ = 6.92 kcal/mol), which gives the site selective at B(9)-position and enantioselective preference (R)-enantiomer at the exohedral carbon-stereocenter, which is a carbon-stereocenter adjacent to cage boron of the carborane. This major II-TS(R)-B(9) leads to the following ground state product complex III (∆G = –37.5 kcal/mol) wherein the desired product is embedded in the dirhodium catalyst pocket (Supplementary Information Fig. S8). A second diazo compound 2d releases the major product (R)-3bd (∆G = –51.1 kcal/mol), as well as molecular nitrogen gas, resulting in regeneration of the dirhodium carbenoid I for the next catalytic cycle. The complete reaction coordinate diagram for this proposed mechanism and the energies are shown in the Supplementary Information Fig. S8.
Previous reports by Houk and Davies hypothesized that the helical arrangement of the phthalimide ligands of the chiral dirhodium catalyst observed in the ground state as important in determining the selectivities of the C–H insertion process in their studies. The conformational complexity and substantial molecular size of this chiral dirhodium-catalyzed carbenoid B–H insertion of carboranes that were challenging to DFT compute also posed significant difficulties to discover and explain where the origins of selectivity arose within the large transition structure complexes. To address these challenges, we employed a Topographical Proximity Surfaces (TPS) visualization to analyze the close contacts that exist in the TSs (Fig. 4c). The intensity of the color on the surface reflects the close contact of the TMS groups of the o-carborane 1b to the rhodium catalyst ligand phthalimides. This approach can reveal close contacts in large, complex transition structures and aid in the rationalization of reaction selectivities. We compared the TPS to noncovalent interaction (NCI) analysis49 and electronic decomposition analysis50 by Shubin Liu (EDA-SBL), and they reveal the same general trends, albeit without the simplicity of the TPS (See Supplementary Information Fig. S10 and Table S3).
In the favored major (R)-B(9)-insertion TS (II-TS(R)-B(9), ∆G‡ = 6.92 kcal/mol), the dirhodium carbenoid insertion occurs at B(9) position of o-carborane 1b to give the (R)-configuration product at the exohedral carbon-stereocenter adjacent to cage boron of the carborane. The unfavored epimeric dirhodium carbenoid insertion results in the minor (S)-B(9)-insertion TS (II-TS(S)-B(9), ∆G‡ = 8.96 kcal/mol, i.e. stereoselectivity of 2.04 kcal/mol), and the unfavored regioisomeric insertion results in the minor (R)-B(8)-insertion TS (II-TS(R)-B(8), ∆G‡ = 9.00 kcal/mol, i.e. site-selectivity of 2.08 kcal/mol). These DFT results agree with the experimental site- and enantioselectivity of 2.00 kcal/mol and 2.87 kcal/mol, respectively. The TPS visualization of the major (R)-B(9)-insertion TS (II-TS(R)-B(9)) reveals a comparatively diminished interaction between TMS groups of the o-carborane 1b and the dirhodium carbenoid complex I (Fig. 4c). This is a result of the o-carborane angle and positioning of the TMS groups into the phthalimide ligand cavity. In contrast, the TPSs of the epimeric (S)-B(9)-insertion TS (II-TS(S)-B(9)) and the regioisomeric (R)-B(8)-insertion TS (II-TS(R)-B(8)) both show greater interaction of the TMS groups against the phthalimide ligands of the dirhodium carbenoid complex I. In the former, in order to achieve the epimeric insertion of the minor (S)-configuration product, it necessitates the angle and positioning of the o-carborane such that the TMS groups clash into the wall of the phthalimide cavity (II-TS(S)-B(9)). Similarly, in the regioisomeric (R)-B(8)-insertion TS (II-TS(R)-B(8)), the rotation of the o-carborane to achieve insertion at the B(8) results in steric interactions between the TMS groups with the phthalimide ligands. These results visually reveal the extent and severity of steric interactions that govern the preference for the favored B–H insertion process by this large and conformationally flexible dirhodium catalyst, Rh2(S-TCPTTL)4.
