
Abstract
Multi-organ biological aging clocks across different organ systems have been shown to predict human disease and mortality. Here, we extend this multi-organ framework to plasma metabolomics, developing five organ-specific metabolome-based biological age gaps (MetBAGs) using 107 plasma non-derivatized metabolites from 274,247 UK Biobank participants. Our age prediction models achieve a mean absolute error of approximately 6 years (0.25<r < 0.42). Crucially, including composite metabolites (e.g. sums or ratios of raw metabolites) results in poor generalizability to independent test data due to multicollinearity. Genome-wide associations identify 405 MetBAG-locus pairs (P < 5 × 10−8/5). Using SBayesS, we estimate the SNP-based heritability (0.09<\({h}_{{SNP}}^{2}\) < 0.18), negative selection signatures (−0.93 < S < −0.76), and polygenicity (0.001<Pi < 0.003) for the 5 MetBAGs. Genetic correlation and Mendelian randomization analyses reveal potential causal links between the 5 MetBAGs and cardiometabolic conditions (e.g., metabolic disorders and hypertension). Integrating multi-organ and multi-omics features improves disease category and mortality predictions. The 5 MetBAGs extend existing biological aging clocks to study human aging and disease across multiple biological scales. All results are publicly available at https://labs-laboratory.com/medicine/.
Similar content being viewed by others


Introduction
Multi-organ biological age gaps (BAG), derived from clinical phenotypes, neuroimaging, and proteomics1,2,3,4,5,6,7,8,9,10 via artificial intelligence and machine learning (AI/ML), have become pivotal in understanding the mechanisms of human aging, disease, and mortality. These aging clocks provide a holistic perspective, capturing functional and structural changes across various organs. Despite the recent advancements, plasma metabolomics – a critical layer of molecular data – remains underexplored in this framework. Unlike proteomics, which focuses on proteins and post-translational modifications, metabolomics examines the downstream products of cellular processes, encompassing the biochemical intermediates and byproducts of metabolic pathways11,12.
Developing plasma metabolome-based BAGs is essential for enhancing the granularity and coverage of multi-organ aging clocks, building upon prior research by our group and others1,2,3,4,5,6,7,8,9,13. Metabolome-based BAGs may provide insights and pose unique challenges compared to plasma proteomics and phenotype-based BAGs due to the complex and dynamic nature of the metabolome. Metabolomics data provide a dynamic and highly responsive view of an individual’s biological state. Its sensitivity to factors such as diet, microbiome composition, environmental exposures, and immediate physiological changes makes it a powerful tool for capturing real-time metabolic shifts. In addition, metabolomics encompasses a diverse array of small molecules, many of which extend beyond direct gene products, offering unique biological insights. While integrating metabolic signatures with genetic and proteomic data requires thoughtful approaches, doing so within a multi-organ framework holds great promise for uncovering aging and disease mechanisms. Unlike phenotype-based BAGs, which rely on observable clinical traits or imaging-derived metrics, metabolome-based BAGs capture aging at a deeper molecular level. This offers a more precise and dynamic reflection of an individual’s metabolic state, providing unique insights into aging processes. While translating molecular data into higher-order outcomes like cognitive decline, disease onset, or lifespan prediction requires sophisticated modeling, this depth of information holds great potential for advancing precision aging biomarkers and therapeutic strategies. By integrating metabolomics data, researchers can achieve a more holistic understanding of aging processes, linking molecular pathways to organ-specific aging and systemic health outcomes14,15,16. This molecular-level insight is crucial for capturing the metabolic state of an organism and bridging the gap between the causal pathway from underlying genotypes to exo-phenotypes, such as cognitive decline, clinical symptoms, or disease onset. In our previous studies, we developed 9 phenome-wide BAGs1,2 (PhenoBAG), 3 brain MRI-based BAGs1, and 11 proteome-wide BAGs (ProtBAG17), representing the causal pathway of aging at multiple scales across different data types. Through genome-wide association studies (GWAS) and subsequent analyses, including genetic correlation18, polygenic risk score19, and causal inference4, we thoroughly evaluated the clinical applicability of these AI/ML-derived aging biomarkers.
In this work, we use 107 plasma non-derived metabolites from 274,247 UK Biobank participants (UKBB) to develop 5 organ-specific metabolome-based BAG (MetBAG), including the digestive, hepatic, immune, endocrine, and metabolic systems. Subsequently, we examine their genetic architecture (i.e., SNP-based heritability, natural selection signature, and polygenicity), genetic correlation, and causal relationships with 525 disease endpoints (DEs) from FinnGen20 and Psychiatric Genomics Consortium (PGC)21. Finally, we assess the clinical promise of the 5 MetBAGs and their PRSs for predicting 14 disease categories and mortality. All results and pre-trained AI/ML models are publicly disseminated at the MEDICINE portal: https://labs-laboratory.com/medicine/.
Results
Biological age prediction performance of the 5 MetBAGs
To rigorously evaluate the performance of biological age prediction models, we partitioned the 34,354 healthy control (CN, without any pathologies) participants into the CN training/validation/test (N = 29,354) and independent test (ind. test; N = 5000) datasets (Method). Supplementary Fig. 1 details this study’s population selection and overall workflow. The CN training set was used for model development and nested cross-validation (Lasso regression) for the two AI/ML models (Lasso regression and neural network (NN)), while the independent test set provided an unbiased assessment of model performance (Supplementary Table 1).
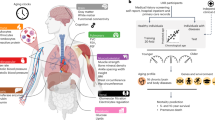
When fitting the organ-specific metabolites (Fig. 1a, Method, and Supplementary Table 2), the two AI/ML models showed marginal variability in model performance, with no single model consistently outperforming the others (Fig. 1b). The optimal model for each organ-specific MetBAG was selected based on a higher degree of generalizability (i.e., a smaller Cohen’s D, as denoted by the # symbol in Fig. 1b). The optimal models of the 5 MetBAGs showed moderate Pearson’s r coefficients (0.25<r < 0.42) in the independent test dataset (Fig. 1c). The metrics after correcting the age bias22,23 are shown in Supplementary Fig. 2. For all subsequent analyses, we used the age bias-corrected23 MetBAGs. Supplementary Table 3 presents detailed statistics for the age prediction tasks before and after the age bias correction.

a We identified organ-specific metabolites for the five human organ systems by mapping the 107 non-derived metabolites to their corresponding plasma proteins using a linear regression model. The most strongly associated protein for each metabolite was then linked to their organ-specific RNA expression profiles using data from the Human Protein Atlas (e.g., S_HDL_P → LCAT→liver: https://www.proteinatlas.org/ENSG00000213398-LCAT/tissue). Method and Supplementary Table 2 detail the metabolite-protein-organ annotations. An interactive graph visualization for this annotation is also accessible at https://labs-laboratory.com/medicine/metabolite_organ_annotation. b We evaluated age prediction performance using the mean absolute error (MAE) on independent test (ind. test) data, employing 2 AI/ML models across 5 organ systems with 107 metabolites. The “#“ symbol indicates the model that generalized best to the independent test data (i.e., smallest Cohen’s D). Data are presented as box plots showing the distribution of MAE across different groups and organs, with the median indicated by a horizontal line inside the box and the interquartile range (IQR) spanning the box from the first to the third quartile. c The scatter plot between the AI/ML-derived biological age and chronological age without applying the age bias correction23. Data are presented with points colored by organ and a linear regression line (black) fit to the data. Pearson’s correlation coefficient (r) and corresponding P-values were computed to assess the strength and significance of the linear relationship, with two-sided p-values provided.
