
Abstract
Biliary tract cancer (BTC) has a poor prognosis with limited treatment options. This phase 2 trial randomized 80 patients with unresectable/metastatic BTC 1:1 to sintilimab, anlotinib, and gemcitabine/cisplatin (SAGC) or chemotherapy alone (GC). At 13.4-month median follow-up, SAGC significantly improved median progression-free survival (8.5 vs. 6.3 months; HR 0.48, 95% CI 0.22–0.64, p = 0.005) and objective response rate (51.4% vs. 29.4%), with higher grade 3/4 adverse events (75.0% vs. 43.6%). Post hoc analysis showed enhanced efficacy with anlotinib 8 mg versus 10 mg (ORR 54.5% vs. 38.8%). In AKT/YAP tumor models, low-dose anlotinib (3 mg/kg) combined with sintilimab improved vascular perfusion, T-cell cytotoxicity, and cytokine secretion compared to high-dose (6 mg/kg). These findings demonstrate improved efficacy and manageable toxicity with SAGC, particularly at the 8 mg anlotinib dose, suggesting low-dose regimens may optimize antitumor response while mitigating adverse effects. Trial registration number ClinicalTrials.gov Identifier: NCT04300959.
Similar content being viewed by others

Introduction
Biliary tract cancers (BTCs) are the second most common liver cancer type after hepatocellular carcinoma, and account for approximately 3% of all gastrointestinal malignancies worldwide1. They are mainly divided into gallbladder carcinoma (GBC), intrahepatic cholangiocarcinoma (ICC), and extrahepatic cholangiocarcinoma (ECC), based on their anatomical primary site. The incidence of BTC is high and has increased over the years, with 6.8 new cases per 100,000 in China in 20222.
Most patients with BTC are at an advanced stage at the time of diagnosis and are unsuitable for surgical resection; systemic chemotherapy is the main treatment option for most patients. The preferred first-line chemotherapy for advanced BTC is gemcitabine plus cisplatin, which provides limited survival benefits with a median overall survival (OS) of 11.7 months and a median progression-free survival (PFS) of 5.7–8.0 months3,4. Target-oriented therapy has progressed and changed the treatment landscape of BTC, with patients harboring FGFR2 fusions or rearrangements benefiting from pemigatinib, infigratinib, and futibatinib, and those with IDH1 mutations and limited to intrahepatic cholangiocarcinoma subtype benefiting from ivosidenib5,6,7,8. Additionally, some patients may be treated based on their molecular profiles of ERBB2, BRAF, and NTRK 9,10,11,12,13. However, the number of patients with these targetable characteristics is small (~7% with FGFR alterations and ~13% with IDH1 mutations in intrahepatic cholangiocarcinoma cases)14; thus, the overall benefits of target-oriented treatment remain limited for most patients with advanced BTC.
The efficacy of immune checkpoint inhibitors (ICIs) in BTC is demonstrated in several studies, either as a single agent or in combination with chemotherapy, including pembrolizumab, nivolumab, and durvalumab, in patients who failed first-line chemotherapy15,16,17 and chemotherapy-naïve patients4,18,19,20,21. However, the improvement in PFS and OS with ICIs plus chemotherapy as the first-line treatment remains modest. Although the TOPAZ-1 trial met its primary endpoint OS in first-line advanced BTC, the incremental median OS and PFS with durvalumab plus gemcitabine and cisplatin versus chemotherapy alone were below 2 months (median OS: 12.8 vs. 11.6 months, median PFS: 7.2 vs. 5.7 months)4. A phase 3 KEYNOTE-966 study also indicated that pembrolizumab plus gemcitabine and cisplatin improve OS compared to chemotherapy as a first-line therapy for advanced BTC21.
Vascular targeting is believed to enhance the antitumor efficacy of ICIs by modulating the tumor microenvironment (TME) and creating positive feedback loops that reinforce each other22. A retrospective real-world study indicated that treatment with an anti-angiogenic agent (lenvatinib) in combination with an ICI showed a trend towards improved survival in patients with advanced BTCs beyond first line treatment with manageable adverse events23. Another anti-angiogenic agent (anlotinib) in combination with ICI showed promising efficacy and a well-tolerated safety profile in advanced BTCs that had failed first-line chemotherapy24. The clinical benefit of chemotherapy in combination with an antiangiogenic agent and ICI was further observed in the first-line treatment of BTC in a prospective single-arm study in 202325. However, there is a lack of validated evidence from randomized controlled clinical studies.
This multicenter, randomized, controlled, and phase 2 SAGC trial was conducted to evaluate the clinical activity and safety of the addition of sintilimab (an anti-PD-1 inhibitor) and anlotinib (an anti-angiogenic VEGF-targeted inhibitor) to chemotherapy as a first-line treatment in patients with advanced BTC. Furthermore, we conducted an animal experiment to investigate the potential optimization of dosage for the drug anlotinib.
Results
Patients and treatment
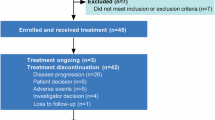
Eighty patients were enrolled between March 26, 2020, and May 25, 2022, with 40 patients in each group (Fig. 1). Overall, most patients were males (61.3%) and had ECOG PS 1 (97.5%), ICC (62.5%), and metastatic disease (100%); 17.5% patients were HBV positive. Baseline characteristics were generally well balanced between the treatment groups (Table 1). Eighteen patients in the SAGC group received a starting dose of 10 mg anlotinib daily under the initial protocol, while 22 patients received 8 mg daily under the amended protocol.

*One patient did not receive assigned treatment after randomization and was subsequently excluded from safety analysis. Abbreviations: SAGC, sintilimab plus anlotinib combined with gemcitabine and cisplatin; GC, gemcitabine plus cisplatin.
Seventy-seven patients discontinued the study treatment at the data cutoff (December 30, 2023), mainly owing to disease progression (33/40 in the SAGC group vs. 32/40 in the GC group); 2 patients continued treatment (1 in the SAGC group and 1 in the GC group) (Fig. 1). Patients in the SAGC group received sintilimab plus anlotinib for a median of seven cycles (range: 1–22) combined with chemotherapy for a median of six cycles (range: 1–8) with a dose intensity of 784.2 mg/m2 for gemcitabine and 18.9 mg/m2 for cisplatin. The median chemotherapy exposure in the GC group was six cycles (range: 1–8) with a dose intensity of 806.1 mg/m2 for gemcitabine and 20 mg/m2 for cisplatin (Supplementary Table S1). Overall, 14 patients in the SAGC group and 23 in the GC group received at least one anti-tumor therapy post study treatment discontinuation, including chemotherapy (11/40 [27.5%] vs. 13/40 [32.5%]), ICI (6/40 [15.0%] vs. 15/40 [37.5%]), and tyrosine kinase inhibitors (0/40 [0%] vs. 8/40 [20.0%]) (Supplementary Table S2).
Efficacy
The median follow-up at the data cutoff was 13.4 months. Thirty-five (87.5%) of 40 patients in the SAGC group and 35 (87.5%) of 40 patients in the GC group had progressive disease or died. The median PFS was 8.5 months (95% CI, 5.6–11.0) in the SAGC group versus 6.3 months (95% CI, 4.4–7.8) in the GC group (HR: 0.48 [95% CI, 0.22–0.64], p = 0.005; Fig. 2A). A post-hoc analysis was conducted using RECIST 1.1, and the PFS survival data demonstrated results similar to those obtained using the mRECIST criteria, as shown in the Supplementary Fig. S1. The 12-month PFS rates were 26.4% and 0% in the SAGC and GC groups, respectively. Consistent improvements in PFS were observed across the most clinically relevant subgroups (Fig. 3). The median PFS was 9.1 months with SAGC for patients with ICC versus 6.3 months with GC (HR: 0.34, 95% CI, 0.15–0.75; Supplementary Fig. S2). Twenty-nine (72.5%) patients died in the SAGC group and 23 (57.5%) in the GC group; the median OS was 13.2 months (95% CI, 8.7–19.0) vs. 13.7 months (95% CI, 10.2–15.3) (HR: 1.04 [95% CI, 0.40–1.49], p = 0.895 Fig. 2B).