Discussion
In summary, effective site- and enantioselective B–H insertion reactions have been developed from the reaction of donor/acceptor carbenes into cage B–H bond of carboranes with chiral rhodium(II) catalyst. This selective B–H functionalization thereby constructs o-, m-, and p-carboranes possessing exohedral carbon-stereocenter, which is adjacent to cage boron of the carborane in excellent site-selectivity (>50:1 r.r.) and enantioselectivity (99% e.e.) in high yields with broad substrate scope under mild reaction conditions. We also report the fully quantum mechanically computed transition structures of the B–H insertion process of carboranes involving the complete large and conformationally flexible chiral dirhodium carbenoids. Gratifyingly, the computed site-selectivity and enantioselectivity (2.08 kcal/mol and 2.04 kcal/mol, respectively) were in good agreement with the experiments (2.00 kcal/mol and 2.87 kcal/mol, respectively). Furthermore, we reveal a tool to visually highlight the structurally subtle, but energetically critical, distribution of the close contact in a topographical fashion. This clearly shows the overall topographical shape created by the large and flexible dirhodium catalyst to which the substrate must bind to undergo reaction. This tool may play a significant role in future computational studies involving large catalyst systems. Ultimately, we discovered that the chiral dirhodium carbenoid is capable of this unique and impressive site- and enantioselectivity on an icosahedral cage substrate because the favored (R)-B(9)-insertion TS is able to angle the di-TMS substituted o-carborane into the phthalimide ligand cavity with minimal steric repulsion. This work opens an efficient way for true site selective transformations of icosahedral complexes and enantioselective functionalization, affording exohedral chirality through the formation of a single, new B–C bond involved in a concerted B–H insertion.
Methods
General procedure for the B–H insertion product of o- and m-Carboranes
An oven dried test tube equipped with a magnetic stirrer was charged with o- and m-carborane 1 (0.2 mmol, 1.0 equiv) and Rh2(S-TCPTTL)4 (1.0 mol %) in PhCF3 (1.5 mL) and stirred at 0 °C under a N2 atmosphere. The diazo 2 (2.0 equiv) in PhCF3 (1.5 mL) was added to the reaction mixture dropwise via syringe pump over 3 min. Then, the reaction mixture was stirred at 0 °C for 10 min and concentrated under reduced pressure for crude 1H NMR. The crude product was purified by column chromatography on silica gel to afford product 3 and 4.
General procedure for the B–H insertion product of p-Carboranes
An oven dried test tube equipped with a magnetic stirrer was charged with p-carborane 1j (0.2 mmol, 1.0 equiv) and Rh2(S-TPPTTL)4 (2.0 mol %) in PhCF3 (1.5 mL) and stirred at 60 °C under a N2 atmosphere. The diazo 2 (2.0 equiv) in PhCF3 (1.5 mL) was added to the reaction mixture dropwise via syringe pump over 3 min. Then, the reaction mixture was stirred at 60 °C for 10 min and concentrated under reduced pressure for crude 1H NMR. The crude product was purified by column chromatography on silica gel to afford product 3.
Data availability
The main data supporting the findings of this study are available within the article and its Supplementary Information files, or from the corresponding author upon request. The X-ray crystallographic data for structures have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2031747, 2085991, 2036998, 2036999, and 2085990 and can be obtained free of charge from https://www.ccdc.cam.ac.uk/structures/. Cartesian coordinates of computationally optimized geometries are available in Supplementary Data.
References
Corey, E. J. & Kürti, L. Enantioselective Chemical Synthesis (Academic Press, 2010).
Christmann, M. & Bräse, S. Asymmetric Synthesis II (Wiley-VCH, 2012).
Zhou, Q.-L. Privileged Chiral Ligands and Catalysts (Wiley-VCH, 2011).
Lin, G.-Q., Li, Y.-M. & Chan, A. S. C. Principles and Applications of Asymmetric Synthesis (John Wiley & Sons, 2002).
Xie, J.-H., Zhu, S.-F. & Zhou, Q.-L. Transition metal-catalyzed enantioselective hydrogenation of enamines and imines. Chem. Rev. 111, 1713–1760 (2011).
Newton, C. G., Wang, S.-G., Oliveira, C. C. & Cramer, N. Catalytic enantioselective transformations involving C−H bond cleavage by transition-metal complexes. Chem. Rev. 117, 8908–8976 (2017).
Grimes, R. M. Carboranes 2nd edn (Academic Press, 2011).
Hosmane, N. S. Boron Science: New Technologies and Applications (CRC, 2012).
Hawthorne, M. F. The role of chemistry in the development of boron neutron capture therapy of cancer. Angew. Chem. Int. Ed. 32, 950–984 (1993).
Issa, F., Kassiou, M. & Rendina, L. M. Boron in drug discovery: carboranes as unique pharmacophores in biologically active compounds. Chem. Rev. 111, 5701–5722 (2011).
Xie, Z. Advances in the chemistry of metallacarboranes of f-block elements. Coord. Chem. Rev. 231, 23–46 (2002).