In our previous study17, we systematically assessed model performance of key modeling components (e.g., the pathology profile of the training population and the degree of organ specificity of the proteins) from both methodological and clinical perspectives, using 11 ProtBAGs as a demonstration. Here, leveraging nested cross-validation and an independent test dataset, we further investigated factors contributing to overfitting and limited generalizability in developing the 5 MetBAGs. Specifically, we identified: i) poor generalizability to independent test datasets in certain models, and ii) substantial overfitting and reduced generalizability when incorporating composite metabolite biomarkers derived from sums or ratios of the 107 non-derived metabolites (Supplementary Fig. 3). A detailed discussion is presented in Supplementary Note 1. We also discussed sex-stratified analyses via the metabolic MetBAG in Supplementary Note 2 and Supplementary Fig. 4.
The genetic architecture of the 5 MetBAGs
We conducted GWAS (Method) for the 5 MetBAGs and identified 405 (P-value < 5 × 10−8/5) genomic locus-BAG pairs. We denoted the genomic loci using their top lead SNPs defined by FUMA (Supplementary Note 3) considering linkage disequilibrium (LD); the genomic loci are presented in Supplementary Table 4. We visually present the shared genomic loci annotated by cytogenetic regions based on the GRCh37 cytoband (Fig. 2a). Manhattan and QQ plots, as well as the genomic inflation factor (λ) of the 5 MetBAG GWASs, are presented in the MEDICINE portal (e.g., the hepatic MetBAG: https://labs-laboratory.com/medicine/hepatic_metbag). The LDSC18 intercept (LDSCb = 1.05 [1.03, 1.08]) of the 5 MetBAG GWASs was close to 1, and the LDSC ratio (an indication of inflation vs. true polygenicity; LDSCr = 0.10 [0.09, 0.13])) was small, indicating that no severe inflation due to population stratification was observed. Supplementary Fig. 5 presents the trumpet plots of the effective allele frequency vs. the β coefficients of the 5 MetBAG GWASs.

a Cytogenetic regions where the genomic region was linked to the 5 MetBAGs. Bonferroni correction was applied to denote significant genomic loci associated with PhenoBAG (P-value < 5 × 10−8/5). SBayesS estimates three genetic parameters to delineate the genetic architecture of the 5 MetBAGs, including (b) the SNP-based heritability (\({h}_{{SNP}}^{2}\)), (c) the relationship between MAF and effect size for the selection signature (S), and (d) polygenicity (Pi). We present the mean estimate of the inferred statistics (\({h}_{{SNP}}^{2}\), S and Pi) along with their standard error bars in the Figure (b–d). We applied fastGWAS to 207,793 participants of European ancestry to generate the GWAS summary statistics (in Figure a), which were subsequently used as input for SBayesS to derive the three key parameters.
We then computed three key genetic parameters of the 5 MetBAGs, including SNP-based heritability estimate (\({h}_{{SNP}}^{2}\)), natural selectional signature (S), and polygenicity (Pi) (Method). We observed small to moderate \({h}_{{SNP}}^{2}\), ranging from 0.09 to 0.18 (Fig. 2b); a detailed comparison for the \({h}_{{SNP}}^{2}\) estimate between the 5 MetBAGs, 11 ProtBAGs, and 9 PhenoBAGs are presented in Supplementary Note 4 and Supplementary Figs. 6. The 5 MetBAGs showed strong negative selection signatures, exemplified by the hepatic MetBAG (−0.94 ± 0.09) (Fig. 2c). The highest polygenic estimate was obtained by the endocrine MetBAG (0.003 ± 0.0002) (Fig. 2d). Supplementary Table 5 presents detailed statistics.
The genetic correlation between the 5 MetBAGs, 107 non-derived metabolites, 9 PhenoBAGs, 11 ProtBAGs, and 525 DEs
We used LDSC to estimate the genetic correlation (gc) of the 5 MetBAGs with three sets of traits (Method). We observed strong genetic correlations among the 5 MetBAGs (0.17 <gc < 0.78), with the highest correlation between the immune and hepatic MetBAGs (gc = 0.78 ± 0.04). In addition, the phenotypic correlations (pc) among the 5 MetBAGs closely reflected the genetic correlations (Fig. 3a). Subsequently, we also observed strong genetic correlations between the 5 MetBAGs and the 107 non-derived metabolites (− 0.44<gc < 0.91; P-value < 0.05/327) (Fig. 3b). We presented two cases where the genetic correlations between organ MetBAGs and metabolites either align or do not align with the metabolite-protein-organ annotations (https://labs-laboratory.com/medicine/metabolite_organ_annotation). For example, we found a negative genetic correlation between the gly metabolite and the endocrine BAG (gc = −0.20 ± 0.03); our metabolite-protein-organ annotation indeed assigned the gly metabolite to the endocrine system (gly→LPL→endocrine) (Fig. 3c). Another example is the acetoacetate metabolite, which was annotated to the endocrine system (Fig. 3c) but showed a positive association with the hepatic MetBAG (gc = 0.24 ± 0.06). Using RNA-seq data from the Human Protein Atlas (HPA), we found that the ANGPTL4 protein was also highly expressed in liver tissue (nTPM = 179.2), slightly lower than its expression in adipose tissue of the endocrine system (nTPM = 189.6). Finally, we also found 18 significant genetic associations (P-value < 0.05/545) between the 5 MetBAGs and 545 traits, including 9 PhenoBAGs2, 11 ProtBAGs, and 525 DEs. For instance, the endocrine MetBAG was associated with the metabolic and hepatic PhenoBAGs, the hepatic ProtBAG, and several cardiometabolic conditions, such as metabolic diseases (FinnGen code: E4_METABOLIA) and hypercholesterolemia (E4_HYPERCHOL). The immune MetBAG was connected to various types of hypertension (Fig. 3d). Supplementary Table 6 and Supplementary Data 1 and 2 present detailed statistics.

a Genetic (gc) and phenotypic correlation (pc) between the 5 MetBAGs. b Genetic correlation between the 5 MetBAGs and the 107 non-derived NMR metabolites. Bonferroni correction (P-value < 0.05/327) was applied to denote the significant metabolites. c The metabolite-protein-organ annotations for the significant metabolites identified in (b). We exemplified cases where the MetBAG-metabolite genetic correlation partially supports this organ-specificity annotation (e.g., gly→LPL→endocrine). d Genetic correlation between the 5 MetBAGs, 11 ProtBAGs, 9 PhenoBAGs2, and 525 DEs. Bonferroni correction (P-value < 0.05/545) was applied to denote statistical significance. LDSC was applied to estimate the genetic correlation between traits, with the correlation coefficients and two-sided P-values reported for each pair of traits (Supplementary Table 6). An interactive network visualization is also available at https://labs-laboratory.com/medicine/metbag_gc. DE: disease endpoint; MetBAG: metabolome-based biological age gap; PhenoBAG: phenotype-based biological age gap; ProtBAG: proteome-based biological age gap.
The causal relationship between the 5 MetBAGs and 525 DEs
Using bi-directional, two-sample Mendelian randomization (Method), we subsequently established two causal networks that linked the 5 MetBAG and 525 DEs (Fig. 4). The MetBAG2DE network found 27 significant causal relationships (P-value < 0.05/500). As anticipated, 4 MetBAGs (immune, hepatic, digestive, and endocrine) were predominantly causally associated with metabolic diseases (FinnGen codes beginning with E4), including the endocrine MetBAG and metabolic diseases (E4_METABOLIA) [P-value = 1.39 × 10−8; OR (95% CI) = 1.30 (1.18, 1.41); number of IVs=90], which aligns with the genetic correlation observed in Fig. 3d. In addition, cardiovascular diseases (FinnGen codes start with I9) were also largely causally linked with the MetBAGs. For example, the digestive MetBAG was causally linked to heart valve disease (I9_VHD) [P-value = 6.00 × 10−6; OR (95% CI) = 1.25 (1.13, 1.37); number of IVs = 79].