A Progression-free survival. B Overall survival. *Per mRECIST. Note: p values are two-sided and are based on log-rank test. Abbreviations: SAGC, sintilimab plus anlotinib combined with gemcitabine and cisplatin; GC gemcitabine plus cisplatin, CI confidence interval, HR hazard ratio.

A Progression-free survival. B Overall survival. Abbreviations: CI confidence interval, ECOG Eastern Cooperative Oncology Group, ECC extrahepatic cholangiocarcinoma, GBC gallbladder carcinoma;ICC intrahepatic cholangiocarcinoma, SAGC sintilimab plus anlotinib combined with gemcitabine and cisplatin, GC gemcitabine plus cisplatin.
Thirty-seven patients in the SAGC group and 34 in the GC group received at least one post-baseline imaging assessment. Nineteen (51.4%) of the 37 patients randomized to SAGC group achieved a partial response (PR), and 16/37 (43.2%) patients had stable disease (SD), contributing to an ORR of 51.4% (95% CI, 34.4%–68.1%) and a DCR of 94.6% (95% CI, 81.8%–99.3%). In addition, 10/34 (29.4%) patients achieved PR, and 19/34 (55.9%) patients achieved SD in the GC group according to their randomized assignments, with an ORR of 29.4% (95% CI, 15.1%–47.5%) and a DCR of 85.3% (95% CI, 68.9%–95.0%) (Table 2, Supplementary Fig. S3). The ORR was significantly higher in the SAGC group than in the GC group (p = 0.033). Response data analysis using RECIST 1.1 showed similar results to those from the mRECIST criteria, as outlined in the Supplementary Table S3. The median time to response was 2.5 (1.7–3.6) months in the SAGC group and 3.0 (2.7–4.0) months in the GC group. A longer median duration of response with SAGC vs. GC (9.1 [4.9, NA] vs. 3.4 [2.0, NA] months) was observed (Table 2).
A post hoc sensitivity analysis was performed to compare the efficacy between patients who received a starting dose of anlotinib of 10 mg daily and those who received an anlotinib dose of 8 mg daily in the SAGC group. The median PFS did not show a significant difference between the 8 mg and 10 mg groups (8.5 vs. 7.6 months, HR: 0.87 [95% CI, 0.30–1.55], p = 0.691) despite a trend of improvement in the 8 mg group (Fig. 4A). A trend towards longer median OS was observed in the 8 mg group compared to the 10 mg group (14.9 vs. 9.3 months, HR: 0.49 [95% CI, 0.14–1.18], p = 0.055) (Fig. 4B), with this difference remaining consistent during long-term follow-up. The ORRs were 38.8% at 10 mg daily and 54.5% at 8 mg daily (Supplementary Table S4). We compared PFS and OS between the SAGC arm with 8 mg anlotinib and the GC group, the median PFS was 8.5 months vs. 6.3 months (HR: 0.45), and the median OS was 14.9 months vs. 13.7 months (HR: 0.77) (Supplementary Fig. S4)

A Progression-free survival. B Overall survival. Note: p values are two-sided and are based on log-rank test. Abbreviations: CI confidence interval, HR hazard ratio, NA not available.
Safety
Seventy-nine of the 80 patients received at least one dose of the assigned treatment and were included in the safety analyses. All patients experienced at least one TRAE;75% had at least one grade 3/4 TRAE in the SAGC group and 43.6% in the GC group. No adverse events in grade 5 were observed in either group. Adverse events led to dose reductions in 28 (70%) and 19 (48.7%) patients in the SAGC and GC groups, respectively. Furthermore, AEs led to treatment discontinuation in 1 (2.5%) and 0 patients in the SAGC and GC groups, respectively.
The most common treatment-related AEs are shown in Table 3 and Supplementary Fig. S5. The most frequent grade 3–4 treatment-emergent AEs (occurring in ≥5% of patients) were leukopenia (9 [22.5%] of 40 patients in SAGC group vs. 11 [28.3%] of 39 patients in GC group), neutropenia (18 [45%] vs. 9 [23.1%]), thrombocytopenia (11 [27.5%] vs. 4 [10.3%]), anemia (6 [15%] vs. 6 [15.4%]), increased γ-glutamyltransferase (5 [12.5%] vs. 1 [2.6%]). Serious hematological toxicities (including neutropenia and thrombocytopenia) were more common in the SAGC group than in the GC group. The incidence of grade 4 AE in the SAGC group decreased from 23.5% to 13.0% after the starting dose of anlotinib was reduced to 8 mg daily (Supplementary Table S5). Potentially immune-mediated AEs occurred in 9 (22.5%) of the 40 participants in the SAGC group, of all were grade 1–2 (Table 3). The GC group was not evaluated for immune-related AEs since this was an open-label trial. Common immune-mediated adverse events included abnormal thyroid function in four patients and rashes in four patients. The particular concern immune-related adverse event was pneumonitis with grade 2, which occurred in one participant in the SAGC group and was treated with corticosteroids.
In vivo validation of the efficacy and safety of anlotinib plus anti-PD-1
Previous observations suggest that dose reduction of anlotinib may lead to improved outcomes and decreased adverse events26,27. To explore the potential mechanisms of these dose-dependent responses, an orthotopic AKT/YAP-induced CCA tumor-bearing model was established. Consistent with expectations, the combination of anlotinib and anti-PD-1 treatment demonstrated greater efficacy in inhibiting tumor growth than monotherapy or control groups. Our study revealed no statistically significant differences in efficacy between high- and low-dose anlotinib monotherapy treatment (Supplementary Fig. S6a-c). However, the combination of low-dose anlotinib with anti-PD-1 (A3 + P group) demonstrated greater efficacy in inhibiting tumor growth compared to high-dose anlotinib combination therapy (A6 + P group) (Fig. 5a-d). Additionally, survival outcomes were similar across treatment groups, with the most favorable prognosis observed in the low-dose anlotinib plus anti-PD-1 subgroup (Fig. 5e).

Low-dose anlotinib combination therapy with anti-PD-1 demonstrated greater efficacy in inhibiting tumor growth than the high-dose group. Representative livers (a) and H&E staining of livers (b) from AKT/YAP-induced CCA tumor-bearing C57BL/6 mice. c Liver weight of tumor-bearing mice. d Quantification of tumor areas per liver section. e Kaplan–Meier survival curves of tumor-bearing mice treated as indicated since the day of tumor injection. Representative immunofluorescent staining (f), quantification of MVD (g) and quantification of perivascular cell coverage (h). Red, CD31 staining; green, α-SMA staining; blue, DAPI staining. Representative immunofluorescent staining (i) and quantification of Ho33342 perfused tumor areas (j). Red, CD31 staining; green, sytogreen staining; blue, Hoechest 33342 staining. Representative immunofluorescent staining (k) and quantification of hypoxyprobe+ areas (l). Red, CD31 staining; green, hypoxyprobe staining; blue, DAPI staining. Representative immunofluorescent staining (m) and quantitation of CD8+ T cells infiltration (n). Red, CD8 staining; green, Ki67 (proliferation marker) staining; blue, DAPI staining. Scale bars: 100 µm (c), 20 µm (f, m) and 500 µm (i, k). n = 5 mice per group (a–d). n = 10 mice per group (e). Data in (f–n) are from one experiment representative (n = 5 mice per group) of two independent experiments. The other independent experiment with similar results are shown in Supplementary Fig. S7a-e. Data are presented as mean ± SEM (c, d, g, h, j, l, n). Statistical significance was determined with one-way ANOVA followed by Holm-Sidak multiple comparison test. ns, no significance, *p < 0.05, **p < 0.01, ***p < 0.001, versus control group (IgG group). Source data are provided as a Source Data file. CCA cholangiocarcinoma; IgG control group; P, anti-PD-1 therapy; A6 high-dose anlotinib (6 mg/kg); A3 low-dose anlotinib (3 mg/kg); A6 + P high-dose anlotinib (6 mg/kg) plus anti-PD-1 therapy; A3 + P low-dose anlotinib (3 mg/kg) plus anti-PD-1 therapy; perivascular cell coverage, define as the ratio of a-SMA/CD31 double-positive area over CD31+ area, MVD tumor microvascular density, define as CD31+ vessel numbers per field.