Xie, Z. Cyclopentadienyl-carboranyl hybrid compounds: a new class of versatile ligands for organometallic chemistry. Acc. Chem. Soc. 36, 1–9 (2003).
Spokoyny, A. M. et al. A coordination chemistry dichotomy for icosahedral carborane-based ligands. Nat. Chem. 3, 590–596 (2011).
Jude, H. et al. Coordination-driven self-assemblies with a carborane backbone. J. Am. Chem. Soc. 127, 12131–12139 (2005).
Wee, K.-R. et al. Carborane-based optoelectronically active organic molecules: wide band gap host materials for blue phosphorescence. J. Am. Chem. Soc. 134, 17982–17990 (2012).
Cioran, A. M. et al. Mercaptocarborane-capped gold nanoparticles: electron pools and ion traps with switchable hydrophilicity. J. Am. Chem. Soc. 134, 212–221 (2012).
Mukherjee, S. & Thilagar, P. Boron clusters in luminescent materials. Chem. Commun. 52, 1070–1093 (2016).
Olid, D., Núñez, R., Viñas, C. & Teixidor, F. Methods to produce B–C, B–P, B–N and B–S bonds in boron clusters. Chem. Soc. Rev. 42, 3318–3336 (2013).
Dziedzic, R. M. et al. B−N, B−O, and B−CN Bond formation via palladium-catalyzed cross-coupling of B‑bromo-carboranes. J. Am. Chem. Soc. 138, 9081–9084 (2016).
Qiu, Z. & Xie, Z. Functionalization of o-carboranes via carboryne intermediates. Chem. Soc. Rev. 51, 3164–3180 (2022).
Yang, L., Zhang, Z.-J., Jei, B. B. & Ackermann, L. Electrochemical cage activation of carboranes. Angew. Chem. Int. Ed. 61, e202200323 (2022).
Quan, Y. & Xie, Z. Controlled functionalization of o-carborane via transition metal catalyzed B–H activation. Chem. Soc. Rev. 48, 3660–3673 (2019).
Qiu, Z. & Xie, Z. A Strategy for selective catalytic B−H functionalization of o‑carboranes. Acc. Chem. Res. 54, 4065–4079 (2021).
Wang, Q., Liu, B., Feng, K. & Hashmi, A. S. K. Recent advance in transition metal-catalyzed carboxylic acid guided B−H functionalization of carboranes. Adv. Synth. Catal. 364, 4174–4188 (2022).
Lebedev, R. V. et al. Pd-catalyzed asymmetric allylation of an o-carborane derivative. Russ. Chem. Bull. 51, 513–516 (2002).
Duan, H.-X., Li, H.-N., Yang, Y., Wu, X.-J. & Wang, Y.-Q. Catalytic asymmetric synthesis of carboranylated diols bearing two adjacent stereocenters located at the α,β-position of o-carborane cage carbon. Dalton Trans. 52, 4077–4085 (2023).
Krasnov, V. P. et al. Enantiomers of 3-amino-1-methyl-1,2-dicarba-closo-dodecaborane. Tetrahedron.: Asymmetry 13, 1833–1835 (2022).
Cheng, R. et al. Enantioselective Synthesis of chiral-at-cage o‑carboranes via Pd-catalyzed asymmetric B−H substitution. J. Am. Chem. Soc. 140, 4508–4511 (2018).
Cheng, R., Zhang, J., Zhang, H., Qiu, Z. & Xie, Z. Ir-catalyzed enantioselective B−H alkenylation for asymmetric synthesis of chiral-at-cage o‑carboranes. Nat. Commun. 12, 7146–7154 (2021).
Zheng, G.-X. & Jones, M. Jr Reaction of (ethoxycarbonyl)carbene with o-carborane. J. Am. Chem. Soc. 105, 6487–6488 (1983).
Yuan, K. & Jones, M. Jr Carbenes do react with p-carborane. Tetrahedron Lett. 33, 7481–7484 (1992).
Sung, D. D., Lee, J. D. & Choi, S. K. A study on the reaction of icosahedral carborane with carbenes. Bull. Korean Chem. Soc. 8, 63–68 (1987).
Tsutsui, H., Abe, T., Nakamura, S., Anada, M. & Hashimoto, S. Practical synthesis of dirhodium(II) tetrakis[N-phthaloyl-(S)-tert-leucinate]. Chem. Pharm. Bull. 53, 1366–1368 (2005).
Tse, E. G. et al. Nonclassical phenyl bioisosteres as effective replacements in a series of novel open-source antimalarials. J. Med. Chem. 63, 11585–11601 (2020).