We constructed a causal network by employing bi-directional two-sample Mendelian randomization, following a rigorous quality control procedure to select exposure and instrumental variables (number of IVs > 7), corrected for multiple comparisons (based on the number of DEs), and performed sensitivity analyses (e.g., horizontal pleiotropy) to scrutinize the robustness of our results. Two causal networks were analyzed: (i) MetBAG2DE and (ii) DE2MetBAG. The arrows indicate the direction of the established causal relationship from the exposure variable to the outcome variable. An interactive network visualization is also available at https://labs-laboratory.com/medicine/metbag_mr. It is crucial to approach the interpretation of these potential causal relationships with caution, despite our thorough efforts in conducting multiple sensitivity checks to assess any potential violations of underlying assumptions. DE: disease endpoint; MetBAG: metabolome-based biological age gap.
For the DE2MetBAG causal network, we found 28 significant signals (P-value < 0.05/179). The involvement of cardiometabolic diseases persisted, exemplified by the causal relationship from hypertension (I9_HYPTENS) to the immune MetBAG and from disorders of lipoprotein metabolism and other lipidaemias (E4_LIPOPROT) to the digestive MetBAG. In addition, Alzheimer’s disease was causally linked to the digestive [P-value = 2.16 × 10−10; OR (95% CI) = 1.08 (1.05, 1.10); number of IVs = 8] and endocrine [P-value = 1.12 × 10−6; OR (95% CI) = 1.04 (1.03, 1.05); number of IVs = 8] MetBAGs (Fig. 4).
Mendelian randomization relies on stringent assumptions that can sometimes be violated. We conducted comprehensive sensitivity analyses for the significant signals identified to scrutinize this. Supplementary Fig. 7 provides an example of our sensitive check analyses, with a detailed discussion in Supplementary Note 5. Detailed statistics and the results of the sensitivity analyses are presented in Supplementary Data 3 and 5.
The clinical promise of the 5 MetBAGs and 5 MetBAG-PRSs
We demonstrate the clinical promise of the 5 MetBAGs and 5 MetBAG-PRSs in predicting various clinical outcomes through binary classification and survival analysis: (i) the classification of 14 systemic disease categories and (ii) the risk of mortality (Method).
We evaluated the predictive ability of a neural network (NN) at the individual level to classify 14 disease categories (“Method”). Due to the large sample size, we selected the NN over support vector machines (SVM), as SVM does not scale well with large sample-sized datasets. We report the balanced accuracy (BA) from the independent test dataset (N = 10,000), with additional metrics and results from the training/test datasets provided in Supplementary Table 7. The MetBAG-PRS alone did not outperform random chance (BA < 0.5). However, combining the MetBAGs with their PRS resulted in a slight increase in classification accuracy, as illustrated by the genitourinary disease category (ICD codes: N) and circulative disease category (ICD codes: I) (Fig. 5a). Adding age and sex further enhanced the classification accuracy (Supplementary Fig. 8). Supplementary Fig. 9 shows the loss of training and validation (at the end of each epoch) during the model training.

a A neural network evaluated the classification balanced accuracy (BA) for 14 ICD-based disease categories using the 5 MetBAGs and MetBAG-PRSs as features. Balanced accuracy (BA) from the independent test dataset (N = 10,000) is presented, with additional metrics provided in the Supplement and training/test dataset results. b The 5 MetBAGs and MetBAG-PRSs show significant associations with the risk of mortality. Age and sex were included as covariates in the Cox proportional hazard model. The symbol * indicates significant results that survived the Bonferroni correction (< 0.05/5). Data are presented as hazard ratios (HR) with 95% confidence intervals (CI), where the point represents the estimated HR and the line indicates the 95% CI range. The horizontal dashed line at y = 1 represents the null value, with group-specific colors and shapes used for distinction. c The 5 MetBAGs and MetBAG-PRSs were cumulatively included as features for mortality risk prediction. The * symbol indicates statistical significance (< 0.05) from a two-sample t-test comparing results between two Cox models. The violin plot shows the data distribution, and the box plot shows the median and interquartile range. The box plot shows the minimum, first quartile, median, third quartile, and maximum, with whiskers extending to 1.5 times the interquartile range. The results were derived after merging all features across all participants (22,672 cases and 236,339 non-cases). DE: disease endpoint; MetBAG: metabolome-based biological age gap; PhenoBAG: phenotype-based biological age gap; ProtBAG: proteome-based biological age gap; CNS: central nervous system.
We also used the 5 MetBAGs and their PRSs to predict mortality risk (“Method”). Our analysis revealed that all 5 MetBAGs, digestive MetBAG-PRS and metabolic MetBAG-PRS, showed significant associations (P-value < 0.05/5) with mortality. The metabolic MetBAG showed the highest mortality risks [HR (95% CI) = 1.16 (1.15, 1.18); P-value = 1.00 × 10−116], followed by the endocrine MetBAG [HR (95% CI) = 1.11 (1.09, 1.12); P-value = 6.77 × 10−53]. The hepatic MetBAG showed a protective association with mortality [HR (95% CI) = 0.93 (0.92, 0.94); P-value = 3.23 × 10−24] (Fig. 5b). We then conducted a cumulative prediction analysis based on the substantial associations. This analysis demonstrated that combining these features provided additional predictive power beyond age and sex, achieving an average concordance index of 0.72 (Fig. 5c). Comprehensive statistics, including HRs, P-values, and sample sizes, are available in Supplementary Table 8. We also assessed the predictive performance of the 5 MetBAGs in predicting the incidence of ICD-based single disease entities (Supplementary Fig. 10 and Supplementary Table 9, and Method).
Discussion
This study explores the clinical potential of 5 multi-organ metabolome-based biological aging clocks, extending previously established aging clocks derived from clinical phenotypes, neuroimaging, and proteomics data. The 5 MetBAGs were developed for 5 key organ systems – immune, endocrine, digestive, hepatic, and metabolic – reflecting the systemic nature of plasma metabolites circulating throughout the body. Given their integral role in these interconnected physiological systems, the 5 MetBAGs were strongly linked to cardiometabolic conditions. Comprehensive genetic analyses illuminated the underlying genetic architecture of these AI/ML-derived aging clocks, uncovering strong genetic correlations and causal relationships that tie each MetBAG to specific cardiometabolic diseases. Furthermore, integrating cross-organ and cross-omics features significantly enhanced the predictive power for 14 systemic disease categories and mortality, underscoring their potential in clinical applications.
Biological age prediction based on plasma metabolites demonstrated comparable performance to previous models using neuroimaging (MAE = 4.92–7.95 years with multi-modal brain MRI features1), plasma proteomics (MAE = 4.33–10.19 years across 11 organ systems), and more broadly-defined phenotype-based aging clocks10 (MAE ~ 5 years). Neuroimaging captures structural and functional brain alterations tied closely to neural aging, making it particularly suited for evaluating central nervous system-related aging processes, such as the brain BAG24. Plasma proteomics offers a window into the dynamic protein landscape and post-translational modifications, reflecting organ-specific aging and systemic responses such as inflammation and metabolic dysregulation25. In contrast, plasma metabolites represent small-molecule intermediates and end products of metabolic pathways, offering maybe a more direct link to cellular metabolism and cross-system integration26, supported by the higher cross-organ correlations (Fig. 3a) than our previously proposed PhenoBAG1,2 and ProtBAGs. The ability of plasma metabolites to predict biological age suggests their potential to serve as accessible and cost-effective biomarkers while complementing proteomics and neuroimaging by providing molecular-level insights into systemic metabolic health and disease processes. Integrating these modalities could yield a more comprehensive understanding of aging, accounting for system-specific and systemic processes.