Furthermore, blood routine test was performed to evaluate drug toxicity. The hematological adverse events in vivo experiments treated with different dosing schedules are shown (Fig. 6a-f, Supplementary Table S6). More obvious hematological toxicities were detected in the A6 + P group than in the A3 + P group, which were consistent with those obtained in clinical studies. The most frequent AEs were leukopenia (6 [100%] in A6 + P group vs. 1 [16.7%] in A3 + P group) and thrombocytopenia (6 [100%] vs. 6[100%]). The values of WBC (white blood cell) and PLT (platelets) declined significantly in the medicated groups, whereas a more pronounced decrease in WBC and PLT counts was observed in the A6 + P group than A3 + P group. Moreover, the mice in the A6 + P group exhibited a notable reduction in small intestinal villus and significant irregular morphologies, suggesting that high-dose anlotinib combination therapy results in noticeable gastrointestinal toxicity (Fig. 6g-j, Supplementary Fig. S7a, b). And there were no significant differences in body weight among the treatment groups at any time point (Fig. 6k).

a-f Major parameters in hematology after analysis of peripheral blood from AKT/YAP-induced CCA tumor-bearing C57BL/6 mice. The number of WBC (a), neutrophils (b), Hb (c), PLT (d) and RBC (e) were measured. Red dashed lines represent the normal ranges of blood routine indexes, which refer to the manufacturer’s instructions (Charles River Laboratories). Summary of hematological adverse events in the experimental animals (f). Intestinal damage was observed in C57BL/6 mice after administration. Histology score (g) and villi height (h) of small intestine were measured based on H&E staining. Representative H&E staining (i) and representative immunofluorescent staining for Ho33342 perfusion (j) of small intestine tissues. Red, CD31 staining; green, sytogreen staining; blue, Hoechest 33342 staining. k Body weight of the orthotopic tumor-bearing mice was measured every 3 days. Scale bars: 200 µm (i, j). n = 6 mice per group (a–f). Data in (g–j) are from one experiment representative (n = 5 mice per group) of two independent experiments. The other independent experiment with similar results are shown in Supplementary Fig. S7f-g. n = 5 mice per group (k). Data are presented as mean ± SEM (a-e, g, h, k). Statistical significance was determined with one-way ANOVA followed by Holm-Sidak multiple comparison test. ns, no significance, *p < 0.05, **p < 0.01, ***p < 0.001, versus control group (IgG group). Source data are provided as a Source Data file. Abbreviations: CCA cholangiocarcinoma, IgG control group, P anti-PD-1 therapy, A6 high-dose anlotinib (6 mg/kg), A3 low-dose anlotinib (3 mg/kg), A6 + P high-dose anlotinib (6 mg/kg) plus anti-PD-1 therapy, A3 + P low-dose anlotinib (3 mg/kg) plus anti-PD-1 therapy, WBC white blood cells, NEUT neutrophils, Hb hemoglobin, PLT platelets, RBC red blood cells.
In vivo exploration of the immune microenvironment mechanisms of low-dose anlotinib combination therapy
Existing research suggests that antiangiogenic agents have the potential to restructure the tumor immunosuppressive microenvironment through the normalization of vascular structure and mitigation of hypoxia in a dose-dependent fashion. Therefore, our initial assessment focused on the alterations in tumor blood vessels. The results demonstrated that high-dose anlotinib resulted in a statistically significant decrease in intratumoral microvascular density (MVD, defined as the number of CD31+ vessels per field) when compared to both the control and low-dose groups (p = 0.0003, p = 0.003, respectively) (Supplementary Fig. S6d, e). In contrast, the low-dose group exhibited a more significant enhancement in perivascular cell coverage (the ratio of α-SMA/CD31 double-positive area to CD31+ area) relative to the high-dose group (p < 0.0001) (Supplementary Fig. S6f). In the context of combined treatment, the combination of low-dose anlotinib with anti-PD-1 therapy significantly increased perivascular cell coverage and improved vascular perfusion compared to the high-dose group ((p = 0.0009, p < 0.0001, respectively), demonstrating enhancements in vascular functionality and the normalization of vascular structure (Fig. 5f-j, Supplementary Fig. S7c-e). The findings depicted in Fig. 5k-l and Supplementary Fig. S7f indicate that the combination of low-dose anlotinib and anti-PD-1 may have a beneficial effect on hypoxia compared to other treatment groups.
Moreover, our analysis focused on the changes in the immune microenvironment. Animal experiment flowchart of CCA tumors was shown in Supplementary Fig. S8a. Flow cytometry was carried out to analyze the infiltrating immune cells after administration of various treatment groups. The proportion of CD8+ T cells and NK (natural killer) cells in lymphocytes (CD45+) in the A3 + P group was significantly increased, especially the CD8+/CD45+ ratios (Fig. 7a). The proliferation of immune cells was evaluated through dual immunofluorescent staining of CD8 and Ki67. Quantitative analysis revealed that the proportion of Ki67+/CD8+ double-positive cells within CD8+ T cells was significantly higher in the treatment group following low-dose anlotinib treatment compared to the other groups (Fig. 5m, n, Supplementary Fig. S6g, h, Supplementary Fig. S7g). We then further examined the subpopulation and composition of the CD8+ T cells. As showcased in Fig. 7b, c, we observed that A3 + P-treated mice had an increased ratio of effector T cells and Tpex (precursor exhausted CD8+ T cells) in their CD8+T cell subtype composition compared to control mice (Fig. 7c, d), while the proportion of Tem (effector memory T cell), naive T cells, Tex (exhausted CD8+ T cells), and Tcm (central memory T cells) remained similar (Supplementary Fig. S8b-e). Meanwhile, the percentages of Treg cells (CD4+ FOXP3+) gated on CD4+ T cells presented a downward trend in mice with low-dose anlotinib combination therapy (Fig. 7d). Additionally, further analysis of the data reveals the co-administration of low-dose anlotinib with anti-PD-1 immune therapy may synergistically enhance activated T cell cytotoxicity. Flow cytometry analysis revealed a significant increase in the secretion of effector cytokines, including GZMB (granzyme B) and perforin in the group receiving the low-dose combination therapy, while a decreased expression of immune suppressors such as TRAIL (TNF-related apoptosis-inducing ligand) and PD-1 (programmed cell death 1) (Fig. 7e-h). A similar trend towards elevated expression of GZMB in NK cells was observed for the low-dose combination group (Fig. 7i). Additional results of effector cytokines in CD8+ T cells and in NK cells are illustrated in Supplementary Fig. S8f-k.

