
Abstract
TRPV3 belongs to the large superfamily of tetrameric transient receptor potential (TRP) ion channels. Recently, using high-speed atomic force microscopy (HS-AFM), we discovered a rare and transient pentameric state for TRPV3 that is in equilibrium with the tetrameric state, and, using cryo-EM, we solved a low-resolution structure of the TRPV3 pentamer, in which, however, many residues were unresolved. Here, we present a higher resolution and more complete structure of the pentamer, revealing a domain-swapped architecture, a collapsed vanilloid binding site, and a large pore. Molecular dynamics simulations and potential of mean force calculations of the pentamer establish high protein dynamics and permeability to large cations. Subunit interface analysis, together with thermal denaturation experiments, led us to propose a molecular mechanism of the tetramer-to-pentamer transition, backed experimentally by HS-AFM observations. Collectively, our data demonstrate that the TRPV3 pentamer is in a hyper-activated state with unique, highly permissive permeation properties.
Similar content being viewed by others


Introduction
Transient receptor potential (TRP) channels are a large superfamily of eukaryotic ion channels responsible for sensing diverse chemical and physical stimuli, such as temperature, pH, pressure, and various natural and synthetic compounds. TRP channels are currently divided into ten families belonging to two large groups: Group 1 consists of TRPV (vanilloid), TRPA (ankyrin), TRPC (canonical), TRPM (melastatin), TRPN (no-mechanoreceptor potential C), TRPVL (vanilloid-like), and TRPS (soromelastatin), and Group 2 consists of TRPML (mucolipin), TRPP (polycystic), and possibly also the newly discovered TRPY/TRPF families1. Since their discovery in 1969, TRP channels have been widely studied both functionally and structurally2: Currently, more than 280 structures have been determined from 23 different TRP channels, all displaying the canonical tetrameric assembly.
However, despite intensive research in the TRP channel field, a topic that is less studied and remains less well understood is the so-called pore-dilation phenomenon, which has been described so far for a few TRP channels3,4,5,6,7,8. This phenomenon is characterized in whole-cell electrophysiology experiments by a slow transition to an increased secondary current, loss of rectification, and permeability to large organic cations such as N-methyl-d-glucamine (NMDG+), tris(hydroxymethyl) aminomethane (Tris+) and 2-(methyl-amino)-ethanol (2-MAE+), following exposure to a high concentration or an extended period of activating stimuli, suggesting pore dilation over time. Although such time-dependent dilation of the pore is somewhat controversial, the ability of these channels to adopt a dilated pore state, which allows permeation of large organic cations, remains undisputed9,10,11. The physiological role of such a pore-dilated state is still unclear, though it could potentially have significant roles in the regulation of Ca2+-sensitive biological processes, such as desensitization of voltage-gated channels, activation of kinases, release of inflammatory peptides, and degeneration of sensory nerve terminals12,13,14. Additionally, pore dilation could be utilized as a therapeutic tool in disease, facilitating the delivery of hydrophilic drugs into cells15,16.
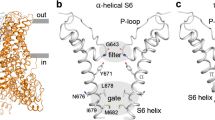
The TRPV3 channel, one of the TRP channels for which pore-dilation has been reported6, belongs to the subfamily of thermo-sensitive TRP channels17, responsible for the measurement of body and environmental temperature. TRPV3 is activated by warm temperatures (above 33 °C)18, as well as by a large range of natural compounds (e.g., camphor, carvacrol, thymol)19 and synthetic compounds (e.g., 2-aminoethoxydiphenylborane (2-APB) and diphenyl boronic anhydride (DPBA))6. The TRPV3 structure has been extensively studied, adopting, like all other TRP channel structures solved to date, a homo-tetrameric assembly. Each monomer consists of an intracellular ankyrin repeat domain (ARD) followed by a transmembrane domain (TMD) composed of six helices (S1–S6). Helices S1–S4 form a voltage sensor-like domain (VSLD), while helices S5–S6, together with the selectivity filter (SF) region, form the pore domain (PD). The VSLD is connected to the PD through the S4–S5 linker. Many of the interactions that stabilize the tetrameric assembly are formed between the VSLD of one subunit and the PD of the neighboring subunit, generating a so-called domain-swapped architecture, as well as by interactions between the ARD of one subunit and the coupling domain (CD) of the neighboring subunit (Fig. 1a). The structure of the tetrameric human TRPV3 has been determined in its closed state20,21,22,23, and recently also in an open and inactivated (or desensitized) states, following binding of the agonist 2-APB to distal ligand binding sites23.

a TRPV3 subunit architecture, with ARD (pink), linker domain and pre-S1 (purple), VSLD (S1–S4, yellow), S4–S5 linker (red), PD (S5–SF–S6, green), TRP helix (blue), and CTD (purple). b Euler diagram showing the orientation distribution of TRPV3 pentamer particles used for cryo-EM 3D-density reconstruction. Note the abundance (red) of particles around 0π elevation corresponding to side-views. c Representative 2D class averages of the TRPV3 pentamer. Note the first two side-view classes. d Cryo-EM 3D-density map and (e) structural model of the TRPV3 pentamer in top (left), side (center), and bottom (right) views, colored according to the domains in (a). Arrows in (d, e) indicate S4–S5-linker.
Recently, we discovered using HS-AFM, that human TRPV3 could self-organize into a rare and transient homo-pentameric assembly21. We observed that the pentamer is in a dynamic equilibrium with the tetramer, transitioning from and to the tetrameric state on the time-scale of seconds to minutes through diffusive exchange of subunits in the membrane. We also observed using HS-AFM that the pentamer population increased by almost 2-fold upon addition of the pore-dilation agent DPBA6. Finally, we validated the existence of this rare pentameric assembly by determining a low-resolution cryo-EM structure of the TRPV3 pentamer. Despite the low resolution, we were able to observe, based on backbone tracing, that the pentamer had a substantially enlarged pore as compared to the tetramer. The large pore of the pentamer that appeared permissive to passage of large cations, the fact that the pentameric state was entered and exited through slow 2D-membrane diffusion—thus matching the structural, functional and kinetic characteristics of what had been proposed for the pore-dilated state4—combined with the findings that the pore-dilation agent DPBA increased the pentameric population at high concentrations21 - led us to suggest that the pentameric state may represent a pore-dilated state of TRPV3 that could allow permeation of large organic cations. However, because of the low resolution of our initial cryo-EM map, we were unable to model most of the sidechains, and certain regions, such as the S4-S5 linker, were missing in the density map. Thus, we could not determine the precise pore-size and pore-properties, nor if the pentamer adopted a domain-swapped architecture like the tetramer24, nor assess the details of the inter-protomer interfaces that enabled the formation of the rare pentameric state as compared to the canonical tetrameric state.
Here, we present a much-improved cryo-EM structure of the human TRPV3 pentamer (Fig. 1 and Supplementary Fig. 1), into which we modeled most side chains. The determination of this structure was enabled through extensive single-particle data processing using machine-learning algorithms that allowed the integration of missing side views. This structure allowed us to analyze the pentameric state in great detail, including its ligand-binding sites, pore profile, and inter-subunit interfaces. Together with complementary molecular dynamics (MD) simulations and nano differential scanning fluorimetry (nano-DSF) experiments, we determine the factors that influence the stability of the pentameric as compared to the tetrameric state and propose a mechanism of pentamer formation.
Results
Structure of the TRPV3 pentamer
When we set out to determine our initial TRPV3 pentamer structure, the only particles that we could unambiguously identify as pentamers were those that were classified in a top-view 2D-class average with 5-fold symmetry21. Splitting these particles into subclasses, and using these in several cycles for particle picking, we succeeded in merging some tilted views into our initial structure determination. However, side-views were missing (Supplementary Fig. 1c), which substantially hampered high-resolution structure determination and rendered the 3D-density anisotropic (Supplementary Fig. 1a, b). To improve structure determination, we reasoned that the pentamer side-view must also be distinguishable from the tetramer side-views regarding two features: (i) a substantially wider micelle and more separated ARD domains, and (ii) while tetramer side-views must in projection have two-fold mirror symmetry, pentamer side-views cannot (Supplementary Fig. 2).