This study provides a comprehensive characterization of the genetic architecture of the 5 MetBAGs through three key genetic parameters. The moderate SNP-based heritability (0.09<\({h}_{{SNP}}^{2}\) < 0.18) estimates indicate that genetic factors significantly contribute to metabolome-based biological aging variation. This underscores the genetic basis of metabolic aging and highlights the influence of inherited genomic variation on age-related metabolic profiles. Notably, we used SBayesS, which relies solely on GWAS summary statistics and an LD reference panel to estimate heritability. Previous studies have demonstrated that this method produces lower heritability estimates than approaches using individual-level data, such as GCTA27; these estimates from different methods showed high correlations28. Strong negative selection signatures indicate that genetic variants influencing MetBAGs are under evolutionary pressure to be less deleterious, consistent with previous estimates29 in various complex human traits. These signatures highlight the evolutionary importance of maintaining stable metabolic-related processes to ensure survival and reproduction, suggesting that maladaptive variants are likely to be eliminated over generations. This also implies that genetic variants associated with unfavorable metabolic aging profiles may contribute to disease susceptibility, particularly in conditions where metabolic dysregulation is a key driver (e.g., cardiometabolic diseases30). Finally, the observed high polygenicity suggests that many genetic variants with small effects collectively shape the metabolic aging process. This supports the complex, multi-genic nature of metabolic regulation and aging and aligns with the involvement of diverse metabolic pathways, reflecting the systemic integration of organ-specific aging processes influenced by genetics12,26,31,32. A previous study33 analyzes 325 NMR biomarkers in 250,341 UKBB participants, identifying 54 aging-related biomarkers linked to all-cause mortality. Using GWAS and Mendelian randomization, the researchers uncover 439 potential causal biomarker-disease relationships. They develop a metabolomic aging/mortality score that improves mortality risk prediction and identify candidate pro-aging and anti-aging biomarkers, highlighting the potential of metabolomics for personalized aging monitoring and early disease intervention.
We further depicted the genetic relationships between the 5 MetBAGs, 20 other omics-based BAGs, and 525 DEs. Consistent with expectations, we identified strong and concordant associations between the 5 MetBAGs and DEs linked to cardiometabolic conditions. This alignment was evident across genetic correlation (Fig. 3d) and Mendelian randomization (Fig. 4) analyses, underscoring the shared genetic and causal pathways. The observed genetic correlations highlight overlapping polygenic architectures, where common genetic variants simultaneously influence metabolomic profiles12,26,31,32 and cardiometabolic diseases30,34,35. Mendelian randomization further supports this link by providing evidence of causality, showing that metabolomic changes directly lead to disease risk and likely vice versa. These findings are likely rooted in the central role of plasma metabolites in systemic physiological processes, such as lipid metabolism, glucose regulation, and inflammation, which are foundational to cardiometabolic health. Interestingly, the strong associations between metabolites and cardiometabolic conditions were also observed in black individuals, where integrative omics approaches harnessed genomic diversity to uncover insights into the complexities of cardiometabolic diseases36.
Finally, we showcased the clinical potential of the 5 MetBAGs through two prediction tasks, highlighting the value of a holistic, integrative approach that combines multi-omics and multi-organ features to predict disease diagnoses, prognoses, and mortality risks. While classification performance across 14 systemic disease categories was modest, reflecting the heterogeneity of disease groups and data, the genitourinary disease category emerged with the highest predictive accuracy. This outcome may stem from the central role of the genitourinary system, including the kidneys and urinary tract, in metabolite filtration, excretion, and homeostasis. Metabolomics provides a direct readout of systemic metabolites interacting with these processes, making it particularly sensitive to genitourinary health and aging37. Furthermore, genitourinary diseases frequently involve metabolic dysregulation, such as in diabetes-related nephropathy or urinary tract dysfunction, which aligns closely with plasma metabolomics38,39. The metabolome’s dynamic nature, reflecting both physiological and pathological states, further enhances its capacity to capture relevant biomarkers of genitourinary conditions, including renal function indicators like creatinine, urea, and urinary metabolites. A notable finding is the unexpected negative association between the hepatic MetBAG and all-cause mortality risk (Fig. 5b). The protective association between hepatic MetBAG and mortality may stem from several factors. First, the hepatic MetBAG captures not only liver aging but also adaptive metabolic responses that may enhance resilience and longevity. Supporting this, a recent metabolomics aging study40 has shown that several hepatic-specific metabolites defined in our study to derive the hepatic MetBAG, such as DHA and ApoA1 (https://labs-laboratory.com/medicine/metabolite_organ_annotation), are negatively associated with mortality. Second, selection bias in the UK Biobank could contribute, as individuals with severe liver dysfunction may be underrepresented due to survival bias. Third, hepatic MetBAG likely reflects broader systemic metabolic profiles, where some aging-related shifts do not necessarily increase mortality risk. Similarly, findings from a recent proteome-based aging clock study41 indicate that some younger biological age profiles in specific organs (e.g., artery) can paradoxically correlate with increased mortality. Collectively, these findings underscore the value of MetBAGs as integrative tools for linking molecular markers of aging to the intricate mechanisms underlying complex diseases and offer AI/ML-derived biomarkers with potential applications in disease monitoring and therapeutic development.
More broadly speaking, the 5 MetBAGs expand our previously proposed aging clcoks based on clinical phenotypes2, neuroimaging1, and plasma proteomics17 data. Recently, Ikram42 suggested that biological aging clocks are flawed concepts in understanding disease. The author challenges the assumption that aging itself is a direct cause of disease and instead suggests that aging (whether measured by calendar age or biological aging) is better understood as a summary measure of accumulated changes over time (i.e., a snapshot within the passage of time). While the author emphasizes that biological aging clocks should be used as a propensity score like calendar age as an additional confounder, but itself not as a causal factor. Subsequently, Ferrucci et al.43 argues that biological aging is not merely a redundant or supplementary measure to calendar age but provides unique insights into aging and disease risk. While Ikram suggests that biological age biomarkers should only be used as covariates alongside chronological age – implying that they do not capture fundamentally distinct information – Ferrucci counters this by asserting that aging is more than just the passage of time. Here, we directly provided support evidence to demonstrate the additional value of these multi-organ and multi-omics aging clocks, on top of chronological age, for predicting mortality and systemic diseases (Supplementary Fig. 11). As shown, each individual BAG among the 23 multi-organ and multi-omics aging clocks contributed only marginally to mortality prediction. The only significant association after Bonferroni correction was observed for the Immune PhenoBAG. However, when all 23 BAGs were combined, they explained an additional 1.52% variance in all-cause mortality. This is expected, as other factors such as genetics, environmental influences, and lifestyle play major roles in mortality. For systemic disease categories, combining all 23 aging clocks accounted for an additional 6.45% variance, beyond chronological age and sex (9.85%). These findings support the use of AI/ML-derived biological aging clocks as clinically relevant biomarkers to enhance our understanding of human aging and disease, with potential implications for developing therapies targeting aging-related conditions.