a-d Flow cytometry was carried out to analyze the population and composition of immune cells after administration of various treatment groups. The proportion of CD4+ T cells, CD8+ T cells, NK cells, and NKT cells in CD45+ immune cells (a). The proportion of effector T cells (b) and Tpex cells (c) in CD8+ T cells. The proportion of Tregs (d) in CD4+ T cells. The production of effector cytokines and the expression of immune suppressors in immune cells were measured by flow cytometry. MFI quantification analyses (left) and representative flow plots (right) of GZMB (e), perforin (f), TRAIL (g) and PD-1(h) in CD8+ T cells. MFI quantification analysis (left) and representative flow plots (right) of GZMB expression in NK cells (i). Additional results of immune cell subsets, effector cytokines in CD8+ T cells and effector cytokines in NK cells are illustrated in Supplementary Fig. S8. Gating procedures are described in Supplementary Figs. S9 and S10. n = 4 mice per group (a, e-i). n = 5 mice per group (b-d). Data are presented as mean ± SEM (a–i). Statistical significance was determined with one-way ANOVA followed by Holm-Sidak multiple comparison test. ns, no significance, *p < 0.05, **p < 0.01, ***p < 0.001, versus control group (IgG group). Source data are provided as a Source Data file. Abbreviations: CCA cholangiocarcinoma, IgG control group, P anti-PD-1 therapy, A6 high-dose anlotinib (6 mg/kg), A3 low-dose anlotinib (3 mg/kg), A6 + P high-dose anlotinib (6 mg/kg) plus anti-PD-1 therapy, A3 + P low-dose anlotinib (3 mg/kg) plus anti-PD-1 therapy, HD injection hydrodynamic tail vein injection, NK cells natural killer cells, NKT natural killer T cell, Tpex precursor exhausted CD8 + T cells, Tregs regulatory T cells, MFI mean fluorescence intensity, GZMB granzyme B, TRAIL TNF-related apoptosis-inducing ligand, PD-1 programmed cell death 1. Source data are provided as a Source Data file.
Discussion
This trial is a randomized controlled trial to evaluate the clinical efficacy and safety of combining anti-PD-1 inhibitor and anti-VEGF therapy with chemotherapy as a first-line treatment in patients with advanced BTC. Indeed, all patients included in the study were diagnosed with metastatic BTC. The addition of sintilimab plus anlotinib to the standard chemotherapy regimen of gemcitabine and cisplatin showed promising clinical benefit and manageable toxicity for patients with metastatic BTC, with significant improvement in median PFS (8.5 months). However, this was a phase 2 study with a small sample size and the results should be interpreted with caution. The median PFS (6.3 months), median OS (13.7 months), and ORR (29.4%) with chemotherapy alone in this study were consistent with previous reports4,28,29.
This study met its primary endpoint, with significantly improved PFS with SAGC versus GC (8.5 months vs. 6.3 months), which was generally consistent with other similar treatment regimens in first-line advanced BTC. The median PFS with gemcitabine and cisplatin plus atezolizumab (an anti-PD-L1 inhibitor) and bevacizumab was 8.3 months in patients with advanced BTC in a phase 2 IMbrave-151 study30. The lower median PFS in the SAGC group in this study than that of the currently reported PFS with gemcitabine plus oxaliplatin combined with toripalimab (an anti-PD-1 inhibitor) plus lenvatinib (10.2 months) in patients with advanced ICC25 may be attributable to patients with older age (median: 62.5 vs. 56.5 years) and relatively poor performance status (ECOG PS 1: 97.5% vs. 0%). The present study enrolled more patients with TNM stage IV (100% vs. 40%), and less patients with ICC (57.5% vs. 100%). Patients with ICC have a better OS than those with biliary tract cancers of different primary origins31. Consistently, patients with ICC in the SAGC group had a higher PFS (9.1 months) than compared with overall patients (8.5 months). In addition, higher ORR and DCR were observed in this study. The ORR of the SAGC group in this study was 51.4%, which was higher than the GC group (29.4%). Previous studies show that the ORR of ICIs combined with chemotherapy is 26.7–29%4,21. Taken together, the present results add evidence for the efficacy and feasibility of combining anti-PD-1 antibody, vascular-targeting tyrosine kinase inhibitor (TKI), and chemotherapy as a first-line treatment in patients with metastatic BTC.
Our study did not demonstrate a statistically significant improvement in OS between the SAGC and GC groups. We have noted some potential factors that might contribute to the OS results. Firstly, the administration of multiple medications of SAGC may limit the choice of second-line treatment for patients in this group. Indeed, we observed that more patients in the GC group than those in the SAGC group received subsequent antitumor treatment after study treatment discontinuation (57.5% vs. 35.0%, p < 0.05), particularly for ICI (37.5% vs. 15.0%, p < 0.05) and TKI (20% vs. 0%, p < 0.05). In addition, the adverse events, in particular for patients treated with higher dose of anlotinib in the SAGC group, might further reduce the patient compliance for subsequent treatment. Secondly, the dose adjustment of anlotinib from 10 mg to 8 mg also play an important role. In vivo studies suggested that administering a higher dose of anlotinib in combination therapy may lead to adverse reactions without improving anti-tumor efficacy. Specifically, the high-dose anlotinib may negatively influence the tumor immune microenvironment by excessive vascular pruning, exacerbation of hypoxia, and hypoperfusion. These changes subsequently impair the cytotoxic capabilities of immune cells and diminish the effectiveness of immunotherapy. Conversely, the low-dose anlotinib combination therapy appeared to enhance anti-tumor efficacy while mitigating adverse reactions. Consequently, future research will focus on optimizing the treatment protocol and identifying more responsive patient populations for further investigation.
This study did not observe any new safety signals in the GC or SAGC groups. Hematological toxicity (such as neutropenia) was the main grade 3 or higher AE in the SAGC group, as well as the most common TRAE for toripalimab combined with lenvatinib and chemotherapy in a phase 2 study on advanced intrahepatic cholangiocarcinoma25. The most significant AE of SAGC with 10 mg anlotinib daily on treatment continuity was the high incidence of grade 4 thrombocytopenia, which is consistent with previous reports32,33,34. SAGC treated with anlotinib 8 mg once daily is generally well tolerated in patients with metastatic BTC. Thus, the starting dose of anlotinib was adjusted from 10 mg to 8 mg after 18 patients were enrolled in the SAGC group, with grade 3/4 thrombocytopenia decreased from 38.9% to 13.6%, and overall safety became more manageable. Notably, the anlotinib dose reduction did not affect the PFS of patients with SAGC and tended to provide better OS. Common immune-mediated AEs included abnormal thyroid function, fever, and rash, which were mainly grade 1 or 2. No new safety concerns were identified. These events were managed with appropriate supportive therapy. The use of systemic corticosteroids to manage immune-mediated AEs was generally low (<10% of the participants).
To validate the effectiveness of low-dose anlotinib in conjunction with anti-PD-1 immunotherapy in mitigating adverse effects and enhancing efficacy in this phase 2 trial, as well as to investigate potential mechanisms, the AKT/YAP-induced CCA tumor-bearing mice model was established for in vivo validation experiments. The results demonstrated that there were no significant differences in efficacy between low-dose and high-dose anlotinib monotherapy in the orthotopic CCA tumor-bearing model. However, the combination of low-dose anlotinib with anti-PD-1 immunotherapy significantly enhanced efficacy compared to both the high-dose anlotinib combination group and the control group. Notably, the low-dose anlotinib combined with anti-PD-1 immunotherapy exhibited a significantly stronger effect on promoting ‘vascular normalization’ as evidenced by reduced neovascularization (CD31+ microvessels) and increased pericyte coverage of tumor vessels, compared to the high-dose treatment. By implementing this enhancement, we may significantly facilitate the TME transition from abnormal structural and functional states to normal states through the increase of vascular perfusion and the decrease of hypoxia area. In addition, our data strongly support the intrinsic role of the combination of low-dose anlotinib with anti-PD-1 therapy in the reprogramming of immunosuppressive environment, as evidenced by the increasing infiltration and activation of CD8+ cytotoxic T cells and NK cells, especially the effector T cells and Tpex, while a reduced infiltration of immunosuppressive Tregs cells. Moreover, we found that low-dose anlotinib drug combinations interact with the immune contexture, triggering the activation of immune effector cytokines (GZMB and perforin) and the inhibition of immunosuppressive components (TRAIL and PD-1). In line with existing experimental results, our findings established a mechanistic association between vasculature and the infiltration of immune cells within tumors, leading us to propose the utilization of a combination therapy involving low-dose anlotinib and anti-PD-1 to mitigate the adverse effects of anticancer drugs while preserving their therapeutic benefits. Our findings mentioned above are consistent with prior research, suggesting that there is a correlation between the dosage of TKI drugs and their impact on vascular normalization35. More specifically, antiangiogenic drugs with the lower doses may potentially ameliorate hypoxia, acidosis, and hypoperfusion in solid tumors by facilitating vascular normalization and enhancing immune cell infiltration, ultimately facilitating the transition of the immunosuppressive microenvironment to an immune-supportive state36,37.