After a lengthy data-analysis workflow to train machine-learning algorithms25 to recognize and parse out pentamer from tetramer side-views (Supplementary Fig. 2), we finally succeeded in integrating the missing pentamer side-views in our 3D-reconstruction (Fig. 1b, c and Supplementary Figs. 1f and 2). The addition of these missing side-views improved our map dramatically, resulting in an isotropic density map (Fig. 1 and Supplementary Fig. 1d–f), determined from 51,588 particles representing ~3% of the total number of particles (Supplementary Table 1). The map corresponded to a smooth FSC curve cutting the resolution limit at 4.07 Å, with many regions at a local resolution better than 4 Å, displaying clear side-chain densities, including the S4–S5-linker region that was missing before (Fig. 1 and Supplementary Figs. 1e and 3)21. The S4–S5-linker region, which is now resolved (Fig. 1d, e and Supplementary Fig. 3b), confirms that the TRPV3 pentamer adopts the canonical domain-swapped architecture characteristic of all TRP channels as we had proposed in our initial report21, and not a non-swapped configuration as has been suggested by others24. Overall, similarly to other TRPV3 tetramer structures20,22, our structural model covers the structured region between residues 118 and 755, without the N-terminal and C-terminal intrinsically disordered regions (IDRs), and without the extracellular loops connecting S1 with S2 and S5 with SF. Comparison of our improved pentamer structure with the previous model reveals general similarity of the backbone trace, although there are notable differences in the positioning of certain helices (the overall backbone root-mean-square deviation (RMSD) amounts to 3.2 Å). Specifically, the positioning of the VSLD and ARD helices is substantially refined, and the TMD helices extend further towards the extracellular face (Supplementary Fig. 1g, h).
To try to further improve the resolution of our structure and to parse out possible flexibility, we performed symmetry expansion, 3D classification, and local refinement for different regions of the protein. Notably, when focusing on the TMD region, we detected conformational variations of the VSLD with respect to the PD (Supplementary Fig. 4a, b). When focusing on the ARD, further conformational variety could be observed, though to a lesser extent (Supplementary Fig. 4c, d). Interestingly, 3D variability analysis demonstrated that these movements were unconcerted and occur in each subunit independently of the other subunits, and in different directions (Supplementary Movies 1 and 2). Despite the different conformational classes that were obtained, the nominal resolutions and actual details of these sub-class maps did not exceed the resolution of the consensus full pentamer map, and therefore, for the sake of simplicity, all subsequent analyzes are conducted on the consensus map, which represents an average conformation of the VSLD and the dominant class of the ARD (Supplementary Fig. 4). Notwithstanding the failure to improve the resolution using these approaches, we learned that the VSLD displays positional variability, which becomes relevant when we later discuss the size of the vanilloid binding pocket that is situated between the VSLD and PD.
Structure of the TRPV3 pentamer pore
Our improved pentamer structure allows us to analyze the pore profile of the pentameric TRPV3 channel more accurately and describe the pore-lining residue structure. Previously, owing to the low resolution of our former pentamer structure in the absence of many side-chains, we only considered the protein backbone when computing a pore-profile21. Now, including all side-chains, we can compare the pore profiles of our improved pentamer structure to the structures of the closed tetramer that we determined previously (at 2.6 Å, PDB 8GKA)21 and to the structure of the open tetramer determined recently (at 2.6 Å, PDB 8V6N)23. From this analysis, it is apparent that the residues forming the constrictions at the SF, the gate, and the pore exit on the intracellular face, are maintained in all structures; namely, G638 at the SF, I674 at the gate, and E682 at the exit (Fig. 2a, b). While pore-lining residues are preserved, the pentamer pore diameter is largely increased: The SF constriction is 6.4 Å in diameter in the pentamer vs 1.2 Å in the closed and 4.1 Å in the open tetramers, and the constriction at the pentamer gate is 10.9 Å in diameter vs 1.5 Å in the closed and 5.7 Å in the open tetramers. When considering all pore-lining residues between the SF (G638) and the pore exit (E682), the average pore-profile diameter is 11.6 Å in the pentamer compared to 4.7 Å and 7.4 Å for the closed and open tetramer structures, respectively (Fig. 2a, b and Supplementary Table 2). Thus, the SF diameter of the pentamer expands 5.3-fold and 1.6-fold compared to the closed and open tetramer structures, respectively; the gate diameter expands 7.3-fold and 1.9-fold, respectively; and the average pore profile expands 2.5-fold and 1.6-fold, respectively, compared to the closed and open tetramers. This pore expansion between pentamer and open tetramer, ~1.6-fold, is in good agreement with our original estimates based on the geometrical transformation of a square into a pentagon with equal side-length, with area A(5) = 1.72 A(4) and D(5) = 1.38 D(4)21.

a Pore profile of the closed tetramer (green, top), open tetramer (blue, middle), and pentamer (pink, bottom). b Surface representation of the pores as in (a). c PMF, w(z), representing the free energy as a function of the distance from the pore center of mass, for the translocation of sodium (green), Tris+ (red), NMDG+ (purple), and 2-MAE+ (yellow) cations through the closed tetramer (top), open tetramer (middle), and pentamer (bottom). The dashed lines represent the average positions of G638 (SF), I674 (gate), and E682 (exit) during the simulations. Error bars represent the variance of the collected forces within each 0.1 Å wide bin of the PMF calculation.
The large pentamer pore led us to suggest21 that the pentameric TRPV3 state may represent the structural correlate of the controversial pore-dilated state3,9,10,11, since such a large pore would permit easy passage of the small-molecule organic cations that have been reported to flow through the pore-dilated state of TRPV channels3,4. The specific organic cations are Tris+, NMDG+, and 2-MAE+5, and their rough dimensions (without considering hydrogens and conformational flexibility) are 6.5 × 5.5 × 2.5 Å for Tris+, 6.8 × 4.4 × 3.0 Å for NMDG+, and 4.3 × 1.5 × 1.4 Å for 2-MAE+ (Supplementary Fig. 5). Considering the dimensions of these molecules and the open tetramer pore-profile constrictions, and taking into account the need for sufficient distance between the peripheral atoms of the small molecules and the pore-lining residues to prevent clashes (at least 2 Å in each direction), these organic cations would not be able to sterically pass through the pore in any orientation (with the possible exception of 2-MAE+) without considerable clashes and/or further widening of the pore. The pentamer, on the other hand, allows passage of all the small molecule cations without steric clashes.
This approximation, however, is made under the assumption that the small molecule cation structures and the protein pore profiles are static. This is, of course, not the case. We therefore set out to investigate further the ability of the small molecular cations to pass through the tetramer and pentamer channels using MD simulations together with potential-of-mean-force (PMF) calculations26,27. Thus, we calculated the PMF, w(z), for the translocation of the Na+, Tris+, NMDG+, and 2-MAE+ ions through the pores of the closed tetramer, the open tetramer, and the pentamer structures (Supplementary Table 3). These simulations provided the free-energy profiles underlying the translocation of the different ions through each structure, allowing us to compare the passage of the molecular cations with that of the permeable sodium through the different pores, together with the associated free-energy barriers.
The PMFs show two or three free-energy barriers that the ions must overcome to flow through all channel pores, corresponding, not surprisingly, to the SF, the gate, and in some cases also the exit region of the pore (Fig. 2c, d). It is immediately apparent that the free-energy barriers against the flow through the closed tetramer are highest, while the barriers against the flow through the pentamer are lowest. Not surprisingly, for the closed and open tetramers, the barriers against the passage of the small molecular cations are higher, while sodium faces the lowest barriers. Interestingly, for the open tetramer, the barriers correlate with the bulk size trend of the small molecular ions, with Tris+ (~30 kcal/mol) > NMDG+ (~22 kcal/mol) > 2-MAE+ (~17 kcal/mol) (Supplementary Fig. 5). Notably, for the pentamer, all barriers are relatively low, ≤10 kcal/mol, and there is no clear preference for sodium. Furthermore, the heights of the barriers for the small molecular ions permeating the pentamer are comparable to that for sodium in the open tetramer (~10 kcal/mol), from which we deduce that the small molecular ions should flow similarly easily through the pore of the pentamer as sodium does through the tetramer. We conclude, therefore, that the larger pore of the pentamer could allow passage of small cations. In contrast to the static pore-profile analysis, the PMFs reflect protein flexibility, which is important to consider in light of the greater flexibility of the pentamer vs the tetramer, as observed in cryo-EM classification analysis (Supplementary Fig. 4) and the equilibrium MD simulations described below.