To summarize, multi-organ biological aging clocks derived from plasma metabolomes provide a dynamic perspective on molecular aging, offering an enhancement to current biological age models1,2,17. These biomarkers have the potential to accelerate clinical applications, extend health span and lifespan, and guide strategies to promote healthy aging. First, we believe MetBAGs could be potentially used in clinical settings for risk stratification of age-related diseases, personalized intervention strategies, and optimizing clinical trial designs. Second, we emphasize the importance of investigating the dynamic shifts in the metabolome throughout the lifespan and integrating these insights into longitudinal models, particularly through multi-omics data, to better capture both programmed and stochastic aging influences. Furthermore, while we acknowledge the limitations of relying on UKBB data, we agree that validating MetBAGs in diverse populations is essential to assess their broader applicability, though platform consistency challenges may arise. Finally, further methodological advancements are needed to enhance the organ specificity of plasma metabolomics data, as these datasets, including plasma proteomics, inherently exhibit pleiotropic effects on traits associated with multiple organ systems.
Methods
The MULTI consortium
The MULTI consortium is an ongoing initiative to integrate and consolidate multi-organ, such as brain MRI, and multi-omics data, including imaging, genetics, proteomics, and metabolomics. Building on existing consortia and studies, MULTI aims to curate and harmonize the data to model human aging and disease across the lifespan. This study used individual-level NMR metabolomics data from UKBB to derive the 5 MetBAGs. Summary-level GWAS summary data from FinnGen and PGC were used for downstream genetic analysis. Supplementary Table 1 details the sample characteristics.
UKBB44 is a population-based research initiative comprising around 500,000 individuals from the United Kingdom between 2006 and 2010. Ethical approval for the UKBB study has been secured, and information about the ethics committee can be found here: https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/governance/ethics-advisory-committee. This study used plasma NMR metabolic biomarkers from Nightingale Health for approximately 280,000 participants in the UK Biobank. Data were collected in Phase 1 (June 2019–April 2020) and Phase 2 (April 2020–June 2022), with measurements performed on EDTA plasma samples using Nightingale’s high-throughput metabolic profiling platform. The biomarkers encompass a wide range of metabolic pathways, including lipoprotein lipids across 14 subclasses, fatty acids, and their compositions, as well as low-molecular-weight metabolites such as amino acids, ketone bodies, and glycolysis-related metabolites, all quantified in molar concentration units. In addition, imputed genotype data covering the populations of the 5 MetBAGs were used for all genetic analyses.
The FinnGen20 study is a large-scale genomics initiative that has analyzed over 500,000 Finnish biobank samples and correlated genetic variation with health data to understand disease mechanisms and predispositions. The project is a collaboration between research organizations and biobanks within Finland and international industry partners. For the benefit of research, FinnGen generously made their GWAS findings accessible to the wider scientific community (https://www.finngen.fi/en/access_results). This research utilized the publicly released GWAS summary statistics (version R9), which became available on May 11, 2022, after harmonization by the consortium. No individual data were used in the current study.
FinnGen published the R9 version of GWAS summary statistics via REGENIE software (v2.2.4)45, covering 2272 DEs, including 2269 binary traits and 3 quantitative traits. The GWAS model encompassed covariates like age, sex, the initial 10 genetic principal components, and the genotyping batch. Genotype imputation was referenced on the population-specific SISu v4.0 panel. We included GWAS summary statistics for 521 FinnGen DEs in our analyses.
PGC21 is an international collaboration of researchers studying the genetic basis of psychiatric disorders. PGC aims to identify and understand the genetic factors contributing to various psychiatric disorders such as schizophrenia, bipolar disorder, major depressive disorder, and others. The GWAS summary statistics were acquired from the PGC website (https://pgc.unc.edu/for-researchers/download-results/), underwent quality checks, and were harmonized to ensure seamless integration into our analysis. No individual data were used from PGC. Each study detailed its specific GWAS models and methodologies, and the consortium consolidated the release of GWAS summary statistics derived from individual studies. In the current study, we included summary data for 4 brain diseases for which allele frequencies were present.
Metabolomics analyses to derive the 5 MetBAGs
We downloaded the original data (Category ID: 220), which were analyzed and made available to the community by Nightingale Health Plc. The original data were (i) calibrated absolute concentrations (or ratios) and not raw NMR spectra, and (ii) before release, had already been subject to quality control (QC) procedures by Nightingale Health Plc46. Details of the protocol is presented online at https://biobank.ndph.ox.ac.uk/ukb/refer.cgi?id=130. The protocol of plasma proteomics is detailed in the original paper47.
Additional quality checks
Following the additional procedures described in Ritchie et al.48, we performed additional quality check steps to remove a range of unwanted technical variations, including shipping batch, 96-well plate, well position, aliquoting robot, and aliquot tip. We focused our analysis on the first instance of the metabolomics data (“instance” = 0). This resulted in 327 metabolites (we used only the 107 non-derived metabolites detailed in Supplementary Table 2 for our main analyses) in 274,247 participants.
Organ-specific profiles of the 107 non-derived metabolites
Plasma NMR metabolites circulate throughout the human body and predominantly influence cardiometabolic diseases. To identify the organ-specific profile of each metabolite, we annotated 107 non-derived metabolites with 2448 Olink plasma proteins and RNA expression data from the Human Protein Atlas (HPA) project (https://www.proteinatlas.org). The 107 non-derived metabolites were selected because composite metabolites, derived by combining two or more from the 107 non-derived biomarkers, often led to model overfitting in our age prediction models, which included all 327 metabolites (Supplementary Fig. 3). The annotation process involved two steps. First, a linear regression model was used to link the 107 metabolites to plasma proteins, assigning the most strong associated protein to the metabolite of interest. Next, the protein’s RNA expression profile was queried in the HPA. The consensus data – combining HPA RNA-seq data and RNA-seq data from the GTEx project – was used to identify the organ or tissue with the highest normalized transcript per million (nTPM) values as the target organ (e.g., S_HDL_P → LCAT→liver: https://www.proteinatlas.org/ENSG00000213398-LCAT/tissue). This workflow resulted in a comprehensive annotation linking plasma metabolites to proteins and their associated organs.
Certain considerations warrant careful attention. Plasma metabolites, by nature, are less organ-specific compared to phenotypic data like brain MRI. As a result, a single metabolite may strongly associate with multiple proteins highly expressed across different tissues. We opted to annotate each metabolite to a single protein and organ, aiming to balance the number of features in each age prediction model and enhance organ specificity across various systems. In our annotation, the 107 metabolites were mapped to 25 unique proteins and 6 organ systems (endocrine, hepatic, immune, digestive, CNS, and heart). An interactive network visualization demonstrating this mapping is accessible at https://labs-laboratory.com/medicine/metabolite_organ_annotation. Notably, metabolites specific to the CNS and heart were excluded due to only one organ-specific metabolite for each; in addition, all 107 metabolites were retained for calculating the metabolic MetBAG. This resulted in organ-specific metabolites for a total of 5 organ systems, as shown in Fig. 1a. Furthermore, the circulative nature of plasma metabolites also echos strong correlations between the 5 MetBAGs and the underlying metabolites (Fig. 3a, b).
Two AI/ML models
Our previous study systematically evaluated age prediction performance across various AI/ML models using multi-modal brain MRI features1 and proteomics data28. Applying the same framework, we assessed the performance of models in deriving the 5 MetBAGs using one linear approach (Lasso regression and SVR) and one non-linear method (neural networks). For linear models, hyperparameter tuning was performed through nested, repeated hold-out cross-validation49 with 50 repetitions (80% training/validation and 20% testing). However, nested cross-validation was not applied to the neural network due to the impracticality of exhaustively testing all hyperparameter combinations. Instead, a 5-fold cross-validation was employed. An independent test dataset was held out for both models. Support vector regressor was not included here because the model does not scale with a large sample size (N > 200,000).