Conversely, there exist risks of excessive pruning of blood vessels and rebound angiogenesis when the doses of TKI drugs are too high in clinical administration, which can exacerbate hypoxia and hypoperfusion, inhibit the killing capacity of immune cells, and reduce the efficacy of immunotherapy38. Consequently, it is very important to select an appropriate dose of TKIs when combined with ICIs to achieve better clinical efficacy, lower side effects and smaller financial burden for BTC patients.
This study has several limitations. The phase II design and safety-based sample size determination indicated that patient numbers were relatively low for efficacy analyses. Although assignment to the treatment arms was randomized, no stratification was applied; thus, the results might have been biased owing to potential confounding factors. Additionally, the open-label design may have influenced the evaluation of PFS, and no blinded review of the imaging was performed. The early design of the study meant that it had only two arms and lacked the ICI plus chemotherapy group, which was recently approved as standard treatment; therefore, it cannot directly explain whether the addition of anti-VEGF or TKI therapy to ICI plus chemotherapy can improve the prognosis of patients with metastatic BTC.
In conclusion, this phase 2 study demonstrated that sintilimab plus anlotinib in combination with gemcitabine plus cisplatin was superior to gemcitabine plus cisplatin alone, with a significant improvement in PFS and manageable toxicity in metastatic BTC. Notably, reducing the dosage of anlotinib to 8 mg may lead to a prolonged overall survival, potentially attributed to the improvement of tumor vascular and immune microenvironment at a lower anlotinib dose. Future studies are needed to evaluate the optimal dose intensity of anti-vascular targeting agents in combination with checkpoint inhibitors and chemotherapy in advanced BTC
Methods
Ethics approval and consent to participate
This trial was approved by the Medical Ethics Committee of Zhejiang Cancer Hospital. All study-related procedures were performed with written informed consent from all patients. This trial was registered at ClinicalTrials.gov. (identifier: NCT04300959) and was performed in accordance with Good Clinical Practice and the Declaration of Helsinki. Approval for all animal experiments was obtained from the Animal Experimental Ethics Committee of Zhejiang Cancer Hospital (zjzlsd2023-08-005), and the experiments were conducted in accordance with guidelines for the care and use of laboratory animals.
Study design and participants
The SAGC trial was a randomized, controlled, open label phase 2 study conducted at three centers in China, including Zhejiang Cancer Hospital, Quzhou People’s Hospital and Taizhou Hospital. The main inclusion criteria were patients aged 18 years or older, with a baseline Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1, with biopsy/pathology-confirmed unresectable, locally advanced, or metastatic BTC (including GBC, ICC, and ECC), who had not received previous systemic therapy for advanced or metastatic disease, had measurable disease according to the Modified Response Evaluation Criteria in Solid Tumors (mRECIST), and had adequate hematological and organ function with a life expectancy exceeding 12 weeks. Patients who received neoadjuvant or adjuvant chemotherapy 6 months or more prior to randomization were eligible.
Key exclusion criteria included active bleeding or clinically significant gastrointestinal (GI) abnormalities that might increase the risk for GI bleeding, immune-related conditions such as autoimmune disease and major inflammatory diseases, clinical evidence of metastatic disease to the brain and clinically significant cardiovascular disease, acute or chronic active hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, HBV DNA greater than 2,000 international units per milliliter (IU/mL) or 104 copies per mL, and HCV RNA greater than 10³ copies per mL. Detailed inclusion and exclusion criteria are seen in the online supplemental material.
The trial was registered at Clinicaltrials. gov (identifier: NCT04300959) and was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. The protocol and amendments were approved by the Institutional Review Board and Ethics Committee of each site. All patients provided written informed consent prior to enrollment.
Randomization and masking
Eligible patients were randomly assigned (1:1) to the sintilimab plus anlotinib plus gemcitabine and cisplatin (SAGC) or gemcitabine and cisplatin (GC) group using a simple randomization method. No masking was performed because this was an open-label study.
Procedures
Patients in the SAGC group received sintilimab (200 mg intravenously administered on day 1 every 3 weeks) and anlotinib (10 mg orally once daily from day 1 to day 14, every 3 weeks) along with chemotherapy (25 mg/m2 cisplatin followed by 1,000 mg/m2 gemcitabine, each administered on days 1 and 8 every 3 weeks) for up to 8 cycles, followed by maintenance sintilimab plus anlotinib. Patients in the GC group received chemotherapy alone (cisplatin 25 mg/m2 followed by gemcitabine 1,000 mg/m2, each administered on days 1 and 8 every 3 weeks) for up to 8 cycles. After 18 patients were enrolled in the SAGC group, the starting dose of anlotinib was modified from 10 to 8 mg daily owing to the observed bone marrow suppression events. Treatment was continued until disease progression, intolerable toxicity, withdrawal of consent, study completion, or study termination occurred, whichever occurred first. Dose adjustments were permitted for chemotherapy and anlotinib but not for sintilimab, according to the protocol. Considering that gemcitabine, cisplatin, and anlotinib are marketed drugs, dose adjustments were made by the investigators according to the treatment-related adverse events determined and medication instructions (details in Supplementary Protocol). Patients who temporarily or permanently discontinued chemotherapy or sintilimab and anlotinib owing to adverse events were allowed to continue taking other agents as long as the investigator determined that there was a clinical benefit.
Assessments
Tumors were assessed by investigators using computed tomography or magnetic resonance imaging (MRI) at baseline and every 6 weeks as per mRECIST and RECIST 1.1 as a post-hoc exploratory analysis. Safety was assessed for the patients who received at least one dose of the assigned treatment. Vital signs, clinical laboratory tests, and other adverse events were continuously monitored throughout the study and assessed according to the National Cancer Institute’s (NCI) Common Terminology Criteria for Adverse Events, version 4.0.
Endpoints
In the initial design phase of this study, the primary endpoint was designated as the 1-year OS rate. However, with the growing application of immune checkpoint inhibitors in the subsequent lines of treatment for advanced biliary tract cancers in clinical practice, it became evident that improvements in PFS did not translate into a benefit in OS. And the 1-year OS rate could not accurately reflect the advantages of the treatment. Therefore, we adjusted the primary endpoint of the study to PFS, defined as the time from randomization to disease progression according to mRECIST, or death from any cause, whichever occurred first. The secondary endpoints were objective response rate (ORR), defined as the percentage of patients with radiologically confirmed complete or partial response according to mRECIST; OS, defined as the time from randomization to death from any cause; and safety, described as the occurrence of treatment-related adverse events (TRAEs) of any grade.
Animal models
All animal experiments in the study were conducted using the C57BL/6N mice (C57BL/6NCrl, female, 6 to 8 weeks old). The mice were procured from two commercial animal vendors: Charles River Laboratories (Zhejiang, China) and Shanghai Laboratory Animal Center, CAS (SLACCAS). Animals were housed in a specific pathogen-free facility at the Experimental Animal Center of Zhejiang Cancer Hospital, where the environmental conditions were maintained at temperatures below 22 °C and humidity levels at 55%. The animals were subjected to a 12-hour light-dark cycle and had unrestricted access to food and water.