The vanilloid binding pocket in the TRPV3 pentamer
We next compared the pentamer and open tetramer protomer structures with that of the closed TRPV3 tetramer. For this, we first assessed the relative angle and radius of the center of mass (CoM) of each helix in each structure, with regard to the symmetry axis of the oligomer, setting the angle of helix S6 to 0° when viewed in top-view (Fig. 3a). Thus, when aligning all structures angularly to the position of S6, it is apparent that the pentamer VSLD is globally rotated to allow the non-canonical interaction with a neighboring subunit positioned at an angle of 108° in the pentamer instead of the 90° angle in the tetramer, as we have reported previously (Fig. 3a, compare pentamer (right, pink) with the position of the helices in the closed (left, green) and open (center, blue) tetramers)21. In detail, averaging the angular position of the VSLD helices S1–S4, we detect a total VSLD rotation of 24° (Fig. 3b, left). This is different from the expected angular difference of 18°, i.e., the difference between the 108° and 90° angles corresponding to the neighboring subunit location in the pentamer and tetramer, respectively. Therefore, the VSLD interacts with the neighboring PD in the pentamer with an angular change of 6° as compared to the same interaction in the closed tetramer, resulting in constriction of the vanilloid binding site, which is located at the interface between VSLD and PD. Thus, the vanilloid binding pocket in the pentamer is substantially smaller than that in the closed tetramer, with respective cavity volumes of V5 = 164 Å3 vs V4(closed) = 250 Å3 (Fig. 3c, right). Interestingly, the open TRPV3 tetramer structure23 VSLD is also rotated when compared to the closed tetramer by roughly ~3° (Fig. 3b, left). This rotation, allowing the opening of the channel23, also results in a constriction of the vanilloid binding site when compared to the closed channel, and in a reduced cavity volume of V4(open) = 220 Å3 vs V4(closed) = 250 Å3 (Fig. 3c, center). Thus, there is no room in the vanilloid pocket of the open tetramer and pentamer structures to host the vanilloid lipid (Fig. 3c, left), as is apparent from the absence of this lipid in the open TRPV3 tetramer structures20,23,28. This finding appears conclusive as the open tetramer structure (PDB 8V6N)23 was solved to equally high resolution, 2.6 Å, as are the closed tetramer structures. While the resolution of the pentamer structure would not necessarily allow to assert the presence or absence of a lipid molecule, the angular rearrangement of the VSLD can be measured accurately, and thus the compaction of the vanilloid binding site. Given that the binding pocket volume in the pentamer is even smaller than that of the open TRPV3, which does not or cannot host the vanilloid lipid, we can confidently infer that the pentamer is also devoid of the vanilloid lipid. When plotting the radial and angular displacement of the CoMs of all 6 helices in the closed and open tetramer and the pentamer structures, we can observe a coherent outward movement of the helices, so that the pentamer subunit is in a hyperactivated state (Fig. 3b, right).

a Analysis of the helix CoM positions (labeled S1–S6) in the pentamer subunit (pink, right), the open tetramer (blue, center), and the closed tetramer (green, left). b Overlay of the helix positions in the three structures, where the central pore axis is used for lateral alignment and the position of pore-helix S6 for angular alignment (arbitrarily set to 0°). In the open tetramer (blue), all helices move outwards, and the VSLD helices (S1–S4) undergo a collective rotation of 3°, with respect to the closed tetramer. In the pentamer (pink), the helices move further outwards, and the VSLD helices undergo a collective rotation of 24°, with respect to the closed tetramer, corresponding to a rotation of 6° with respect to the next subunit in the pentamer. c Close-up view of the vanilloid pocket, located at the interface between the VSLD (color) of one subunit and the PD (gray) of a neighboring subunit, of the closed tetramer (green, left), open tetramer (blue, center), and pentamer (pink, right). In the closed tetramer, the vanilloid lipid (yellow) is present in the vanilloid binding pocket, ‘glueing’ the two adjacent subunits together via five hydrophobic interactions from each subunit. In the open tetramer and pentamer, the pocket volumes are constricted, leading to clashes of the vanilloid lipid (shown semitransparent) with the surrounding protein structures and ejection of the lipid from the vanilloid pocket.
The presence of the vanilloid lipid in the vanilloid binding site in the closed TRPV3 tetramer structure assists in ‘glueing’ neighboring subunits together through five lipid-mediated hydrophobic interactions between the PD of one subunit and the VSLD of its neighboring subunit (Fig. 3c, left). Thus, the residue pairs F656 (PD) and L557 (VSLD), F601 (PD) and L557 (VSLD), T660 (PD) and A560 (VSLD), L664 (PD) and I583 (VSLD), and F597 (PD) and I583 (VSLD), which would otherwise be too far apart to interact with one another, are connected through mutual hydrophobic interactions with the vanilloid lipid (Supplementary Fig. 6 and Supplementary Tables 4 and 5). In the open tetramer (Fig. 3c, center) and pentamer structures (Fig. 3c, right), however, these interactions are missing owing to a constriction of the vanilloid binding site and ejection of the vanilloid lipid from the cavity due to steric clashes.
Inter-subunit protein-protein interactions in the TRPV3 pentamer
The stabilizing lipid-mediated interactions between the PD of one subunit and the VSLD of the neighboring subunit in the closed tetramer are naturally not the only interactions connecting TRPV3 subunits. In all domain-swapped TRPV3 structures, there are many protein-protein interactions at the subunit interfaces, which can be divided into four main groups. (i) PD–PD: interactions stabilizing the PD of one subunit to the PD of another subunit, (ii) PD–linker: interactions stabilizing the PD of one subunit to the S4–S5 linker of another subunit, (iii) PD–VSLD: interactions stabilizing the PD of one subunit to the VSLD of another subunit, and (iv) ARD–CD: interactions stabilizing the ARD of one subunit to the CD of another subunit (Fig. 4 and Supplementary Table 5).

a–d Open book representation of the closed tetramer (a, green), open tetramer (b, blue), inactivated tetramer (c, yellow), and pentamer (d, pink), inter-subunit contact areas. Residues forming contacts are shown as spheres and colored according to domain interaction groups PD–VSLD (yellow), PD–linker (red), PD–PD (green), ARD–CD (purple), and, in the case of the open tetramer only, ARD-IDR (orange). e–h Inter-subunit interactions of the PD of one subunit (gray) with the PD, VSLD, and S4–S5 linker of an adjacent subunit in the closed tetramer (e, green), open tetramer (f, blue), inactivated tetramer (g, yellow), and pentamer (h, pink). Solid and dashed arrows indicate, respectively, many (solid) and lesser (dashed) interactions between two adjacent helices. i–j Plots of the total (i) or TMD (j) interactions, formed by one subunit with the adjacent subunit, for the closed tetramer (green circles), open tetramer (blue triangles), inactivated tetramer (yellow crosses), and pentamer (red dots). The background coloring in (j) indicates the VSLD (yellow), S4/S5 linker (red), and PD (green) regions of the adjacent subunit (chain B). k Bar graph comparing the number of ARD–CD, PD–VSLD, PD–linker, PD–PD, and total number of interactions one subunit forms with an adjacent subunit (left), as well as the total number of inter-subunit interactions per oligomer (right), for the closed tetramer (green), open tetramer (blue), inactivated tetramer (yellow), and pentamer (pink). l Energy of interaction per two adjacent subunits (left) or per the total oligomer (right), as computed by equilibrium MD simulations, for the closed tetramer (green), open tetramer (blue), and pentamer (pink).
When comparing these interaction groups between the different TRPV3 structures, it is evident, perhaps not surprisingly, that the closed tetramer structure engages in the most protein-protein interactions in every category (in addition to the five unique lipid-mediated interactions in the vanilloid site at the PD–VSLD interface). In the TMD region the closed tetramer forms primarily hydrophobic interactions, specifically 14 PD–VSLD interactions mediated by helix S5 with helices S1 and S4 of the neighboring subunit (S5–S1 and S5–S4 interactions) and helix S6 with helix S4 of the neighboring subunit (S6–S4 interactions), 8 PD–linker interactions mediated by the S5 and S6 helices of one subunit with the S4/S5-linker region of the neighboring subunit (S5-linker and S6-linker interactions), and 14 PD–PD protein-protein interactions mediated by SF–SF and S6–S6 interactions. Additionally, in the closed tetramer, a combined 19 interactions (hydrophobic, π, and H-bond) are formed at the ARD–CD interface (Fig. 4a, e, and i–k and Supplementary Table 5).