Population splits
To rigorously train the AI/ML models, we have split the CN (without any pathologies based on ICD code and inpatient history) data (N = 34,354, varying across the 5 organs) into the following datasets (Supplementary Table 1):
-
CN independent test dataset: 5000 participants were randomly drawn from the CN population;
-
CN training/validation dataset: 80% of the remaining 29,354 CN were used for the inner loop 10-fold CV for hyperparameter selection;
-
CN cross-validation test dataset: 20% of the remaining 29,354 CN were used for the outer loop 50 repetitions;
-
PT dataset: 239,893 patients that have at least one ICD-10-based diagnosis.
Model evaluation metrics included mean absolute error (MAE) and Pearson’s r. Importantly, consistent with our prior studies, only healthy control (CN) participants were included in the training/validation dataset. At the same time, individuals with any disease diagnosis were reserved for the independent PT dataset.
The BAG was determined by subtracting the participant’s chronological age from the AI/ML-predicted age, and age bias correction was applied using the approach outlined by Beheshti et al.23.
Genetic analyses
We used the imputed genotype data from UKBB for all genetic analyses. Our quality check pipeline focused on European ancestry in UKBB (6,477,810 SNPs passing quality checks), and the quality-checked genetic data were merged with respective organ-specific populations for GWAS. We summarize our genetic quality check steps. First, we skipped the step for family relationship inference50 because the linear mixed model via fastGWA51 inherently addresses population stratification, encompassing additional cryptic population stratification factors. We then removed duplicated variants from all 22 autosomal chromosomes. Individuals whose genetically identified sex did not match their self-acknowledged sex were removed. Other excluding criteria were: (i) individuals with more than 3% of missing genotypes; (ii) variants with minor allele frequency (MAF; dosage mode) of less than 1%; (iii) variants with larger than 3% missing genotyping rate; (iv) variants that failed the Hardy-Weinberg test at 1 × 10−10. To further adjust for population stratification52, we derived the first 40 genetic principle components using the FlashPCA software53. Details of the genetic quality check protocol are described elsewhere1,2,54,55,56.
GWAS
We applied a linear mixed model regression to the European ancestry populations using fastGWA51 implemented in GCTA57. We used fastGWA to perform the 5 MetBAG GWASs, adjusting age, dataset status (training/validation/test or independent test), age-squared, sex, interactions of age with sex, systolic/diastolic blood pressure, BMI, waist circumstance, standing height, weight, and the first 40 genetic principal components. We applied a genome-wide significance threshold (5 × 10−8/5) to annotate the significant independent genomic loci. In addition, we conducted GWAS for the 327 NMR metabolites and used the resulting summary statistics for our genetic correlation analyses.
Annotation of genomic loci
For all GWASs, genomic loci were annotated using FUMA58. For genomic loci annotation, FUMA initially identified lead SNPs (correlation r2 ≤ 0.1, distance < 250 kilobases) and assigned them to non-overlapping genomic loci. The lead SNP with the lowest P-value (i.e., the top lead SNP) represented the genomic locus. Further details on the definitions of top lead SNP, lead SNP, independent significant SNP, and candidate SNP can be found in Supplementary Note 3. For visualization purposes in Fig. 3, we have mapped the top lead SNP of each locus to the cytogenetic regions based on the GRCh37 cytoband.
Three key genetic parameters
We used SBayesS29 to estimate three key genetic parameters that characterize the genetic architecture of the 5 MetBAGs. SBayesS is an expanded approach capable of estimating three essential parameters characterizing the genetic architecture of complex traits through a Bayesian mixed linear model59. This method only requires GWAS summary statistics of the SNPs and LD information from a reference sample. These parameters include SNP-based heritability (\({h}_{{SNP}}^{2}\)), polygenicity (Pi), and the relationship between minor allele frequency (MAF) and effect size (S) as nature selection signatures. We used the software pre-computed sparse LD correlation matrix derived from the European ancestry by Zeng et al.29. More mathematical details can be found in the original paper from Zeng et al.29. We ran the gctb command59 using the argument –sbayes S, and left all other arguments by default.
Genetic correlation
We estimated the genetic correlation (gc) using the LDSC18 software. We employed precomputed LD scores from the 1000 Genomes of European ancestry, maintaining default settings for other parameters in LDSC. It’s worth noting that LDSC corrects for sample overlap, ensuring an unbiased genetic correlation estimate60. We also computed the pairwise Pearson’s r correlation coefficient to understand whether the genetic correlation largely mirrors the phenotypic correlation (pc). Statistical significance was determined using Bonferroni correction (0.05/N). Three sets of analyses were performed: (i) pair-wise gc and pc among the 5 MetBAGs (Fig. 3a), (ii) the genetic correlation between the 5 MetBAGs and 107 non-derived raw metabolites (Fig. 3b), and (iii) the genetic correlation between the 5 MetBAGs, and 9 PhenoBAGs2, 11 ProtBAGs, and 525 DEs (Fig. 3d).
Two-sample bidirectional Mendelian randomization
We constructed a causal network linking the 5 MetBAGs, and 525 DEs using a bi-directional Mendelian randomization approach. In total, 2 bi-directional causal networks were established: (i) MetBAG2DE and (ii) DE2MetBAG. These networks used summary statistics from our MetBAG GWAS in the UKBB, the FinnGen20, and the PGC21 study for the 525 DEs. The systematic quality-checking procedures to ensure unbiased exposure/outcome variable and instrumental variable (IVs) selection are detailed below.
We used a two-sample Mendelian randomization approach implemented in the TwoSampleMR package61 to infer the causal relationships within these networks. We employed five distinct Mendelian randomization methods, presenting the results of the inverse variance weighted (IVW) method in the main text and the outcomes of the other four methods (Egger, weighted median, simple mode, and weighted mode estimators) in the supplement. The STROBE-MR Statement62 guided our analyses to increase transparency and reproducibility, encompassing the selection of exposure and outcome variables, reporting statistics, and implementing sensitivity checks to identify potential violations of underlying assumptions. First, we performed an unbiased quality check on the GWAS summary statistics. Notably, the absence of population overlapping bias63 was confirmed, given that FinnGen and UKBB participants largely represent populations of European ancestry without explicit overlap with UKBB. PGC GWAS summary data were ensured to exclude UKBB participants. Furthermore, all consortia’s GWAS summary statistics were based on or lifted to GRCh37. Subsequently, we selected the effective exposure variables by assessing the statistical power of the exposure GWAS summary statistics in terms of instrumental variables (IVs), ensuring that the number of IVs exceeded 7 before harmonizing the data. Crucially, the function “clump data” was applied to the exposure GWAS data, considering LD. The function “harmonize data” was then used to harmonize the GWAS summary statistics of the exposure and outcome variables. Bonferroni correction was applied to all tested traits based on the number of effective DEs.
Finally, we conducted multiple sensitivity analyses. First, we conducted a heterogeneity test to scrutinize potential violations in the IV’s assumptions. To assess horizontal pleiotropy, which indicates the IV’s exclusivity assumption64, we utilized a funnel plot, single-SNP Mendelian randomization methods, and the Egger estimator. Furthermore, we performed a leave-one-out analysis, systematically excluding one instrument (SNP/IV) at a time, to gauge the sensitivity of the results to individual SNPs.
PRS calculation
PRS was computed using split-sample GWASs (split1 and split2) for the 5 MetBAG GWASs. The PRS weights were established using split1/discovery GWAS data as the base/training set, while the split2/replication GWAS summary statistics served as the target/testing data. Both base and target data underwent rigorous quality control procedures involving several steps: (i) excluding duplicated and ambiguous SNPs in the base data; (ii) excluding high heterozygosity samples in the target data; and (v) eliminating duplicated, mismatching, and ambiguous SNPs in the target data.