The orthotopic cholangiocarcinoma (CCA) tumor-bearing models were established through hydrodynamic tail vein injection (HD inj) of 30 μg YapS127A, 20 μg AKT, and 2 μg SB100 transposase plasmids diluted in 2 ml of 0.9% NaCl39. Anlotinib (kindly provided by Chia Tai Tianqing Pharmaceutical Group Co. Ltd) treatment at varying doses40,41,42(low-dose: 3 mg/kg, high-dose: 6 mg/kg) was initiated 7 days post tumor cell inoculation and administered orally via gavage daily. Anti-mouse PD-1 antibody (CD279, BE0146, BioXcell) was administered at a dose of 200 mg per mouse through intraperitoneal injection (i.p.) every 3 days. The control group received intragastric administration of 0.9% saline and intraperitoneal injection of IgG, while tumor-bearing mice were euthanized at the conclusion of the experiment. Additional independent experiments were conducted to compare the survival times of mice in each group.
At the experiment endpoint, blood samples were obtained from the retro-orbital plexus and collected into EDTA-anticoagulant tubes for subsequent routine blood tests using a BC-5000vet Automatic animal blood cell analyzer (Mindray) at Huanyun Biological Technology (Hangzhou, China). After euthanizing the animals by cervical dislocation, the liver and small intestine were harvested. To adhere to ethical standards, mice were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) before euthanasia to minimize pain and stress. In accordance with the humanitarian endpoint criteria, all mice were continuously monitored, and euthanasia was performed if they met predefined conditions like rapid weight loss (≥20%) or weight gain (≥20% due to ascites) and labored respiration. In accordance with the specifications by the ethics committee, the protocol for orthotopic tumors allowed a maximum tumor burden for growing up to two months or until the liver weight increased by no more than 10% of the mice’s body weight. This study adhered to these limitations.
Approval for all animal experiments was obtained from the Animal Experimental Ethics Committee of Zhejiang Cancer Hospital (zjzlsd2023-08-005), and the experiments were conducted in accordance with guidelines for the care and use of laboratory animals.
Isolation of murine immune cells
Liver immune cells were isolated and purified using a previously described method43, with some modifications. The liver tumor tissues were sectioned into 1 mm fragments utilizing ophthalmic scissors, followed by incubation in complete RPMI-1640 medium prior to mechanical dissociation with the gentleMACS Dissociator (Miltenyi Biotec). The resulting cell suspension was filtered through a 70 µm nylon strainer and centrifuged at 300 × g for 5 min to obtain a single-cell population. Subsequently, immune cells were isolated by 40% and 70% Percoll (Absin, abs9102). Red blood cells (RBCs) were lysed using Red Blood Cell Lysis Buffer (Absin, abs9101) for 2 min at room temperature and then resuspended in PBS at a concentration of 1 × 10^7 cells/mL.
Flow cytometry analysis
Cell suspensions (1 × 10^6 cells for each staining panel) were used for flow cytometry according to the following procedure44,45. For detection of intracellular cytokines, cytokine-producing immune cells were activated using the Leukocyte Activation Cocktail (BD Biosciences, 550583) for a duration of 6 h before antibody staining for flow cytometric analysis following the manufacturer’s protocol. Subsequently, the cells underwent two washes with FACS buffer and were incubated with Fc-blocking reagent (BD Pharmingen, 553141) for 10 min at ambient temperature. This was followed by incubation with surface antibodies for 30 minutes at 4 °C. For intracellular staining, the Transcription Factor Buffer Set (BD Pharmingen, 562574) was employed. The cells were incubated in Fix/Perm solution for 45 min at 4 °C and subsequently washed twice with Perm buffer at 300 × g for 5 min. Cells were resuspended in a staining buffer containing the appropriate dilutions of intracellular antibodies and incubated for 50 minutes at 4 °C, shielded from light. Post-staining, the immune cells were analyzed using a flow cytometer. Unstained cells served as blank controls, while single-stained tubes were utilized for compensation controls. Fluorescence Minus One (FMO) controls were employed by excluding one antibody from the staining process to validate the gating strategy. The flow cytometry gating strategy is illustrated in Supplementary Figs. S9 and S10. The counts of each cell subset were determined based on frequency, while the expression levels of individual molecules were assessed using mean fluorescence intensity (MFI), analyzed with FlowJo v10.8 software.
The flow cytometry antibodies utilized and reagents in this study included L/D-FVS620 (BD Pharmingen, 564996, 1:1000), anti-Mouse CD45-APC/CY7 (BD Pharmingen, 557659, 1:50), anti-Mouse CD3e- BUV395 (BD Pharmingen, 563565, 1:50), anti-Mouse CD4- BUV605 (BD Pharmingen, 563151, 1:50), anti-Mouse CD8a (BD Pharmingen, 557959, 1:50), anti-Mouse NK1.1-BV421 (BD Pharmingen, 562921, 1:50), anti-Mouse TRAIL-APC (Biolegend, 109310, 1:50), anti-Mouse GZMB-FITC (Thermo, 11-8898-82, 1:50), anti-Mouse TNF-BB700 (BD Pharmingen, 566510, 1:50), anti-Mouse Perforin-PE ((Thermo, 12-9392-82, 1:50), anti-Mouse IFN-γ-BV480 (BD Pharmingen, 566097, 1:50), anti-Mouse FOXP3-PE/CY7 ((Thermo, 25-5773-82, 1:50), anti-Mouse PD-1-BV650 (BD Pharmingen, 744546, 1:50), anti-Mouse CD44-BV510 (BD Pharmingen, 563114, 1:50), anti-Mouse CD62L-FITC (Biolegend, 104405, 1:50), anti-Mouse CD127-PE/CY7 (BD Pharmingen, 560733, 1:50), anti-Mouse PD-1-BB700 (BD Pharmingen, 566514, 1:50), anti-Mouse TIM-3-BV421 (Biolegend, 119723, 1:50), anti-Mouse TCF-1-AF647 (BD Pharmingen, 566693, 1:50).
Hematoxylin and eosin (H&E) and Immunofluorescence (IF) staining
The following protocol was adapted from reference46. Mouse tissues were collected and fixed in 4% paraformaldehyde (PFA, Biosharp, BL539A) for 16 to 24 h at room temperature, followed by further processing and embedding in paraffin. The paraffin-embedded tissue sections were deparaffinized, rehydrated, and subjected to heat-induced antigen retrieval using a citrate buffer (pH 6.0) for 10 minutes. Three-micrometer paraffin sections were used for H&E staining. H&E staining was conducted according to routine protocols. Briefly, after deparaffinization and rehydration, the tissue sections were stained with hematoxylin (Zhongshan Goldenbridge Biotechnology) for 5 min, soaked in 1% acid ethanol (1% HCl in 75% ethanol), and then rinsed in distilled water. Subsequently, the sections were stained using an eosin solution (Zhongshan Goldenbridge Biotechnology) for a duration of three minutes, followed by a dehydration process utilizing graded alcohols and a clearing step in xylene.
Five-micrometer paraffin sections were employed for IF staining. IF staining was performed on both paraffin-embedded and frozen sections of murine tissues to analyze and characterize the tumor microenvironment, as previously documented. After incubation with primary antibodies at 4 °C overnight, the sections were stained with fluorophore-conjugated secondary antibodies (Alexa Fluor® 488-conjugated Goat Anti-Rabbit IgG (H + L), Servicebio, GB25303, 1:500; Alexa Fluor® 594-conjugated Goat Anti-Mouse IgG (H + L), Servicebio, GB28303, 1:500; Cy3-conjugated AffiniPure Goat Anti-Rat IgG (H + L), Servicebio, GB21302, 1:500) for one hour at room temperature. Additionally, DAPI was used for counterstaining cell nuclei. Vascular analysis was conducted using the CD31/α-SMA ratio, while the proliferation of CD8 + T cells was assessed through the CD8/Ki67 ratio (anti-CD31, 550274, BD Biosciences, 1:50; anti-α-SMA, 14395-1-AP, Proteintech, 1:1000; anti-CD8, #14-0808-82, Thermofisher, 1:200; anti-Ki67, 28074-1-AP, Proteintech, 1:800). To identify hypoxic regions within the tumor, a solution of Hypoxyprobe-1 (60 mg/kg, Hypoxyprobe-1 Plus Kit, Hypoxyprobe, Inc.) was administered via tail-vein injection 60 min before the collection and fixation of tumor samples with 4% PFA. Subsequently, the tumor tissues were sectioned and subjected to staining with the primary antibody (FITC-Mab1) and secondary antibody (HRP linked to rabbit anti-FITC IgG) following the manufacturer’s guidelines. Immunofluorescence was visualized using a confocal laser scanning microscope (Olympus VS200, Japan).