In contrast, the TRPV3 pentamer structure is stabilized by much fewer protein-protein interactions, particularly in the TMD region. The reduced number of interactions in the TMD is a result of the 24° rotation of the VSLD, 6° with respect to the neighboring PD, reducing substantially the ability to form the PD–VSLD interactions that are found in the tetramer; specifically, the S5-S1 and S6-S4 interactions are essentially abolished. Given that the pentameric oligomeric state implies an overall outwards-displacement of the protomers, PD–PD interactions are also dramatically reduced, specifically, the SF–SF and S6–S6 interactions of the PD–PD interactions are essentially abolished, though new S6–SF interactions are formed. However, the PD–linker interactions are largely maintained. Similarly, on the intracellular side, the bulk exposed ARD–CD interactions are generally unchanged in the pentamer (Fig. 4d, h, and i–k and Supplementary Table 6). Notably, the fewer interactions at the PD–VSLD interface are in line with the increased conformational flexibility of the VSLD with respect to the PD, as detected by cryo-EM 3D classification analysis (Supplementary Fig. 4).
The open TRPV3 tetramer also features a rotated VSLD as compared to the closed tetramer. Though the rotation is smaller than in the pentamer, 3° vs 6°, this rearrangement also affects the number of inter-subunit interactions. Specifically, in the open tetramer, the PD–PD interactions are substantially impacted, with only a few S6–S6 and SF–SF interactions remaining, and there are also fewer PD–linker interactions. On the other hand, in the open tetramer the PD–VSLD interactions are identical in number to the closed tetramer, the ARD–CD interactions are similar in number, while there are a few additional unique interactions between the ARD of one subunit with the IDR of another subunit that appear so far only in the open tetramer structure (Fig. 4b, f, and i–k and Supplementary Table 5).
The open TRPV3 tetramer structure that we analyzed here (PDB 8V6N) was obtained alongside another TRPV3 tetramer conformation assigned as an inactivated state (PDB 8V6O)23. Both these states were determined following binding of the agonist 2-APB to distal ligand binding sites. In the inactivated state, the pore is closed, though many other structural similarities to the open state remain23. Interestingly, however, when analyzing the subunit interactions of the inactivated state, it is apparent that the inactivated tetramer state makes more inter-subunit interactions than the open tetramer and the pentamer states, though less than the closed tetramer. Although the number of PD–VSLD interactions decreased, its PD–PD interactions increased substantially relative to the open tetramer state, specifically the S6–S6 and S6–SF interactions, owing to the constriction of the pore diameter (Fig. 4c, g, and i–k and Supplementary Table 5).
When considering all these different interactions, it is evident that the closed tetramer state engages in most inter-subunit interactions (including the five unique lipid-mediated interactions), the inactivated tetramer state is second in number, followed by the open tetramer, and finally by the pentamer with least inter-subunit interactions (Fig. 4k, left). We should note, however, that our interaction analysis is limited by the precision with which we can place the amino acid side chains in the pentamer structure, and by the conformational flexibility of the protein, which is apparently higher in the pentamer as compared to the tetramer. Also, in this comparison we did not consider the ARD-IDR interactions in the open tetramer which are absent in all other structures, since, owing to the high flexibility of the IDR, it is well possible that these may also be present in other TRPV3 states but are missing in the density maps due to the experimental conditions or data quality. Still, since the pentameric complex forms five subunit–subunit interactions vs four subunit–subunit interactions in the tetramers, the pentamer totals more inter-subunit interactions than the open and inactivated tetramer states, though still less than the closed tetramer (Fig. 4k, right). Since the number of interactions in a complex is directly related to the total energy of interactions stabilizing the complex, we suggest that among the TRPV3 structures, the closed tetramer is most stable, followed by the pentamer, the inactivated tetramer, and finally the open state. This order is consistent with the observation that the pentameric state has a lifetime of ~3 min, certainly longer than the open and inactivated tetramer states, though this is a comparison between different oligomeric and conformational states. Indeed, while the pentamer accumulates a great number of interactions over its entire complex, its inter-subunit interfaces are the weakest, and thus, while the open and inactivated tetramers typically decay to the closed tetrameric state, the pentamer tends to shed a subunit.
Interface dynamics by equilibrium MD simulations
To gain further insights into TRPV3 assembly flexibility and stability, we set up equilibrium MD simulations of the closed tetramer, the open tetramer, and the pentamer over a period of 1 μs. Analysis of the RMSD of the different states over the length of the MD simulations, after adequate structure equilibration, revealed that the pentamer fluctuated more than the tetramers, with particularly high RMSD in the intracellular domains (Supplementary Fig. 7). We then compared the inter-subunit interaction energy between neighboring subunits over the length of the MD simulation runs and found that indeed the interaction energy of the pentamer interfaces was lesser than that of the tetramers (Fig. 4l, left), similar to the results we obtained from our static structural analysis (Fig. 4k). While our structural analysis may be plagued with uncertainties of side chain placement and rotamer assignment, the 1 µs equilibrium MD simulations probe side chain fluctuations and rotamers over time, give a more dynamic picture of the interactions, and/or allow interfaces to equilibrate into their most favorable interactions.
Interestingly, the inter-subunit interaction energy of the open tetramer simulation was very similar to that of the closed tetramer (Fig. 4l), unlike the structural interaction analysis, where the open tetramer appeared less stable (Fig. 4k). This result is explained, however, upon closer examination of the open tetramer trajectory: When examining the evolution of the open tetramer pore over time, we found that early in the simulation, either during the equilibration steps or the first 100 ns of the simulation, the open pore of the tetramer collapsed into a closed pore conformation. This was evident from the sudden decrease in diameter between the residues forming the SF, gate, and exit of the pore as compared to the initial open tetramer structure, resulting in a diameter that is quite similar to that of the closed tetramer structure. In other words, the open tetramer structure closes or inactivates during the equilibrium simulation (Supplementary Fig. 8). Such a collapse of the open to the closed tetramer after <100 ns is also an indicator of the lesser stability of the open tetramer conformation, confirming our previous conclusion regarding its lesser interface stability from our static open tetramer structure analysis. This finding also highlights the complementary power of MD simulations for such analyses, as we find a structure equilibrating into a lower energy state, engaging in tighter interface interactions.
However, when considering the total interface energy per assembly (4 inter-subunit interface interactions in the tetramers vs the five interfaces in the pentamer), there is not much of an energy difference during the MD simulations between the pentamer and closed tetramer structures (Fig. 4l, right). Thus, our MD simulations analysis showed, akin to the static analysis, that though the inter-subunit interaction energy per inter-subunit interface in the pentamer is lesser than that of the closed tetramer structure, leading to fragile protomer interfaces, the total interaction energy within the noncanonical pentamer is comparable to that of the closed tetramer state.
Channel activators destabilize TRPV3
We previously demonstrated using HS-AFM imaging that pentamer formation occurred through a break of the TRPV3 tetramer inter-subunit interface and membrane-diffusive insertion of a protomer, which must have originated from an oligomer that previously dissociated from a tetramer21. We consequently hypothesized that factors decreasing the stability of TRPV3 would favor pentamer formation. Interestingly, when using nanoDSF to examine the thermal denaturation of TRPV3 in the absence and presence of the activator and reported pore-dilation agent DPBA, we discovered that DPBA destabilized TRPV3 significantly when applied at a concentration that was shown to lead to pore-dilation.
To investigate further the effect of agonists on the stability of TRPV3, we examined whether there were other TRPV3 ligands that might destabilize TRPV3 similarly as did DPBA21, and if so, at what concentrations. The ligands that we tested are the known agonists 2-APB and camphor, which have been reported to activate TRPV3 at 32–320 μM6,29 and 1–10 mM30,31,32, respectively, and molecules that have been shown to interact with TRPV1 such as capsaicin and the general anesthetic propofol at concentrations 0.1–1.0 μM33 and 10–300 μM34,35, respectively. When examining the nanoDSF thermal denaturation curves of TRPV3 in the presence of these ligands at these concentrations (Supplementary Table 7), consistently two melting temperatures (Tm1 and Tm2) were observed, akin to what we previously observed for DPBA21. Notably, for the two ligands, 2-APB and camphor, and the general anesthetic propofol, we found specific concentrations above which the melting temperatures decreased significantly, specifically, 100 μM 2-APB (Fig. 5a), 5 mM camphor (Fig. 5b), and 100 μM propofol (Fig. 5c). These results match electrophysiology studies where 2-APB and camphor substantially increased whole-cell currents at these concentrations29,32.