After completing the QC procedures, PRS for the split2 and split1 groups was calculated using the PRS-CS65 method. PRC-CS applies a continuous shrinkage prior, which adjusts the SNP effect sizes based on their LD structure. SNPs with weaker evidence are “shrunk” toward zero, while those with stronger evidence retain larger effect sizes. This avoids overfitting and improves prediction performance. The shrinkage parameter was not set, and the algorithm learned it via a fully Bayesian approach. The predictive power of the PRS for the MetBAG was evaluated using its incremental R2 (Supplementary Table 10).
Prediction analyses for 14 systemic disease categories, the risk of mortality, and single disease endopoints
We investigated the clinical promise of the 5 MetBAGs and their PRSs (MetBAG-PRS) in two sets of prediction analyses: (i) classification tasks for predicting 14 systemic disease categories based on the ICD-10 code, (ii) survival analysis for the risk of all-cause mortality, and (iii) ICD-based single disease endpoints.
Neural networks to classify patients of disease categories vs. controls
We applied a neural network with MetBAG and their PRS. 5-fold cross-validation was used for the training/test dataset (N varies across disease categories and the MetBAG), and the model was then applied to an independent test dataset (N = 10,000) with the same distribution of PT/CN ratios. Patients for each disease category were defined by the ICD-10 code (Field-ID: 41270). Figure 5a reports the balanced accuracy (BA) obtained from the independent test data. Supplementary Table 7 provides detailed metrics, including balanced accuracy, sensitivity, specificity, negative predictive value, positive predictive value, and sample sizes for the training/test datasets.
Survival analysis for mortality risk
we employed a Cox proportional hazard model while adjusting for covariates(i.e., age and sex) to test the associations of the 5 MetBAGs and 5 MetBAG-PRSs with all-cause mortality. The covariates were included as additional right-side variables in the model. The hazard ratio (HR), exp(βR), was calculated and reported as the effect size measure that indicates the influence of each biomarker on the risk of mortality. To train the model, the “time” variable was determined by calculating the difference between the date of death (Field ID: 40000) for cases (or the censoring date for non-cases) and the date attending the assessment center (Field ID: 53). Participants who passed away after enrolling in the study were classified as cases.
Survival analysis for ICD-based single disease endpoint
We employed a Cox proportional hazard model while adjusting for covariates(i.e., age and sex) to test the associations of the 5 MetBAGs with the incidence of ICD-based single disease entity. The covariates were included as additional right-side variables in the model. To train the model, the “time” variable was determined by calculating the difference between the date of death (Field ID: 40000) for cases (or the censoring date for non-cases) and the date attending the assessment center (Field ID: 53). Participants who were diagnosed for a specific disease of interest after enrolling in the study were classified as cases.
Statistics & Reproducibility
No statistical method was used to predetermine sample size. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GWAS summary statistics and pre-trained AI models from this study are publicly accessible via the MEDICINE Knowledge Portal (https://labs-laboratory.com/medicine/) and Synapse (https://www.synapse.org/Synapse:syn64923248/wiki/63099266). Our study used data generated by the Human Protein Atlas and can be accessed at: https://www.proteinatlas.org. GWAS summary data for the DEs were downloaded from the official websites of FinnGen (R9: https://www.finngen.fi/en/access_results) and PGC. Individual data, including plasma proteomics and metabolomics, from UKBB can be requested with proper registration at https://www.ukbiobank.ac.uk/. All original data for the main figures, along with the large-sized tables for Supplementary Data 1–5, are available on Zenodo (https://zenodo.org/records/1512818567). Although allele frequency information (i.e., one column from the GWAS summary statistics) based on the UK Biobank are not considered sensitive individual-level patient information, access is restricted to academic research use and must not be used to re-identify individual participants. Requests for access should be directed to the corresponding author, Dr. Junhao Wen, and should include the requestor’s name, academic institution, and the intended research purpose. A response will typically be provided within 2–3 business days.
Code availability
The software and resources used in this study are all publicly available:
• MLNI68 (v0.1.4): https://github.com/anbai106/mlni, Age prediction
• Ukbnmf (v3.2.0): https://github.com/sritchie73/ukbnmr, Quality checks for the raw metabolites;
• FUMA (v1.5.0): https://fuma.ctglab.nl/, Gene mapping, genomic locus annotation;
• GCTA (v1.94.1): https://yanglab.westlake.edu.cn/software/gcta/#Overview, fastGWA;
• LDSC (git version aa33296): https://github.com/bulik/ldsc, genetic correlation
• TwoSampleMR (v0.5.6): https://mrcieu.github.io/TwoSampleMR/index.html, Mendelian randomization;
• PRScs (release date: Aug 10, 2023): https://github.com/getian107/PRScs, PRS calculation;
• Lifelines (v0.27.8): https://lifelines.readthedocs.io/en/latest/, Survival analysis;
• GCTB (v2.0.5): https://cnsgenomics.com/software/gctb/; SBayesS.
References
Wen, J. et al. The genetic architecture of multimodal human brain age. Nat. Commun. 15, 2604 (2024).
Wen, J. et al. The genetic architecture of biological age in nine human organ systems. Nat. Aging 4, 1290–1307 (2024).
McCracken, C. et al. Multi-organ imaging demonstrates the heart-brain-liver axis in UK Biobank participants. Nat. Commun. 13, 7839 (2022).
Wen, J. et al. Neuroimaging endophenotypes reveal underlying mechanisms and genetic factors contributing to progression and development of four brain disorders. Nature Biomedical Engineering https://doi.org/10.1038/s41551-025-01412-w (2025).
Nie, C. et al. Distinct biological ages of organs and systems identified from a multi-omics study. Cell Rep. 38, 110459 (2022).
Liu, Y. et al. Genetic architecture of 11 organ traits derived from abdominal MRI using deep learning. Elife 10, e65554 (2021).
Oh, H. S.-H. et al. Organ aging signatures in the plasma proteome track health and disease. Nature 624, 164–172 (2023).
Jaggi, A. et al. A structural heart-brain axis mediates the association between cardiovascular risk and cognitive function. Imaging Neurosci. 2, 1–18 (2024).
Wen, J. Multiorgan biological age shows that no organ system is an island. Nat. Aging 4, 1182–1183 (2024).
Tian, Y. E. et al. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat. Med. 29, 1221–1231 (2023).
Julkunen, H. et al. Atlas of plasma NMR biomarkers for health and disease in 118,461 individuals from the UK Biobank. Nat. Commun. 14, 604 (2023).
Karjalainen, M. K. et al. Genome-wide characterization of circulating metabolic biomarkers. Nature 628, 130–138 (2024).
Zhao, B. et al. Heart-brain connections: Phenotypic and genetic insights from magnetic resonance images. Science 380, abn6598 (2023).
Wang, F. et al. Plasma metabolomic profiles associated with mortality and longevity in a prospective analysis of 13,512 individuals. Nat. Commun. 14, 5744 (2023).
Cheng, S. et al. Distinct metabolomic signatures are associated with longevity in humans. Nat. Commun. 6, 6791 (2015).
Huang, J. et al. Serum metabolomic profiling of all-cause mortality: A prospective analysis in the alpha-tocopherol, beta-carotene cancer prevention (ATBC) study cohort. Am. J. Epidemiol. 187, 1721–1732 (2018).
Wen, J. Multi-organ and multi-omics biological aging clocks: generation, interpretation, and application. Preprint at https://www.medrxiv.org/content/10.1101/2025.02.06.25321803v4 (2025).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Kachuri, L. et al. Principles and methods for transferring polygenic risk scores across global populations. Nat. Rev. Genet. 25, 8–25 (2024).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
O’Donovan, M. C. What have we learned from the Psychiatric Genomics Consortium. World Psychiatry 14, 291–293 (2015).
de Lange, A.-M. G. & Cole, J. H. Commentary: Correction procedures in brain-age prediction. Neuroimage Clin. 26, 102229 (2020).