Tumor vessel perfusion analysis
Tumor blood perfusion was assessed using a previously established protocol36. In brief, mice were subjected to systemic perfusion with PBS following intravenous administration of a 200 μl PBS solution containing 10 mg/kg of Hoechst 33342 (Sigma, USA) for 5 min. Subsequently, tumors were excised and fixed in 4% paraformaldehyde (PFA) for 2–3 h. The method involved staining perfused blood vessels within the tumor tissues with Hoechst 33342, followed by counterstaining of the slides using Sytox Green (Catalog S7020, Molecular Probes, USA) for nonspecific nuclear staining. The images were acquired utilizing a confocal laser scanning microscope (Olympus, VS200, Japan), and the intensities of CD31 and Hoechst 33342-positive areas in each field were calculated using an Image-Pro plus software (version 6.0).
Morphometric analysis of intestinal villi
Morphometric analysis of intestinal villi was performed referring to literature concerned35. Three-micrometer paraffin-embedded sections of the intestinal tissue were utilized for hematoxylin and eosin (H&E) staining, while 5-micrometer paraffin-embedded sections were employed for immunofluorescence staining. All images were acquired using the OLYMPUS VS200 microscope. Histological scoring and villus height measurements were conducted based on analyses of the small intestine. A minimum of 30 crypts or 30 visual fields were evaluated per mouse. The histological scoring system ranged from 0 to 13, as delineated in the following criteria: ① villus aspect, normal = 0, short = 1, extremely short = 2; ② villus tops, normal = 0, damaged = 1, severely damaged = 2; ③ epithelium, normal = 0, flattened = 1, damaged = 2, severely damaged = 3; ④ inflammation, no infiltration = 0, mild infiltration = 1, severe infiltration = 2; ⑤ crypts, normal = 0, mild crypt loss = 1, severe crypt loss = 2; ⑥ crypt abscesses, none = 0, present = 1; ⑦ bleeding, none = 0, present = 1.
Statistical analysis
The sample size was determined based on the primary endpoint of PFS under the assumption that the GC group had a median PFS of 6.5 months. Due to a different tumor assessment interval was applied in ABC-023 than current study design (i.e., every 12 weeks vs every 6 weeks), a midpoint method47 was used to re-estimate the potential median PFS from ABC-02 study if following an every 6 weeks assessment interval, and that SAGC would improve PFS with a hazard ratio (HR) of 50%; approximately 51 PFS events would be needed to provide 80% power to detect this difference under a type I error controlled at 10% (two-sided). Eighty subjects were randomized in a 1:1 ratio to achieve 51 PFS events to complete the study in a reasonable time, and considering the possible loss to follow-up.
Patients who were lost to follow-up were censored at the randomization date for PFS analysis and at the last known survival date for OS analysis. The efficacy analysis set included patients randomly assigned to each treatment group, regardless of whether they had received treatment. The safety analysis set included patients who received at least one dose of the study treatment.
Quantitative data were displayed as the median (range) or number of patients (percentage). Proportional comparisons between groups were performed using Fisher’s exact test. Survival analyses (PFS and OS) were performed using Kaplan-Meier curves, P values were determined using the log-rank test, and HRs were calculated using the Cox proportional hazards model. A two-sided P value lower than 0.05 was considered statistically significant for all tests except for the primary endpoint, unless indicated otherwise. A two-sided P value ≤ 0.1 was used to determine statistical significance for the primary endpoint PFS. Univariate analysis was used to explore the associations between different variables and PFS, and the results are presented as HRs with 95% confidence intervals (CIs). All analyses were performed using R 3.4.0 [Vienna, Austria].
Quantification and statistical analysis data from in vivo experiments are presented as mean ± SEM (Standard Error of the Mean), and n represents the number of experiments or animals. Statistical significance was determined with one-way ANOVA followed by Holm-Sidak multiple comparison test. Statistical analysis was performed with GraphPad Prism (GraphPad Prism v9.0).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The study protocol is presented in Appendix A. The datasets generated during this study are not publicly available due to patient privacy protections. De-identified data will be made available to qualified researchers upon reasonable request, subject to approval by the Ethics Committee of Zhejiang Cancer Hospital and a signed data access agreement. Requests should be directed to the corresponding authors (Jieer Ying: jieerying@aliyun.com).Source data are provided with this paper.
Change history
18 July 2025
In this article the affiliation 'Department of Hepato-Pancreato-Biliary & Gastric Medical Oncology, Zhejiang Cancer Hospital, Hangzhou, Zhejiang, PR China' was incorrectly assigned to Jieer Ying and has been removed. In addition, affiliation 1 was given as 'Wenzhou Medical University, Wenzhou, Zhejiang, PR China' and affiliation 2 was given as 'Department of Hepato-Pancreato-Biliary & Gastric Medical Oncology, Zhejiang Cancer Hospital, Hangzhou, Zhejiang, PR China', but should have been affiliations 2 and 1, respectively. The original article has been corrected.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Zheng, R. S. et al. Cancer incidence and mortality in China, 2022. Zhonghua Zhong Liu Za Zhi 46, 221–231 (2024).
Valle, J. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362, 1273–1281 (2010).
Oh, D.-Y. et al. A phase 3 randomized, double-blind, placebo-controlled study of durvalumab in combination with gemcitabine plus cisplatin (GemCis) in patients (pts) with advanced biliary tract cancer (BTC): TOPAZ-1. J. Clin. Oncol. 40, 378 (2022).
Javle, M. et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol. Hepatol. 6, 803–815 (2021).
Goyal, L. et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N. Engl. J. Med. 388, 228–239 (2023).
Zhu, A. X. et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma With IDH1 mutation: the phase 3 randomized cLINical ClarIDHy trial. JAMA Oncol. 7, 1669–1677 (2021).
Abou-Alfa, G. K. et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 21, 671–684 (2020).
Lee, C. K. et al. Trastuzumab plus FOLFOX for HER2-positive biliary tract cancer refractory to gemcitabine and cisplatin: a multi-institutional phase 2 trial of the Korean Cancer Study Group (KCSG-HB19-14). Lancet Gastroenterol. Hepatol. 8, 56–65 (2023).
Subbiah, V. et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 21, 1234–1243 (2020).
Doebele, R. C. et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. 21, 271–282 (2020).
Harding, J. J. et al. Zanidatamab for HER2-amplified, unresectable, locally advanced or metastatic biliary tract cancer (HERIZON-BTC-01): a multicentre, single-arm, phase 2b study. Lancet Oncol. 24, 772–782 (2023).
Ostwal, V. et al. Trastuzumab plus gemcitabine-cisplatin for treatment-naive human epidermal growth factor receptor 2-positive biliary tract adenocarcinoma: a multicenter, open-label, phase II study (TAB). J. Clin. Oncol. 42, 800–807 (2024).
Valle, J. W., Kelley, R. K., Nervi, B., Oh, D. Y. & Zhu, A. X. Biliary tract cancer. Lancet 397, 428–444 (2021).
Ueno, M. et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: a non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol. Hepatol. 4, 611–621 (2019).
Piha-Paul, S. A. et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: results from the KEYNOTE-158 and KEYNOTE-028 studies. Int. J. Cancer 147, 2190–2198 (2020).