NanoDSF thermal denaturation data of TRPV3 in the presence of different concentrations of (a) 2-APB, b camphor, c propofol, d DPTHF, e capsaicin, and (f) DMSO. Each panel shows the raw data NanoDSF curves (top left), their first derivative (bottom left; Tm1 and Tm2 are indicated by downwards and upwards arrowheads, respectively, in (a–c) and by shaded bars in (d–f) where no Tm1/Tm2 changes were observed), bar graph (top right) and curve plot (bottom right) comparing Tm1 and Tm2 following addition of different concentrations of additive. Tm values are presented as mean ± s.e.m. obtained from n = 3 to 5 biologically independent repeats. Statistical significance was assessed with the one-tailed Welch’s t-test. **P < 0.025; ***P < 0.005.
To assess whether these results truly reflected activity-related changes and not protein destabilization from exposure of the channel to high concentrations of such hydrophobic molecules, we tested as a negative control the inhibitor 2,2-diphenyltetrahydrofuran (DPTHF), which is structurally similar to DPBA and 2-APB. Notably, the addition of the inhibitor DPTHF in the same concentration range, up to 1 mM as used for the DPBA experiments, resulted in an absence of change in the melting temperatures (Fig. 5d). Neither was any significant melting temperature change observed when adding capsaicin, which activates TRPV1 but not TRPV3 (Fig. 5e). Finally, to assess whether the destabilizing effect of the ligands may have resulted from the presence of dimethyl sulfoxide (DMSO), which was used to solubilize the hydrophobic compounds (Methods), we tested the stability of TRPV3 in the presence of DMSO alone, at all concentrations employed throughout the experiments with the various compounds. No decrease in TRPV3 stability was observed upon addition of DMSO (Fig. 5f). We therefore conclude that the activators 2-APB, camphor, and perhaps also the general anesthetic propofol, destabilize TRPV3 similarly to the pore-dilation agent DPBA.
Discussion
We present here a substantially improved TRPV3 pentamer structure as compared to the cryo-EM structure that we had previously reported as support by a secondary technique for the HS-AFM discovery of the pentameric TRPV3 state21. The improved pentamer structure, in which most residues were modeled with reasonable confidence, allowed us to analyze the pore profile and inter-subunit interactions of the TRPV3 pentamer, and compare them to structures of the TRPV3 tetramer in various states. This comparison revealed that the pentamer possesses indeed a substantially enlarged pore, ~1.9-fold wider at the SF and ~1.6-fold wider in the gate region than the open tetramer. MD simulations confirmed that such a pore enlargement is sufficient to allow passage of small molecular cations (Fig. 2)21. Of course, ultimately, the channel properties of the pentamer must be studied experimentally, and under well-controlled conditions. However, in order to achieve that, environmental conditions, a drug, or mutations that stabilize the pentameric state, would have to be found to perform single-channel recordings and/or whole-cell recordings. As such tools are not yet available, MD simulations and PMF calculations provide us with initial evidence regarding the permeability of the TRPV3 pentamer.
We also assessed the stability of the pentameric vs the tetrameric TRPV3 by comparing the interacting residues at the subunit interfaces both in static structures and dynamically in MD simulations. Analysis of these interactions revealed that the pentamer engaged in fewer inter-subunit interactions compared to all the closed, open, and inactivated, tetramer structures. A similar conclusion was obtained from MD simulation trajectories. However, when considering the total inter-subunit interactions per full oligomer, the pentamer forms more inter-subunit interactions than the open and inactivated tetramers, though still less than the closed tetramer (Fig. 4). It would therefore appear that the pentamer has fragile interfaces, but is overall more stable than the open and inactivated tetramer structures, though less stable than the closed tetramer. This conclusion is perhaps counterintuitive when considering that the pentamer is a rare state with a relatively short lifetime of ~3 min, as imaged by HS-AFM21. Yet, when taking into account the difficulty to capture cryo-EM structures of the open and inactive states, as well as the short lifetime of the tetramer in its open state—as corroborated by MD simulations where the open tetramer rapidly decayed into the closed state in equilibrium simulations (Supplementary Fig. 8)—it is not surprising that the open and inactivated tetramer states are high-energy states. The pentamer may be additionally stabilized by an entropic gain following its formation, resulting from the release of lipids that had been forming tight interactions in the inter-subunit regions of the tetramer and from the increased mobility of the water that can now flow more easily through the pentamer channel pore. We therefore conclude that the closed tetramer is the most stable state, followed by the pentameric state, then the inactivated state, and finally by the open tetramer state with the least inter-subunit interactions in the full oligomer.
Despite the relative stability of the pentamer at equilibrium, there is obviously a very high energy barrier that must be overcome in order to transition from the tetrameric to the pentameric state. This barrier results primarily from the need to disrupt the many interactions of a subunit interface in order to allow the entry of a fifth subunit. Notwithstanding the high energetic cost to overcome this barrier, evidently it can be overcome when considering that the pentamers constitute 3–10% of the TRPV3 population in and under the conditions of our cryo-EM and HS-AFM experiments21. So, how is this energy barrier overcome in practice?
We propose the following mechanism for pentamer formation (Fig. 6). Initially, as has been put forth23, agonists bind to TRPV3 and activate the channel, leading to displacement of the vanilloid lipid from the vanilloid pocket and subsequent opening of the channel. The displacement of the vanilloid lipid, as well as the conformational change the channel undergoes to reach the open state, leads to reduced inter-subunit interface interactions. Most significantly reduced are the PD–PD interactions, as the pore helices move outwards and thus apart, destabilizing the open TRPV3 tetramer. The unstable open tetramer then relaxes to the inactivated (or desensitized) state, which is lower in energy than the open tetramer. Transition to the inactivated state leads to renewed formation of PD–PD interactions but to substantially less VSLD–PD and ARD–CD interactions (Fig. 4c, g, and i–k). The inactivated tetramer state can then either further relax into the closed tetramer state, or, if a free protomer is available in proximity, form the pentameric state (Fig. 6c). We hypothesize that the transition to the pentamer occurs from the inactivated state rather than the closed or open tetramer states, because the inactivated state has reduced PD–VSLD interactions, similar to the pentamer, and is similar also in interface interaction energy. Furthermore, the inactivated state should accumulate under conditions of activator abundance, and so does the pentamer21. Thus, we propose, partly based on HS-AFM imaging evidence, that the transition to the pentameric state goes through a series of intermediates, thereby reducing the energetic cost of each step. Indeed, based on our HS-AFM videos of tetramer-pentamer transitions21, we suggest that in the first transition step, a canonical tetramer (Fig. 6a, stage 1) subunit’s VSLD and ARD domains dissociate from their respective neighbor subunit’s PD and CD (Fig. 6a, stage 2). This is most likely to happen from the inactivated tetramer, since it has the least PD–VSLD and ARD–CD interactions amongst the tetramer states, followed by a transition state where the free VSLD captures a free PD from an isolated subunit in the membrane (Fig. 6a, stage 3). Assuming that the VSLD and ARD rotate together with respect to the PD, similarly to the conformational change between the tetramer and pentamer subunits (see Fig. 3a, b), the displacement of the ARD of one subunit may actually be observed in some HS-AFM video sequences (channels viewed from the intracellular side, Fig. 6b). Similarly, in some videos, the entrant protomer can be observed ‘dancing’ around the tetramer before inserting the complex. We think that these protomers, of which we monitor the position of the ARD, may represent protomers that already interact in the membrane with the distorted tetramer (Fig. 6a, stage 4). Finally, the VSLD of the entrant protomer forms interactions with the free PD, leading to pentamer formation (Fig. 6a, stage 5). Interestingly, a recent report demonstrated an alternative tetrameric assembly of a pentameric ligand-gated ion channel family member36. Similarly, as tetramers and pentamers coexisted in the cell membrane, a mechanism involving the insertion of a subunit in the membrane into the tetramer has been proposed for the formation of the pentamer36.

a Schematic of the proposed tetramer-to-pentamer transition mechanism implicating dissociation of a subunit–subunit interface in the tetramer, in order to accommodate the insertion of a fifth subunit. VSLDs are represented by the outer (darker colors) while PDs are represented by the inner and smaller (lighter colors) moon shapes. Each subunit is colored in a different color. b HS-AFM video sequences capturing proposed intermediate states of the tetramer-to-pentamer transition, viewed from the intracellular side, displaying the ARD domains. c Proposed reaction coordinate for pentamer formation from the closed tetramer state through passage of many intermediate states, including the open tetramer, inactivated tetramer, and various transition states, as indicated in (a).