Beheshti, I., Nugent, S., Potvin, O. & Duchesne, S. Bias-adjustment in neuroimaging-based brain age frameworks: A robust scheme. NeuroImage Clin. 24, 102063 (2019).
Gaser, C., Kalc, P. & Cole, J. H. A perspective on brain-age estimation and its clinical promise. Nat. Comput. Sci. 4, 744–751 (2024).
Duggan, M. R. & Walker, K. A. Organ-specific aging in the plasma proteome predicts disease. Trends Mol. Med. 30, 423–424 (2024).
Chen, Y. et al. Genomic atlas of the plasma metabolome prioritizes metabolites implicated in human diseases. Nat. Genet. 55, 44–53 (2023).
Evans, L. M. et al. Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat. Genet. 50, 737–745 (2018).
Boquet-Pujadas, A. et al. MUTATE: a human genetic atlas of multiorgan artificial intelligence endophenotypes using genome-wide association summary statistics. Brief. Bioinforma. 26, bbaf125 (2025).
Zeng, J. et al. Widespread signatures of natural selection across human complex traits and functional genomic categories. Nat. Commun. 12, 1164 (2021).
Jurgens, S. J. et al. Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank. Nat. Genet. 54, 240–250 (2022).
Shin, S.-Y. et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 46, 543–550 (2014).
He, Q. et al. A genome-wide association study of neonatal metabolites. Cell Genom. 4, 100668 (2024).
Zhang, S. et al. A metabolomic profile of biological aging in 250,341 individuals from the UK Biobank. Nat. Commun. 15, 8081 (2024).
Koplev, S. et al. A mechanistic framework for cardiometabolic and coronary artery diseases. Nat. Cardiovasc. Res. 1, 85–100 (2022).
Kiiskinen, T. et al. Genetic predictors of lifelong medication-use patterns in cardiometabolic diseases. Nat. Med. 29, 209–218 (2023).
Tahir, U. A. et al. Whole genome association study of the plasma metabolome identifies metabolites linked to cardiometabolic disease in black individuals. Nat. Commun. 13, 4923 (2022).
Mitchell, C. M. & Waetjen, L. E. Genitourinary changes with aging. Obstet. Gynecol. Clin. North Am. 45, 737–750 (2018).
Wei, T. et al. Metabonomic analysis of potential biomarkers and drug targets involved in diabetic nephropathy mice. Sci. Rep. 5, 11998 (2015).
Luo, Y., Zhang, W. & Qin, G. Metabolomics in diabetic nephropathy: Unveiling novel biomarkers for diagnosis (Review). Mol. Med. Rep. 30, 156 (2024).
Mutz, J., Iniesta, R. & Lewis, C. M. Metabolomic age (MileAge) predicts health and life span: A comparison of multiple machine learning algorithms. Sci. Adv. 10, eadp3743 (2024).
Oh, H. S.-H. et al. Plasma proteomics in the UK Biobank reveals youthful brains and immune systems promote healthspan and longevity. Nat. Med. https://doi.org/10.1101/2024.06.07.597771 (2024). in press.
Ikram, M. A. The use and misuse of ‘biological aging’ in health research. Nat. Med. 30, 3045 (2024).
Ferrucci, L., Barzilai, N., Belsky, D. W. & Gladyshev, V. N. How to measure biological aging in humans. Nat. Med. 31, 1057 (2025).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Würtz, P. et al. Quantitative serum nuclear magnetic resonance metabolomics in large-scale epidemiology: A primer on -Omic technologies. Am. J. Epidemiol. 186, 1084–1096 (2017).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Ritchie, S. C. et al. Quality control and removal of technical variation of NMR metabolic biomarker data in ~120,000 UK Biobank participants. Sci. Data 10, 64 (2023).
Wen, J. et al. Convolutional neural networks for classification of Alzheimer’s disease: Overview and reproducible evaluation. Med. Image Anal. 63, 101694 (2020).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 51, 1749–1755 (2019).
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11, 459–463 (2010).
Abraham, G., Qiu, Y. & Inouye, M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics 33, 2776–2778 (2017).
Wen, J. et al. Genomic loci influence patterns of structural covariance in the human brain. Proc. Natl. Acad. Sci. USA 120, e2300842120 (2023).
Wen, J., et al. Characterizing heterogeneity in neuroimaging, cognition, clinical symptoms, and genetics among patients with late-life depression. JAMA Psychiatry 79, 464–474 (2022).
Wen, J. et al. Genetic and clinical correlates of two neuroanatomical AI dimensions in the Alzheimer’s disease continuum. Transl. Psychiatry 14, 1–14 (2024).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Zeng, J. et al. Signatures of negative selection in the genetic architecture of human complex traits. Nat. Genet. 50, 746–753 (2018).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, e34408 (2018).
Skrivankova, V. W. et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA 326, 1614–1621 (2021).
Sanderson, E. et al. Mendelian randomization. Nat. Rev. Methods Prim. 2, 1–21 (2022).
Bowden, J. et al. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat. Med. 36, 1783–1802 (2017).
Ge, T., Chen, C.-Y., Ni, Y., Feng, Y.-C. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Wen, J. MEDICINE: AI-derived GWAS summary statistics sharing portal. https://doi.org/10.7303/syn64923248 (2025).
Wen, J. Figures and Supplementary Data for the 5 metabolome-based aging clocks in humans. https://doi.org/10.5281/zenodo.15128185 (2025).
Wen, J. MLNI (Version 0.1.4) [Computer software]. https://github.com/anbai106/mlni (2025).
Acknowledgements
Dr. Junhao Wen and the Laboratory of AI and Biomedical Science (LABS) are supported by the start-up funding from Columbia University. The MULTI consortium (J.W.; UK Biobank Application Number: 647044) aims to integrate multi-organ imaging and multi-omics data to advance our understanding of human aging and disease mechanisms. This study used the UK Biobank resource under Application Number 35148. We want to express our sincere gratitude to the UK Biobank team for their invaluable contribution to advancing clinical research in our field (https://www.ukbiobank.ac.uk/). We want to acknowledge the participants and investigators of the FinnGen study and the PGC consortium, and we thank FinnGen (https://www.finngen.fi/en) and PGC for their generosity in sharing the GWAS summary statistics with the scientific community.
Author information
Authors and Affiliations
Consortia
Contributions
Dr. Wen (W.JH) has full access to all study data and is responsible for ensuring its integrity and accuracy. W.JH refers to Dr. Junhao Wen, and W.JY refers to Miss Jingyue Wang. Study concept and design: W.JH. Drafting of the manuscript: W.JH. Critical revision of the manuscript for important intellectual content: W.JH, A.F., K.S., S.M., W.JY, and D.C. Statistical analysis: W.JH.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Julian Mutz, Xia Jiang, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
The MULTI consortium., Anagnostakis, F., Ko, S. et al. Multi-organ metabolome biological age implicates cardiometabolic conditions and mortality risk. Nat Commun 16, 4871 (2025). https://doi.org/10.1038/s41467-025-59964-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59964-z
This article is cited by
-
Multi-organ AI endophenotypes chart the heterogeneity of brain, eye and heart pan-disease
Nature Mental Health (2026)
-
Refining the generation, interpretation and application of multi-organ, multi-omics biological aging clocks
Nature Aging (2025)
-
Brain–heart–eye axis revealed by multi-organ imaging genetics and proteomics
Nature Biomedical Engineering (2025)