Delaye, M. et al. Durvalumab (D) plus tremelimumab (T) immunotherapy in patients (Pts) with advanced biliary tract carcinoma (BTC) after failure of platinum-based chemotherapy (CTx): Interim results of the IMMUNOBIL GERCOR D18-1 PRODIGE-57 study. J. Clin. Oncol. 40, 4108–4108 (2022).
Oh, D. Y. et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: an open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 7, 522–532 (2022).
Chen, X. et al. Camrelizumab plus gemcitabine and oxaliplatin (GEMOX) in patients with advanced biliary tract cancer: a single-arm, open-label, phase II trial. J. Immunother. Cancer 8, e001240 (2020).
Feng, K. et al. Efficacy and biomarker analysis of nivolumab plus gemcitabine and cisplatin in patients with unresectable or metastatic biliary tract cancers: results from a phase II study. J. Immunother. Cancer 8, e000367 (2020).
Kelley, R. K. et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 401, 1853–1865 (2023).
Liu, Z. et al. Tumor vasculatures: a new target for cancer immunotherapy. Trends Pharm. Sci. 40, 613–623 (2019).
Shi, C. et al. Lenvatinib plus programmed cell death protein-1 inhibitor beyond first-line systemic therapy in refractory advanced biliary tract cancer: a real-world retrospective study in China. Front. Immunol. 13, 946861 (2022).
Zhou, J. et al. Phase Ib study of anlotinib combined with TQB2450 in pretreated advanced biliary tract cancer and biomarker analysis. Hepatology 77, 65–76 (2023).
Shi, G. M. et al. Toripalimab combined with lenvatinib and GEMOX is a promising regimen as first-line treatment for advanced intrahepatic cholangiocarcinoma: a single-center, single-arm, phase 2 study. Signal Transduct. Target Ther. 8, 106 (2023).
Fan, P. et al. Effective low-dose anlotinib induces long-term tumor vascular normalization and improves anti-PD-1 therapy. Front. Immunol. 13, 937924 (2022).
Yuan, S. et al. Low-dose anlotinib confers improved survival in combination with immune checkpoint inhibitor in advanced non-small cell lung cancer patients. Cancer Immunol. Immunother. 72, 437–448 (2023).
Morizane, C. et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann. Oncol. 30, 1950–1958 (2019).
Okusaka, T. et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br. J. Cancer 103, 469–474 (2010).
El-Khoueiry, A. B. et al. IMbrave151: A phase 2, randomized, double-blind, placebo-controlled study of atezolizumab with or without bevacizumab in combination with cisplatin plus gemcitabine in patients with untreated, advanced biliary tract cancer. J. Clin. Oncol. 41, 491–491 (2023).
McNamara, M. G. et al. Landmark survival analysis and impact of anatomic site of origin in prospective clinical trials of biliary tract cancer. J. Hepatol. 73, 1109–1117 (2020).
Kuter, D. J. Managing thrombocytopenia associated with cancer chemotherapy. Oncology29, 282–294 (2015).
Li, Q. et al. A real-world study of optimal treatment with anlotinib first-line therapy in advanced hepatocellular carcinoma. Cancer Manag. Res. 14, 3037–3046 (2022).
Zeng, T. M. et al. Clinical and biomarker analyses of sintilimab plus gemcitabine and cisplatin as first-line treatment for patients with advanced biliary tract cancer. Nat. Commun. 14, 1340 (2023).
Mpekris, F. et al. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc. Natl. Acad. Sci. USA 117, 3728–3737 (2020).
Huang, Y. et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 109, 17561–17566 (2012).
Li, Q. et al. Low-dose anti-angiogenic therapy sensitizes breast cancer to PD-1 blockade. Clin. Cancer Res. 26, 1712–1724 (2020).
Huang, Y., Stylianopoulos, T., Duda, D. G., Fukumura, D. & Jain, R. K. Benefits of vascular normalization are dose and time dependent-letter. Cancer Res. 73, 7144–7146 (2013).
Zhang, Q. et al. Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov. 11, 1248–1267 (2021).
Song, F. et al. Anlotinib suppresses tumor progression via blocking the VEGFR2/PI3K/AKT cascade in intrahepatic cholangiocarcinoma. Cell Death Dis. 11, 573 (2020).
Li, H. et al. CircHAS2 activates CCNE2 to promote cell proliferation and sensitizes the response of colorectal cancer to anlotinib. Mol. Cancer 23, 59 (2024).
Liang, L. et al. Autophagy inhibition potentiates the anti-angiogenic property of multikinase inhibitor anlotinib through JAK2/STAT3/VEGFA signaling in non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 38, 71 (2019).
Sbierski-Kind, J. et al. Effects of caloric restriction on the gut microbiome are linked with immune senescence. Microbiome 10, 57 (2022).
Xiong, X. et al. Cannabis suppresses antitumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct. Target Ther. 7, 99 (2022).
Reis, B. S., Rogoz, A., Costa-Pinto, F. A., Taniuchi, I. & Mucida, D. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nat. Immunol. 14, 271–280 (2013).
Maller, O. et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat. Mater. 20, 548–559 (2021).
Panageas, K. S., Ben-Porat, L., Dickler, M. N., Chapman, P. B. & Schrag, D. When you look matters: the effect of assessment schedule on progression-free survival. J. Natl. Cancer Inst. 99, 428–432 (2007).
Acknowledgements
We thank the patients, their families, and all participating clinical teams for making this study possible. This trial was funded by Innovent Biologics (Suzhou) Co., Ltd. and Chia Tai Tianqing Pharmaceutical Group, which provided sintilimab and anlotinib hydrochloride, respectively. The study was supported by grants from Natural Science Fund of Zhejiang Province (LTGY23H160010, LQ20H160005), Medical and Health Science and Technology Program of Zhejiang Province (2021KY087), Quzhou Phoenix Medical Scholar Talent Training Fund (258 Key Discipline Talent Training Project)(2021ZYC-B31), Key Laboratory of Prevention, Diagnosis and Therapy of Upper Gastrointestinal Cancer of Zhejiang Province (2022E10021), Medical Process Advancement Medical Scientific Research Project of Zhongguancun Precision Medicine Foundation (GLYZH35) and National Natural Science Foundation of China (82303080, 82303415). The funders played no role in the study design, data collection, data analysis, data interpretation, or manuscript writing.
Author information
Authors and Affiliations
Contributions
J.L. (Jingjing Li), W.S. (Weijing Sun), and J.Y. (Jieer Ying) designed the study. J.L., Q.Z. (Qinhong Zheng), X.X. (Xiaoqing Xu), F.Z. (Fabiao Zhang), Z.H. (Zhe Han), W.W. (Wei Wu), X.W. (Xinbao Wang), Y.Z. (Yuhua Zhang), B.W. (Bingchen Wu), Q.W. (Qing Wei), D.L. (Da Li), X.X. (X. Xu), and J.Y. recruited patients and collected data. J.L. and Q.X. (Qi Xu) managed patient randomization. J.L., X.S. (Xing Sun), W.S., X.W., and J.Y. verified data integrity. X.S., J.Y.and J.Yan. (Junrong Yan) performed statistical analyses. S.Z. (Shurui Zhou) and Y.Z. (Yiwen Zhou) conducted in vivo experiments. J.L., S.Z., and J.Y. drafted the manuscript. Y.S. (Yang Shao) assisted with data visualization. All authors (J.L., S.Z., X.X., Q.Z., F.Z., C.L., D.L., X.S., Z.H., W.W., J.Yan., Y.S., Y.Z., B.W., Q.W., X.W., Y.Z., W.S., Q.X., J.Y.) critically reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Consent for publication Not applicable.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, J., Zhou, S., Xu, X. et al. Sintilimab and anlotinib with gemcitabine plus cisplatin in advanced biliary tract cancer: SAGC a randomized phase 2 trial. Nat Commun 16, 5559 (2025). https://doi.org/10.1038/s41467-025-60119-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60119-3