Finally, we show here that besides the documented pore-dilation agent DPBA, other agonists, i.e., 2-APB, camphor, and also the general anesthetic propofol, destabilize TRPV3 similarly to DPBA, at relatively high concentrations (Fig. 5). We propose therefore the following mechanism for agonist activation of TRPV3. At low concentrations, agonists shift the equilibrium towards the open and, as a consequence, the inactivated state, similarly to what has been observed upon binding of 2-APB23, thereby destabilizing the protein temporarily. However, when these ligands are present at high concentrations, which have been shown to induce pore-dilation in the case of DPBA5,6, the tetramer population is shifted towards the unstable open and inactivated states, and therefore, the drop in stability is significant, as reported by nanoDSF. Once many tetramers are in either the open or inactivated conformation and destabilized, the transition to the pentameric state could potentially occur with higher probability and frequency, thereby increasing the probability of the pentameric state. In other words, the fact that (i) the 2-APB activator-bound open and inactivated states make lesser inter-subunit contacts as analyzed in static structures and in MD simulations, (ii) activators 2-APB, DPBA, and camphor, decrease the protein thermal stability in nanoDSF experiments, (iii) DPBA-addition induces increased percentiles of free subunits and pentameric channels in HS-AFM21, and (iv) DPBA-addition induces increased and non-rectified currents in whole-cell functional experiments6, are all signatures underpinning our tetramer-to-pentamer transition model whereby the pentamer is the result of hyper-activation. However, many more experiments and experimental methods are needed to complete this large matrix of activator conditions to bolster this hypothesis.
In conclusion, we present here a cryo-EM structure that allowed us to build a much more complete and reliable structural model of the TRPV3 pentamer. This structure determination, together with complementary MD simulations and nanoDSF experiments, as well as further analysis of our HS-AFM data, allows us to propose a mechanism through which the high energy barrier of the tetramer-to-pentamer transition is overcome: The stable low-energy closed tetramer state transitions, with the help of agonists, to higher energy intermediate states, thereby dividing the barrier of the tetramer-to-pentamer transition into several smaller energy barriers that are more easily overcome. Finally, notwithstanding its rather weak inter-subunit interactions, the fact that the pentamer forms five interfaces lowers the energy level of the state through the integration of an additional subunit. The details of this mechanism, however, are still highly hypothetical, and many more experiments are needed to investigate and understand under what conditions the pentamer can be stabilized. Our results and analyses presented here lay the foundation for research in this direction.
Methods
Cryo-EM image processing
Cryo-EM sample preparation, data collection, frame alignment, and contrast transfer function (CTF) estimation of the data have been described in detail in ref. 21. Extensive data reprocessing was performed in order to parse out missing side-views of the TRPV3 pentamer (Supplementary Fig. 2). Unless otherwise stated, all calculations were performed in CryoSparc version 4.4.137. Specifically, side-view 2D classes from the total particle stack (tetramer and pentamer particles) were further sub-classified until asymmetric side views with larger detergent micelles and more separated ARD domains could be detected by eye (3018 particles). These particles were then combined with the best pentamer stack obtained from previous processing (before Bayesian polishing; 41,737 particles)21 together with removal of duplicate particles. The particles were then subjected to ab initio 3D classification (2 classes, multi-class ab initio reconstruction). This classification resulted in a class that possessed a small amount of side views (based on the particle orientation distribution), which, despite the relatively low nominal resolution (6.11 Å), demonstrated new structural features such as elongated TMD densities and clearer TRP helix densities that were missing in our previous structure. This map was then used as a reference map for heterogenous refinement, together with five other reference maps corresponding to the best tetramer map obtained previously21 and 4 decoy noise maps38. The particle stack that was used as input for the heterogenous refinement comprised all sideview 2D classes (272,177 particles, before sub-classifications) together with the best pentamer stack obtained previously21, after removal of duplicate particles. After five rounds of such heterogenous refinement cycles, where in each new cycle only the tetramer and pentamer particles were selected as input, and the pentamer map was updated to the best map available, an improved pentamer map was obtained (38,582 particles; FSC resolution of 5.46 Å), containing significantly more side views and displaying even more TMD features (much clearer TMD helices and presence of the S4–S5 linker). 2D classification of this pentamer stack revealed that indeed it contained side-view 2D classes with larger detergent micelles and wider ARD domains (5560 particles). These particles were then used for training a Topaz model25 that could be used to selectively repick more large-micelle side-views. The Topaz picked particles (1,801,742 particles) were then used as input, together with the previous best large-micelle side-views particle stack and the original pentamer stack21, after removal of duplicate particles, for 10 rounds of iterative heterogeneous refinement cycles. In each new cycle, the most recent pentamer map was used as the pentamer reference map, while all other maps (i.e., tetramer and decoy noise maps) remained the same. These iterative rounds of heterogeneous refinement resulted in an improved map that was only slightly better in terms of resolution, but that contained many more pentamer side-views and tilted views (10,385 particles). The side-views and tilted views were used yet again for an additional round of Topaz training and particle picking. The repicked particles (2,566,939 particles), together with the pentamer particle stacks from the previous steps, after removal of duplicates, were then used for 29 rounds of heterogeneous refinement cycles, conducted similarly as described above. The process ended this time with a substantially improved map at 4.12 Å (61,633 particles), containing many more side-views and tilted views. After Bayesian Polishing in Relion39, another round of heterogenous refinement, and density modification40 in Phenix41, the map reached a nominal FSC resolution of 4.07 Å, in which many sidechains were clearly resolved (Supplementary Figs. 1 and 3). Further Topaz training of the data was attempted using the more abundant side-views and tilted views, followed by similar heterogeneous refinement iterative cycles, however, no further improvement in resolution was observed. The 4.07 Å map was therefore the final map used for subsequent model building and analysis.
Symmetry expansion, 3D classification, and variability analysis
For symmetry expansion and 3D classification, soft masks were generated from the 4.07 Å consensus map as follows: Map regions, corresponding to the VSLD, PD, TMD, and ARD regions of one subunit, were selected using Segger42 as implemented in Chimera (v.1.13.1)43, and then processed into masks (dilation 4 pixels, padding 40 pixels) using Volume Tools of CryoSparc37. For symmetry expansion, we used the particles from our best consensus TRPV3 pentamer map (C5 symmetry), followed by 3D classification without alignment within the different mask regions, searching for 2–10 different classes with O-EM learning rates of 0.5–1. Most notably separated classes were obtained when using the TMD and ARD masks, corresponding to three and two different classes, respectively (Supplementary Fig. 4). After local refinement, the three TMD classes corresponded to FSCs with nominal resolutions of 4.46 Å, 4.46 Å, and 4.72 Å resolution, respectively, and the two ARD classes had nominal resolutions of 4.25 Å and 4.20 Å resolution, respectively. 3D variability analysis (C1 symmetry) was then performed on the original pentamer particle stack (before symmetry expansion), using masks covering the five TMD subunits or the five ARD domains, and searching for 3–5 modes of variability (eigenvectors), with a filter resolution of 10 Å. Visualization of the resulting five modes of variability was conducted with ‘3D variability display’, using five frames at a filter resolution of 6 Å (Supplementary Videos 1 and 2).
Cryo-EM structure modeling and analysis
To build the TRPV3 pentamer structural model into the consensus pentamer map, a monomer from the human TRPV3 tetramer structure that we previously published21 (PDB 8GKA) was docked five times into the density-modified pentamer map using Chimera (v.1.13.1)43, and then relaxed and further refined into the density map using real space refinement in PHENIX (v.1.20.1-4487)41 with default parameters. Coot (v.0.9.8.1)44 was used to manually refine the positions of the sidechains. Residue positions were further adjusted to the different local refinement maps. To maintain good geometry, MolProbity (v.4.2)45 was used for geometrical validation, and iterative cycles of real space refinement in PHENIX (v.1.20.1-4487)41 and manual adjustments in Coot (v.0.9.8.1)44 were performed until good geometrical parameters were achieved (Supplementary Table 1). Structure analysis was conducted with Pymol (v.2.3.3), Chimera (v.1.13.1)43, and VMD (v.1.9.4)46. Additionally, PDBsum47 was used to analyze the interactions at the subunit interfaces, Mole 2.548 was used to calculate binding pocket cavities, and Hole49 was used to analyze the pore profiles.