Ogawa, A. & Curran, D. P. Benzotrifluoride: a useful alternative solvent for organic reactions currently conducted in dichloromethane and related solvents. J. Org. Chem. 62, 450–451 (1997).
Guptill, D. M. & Davies, H. M. L. 2,2,2-Trichloroethyl aryldiazoacetates as robust reagents for the enantioselective C−H functionalization of methyl ethers. J. Am. Chem. Soc. 136, 17718–17721 (2014).
Müller, P. & Tohill, S. Intermolecular cyclopropanation versus CH insertion in RhII-catalyzed carbenoid reactions. Tetrahedron 56, 1725–1731 (2000).
Liao, K., Negretti, S., Musaev, D. G., Bacsa, J. & Davies, H. M. L. Site-selective and stereoselective functionalization of unactivated C–H bonds. Nature 533, 230–234 (2016).
Liao, K. et al. Site-selective and stereoselective functionalization of non-activated tertiary C–H bonds. Nature 551, 609–613 (2017).
Fu, J., Ren, Z., Bacsa, J., Musaev, D. G. & Davies, H. M. L. Desymmetrization of cyclohexanes by site- and stereoselective C–H functionalization. Nature 564, 395–399 (2018).
Pang, Y. et al. Rhodium-catalyzed B–H bond insertion reactions of unstabilized diazo compounds generated in situ from tosylhydrazones. J. Am. Chem. Soc. 140, 10663–10668 (2018).
Liao, K. et al. Design of catalysts for site-selective and enantioselective functionalization of non-activated primary C–H bonds. Nat. Chem. 10, 1048–1055 (2018).
Liu, W. et al. Catalyst-controlled selective functionalization of unactivated C−H bonds in the presence of electronically activated C−H bonds. J. Am. Chem. Soc. 140, 12247–1225 (2018).
Pons, A. et al. Catalytic enantioselective cyclopropanation of α‑fluoroacrylates: an experimental and theoretical study. ACS Catal. 9, 2594–2598 (2019).
Lee, M., Ren, Z., Musaev, D. G. & Davies, H. M. L. Rhodium-stabilized diarylcarbenes behaving as donor/acceptor carbenes. ACS Catal. 10, 6240–6247 (2020).
Garlets, Z. J. et al. Enantioselective C–H functionalization of bicyclo[1.1.1]pentanes. Nat. Catal. 3, 351–357 (2020).
Bergstrom, B. D., Nickerson, L. A., Shaw, J. T. & Souza, L. W. Transition metal catalyzed insertion reactions with donor/donor carbenes. Angew. Chem. Int. Ed. 60, 6864–6878 (2021).
Guo, W., Hare, S. R., Chen, S. S., Saunders, C. M. & Tantillo, D. J. C−H Insertion in dirhodium tetracarboxylate-catalyzed reactions despite dynamical tendencies toward fragmentation: implications for reaction efficiency and catalyst design. J. Am. Chem. Soc. 144, 17219–17231 (2022).
Johnson, E. R. et al. Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010).
Liu, S. Steric effect: A quantitative description from density functional theory. J. Chem. Phys. 126, 244103 (2007).
Acknowledgements
We thank CTK (Chiral Technology Korea) for the chiral HPLC analysis and Professor Mu-Hyun Baik (KAIST) and Dr. Euijae Lee (KAIST) for helpful discussion. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (RS-2024-00342573 and RS-2023-00271205 (P.H.L.)). PHYC is the Bert and Emelyn Christensen Professor and gratefully acknowledges financial support from the Vicki & Patrick F. Stone family and the computing infrastructure in part provided by the National Science Foundation (CHE-1352663 and NSF Phase-2 CCI, Center for Sustainable Materials Chemistry CHE-1102637 (P.H.-Y.C.)).
Author information
Authors and Affiliations
Contributions
Conceptualization: P.H.L.; Project administration & supervision: P.H.L. and P.H.-Y.C.; Experimental study: K.L., H.E., T.H.K. and H.C.N.; Computational study: G.A.G.-M., A.O.F. and H.R.W.; Crystallographic analysis: D.K.; Writing – original draft: P.H.L., P.H.-Y.C., K.L. and G.A.G.-M.; Writing – review & editing: P.H.L., P.H.-Y.C., K.L., and G.A.G.-M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Trevor Hamlin, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, K., González-Montiel, G.A., Eom, H. et al. Site- and enantioselective B−H functionalization of carboranes. Nat Commun 16, 4182 (2025). https://doi.org/10.1038/s41467-025-59410-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59410-0