Computational systems
Three distinct computational systems were constructed for TRPV3 in its open (a) and closed (b) tetrameric forms, and in its pentameric (c) form, using the CHARMM-GUI utility50. Each assay consisted of the protein embedded in a fully hydrated lipid bilayer composed of 80% of 1,2-palmitoyl-oleyl-sn-glycero-3-phosphatidylcholine (POPC), 10% of 1,2-dioleyl-sn-glycero-3-phosphatidylserine (DOPS), and 10% of cholesterol, in line with the experimental conditions21. To ensure electric neutrality of each assay, and mimic physiological conditions, sodium chloride was added at a concentration of 150 mM. The dimensions of computational systems (a), (b), and (c), after suitable thermalization, were approximately 152 × 152 × 165 Å3, 151 × 151 × 166 Å3, and 172 × 172 × 157 Å3, corresponding, respectively, to 392,171, 386,456, and 473,976 atoms. Moreover, to probe the influence of the size of the simulation cell on the dynamics of the pentamer, a fourth, larger assay (d), of dimensions 186 × 186 × 147 Å3, was built, corresponding to 521,715 atoms. To investigate ion permeation of the TRPV3 in its different forms, 12 additional computational systems were constructed by inserting in either (a), (b), or (c) adequately thermalized Na+, 2-MAE+, NMDG+, or Tris+ ions (Supplementary Table 3).
MD simulations
All MD simulations were carried out using NAMD 3.051, with the CHARMM36 force field for proteins and lipids52,53, and the TIP3P water model54. Periodic boundary conditions were applied for all computational systems. The temperature was maintained at 310 K by means of a stochastic velocity rescaling thermostat55 with a timescale of 2 ps−1, and pressure at 1 atm, using the Langevin piston algorithm56. All covalent bonds between heavy and hydrogen atoms were constrained to their equilibrium length with the Rattle algorithm57. Water molecules were constrained to their equilibrium geometry with the Settle algorithm58. Long-range electrostatic forces were computed with the particle-mesh Ewald algorithm59, and a grid spacing of 1.2 Å. Short-range van der Waals and electrostatic interactions were smoothly truncated with a 12-Å spherical cutoff. Hydrogen-mass repartitioning was applied to the protein and surrounding lipids to allow longer integration time steps to be utilized60,61. The Verlet-I/r-RESPA multiple time-stepping algorithm was employed to integrate the equations of motion with an effective time step of 4 fs for short-range interactions, and 8 fs for long-range interactions62.
Equilibrium simulations
As a preamble to their equilibrium simulation, each computational assay underwent a suitable energy minimization, followed by 20 ns of thermalization, whereby the protein was tethered to its initial position, allowing lipids to relax and form contacts with it, and an additional 20 ns of thermalization bereft of geometrical restraints. Each equilibrium simulation was carried out over a period of 1 µs. Analysis of the MD trajectories was performed with the visualization program VMD (v.1.9.4)46.
Free-energy calculations
The PMF w(z), underlying the translocation of each one of the four permeant ions across the pore of the channel in its open and closed tetrameric forms, and in its pentameric form, were determined with the well-tempered metadynamics extended adaptive biasing force (WTM-eABF) algorithm26,27. In a nutshell, the free-energy profiles were obtained by integrating the average force acting along a model of the reaction coordinate, chosen here as the projection onto the z-axis of Cartesian space of the Euclidean distance between the CoM of the pore and that of the permeant. The total permeation pathway spanned 40 Å, i.e., −20 ≤ z ≤ +40 Å, and was discretized in bins 0.1 Å wide, where samples of the local force acting along z were accrued. To mitigate the risk of deleterious nonequilibrium effects, no time-dependent bias was applied until a threshold of 10,000 samples was reached63. Convergence of the PMF calculation was assessed by monitoring the time evolution of the free-energy gradients, RMSD profiles, and determining the time point when these profiles plateaued.
The measure of statistical error was calculated thus: In the ABF framework, free-energy differences were computed by integrating the mean force of the system along the reaction-coordinate model, \(z\). Here, \(z\) represents the Euclidean distance between the CoM of the permeating ion and that of the pore, projected along the \(z\)–axis. To assess the statistical error in the free-energy difference, it is essential to analyze the statistical properties of the mean force. The reaction-coordinate model, \(z\), is typically discretized into bins, and instantaneous force values obtained during the simulation are accumulated within these bins. A common approach to estimate the error in the mean force for a given bin, i, involves quantifying the variance of the collected forces within that bin,
where ΔFz(xt) = Fz(xt) − 〈Fz〉i is the random component of the instantaneous force, \({n}_{i}\) is the number of samples accrued in bin \(i\), \(\Delta t\) is the time step of the simulation, and \({\tau }_{i}\) and \({\left\langle {\Delta F}_{z}^{2}\right\rangle }_{i}\) are the autocorrelation time and variance of ΔFz(xt) in bin \(i\). With a reliable estimate of the error of the mean force for each bin, we can proceed to examine how these uncertainties propagate to yield free-energy differences. On a discrete grid along the reaction-coordinate model, the free-energy between state \(a\) and states \(b\) writes,
where \({i}_{a}\) and \({i}_{b}\) are bin indices delimiting the \(z\)–interval [\({z}_{a}\), \({z}_{b}\)]. Assuming an independent behavior in each bin, the error of a sum of mean forces is approximated by means of the Bienaymé formula as equal to the square root of the sum of squares of the errors of these mean forces,
NanoDSF experiments
NanoDSF thermal denaturation profiles were obtained of purified TRPV3, utilizing the peak fraction from the size-exclusion chromatography (SEC) indicative of a pure tetramer. The final concentration of TRPV3 was 3.2 μM (in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 0.01% (w/v) GDN). Measurements were conducted in apo conditions and upon the addition of various ligands, including camphor, propofol, DPBA, and 2-APB, as well as a mixed ligand solution containing 2 mM camphor and 320 μM DPBA. Capsaicin and DPTHF, which are known not to activate TRPV3, were chosen as controls6,64. Additionally, the impact of DMSO, used as the solvent of the ligands, was evaluated by incorporating it at concentrations ranging from 0.01% to 1% in the protein samples. The presence of DMSO in all NanoDSF measurements was maintained below 1%. The concentrations of all chosen ligands and DMSO concentrations are detailed in Supplementary Table 7. Statistical significance of changes in melting temperatures (Tm1 and Tm2) was assessed with the unequal variance (Welch’s) t-test under apo and various agonist conditions. Significant reductions in melting temperatures are labeled in Fig. 5 with ** representing a 95% confidence level and *** denoting a 99% confidence level.
HS-AFM experiments
Data collection of the HS-AFM data described here is described in detail in Ref. 21. Briefly, TRPV3 reconstituted vesicles were deposited on a freshly cleaved mica surface and then imaged in a fluid cell containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl in the presence of 150 mM NaCl. Images were taken at room temperature with an HS-AFM (SS-NEX, RIBM), operated in amplitude modulation mode with optimized scan and feedback parameters, using the IgorPro RIBM software (Ibis v.1.1.0, IgorPro v.6.3.7.2). Ultrashort (8 μm) cantilevers (USC-F1.2-k0.15, NanoWorld) with a nominal spring constant of 0.15 N m−1, a resonance frequency of roughly 650 kHz, and a quality factor of roughly 1.5 in buffer, were used. IgorPro v.8 (WaveMetrics) was used for HS-AFM data collection. HS-AFM images were taken at 0.5–3 frames per second at pixel-sampling ratios of 1.25–8 Å per pixel.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data and materials to draw the conclusions in this paper are presented in the main text, Figures and data Figures and tables, and supplementary material. The cryo-EM map of the TRPV3 pentamer has been deposited in the Electron Microscopy Data Bank (EMDB) with accession code EMD-46907, and the structural model has been deposited in the Protein Data Bank (PDB) with accession code 9DIJ. Additional structural models analyzed in this study are the TRPV3 tetramers in their closed (8GKA), open (8V6N), and inactivated state (8V6O). For transparency and reproducibility of the MD simulations data, relevant files were uploaded to Zenodo, https://doi.org/10.5281/zenodo.15213718. Further data can be received from the authors upon reasonable request.
References
Himmel, N. J. & Cox, D. N. Transient receptor potential channels: current perspectives on evolution. Proc. R. Soc. B Biol. Sci. 287, 1–9 (2020).
Cosens, D. J. & Manning, A. Abnormal electroretinogram from a Drosophila mutant. Nature 224, 285–287 (1969).
Ferreira, L. G. B. & Faria, R. X. TRPing on the pore phenomenon: What do we know about transient receptor potential ion channel-related pore dilation up to now? J. Bioenerg. Biomembr. 48, 1–12 (2016).
Zheng, J. & Ma, L. Structure and function of the thermoTRP channel pore. Curr. Top. Membr. 74, 233–257 (2014).
Chung, M. K., Güler, A. D. & Caterina, M. J. TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nat. Neurosci. 11, 555–564 (2008).
Chung, M. K., Güler, A. D. & Caterina, M. J. Biphasic currents evoked by chemical or thermal activation of the heat-gated ion channel, TRPV3. J. Biol. Chem. 280, 15928–15941 (2005).
Chen, J. et al. Pore dilation occurs in TRPA1 but not in TRPM8 channels. Mol. Pain 5, 2–7 (2009).
Bautista, D. & Julius, D. Fire in the hole: pore dilation of the capsaicin receptor TRPV1. Nat. Neurosci. 11, 528–529 (2008).
Huffer, K., Tan, X.-F., Fernández-Mariño, A. I., Dhingra, S. & Swartz, K. J. Dilation of ion selectivity filters in cation channels. Trends Biochem. Sci. 49, 417–430 (2024).
Peverini, L., Beudez, J., Dunning, K., Chataigneau, T. & Grutter, T. New insights into permeation of large cations through ATP-gated P2X receptors. Front. Mol. Neurosci. 11, 1–12 (2018).
Bean, B. P. Pore dilation reconsidered. Nat. Neurosci. 18, 1534–1535 (2015).
Bleakman, D., Broroson, J. R. & Miller, R. J. The effects of capsaicin on voltage-gated calcium currents and calcium signals in cultured dorsal root ganglion cells. Br. J. Pharmacol. 101, 423–431 (1990).
Evans, A. R., Nicol, G. D. & Vasko, M. R. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res 712, 265–273 (1996).
Jancso, G. Pathobiological reactions of C‐fibre primary sensory neurones to peripheral nerve injury. Exp. Physiol. 77, 405–431 (1992).
Puopolo, M. et al. Permeation and block of TRPV1 channels by the cationic lidocaine derivative QX-314. J. Neurophysiol. 109, 1704–1712 (2013).
Nakagawa, A. & Hiura, A. Comparison of the transport of QX-314 through TRPA1, TRPM8, and TRPV1 channels. J. Pain Res. https://doi.org/10.2147/jpr.s41614 (2013).
Kashio, M. & Tominaga, M. TRP channels in thermosensation. Curr. Opin. Neurobiol. 75, 102591 (2022).
Peier, A. M. et al. A heat-sensitive TRP channel expressed in keratinocytes. Science 296, 2046–2049 (2002).
Xu, H., Delling, M., Jun, J. C. & Clapham, D. E. Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat. Neurosci. 9, 628–635 (2006).
Deng, Z. et al. Gating of human TRPV3 in a lipid bilayer. Nat. Struct. Mol. Biol. 27, 635–644 (2020).
Lansky, S. et al. A pentameric TRPV3 channel with a dilated pore. Nature 621, 206–214 (2023).
Zubcevic, L. et al. Conformational ensemble of the human TRPV3 ion channel. Nat. Commun. 9, 1–12 (2018).
Nadezhdin, K. D. et al. TRPV3 activation by different agonists accompanied by lipid dissociation from the vanilloid site. Sci. Adv. 10, eadn2453 (2024).
Neuberger, A. & Sobolevsky, A. I. Pentameric TRPV3: An artifact or a clue to channel function? Cell Calcium 116, 102812 (2023).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Fu, H. et al. Zooming across the free-energy landscape: shaving barriers, and flooding valleys. J. Phys. Chem. Lett. 9, 4738–4745 (2018).
Fu, H., Shao, X., Cai, W. & Chipot, C. Taming rugged free energy landscapes using an average force. Acc. Chem. Res. 52, 3254–3264 (2019).
Nadezhdin, K. D. et al. Structural mechanism of heat-induced opening of a temperature-sensitive TRP channel. Nat. Struct. Mol. Biol. 28, 564–572 (2021).
Hu, H.-Z. et al. 2-Aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 279, 35741–35748 (2004).
Moqrich, A. et al. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 307, 1468–1472 (2005).
Macpherson, L. J. et al. More than cool: p[romiscuous relationships of menthol and other sensory compounds. Mol. Cell. Neurosci. 32, 335–343 (2006).
Xu, H., Blair, N. T. & Clapham, D. E. Camphor activates and strongly desensitizes the transient receptor potential vanilloid subtype 1 channel in a vanilloid-independent mechanism. J. Neurosci. 25, 8924–8937 (2005).
Caterina, M. J. et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997).
Fischer, M. J. M. et al. The general anesthetic propofol excites nociceptors by activating TRPV1 and TRPA1 rather than GABAA receptors. J. Biol. Chem. 285, 34781–34792 (2010).
Nishimoto, R., Kashio, M. & Tominaga, M. Propofol-induced pain sensation involves multiple mechanisms in sensory neurons. Pflüg. Arch. Eur. J. Physiol. 467, 2011–2020 (2015).
Introini, B. et al. Structure of tetrameric forms of the serotonin-gated 5-HT3A receptor ion channel. EMBO J. https://doi.org/10.1038/s44318-024-00191-5 (2024).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Paknejad, N., Sapuru, V. & Hite, R. K. Structural titration reveals Ca2+-dependent conformational landscape of the IP3 receptor. Nat. Commun. 14, 6897 (2023).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 478, 4169–4185 (2021).
Terwilliger, T. C., Sobolev, O. V., Afonine, P. V., Adams, P. D. & Read, R. J. Density modification of cryo-EM maps. Acta Crystallogr. Sect. Struct. Biol. 76, 912–925 (2020).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Pintilie, G. & Chiu, W. Comparison of Segger and other methods for segmentation and rigid‐body docking of molecular components in Cryo‐EM density maps. Biopolymers 97, 742–760 (2012).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. https://doi.org/10.1002/pro.3235 (2017).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Laskowski, R. A., Jabłońska, J., Pravda, L., Vařeková, R. S. & Thornton, J. M. PDBsum: structural summaries of PDB entries. Protein Sci. 27, 129–134 (2018).
Sehnal, D. et al. MOLE 2.0: advanced approach for analysis of biomacromolecular channels. J. Cheminformatics 5, 39 (2013).
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A. & Sansom, M. S. P. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360 (1996).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020).
Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ 1 and χ 2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Feller, S. E., Zhang, Y., Pastor, R. W. & Brooks, B. R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103, 4613–4621 (1995).
Andersen, H. C. Rattle: a “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 52, 24–34 (1983).
Miyamoto, S. & Kollman, P. A. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N ⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Feenstra, K. A., Hess, B. & Berendsen, H. J. C. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20, 786–798 (1999).
Hopkins, C. W., Le Grand, S., Walker, R. C. & Roitberg, A. E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015).
Tuckerman, M., Berne, B. J. & Martyna, G. J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 97, 1990–2001 (1992).
Miao, M. et al. Avoiding non-equilibrium effects in adaptive biasing force calculations. Mol. Simul. 47, 390–394 (2021).
Nilius, B., Vennekens, R. & Owsianik, G. Vanilloid transient receptor potential cation channels: an overview. Curr. Pharm. Des. 14, 18–31 (2008).
Acknowledgements
Cryo-EM data were collected at the Simons Electron Microscopy Center at the New York Structural Biology Center, with support from the Simons Foundation (grant no. SF349247). Work in the Scheuring laboratory was supported by grants from the National Institute of Health (NIH), National Center for Complementary and Integrative Health (NCCIH), DP1AT010874, and National Institute of Neurological Disorders and Stroke (NINDS), R01NS134559. S.L. is an awardee of the Weizmann Institute of Science Women’s Postdoctoral Career Development Award.
Author information
Authors and Affiliations
Contributions
S.L., C.C., and S.S. designed the study. S.L. and O.B.C. analyzed the cryo-EM data. S.L. solved and analyzed the cryo-EM structure. C.C. performed the MD simulations and PMF calculations. C.C. and S.L. analyzed the MD simulations' trajectories. Z.W. performed the nanoDSF experiments. S.L. and S.S. wrote the paper. All authors edited the manuscript. S.S. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lansky, S., Wang, Z., Clarke, O.B. et al. Structural dynamics and permeability of the TRPV3 pentamer. Nat Commun 16, 4520 (2025). https://doi.org/10.1038/s41467-025-59798-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59798-9
